

Abstract
Introduction
Amyloid beta (Aβ), tau, and neurodegeneration jointly with the Alzheimer's disease (AD) risk factors affect the severity of clinical symptoms and disease progression.
Methods
Within 248 Aβ‐positive elderly with and without cognitive impairment and dementia, partial least squares structural equation pathway modeling was used to assess the direct and indirect effects of imaging biomarkers (global Aβ‐positron emission tomography [PET] uptake, regional tau‐PET uptake, and regional magnetic resonance imaging–based atrophy) and risk‐factors (age, sex, education, apolipoprotein E [APOE], and white‐matter lesions) on cross‐sectional cognitive impairment and longitudinal cognitive decline.
Results
Sixteen percent of variance in cross‐sectional cognitive impairment was accounted for by Aβ, 46% to 47% by tau, and 25% to 29% by atrophy, although 53% to 58% of total variance in cognitive impairment was explained by incorporating mediated and direct effects of AD risk factors. The Aβ–tau–atrophy pathway accounted for 50% to 56% of variance in longitudinal cognitive decline while Aβ, tau, and atrophy independently explained 16%, 46% to 47%, and 25% to 29% of the variance, respectively.
Discussion
These findings emphasize that treatments that remove Aβ and completely stop downstream effects on tau and neurodegeneration would only be partially effective in slowing of cognitive decline or reversing cognitive impairment.
Keywords: Alzheimer's Disease Assessment Scale–Cognitive Subscale, amyloid beta, atrophy, cognition, magnetic resonance imaging, positron emission tomography, Preclinical Alzheimer Cognitive Composite, tau, white matter lesions
1. BACKGROUND
Although deposition of fibrillar amyloid beta (Aβ) in the brain was identified as one of the earliest pathological changes occurring at least a decade before the clinical diagnosis of Alzheimer's disease (AD), treatments targeting Aβ have been, thus far, largely ineffective in slowing cognitive decline, with modest beneficial clinical effects being reported recently. 1 , 2 There have been many explanations suggested for AD therapeutic trial failure, especially that the treatments were administered too late in the disease process, aimed at the wrong targets, or that the treatments failed to properly engage with the targets. It is also likely that not all cognitive decline, even if diagnosed with AD biomarkers, is due to AD pathology, defined as Aβ and tau leading to neurodegeneration. That being the case, the extent to which AD pathology accounts for clinical symptoms and disease progression in living individuals in the AD continuum is indeed of great interest for the development and targeting of effective therapies.
There is strong evidence from prior clinicopathology studies suggesting that the age‐related neuropathologies account for 40% to 50% of the variation in late life cognitive decline in which the pathological markers of AD accounted for 30% to 36% of the variation. 3 , 4 Furthermore, the proportion of the observed cognitive loss accounted for by AD pathology at the individual level ranges widely from 22% to 100%. 4 These findings highlight the complexity of cognitive aging and have important implications for the ongoing effort to develop effective therapeutics, yet only partially elucidate the precise pathological sequence and its impact on cognitive impairment and decline in living humans.
With advances in AD pathology biomarkers for in vivo assessments we now have compelling evidence that AD in its continuum is a complex disease in nature; that Aβ, tau, and neurodegeneration impact cognition in concert as dynamic neuropathological factors; and that different pathology positivity stages throughout the disease continuum might have distinct mechanisms affecting the clinical disease progression. 5 Leveraging multimodal neuroimaging, our objective was to determine the extent to which cognitive impairment and decline is accounted for by the level of Aβ and tau pathologies and neurodegeneration detected by imaging markers, in particular florbetapir 6 or florbetaben 7 positron emission tomography (PET) for global Aβ burden, flortaucipir PET 8 for the burden and the anatomical distribution of tau, and structural magnetic resonance imaging (MRI) for the anatomical distribution of neurodegeneration. The study cohort consists of older individuals with biomarker evidence of Aβ‐positivity with and without clinical symptoms from a multicenter observational study, the Alzheimer's Disease Neuroimaging Initiative (ADNI). Combined AD biomarker (Aβ, tau, and neurodegeneration) pathways mediating the effects of AD risk factors (age, sex, education, apolipoprotein E [APOE], and vascular brain lesion burden) on Preclinical Alzheimer Cognitive Composite (PACC) and Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS‐Cog) were assessed to determine how much of the variance of baseline cognition and longitudinal cognitive decline was accounted for by AD imaging biomarkers.
2. METHODS
2.1. Study design
Data were obtained from the ADNI database (adni.loni.usc.edu). The National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies, and nonprofit organizations launched ADNI in 2004 as a public–private partnership. ADNI is a longitudinal multicenter natural history study designed to characterize clinical, neuropsychological, MRI and PET, genetic, and biochemical biomarkers for early detection and tracking of AD. 9 The principal investigator of ADNI is Michael Weiner, MD, VA Medical Center and University of California, San Francisco. For current information on ADNI, see www.adni‐info.org.
2.2. Study participants
The main study cohort included ADNI participants who (1) had PET evidence for Aβ‐positivity; (2) underwent multimodality neuroimaging for flortaucipir‐PET, florbetapir‐ or florbetaben‐PET, structural MRI, and fluid‐attenuated inversion recovery (FLAIR) MRI, all performed no longer than 6 months apart; and (3) had clinical and cognitive assessments cross‐sectionally within 6 months of neuroimaging and longitudinally up to 4 years after neuroimaging assessments.
2.3. Cognitive outcome measures
A modified version of PACC (mPACC) 10 and the ADAS‐Cog 13‐item score were used as the cognitive outcome measures as these are tools typically used in AD clinical trials. 11 ADAS‐Cog and mPACC scores were corrected for normal confounding effects of age, sex, and education based on scores of ADNI individuals who were cognitively unimpaired (CU), APOE ε4‐noncarriers, and Aβ‐negative based on Aβ‐PET.
Rates of cognitive decline were estimated from the longitudinal assessments within 4 years of flortaucipir‐PET (cf. supporting information), prospectively, allowing for linear approximation of decline rates, known to be nonlinear in the disease spectrum. 12
2.4. Image processing
A threshold of global cortical Aβ load ≥1.11 for florbetapir 13 and ≥ 1.08 for florbetaben was used to determine Aβ‐positivity. Global cortical Aβ load in Centiloid units was estimated using the ADNI pipeline (http://adni.loni.usc.edu/wp‐content/themes/freshnews‐dev‐v2/documents/pet/ADNI%20Centiloids%20Final.pdf). Flortaucipir‐PET were quantitatively evaluated for estimation of a tau standardized uptake value ratio (SUVR) for 31 bilateral cerebral regions of interest (ROIs) according to published methods. 14 Atrophy within each ROI was estimated using the DiReCT method. 15 Total volume of cerebral white matter hyperintensities (WMH) detected in FLAIR‐MRI normalized to total intracranial volume was estimated as a measure of vascular white matter lesion (WML) burden for each participant. 16 For detailed information on image processing methods, see the supporting information.
2.5. Statistical analysis
We assessed the direct and indirect effects of observed and latent variables of AD imaging biomarkers (global Aβ burden, regional tau burden from all ROIs, and regional atrophy from all ROIs), and AD risk factors (age, sex, years of education, APOE genotype, and WML burden) on cognitive outcome measures using partial least squares structural equation modeling (PLS‐SEM), 17 testing a priori hypothesized biomarker pathways illustrated in Figure 1 (cf. supporting information).
FIGURE 1.
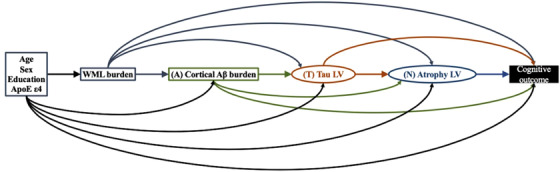
A priori hypothesized biomarker pathways by which amyloid beta (Aβ)–tau–atrophy biomarkers might mediate the association of Alzheimer's disease (AD) risk factors and cognition. Rectangles represent manifest variables and ellipses represent latent variables. Each single‐headed arrow denotes a hypothesized unidirectional effect of one variable on another. For graphical simplicity, age, sex, education, and apolipoprotein E (APOE) ε4 is grouped although each AD risk factor is separately hypothesized to have unidirectional effect on white matter lesion (WML), cortical Aβ burden, tau latent variable (LV), atrophy LV, and cognitive outcome. Our analysis is premised on a conceptual Aβ–tau–atrophy pathologic pathway thought to mediate the association of AD risk factors and cognition. A priori, age, sex, years of education, and presence of APOE ε4 allele were specified to have direct effects on global Aβ, regional tau, regional atrophy, and WML, in addition to their direct effects on cognition. WML was hypothesized to have a direct effect on global Aβ, regional tau, and regional atrophy, in addition to its direct effect on cognition. Global Aβ was hypothesized to have a direct effect on regional tau and regional atrophy, in addition to its direct effect on cognition. In turn, the regional tau was hypothesized to have direct effect on regional atrophy, together with the direct effects of regional tau and regional atrophy on cognition. We note that the regional specificity of Aβ pathology was examined by including regional Aβ burden from all 31 ROIs instead of limiting the Aβ construct to the global cortical Aβ burden in the partial least squares structural equation modeling (PLS‐SEM). The estimated latent construct for the regional Aβ burden in the final PLS‐SEM involved all but bilateral entorhinal, amygdala, and hippocampus regions, suggesting the effect of Aβ being distributed across the cortex rather than localized in specific cortical regions in this cohort of all Aβ‐positive individuals. Therefore, Aβ construct of all PLS‐SEMs in this study was limited to global cortical Aβ burden.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts. While the extent to which Alzheimer's disease (AD) imaging biomarkers account for the clinical symptoms and progression in living individuals is not widely studied, there have been several clinicopathology publications describing the degree to which late life cognitive decline is driven by age‐related neuropathologies.
Interpretation: Only a limited percentage of the variance in cognitive decline can be explained by the currently available imaging biomarkers (amyloid beta positron emission tomography (PET), tau‐PET, structural magnetic resonance imaging (MRI), and fluid‐attenuated inversion recovery MRI). This is consistent with the previous clinicopathology studies reporting that ≤ 50% of variance in cognitive decline before death can be accounted for by the indices of AD, cerebrovascular disease, and Lewy body pathologies, even after considering hippocampal sclerosis, TDP‐43, and atherosclerosis.
Future directions: Our findings support the strategy for biomarkers and disease‐modifying therapies that target non‐AD pathologies that are highly comorbid in AD for effective slowing of cognitive decline and ideally reversing cognitive impairment.
3. RESULTS
Based on the clinical assessment closest in time to neuroimaging visit, the study cohort included 120 CU, 83 with mild cognitive impairment (MCI), and 45 with dementia (Table 1). Seventy‐three percent of CU, 80% of MCI, and 56% of dementia participants in the main study cohort had longitudinal cognitive outcome measures available. The baseline characteristics of the main study cohort and those of the longitudinal subcohort did not differ statistically (cf. supporting information).
TABLE 1.
Baseline demographic and clinical characteristics of the study participants by cognitive impairment and dementia status
| All | Cognitively unimpaired (CU) | Mild cognitive impairment (MCI) | Dementia | CU vs. MCI a | MCI vs. dementia a | CU vs. dementia a | |
|---|---|---|---|---|---|---|---|
| Main cohort, N | 248 | 120 | 83 | 45 | |||
| Age (years, mean ± std), baseline | 75.3 ± 5.6 | 74.7 ± 5.4 | 75.8 ± 5.7 | 76.2 ± 5.8 |
t = 1.31 P = .19 |
t = 0.26 P = .79 |
t = 1.36 P = .175 |
| Sex (F %) | 51% | 59% | 41% | 47% | P = .016 | P = .71 | P = .16 |
| Education (years, mean ± std) | 16.4 ± 2.4 | 16.8 ± 2.1 | 16.1 ± 2.6 | 16.1 ± 2.4 |
t = ‐2.16 P = .032 |
t = 0.11 P = .91 |
t = ‐1.74 P = .084 |
| APOE ε4 (carrier, %) | 61% | 53% | 63% | 78% | P = .0057 | P = .17 | P < 10−4 |
| Aβ centiloid (mean ± std), baseline | 72.6 ± 37.7 | 60.0 ± 32.6 | 77.3 ± 38.0 | 97.4 ± 36.3 |
t = 3.48 P = .00063 |
t = 2.89 p = 0.0045 |
t = 6.31 p < 10−8 |
| WML (ICV %), baseline | 0.39 ± 0.69 | 0.29 ± 0.37 | 0.49 ± 1.04 | 0.46 ± 0.47 |
t = 1.98 P = .049 |
t = ‐0.23 P = .82 |
t = 2.36 P = .019 |
| ADAS–Cog (mean ± std), baseline | 19.1 ± 10.1 | 12.9 ± 4.8 | 19.9 ± 6.8 | 34.3 ± 9.0 |
t = 8.56 P < 10−14 |
t = 10.14 P < 10−16 |
t = 19.41 P < 10−16 |
| ADAS–Cog (mean ± std), rate of change | 1.53 ± 1.04 | 0.91 ± 0.48 | 1.79 ± 0.87 | 3.14 ± 0.92 |
t = 8.17 P < 10−12 |
t = 6.63 P < 10−8 |
t = 16.63 P < 10−16 |
| mPACC (mean ± std), baseline | −4.85 ± 7.20 | 0.02 ± 2.8 | −5.89 ± 4.34 | −15.9 ± 6.5 |
t = ‐11.59 P < 10−16 |
t = ‐10.27 P < 10−16 |
t = ‐21.45 P < 10−16 |
| mPACC (mean ± std), rate of change | −0.79 ± 1.02 | −0.11 ± 0.40 | −1.05 ± 0.69 | −2.50 ± 0.97 |
t = ‐10.54 P < 10−16 |
t = ‐8.02 P < 10−11 |
t = ‐18.37 P < 10−16 |
| CDR‐SB (mean ± std), baseline | 1.59 ± 2.42 | 0.09 ± 0.29 | 1.58 ± 1.21 | 5.6 ± 2.7 |
t = 12.95 P < 10−16 |
t = 11.46 P < 10−16 |
t = 21.66 P < 10−16 |
| MMSE (mean ± std), baseline | 27.0 ± 3.54 | 28.9 ± 1.3 | 27.3 ± 2.3 | 21.6 ± 3.9 |
t = ‐6.46 P < 10−9 |
t = ‐10.24 P < 10−16 |
t = ‐17.76 P < 10−16 |
| Logical memory – delayed (mean ± std), baseline | 9.09 ± 5.98 | 13.6 ± 3.5 | 6.69 ± 4.40 | 1.4 ± 2.7 |
t = ‐12.26 P < 10−16 |
t = ‐7.06 P < 10−9 |
t = ‐20.29 P < 10−16 |
| RAVLT – immediate (mean ± std), baseline | 37.4 ± 13.7 | 46.0 ± 11.0 | 33.1 ± 10.2 | 22.1 ± 7.7 |
t = ‐8.46 P < 10−14 |
t = ‐6.06 P < 10−7 |
t = ‐12.97 P < 10−16 |
| RAVLT – learning (mean ± std), baseline | 4.47 ± 2.86 | 6.0 ± 2.3 | 3.6 ± 2.8 | 2.0 ± 2.0 |
t = ‐6.52 P < 10−9 |
t = ‐3.32 P = .0012 |
t = ‐9.82 P < 10−16 |
| RAVLT – forgetting (mean ± std), baseline | 4.02 ± 3.98 | 3.8 ± 2.8 | 4.0 ± 5.8 | 4.7 ± 1.8 |
t = 0.54 P = .59 |
t = 0.79 P = .43 |
t = 2.32 P = .022 |
| Trail Making Test – B (mean ± std), baseline | 109.6 ± 73.2 | 77.7 ± 36.6 | 118.3 ± 71.1 | 191.6 ± 94.2 |
t = 5.25 P < 10−6 |
t = 4.85 P < 10−5 |
t = 11.00 P < 10−16 |
| Conversion to MCI within 4 years; N (%) | 14 (12%) | ||||||
| Conversion to dementia within 4 years; N (%) | 17 (20%) |
Notes: All study participants had PET imaging evidence for Aβ‐positivity b . The study cohort was limited to individuals of age ≥65 years and ≤85 years. Selection was made a priori from all ADNI subjects based on the availability of complete data including cross‐sectional neuroimaging, longitudinal clinical and cognitive measures, and APOE genotyping as of February 28, 2021. Because previous studies have shown that neuropathological features of dementia in the oldest‐old could differ from those of cognitively impaired younger‐old individuals, the study cohort was limited to individuals of age ≥65 years and ≤85 years. 56 Only one of the participants in this study cohort had autopsy findings, which confirmed AD neuropathologic changes of frequent neuritic plaques and neurofibrillary degeneration at Braak & Braak stage 5, and additionally identified regional TDP‐43 pathological inclusions, Lewy‐body pathology, and mild cerebral amyloid angiopathy.
Abbreviations: Aβ, amyloid beta; ADAS‐Cog, Alzheimer's Disease Assessment Scale–Cognitive subscale; ADNI, Alzheimer's Disease Neuroimaging Initiative; APOE, apolipoprotein E; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; MMSE, Mini‐Mental State Examination; PACC, Preclinical Alzheimer Cognitive Composite; PET, positron emission tomography; RAVLT, Rey Auditory Verbal Learning Test; ROI region of interest; std, standard deviation; SUVR, standardized uptake value ratio; WML, white matter lesion.
Descriptive statistics for continuous variables were compared between groups with an independent‐samples t‐test, and categorical variables with the chi‐square test.
A composite ROI consisting of middle frontal, anterior cingulate, posterior cingulate, inferior parietal, precuneus, supramarginal, middle temporal, and superior temporal was used to compute a global SUVR for florbetapir and florbetaben. A threshold of SUVR ≥ 1.11 for florbetapir and ≥ 1.08 for florbetaben was then used to determine Aβ‐positivity status.
Full constructs of the PLS‐SEMs considering mediation by the Aβ–tau–atrophy biomarker pathway of the effects of AD risk factors on baseline mPACC and ADAS‐Cog measures and the parameter estimates for the final models are shown in Figure 2.
FIGURE 2.
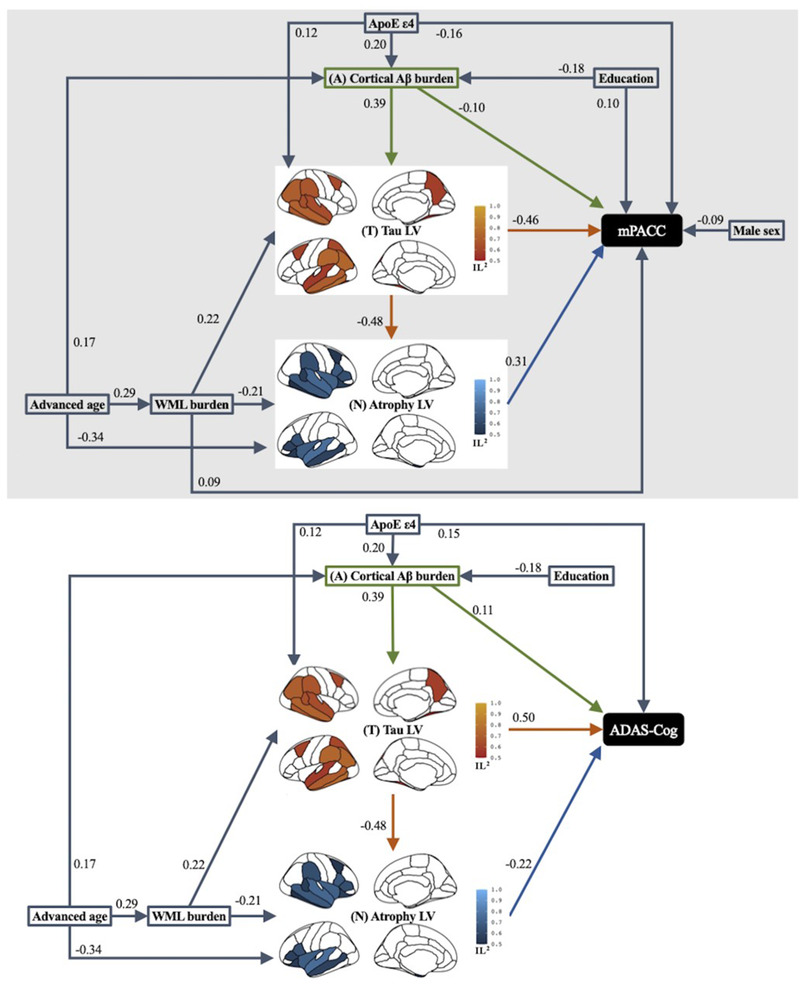
Results of path analysis of combined Alzheimer's disease (AD) imaging biomarker pathways mediating the effect of AD risk factors on baseline cognitive outcome measure of modified Preclinical Alzheimer Cognitive Composite (mPACC) and Alzheimer's Disease Assessment Scale–Cognitive subscale (ADAS‐Cog) across the AD continuum. Goodness‐of‐fit was 0.43 for mPACC modeling and 0.42 for ADAS‐Cog modeling. Squares or rectangles represent manifest variables and brain maps represent latent variables (LV). Tau LV involved fusiform, inferior temporal, middle temporal, superior temporal, supramarginal, inferior parietal, superior frontal, and caudal middle frontal bilaterally, and left posterior cingulate, left superior parietal, right banks of superior temporal sulcus, and right precuenus. Atrophy LV involved amygdala, middle temporal, superior temporal, lateral orbitofrontal, parsopercularis, parstriangularis, supramarginal, and insula bilaterally, and left hippocampus, left entorhinal, right banks of superior temporal sulcus, right caudal middle frontal, right inferior temporal. Each single‐headed arrow denotes a hypothesized unidirectional effect of one variable on another. Numbers associated with effects are standardized regression coefficients or standardized factor loadings (i.e., from a latent variable to its indicators). Only the paths that were statistically significant at P < .05 are represented. Paths that were hypothesized but were not statistically significant at P < .05 are excluded. All AD imaging markers considered in this study, specifically greater global Aβ burden, tau LV with greater burden in the parietotemporal neocortical regions, and atrophy LV within the frontotemporal as well as parietal regions, together with presence of APOE ε4 allele had significant direct effects on greater baseline cognitive impairment measured by either mPACC or ADAS‐Cog. Fewer years of education, male sex, and greater white matter lesion (WML) had significant direct effects on worse baseline mPACC but not ADAS‐Cog. In addition to these direct effects on cognitive impairment, we also observed that advanced age had significant direct effects on greater cortical amyloid beta (Aβ), WML, and the atrophy LV, but not on the tau LV or baseline cognitive outcome measures, suggesting that the biomarker model mediated the effect of age on cognitive impairment. Fewer years of education had a significant direct effect on greater global Aβ, even though its direct effect on baseline cognitive impairment was only significant in the mPACC model. Similarly, presence of APOE ε4 allele had significant direct effects on greater global Aβ and neocortical tau LV, but not on the WML or atrophy LV. Greater WML had significant direct effects on both tau and atrophy LVs, but not on global Aβ, and its direct effect on baseline cognitive impairment was significant only for mPACC but not ADAS‐Cog. Greater global Aβ had significant direct effects on tau LV but not atrophy LV. Tau LV had a significant direct effect on atrophy LV, suggesting mediation of the effects of Aβ on atrophy by tau. IL2, indicator loading squared.
Overall, the final PLS‐SEMs (Figure 2) explained 58% of variance in baseline mPACC and 53% of the variance in baseline ADAS‐Cog. Aβ, tau, and atrophy each independently explained 16%, 46% to 47%, and 25% to 29% of variance in baseline cognition, respectively. Estimated direct and indirect effects of AD‐biomarkers on cognitive impairment are plotted in Figure 3.
FIGURE 3.
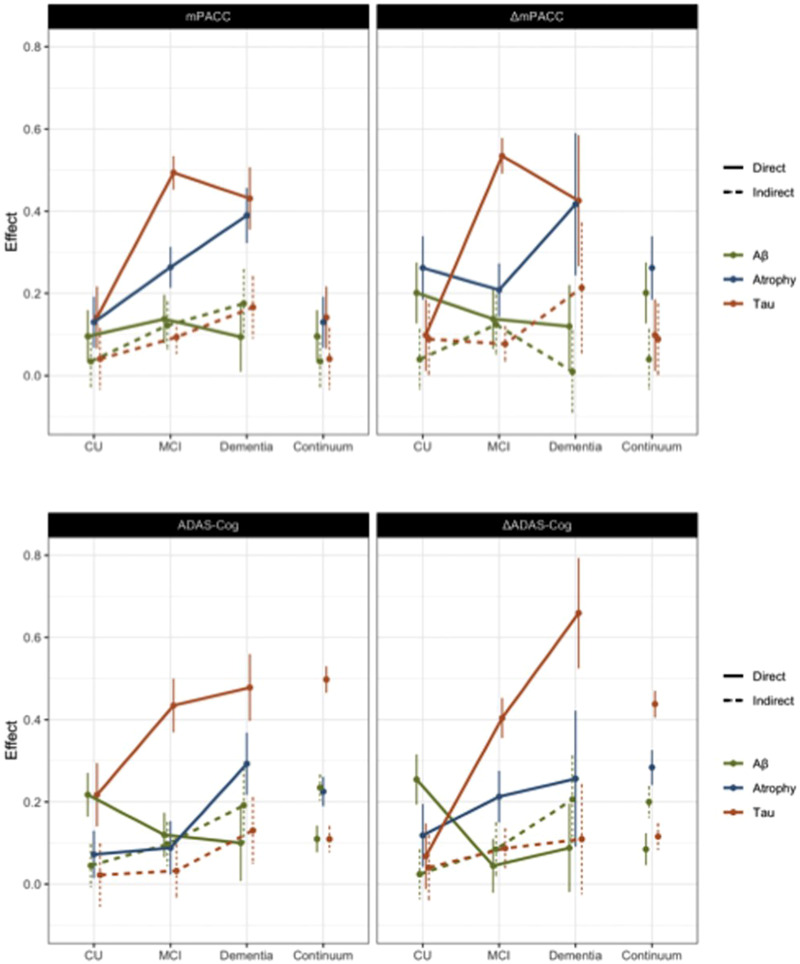
Direct and indirect effects of Alzheimer's disease (AD) imaging biomarkers (Aβ: global cortical amyloid beta burden, Tau: latent construct of the regional tau burden, and Atrophy: latent construct of the regional atrophy) on baseline cognitive impairment and longitudinal cognitive decline operationalized with modified Preclinical Alzheimer Cognitive Composite (mPACC) and Alzheimer's Disease Assessment Scale–Cognitive subscale (ADAS‐Cog). Confidence intervals were based on a bootstrapping procedure with 100 repetitions.
Fifty‐six percent of the variance in ΔmPACC and 50% of the variance in ΔADAS‐Cog were accounted for by Aβ–tau–atrophy pathway partially mediating the AD risk factor and cognitive decline relations, as illustrated in Figure 4. To a great extent, the significant pathways identified in cognitive decline modeling were similar to ones in baseline cognitive impairment modeling, with the exception of male sex having a significant direct effect on tau latent variable (LV) but not on either cognitive decline measures, and of greater WML having a significant direct effect on ΔADAS‐Cog but not ΔmPACC. Overall, Aβ, tau, and atrophy independently explained 12% to 13%, 39% to 41%, and 23% to 30% of the variance in longitudinal cognitive decline, respectively. ,
FIGURE 4.
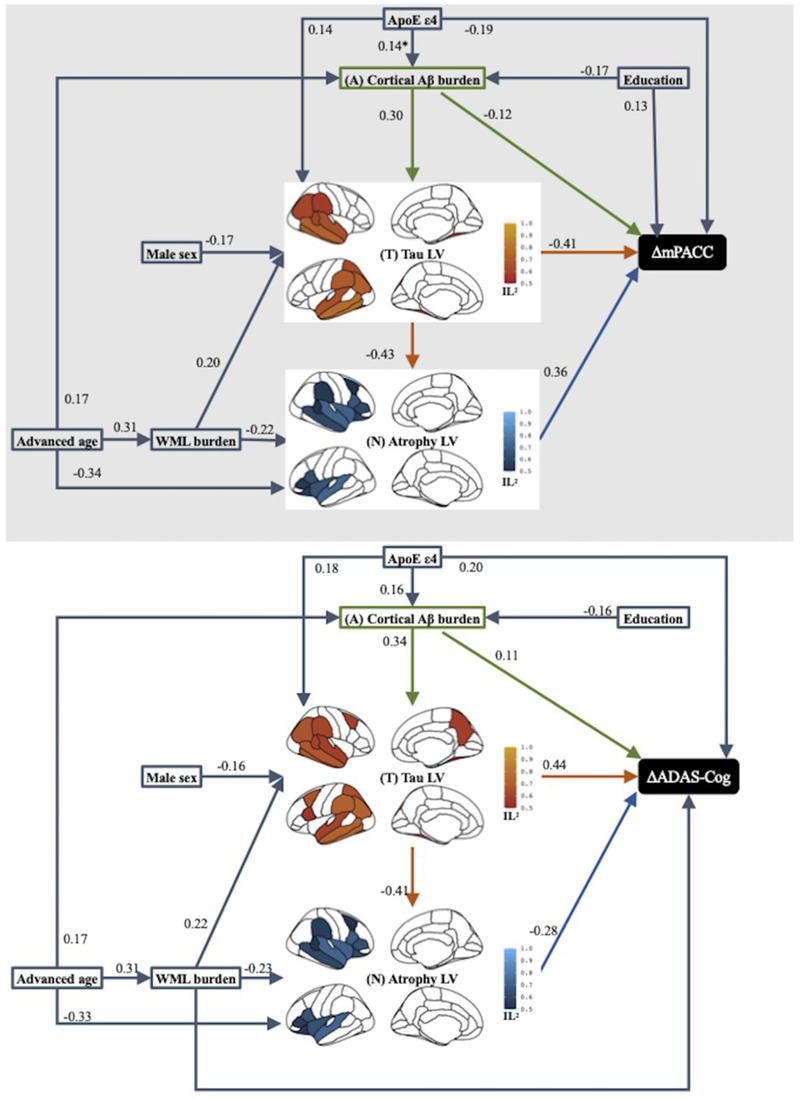
Results of path analysis of combined Alzheimer's disease (AD) imaging biomarker pathways mediating the effect of AD‐risk factors on longitudinal cognitive decline measure of modified Preclinical Alzheimer Cognitive Composite (ΔmPACC) and Alzheimer's Disease Assessment Scale–Cognitive subscale (ΔADAS‐Cog) across the AD continuum. Goodness‐of‐fit was 0.41 for both ΔmPACC modeling and ΔADAS‐Cog modeling. Squares or rectangles represent manifest variables and brain maps represent latent variables (LV). Tau LV involved fusiform, inferior temporal, middle temporal, superior temporal, supramarginal, inferior parietal, and posterior cingulate bilaterally, and left superior parietal, left superior frontal, right banks of superior temporal sulcus. Tau LV further involved bilateral caudal middle frontal, left pars opercularis, and right precuneus in ΔADAS‐Cog modeling. Atrophy LV involved hippocampus, amygdala, middle temporal, superior temporal, lateral orbitofrontal, pars opercularis, pars triangularis, and insula bilaterally, and right banks of superior temporal sulcus, right caudal middle frontal, and right supramarginal. Each single‐headed arrow denotes a hypothesized unidirectional effect of one variable on another. Numbers associated with effects are standardized regression coefficients or standardized factor loadings (i.e., from a latent variable to its indicators). Only the paths that were statistically significant at P < .05 are represented. Paths that were hypothesized but were not statistically significant at P < .05 are excluded. IL2, indicator loading squared.
Stratifying the main study cohort into those with and without cognitive impairment and dementia and allowing the path coefficients to be estimated separately for each cognitive diagnostic group, the Aβ–tau–atrophy model together with the AD risk factors explained a smaller percent of the variance in baseline cognitive impairment and longitudinal cognitive decline within CU compared to MCI and dementia (i.e., 10% vs. 49% vs. 53% variance in baseline mPACC; 17% vs. 34% vs. 47% variance in baseline ADAS‐Cog; 16% vs. 51% vs. 59% variance in ΔmPACC; and 12% vs. 37% vs. 59% variance in ΔADAS‐Cog, respectively), although the models had excellent global fit (goodness‐of‐fit of 0.37–0.48). The sensitivity analyses on cognitive impairment outcome measures within each cognitive diagnostic group suggested greater direct effect of global Aβ (a standardized coefficient of β = 0.22 and standard error [SE] of 0.10) but lower direct effect of tau LV (β = 0.22; SE = 0.11) with a lack of a direct effect of atrophy in CU (Figure 3). In contrast, only tau LV (β = 0.43; SE = 0.11) but not global Aβ or atrophy had a direct effect on baseline cognitive impairment in MCI. Greater direct effect of atrophy LV (β = 0.29; SE = 0.16) with tau LV (β = 0.48; SE = 0.17) but not global Aβ was observed in dementia. Finally, only global Aβ, but neither tau nor atrophy LVs, had a greater direct effect (β = 0.25; SE = 0.12) on longitudinal cognitive decline in CU, although tau LV had a greater direct effect on cognitive decline in MCI and dementia (β = 0.40; SE = 0.13 and β = 0.66; SE = 0.26, respectively), while direct effects of global Aβ and atrophy LV were not significant in these models.
4. DISCUSSION
Our study was conducted to determine the contribution of Aβ and tau pathologies and atrophy, jointly with AD risk factors of age, sex, education, APOE, and WMLs to the cognitive impairment and cognitive decline in older individuals in the AD continuum. Our major findings were that across the AD continuum in individuals with PET biomarker evidence for Aβ‐positivity (1) 16% of variance in cross‐sectional cognitive impairment, measured by mPACC or ADAS‐Cog, was accounted for by Aβ, 46% to 47% by tau, and 25% to 29% by atrophy, although about 53% to 58% of total variance in cognitive impairment was explained by incorporating mediated and direct effects of AD risk factors; and (2) the Aβ–tau–atrophy pathway accounted for 50% to 56% of variance in longitudinal cognitive decline while Aβ, tau, and atrophy each independently explained 16%, 46% to 47%, and 25% to 29% of the variance, respectively.
Among all neurodegenerative imaging markers considered in this study, tau and atrophy had the greatest and most consistent relationship to cognitive decline. Recent studies suggested that tau, not Aβ, burden and topography in PET predicts atrophy, and might be the key driver of atrophy and subsequent neurodegenerative processes. 5 , 18 We observed that greater tau accumulation in a stereotypical brain pattern affected cognitive impairment and decline. Overall, the topography of significant regional tau accumulation overlapped with the locational sensitivity to the cognitive tests, such as logical and verbal memory function being localized mostly in the parietal‐temporal brain regions 19 and deficits in semantic fluency involving the parietal lobe in addition to the temporal lobe, frontal lobe, and anterior cingulate. 20 Consistent with neuropathological reports, 21 our findings suggested an impact of significant tau accumulation in the Braak & Braak III–IV brain regions on detectable cognitive impairment, with an increased regional burden and spread expanding to frontal regions. Persistent direct tau‐cognition association through different clinical stages suggested that tau pathology may lead to cognitive impairments through a variety of mechanisms, including, but not limited to, atrophy. 18 , 22 We also observed that cognitive impairment and decline was directly associated with a specific atrophy pattern particularly in advancing clinical stages, consistent with the view that neurodegeneration is the strongest driver of future cognitive decline. 22 , 23 The atrophy pattern was similar, but not identical, to the spatial spread of tau. We should note that gray matter tissue volume as a measure of atrophy reflects a cumulative effect of diverse neurodegenerative processes including not only AD pathologies but also the effects of aging, 24 vascular pathologies, 25 α‐synuclein, 26 and TDP‐43. 27 , ,
In contrast to tau burden and atrophy, the effect of global Aβ burden within Aβ‐positive individuals on cognitive outcome measures shifted from being largely direct at the CU stage to indirect effect mediated through greater tau accumulation at the later clinical stages. The observed indirect effects of Aβ are consistent with previous reports of indirect effects of Aβ on memory function, 18 , 19 and closely conform to the AD Aβ cascade hypothesis. 28 These findings also support the widely accepted AD biomarker model positing that Aβ has an initiating role in early stages of AD pathophysiological changes by facilitating spread and accumulation of tau pathology. 29 In a separate PLS‐SEM (results not shown) that included regional Aβ burden from 31 ROIs, we observed that the effect of Aβ was distributed across the cortex rather than localized in specific regions. This may indicate that local Aβ burden did not convey additional information beyond the global Aβ burden in explaining variance in concurrent impairment and decline in cognition, or that we lacked the statistical power to detect regional specificity of Aβ burden due to its limited within‐subject variance once individuals are Aβ phenotype converted.
Risk factors beyond the Aβ–tau–atrophy axis affected cognitive impairment and decline. We observed both direct and mediated, by greater regional tau and atrophy, effects of greater WML burden, supporting a role for cerebrovascular disease. Most elderly individuals, including those with clinical AD diagnoses, show comorbid cerebrovascular brain pathologies with a prevalence as high as 32% to 48%, in addition to AD‐related Aβ and tau pathologies. 4 Together with the emerging evidence that the threshold at which AD pathology becomes symptomatic might be lowered by cerebrovascular disease, 4 our findings support the idea that, in addition to Aβ or tau targeting therapies, vascular protective strategies should be considered as the effects of WM disease on cognition might be independent of Aβ and tau pathologies.
Our results demonstrated that advanced age was an important risk factor for both cognitive impairment and decline, acting predominantly indirectly via greater Aβ burden, greater atrophy, and greater WML burden, as has been repeatedly reported. 30 , 31 We observed that female sex was associated with greater tau burden. Neuropathological studies suggest that women have a 3‐year acceleration in tau tangle neuropathology 32 and this sex difference is largely attributable to APOE ε4 status. 33 Recent in vivo neuroimaging studies replicated some of these neuropathological findings, reporting that females had greater brain resilience to pathological tau. 34 These sex effects may be explained by risk factors such as cardiometabolic disease, depression, sleep cycle abnormalities, and menopause, as well as sociocultural factors such as education, exercise, and caregiving status. 35 The impact of these potentially modifiable factors on the clinical expression of AD pathology and neurodegeneration warrants future studies. Finally, education was a protective factor on cognitive functioning as well as on cognitive decline. Epidemiological studies suggest that education, in addition to other lifelong experiences, is associated with lower prevalence of AD. 34 , 36 Education may increase cognitive reserve, the ability to harbor greater AD pathology without experiencing cognitive decline in their cognitive functioning. 37
Our observation that APOE ε4 is associated with greater Aβ burden is consistent with APOE being the most replicated risk factor for AD after advanced age. 38 , 39 Most neuropathological studies suggest the lack of an independent effect of APOE on neurofibrillary tau tangle formation, 40 , 41 , 42 but an association of APOE with tau tangle pathology only in the presence of Aβ. 43 Our observation of this expected mediated effect of APOE on cognitive impairment and decline through greater Aβ burden in addition to the direct effect of APOE on tau‐mediated neurodegeneration is consistent with the evidence from animal models. 44
Only a limited percentage of the variance in cognitive decline was explained by the currently available imaging biomarkers or by incorporating AD risk factors, consistent with the previous neuropathological studies repeatedly reporting that ≤50% of the variance in cognitive decline before death can be accounted for even after considering hippocampal sclerosis, TDP‐43, and atherosclerosis in addition to commonly considered demographics and indices of AD, cerebrovascular disease, and Lewy body pathologies. 3 , 23 in vivo imaging studies have reached similar conclusions. 5 , 19 , 45 , 46 , 47 , 48 A combination of cortical thickness, structural connectivity, and WMHs accounted for only 20% of total variance, 45 and in MCI, temporal lobe atrophy rates explained 9.5% to 16% of the variance in decline in various cognitive domains. 46 Similarly, up to 38% of decline in memory could be explained by interactions between measures of Aβ‐PET, fluorodeoxyglucose‐PET, and structural‐MRI, 47 and tau‐PET tracer binding in the early Braak & Braak stage regions accounted for only 20% of the variance in memory decline, while measures of atrophy, Aβ burden, age, sex, or education did not explain additional variance in memory performance in a CU cohort. 19 In MCI/AD, microglial activation, tau, and atrophy accounted for up to 52% of the variance in memory decline. 48 Despite the variability in the magnitude of estimated associations, which might be explained by the differences in cohort composition, sensitivity of the considered imaging modalities, or single versus multiple cognitive domain considerations, both autopsy studies and in vivo imaging biomarker studies including ours emphasize the need to consider other neuropathology causes, such as TDP‐43 aggregation, hippocampal sclerosis, or neuroinflammation, to better explain cognition across the AD continuum.
Most clinical trials using treatments aimed at Aβ pathology accumulation target individuals with MCI and mild‐to‐moderate AD clinical diagnoses, similar to participants recruited in ADNI. Based on our results, treatments that remove Aβ and completely stop downstream effects on tau and neurodegeneration would only partially affect cognitive decline in these individuals, consistent with previous reports. 3 , 4 , 5 Furthermore, our findings that 16% of the total variance in cognitive decline of CU was explained by the PLS‐SEM and that only 14% of the variance was explained by Aβ, tau, and atrophy biomarkers raise concerns about the effectiveness of Aβ removing treatments to prevent decline in CU, an approach being tested in several treatment trials. One implication of this study could be that disease‐modifying therapies that target Aβ, tau, and neurodegeneration according to the clinical disease stage may be more effective in slowing cognitive decline and ideally reverting cognitive impairment. The substantial spatial overlap between tau burden and atrophy signatures in our results could also provide important support for promising anti‐tau therapies.
It is worth noting that within the hypothesized structural pathway model construct, the neurobiological basis of cognitive impairment and decline measured by either mPACC or ADAS‐Cog was similar. This suggests that the neural basis of cognition may be specific to the disease but a continuum in the disease spectrum, and robust to the tools used to measure the cognitive outcome even though the direct effects of AD biomarkers on these cognitive outcomes showed clinical state–specific differences (cf. Figure 3). A great similarity was observed between models explaining variance in concurrent cognitive impairment and decline in cognition. This might be due to the current level of cognition being a good predictor of cognitive decline, 49 , 50 reflecting the fact that people who are declining are likely to already show some impairment, and people with more severe impairments are more likely to be declining more rapidly.
The cross‐sectional nature of the imaging biomarker data assessed in this study makes it impossible to speculate about longitudinal pathophysiological changes potentially characteristic of future clinical progression at different disease stages. It is possible that the relative contribution of AD risk and AD biomarkers to clinical progression in this sample might increase over time as underlying neurodegenerative processes progress. Additionally, the current study is based on a convenience cohort in which the degree of true population representation is not known. Most notably, WML burden was overall low in our study cohort compared to the general population due to strict exclusion of participants with vascular pathology etiologies in ADNI. The WML measure does not cover all vascular pathology and a large proportion of unexplained variance might still be related to a vascular origin. Although standardized ADNI‐3 specific neuroimaging protocols were used at each ADNI site to minimize the non‐biological variance in biomarker measures, we acknowledge potential residual scanner variability in multisite studies. Various statistical and deep learning approaches for neuroimaging data harmonization have been extensively applied to data from ADNI studies. In many, 51 except Fortin et al., 52 imaging measures were harmonized by removing non‐biological variances estimated across different ADNI study phases by pooling data from multiple sites within each study phase, an approach that relys on the effectiveness of standardized study phase specific imaging protocols deployed at each ADNI site. Although effectiveness and robustness of neuroimaging data harmonization has been shown in small sample sizes using an empirical Bayesian framework, 53 we believe that the current study cohort, which is limited to the ADNI‐3 cases, does not provide enough samples per site for proper site‐level data harmonization. Specifically, T1‐weighted images using the ADNI‐3 acquisition protocol are available from 59 different ADNI sites. The number of subjects scanned at each site varies between 3 and 51, with an average of 16.7 cases per site. When limited to CU, the subgroup typically used for estimating empirical distributions, 22 sites have a sample size < 10. Finally, stage‐specific genetic contributions, other than APOE, in relation to the differential stage‐related pathophysiological mechanisms warrant further studies. 54
Our results recapitulated the previously proposed mediating effects of Aβ burden on cognitive impairment through cortical tau and cortical atrophy, closely conforming to the AD Aβ cascade hypothesis 28 and consistent with recently reported one‐direction‐only sequence of pathological biomarker changes beginning with Aβ deposition, through tau deposition, neurodegeneration, and cognitive decline. 55 We also observed that at every clinical stage, tau was a major contributor to cognitive decline and that while tau mediated the effects of Aβ burden on cognitive decline in all clinical stages, Aβ burden had a stronger independent direct effect on cognitive decline in CU, whereas cognitive decline in MCI and dementia was largely accounted for by tau and atrophy. Our findings that a substantial proportion of the variance in cognition and cognitive decline was not explained by combinations of Aβ, tau, and atrophy together with risk factors suggests that other pathological aspects such as cerebrovascular, α‐synuclein, or TDP‐43 may contribute to variance.
CONFLICTS OF INTEREST
Dr. Weintraub reports grants from NIA, Department of Veterans Affairs, Fox Foundation, Acadia Pharmaceuticals, and IPMDS. All payments made to U. Pennsylvania, compensation for licensing of QUIP, QUIP‐RS, and PDAQ‐15 through the University of Pennsylvania. He received personal consulting compensation from Acadia, Aptinyx, CHDI Foundation, Clintrex LLC (Otsuka), Eisai, Great Lake Neurotechnologies, Janssen, Sage, Scion, Signant Health, Sunovion, and Vanda. He serves as Chair DSMB for ATRI and ADCS. Dr. Aisen has received support over the last 36 months from payments to his institution from NIA, FNIH, Alzheimer's Association, Janssen, Lilly, Merck, and Eisai. He has served on the advisory boards of Biogen, Merck, Roche, Abbvie, Rainbow Medical, ImmunoBrain Checkpoint, Shionogi, and has no other support or conflicts of interest to declare. Dr. Jack was supported in the current work by NIH funding. Over the last 36 months, he has received support from NIH grants and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic to his institution and has served on the advisory boards of iDMC and Roche with no payments made. He serves on an independent data monitoring board for Roche, has consulted for and served as a speaker for Eisai, and consulted for Biogen, but he receives no personal compensation from any commercial entity. He has no other support or conflicts of interest to declare. Dr. Jagust has received support over the last 36 months from grants to his institution (NIH grants R01 AG034570 [Dr. Jagust], R01AG062542 [Dr. Jagust], U24 AG067418 [Dr. Jagust], P01AG019724 [Dr. Bruce Miller], R01 AG031164 [Dr. Matthew Walker], RF1 AG054019 [Dr. Matthew Walker], U01 AG024904 [Dr. Weiner], RF1 AG054106‐01A1 [Dr. Matthew Walker], R44AG046025‐03 [Dr. Daojing Wang], R01 AG061303 [Dr. Lexin Li], MH112775 [Dr. Ming Hsu], 1R01AG062689‐01 [Dr. Landau], AG062624 [Dr. José Luchsinger], and R01AG069090), direct consulting fees (Biogen, Bioclinica, Genentech/Roche, CuraSen, Grifols), and has served on the advisory board of the Alzheimer's Prevention Initiative. He has no other support or conflicts of interest to declare. Dr. Petersen over the past 36 months received grants through his institution (P30 AG062677, U01 AG006786); licenses or royalties from Oxford University Press and UpToDate; and consulting fees from Roche, Merck, Biogen, Genentech, and Eisai. He served on the advisory board of Genentech and has no other support or conflicts of interest to declare. Dr. Saykin was supported in this work by grants from NIH and Department of Defense (NIH grants U01 AG024904, P30 AG010133, R01 AG019771, R01 LM013463, R01 LM011360 and DoD grants W81XWH‐13‐1‐0259 and W81XWH‐12‐2‐0012), and over the past 36 months received support from grants to his institution (as detailed above). He served on the Bayer Oncology Advisory Board and received PET tracer precursor from Eli Lilly/Avid Radiopharmaceuticals. He reports personal fees from Arkley BioTek and Springer Nature, outside the submitted work. He has no other support or conflicts of interest to declare. Dr. Shaw has received over the past 36 months grants through his institution (NIH grants U01 AG024904 (ADNI3), UPenn ADRC NIA grant for Biomarker Core; Michael J. Fox Foundation for Parkinson's Research for AD biomarker studies; Roche IIS for AD biomarker studies), fees for the Biogen Teaching program on AD Biomarkers, and travel funds from NIA ADNI3 Biomarker Core. He has served on the Roche Advisory Board, LEADS Advisory Board, and Fujirebio Advisory Board. He received in kind support from Roche (immunoassay reagents and equipment) for ADNI3. He has no other support or conflicts of interest to declare. Dr. Trojanowski has received over the past 36 months grants through his institution (AG10124). He has no other support or conflicts of interest to declare. Dr. Weiner is the Principal Investigator of NIH funded grants. Over the past 36 months he received funding administered through his institutions (NIH grants: 1RF1AG059009‐01 and 1R01AG058676‐01A1; CA Dept. of Health grant: 19‐10616; NIH Subaward from Dr. Richard Gershon: 1U2CA060426‐01), consulting fees (Cerecin/Accera, Inc., BioClinica, Nestle/Nestec, Roche, Genentech, NIH, The Buck Institute for Research on Aging, FUJIFILM‐Toyama Chemical [Japan], Garfield Weston, Baird Equity Capital, University of Southern California [USC], Cytox, and Japanese Organization for Medical Device Development, Inc. [JOMDD] and T3D Therapeutics), and payment for lecturing (The Buck Institute for Research on Aging). He holds stock options in Anven, Alzheon, and Aleca. He receives other grant support for his work (NIH: 5U19AG024904‐14; 1R01AG053798‐01A1; R01 MH098062; U24 AG057437‐01; 1U2CA060426‐01; 1R01AG058676‐01A1; and 1RF1AG059009‐01, DOD: W81XWH‐15‐2‐0070; 0W81XWH‐12‐2‐0012; W81XWH‐14‐1‐0462; W81XWH‐13‐1‐0259, PCORI: PPRN‐1501‐26817, California Dept. of Public Health: 16‐10054, U. Michigan: 18‐PAF01312, Siemens: 444951‐54249, Biogen: 174552, Hillblom Foundation: 2015‐A‐011‐NET, Alzheimer's Association: BHR‐16‐459161; The State of California: 18‐109929). He also receives support from Johnson & Johnson, Kevin and Connie Shanahan, GE, VUmc, Australian Catholic University (HBI‐BHR), The Stroke Foundation, and the Veterans Administration. He has served on advisory boards for Eli Lilly, Cerecin/Accera, Roche, Alzheon, Inc., Merck Sharp & Dohme Corp., Nestle/Nestec, PCORI/PPRN, Dolby Family Ventures, National Institute on Aging (NIA), Brain Health Registry, and ADNI. He has no other support or conflicts of interest to declare.
Supporting information
Supporting information
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
This work is funded by the National Institutes of Health Grants U01 AG024904, P30 AG062677, U01 AG006786, P30 AG010133, R01 AG019771, and U19 AG062418.
Tosun D, Demir Z, Veitch DP, et al. Contribution of Alzheimer's biomarkers and risk factors to cognitive impairment and decline across the Alzheimer's disease continuum. Alzheimer's Dement. 2022;18:1370–1382. 10.1002/alz.12480
REFERENCES
- 1. Budd Haeberlein S, O'Gorman J, Chiao P, et al. Clinical development of aducanumab, an anti‐aβ human monoclonal antibody being investigated for the treatment of early Alzheimer's disease. J Prev Alzheimers Dis. 2017;4:255‐263. [DOI] [PubMed] [Google Scholar]
- 2. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 3. Boyle PA, Wang T, Yu L, et al. To what degree is late life cognitive decline driven by age‐related neuropathologies?. Brain. 2021;144:2166‐2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person‐specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83:74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jack CR Jr, Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321:2316‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir‐PET for imaging beta‐amyloid pathology. JAMA. 2011;305:275‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Villemagne VL, Ong K, Mulligan RS, et al. Amyloid imaging with (18)F‐florbetaben in Alzheimer disease and other dementias. J Nucl Med. 2011;52:1210‐1217. [DOI] [PubMed] [Google Scholar]
- 8. Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Des. 2013;34:457‐468. [DOI] [PubMed] [Google Scholar]
- 9. Weiner MW, Veitch DP, Aisen PS, et al. 2014 Update of the Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement (Amst). 2015;11:e1‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donohue MC, Sperling RA, Salmon DP, et al. The Preclinical Alzheimer Cognitive Composite: measuring amyloid‐related decline. JAMA Neurol. 2014;71:961‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6:e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilson RS, Capuano AW, Bennett DA, Schneider JA, Boyle PA. Temporal course of neurodegenerative effects on cognition in old age. Neuropsychology. 2016;30:591‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Landau SM, Breault C, Joshi AD, et al. Amyloid‐β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med. 2013;54:70‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Southekal S, Devous MD Sr, Kennedy I, et al. Flortaucipir F 18 quantitation using parametric estimation of reference signal intensity. J Nucl Med. 2018;59:944‐951. [DOI] [PubMed] [Google Scholar]
- 15. Tustison NJ, Cook PA, Klein A, et al. Large‐scale evaluation of ANTs and FreeSurfer cortical thickness measurements. Neuroimage. 2014;99:166‐179. [DOI] [PubMed] [Google Scholar]
- 16. Schwarz C, Fletcher E, DeCarli C, Carmichael O. Fully‐automated white matter hyperintensity detection with anatomical prior knowledge and without FLAIR. Inf Process Med Imaging. 2009;21:239‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hair JF. When to use and how to report the results of PLS‐SEM. Eur Bus Rev. 2019;31:2‐24. [Google Scholar]
- 18. Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140:3286‐3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, et al. Entorhinal Tau pathology, episodic memory decline, and neurodegeneration in aging. The J Neurosci. 2018;38:530‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McDonald CR, Gharapetian L, McEvoy LK, et al. Relationship between regional atrophy rates and cognitive decline in mild cognitive impairment. Neurobiol Aging. 2012;33:242‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960‐969. [DOI] [PubMed] [Google Scholar]
- 22. Hammond TC, Xing X, Wang C, et al. β‐amyloid and tau drive early Alzheimer's disease decline while glucose hypometabolism drives late decline. Commun Biol. 2020;3:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dawe RJ, Yu L, Arfanakis K, Schneider JA, Bennett DA, Boyle PA. Late‐life cognitive decline is associated with hippocampal volume, above and beyond its associations with traditional neuropathologic indices. Alzheimers Dement. 2020;16:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fox NC, Schott JM. Imaging cerebral atrophy: normal ageing to Alzheimer's disease. Lancet North Am Ed. 2004;363:392‐394. [DOI] [PubMed] [Google Scholar]
- 25. Villeneuve S, Wirth M, La Joie R. Are AD‐typical regions the convergence point of multiple pathologies?. Front Aging Neurosci. 2015;7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging. 2012;33:2091‐2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bejanin A, Murray ME, Martin P, et al. Antemortem volume loss mirrors TDP‐43 staging in older adults with non‐frontotemporal lobar degeneration. Brain. 2019;142:3621‐3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reitz C. Alzheimer's disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis. 2012;2012:369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pooler AM, Polydoro M, Maury EA, et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer's disease. Acta Neuropathol Commun. 2015;3:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog Neurobiol. 2014;117:20‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oh H, Madison C, Baker S, Rabinovici G, Jagust W. Dynamic relationships between age, amyloid‐β deposition, and glucose metabolism link to the regional vulnerability to Alzheimer's disease. Brain. 2016;139(8), 2275‐2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci. 2004;1019:24‐28. [DOI] [PubMed] [Google Scholar]
- 33. Liu M, Paranjpe MD, Zhou X, et al. Sex modulates the ApoE ε4 effect on brain tau deposition measured by (18)F‐AV‐1451 PET in individuals with mild cognitive impairment. Theranostics. 2019;9:4959‐4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ossenkoppele R, Lyoo CH, Jester‐Broms J, et al. Assessment of demographic, genetic, and imaging variables associated with brain resilience and cognitive resilience to pathological Tau in patients with Alzheimer disease. JAMA Neurol. 2020;77:632‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nebel RA, Aggarwal NT, Barnes LL, et al. Understanding the impact of sex and gender in Alzheimer's disease: a call to action. Alzheimers Dement (Amst). 2018;14:1171‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharp ES, Gatz M. Relationship between education and dementia: an updated systematic review. Alzheimer Dis Assoc Disord. 2011;25:289‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arenaza‐Urquijo EM, Bejanin A, Gonneaud J, et al. Association between educational attainment and amyloid deposition across the spectrum from normal cognition to dementia: neuroimaging evidence for protection and compensation. Neurobiol Aging. 2017;59:72‐79. [DOI] [PubMed] [Google Scholar]
- 38. Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid‐β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci. 2009;106(16), 6820‐6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer's disease: progress to date and the path gorward. Neuron. 2019;101:820‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu L, Boyle PA, Leurgans S, Schneider JA, Bennett DA. Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol Aging. 2014;35:819‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrano‐Pozo A, Qian J, Monsell SE, Betensky RA, Hyman BT. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. 2015;77:917‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mufson EJ, Malek‐Ahmadi M, Perez SE, Chen K. Braak staging, plaque pathology, and APOE status in elderly persons without cognitive impairment. Neurobiol Aging. 2016;37:147‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA. Association of APOE with tau‐tangle pathology with and without β‐amyloid. Neurobiol Aging. 2016;37:19‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau‐mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hedden T, Schultz AP, Rieckmann A, et al. Multiple brain markers are linked to age‐related variation in cognition. Cereb Cortex. 2016;26:1388‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McDonald CR, Gharapetian L, McEvoy LK, et al. Relationship between regional atrophy rates and cognitive decline in mild cognitive impairment. Neurobiol Aging. 2012;33:242‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wirth M, Oh H, Mormino EC, Markley C, Landau SM, Jagust WJ. The effect of amyloid β on cognitive decline is modulated by neural integrity in cognitively normal elderly. Alzheimers Dement (Amst). 2013;9:687‐698.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malpetti M, Kievit RA, Passamonti L, et al. Microglial activation and tau burden predict cognitive decline in Alzheimer's disease. Brain. 2020;143:1588‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schaeverbeke JM, Gabel S, Meersmans K, et al. Baseline cognition is the best predictor of 4‐year cognitive change in cognitively intact older adults. Alzheimer's Res Ther. 2021;13:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Shea DM, Thomas KR, Asken B, et al. Adding cognition to AT(N) models improves prediction of cognitive and functional decline. Alzheimers Dement (Amst). 2021;13:e12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pomponio R, Erus G, Habes M, et al. Harmonization of large MRI datasets for the analysis of brain imaging patterns throughout the lifespan. Neuroimage. 2020;208:116450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fortin J‐P, Cullen N, Sheline YI, et al. Harmonization of cortical thickness measurements across scanners and sites. Neuroimage. 2018;167:104‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118‐127. [DOI] [PubMed] [Google Scholar]
- 54. Apostolova LG, Risacher SL, Duran T, et al. Associations of the top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol. 2018;75:328‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guo T, Korman D, Baker SL, Landau SM, Jagust WJ. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer's disease pathophysiology. Biol Psychiatry. 2021;89(8), 786‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Middleton LE, Grinberg LT, Miller B, Kawas C, Yaffe K. Neuropathologic features associated with Alzheimer disease diagnosis: age matters. Neurology. 2011;77:1737‐1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information