

SUMMARY
Several mental illnesses, characterized by aberrant stress reactivity, often arise after early-life adversity (ELA). However, it is unclear how ELA affects stress-related brain circuit maturation, provoking these enduring vulnerabilities. We find that ELA increases functional excitatory synapses onto stress-sensitive hypothalamic corticotropin-releasing hormone (CRH)-expressing neurons, resulting from disrupted developmental synapse pruning by adjacent microglia. Microglial process dynamics and synaptic element engulfment were attenuated in ELA mice, associated with deficient signaling of the microglial phagocytic receptor MerTK. Accordingly, selective chronic chemogenetic activation of ELA microglia increased microglial process dynamics and reduced excitatory synapse density to control levels. Notably, selective early-life activation of ELA microglia normalized adult acute and chronic stress responses, including stress-induced hormone secretion and behavioral threat responses, as well as chronic adrenal hypertrophy of ELA mice. Thus, microglial actions during development are powerful contributors to mechanisms by which ELA sculpts the connectivity of stress-regulating neurons, promoting vulnerability to stress and stress-related mental illnesses.
In brief
Early-life adversity (ELA) promotes lifelong aberrant stress responses and vulnerability to mental illnesses. Bolton et al. identify poor dynamics and hypothalamic CRH neurons’ excitatory synapse pruning of ELA microglia, implicating microglial MerTK. Chronic chemogenetic activation of ELA microglia normalized process dynamics, synapse density, and adult hormonal and behavioral stress responses.
Graphical Abstract

INTRODUCTION
Brain development is governed by both genetics and early-life experiences (Farah, 2018; Klengel and Binder, 2015; Luby et al., 2020; McLaughlin et al., 2019). Specifically, adverse early-life experiences such as stress or poverty are associated with altered trajectories of structural and functional brain maturation (Luby et al., 2020; Tooley et al., 2021) and significant cognitive and emotional vulnerabilities (Bick and Nelson, 2016; Gee, 2021; Luby et al., 2017; McLaughlin et al., 2019; Short and Baram, 2019). Importantly, early-life adversity (ELA)/stress is a robust risk factor for neuropsychiatric disorders such as major depression, which are characterized by abnormal stress responses and a dysregulation and aberrant reactivity of the hypothalamic pituitary adrenal axis (Bick and Nelson, 2016; Danese and McEwen, 2012; Heim and Binder, 2012; Insel, 2009; Luby et al., 2020; McLaughlin et al., 2019; Stout and Nemeroff, 1994; Watson and Mackin, 2006).
A key aspect of brain maturation involves the formation and sculpting of neuronal circuits that execute complex behaviors. This process involves axonal and dendritic growth and synaptic formation, stabilization, and pruning (Emoto et al., 2021; Espinosa and Stryker, 2012; Hensch et al., 1998; Huh et al., 2020; Schafer and Stevens, 2013; Vainchtein and Molofsky, 2020). These take place during sensitive developmental periods and are modulated by experiences, including adversity (Bale et al., 2010; Birnie et al., 2020; Bolton et al., 2019; Emoto et al., 2021; Short and Baram, 2019; Takesian et al., 2018). However, the mechanisms by which ELA may influence and disrupt brain circuit maturation are unclear. Microglia play a key role in synaptic pruning in the developing visual and somatosensory systems (Hoshiko et al., 2012; Miyamoto et al., 2016; Schafer et al., 2012; Sipe et al., 2016), as well as in developing hippocampus (Paolicelli et al., 2011; Weinhard et al., 2018). These professional phagocytes contact and engulf synaptic elements (Lenz et al., 2013; Schafer et al., 2012; Vainchtein et al., 2018; Weinhard et al., 2018), contributing to circuit sculpting. However, a potential role for microglial dysfunction in the influence of ELA on salient brain circuit maturation has not been identified.
Here, we capitalize on a robust, well-characterized model of ELA (Gilles et al., 1996; Molet et al., 2014; Walker et al., 2017), in which exposure to an impoverished environment and unpredictable maternal care during a sensitive developmental period provokes deficits in emotional (Bolton et al., 2018; Levis et al., 2019; Molet et al., 2016) and cognitive (Brunson et al., 2005; Ivy et al., 2010) functions, such as anhedonia and spatial memory problems, respectively. Importantly, this ELA induces enduring alteration of the responses to stress and is associated with aberrant maturation of the hypothalamic stress-related circuit (Chen and Baram, 2016; Gilles et al., 1996; Gunn et al., 2013; Rice et al., 2008). Focusing on the stress-sensitive corticotropin-releasing hormone (CRH)-expressing neurons in the paraventricular nucleus (PVN) of the hypothalamus that govern the neuroendocrine responses to stress, we find increased numbers of functional excitatory synapses (Gunn et al., 2013). The augmented excitatory input promotes the excitability of these neurons that may mediate ELA-induced aberrant stress responses (Gilles et al., 1996; Gunn et al., 2013; Rice et al., 2008), in analogy to changes accompanying stress-related mental illnesses. Given the involvement of microglia in sculpting developing brain circuits elsewhere, we test here the hypothesis that ELA impairs microglia-dependent synapse pruning, resulting in augmented excitatory innervation of stress-sensitive PVN-CRH+ neurons and subsequent aberrant responses to stress. We find deficits in microglial process dynamics and microglial engulfment of excitatory synapses in microglia abutting CRH neurons of ELA mice, related to deficient microglial Mer tyrosine kinase (MerTK) receptor activity. Selective chemogenetic activation of ELA microglia during the developmentally sensitive period restores synapse numbers and prevents the lifelong aberrant behavioral and neuroendocrine stress responses orchestrated by hypothalamic CRH neurons.
RESULTS
ELA augments the number of excitatory synapses on CRH-expressing stress-sensitive neurons
We reared neonatal mice in either typical cages or in cages with limited bedding and nesting materials (Figure S1A), an ELA environment, between postnatal days (P)2 and 10. We then compared the numbers of excitatory synapses and the role of microglia in the pruning of these synapses at several timepoints during and immediately following the ELA epoch. The number and function of excitatory synapses on CRH+ neurons in the mediodorsal parvocellular (mpd) of the PVN of the hypothalamus were augmented after 1 week of ELA. Specifically, the number and density of excitatory synapses, defined by the colocalization of vesicular glutamate transporter (vGlut)2 and the postsynaptic density (PSD95) per volume of CRH+ neurons, were higher in ELA pups at P10 relative to those reared in control (CTL) conditions (t12.44 = 2.95, p = 0.01; Welch’s t test; Figures 1A and 1B). In contrast, synapse density did not differ in the non-CRH-expressing neuron-containing region just ventral to the mpd (anterior hypothalamic nucleus; p > 0.2; Figure 1B, inset). The significant increase in excitatory synapses was not a result of changes in CRH+ neuron number (p > 0.7; CTL = 2,475 ± 271 cells/mm2; ELA = 2,568 ± 110 cells/mm2) or volume (p > 0.5; CTL = 31,050 ± 1,773 μm3; ELA = 32,710 ± 1,741 μm3). Notably, the increased synapse density endured, remaining elevated on P24–25 (t8.96 = 3.69, p = 0.005; Welch’s t test; Figure 1C). Buttressing the neuroanatomical results, electrophysiology revealed an increased frequency of spontaneous excitatory postsynaptic currents (sEPSCs; t19 = 2.39, p = 0.03; unpaired t test; Figures 1D and 1E), in mpd PVN neurons of ELA mice, with no change in amplitude (p > 0.8; Table S1). These findings are in accord with prior work that additionally demonstrated a similar increased frequency of miniature EPSCs (Gunn et al., 2013). Changes were specific to excitatory synapses, as neither frequency nor amplitude of inhibitory PSCs (sIPSCs; p > 0.8; Figures 1D and 1F; Table S1) were altered. Together, these data are consistent with an increased number of functional excitatory inputs onto CRH+ neurons in the PVN of ELA mice.
Figure 1. Early-life adversity (ELA) augments the number and function of excitatory synapses on CRH-expressing hypothalamic neurons.

(A) Top: A representative confocal composite image of excitatory synaptic puncta and CRH-tdTomato+ neurons in mediodorsal parvocellular (mpd) paraventricular hypothalamic nucleus (PVN). Inset: puncta colocalizing vGlut2 (magenta) and PSD95 (green) on a CRH-tdTomato+ neuron (red). These puncta satisfied the criteria for synapses (Imaris software). Scale bar, 10 μm. Bottom: Representative wide-field composite image of the overall PVN source of the mpd image (yellow, ROI); DAPI (blue), CRH-tdTomato+ cells (red), followed by 3D reconstruction of the selected CRH-tdTomato+ neuron (white inset), high-resolution confocal images masking PSD95 and vGlut2 channels on the 3D volume, their overlap, and remaining puncta that lie within the colocalization threshold. White arrowhead marks a synapse through all of the insets. Scale, 50 μm.
(B) ELA increases the number of excitatory synapses onto PVN CRH+ neurons at postnatal day (P)10 in male mice (t12.44 = 2.95, p = 0.01; Welch’s t test; see Figure S5A for female data). The same result is obtained by Imaris or manual counts using FIJI (r = 0.90, p < 0.0001; Pearson correlation). Synapse density in the non-CRH-dense region below the mpd (anterior hypothalamic nucleus) did not distinguish ELA and control mice (p > 0.2; inset).
(C) The ELA-induced increase in excitatory synapse number endured on P24–25 (t8.96 = 3.69, p = 0.005; Welch’s t test; see Figure S5B for female data).
(D) ELA functionally changes excitatory synapses of presumed CRH-expressing neurons: Traces are epochs (10 s) of whole-cell voltage-clamp recordings of spontaneous inhibitory synaptic currents (sIPSCs, top) and spontaneous excitatory synaptic currents (sEPSCs, bottom) from mpd PVN neurons derived from CTL (left) and ELA (right) mice (also see Table S1). A subsection (shaded area; 1 s) of the recorded epoch is displayed on an expanded timescale. Scale bars, sIPSC: y = 50 pA, sEPSC: y = 10 pA, x = 2 s, and 200 ms for top and bottom traces, respectively.
(E) ELA increases the frequency of sEPSCs in mpd PVN neurons of P18–26 male and female mice (t14 = 2.28, p = 0.04; unpaired t test).
(F) ELA does not alter sIPSCs frequency in the same mice (p > 0.8).
Data are means ± SEMs; *p < 0.05.
Process dynamics of microglia abutting PVN-CRH+ cells are impaired by ELA
Microglia contribute to the pruning of synapses in the developing brain, a prerequisite for the formation of stable, functional brain circuits (Paolicelli et al., 2011; Schafer et al., 2012; Zhan et al., 2014). Here, we tested for the presence of these cells in the proximity of CRH+ neurons in developing mouse PVN. Microglia were abundant already at P4 (near the onset of the differential rearing epoch; Figure S2A); their neuroanatomical interactions with CRH+ neurons increased during the first week of life (t13 = 3.71, p = 0.003; unpaired t test; Figure S2B). ELA did not change the number or density of microglia abutting CRH+ cells (p > 0.1; Figures 2A and 2B), nor their morphology (not shown). We next interrogated microglial surveillance and phagocytic functions in ELA and CTL mice during the differential rearing period (P8). To assess microglial process dynamics, we used 2-photon live imaging of acute slices of PVN from dual-reporter mice (CRH-Cre+/−:tdTomato+/−; CX3CR1-GFP+/−) enabling the visualization of both CRH+ neurons and microglia (Figure 2C; Video S1), and compared microglial dynamics in slices from ELA and CTL mice. We used two independent quantification methods of microglial process excursions onto neurons (Dumas et al., 2021): First, a manual kymograph method (Noam et al., 2010) tracked the movements of the tip of a microglial process (Figure 2D), showing that ELA significantly attenuated microglial process dynamics measured by total distance traveled over 30 min (t16 = 2.79, p = 0.01; unpaired t test; Figure 2E). Second, a novel, automated Python-based algorithm tracked multiple microglia processes simultaneously (Figure 2F) and established that ELA significantly decreased microglial process velocity (t12 = 2.26, p = 0.04; unpaired t test; Figure 2G). Together, these independent, convergent findings suggest that microglial process dynamics, a reliable indicator of microglial function (Badimon et al., 2020; Davalos et al., 2005; Eyo et al., 2014; Nimmerjahn et al., 2005; Weinhard et al., 2018), was impaired in ELA mice PVN.
Figure 2. Process dynamics of microglia abutting CRH-expressing neurons in PVN are impaired by ELA.
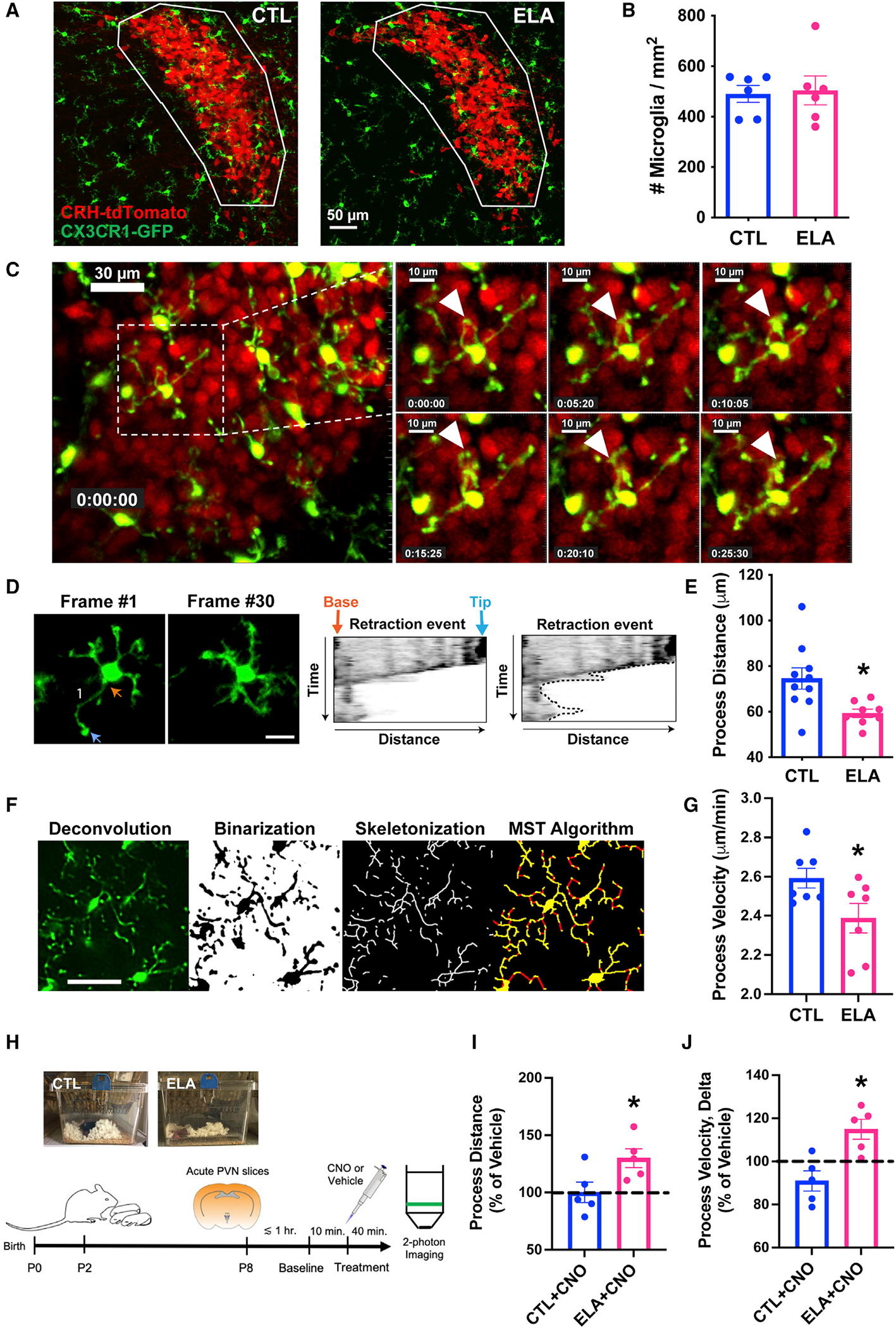
(A) Representative confocal images of microglia (green; CX3CR1-GFP) and CRH+ neurons (red; CRH-tdTomato) in PVN (white ROI) of P10 CTL (left) and ELA (right) male mice. Scale bar, 50 μm.
(B) ELA does not alter the density of microglia (number per ROI area, mm2) in the PVN.
(C) Still frames in ~5-min increments from a representative 2-photon video of microglia interacting with CRH+ neurons in the mpd PVN of a P8 male mouse. White arrowhead points to a microglial process, highlighting that its position shifts across frames. Scale bar, 30 μm (left image); 10 μm in insets.
(D) Example of a kymograph analysis of a microglia from a 2-photon imaging video, tracking retraction of a microglial process over time. In frame 1, the orange arrow points to the base of the microglial process; the blue arrow points to the process tip. The dotted line around the edges of the right kymograph represents the total distance traveled by process tip. Scale bar, 10 μm.
(E) ELA significantly attenuates microglial process dynamics, measured by the total distance traveled by process tips over 30 min (t16 = 2.79, p = 0.01; unpaired t test).
(F) Sequential steps for processing microglial images in real time using an automated Python-based algorithm tracking concurrently movements of all of the process tips from multiple microglia. From left to right: deconvolution, binarization, skeletonization, and minimum spanning tree (MST) algorithm to fill any gaps (red) in skeletons (yellow). Scale bar, 30 μm.
(G) ELA reduces microglial process velocity as measured by this novel technique (t12 = 2.26, p = 0.04; unpaired t test).
(H) Schematic of the chemogenetic activation: Litters of CX3CR1-Cre+:Gq-DREADD+ mice are randomly assigned to CTL or ELA conditions on P2. On P8, acute hypothalamic slices are prepared, recover for ~1 h, and used for 2-photon live imaging in superfusion chambers.
(I) Slices are imaged for a 10-min baseline period, then are exposed to CNO (to activate microglia-specific Gq-DREADDs) or vehicle for 40 min. The addition of CNO to microglia expressing excitatory Gq-DREADDs significantly augments the distance traveled by process tips in ELA slices compared to vehicle treatment (t4 = 3.63, p = 0.02; 1-sample t test) and CTL slices (t8 = 2.45, p = 0.04; unpaired t test; see Figure S2H for raw data). Slices were sampled at 30 min after drug onset.
(J) In slices from ELA mice, CNO significantly increases microglial process velocity measured from the onset to end of the incubation period, compared to vehicle (t4 = 3.23, p = 0.03; 1-sample t test) and to CTL slices (t8 = 3.64, p = 0.007; unpaired t test).
Means ± SEMs; *p < 0.05.
Aberrant microglia function is important because their selective pruning of synapses during development may be pivotal for the formation of refined, functional brain circuits (Paolicelli et al., 2011; Schafer et al., 2012; Zhan et al., 2014). Therefore, we examined whether chemogenetic activation of ELA microglia during the ELA epoch reversed microglial process dynamics deficits. We prepared acute hypothalamic slices from CTL and ELA P8 mice expressing excitatory Gq-designer receptors exclusively activated by designer drugs (DREADDs) specifically in microglia (CX3CR1-Cre+:Gq-DREADD+), superfused clozapine N-oxide (CNO) or vehicle, and used 2-photon imaging to quantify process dynamics (Figure 2H). The addition of CNO to microglia expressing the excitatory (Gq)-DREADD significantly augmented the distance traveled by process tips in ex vivo ELA slices relative to vehicle treatment by 30 min after drug addition (t4 = 3.63, p = 0.02; one-sample t test; Figure 2I), and this effect was significantly larger than that observed in CTL slices (t8 = 2.45, p = 0.04; unpaired t test; Figure 2I; see Figure S2H for raw data). Similarly, CNO significantly increased microglial process velocity between the onset and end of drug incubation in ELA slices relative to vehicle treatment (t4 = 3.23, p = 0.03; one-sample t test; Figure 2J) and to the effect in CTL slices (t8 = 3.64, p = 0.007; unpaired t test; Figure 2J). Together, these findings show that ELA leads to reduced microglial process dynamics not attributable to non-specific toxicity as it was reversed using selective chemogenetic activation of the microglia.
Synapse engulfment is impaired in microglia interacting with CRH+ cells of ELA-experiencing mice
Whereas reduced microglial process dynamics has been associated with diminished “surveillance” of neurons and blunted microglial function (Abiega et al., 2016; Damani et al., 2011; Krabbe et al., 2013), it remains unclear whether these deficits in ELA microglia abutting CRH+ neurons result in deficient synapse engulfment. To probe this question, we used confocal microscopy and scanning electron microscopy (SEM) to visualize microglial engulfment of excitatory synaptic elements within PVN (Figures 3A, 3B, and S2C–S2E). We identified P8 as a period of active synapse engulfment, with a significantly higher number and density of engulfed vGlut2+ synaptic puncta in microglial elements on P8 compared to P30 in CTL mice (Figure S2F). Then, we identified in ELA mice fewer vGlut2+ synaptic puncta engulfed by microglia compared with CTLs (t13.87 = 2.22, p = 0.04; Welch’s t test; Figure 3C). This reduction did not result from an ELA-induced reduction in total volumes occupied by microglia in the PVN (p > 0.1; CTL = 12,754 ± 674 μm3; ELA = 14,079 ± 705 μm3). Notably, reduced synapse engulfment was selective to ELA microglia abutting CRH+ neurons; synapse engulfment in microglia residing at the PVN periphery, which were not in direct contact with CRH+ neurons, did not differ between CTL and ELA mice (p > 0.2; Figure S2G). These data suggest that reduced microglial process dynamics associates with, and likely contributes to, poor synapse engulfment in microglia abutting CRH+ neurons in ELA mice.
Figure 3. Microglial mechanisms for augmented excitatory synapses on CRH+ cells in ELA mice.
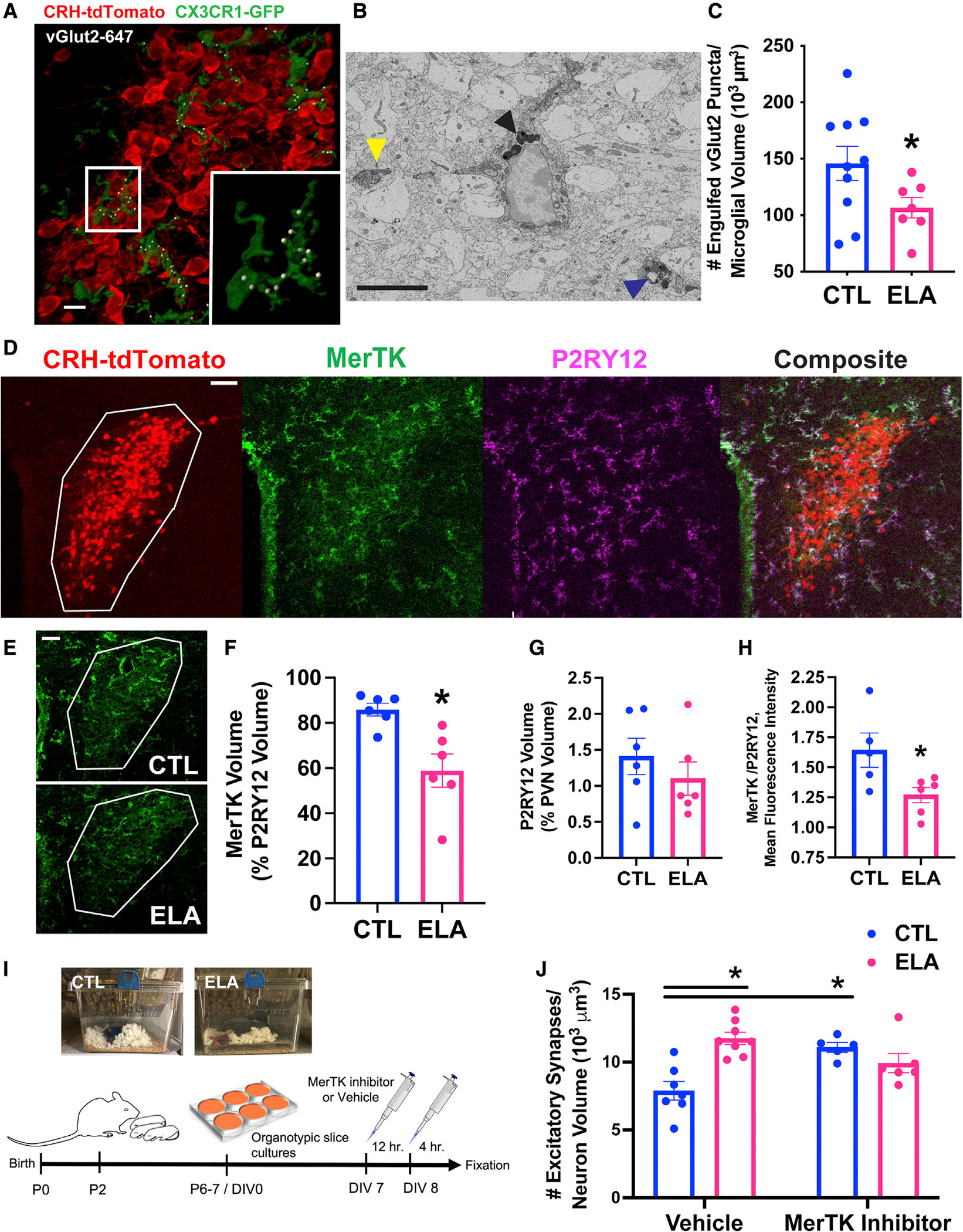
(A) Representative confocal image of microglia (green; CX3CR1-GFP) abutting CRH+ neurons (red; CRH-tdTomato), engulfing vGlut2+ excitatory presynaptic puncta (white; vGlut2 puncta identified as inside microglia by Imaris 3D reconstruction) in mpd PVN of a P8 male mouse. Scale bar, 10 μm (raw images in Figures S2C–S2E).
(B) Representative electron micrograph of a microglia labeled with ionized calcium binding adaptor molecule (Iba)1 and 3′,3-diaminobenzidine (DAB; electron-dense soma and processes) in P8 PVN. The yellow arrowhead points to the postsynaptic density of an excitatory synapse directly abutting a labeled microglial process. The black arrowhead indicates multiple lysosomes (electron dense) in microglial soma; these degrade engulfed material. The blue arrowhead points to an engulfed endosome of synaptic elements, including a putative postsynaptic density, surrounded by lysosomes in a microglia process. Scale bar, 5 μm.
(C) ELA reduces the number of vGlut2+ synaptic puncta engulfed by microglia abutting CRH+ neurons in the P8 mpd PVN of male mice (t13.87 = 2.22, p = 0.04; Welch’s t test; see Figure S5C for female data), as quantified in confocal 3D reconstructions.
(D) Representative confocal image of the microglial phagocytic receptor Mer tyrosine kinase (MerTK) expression. MerTK is primarily in microglia in the P8 PVN (white ROI; see also Figure S3D). Red = CRH-tdTomato+ neurons; green = MerTK immunoreactivity (IR); magenta = P2RY12 IR (microglia); white = overlap of green and magenta in composite image. Scale bar, 50 μm.
(E) Representative confocal images of MerTK IR in CTL (top) compared to ELA (bottom) P8 PVN (white ROI). Scale bar, 50 μm.
(F) MerTK volume per unit volume of P2RY12+ microglia (measured using Imaris) was lower in P8 ELA male PVN than in controls (t6.46 = 3.44, p = 0.01; Welch’s t test).
(G) Notably, P2RY12 volume and its percentage of PVN volume did not differ in ELA compared to control mice.
(H) The ratio of MerTK mean fluorescence intensity (MFI; measured using Imaris) to P2RY12 MFI was significantly reduced in ELA mice PVN (t9 = 2.54, p = 0.03; unpaired t test).
(I) Schematic of the MerTK inhibition experiment: Litters of CRH-tdTomato+ mice were randomly assigned to CTL or ELA conditions on P2. On P6–7, organotypic hypothalamic slice cultures were prepared and maintained for 7 d in vitro (DIV). On DIV7, cultures were treated with 20 nM of a small-molecule MerTK inhibitor (UNC2025: ~40-fold greater selectivity for MerTK over the Axl and Tyro3 TAM receptors [McDaniel et al., 2018; Zhang et al., 2014a]) or vehicle. Twelve hours later, the medium was refreshed with new drug, and the cultures were fixed 4 h later (Figure S3E).
(J) MerTK inhibition increased the number of excitatory synapses on CRH+ neurons in PVN cultures from control mice but not in ELA mice (significant interaction of ELA × drug, F1,22 = 17.89, p = 0.0003; 2-way ANOVA; p < 0.05; post hoc Tukey’s test).
Means ± SEMs; *p < 0.05.
Mechanisms of impaired synapse pruning in ELA microglia involve the phagocytic receptor MerTK
Microglia prune synapses in response to several cellular and molecular triggers, including activation of the phagocytic receptor MerTK (Abiega et al., 2016; Diaz-Aparicio et al., 2020; Fourgeaud et al., 2016). Hence, we tested whether this receptor was expressed in neonatal mouse PVN and whether it contributed to the microglial failure to prune CRH+ neuronal synapses in ELA mice. MerTK was expressed in the immature PVN, predominantly in microglia (Figures 3D and S3D). In our hands, MerTK expression overlapped neither with the astrocytic markers glial fibrillary acidic protein (GFAP) and S100b (Zhang et al., 2019) nor with the neuronal marker NeuN (Figures S3A–S3C). In addition, MerTK immunoreactivity (IR) relative to microglial volume was lower in PVN of P8 ELA mice compared with CTLs (Figures 3E and 3F; t6.46 = 3.44, p = 0.01; Welch’s t test). Importantly, neither P2RY12 IR nor PVN volume were altered by ELA (p > 0.3; P2RY12 volume: Figure 3G; PVN volume: CTL = 330,833 ± 19,493 μm3; ELA = 355,500 ± 22,113 μm3). To enhance the volumetric data, we analyzed the mean fluorescence intensity (MFI) of MerTK relative to P2RY12 and found that it was significantly reduced in ELA mice (t9 = 2.54, p = 0.03; unpaired t test; Figure 3H). We then tested whether deficient MerTK signaling may explain the attenuated synapse engulfment by ELA microglia. This hypothesis predicts that blocking MerTK activity would increase the synapse number in CTL CRH+ cells, but not in ELA neurons, in which the receptor is already less active. We tested this prediction in hypothalamic organotypic slice cultures containing PVN using a small-molecule selective MerTK inhibitor, UNC2025, which has a ~40-fold greater selectivity for MerTK over other TAM receptors (Axl and Tyro3) (McDaniel et al., 2018; Zhang et al., 2014a). As shown in Figures 3I and S3E, a 16-h incubation with the inhibitor but not the vehicle increased the number of excitatory synapses on CRH+ neurons in CTL mice but not in ELA mice (significant interaction of ELA × drug, F1,22 = 17.89, p = 0.0003; two-way ANOVA; p < 0.05, post hoc Tukey’s test; Figure 3J). These data support the notion that inhibited microglial MerTK signaling may underlie the increase in excitatory synapses on CRH+ neurons in ELA mice.
Functional significance of ELA-induced microglial deficits
ELA often precedes several prevalent neuropsychiatric disorders that are associated with aberrant stress responses involving the hypothalamic CRH+ cells (Stout and Nemeroff, 1994; Watson and Mackin, 2006). To determine whether ELA-induced transient microglial deficits contribute to long-lasting stress-related outcomes, we chemogenetically activated microglia in vivo in developing CTL and ELA mice and determined the hormonal and behavioral responses of these mice to acute stress during adulthood. We also assessed the presence of chronic, aberrant stress responses by measuring the weight of adrenal glands, which enlarge under such conditions (Armario, 2006; Harper and Austad, 2015; Ulrich-Lai et al., 2006). We tested behavioral responses to acute stress/threat using the looming shadow task, which involves the activity of CRH+ PVN cells (Daviu et al., 2020). We crossed CX3CR1-Cre+:Gq-DREADD+ mice with Gq-DREADD+ mice, generating litters in which ~50% of pups express Gq-DREADDs exclusively in their microglia (Figure 4A) and the rest serve as littermate controls. After validating the exclusive microglial expression of the Gq-DREADD (Figures S4A and S4B), we provided all of the mice with subcutaneous small, sustained-release CNO- or placebo-containing pellets on P3, before their randomization, to avoid the need for daily stressful injections. Following randomization (Figure 4B), outcome measures included the number of excitatory synapses onto presumed CRH+ PVN neurons at the end of the week (P10) and, in separate cohorts, hormonal and behavioral responses to adult stresses.
Figure 4. Enduring functional significance of ELA-related microglial deficits.
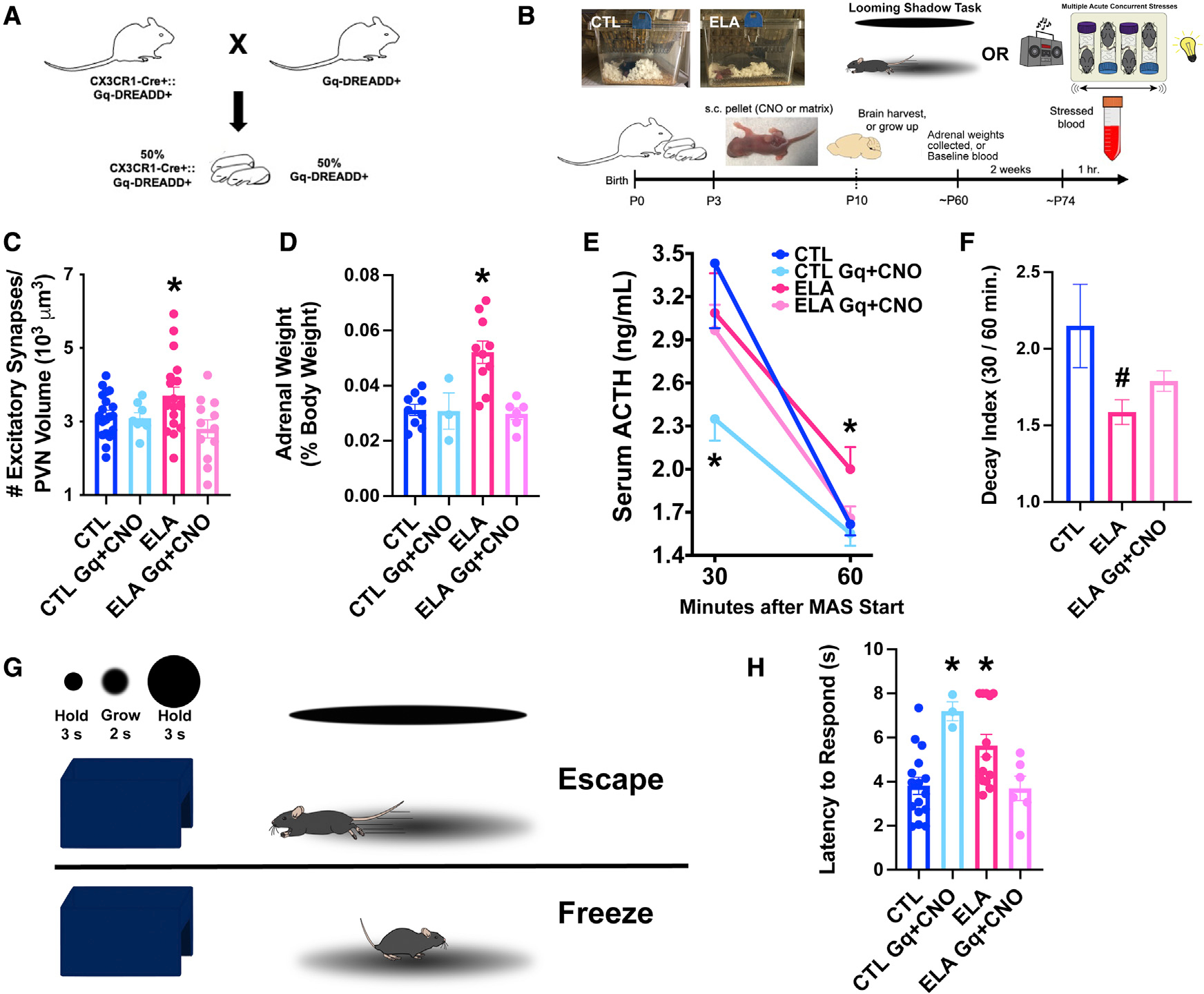
(A) Breeding strategy for the chemogenetic studies: CX3CR1-Cre+:Gq-DREADD+ mice were crossed with Gq-DREADD+ mice to generate ~50% pups expressing Gq-DREADDs exclusively in microglia (see Figures S4A and S4B).
(B) Schematic of in vivo chemogenetic activation experiment: Male mice of CX3CR1-Cre+:Gq-DREADD+ litters, randomly assigned to CTL or ELA conditions (P3) received small, sustained-release CNO- or placebo-containing pellets subcutaneously (s.c.). A cohort was perfused on P10 for the quantification of excitatory synapses on mpd-PVN cells. A second cohort were behaviorally tested as adults in the looming-shadow threat (Daviu et al., 2020) and provided adrenal gland weights, a measure of lifetime stress responses. A third cohort provided baseline and stress-induced ACTH and corticosterone (CORT) levels. After an adult acute, complex stress experience (Hokenson et al., 2020), blood was collected at 30 and 60 min from stress onset.
(C) Chronic chemogenetic microglial activation during P3–10 in microglia-specific Gq-DREADDs in ELA male mice decreased excitatory synapse numbers on mpd PVN neurons to control levels; this was not observed in mice lacking the microglial expression of Gq-DREADDs or in placebo-receiving mice (F2,23.8 = 3.76, p = 0.04; Welch’s 1-way ANOVA; p < 0.05, Dunnett’s T3 multiple comparisons test; also see Figure S6). As CNO alone did not alter the synapse number in microglial Gq-DREADD-expressing CTL mice (p > 0.6), CTL groups were combined for analysis.
(D) Adrenal weights of ELA-adult male mice were higher than those of control mice, indicative of lifelong exposure to augmented stress-hormone release. Chemogenetic microglial activation during the ELA epoch prevented adrenal weight increases (F3,24 = 11.11, p < 0.0001; 1-way ANOVA; p < 0.05, Holm-Sidak’s multiple comparisons test).
(E) At 30 min from onset, stress elicited a robust elevation of ACTH in both CTL and ELA male mice. Chemogenetic activation of microglia in CTL mice blunted this response (F3,20.6 = 3.19, p = 0.04; Welch’s 1-way ANOVA; p < 0.05, Dunnett’s T3 test). At 60 min from stress onset, ACTH levels dropped to 50% of peak values in CTL but not ELA mice (F2,53 = 3.45, p = 0.04; 1-way ANOVA; p < 0.05, Holm-Sidak’s test). Chemogenetic microglial activation in ELA mice ameliorated the prolonged surge of ACTH, which returned to control levels by 60 min (p > 0.8, Holm-Sidak’s test).
(F) The ACTH decay index (30 min/60 min levels) differed in ELA mice (F3,21.5 = 3.19, p = 0.04; Welch’s 1-way ANOVA; p = 0.06, Dunnett’s T3 test), and was partially restored in chemogenetically activated ELA mice (p > 0.2 compared to CTL, same post hoc test). The data show an aberrant prolongation of the hormonal stress response in ELA mice, and the restoration of normal “shutoff” mechanisms by early-life microglial activation (CTL Gq+ CNO mice were not included in the decay index analysis because their stress response was already blunted).
(G) Schematic of the looming-shadow threat task: mice experienced 5 looming-shadow stimulus trials, and their response was scored as escape (top), freezing (bottom), or none.
(H) ELA significantly prolonged latencies of responses to the threat (F3,35 = 6.25, p = 0.002; 1-way ANOVA; p < 0.05, Holm-Sidak’s test). Chemogenetic activation of microglia prevented ELA-induced aberrant threat responses (p > 0.9, Holm-Sidak’s test). Chemogenetic activation of microglia in CTL mice led to delayed responses to the threat, suggesting that normal responses require graded microglia-related synapse pruning (p < 0.05).
Means ± SEMs; *p < 0.05.
In ELA mice expressing microglia-specific Gq-DREADDs, CNO reduced the number of excitatory synapses on presumed PVN CRH+ cells to control levels (F2,23.8 = 3.76, p = 0.04; Welch’s one-way ANOVA; p < 0.05, Dunnett’s T3 multiple comparisons test; Figure 4C). CNO had no effect in ELA mice lacking microglial expression of Gq-DREADDs (p > 0.9). In addition, placebo-containing pellets did not alter synapse numbers (p > 0.5). The effect of CNO was specific to ELA mice: CNO had no effect in microglial Gq-DREADD-expressing CTL mice (p > 0.6). Thus, artificial activation of microglia during ELA, a sensitive developmental period, eliminates the increase in synapse number provoked by ELA without changing synapse numbers in CTLs, supporting the notion that ELA-induced impaired microglial function is the mechanism for the increased excitatory synapses on PVN CRH+ neurons.
We next determined the enduring consequences of ELA and of activating microglia early in life on the hormonal responses to stress of adult control and ELA mice. ELA augmented adrenal weights, in accord with our prior work (Avishai-Eliner et al., 2001; Brunson et al., 2005; Short et al., 2021). Remarkably, chemogenetic microglial activation prevented the ELA-induced increase in adrenal weight (F3,24 = 11.11, p < 0.0001; one-way ANOVA; p < 0.05, Holm-Sidak’s multiple comparisons test; Figure 4D). Acute stress (Chen et al., 2016; Hokenson et al., 2020) elicited a robust elevation of the stress hormones adrenocorticotropic hormone (ACTH) and corticosterone (CORT), governed by CRH+ PVN cells, equally in CTL and ELA-reared adult mice (ACTH, Figure 4E; CORT, Figure S4C) at 30 and 60 min (CORT, Figure S4C) after stress onset. However, the decline kinetics of ACTH, reflecting the termination of the hormonal response to stress, differed significantly among groups: At 60 min after stress onset, ACTH levels were 50% lower than their peak in CTL mice, and this decline was attenuated in ELA mice (F2,53 = 3.45, p = 0.04; one-way ANOVA; p < 0.05, Holm-Sidak’s multiple comparisons test; Figure 4E). This aberrant prolongation of the stress response, typical of ELA (Gilles et al., 1996), was also apparent as a lower ACTH decay index (30 min/60 min F3,21.5 = 3.19, p = 0.04; Welch’s one-way ANOVA; p = 0.06, Dunnett’s T3 multiple comparisons test; Figure 4F) in ELA mice. Notably, early-life chemogenetic microglial activation in ELA mice prevented protracted stress-induced elevations of ACTH levels in adulthood: ACTH levels at 60 min were similar to those in CTL mice (p > 0.8, Holm-Sidak’s multiple comparisons test; Figure 4E), accompanied by partial restoration of the decay index (p > 0.2 compared with CTL, Dunnett’s T3 multiple comparisons test; Figure 4F).
Adult behavioral responses to threat/stress cues were also affected by ELA and were rescued by microglial activation during the ELA period. In the looming shadow task (Figure 4G), ELA provoked an aberrant threat response, apparent as a prolonged latency to either freezing or escape, the two typical behavioral responses of mice in this task (F3,35 = 6.25, p = 0.002; one-way ANOVA; p < 0.05, Holm-Sidak’s multiple comparisons test; Figure 4H). Indeed, several ELA mice never responded to the stressor. Strikingly, chemogenetic microglial activation during the ELA period rescued behavioral stress responses such that the latency to respond in ELA mice resembled that of CTL mice (p > 0.9; Holm-Sidak’s multiple comparisons test). Together, these results demonstrate that ELA induces microglial dysfunction in the developing PVN, which, in turn, provokes enduring disruption of adult neuroendocrine and behavioral responses to stress/threat.
DISCUSSION
The principal findings described here are as follows: (1) Microglia are critical for normal synapse pruning and the maturation of structural and functional connectivity of stress-regulating hypothalamic CRH+ neurons; (2) microglial process dynamics may be a potent indicator and a potential mechanism of their synapse pruning capacity during development; (3) ELA provokes microglial dysfunction during development, which results in augmented excitatory synapses on PVN CRH+ neurons and consequent aberrant responses to future stress/threats; (4) the mechanism for impaired synapse engulfment during ELA involves differential expression and activity of the microglial phagocytic receptor MerTK and a likely derangement of neuronal-microglial cross-talk. These discoveries identify an important role for microglial actions during a sensitive period in ELA-induced disruption of stress responses, a hallmark of stress-related affective disorders.
These studies demonstrate that microglia are critical for the normal, selective synapse pruning during a sensitive developmental period in a stress-related circuit, and specifically in a population of stress-sensitive CRH+ neurons in the PVN. Previous work has demonstrated that microglia engulf synapses in other developing brain regions, including the thalamus (Schafer et al., 2012), hippocampus (Paolicelli et al., 2011), and cortex (Sipe et al., 2016; Tremblay et al., 2010). However, most prior analyses did not consider cell-type specificity of the neuronal targets of microglial synapse pruning. Recent data have supported the notion that microglia are heterogeneous (Ayata et al., 2018; Hammond et al., 2019), differing across brain regions and even within a brain region (Ayata et al., 2018; Mondo et al., 2020)—for example, subpopulations of GABA-receptive microglia in the developing cortex prune inhibitory synapses (Favuzzi et al., 2021). Here, we demonstrate that as a result of ELA, microglia abutting CRH+ neurons in the PVN engulf fewer synaptic elements compared with controls (Figure 3), whereas synapse engulfment by microglia not in contact with CRH+ neurons is unchanged (Figure S2G). These results suggest a selectivity of the neuronal targets of microglia within PVN, which may derive from altered signaling of CRH+ cells to adjacent microglia as a result of ELA (Bolton et al., 2020; Singh-Taylor et al., 2018). Alternatively, distinct microglia populations may be assigned to synapse pruning of specific neuronal populations within PVN. Distinguishing between these two possibilities will require future studies.
Notably, microglial synapse pruning of PVN-CRH+ neurons is required for the proper function of the neuroendocrine and behavioral responses to stress later in life, because the augmented number of excitatory synapses observed after ELA associated with higher adrenal weights, indicative of lifelong chronic stress, as well as prolonged secretion of stress hormones and disrupted behavioral responses to acute stress. All of these indicators of aberrant functions of the “stress circuit” were normalized by selective chemogenetic activation of microglia in vivo during the ELA period (Figure 4), providing a causal link between microglial function during development and lifelong stress reactivity.
We quantified microglial process dynamics in real time. Conventional measures of microglial activation, such as microglial density and shape, provide limited insight into microglial function during development, when these measures are in flux (Schwarz et al., 2012). We did not find differences in microglial density or shape in ELA hypothalamus and instead obtained direct measures of microglial function using two salient, independent parameters—microglial process dynamics and synapse engulfment. ELA inhibited both, suggesting that they may be related; process dynamics (velocity and excursion distance) may predict and perhaps underlie synapse engulfment efficiency.
Mechanistically, we discovered that the regulation of excitatory synapse number on PVN CRH+ neurons by microglia depended on the phagocytic receptor MerTK. Ex vivo treatment of PVN organotypic slices with a small-molecule MerTK inhibitor augmented excitatory synapses on CRH+ neurons from control mice, indicating that MerTK contributes to synapse pruning. MerTK is expressed by microglia (Diaz-Aparicio et al., 2020; Fourgeaud et al., 2016) and astrocytes (Chung et al., 2013) in the brain. In the immature PVN, we localized MerTK expression exclusively to microglia. Recent work implicates astrocytes in synaptic pruning (Chung et al., 2013; Lee et al., 2020), likely in concert with microglia (Vainchtein and Molofsky, 2020; Vainchtein et al., 2018), and these interactions merit future studies. Here, ELA reduced MerTK expression in microglia of the developing PVN. In addition, whereas MerTK inhibition increased synapse numbers in control PVN cultures, it did not influence PVN cultures from ELA mice, consistent with ELA-induced inactivity of MerTK as a mechanism for the augmented synapse number observed in ELA mice. MerTK interacts with a neuronal ligand, phosphatidylserine, an intracellular molecule that is externalized during cell death (Diaz-Aparicio et al., 2020; Fourgeaud et al., 2016), providing a phagocytic signal. Recently, selective phosphatidylserine externalization at synaptic elements has been identified and implicated in developmental synaptic pruning (Park et al., 2021; Scott-Hewitt et al., 2020). It is not known whether the externalization of phosphatidylserine at synapses on CRH+ neurons is reduced by ELA, attenuating the signal for microglial synaptic pruning. The data presented here suggest that the expression and function of MerTK is inhibited in ELA hypothalamus. This, potentially in concert with diminished externalization of its ligand, results in the diminished microglial synapse pruning observed in the PVN during ELA.
Here, the use of selective chemogenetic activation of microglia via Gq-DREADDs restored microglial pruning of synapses in immature ELA mice and rescued behavioral and neuroendocrine responses to stresses later in life. The manipulation likely activated microglia downstream of MerTK, rather than via reversing MerTK deficits: MerTK activates engulfment via phospholipase C (PLC) and Rac1 signaling in peripheral macrophages phagocytosing apoptotic cells (Linger et al., 2008). Whereas this mechanism remains to be confirmed in microglial synaptic pruning, brain microglial engulfment requires process motility and actin cytoskeletal dynamics. Activation of Gq-DREADDs increases intracellular calcium concentrations via inositol triphosphate/diacylglycerol (IP3/DAG) in multiple cell types, including neurons and astrocytes (van den Herrewegen et al., 2021). Increased intracellular calcium, in turn, augments microglial process extension and dynamics, facilitating synapse engulfment (see Figure S6). Of note, such intracellular calcium increases occur during insults such as epilepsy, focal damage, and lipopolysaccharides (LPS) (Pozner et al., 2015; Umpierre et al., 2020). Activating Gq-DREADDs in microglia with CNO also increases markers of microglial activation such as Iba-1 and CD11b and their production of proinflammatory cytokines such as interleukin-1 β (IL-1β) and tumor necrosis factor α (TNF-α) (Saika et al., 2021; Grace et al., 2018), further supporting the approach taken here.
How might ELA influence microglial activity? Stress, including early in life, is associated with the chronic elevation of plasma glucocorticoids (Brunson et al., 2005; Gilles et al., 1996; Rice et al., 2008) and increased brain levels of both glucocorticoids and CRH (Bolton et al., 2018; Chen et al., 2016). Microglia express glucocorticoid receptor (GR), a key corticosterone receptor (Li et al., 2007; Nichols et al., 2005; Picard et al., 2021; Zhang et al., 2014b) and potent transcription factor that regulates microglial MerTK expression (Picard et al., 2021). Thus, ELA may directly downregulate MerTK expression and function in microglia. In addition to altered transcription, corticosterone impairs a number of microglial activities (Li et al., 2007; Nichols et al., 2005). Microglia also express CRH receptors (Stevens et al., 2003; Wang et al., 2002; Zhang et al., 2014b) and respond to the peptide via changes in gene expression and the release of factors such as brain-derived neurotrophic factor (BDNF), which, in turn, may influence neurons (Fan et al., 2014; Yang et al., 2005). Thus, ELA may directly modify the function of developing hypothalamus microglia via several complementary mechanisms.
The intriguing specificity of the deficits in microglial synapse phagocytosis to microglia that abut hypothalamic CRH+ neurons raises the possibility that the primary site of action of ELA lies within these neurons. As noted above, reduced surface expression of phosphatidylserine will reduce neuronal synapse interaction with microglial MerTK, leading to attenuated synapse engulfment (Park et al., 2021; Scott-Hewitt et al., 2020). In support of a neuron-to-glia information flow induced by ELA, a recent single-cell transcriptomics analysis of these hypothalamic CRH cells identified profound effects of ELA, which were apparent already by the end of the ELA period (Short et al., 2021). Importantly, these ELA-induced changes were specific to CRH-expressing neurons residing in the dorsomedial PVN, in accord with the specificity of microglial deficits reported here. Thus, a likely mechanism for the remarkable deficits in microglial dynamics and synapse engulfment identified in the present study may involve the combined effects of ELA on microglia and neurons as a result of the concerted actions of GR and corticotropin releasing hormone receptor 1 (CRHR1), as reported elsewhere in the brain in response to stress (Chen et al., 2016). Our ability to rescue a normal stress-response phenotype by chemogenetically activating microglia in ELA mice suggests that the ELA-provoked functional deficits can be overcome by interventions delivered selectively to microglia, holding promise for future therapeutic strategies.
Does ELA affect both sexes equally, and do microglial disruptions occur differentially in males and females? To address sex as a biological variable, both sexes were used for all of the initial experiments (synapse counts and electrophysiology). These studies demonstrated an additional effect of sex. Specifically, in contrast to males, increased excitatory synapse number and function on PVN CRH+ neurons was not observed in P10 ELA females, although both sexes were affected later in life (P24–25; Figures S5A and S5B). Notably, microglial synapse engulfment at P8 was not altered in ELA females (Figure S5C). Because these data suggested sex-modulated time course and potential mechanisms of synapse development and the contributions of ELA, we elected to focus the subsequent studies reported here on males, and plan to devote future work to studies of females.
Limitations of the study
The MerTK mechanistic experiment was performed in organotypic slice cultures, enabling isolating the pharmacological effects of the MerTK inhibitor to the PVN, rather than the whole brain or whole body. Organotypic slice cultures are not a perfect reflection of the in vivo context, as they do involve initial microglial activation and the loss of extrahypothalamic synaptic connections (Czapiga and Colton, 1999; Delbridge et al., 2020). However, most inflammatory markers and neuronal death rates stabilize at very low levels by 1 week in culture, the time point at which we begin pharmacological treatments. Importantly, organotypic slice cultures are far superior to dissociated cultures in approximating the in vivo condition, because of the relative preservation of tissue architecture and cellular composition (Czapiga and Colton, 1999; Delbridge et al., 2020; Weinhard et al., 2018). Off-target actions of the MerTK inhibitor should be considered: UNC2025 has a ~40-fold greater selectivity for MerTK over Axl and Tyro3. At the low dose we used (20 nM; half-maximal inhibitory concentration [IC50] = 2.7 nM),the possibility of actions on these other receptors is thus very low (McDaniel et al., 2018). We propose that the inhibitor influenced MerTK specifically in microglia. Whereas we did not completely exclude the possibility of low-level non-microglial expression of MerTK, the extensive overlap of MerTK with microglial markers (P2RY12, CX3CR1) and lack of overlap with astrocytic (S100β, GFAP) and neuronal (NeuN) markers suggests that any non-microglial expression of MerTK is very low and likely insignificant in our pharmacological experiments. Future studies should investigate the impact of the microglia-specific manipulation of MerTK in vivo (e.g., MerTK conditional deletion), and additional experiments interrogating the impact of microglia-specific chemogenetic activation on CRH+ neuronal excitability would strengthen the mechanistic linkages between early microglial activity and later PVN function.
CONCLUSIONS
The findings described here establish microglia abutting stress-sensitive hypothalamic neurons as powerful contributors to the enduring consequences of ELA on responses to stress later in life. ELA provokes microglial dysfunction during a sensitive period of development, characterized by diminished microglial process dynamics and deficits in microglial synapse engulfment, and resulting in an augmented number and function of excitatory synapses on CRH+ neurons in PVN. These changes are critical, because they lead to the chronically augmented release of stress hormones and aberrant behavioral responses to stress/threat, an important factor in determining the risk for stress-related mental illnesses. The manipulation of microglial function during development may provide novel targets for the prevention of stress-related disorders.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Tallie Z. Baram (tallie@uci.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
The code generated during this study is openly available on GitHub at https://github.com/jaclynrbeck/BaramLabMicrogliaTracking.
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were performed in accordance with National Institutes of Health (NIH) guidelines and were approved by the UC Irvine animal care and use committee. All analyses were performed without knowledge of treatment group.
Animals
Mice of both sexes were housed in temperature-controlled, quiet, uncrowded conditions on a 12-hr light, 12-hr dark schedule (lights on at 0630 hr., lights off at 1830 hr.) with free access to food and water. The transgenic mouse lines employed were all on a C57BL/6 genetic background bred in-house from the following founders: CRH-IRES-Cre+/+ (Jax #: 012704; Jackson Laboratory, Bar Harbor, ME), tdTomato+/+ (stop-floxed; Ai14; Jax #: 007914), CX3CR1-GFP+/+ (Jax #: 005582), Gq-DREADD+/− (stop-floxed; Jax #: 026220), CX3CR1-BAC-Cre+ (GENSAT; MMRRC stock #036395-UCD; obtained from Dr. Staci Bilbo, Duke University), and MerTK-fl/fl [floxed; obtained from Dr. Carla Rothlin, Yale University (Fourgeaud et al., 2016)]. Wild-type C57BL/6J mice of both sexes used for the electrophysiology experiments were bred in-house from founders obtained from Jax (#000664). All experiments were performed in accordance with National Institutes of Health guidelines and were approved by the University of California, Irvine Animal Care and Use Committee.
Limited bedding and nesting (LBN) model of early-life adversity (ELA)
Dams were checked for copulatory plugs daily while paired with a male, housed singly on embryonic day (E)17, and monitored every 12 h for the birth of pups. The day of birth was termed postnatal day (P)0. On the morning of P2, litters were culled to 8 pups if needed (5 pups minimum) and included both sexes. Also on P2, the ELA paradigm was initiated as in previous studies (Molet et al., 2014; Rice et al., 2008): Control (CTL) dams were placed in cages with standard amounts of corn husk bedding (650 mL) and nesting material, i.e. one square piece of cotton material measuring 5 × 5 cm (nestlet). This material was shredded by the dam to create a nest area. In contrast, ELA dams received one-half nestlet placed on a fine-gauge plastic-coated aluminum mesh platform (mesh dimensions 0.4 × 0.9 cm, cat# 57,398; McNichols Co., Tampa, FL), approximately 2.5 cm above the cage floor. The cage floor was covered with a small amount of corn husk bedding (~60 mL). This setup permitted mouse droppings to fall below the platform without trapping the pups. In addition, all cages were housed in a room with robust ventilation, avoiding the accumulation of ammonia. Figure S1A shows examples of the ELA and standard cage setups. Both groups were completely undisturbed from the morning of P2 to the morning of P10, at which point the dams and litters were transferred to standard cages.
The impoverished cage environment in mice results in marked alterations in maternal behavior, such as increased sorties (i.e., exits) from the nest during P2–8 (F1,19 = 25.15, p < 0.0001; 2-way repeated measures ANOVA; Figure S1B) and shorter, fragmented bouts of licking and grooming the pups (t18 = 5.26, p < 0.0001; unpaired t test; Figure S1C), without changes in the total amount of licking and grooming behaviors (Figure S1D), in accord with our previous studies (Molet et al., 2016; Rice et al., 2008). This environment provokes chronic stress in the mouse pups, including reduced weight gain on P10 (t101 = 7.05, p < 0.0001; unpaired t test; Figure S1E) and P21 (t41 = 3.45, p = 0.001; unpaired t test; Figure S1F). These modest changes disappear by adulthood (P60; Figure S1G). We have also previously identified increased adrenal weights and serum corticosterone levels during the ELA period (Brunson et al., 2005; Ivy et al., 2008; Rice et al., 2008).
For short-term studies, pups were euthanized with sodium pentobarbital and transcardially perfused with ice-cold phosphate-buffered saline (PBS; pH = 7.4) followed by 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH = 7.4) on P8 (for microglial synapse engulfment studies) or P10 (for synapse counts). For 2-photon imaging studies, pups were euthanized via rapid decapitation and acute slices of the paraventricular hypothalamic nucleus (PVN) prepared. For long-term studies in the juvenile period (i.e., electrophysiology, synapse counts) and adulthood (i.e., functional studies of stress response), pups were weaned on P21 into standard, same-sex cages of 2–5 mice.
METHOD DETAILS
Assessment of maternal behaviors in CTL vs. ELA cages
Maternal behaviors and interactions with pups were evaluated on P2–8. Control (n = 9 litters) and ELA dams (n = 11) were observed daily in the morning (between 0700–0900 hr.). Each maternal observation session consisted of 50 min, during which maternal behaviors and dam-pup interactions were scored continuously, allowing for an accurate analysis of the duration of each bout of behavior (Molet et al., 2016). The behaviors recorded included: licking and grooming pups (LG), nursing pups, self-grooming, nest-building, off-nest activity, and eating/drinking. The number of times the dam left the nest (sorties) was also recorded (Rice et al., 2008). To optimize visibility into the nest and minimize interference with normal cage activities, the observer used mirrors placed beside the cages.
Immunohistochemistry (IHC) for excitatory synaptic markers
Perfused brains of CRH-Cre+/−::tdTomato+/− mice (P10 or P24–5) were post-fixed in 4% paraformaldehyde in 0.1 M PBS (pH = 7.4) for 4–6 hr. before cryoprotection in a 25% sucrose solution. Brains were frozen, then sectioned coronally into 25-μm-thick slices (1:4 series of the PVN) using a Leica CM1900 cryostat (Leica Microsystems, Wetzlar, Germany). Sections were subjected to IHC using standard methods, as described previously (Chen et al., 2015). Briefly, after several washes with PBS containing 0.3% Triton X-100 (PBS-T, pH 7.4), sections were blocked with 5% normal donkey serum (cat# 017–000-121, Jackson ImmunoResearch, West Grove, PA, USA) for 1 hr. to prevent non-specific binding. Sections were then incubated overnight at 4°C with rabbit anti-PSD95 antiserum (1:1,000; cat# 51–6900, Invitrogen/ThermoFisher, Waltham, MA) and guinea pig anti-vGlut2 antiserum (1:12,000, cat# AB2251–1, Millipore Sigma, Temecula, CA, USA) in PBS-T containing 2% normal donkey serum. The next morning, sections were rinsed in PBS-T (3 × 5 min), and then incubated with donkey-anti-rabbit IgG-488 (1:1,000; A-21206, ThermoFisher) and donkey-anti-guinea pig IgG-647 (1:1,000; cat# 706–605-148, Jackson Immunoresearch) for 3 hr. at room temperature. After washing (3 × 5 min), sections were mounted onto gelatin-coated slides and coverslipped with Vectashield containing DAPI (cat. #H-1200, Vector Laboratories, Burlingame, CA, USA).
Confocal imaging
Confocal images of the mediodorsal parvocellular (mpd) paraventricular hypothalamic nucleus (PVN) were collected using an LSM-510 confocal microscope (Zeiss, Dublin, CA, USA) with an Apochromat ×63 oil objective. 11 z-stack images of 142.86 × 142.86 μm were taken at 1-μm intervals. Image frame was digitized at 12-bit using a 1024 × 1024-pixel frame size. CRH+ neuron number was counted manually in FIJI, and CRH neuronal volume was automatically calculated using Imaris 3D reconstruction software (Bitplane, Zurich, Switzerland). Excitatory synapses onto CRH+ neurons were identified as colocalized puncta of vGlut2+PSD95 within the CRH-tdTomato+ volume using Imaris’ colocalization function (threshold = 1.0). This methodology agreed well with manually counting synapses (r = 0.90, p < 0.0001) using FIJI [https://imagej.net/software/fiji/; (Schindelin et al., 2012)].
Electrophysiology
Slice preparation
Wild-type control and ELA mice (P18–26) were killed by cervical dislocation in accordance with Schedule I of the UK Government Animals (Scientific Procedures) Act, 1986. Coronal hypothalamic slices containing the PVN were prepared as previously described (Gunn et al., 2013). The brain was rapidly dissected and placed in ice cold (0–4°C), oxygenated (95% O2) artificial cerebro-spinal fluid (aCSF) containing the following (in mM): 135 NaCl, 2.5 KCl, 1.25 NaH2PO4, 10 MgCl2, 0.5 CaCl2, 26 NaHCO3, 10 glucose (320–335 mOsm, pH ~7.4). Coronal hypothalamic slices were then cut (300–320 μm) using a vibratome (Leica) at 0–4°C. Slices were subsequently incubated for at least 1 hour at room temperature in a holding chamber containing oxygenated aCSF (as above) that additionally contained 1 mM ascorbic acid and 3 mM sodium pyruvate. Slices were then transferred to the recording chamber and recordings were performed on slices perfused (3–6 mL/min) with extracellular solution (ECS) containing (in mM): 126 NaCl, 26 NaHCO3, 2.95 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 10 glucose (306–309 mOsm, pH ~7.4).
Whole-cell recordings
All recordings were performed using an Axopatch 1D amplifier (Molecular Devices, Wokingham, UK), stored directly to a PC using a NI-DAQmx interface (National Instruments, Newbury, UK) for analysis offline.
Recordings of sEPSCs and sIPSCs
Whole-cell voltage-clamp recordings of sEPSCs and sIPSCs were made at 35°C in ECS using a low Cl− (12 mM) intracellular solution. In these recordings, patch pipettes (4–6 MΩ) were filled with an intracellular solution containing (in mM): 135 CH3O3SCs, 8 CsCl, 10 HEPES, 10 EGTA, 1 MgCl2, 1 CaCl2 (pH 7.2–7.3 with CsOH, 300–310 mOsm). Under such recording conditions, the calculated (pClamp v 8.2) and experimentally verified EGABA and Eglutamate were −63 mV and 0 mV respectively. Synaptic currents were filtered at 2 kHz using an 8 pole low-pass Bessel filter. The series resistance was between 8 and 20 MΩ with up to 80% compensation. Only cells with a stable access resistance were used, and experiments were aborted if series resistance changes >20% occurred.
Data analysis
All recordings were analyzed offline using the Strathclyde Electrophysiology Software (Electrophysiology Data Recorder [WinEDR] and Whole-Cell analysis Program [WinWCP]; courtesy of Dr J. Dempster, University of Strathclyde).
Analysis of sEPSCs & sIPSCs
Individual events were detected in WinEDR using an amplitude threshold detection algorithm (−4 pA threshold, 3 ms duration) and visually inspected for validity. Any noise that reached the threshold, or traces that contained multiple events were rejected from analysis. Accepted events (a minimum of 40 for each experimental condition) were digitally averaged. Synaptic currents (i.e. EPSCs and IPSCs) were analyed with regard to peak amplitude, rise time and decay kinetics. Events with rise times >1 ms (representing <1% of total) were discarded in order to eliminate from the analysis events subject to dendritic filtering. The decay phase of the digitally averaged event was then fitted with single exponential, [Y(t) = A*exp(−t/τ)] and bi-exponential functions, [Y(t) = A1*exp(−t/τ1)+ A2*exp(−t/τ 2)]. An F test was then used to establish whether the decay was best described by a mono- or bi-exponential fit, as indicated by a decrease in the standard deviation of the residuals. The frequency of sEPSCs and sIPSCs was determined by counting the number of events in 20-second bins over a minimum of 2 separate 1-min. periods (first and last minute) of the recording. In WinEDR events were detected (offline) using a rate of rise time threshold that was specific for individual cells and that allows detection of the slowest events (~40 pA/ms). Recordings were then visually inspected to ensure all events were included and any detected spurious noise removed. A mean frequency and inter-event interval were then calculated.
Confocal imaging of microglial density in the PVN
Perfused brains of CRH-Cre+/−::tdTomato+/−; CX3CR1-GFP+/+ mice (P10) were cryoprotected and frozen, then sectioned coronally into 25-μm-thick slices using a cryostat (1:4 series of the PVN). Unstained sections were mounted on gelatin-subbed slides and coverslipped with Vectashield containing DAPI. Confocal images of the PVN were collected with an LSM-510 confocal microscope (Zeiss) with an Apochromat ×20 oil objective. 11 z-stack images of 450 × 450 μm were taken at 1-μm intervals. Image frame was digitized at 12-bit using a 1024 × 1024-pixel frame size. An ROI was manually drawn around the perimeter of the CRH-tdTomato+ neurons of the PVN, then microglia number in this region was counted manually in FIJI, and microglial volume was automatically calculated using Imaris’ 3D reconstruction (Bitplane, Zurich, Switzerland).
Two-photon microscopy
Slice preparation
Acute PVN slices from P8 CRH-Cre+/−::tdTomato+/−; CX3CR1-GFP+/+ pups were prepared in the same manner as described above for Electrophysiology. Briefly, the brain was rapidly dissected and placed in ice-cold (0–4°C), oxygenated (95% O2) aCSF, and 320-μm coronal hypothalamic slices were collected using a vibratome (Ted Pella, Redding, CA, USA). Slices were subsequently incubated for 30 min.−1 hr. at room temperature in a holding chamber containing oxygenated imaging media (50% MEM, 50% HBSS, 6 mg/mL D-glucose, and 25 mM HEPES; all from Invitrogen/ThermoFisher; adapted from (Dailey et al., 2013) to be serum-free).
Imaging
Two-photon (2p) imaging of hypothalamic slices was performed using a multiphoton microscope built on an Olympus BX51 upright microscope frame, fitted with a motorized Z-Deck stage (Prior Scientific, Cambridge, UK) and Nikon 25×, water dipping objective (CFI75 Apo L, NA = 1.1, WD = 2.0 mm). 2p excitation was generated by a tunable femtosecond laser (Chameleon Ultra-II or Vision-II, Coherent, Santa Clara, CA, USA) set to 920 nm to excite EGFP and TdTomato as described previously (Othy et al., 2020). Fluorescence emission was split by 484 nm and 538 nm dichroic mirrors into three non-descanned PMT detectors (R3896, Hamamatsu Photonics, Bridgewater, NJ, USA) to detect second-harmonic signal generated from collagen in blue; EGFP signal in green; and TdTomato signal in red. Supported brain slices were anchored in a RC-27L bath (Warner Instruments, Holliston, MA, USA) assembled into a custom-built heated imaging chamber maintained at 37° ± 0.5°C using continuous perfusion of oxygenated imaging media. 3D image stacks of x = 250 μm, y = 250 μm, and z = 56 μm (XYZ voxel size 0.244 μm × 0.244 μm × 4 μm) were acquired every 37 s interval using image acquisition software Slidebook (Intelligent Imaging Innovations, Denver, CO, USA), up to 50 min to create a 4D data set. Imaris version 9.2.1 or later (Bitplane) was used for rendering and image processing for figures.
Automated python-based algorithm for analysis of microglial process dynamics
Two-photon video pre-processing
All video files were converted from z-stacks to max projections using Slidebook software. The resulting videos had one 3-channel image per time point. All other processing code was written in Python. Videos were truncated to span 30 minutes of capture time for CTL vs. ELA experiments, or 50 min for Gq-DREADD/CNO experiments, and only the green channel (containing microglia) was used for processing. To account for drift of tissue specimens during imaging, each frame in a given video was registered to the previous frame in the series using OpenCV’s (Bradski, 2000) findTransformECC and warpAffine functions.
Noise and point spread were reduced in each image using deconvolution (Figure 2F). A measured point spread function for the microscope was unavailable, so one was generated with the FIJI (National Institutes of Health, Bethesda, MD, USA) plugin “PSF Generator” (http://bigwww.epfl.ch/algorithms/psfgenerator) using the following parameters:
The plugin generates a z-stack of 3 PSF images, and only the first slice of the z stack was used for deconvolution of the 2D max projection image. The values in the PSF image were squared, as is necessary for two-photon images. Using the generated PSF image, each frame of the video was deconvolved using the Richardson-Lucy method (provided by the scikit-image (van der Walt et al., 2014) function skimage.restoration.richarson_lucy).
After deconvolution, an optimal threshold for each video was determined manually in FIJI. Thresholds ranged from the top 10–15% of pixel values in the video and were chosen to remove background noise while maintaining microglia shape and branching. The thresholds were recorded but not applied to the videos at this point.
Soma finding
Microglial somas were located by thresholding the videos with a threshold that was 50% higher than the threshold determined by preprocessing. Erosion, dilation, and close operations were performed on the resulting binary image (using OpenCV’s (Bradski, 2000) morphologyEx function) to remove processes and fill in small gaps in the somas (Figure 2F). Connected regions of non-zero pixels with an area greater than 281 pixels (approximately 17 microns2) were identified as somas.
Somas were tracked across frames in a video by centroid matching. Each soma in a frame is “matched” to the closest soma in the next frame if their centroids are less than 20 pixels apart. This process is repeated for each time point in a video to create a chain of somas across timepoints that belong to the same microglia. Occasionally, thresholding or processing criteria in previous steps that would result in a frame with a “missing” soma, where frames on either side have an identified soma in the same location but no soma was identified in the current frame. To account for this, somas in these “missing soma” frames were interpolated by overlapping (via binary “and” operation) the current thresholded image with the previous frame. Any connected region larger than 10 pixels that coincides with the location of a soma in the previous frame is labeled as a soma in the current frame.
Microglia tracing
Microglia structure was estimated by skeletonization (Figure 2F). Deconvolved images were thresholded using the values determined during pre-processing, and connected regions of non-zero pixels with an area less than 20 pixels were removed from the thresholded image by setting their values to 0. The filtered binary image was skeletonized using the scikit-image (van der Walt et al., 2014) function skimage.morphology.skeletonize.
To connect branches that became disconnected due to thresholding, each skeletonized image was turned into a graph using the minimum spanning tree (MST) algorithm (scipy.sparse.csgraph.minimum_spanning_tree (Virtanen et al., 2020)), which connects all points using the shortest path possible, with no cycles in the graph (Figure 2F). Some modifications were made prior to running the algorithm. Briefly, a nearest-neighbor graph was generated for all non-zero points in the skeletonized image (using scikit-learn’s (Pedregosa et al., 2001) sklearn.neighbors.kneighbors_graph). The graph was modified to force all soma centroids to appear closer to the perimeter points of the soma than to any other point, which forced the MST algorithm to treat each soma as a tree center. Additionally, the original pixel values of the deconvolved image were used as weights for the algorithm. The weight for each pair of points was calculated as the maximum (darkest) value between the points, which causes brighter pairs of pixels to be connected first. The end result was a graph describing how all skeletonized points connected to each other in the image. Prior to running MST, cycles in the original skeleton were identified. MST can break cycles in inconsistent places from image to image, so any “process tip” or branch that resulted from breaking a cycle was discarded from analysis. Tips that fell on the soma border were also discarded as they were artifacts of tracing outward from the soma center.
To identify individual microglia from the MST graph, all connections were broken between points that were greater than 50 pixels apart, as these were unlikely to belong to the same branch or microglia. If two or more somas were still connected to each other by a path, the path was broken at the longest gap between pixels in the skeletonized image. “Process tips” and branches resulting from these broken paths were discarded from analysis. This process resulted in individual graphs centered at each soma. Lastly, to reduce noise from skeletonization, individual branches that were less than 10 pixels long were pruned from the graph.
Similar to soma tracking, individual processes were tracked through frames in the video. Each process tip was “matched” to the closest process tip in the next frame, if it met three conditions. One, both tips must belong to the same microglia as identified by soma matching. Two, the tips must be less than 50 pixels away. Three, the tips must belong to the same branch. We determine this by tracing from each process tip to the nearest fork in its branch in each image. The branches must at some point in this path be less than 5 pixels away from each other to be considered the same. To reduce noise, only process tips that appear in 5 or more frames are considered for analysis.
Analysis
In addition to basic information about microglia location in each image in a video, information characterizing microglial process dynamics was also calculated. We define tip velocity between two frames as d/t, where d is equal to the Euclidean distance between matched process tips in two frames, and t is the time elapsed between frames.
Code availability
The Python tracking code is openly available on GitHub at https://github.com/jaclynrbeck/BaramLabMicrogliaTracking.
Manual kymograph analysis of microglial process dynamics
Pre-processing of 2-photon videos was similar to that described above for the automated method. The ProMoIJ plug-in for FIJI was used for drift correction/registration of the maximum projection, single-channel, time-lapse videos (Paris et al., 2017), and the MultipleKymograph plug-in for FIJI (https://www.embl.de/eamnet/html/body_kymograph.html) was utilized to generate the kymographs themselves. In each video, 4 clearly delineated (located completely within the frame throughout the video), non-overlapping microglial cells were selected and cropped, and the brightest, most active process on each cell was selected for further analysis, resulting in 4 processes from 4 different microglia analyzed for each video. Next, an ROI was drawn as a line along the length of each microglial process, extending from the base where the process connects to the cell body, to slightly past the process tip. The kymograph analysis measured the total distance traveled by the tip of the microglial process (Figure 2D), which was averaged across the 4 analyzed processes to attain one average value for each animal.
Ex vivo chemogenetic activation 2p imaging experiment
The preparation of acute hypothalamic slices from P8 CX3CR1-Cre+:Gq-DREADD + mice was conducted as described above. Rather than EGFP, mCitrine-labeled Gq-DREADDs labeled microglia in this strain and were used to track microglial process dynamics. A baseline period of 10 min was imaged using the same parameters as before. At 10 min, the perfusion tubing was switched to imaging media containing 10 μM CNO (clozapine N-oxide; CAS# 34,233–69-7, MH# C-929, National Institute of Mental Health, Bethesda, MD, USA) in 0.05% dimethyl sulfoxide (DMSO; cat# D8418–100, Millipore Sigma) or 0.05% DMSO alone (Vehicle) for 40 more min. of imaging to determine the effect of CNO vs. Vehicle. 2p videos were analyzed using the automated Python-based algorithm and manual kymograph method as described above.
Microglial synapse engulfment analysis
Coronal sections of the PVN from P8 CRH-Cre+/−::tdTomato+/−; CX3CR1-GFP+/+ mice were subjected to vGlut2 IHC using a similar procedure as described above for the synapse IHC. Briefly, after several washes in PBS containing 0.3% Triton X-100 (PBS-T, pH 7.4), sections were blocked with 5% normal donkey serum (Jackson ImmunoResearch) for 1 hr. to prevent non-specific binding. Sections were then incubated overnight at 4°C with guinea pig anti-vGlut2 antiserum (1:12,000, cat# AB2251–1, Millipore Sigma) in PBS-T containing 2% normal donkey serum. The next morning, sections were rinsed in PBS-T (3 × 5 min), and then incubated with donkey-anti-guinea pig IgG-647 (1:1,000; cat# 706–605-148, Jackson Immunoresearch) for 3 hr. at room temperature. Sections were washed several times in PBS-T, mounted on gelatin-coated slides, and coverslipped with Vectashield mounting medium with DAPI (Vector Laboratories, H-1200). Confocal images of the mpd PVN were collected with an LSM-510 confocal microscope (Zeiss) with an Apochromat ×63 oil objective. 11 z-stack images of 142.86 × 142.86 μm were taken at 1-μm intervals. Image frame was digitized at 12-bit using a 1024 × 1024 pixel frame size. An ROI was manually drawn around the perimeter of the CRH-tdTomato+ neurons of the mpd PVN, and microglial volume within the ROI was automatically calculated using Imaris’ 3D reconstruction. Excitatory vGlut2+ synaptic puncta located inside of the 3D microglial volume were automatically identified and counted using Imaris’ spot detection function.
Microglial ultrastructure analysis
Two PFA/acrolein-perfused brain sections containing the PVN were selected from a P8 CRH-Cre+/−:tdTomato+/− male mouse (PVN sections were selected based on the presence of densely packed CRH-tdTomato+ neurons around the 3rd ventricle). Sections were washed in PBS, then quenched 10 min. in 0.3% H2O2 in PBS and permeabilized 30 min. in 0.1% NaBH4 in PBS. Sections were first incubated 1 hr. at room temperature in blocking solution (10% fetal bovine serum, 3% bovine serum albumin, 0.01% Triton X-100 in [50 mM] tris-buffered saline [TBS]). Afterwards, sections were incubated overnight at 4°C with rabbit anti-IBA1 polyclonal primary antibody (1:1,000; cat# 019–19741, FUJIFILM Wako Chemical, Osaka, Japan) in blocking solution. The following day, antibody was washed off, and the sections were incubated with biotinylated goat anti-rabbit polyclonal secondary antibody (cat#111–066-046, Jackson ImmunoResearch) in TBS for 1.5 hr., followed by avidin-biotin complex solution (1:1:100 in TBS; cat# PK-6100, Vector Laboratories) for 1 hr. at room temperature. The staining was revealed in 0.05% diaminobenzidine (DAB; cat# D5905–50TAB, Millipore Sigma) with 0.015% H2O2 in TBS for 4.5 min. at room temperature.
The immunostained sections were next post-fixed flat in osmium-thiocarbohydrazide-osmium for scanning electron microscopy (SEM). In particular, sections were incubated in 3% ferrocyanide (cat# PFC232.250, BioShop, Burlington, ON, Canada) diluted in water combined (1:1) with 4% aqueous osmium tetroxide (cat#19170, Electron Microscopy Sciences, Hatfield, PA, USA) for 1 hr. in 1% thiocarbohydrazide diluted in water (cat# 2231–57-4, Electron Microscopy Sciences) for 20 min., in 2% osmium tetroxide diluted in water, then dehydrated in ascending concentration of ethanol (2 × 35%, 50%, 70%, 80%, 90%, 3 × 100%) followed by propylene oxide (3×) for 5 min. each. After post-fixation, tissues were embedded in Durcupan ACM resin (cat# 44611–44614, Millipore Sigma) for 24 hr. and carefully placed between two ACLAR® embedding films (cat# 50425–25, Electron Microscopy Sciences), and the resin was let to polymerize at 55°C for 72 hr. Regions of selection—PVN—were excised from the embedded sections on ACLAR® sheets and re-embedded on top of a resin block for ultrathin sectioning (Ultracut UC7 ultramicrotome, Leica Microsystems). Ultrathin sections (~75 nm thickness) were collected and placed on a silicon nitride chip and glued on specimen mounts for SEM. Representative microglial cell bodies and processes in the PVN were imaged at 5 nm of resolution using a Crossbeam 540 field emission SEM with a Gemini column (Zeiss).
MerTK IHC
Coronal sections of the PVN from P8 CRH-Cre+/−::tdTomato+/− male mice were subjected to MerTK IHC using a similar procedure as described above for the synapse IHC. Briefly, after several washes in PBS containing 0.3% Triton X-100 (PBS-T, pH 7.4), free-floating sections were incubated in 0.5% Triton X-100 in PBS for 10 minutes, followed by 2 additional washes in PBS-T. Sections were blocked in 5% normal donkey serum (NDS) and 5% normal goat serum (NGS) in PBS-T, followed by incubation overnight at 4°C with rat anti-MerTK (1:1000, eBioscience, cat# 14–5751-82) and rabbit anti-P2RY12 (1:2000, AnaSpec, cat# AS-55043A) in 2% NGS and 2% NDS in PBS-T. Following several washes in PBS-T, sections were incubated with goat anti-rat IgG conjugated to Alexa-Fluor 488 (1:500, Invitrogen Life Technologies, cat# A11055) and donkey anti-rabbit IgG conjugated to Alexa-Fluor 647 (1:500, Jackson ImmunoResearch Inc., cat# 711–605-152) in 2% NDS and 2% NGS in PBS-T for 3 hr. at room temperature. Sections were washed several times in PBS-T, mounted on gelatin-coated slides, and coverslipped with Vectashield mounting medium with DAPI (Vector Laboratories, H-1200). Confocal images of the PVN were collected with an LSM-510 confocal microscope (Zeiss) with an Apochromat ×20 oil objective. 11 z-stack images of 450 × 450 μm were taken at 1-μm intervals. Image frame was digitized at 12-bit using a 1024 × 1024 pixel frame size. An ROI was manually drawn around the perimeter of the CRH-tdTomato+ neurons of the PVN, then MerTK volume and microglial volume (based on P2RY12+ immunoreactivity) were automatically calculated using Imaris’ 3D reconstruction.
MerTK inhibitor treatment of organotypic PVN slice cultures
P6–7 male CRH-Cre+/−:tdTomato+/− mouse pups were rapidly decapitated, brains were removed from the skull, and hypothalamic blocks were dissected and cut into 350-μm coronal sections on a McIlwain tissue chopper. To create organotypic PVN cultures, up to 6 slices were collected posterior to the anterior commissure. These slices were explanted onto Millicell cell culture inserts (pore size 0.4 μm, diameter 30 mm, Merck Millipore Inc., cat# PICMORG50). Membrane inserts were placed into a six-well plate with 1 mL of culture medium. Culture medium consisted of 52% modified Eagle’s medium [MEM; cat# 11700, Invitrogen], 25% Hanks Balanced Salt Solution [HBSS; cat# 24020, Invitrogen], 20% Heat Inactivated Horse Serum (added post-filtration) supplemented with 3 mM L-Glutamine (Gibco, cat# 25030081), 25 mM D-Glucose (Sigma, cat# G7528), 1.9 mM NaHCO3 (ThermoFisher, cat# 25080094), 12.5 mM HEPES (Gibco, cat# 15630080), 0.6 mM L-Ascorbic acid (Sigma-Aldrich, cat# A4403), 1 μg/mL Insulin (Sigma-Aldrich, cat# I0516) and 25 μg/mL containing penicillin-streptomycin (Gibco, cat# 15140122). Slices were cultured at 37°C in 5% CO2 enriched air for 48 hr., at which point the medium was refreshed. After an additional 48 hours, the medium was refreshed with medium without antibiotics. 6 days after initial culture, each membrane was washed twice with 2 mL of antibiotic-free, serum-free medium (97% MEM supplemented with 3 mM L-Glutamine, 10 mM D-Glucose,1.9 mM NaHCO3, 12.5 mM HEPES, 0.6 mM L-Ascorbic acid, 1 μg/mL Insulin) and treatment with 20 nM of a small-molecule MerTK inhibitor as previously described (UNC2025, Selleck Chemicals, cat# S7576) (Zhang et al., 2014a) or sterile culture-grade water as vehicle was commenced. UNC2025 has IC50 = 2.7 nmol/L and ~40-fold greater selectivity for MerTK over Axl and Tyro3 (McDaniel et al., 2018). UNC2025 also acts on FLT3, but this molecule is mostly expressed in hematopoietic stem cells with no expression in the neonatal mouse brain (Ito et al., 1993). Media containing either vehicle or MerTK inhibitor was refreshed 12 hr. later. After an additional 4 hr., cultures were fixed in 4% paraformaldehyde in 0.1 M phosphate-buffer (PB) on ice for 30 min. rinsed in PB, and cryoprotected in a 25% sucrose solution for 4–6 hr.
Organotypic PVN slice cultures IHC and imaging
Following cryoprotection, each slice confirmed to contain PVN was individually frozen on dry ice while still attached to the membrane. Slices were sectioned to 14-μm thickness using a Leica CM1900 cryostat and were immediately mounted onto gelatin-coated slides. Slides were stored at −20°C until further processing. Upon thawing, slides were rinsed in PBS-T, followed by treatment in 0.3% H2O2 in 0.01 M PBS for 20 min. Sections were blocked in PBS-T containing 5% NDS and then incubated overnight at 4°C with rabbit anti-PSD95 (1:1000, Invitrogen/ThermoFisher) and guinea pig anti-vGlut2 (1:10,000, Millipore Sigma) in PBS-T containing 2% NDS. Sections were washed in PBS-T and then incubated in donkey anti-guinea pig IgG conjugated to Alexa-Fluor 647 (1:500, Jackson ImmunoResearch) and donkey anti-rabbit pig IgG conjugated to Alexa-Fluor 488 (1:500, ThermoFisher) in PBS-T containing 2% NDS for 3 hr. at room temperature. Sections were washed with PBS-T and coverslipped with Vectashield mounting medium with DAPI (Vector Laboratories). Confocal images of the mpd PVN were collected with an LSM-510 confocal microscope (Zeiss) with an Apochromat ×63 oil objective. 11 z-stack images of 142.86 × 142.86 μm were taken at 1-μm intervals. Image frame was digitized at 12-bit using a 1024 × 1024-pixel frame size (Figure S3E). CRH neuronal volume was automatically calculated using Imaris’ 3D reconstruction function. Excitatory synapses onto CRH+ neurons were identified as colocalized puncta of vGlut2+PSD95 within the CRH-tdTomato+ volume using Imaris’ colocalization function (threshold = 1.0).
In vivo chemogenetic activation experiments
CX3CR1-Cre+::Gq-DREADD+ mice were crossed with Gq-DREADD+ mice to generate ~50% of pups expressing excitatory Gq-DREADDs specifically in their microglia (Figures 4A and S4), and the rest served as littermate controls. On P3, small, sustained-release CNO- or placebo-containing pellets (CNO: cat# X-999, 0.025 mg/pellet, 8-day release; Placebo: cat# C-111; Innovative Research of America, Sarasota, FL, USA) were inserted under the skin (s.c.) of male pups to obviate the stress of daily injections. Litters were culled to 6 pups maximum, with at least 1 female. Also on P3, the litters were randomized to CTL or ELA rearing for a week (Figures 4B and S1A). One cohort of mice were transcardially perfused on P10 (as described above) to measure the number of excitatory synapses onto PVN mpd neurons after CTL vs. ELA rearing. A separate cohort was transferred to standard cages at P10 and grown up until adulthood, behaviorally tested in the looming-shadow threat task, and >1 week later, transcardially perfused and adrenal glands collected and weighed as a measure of lifetime chronic stress. In another cohort, adult mice were subjected to acute, complex stresses (Chen et al., 2016; Hokenson et al., 2020) in order to assess their response to stress in adulthood.
IHC for excitatory synaptic markers and confocal imaging in P10 Gq-DREADD brains
Perfused brains of P10 CX3CR1-Cre+::Gq-DREADD+ mice were post-fixed, cryoprotected, and cryosectioned as described above. PVN-containing sections were subjected to IHC protocols similar to those described above. Briefly, after several washes with PBS containing 0.3% Triton X-100 (PBS-T, pH 7.4), sections were blocked with 5% normal donkey serum (Jackson ImmunoResearch) for 1 hr. to prevent non-specific binding. Sections were then incubated overnight at 4°C with rabbit anti-PSD95 antiserum (1:1,000, Invitrogen/ThermoFisher) and guinea pig anti-vGlut2 antiserum (1:12,000, Millipore) in PBS-T containing 2% normal donkey serum. The next morning, sections were rinsed in PBS-T (3 × 5 min.), and then incubated with donkey-anti-rabbit IgG-568 (1:1,000; cat# A10042, ThermoFisher) and donkey-anti-guinea pig IgG-647 (1:1,000; Jackson ImmunoResearch) for 3 hr. at room temperature. After washing (3 × 5 min), sections were mounted onto gelatin-coated slides and coverslipped with Vectashield containing DAPI (Vector Laboratories). Confocal images of the mpd PVN were collected using an LSM-780 confocal microscope (Zeiss) with an Apochromat ×63 oil objective. 11 z-stack images of 142.86 × 142.86 μm were taken at 1-μm intervals. Image frame was digitized at 12-bit using a 1024 × 1024-pixel frame size. An ROI was manually drawn around the perimeter of the densely packed DAPI+ nuclei of the mpd PVN. Excitatory synapses in this region were identified as colocalized puncta of vGlut2+PSD95 using Imaris’ colocalization function.
Acute, complex stress paradigm in adulthood
The acute, complex stress consists of concurrent restraint, bright lights, loud noise, physical jostling and awareness of peer discomfort [as described in (Hokenson et al., 2020)], and has been previously shown to elicit a robust stress response that are greater than a single stressor alone (Maras et al., 2014). Briefly, adult male mice were put in a restrainer made from a 50-mL plastic tube (Corning, Corning, NY, USA), placed next to unfamiliar conspecifics (social stress) on a laboratory shaker, and jostled in a brightly lit room with loud rap music playing (dB level = 95) for 1 h. Small blood samples (~100 μL) were collected from the facial vein of each mouse at baseline, 30 min and 60 min after stress initiation, and serum was collected after clotting for ~30 min at room temperature and centrifugation at 1,200×g for 15 min. Serum was stored at −20°C until later assayed for ACTH and CORT using commercially available enzyme-linked immunosorbent assay kits according to the manufacturers’ instructions (ACTH kit: cat# EK-001–21, Phoenix Pharmaceuticals, Burlingame, CA; CORT kit: cat# 501320, Cayman Chemical, Ann Arbor, MI). Due to the rapid release of ACTH relative to CORT, commencing upon the moment we disturbed the cage, the baseline ACTH samples were somewhat elevated in all groups (and not significantly different from each other), and thus were not included in Figure 4.
Looming-shadow threat task
The looming-shadow task has been shown to be dependent on the CRH+ neurons in the PVN (Daviu et al., 2020). The task was performed in the dark, active phase as described previously (Daviu et al., 2020). Briefly, adult male mice were initially habituated to an arena containing a shelter for 15 min. Following habituation, a stimulus was presented to the mouse from above, consisting of an expanding disk (holding for 3 s small, growing for 2 s, then holding for 3 s fully expanded), which was repeated 5 times with at least 1 minute between stimuli. The mouse’s response to the looming stimulus was scored as no response, freezing, or escape, both live and on recording by an independent experimenter, and the latency to respond during the 8-s stimulus was measured on the recording (the latency was recorded as 8 s if no response was observed during the stimulus).
QUANTIFICATION AND STATISTICAL ANALYSIS
Differences between CTL and ELA groups were assessed using unpaired t-tests, with Welch’s correction for unequal variance as necessary. One-sample t-tests were used for comparing CNO to Vehicle (i.e., 100%) in the ex vivo chemogenetics experiment. 2-way ANOVA was used to analyze the effects of ELA and MerTK inhibition separately and in interaction (ELA X Drug), followed by Tukey’s post hoc tests. Differences after ELA and the in vivo chemogenetics intervention were analyzed by one-way ANOVA or Welch’s ANOVA (corrected for unequal variance) as necessary, followed by the appropriate post hoc tests (specified for each test in Results section). Because there were no significant differences detected between the CTL/Placebo/Cre−, CTL/Placebo/Cre+, and CTL/CNO/Cre− groups (and the equivalent for ELA), these 3 different varieties of control groups were combined into one CTL group and one ELA group for further analysis. Thus, there were no off-target effects of CNO detected (i.e., in the absence of Gq-DREADD expression). Significance levels were set at 0.05, and data are presented as mean ± SEMs. Grubbs’ test was used to remove statistical outliers from the data. Statistical analyses were performed using GraphPad Prism 9.0 software (GraphPad, San Diego, CA, USA). All experiments were assessed blindly without prior knowledge of the experimental group.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Guinea pig anti-vGlut2 antiserum | Millipore | Cat# AB2251 -I; RRID: AB_2665454 |
| Rabbit anti-PSD95 antiserum | Invitrogen/ThermoFisher | Cat# 51–6900; RRID: AB_2533914 |
| Rabbit anti-Ibal antiserum | Wako Chemical | Cat# 019–19741; RRID: AB_839504 |
| Rat anti-MerTK antiserum | eBioscience | Cat# 14–5751-82; RRID: AB_2688282 |
| Rabbit anti-P2RY12 antiserum | AnaSpec | Cat# 55043A; RRID: AB_2298886 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Clozapine-N-oxide (CNO) | National Institute of Mental Health | CAS# 34233–69-7, MH# C-929 |
| MerTK small-molecule inhibitor, UNC2025 | Selleck Chemicals | Cat# S7576 |
| CNO-containing, sustained-release pellets (s.c.) | Innovative Research of America | Cat# X-999 |
| Placebo-containing, sustained-release pellets (s.c.) | Innovative Research of America | Cat# C-111 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ACTH ELISA kit | Phoenix Pharmaceuticals | Cat# EK-001 –21 |
| CORT ELISA kit | Cayman Chemicals | Cat# 501320 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: CRH-IRES-Cre+/+ | Jackson Laboratory | JAX: 012704 |
| Mouse: tdTomato+/+ Ai14 | Jackson Laboratory | JAX: 007914 |
| Mouse: CX3CR1-GFP+/+ | Jackson Laboratory | JAX: 005582 |
| Mouse: CX3CR1-BAC-Cre+ [Tg(Cx3cr1-cre)MW126Gsat] | GENSAT BAC Transgenic Project (Rockefeller University) | MMRRC: 036395-UCD; MGI: 5311737 |
| Mouse: Gq-DREADD+/− (stop-floxed) | Jackson Laboratory | JAX:026220 |
| Mouse: Mertkfl/fl | Dr. Carla Rothlin (Yale University); Fourgeaud et al., 2016 | N/A |
| Mouse: C57BL/6J (wild-type) | Jackson Laboratory | JAX: 000664 |
|
| ||
| Software and algorithms | ||
|
| ||
| FIJI | https://imagej.net/software/fiji/; Schindelin et al., 2012 | N/A |
| Imaris 3D reconstruction software | Bitplane | N/A |
| Strathclyde Electrophysiology Software (Electrophysiology Data Recorder [WinEDR] and Whole-Cell analysis Program [WinWCP]) | Courtesy of Dr J. Dempster, University of Strathclyde | N/A |
| Slidebook microscopy software | 3i (Intelligent Imaging Innovations) | N/A |
| Python-based microglial process dynamics analysis algorithm | This paper, available: https://github.com/jaclynrbeck/BaramLabMicrogliaTracking | N/A |
| ProMoIJ plug-in for FIJI | Paris et al., 2017 | N/A |
| MultipleKymograph plug-in for FIJI | http://www.embl.de/eamnet/html/body_kymograph.html | N/A |
|
| ||
| Other | ||
|
| ||
| Fine-gauge plastic-coated aluminum mesh platform (mesh dimensions 0.4 X 0.9 cm) | McNichols Co. | Cat# 57398 |
| Model | Gibson & lanni 3D optical model |
|---|---|
| Refractive index immersion | 1 |
| Refractive index sample | 1 |
| Working distance (ti) | 2,000 um |
| Particle position Z | 2,000 nm |
| Accuracy computation | Good |
| Wavelength | 920 nm |
| NA | 1.1 |
| Pixelsize XY | 244.140625 nm |
| Z-step | 4,000 nm |
| Size XYZ | 256/256/3 |
| Display | Linear/32-bits/Grays |
Highlights.
Early-life stress (ELA) augments excitatory synapses on hypothalamic CRH cells
During ELA, the adjacent microglia poorly survey and prune synapses on CRH neurons
Mechanisms involve MerTK and yield aberrant hormonal and behavioral stress response
Chemogenetic microglial activation throughout ELA normalizes adult stress responses
ACKNOWLEDGMENTS
This work was supported by NIH grants K99 MH120327 (to J.L.B.), P50 MH096889 (to T.Z.B.), R01 MH73136 (to T.Z.B.), R01 NS14609 (to M.D.C.), and R01 AI121945 (to M.D.C.); the Brain & Behavior Research Foundation NARSAD Young Investigator Grant (to J.L.B.); and the Hewitt Foundation for Biomedical Research (to J.L.B. and to T.Z.B.). We thank the Optical Biology Core supported by CA-62203 and GM-076516 at University of California, Irvine. We thank Dr. Ian Parker for access to the custom-built 2-photon microscope used for the live imaging. We thank Dr. Staci Bilbo (Duke University) and Dr. Carla Rothlin (Yale University) for the CX3CR1-BAC-Cre and the MerTK-fl/fl mice, respectively. We appreciate the excellent technical assistance of Emily Majorkiewicz, Andrew Quan Dong, Xinwen Li, Yanan Wu, Catherine Chiou, Keshav Suresh, Kathleen Guangying Zhou, Pouria Vadipour, Graciella Angeles, Johnathan Ho, Erin Card, and Jennifer Daglian, and we also thank Dr. Yoav Noam for his insight and support with kymograph analyses. The graphical abstract was created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110600.
REFERENCES
- Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Layé S, Leyrolle Q, Gómez-Nicola D, Domercq M, Pérez-Samartín A, Sánchez-Zafra V, et al. (2016). Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 14, 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armario A (2006). The Hypothalamic-Pituitary-Adrenal Axis: What can it Tell us About Stressors? CNS & Neurological Disorders - Drug Targets 5, 485–501. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Gilles EE, Eghbal-Ahmadi M, Bar-El Y, and Baram TZ (2001). Altered regulation of gene and protein expression of hypothalamic-pituitary-adrenal Axis components in an immature rat model of chronic stress. J. Neuroendocrinol. 13, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh Y-HE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, et al. (2018). Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 21, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO, et al. (2020). Negative feedback control of neuronal activity by microglia. Nature 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, et al. (2010). Early life programming and neurodevelopmental disorders. Biol. Psychiat. 68, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick J, and Nelson CA (2016). Early adverse experiences and the developing brain. Neuropsychopharmacol 41, 177–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnie MT, Kooiker CL, Short AK, Bolton JL, Chen Y, and Baram TZ (2020). Plasticity of the reward circuitry after early-life adversity: mechanisms and significance. Biol. Psychiat. 87, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Molet J, Regev L, Chen Y, Rismanchi N, Haddad E, Yang DZ, Obenaus A, and Baram TZ (2018). Anhedonia following early-life adversity involves aberrant interaction of reward and anxiety circuits and is reversed by partial silencing of amygdala corticotropin-releasing hormone gene. Biol. Psychiat. 83, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Short AK, Simeone KA, Daglian J, and Baram TZ (2019). Programming of stress-sensitive neurons and circuits by early-life experiences. Front. Behav. Neurosci. 13, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Schulmann A, Garcia-Curran MM, Regev L, Chen Y, Kamei N, Shao M, Singh-Taylor A, Jiang S, Noam Y, et al. (2020). Unexpected transcriptional programs contribute to hippocampal memory deficits and neuronal stunting after early-life adversity. Cell Rep. 33, 108511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradski G (2000). The OpenCV library. Dr. Dobb’s J. Softw. Tools Prof. Program 25, 120–123. [Google Scholar]
- Brunson KL, Kramár E, Lin B, Chen Y, Colgin LL, Yanagihara TK, Lynch G, and Baram TZ (2005). Mechanisms of late-onset cognitive decline after early-life stress. J. Neurosci. 25, 9328–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Baram TZ (2016). Toward understanding how early-life stress reprograms cognitive and emotional brain networks. Neuropsychopharmacology 41, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Molet J, Gunn BG, Ressler K, and Baram TZ (2015). Diversity of reporter expression patterns in transgenic mouse lines targeting corticotropin-releasing hormone-expressing neurons. Endocrinology 156, 4769–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Molet J, Lauterborn JC, Trieu BH, Bolton JL, Patterson KP, Gall CM, Lynch G, and Baram TZ (2016). Converging, synergistic actions of multiple stress hormones mediate enduring memory impairments after acute simultaneous stresses. J. Neurosci. 36, 11295–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czapiga M, and Colton CA (1999). Function of microglia in organotypic slice cultures. J. Neurosci. Res. 56, 644–651. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Eyo U, Fuller L, Hass J, and Kurpius D (2013). Imaging microglia in brain slices and slice cultures. Cold Spring Harb. Protoc. 2013, 1142–1148. [DOI] [PubMed] [Google Scholar]
- Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, and Wong WT (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, and McEwen BS (2012). Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol. Behav. 106, 29–39. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, and Gan W (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Daviu N, Füzesi T, Rosenegger DG, Rasiah NP, Sterley T-L, Peringod G, and Bains JS (2020). Paraventricular nucleus CRH neurons encode stress controllability and regulate defensive behavior selection. Nat. Neurosci. 23, 398–410. [DOI] [PubMed] [Google Scholar]
- Delbridge ARD, Huh D, Brickelmaier M, Burns JC, Roberts C, Challa R, Raymond N, Cullen P, Carlile TM, Ennis KA, et al. (2020). Organotypic brain slice culture microglia exhibit molecular similarity to acutely-isolated adult microglia and provide a platform to study neuroinflammation. Front. Cell. Neurosci. 14, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Aparicio I, Paris I, Sierra-Torre V, Plaza-Zabala A, Rodríguez-Iglesias N, Márquez-Ropero M, Beccari S, Huguet P, Abiega O, Alberdi E, et al. (2020). Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J.Neurosci. 40, 1453–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas AA, Borst K, and Prinz M (2021). Current tools to interrogate microglial biology. Neuron 109, 2805–2819. [DOI] [PubMed] [Google Scholar]
- Emoto K, Hensch TK, and Yuzaki M (2021). “Scrap & build” functional circuits: molecular and cellular basis of neural remodeling. Neurosci. Res. 167, 1–2. [DOI] [PubMed] [Google Scholar]
- Espinosa JS, and Stryker M (2012). Development and plasticity of the primary visual cortex. Neuron 75, 230–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, and Wu L-J (2014). Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. 34, 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Chen J, Ye J, Yan H, and Cai Y (2014). Proteinase-activated receptor 2 modulates corticotropin releasing hormone-induced brain-derived neurotrophic factor release from microglial cells. Cell Biol. Int. 38, 92–96. [DOI] [PubMed] [Google Scholar]
- Farah MJ (2018). Socioeconomic status and the brain: prospects for neuroscience-informed policy. Nat. Rev. Neurosci. 19, 428–438. [DOI] [PubMed] [Google Scholar]
- Favuzzi E, Huang S, Saldi GA, Binan L, Ibrahim LA, Fernández-Otero M, Cao Y, Zeine A, Sefah A, Zheng K, et al. (2021). GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 184, 4048–4063.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Través PG, Tufail Y, Leal-bailey H, Lew ED, Burrola PG, Nimmerjahn A, Lemke G, Callaway P, Zagórska A, et al. (2016). TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee DG (2021). Early adversity and development: parsing heterogeneity and identifying pathways of risk and resilience. Am. J. Psychiat. 178, 998–1013. [DOI] [PubMed] [Google Scholar]
- Gilles EE, Schultz L, and Baram TZ (1996). Abnormal corticosterone regulation in an immature rat model of continuous chronic stress. Pediatr. Neurol. 15, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Wang X, Strand KA, Baratta M.v., Zhang Y, Galer EL, Yin H, Maier SF, and Watkins LR (2018). DREADDed microglia in pain: Implications for spinal inflammatory signaling in male rats. Experimental Neurology 304, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn BG, Cunningham L, Cooper MA, Corteen NL, Seifi M, Swinny JD, Lambert JJ, and Belelli D (2013). Dysfunctional astrocytic and synaptic regulation of hypothalamic glutamatergic transmission in a mouse model of early-life adversity: relevance to neurosteroids and programming of the stress response. J. Neurosci. 33, 19534–19554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JM, and Austad SN (2015). Fecal glucocorticoids: a noninvasive method of measuring adrenal activity in wild and captive rodents. Physiological and Biochemical Zoology : PBZ 73, 12–22. [DOI] [PubMed] [Google Scholar]
- Heim C, and Binder EB (2012). Current research trends in early life stress and depression: review of human studies on sensitive periods, gene–environment interactions, and epigenetics. Exp. Neurol. 233, 102–111. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, and Kash SF (1998). Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 282, 1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokenson R, Oijala M, Short A, Bolton J, Chen Y, Molet J, Maras P, Baram T, and Lur G (2020). Multiple simultaneous acute stresses in mice: single or repeated induction. Bio. Protoc. 10, e3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko M, Arnoux I, Avignone E, Yamamoto N, and Audinat E (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh CYL, Abdelaal K, Salinas KJ, Gu D, Zeitoun J, Figueroa Velez DX, Peach JP, Fowlkes CC, and Gandhi SP (2020). Long-term monocular deprivation during juvenile critical period disrupts binocular integration in mouse visual thalamus. J. Neurosci. 40, 585–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR (2009). Translating scientific opportunity into public Health impact. Arch. Gen. Psychiat. 66, 128. [DOI] [PubMed] [Google Scholar]
- Ito A, Hirota S, Kitamura Y, and Nomura S (1993). Developmental expression offlt3 mRNA in the mouse brain. J. Mol. Neurosci. 4, 235–243. [DOI] [PubMed] [Google Scholar]
- Ivy AS, Rex CS, Chen Y, Dubé C, Maras PM, Grigoriadis DE, Gall CM, Lynch G, and Baram TZ (2010). Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J. Neurosci. 30, 13005–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy ASS, Brunson KLL, Sandman C, and Baram TZZ (2008). Dysfunctional nurturing behavior in rat dams with limited access to nesting material: a clinically relevant model for early-life stress. Neuroscience 154, 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, and Binder EB (2015). Epigenetics of stress-related psychiatric disorders and gene 3 environment interactions. Neuron 86, 1343–1357. [DOI] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, and Heppner FL (2013). Functional impairment of microglia coincides with beta-amyloid deposition in mice with alzheimer-like pathology. PLoS One 8, e60921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, Kim J, Noh S, Lee H, Lee SY, Mun JY, Park H, and Chung W-S (2020). Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 570, 612–617. [DOI] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM, Haliyur R, and McCarthy MM (2013). Microglia are essential to masculinization of brain and behavior. J. Neurosci. 33, 2761–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis SC, Bentzley BS, Molet J, Bolton JL, Perrone CR, Baram TZ, and Mahler SV (2019). On the early life origins of vulnerability to opioid addiction. Mol. Psychiatr. 26, 4409–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang Y, Guo R, Bai Y, and Yu Z (2007). Glucocorticoids impair microglia ability to induce T cell proliferation and Th1 polarization. Immunol. Lett. 109, 129–137. [DOI] [PubMed] [Google Scholar]
- Luby JL, Barch D, Whalen D, Tillman R, and Belden A (2017). Association between early life adversity and risk for poor emotional and physical Health in adolescence. JAMA Pediatr. 171, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linger RMA, Keating AK, Earp HS, and Graham DK (2008). TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. Advances in Cancer Research 100, 35–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby JL, Baram TZ, Rogers CE, and Barch DM (2020). Neurodevelopmental optimization after early-life adversity: cross-species studies to elucidate sensitive periods and brain mechanisms to inform early intervention. Trends Neurosci. 43, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras PM, Molet J, Chen Y, Rice C, Ji SG, Solodkin A, and Baram TZ (2014). Preferential loss of dorsal-hippocampus synapses underlies memory impairments provoked by short, multimodal stress. Mol. Psychiatr. 19, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel NK, Cummings CT, Iida M, Hülse J, Pearson HE, Vasileiadi E, Parker RE, Orbuch RA, Ondracek OJ, Welke NB, et al. (2018). MERTK mediates intrinsic and adaptive resistance to AXL-targeting agents. Mol. Cancer Ther. 17, 2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin KA, Weissman D, and Bitrán D (2019). Childhood adversity and neural development: a systematic review. Annu. Rev. Dev. Psychol. 1, 277–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, Koizumi S, Moorhouse AJ, Yoshimura Y, and Nabekura J (2016). Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 7, 12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molet J, Maras PM, Avishai-Eliner S, and Baram TZ (2014). Naturalistic rodent models of chronic early-life stress. Dev. Psychobiol. 56, 1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molet J, Heins K, Zhuo X, Mei YT, Regev L, Baram TZ, and Stern H (2016). Fragmentation and high entropy of neonatal experience predict adolescent emotional outcome. Transl. Psychiat 6, e702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondo E, Becker SC, Kautzman AG, Schifferer M, Baer CE, Chen J, Huang EJ, Simons M, and Schafer DP (2020). A developmental analysis of juxtavascular microglia dynamics and interactions with the vasculature. J. Neurosci. 40, 6503–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NR, Agolley D, Zieba M, and Bye N (2005). Glucocorticoid regulation of glial responses during hippocampal neurodegeneration and regeneration. Brain Res. Rev. 48, 287–301. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1319. [DOI] [PubMed] [Google Scholar]
- Noam Y, Zha Q, Phan L, Wu RL, Chetkovich DM, Wadman WJ, and Baram TZ (2010). Trafficking and surface expression of hyperpolarization-activated cyclic nucleotide-gated channels in hippocampal neurons. J. Biol. Chem. 285, 14724–14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othy S, Jairaman A, Dynes JL, Dong TX, Tune C, Yeromin A, Zavala A, Akunwafo C, Chen F, Parker I, et al. (2020). Regulatory T cells suppress Th17 cell Ca2+ signaling in the spinal cord during murine autoimmune neuroinflammation. Proc. Natl. Acad. Sci. U S A 117, 20088–20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Paris I, Savage JC, Escobar L, Abiega O, Gagnon S, Hui C-W, Tremblay M-È, Sierra A, and Valero J (2017). ProMoIJ: a new tool for automatic three-dimensional analysis of microglial process motility. Glia 66, 828–845. [DOI] [PubMed] [Google Scholar]
- Park J, Choi Y, Jung E, Lee S, Sohn J, and Chung W (2021). Microglial MERTK eliminates phosphatidylserine-displaying inhibitory post-synapses. EMBO J. 40, e107121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2001). Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- Picard K, Bisht K, Poggini S, Garofalo S, Golia MT, Basilico B, Abdallah F, Ciano Albanese N, Amrein I, Vernoux N, et al. (2021). Microglial-glucocorticoid receptor depletion alters the response of hippocampal microglia and neurons in a chronic unpredictable mild stress paradigm in female mice. Brain Behav. Immun. 97, 423–439. [DOI] [PubMed] [Google Scholar]
- Pozner A, Xu B, Palumbos S, Gee JM, Tvrdik P, and Capecchi MR (2015). Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Frontiers in Molecular Neuroscience 8, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CJ, Sandman CA, Lenjavi MR, and Baram TZ (2008). A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology 149, 4892–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, and Stevens B (2013). Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 23, 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saika F, Matsuzaki S, Kishioka S, and Kiguchi N (2021). Chemogenetic Activation of CX3CR1-Expressing Spinal Microglia Using Gq-DREADD Elicits Mechanical Allodynia in Male Mice. Cells 10, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer D, Lehrman E, Kautzman A, Koyama R, Mardinly A, Yamasaki R, Ransohoff R, Greenberg M, Barres B, and Stevens B (2012). Microglia sculpt postnatal neuronal circuits in an activivty and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Sholar PW, and Bilbo SD (2012). Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 120, 948–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, et al. (2020). Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 39, e105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short AK, and Baram TZ (2019). Early-life adversity and neurological disease: age-old questions and novel answers. Nat. Rev. Neurol. 15, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short AK, Thai CW, Chen Y, Kamei N, Pham AL, Birnie MT, Bolton JL, Mortazavi A, and Baram TZ (2021). Single-cell transcriptional changes in hypothalamic corticotropin-releasing factor expressing neurons after early-life adversity inform enduring alterations in vulnerabilities to stress. Biol. Psychiat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Taylor A, Molet J, Jiang S, Korosi A, Bolton JL, Noam Y, Simeone K, Cope J, Chen Y, Mortazavi A, et al. (2018). NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol. Psychiatr. 23, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe GO, Lowery RL, Tremblay MÈ, and Kelly EA (2016). Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 7, 10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL, Shaw TE, Dykhuizen E, Lessov NS, Hill JK, Wurst W, and Stenzel-Poore MP (2003). Reduced cerebral injury in CRH-R1 deficient mice after focal ischemia: a potential link to microglia and atrocytes that express CRH-R1. J. Cerebr. Blood Flow Metabol. 23, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Stout SC, and Nemeroff CB (1994). Stress and psychiatric disorders. Semin. Neurosci. 6, 271–280. [Google Scholar]
- Takesian AE, Bogart LJ, Lichtman JW, and Hensch TK (2018). Inhibitory circuit gating of auditory critical-period plasticity. Nat. Neurosci. 21, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley UA, Bassett DS, and Mackey AP (2021). Environmental influences on the pace of brain development. Nat. Rev. Neurosci. 22, 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MÈ, Lowery RL, and Majewska AK (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8, e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Figueiredo HF, Ostrander MM, Choi DC, Engeland WC, and Herman JP (2006). Chronic stress induces adrenal hyperplasia and hypertrophy in a subregion-specific manner. Am. J. Physiol. Endocrinol. Metab. 291, E965–E973. [DOI] [PubMed] [Google Scholar]
- Vainchtein ID, and Molofsky AV (2020). Astrocytes and microglia: in sickness and in Health. Trends Neurosci. 43, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umpierre AD, Bystrom LL, Ying Y, Liu YU, Worrell G, and Wu L-J (2020). Microglial calcium signaling is attuned to neuronal activity in awake mice. ELife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C-D, Bath KG, Joels M, Korosi A, Larauche M, Lucassen PJ, Morris MJ, Raineki C, Roth TL, Sullivan RM, et al. (2017). Chronic early life stress induced by limited bedding and nesting (LBN) material in rodents: critical considerations of methodology, outcomes and translational potential. Stress 20, 421–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Herrewegen Y, Sanderson TM, Sahu S, de Bundel D, Bortolotto ZA, and Smolders I (2021). Side-by-side comparison of the effects of Gq- and Gi-DREADD-mediated astrocyte modulation on intracellular calcium dynamics and synaptic plasticity in the hippocampal CA1. Molecular Brain 14, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, and Yu T (2014). Scikit-image: image processing in Python. PeerJ 2, e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ji P, Riopelle RJ, and Dow KE (2002). Functional expression of corticotropin-releasing hormone (CRH) receptor 1 in cultured rat microglia. J. Neurochem. 80, 287–294. [DOI] [PubMed] [Google Scholar]
- Watson S, and Mackin P (2006). HPA axis function in mood disorders. Psychiatry 5, 166–170. [Google Scholar]
- Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A, et al. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 9, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hahm E, Kim Y, Kang J, Lee W, Han I, Myung P, Kang H, Park H, and Cho D (2005). Regulation of IL-18 expression by CRH in mouse microglial cells. Immunol. Lett. 98, 291–296. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. [DOI] [PubMed] [Google Scholar]
- Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, Cummings CT, Lee M, Glaros TG, Newton DL, et al. (2014a). UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 57, 7031–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, Sean O, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014b). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ma Z, Zou W, Guo H, Liu M, Ma Y, and Zhang L (2019). The appropriate marker for astrocytes: comparing the distribution and expression of three astrocytic markers in different mouse cerebral regions. Biomed. Res. Int. 2019, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
The code generated during this study is openly available on GitHub at https://github.com/jaclynrbeck/BaramLabMicrogliaTracking.
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.