

Abstract
Purpose.
To investigate tolerability, efficacy, and pharmacokinetics/-dynamics (PK/PD) of Debio 1347, a selective fibroblast growth factor receptor (FGFR) inhibitor.
Patients and Methods.
This was a first-in-human, multicenter, open-label study in patients with advanced solid tumors harboring FGFR1–3 gene alterations. Eligible patients received oral Debio 1347 at escalating doses once daily until disease progression or intolerable toxicity. Dose limiting toxicities (DLTs) were evaluated during the first 4 weeks on treatment, PK/PD post-first dose and after 4 weeks.
Results.
Seventy-one patients were screened and 58 treated with Debio 1347 at doses from 10 to 150 mg/day. Predominant tumor types were breast and biliary duct cancer, most common gene alterations were FGFR1 amplifications (40%) and mutations in FGFR2 (12%) and FGFR3 (17%); 12 patients (21%) showed FGFR fusions. Five patients at three dose levels had 6 DLTs (dry mouth/eyes, hyperamylasemia, hypercalcemia, hyperbilirubinemia, hyperphosphatemia, stomatitis). The maximum tolerated dose was not reached, but dermatological toxicity became sometimes dose-limiting beyond the DLT period at ≥80 mg/day. Adverse events required dose modifications in 52% of patients, mostly due to dose-dependent, asymptomatic hyperphosphatemia (22%). RECIST responses were seen across tumor types and mechanisms of FGFR activation. Six patients, three with FGFR fusions, demonstrated partial responses, 10 additional patients tumor size regressions of ≤30%. Plasma half-life was 11.5 h. Serum phosphate increased with Debio 1347 plasma levels and confirmed target engagement at doses ≥60 mg/day.
Conclusion.
Preliminary efficacy was encouraging and tolerability acceptable up to 80 mg/day, which is now used in an extension part of the study.
Keywords: Debio 1347, fibroblast growth factor, FGFR inhibitor, first-in-human, targeted therapy
INTRODUCTION
Signaling mediated by fibroblast growth factor receptors (FGFR) is upregulated in many cancers (1). The FGFR family comprises 5 members, 4 of which are receptor-type tyrosine kinases (2). By alternative splicing, they form 7 isoforms, i.e. FGFR 1b, 1c, 2b, 2c, 3b, 3c, and 4, which are expressed at tissue-specific levels and vary in their specificity for 22 different known FGF ligands. However, despite this variability in receptors and ligands, downstream signaling mainly occurs through two pathways, i.e. the Ras-dependent mitogen-activated protein kinase (MAPK) and the Ras-independent phosphoinositide 3-kinase (PI3K) pathway (3). Accordingly, interruption of signaling through these pathways with isoform-specific inhibitors may more readily be bypassed as opposed to use of pan-FGFR tyrosine kinase inhibitors (TKIs). However, unspecific pan-FGFR TKIs also block FGF19 signaling through FGFR 4 and by this may increase the risk of liver toxicity (4). Thus, FGFR 1–3 inhibitors might be a valuable treatment option in cancer patients with genetic alterations of FGFR 1 to 3, particularly in those with liver dysfunction.
A variety of genetic alterations of FGFRs have been found in almost all types of tumors (5–14). Gene amplification or aberrant transcriptional regulation can result in receptor overexpression, whilst a number of point mutations render receptors either constitutively active or less dependent on ligand binding for activation. In addition, chromosomal translocation can result in the expression of FGFR-fusion proteins with constitutive FGFR kinase activity (15). Finally, isoform switching alters the ligand binding specificity of resulting receptors and sensitizes cells to FGFs that they would normally not respond to (16, 17). Aberrant expression, amplification, and overexpression of FGF ligand proteins, as well as altered gene splicing of FGFRs represent other mechanisms through which FGFR signaling can become dysregulated in cancer (16, 18, 19). There is some early clinical evidence that inhibition of FGFRs is effective in patients with FGFR-dependent cancers (20, 21). Moreover, aberrant activation of FGFR-signaling was shown to result in poor prognosis and amplification of FGFR1 to be the strongest independent predictor of poor outcome in breast cancer (5).
Several FGFR TKIs are currently in clinical development (22). Most of them also inhibit receptors of vascular endothelial or platelet-derived growth factors due to the structural similarity of the kinase domains, bearing a broader risk of side effects, in particular of cardiovascular complications. In view of the multiple FGFR isoforms that might be involved, monoclonal antibodies specific to one isoform might not be able to switch off tumorigenic signaling.
The small molecule Debio 1347 (CH5183284), an ATP-competitive, highly selective inhibitor of FGFRs 1–3 at low nanomolar concentrations in vitro, was shown to be effective in several tumor models with FGFR alterations in vivo (23). Simultaneous targeting of FGFR1–3 may broaden indications and maintain activity against drug-resistant mutations. In the present phase I trial (NCT01948297), we embarked on a targeted approach by using Debio 1347 in patients prospectively tested positive for FGFR1–3 alterations only.
METHODS
Patients
Eligible were adult patients with advanced solid tumors harboring alterations in FGFR1–3 genes as assessed at participating sites using a number of locally approved diagnostic molecular pathology assays including fluorescence in situ hybridization (FISH), DNA and RNA based; next generation sequencing (NGS; see Supplemental methods). Patients had previously progressed on standard treatment, had radiologically measurable or clinically evaluable tumor, an Eastern Cooperative Oncology Group (ECOG) performance status ≤2, an estimated life-expectancy ≥16 weeks, and adequate bone marrow, liver, and renal function. Patients were excluded if they had prior FGFR treatment, symptomatic or unstable brain tumors or metastases, a history of endocrine alteration of calcium-phosphate homeostasis or ectopic mineralization/calcification, or corneal disease. Therapy with anticoagulants, systemic steroids, chronic immune-suppressants, any drug affecting calcium and phosphorus metabolism or with known risk of QTc prolongation had to be stopped for the duration of the trial.
Prior to enrolment, all patients provided written informed consent. The protocol was approved by institutional review boards/ethics committees and the study conducted according to the Declaration of Helsinki and all applicable legal regulatory requirements.
Study design
This was a first-in-human, multicenter, open-label, dose-escalation study to determine dose-limiting toxicities (DLTs) and the maximum tolerated dose (MTD) of Debio 1347 after 4 weeks of treatment (although it has meanwhile been acknowledged that toxicities of FGFR inhibitors may develop beyond the first month of use and that tolerable doses in clinical practice might be determined through monitoring of serum phosphate levels (24)). Secondary objectives were to determine the (1) recommended phase 2 dose (RP2D); (2) preliminary anti-tumor activity and (3) pharmacokinetics (PK) of Debio 1347, and (4) to explore pharmacodynamic (PD) biomarkers and PK/PD relationships.
As 4 mg/kg/day was the highest non-severely toxic dose in 4-week repeated-dose toxicity studies with monkeys, the most sensitive and relevant animal model, 10 mg/day was selected as starting dose. An allometric scaling model of single dose PK data from mice, rats, and monkeys predicted a plasma half-life of 9 hours in humans. Still, the study was initiated with a once-daily dosing regimen, which was maintained after a preliminary analysis revealed sustained plasma levels over a 24-hour dosing interval.
Per the original protocol, dose-escalation followed a modified 3+3 design which was later amended to a 3+3+3 design due to difficulties with a DLT assessment in one patient at the 80 mg level who developed asymptomatic pancreatitis after changing several medications simultaneously. According to the 3+3+3 design, doses were escalated if no DLT was observed during the first treatment course in the initial 3 patients, in 1 out of the initial 3, but in none of further 3 patients to be treated at the same dose level; in 2 out of 3+3 patients, but in none of another 3 patients to be treated at the same dose level, until the highest dose level planned in the protocol was attained. The MTD was defined as the highest dose level at which at least 2 out of 3 or 3 out of 6 to 9 patients, experienced a DLT during the first 28 days of treatment. Otherwise, the decision for the RP2D had to be taken on the basis of safety, PK/PD, and efficacy data. Another amendment concerned a switch of the drug formulation from capsules to tablets with determination of the relative bioavailability after a single dose of 40 mg in a cross-over design, as published previously (25).
During dose-escalation, sequential patient cohorts received Debio 1347 once daily in treatment courses of 28 days until disease progression, unacceptable toxicity, or the decision to discontinue by either investigator or patient. As supportive care measures, enrolled patients were advised to restrict dietary phosphate to prevent hyperphosphatemia, a suspected drug class-effect. For gastrointestinal disorders, hematological support, infections, and pain, the usual standard of care was permitted.
After the first 28 days of treatment, patients were evaluated for DLT, tolerability, and PK/PD, and thereafter monthly for safety and disease status. Patients were enrolled onto subsequent cohorts per consensus decision among investigators and sponsor after they had reviewed all available cases. Patients not completing the first treatment course for reasons other than DLT were replaced. Patients experiencing a DLT were able to continue after dose reduction, if deemed appropriate.
Outcome measures
Safety/Tolerability
Primary endpoint was the occurrence of DLTs. A DLT was defined as possibly treatment-related (a) neutropenia (absolute neutrophil count, ANC <1.0/nL) along with fever ≥38.2°C, of CTC-grade ≥3 with infection, or of grade 4 persisting for >7 days; (b) thrombocytopenia of grade 3 requiring platelet transfusion or of grade 4 persisting for >7 days; (c) diarrhea, constipation, nausea, vomiting or skin toxicity of grade 4 or of grade 3, if lasting >72 h despite optimal symptomatic therapy; (d) non-hematologic toxicity of grade ≥3 (except electrolyte abnormalities lasting <48 h, hepatotoxicity resolving to grade 1 [or 2 in patients with liver metastases] within 7 days, alkaline phosphatase increase if related to bone metastases); (e) serum hyperphosphatemia, if >7.0 mg/dL and lasting for >7 consecutive days or if >9.0 mg/dL, both despite phosphorus lowering therapy for ≥14 days, or if >10.0 mg/dL; (f) adverse events (AEs) causing treatment delays >7 days; or (g) other life-threatening toxicity.
Secondary safety endpoints were the incidence of treatment-emergent (i.e. until 28 days from last dose) AEs (TEAEs), associated treatment discontinuations/modifications, the change in vital signs, electrocardiograms, safety laboratory, and ophthalmological exams. TEAEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE version 4.0). As there is no CTCAE grading for hyperphosphatemia, serum phosphate levels were graded according to ad hoc criteria (grade 1, >upper limit of normal (ULN) to 5.4 mg/dL; grade 2: 5.5 to 6.9 mg/dL; grade 3: 7.0 to 9.9 mg/dL, as soft tissue calcifications may develop; grade 4: >10 mg/dL regardless of renal impairment) to implement specific hyperphosphatemia management guidelines (Supplemental methods).
Efficacy
Tumor response was assessed as secondary endpoint according to RECIST (version 1.1) every 6 weeks from baseline until treatment course 6 and every 3 courses thereafter. An independent radiological review of all responses and a post-hoc analysis to confirm the FGFR genetic alterations were performed centrally (the post-hoc FGFR analysis was performed at the MGH Translational Research laboratory using CLIA validated, locally developed tests such as an DNA-based oncopanel for mutation screening, an RNA-based panel for fusion detection and FISH for amplification level assessment).
PK
Serial blood samples were collected pre- and post-first (0.5, 1, 1.5, 2, 3, 4, 6, 8, 24, 32, 48 h) and last dose (same schedule until 24 h post-dose) of the first treatment course as well as pre-dose in weekly intervals in-between (on days 8, 15, 22), and at the beginning of the 3rd course for the determination of Debio 1347 plasma levels using a validated LC-MS/MS assay (Supplemental methods). Standard PK parameters were calculated in a non-compartmental analysis using Phoenix WinNonlin version 7.0 (Certara).
PD
Blood samples for the determination of phosphate and FGF23 were collected at screening, pre- and post-first dose (2, 4, 8, 24 h) as well as after the last dose of the first treatment course. Both were measured with standard commercial assays.
Optional skin and tumor biopsies were collected at baseline and after 8 days of treatment to determine potential markers of FGFR inhibition, i.e. phosphorylated FRS2, ERK and S6 in skin biopsies by immunohistochemistry, and dual-specificity phosphatase 6 (DUSP6) in tumor biopsies using RNAscope® technology (Supplemental methods). DUSP6 might be less prone to pre-analytical bias and proved to be a marker of Debio 1347 efficacy in FGFR-addicted cancers (26).
Statistical analysis
Based on the original design (3+3), the total number of patients for the MTD evaluation was expected to be 49. After switch to the 3+3+3 design, this was adjusted to 58. For safety, the incidence of TEAEs was calculated overall, by dose, severity, and relatedness. For efficacy, best overall response, change in tumor size by time, the best change in tumor size, and the number and percentage of patients with disease control were calculated. PK parameters and for biomarkers mean and maximum changes through treatment were calculated by dose group. In addition, Loess curve fitted by polynomials of degree 1 and cubic interpolation was presented graphically to explore the association between PK/PD data using SAS version 9.4.
RESULTS
From August 2013 to March 2017, 71 patients were screened and 58 enrolled at four sites in the United States and at one site in Europe. All patients received at least one Debio 1347 dose in 8 subsequent dose cohorts of 10, 20, 30, 40 (capsule formulation), 40, 60, 80, 110, and 150 mg daily (tablet formulation) (Supplement 1).
The most frequent primary tumors were breast and biliary duct cancer (Table 1). Various mechanisms of pathway activation were represented, including 31 patients with FGFR amplification (54%), 19 patients harboring FGFR mutations in genomic regions of interest (33%) and 12 patients with FGFR fusions (21%). One patient had high-level amplification in FGFR2 with an activating mutation on the amplicon. Overall, FGFR1 gene amplification was predominant; FGFR2 and FGFR3 genes showed mutations as the most frequent type of alteration (Table 1). Noteworthy, some patients also displayed combinations of amplifications with fusions or mutations. The post-hoc analysis did not confirm local FGFR results in 9 patients (4 amplifications; 3 mutations; and 2 fusions). For 15 patients no post-hoc analysis was performed due to missing archival biopsy or poor quality of the remaining biopsy material.
Table 1.
Patient baseline characteristics (n=58)
| Age, years | Median ± SD (range) | 58.5 ± 11.4 (26 – 79) | |
| Sex | Female | 36 (62%) | |
| Male | 22 (38%) | ||
| Race | White | 51 (88%) | |
| Black or African American | 2 (3%) | ||
| Asian | 2 (3%) | ||
| Other | 3 (5%) | ||
| ECOG | 0 | 20 (35%) | |
| 1 | 37 (68%) | ||
| 2 | 1 (2%) | ||
| Tumor type | Breast cancer | 12 (21%) | |
| Biliary tract cancer | 8 (14%) | ||
| Urothelial carcinoma | 6 (10%) | ||
| Uterine neoplasm | 5 (9%) | ||
| sqNSCLC | 4 (7%) | ||
| Gastric cancer | 4 (7%) | ||
| Prostate cancer | 2 (3%) | ||
| Cervical cancer | 2 (3%) | ||
| Others (each with only one occurrence) | 15 (26%) | ||
| FGFR gene alteration status* | Local | Central* | |
|---|---|---|---|
| FGFR1 | Amplification | 23 (40%) | 15 (26%) |
| Fusion | 1 (2%) | 0 | |
| Mutation | 2 (3%) | 0 | |
| FGFR2 | Amplification | 5 (9%) | 5 (9%) |
| Fusion | 6 (10%) | 5 (9%) | |
| Mutation | 7 (12%) | 3 (5%) | |
| FGFR3 | Amplification | 4 (7%) | 1 (2%) |
| Fusion | 5 (9%) | 4 (7%) | |
| Mutation | 10 (17%) | 3 (5%) | |
Patients with multiple concurrent alterations were counted for each alteration. A post-hoc central analysis did not confirm any local FGFR results in 9 patients (4 amplifications; 3 mutations; and 2 fusions). Among those 9 patients, one patient had a concomitant alteration not tested in post-hoc analysis for technical reasons. For 15 patients no post-hoc analysis was performed due to the lack or poor quality of remaining biopsy material.
Safety
All 58 treated patients had at least one TEAE, of which the highest severity was of grade 1 in 4 (7%), grade 2 in 17 (29%), grade 3 in 28 (48%), grade 4 in 2 (3%) and grade 5 in 7 (12%) patients. Overall, 21 patients died from disease progression; no death was considered drug-related. The most common TEAEs with an incidence >25% were hyperphosphatemia, diarrhea, nausea, fatigue, constipation, decreased appetite, nail changes, and dry mouth (Table 2). Most often severe (i.e. of grade >2) were hyperphosphatemia, anemia, hyponatremia, and dyspnea. In total, 20 patients experienced 39 SAEs, most commonly dyspnea, but only two SAEs were deemed possibly study drug related and thus DLTs, i.e. hyperamylasemia (80 mg) and stomatitis (110 mg). DLTs occurred in 3 other patients, i.e. grade-2 dry mouth and eyes (60 mg); grade-3 asymptomatic hypercalcemia (80 mg); grade-3 bilirubin increase and hyper-phosphatemia (110 mg) (Supplement 2). The patient with hyperamylasemia died from disease progression. The remaining 4 patients recovered after Debio 1347 interruption and/or dose modification. As there were only ≤2 DLTs at the same dose level, the MTD was not formally reached. Dose modifications due to AEs were required by 30 (52%) patients (Supplement 3). At ≥110 mg, 7 out of 9 patients required a dose reduction after completion of the 28-day DLT period, predominantly due to hyperphosphatemia, stomatitis, skin, and nail toxicities. At 60 and 80 mg, these were only required in 20% of patients and up to 40 mg, no dose reduction was required at all.
Table 2.
Number (%) of patients with most common TEAE (incidence ≥10%) irrespective of relatedness to study drug
| Dose level [mg] | 60 (n= 10) | 80 (n= 10) | 110 (n=9) | 150 (n=3) | all (n = 58) | |
|---|---|---|---|---|---|---|
| Grade | all | all | all | all | all | ≥3 |
| Any | 10 (100) | 10(100%) | 9 (100%) | 3 (100%) | 58 (100%) | 37 (64%) |
| Any leading to | ||||||
| treatment interruption | 6 (60) | 5 (50) | 8 (89) | 3 (100) | 26 (44.8) | - |
| dose reduction | 2 (20) | 2 (20) | 7 (78) | 2 (68) | 13 (22.4) | - |
| discontinuation | 1 (10) | 3 (30) | 1 (11) | 0 (0) | 7 (12.1) | - |
| Serious | 2 (20) | 3 (30) | 4 (44) | 2 (67) | 20 (34.5) | - |
| Hyperphosphatemia | 9 (90) | 9 (90) | 9 (100) | 3 (100) | 44 (76) | 12 (21) |
| Diarrhea | 7 (70) | 5 (50) | 3 (33) | 1 (33) | 24 (41) | 2 (3) |
| Nausea | 3 (30) | 3 (30) | 6 (67) | 2 (67) | 23 (40) | 0 |
| Fatigue | 5 (50) | 5 (50) | 5 (55) | 0 | 22 (38) | 0 |
| Constipation | 2 (20) | 3 (30) | 3 (33) | 1 (33) | 19 (33) | 1 (2) |
| Decreased appetite | 3 (30) | 5 (50) | 3 (33) | 1 (33) | 18 (31) | 1 (2) |
| Nail changesa | 2 (20) | 3 (30) | 8 (81) | 3 (100) | 17 (29) | 0 |
| Dry mouth | 4 (40) | 5 (50) | 4 (44) | 1 (33) | 15 (26) | 0 |
| Stomatitis | 1 (10) | 3 (30) | 7 (78) | 1 (33) | 12 (21) | 2 (3) |
| Abdominal pain | 3 (30) | 3 (30) | 2 (22) | 1 (33) | 12 (21) | 1 (2) |
| Anemia | 4 (40) | 2 (20) | 3 (33) | 0 | 12 (21) | 7 (12) |
| Dyspnea | 3 (30) | 1 (10) | 3 (33) | 1 (33) | 12 (21) | 3 (5) |
| Vomiting | 2 (20) | 1 (10) | 3 (33) | 1 (33) | 11 (19) | 0 |
| Myalgia | 1 (10) | 3 (30) | 3 (33) | 0 | 12 (21) | 0 |
| Alopecia | 1 (10) | 1 (10) | 4 (44) | 2 (67) | 11 (19) | 0 |
| Dry skin | 0 | 2 (20) | 5 (55) | 1 (33) | 10 (17) | 0 |
| Back pain | 0 | 0 | 1 (11) | 1 (33) | 9 (16) | 1 (2) |
| Dysgeusia | 1 (10) | 2 (20) | 4 (44) | 2 (67) | 10 (17) | 0 |
| Dry eye | 2 (20) | 0 | 4 (44) | 1 (33) | 8 (14) | 0 |
| Hypokalemia | 0 | 2 (20) | 1 (11) | 0 | 8 (14) | 1 (2) |
| Creatinine increasedb | 2 (20) | 2 (20) | 0 | 0 | 8 (14) | 0 |
| Mucosal inflammation | 1 (10) | 3 (30) | 2 (22) | 1 (33) | 8 (14) | 1 (2) |
| Edema peripheral | 1 (10) | 2 (20) | 2 (22) | 1 (33) | 8 (14) | 0 |
| Hypomagnesemia | 2 (20) | 1 (10) | 0 | 0 | 7 (12) | 0 |
| Hypertension | 1 (10) | 0 | 0 | 0 | 7 (12) | 0 |
| Blurred vision | 1 (10) | 2 (20) | 2 (22) | 0 | 6 (10) | 0 |
| ALT increased | 1 (10) | 1 (10) | 1 (11) | 0 | 6 (10) | 2 (3) |
| AST increased | 2 (20) | 1 (10) | 1 (11) | 0 | 6 (10) | 1 (2) |
ALT, alanine transaminase; AST, aspartate transaminase;
include onychomadesis, onychoclasis, onychalgia, nail dystrophy, nail bed disorder, and nail discoloration;
in blood
Percentages were calculated using the number of patients in the safety population of each group as denominator. Recurring events are counted only once for each patient with highest grade
The incidence and severity of hyperphosphatemia were both dose-dependent (Figure 1). Increases occurred already at 20 mg and typically in-between the 1st and 3rd week of dosing, were always asymptomatic, without clinical complications, and generally resolved with the use of a phosphate chelator (sevelamer, depending on severity of hyperphosphatemia either alone or in combination with acetazolamide as instructed by study specific management guidelines, Supplemental methods) and/or interruption of the investigational product, which was required in 13 (22%) patients. Dermatological toxicity became clinically relevant at doses ≥80 mg with 11 out of 12 patients suffering from “nail changes”, i.e. nail bed disorders, nail discoloration, nail dystrophy, onychalgia, onychoclasis, and onychomadesis (Table 2). Severity never exceeded grade 2, but 3 patients with nail changes and 3 with palmar-plantar erythrodysesthesia syndrome required dose adjustment. Among eye disorders, another potential drug class effect, blurred vision was reported in 6 patients (at 30–150 mg), however severity was mainly of grade 1; there were no findings on ocular exams, in particular none compatible with central serous retinal detachment (27). Three patients experienced QTc prolongations of grade 1 or 2. No complications were observed during biopsy sampling with 2 patients reporting AEs of grade 1 and 2 pain only.
Figure 1.
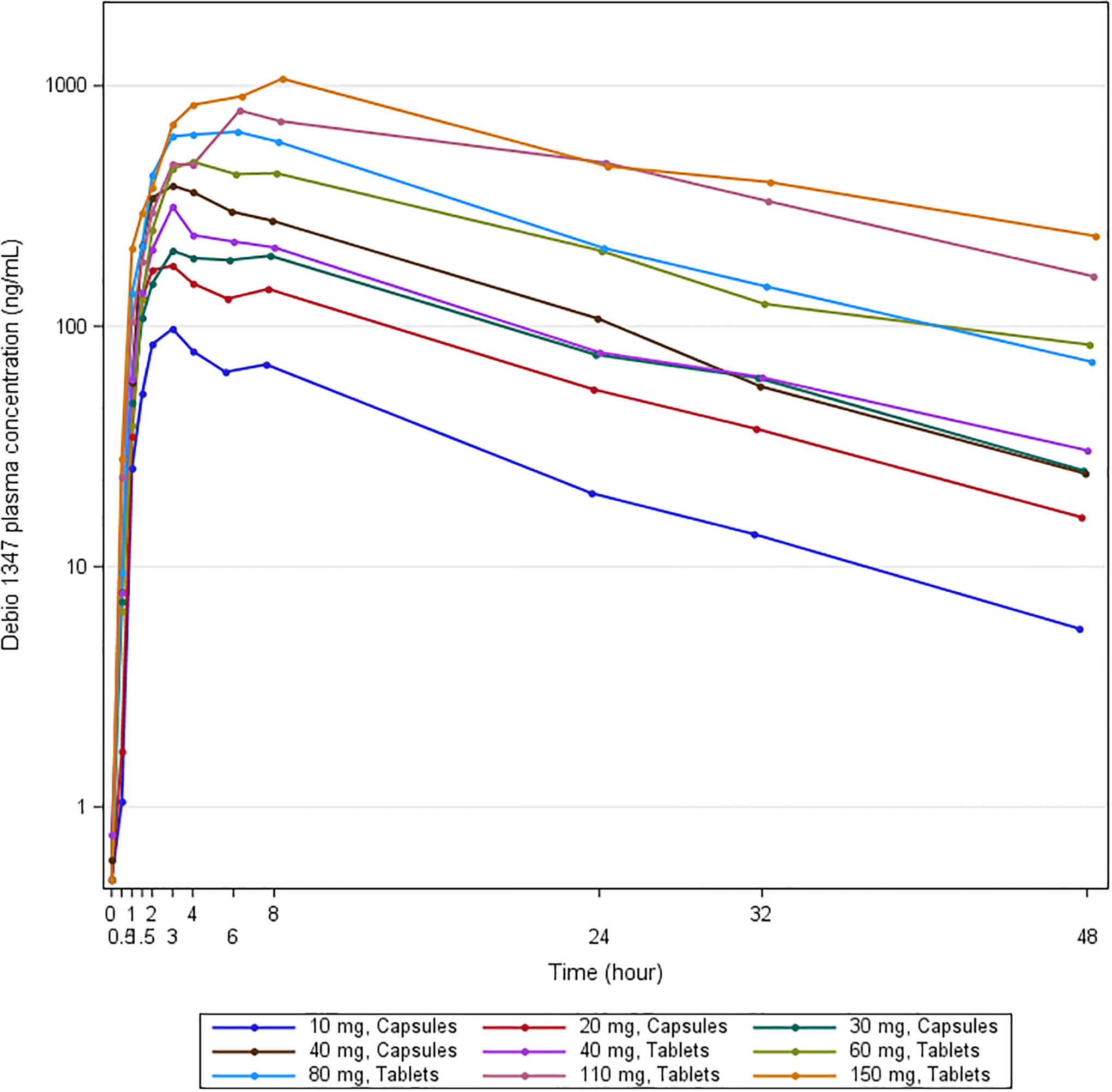
Geometric means of Debio 1347 plasma concentrations post-first dose
Efficacy
A total of 57 patients were evaluable for tumor response. One patient had no measurable disease. Overall, 6 patients achieved PRs (2 unconfirmed; Figure 2, Supplement 4); for 16 patients best objective response was stable disease, of whom 10 showed reduced target lesion size at least once post-dose; the remaining 35 patients had progressive disease, 13 per clinical course rather than radiographic assessment. Underlying malignancy for the 6 PRs included urothelial carcinoma (two patients: 80, 150 mg), endometrial carcinoma (one patient: 30 mg), cervical carcinoma (one patient: 80 mg), IBD-associated colorectal cancer or cholangiocarcinoma (each one patient; each 110 mg). For the latter the independent central radiological review changed response from partial to confirmed complete response (CR). All 6 patients with PR had confirmed FGFR alterations on central assessment. Among the 16 patients with stable disease, post-hoc analysis confirmed FGFR alterations in 11 patients, it was not performed due to insufficient biopsy material in 2 patients, and did not confirm the presence of the FGFR alteration in 3 patients (no amplification twice and no mutation once). In 18 patients with early disease progression, the post-hoc analysis did not confirm the presence of FGFR alterations (6 patients) or it could not be done (12 patients). Among 10 patients with confirmed FGFR fusions treated at doses ≥60 mg/day, 3 achieved PRs and 5 disease stabilization (Figure 3). This represents a disease control rate (DCR) in this subpopulation of 80% (versus 39% overall). The median treatment duration in patients with disease control was 34 weeks (range: 24–47).
Figure 2.
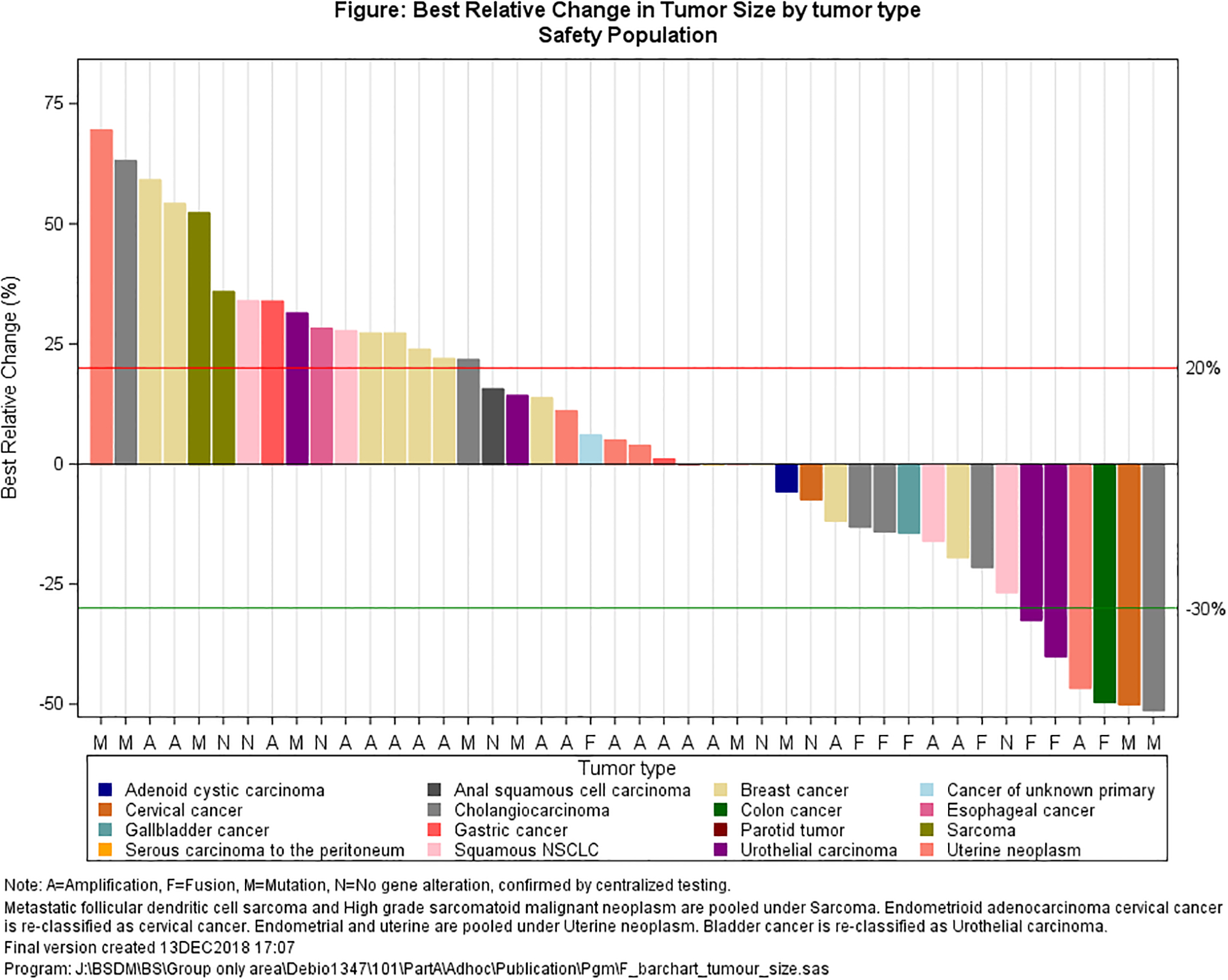
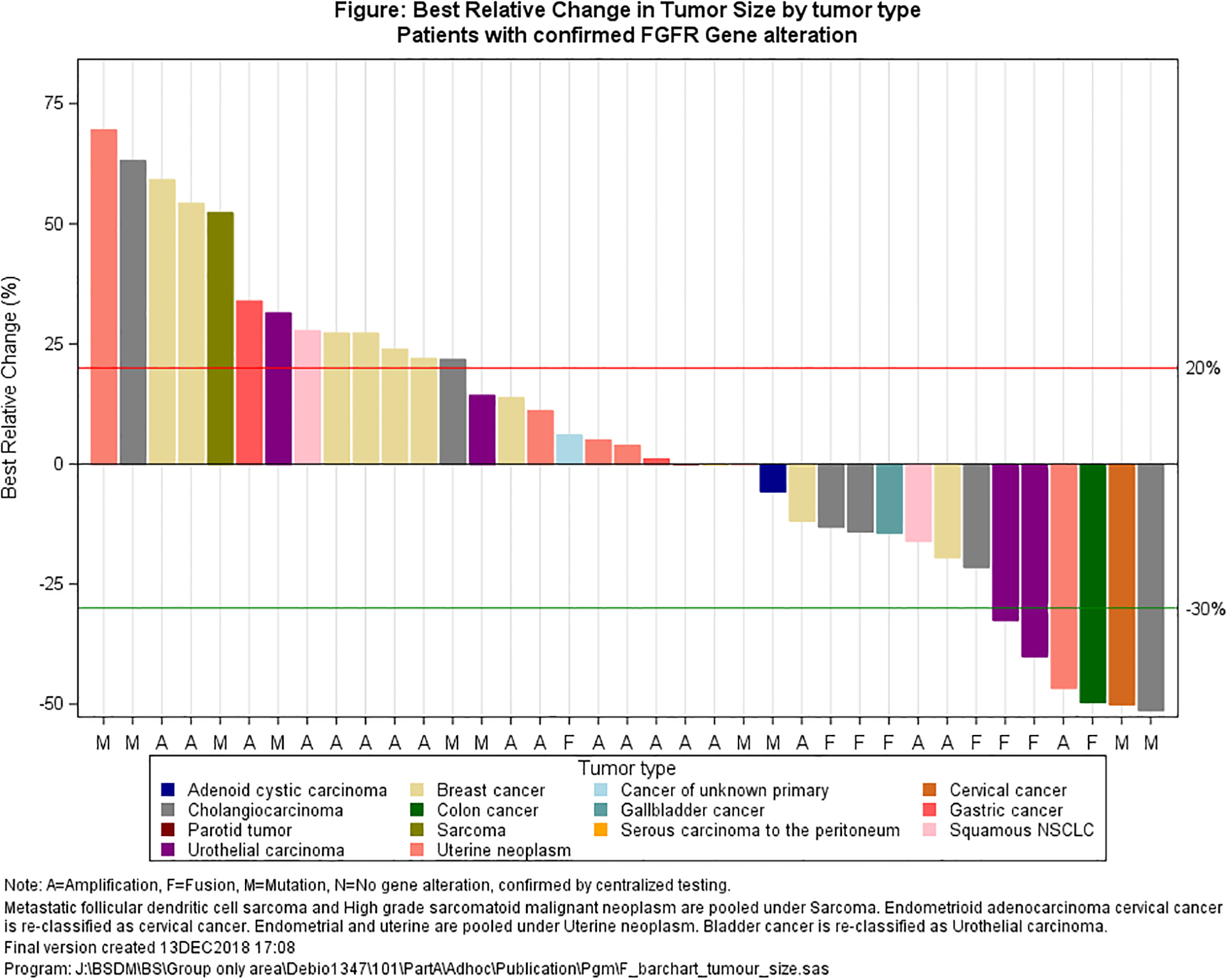
Best target lesion change from baseline (waterfall plot): a. all patients; b. only patients with confirmed genetic alteration
Figure 3.
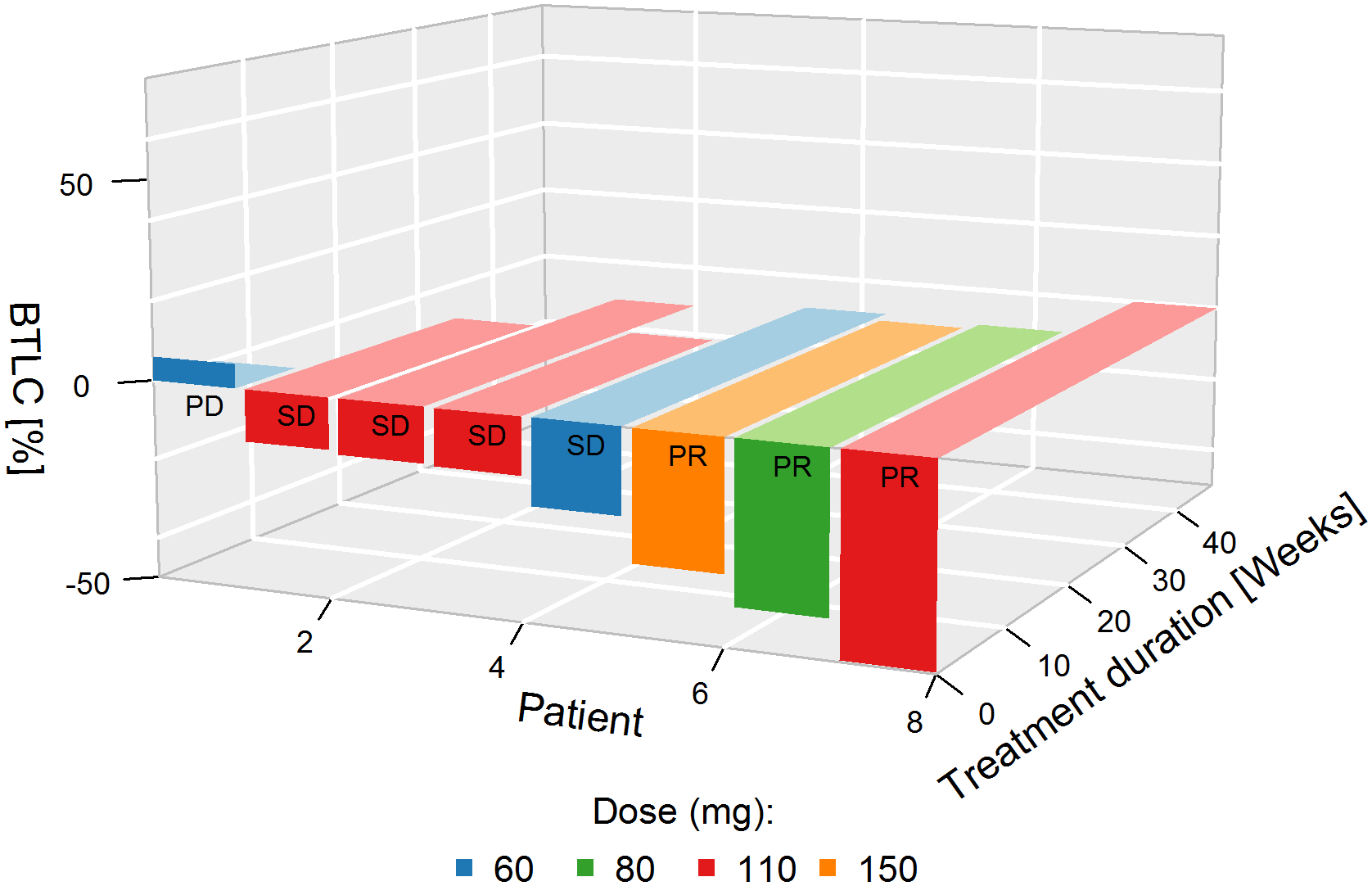
Best target lesion change from baseline in patients with FGFR fusions irrespective of histology and treated at ≥ 60 mg (3D plot)
Patient 1: Unknown – FGFR2-BICC1;
Patient 2: Cholangiocarcinoma –FGFR2-DDX21;
Patient 3: Cholangiocarcinoma – FGFR2-KIAA1217;
Patient 4: Gallbladder ca. – FGFR3-TACC3;
Patient 5: Cholangiocarcinoma – FGFR2-ROCK1;
Patient 6: Urothelial ca. – FGFR3-TACC3;
Patient 7: Urothelial ca. – FGFR3-TACC3;
Patient 8: Colon cancer –FGFR2-INA
Pharmacokinetics
In a majority of patients, Debio 1347 was detectable in plasma within 30 minutes post-first dose, but median tmax was 3 h (range: 1.5–24). Plasma levels decreased mono-exponentially with a mean half-life of 11.5 h (range: 4.7–20.6; Figure 1, Supplement 5). Mean apparent oral clearance was 7.0 L/h (range: 2.7–13.9) and mean volume of distribution 110 L (range: 57–193). At the 40 mg dose level, PK assessments were separately performed for tablet and capsule formulations which proved to be bioequivalent (mean relative oral bioavailability of tablets versus capsules: 0.88; 90% CI 0.73–1.05, n=9). Although the graphical representation of Debio 1347 plasma exposure (Cmax, AUC) against dose level suggested a potential dose-proportionality (Figure 1, Supplement 2), a power model used to test dose-proportionality was inconclusive because of the limited sample size and marked inter-individual variability (Supplements 2 and 5). No clear relationship could be recognized between exposure and the occurrence of DLTs (Supplement 2). In line with the half-life and 24-h dosing intervals, a limited accumulation was observed at the end of the first treatment course, with on average 1.9 and 1.7 times higher AUCtau and Cmax after 28 days of repeated once-daily dosing, respectively. Trough levels (Ctrough) indicated that steady-state was achieved in the majority of patients during the first week of treatment.
Pharmacodynamics
Plasma biomarkers
Hyperphosphatemia, an on-target effect of FGFR inhibition, correlated with Debio 1347 exposure (Figure 4). Accordingly, plasma FGF23, an inhibitor of phosphate reabsorption, demonstrated an overall increase at steady-state, but without clear relationship to the dose (Supplement 6).
Figure 4.
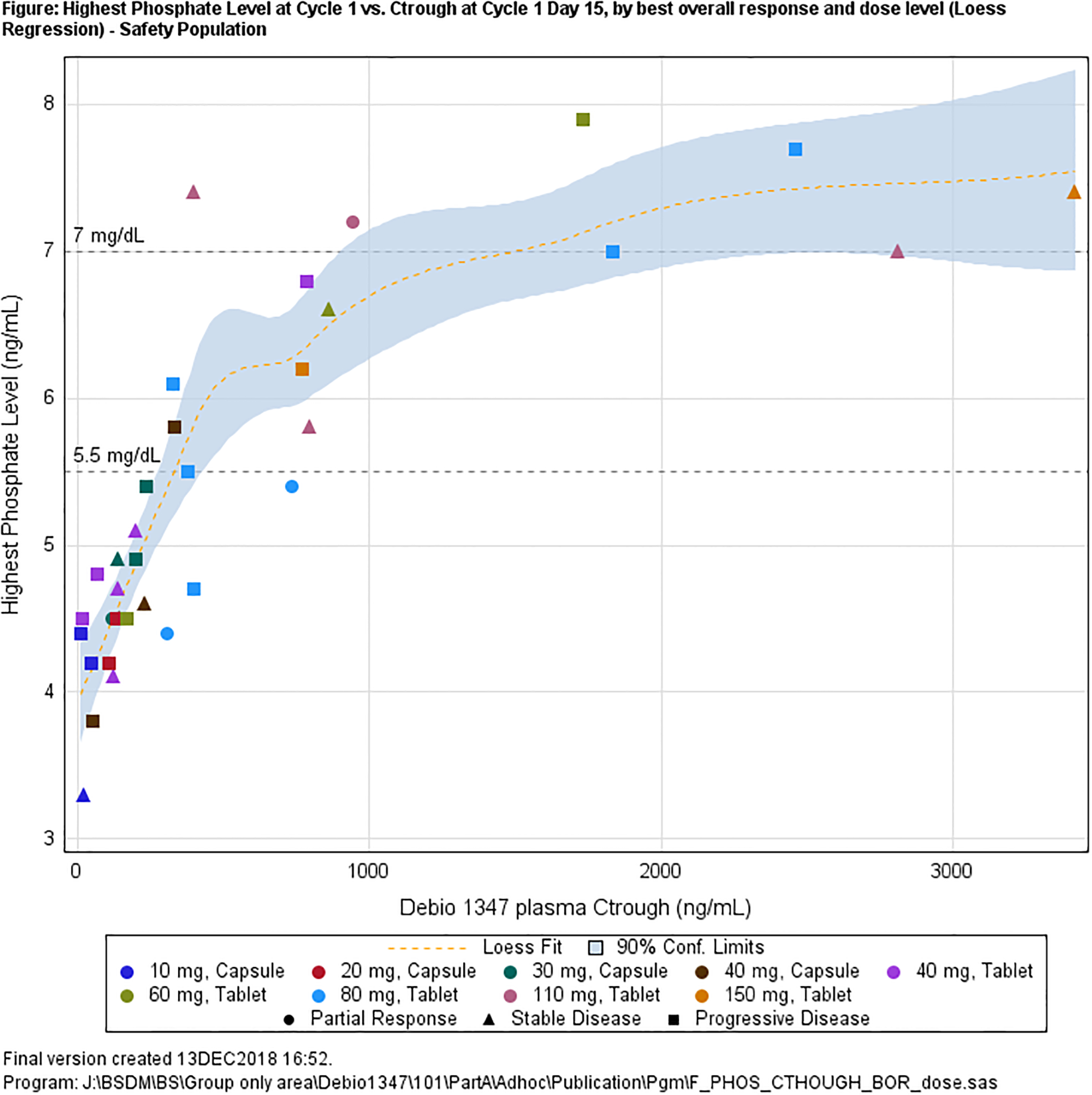
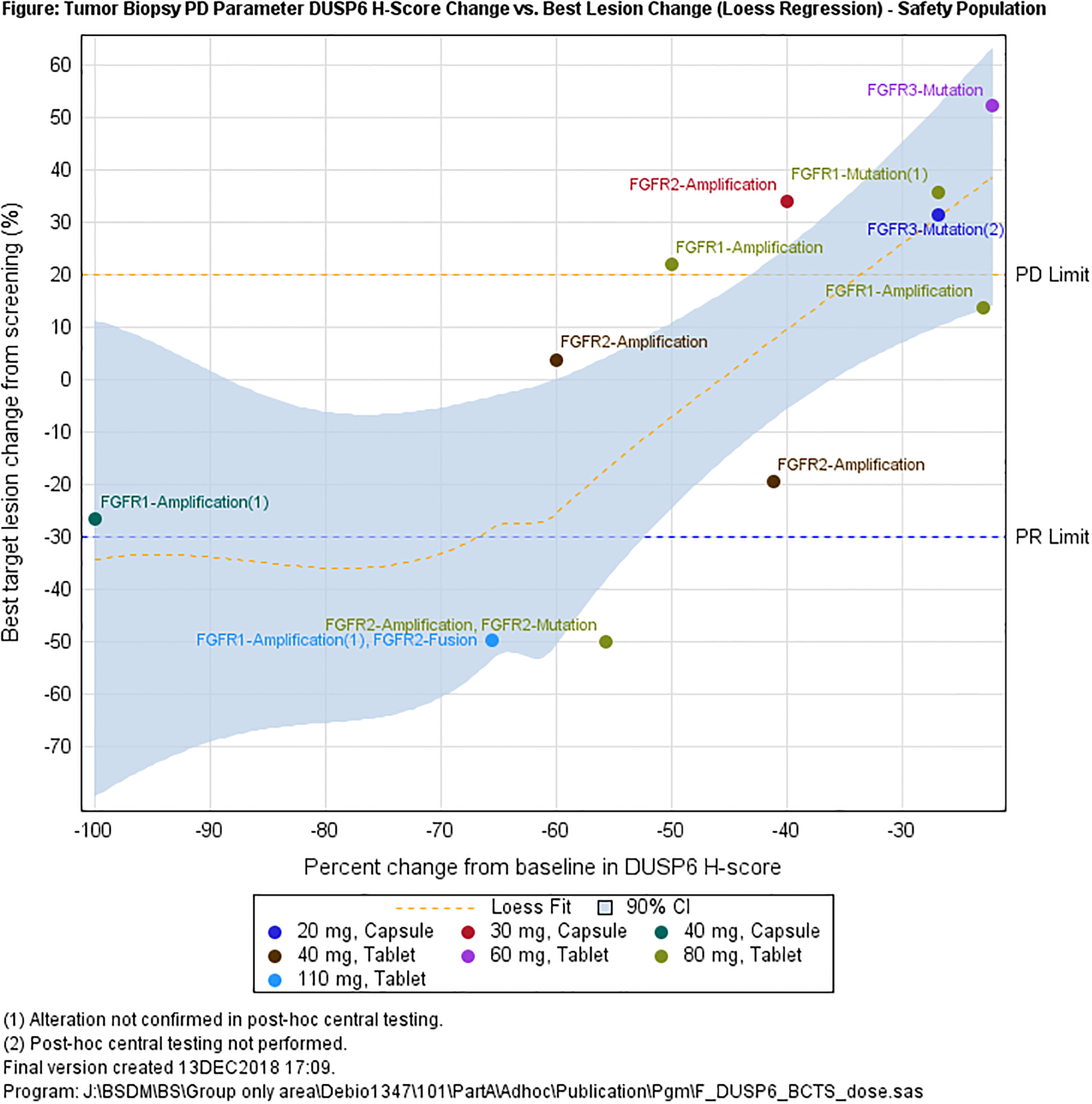
Biomarker in blood and biopsies: a. maximum serum phosphate levels observed during cycle 1 versus Debio 1347 plasma trough levels; b. DUSP6 % change from baseline versus best lesion change.
Tissue biomarkers
In total, 48 and 33 skin biopsies were collected pre- and post-treatment, respectively. One sample was lost and another one had insufficient material, so that for 31 biopsy pairs immunohistochemistry results were available. Baseline values were rather low so that no clear decrease of phosphorylated markers (pERK, pFRS2, pS6) from predose to day 8 of treatment could be demonstrated (Supplement 7).
At the same time points, 42 and 25 tumor biopsies were collected pre- and post-dose, respectively. Of these samples, 8 contained no tumor cells, 5 insufficient material, and 2 did not pass staining quality control, leaving 14 paired biopsies for analysis of DUSP6 mRNA expression. Overall, a decrease in DUSP6 was observed on treatment; two out of three patients with a decrease >50% achieved a PR on treatment (Figure 4, Supplement 9).
DISCUSSION
This first-in human study of Debio 1347 demonstrated overall acceptable tolerability. The MTD was not reached as per 3+3+3 design, and ultimately the RP2D was determined based on (i) toxicities limiting tolerance beyond the first 4 weeks of treatment, (ii) antitumor activity at each dose level, (iii) the relationship between treatment-effect and phosphate level increases, and (iv) observed pharmacokinetics (Ctrough).
The incidence and severity of hyperphosphatemia was dose-dependent and occurred in almost all patients treated at doses ≥80 mg. Other common TEAEs deemed related to Debio 1347 included diarrhea, nausea, fatigue, constipation, decreased appetite, dry mouth, and stomatitis. After completion of the 28-day DLT period, dose modifications due to TEAEs became more frequent at higher doses, recurrent dose-limiting events included hyperphosphatemia, mucositis, skin and nail toxicities. In contrast, toxicities remained manageable beyond the first month at the 80 mg/day dose level. On the other hand, 5 out of the 6 PRs occurred at doses ≥80 mg and 8 out of 12 patients reached at least stable disease as BOR at doses ≥110 mg.
Elevated serum phosphate levels, a key biomarker of FGFR signaling (24), was previously determined a sensitive indicator of on-target activity of other FGFR1–3 inhibitors at increases of 50% from baseline (20). Such increases were achieved at Debio 1347 Ctrough values of about 400 ng/mL, which were observed in approximatively half of the patients dosed with 60 mg/day and in the majority of patients dosed with ≥80 mg/day. As noted above, the majority of patients treated with doses ≥110 mg required dose reductions and long-term tolerance was limited. Based on this data, the Study Safety Committee formally endorsed an RP2D of 80 mg/daily and a dose reduction to 60 mg/day in case of intolerable toxicity.
Based on recently published phase-I data on other selective FGFR antagonists (20, 21, 28), the observed toxicity profile, predominated by dose-dependent asymptomatic hyperphosphatemia, appears to be typical for this new class of drug, despite varying dosing schedules, with BGJ398 (20) and JNJ-42756493 (21) given intermittently and LY2874455 given twice daily (28). The observed PK profile of Debio 1347 with steady-state rapidly being achieved and only limited plasma accumulation in this study supports continuous once-daily dosing.
So far, efficacy of the selective FGFR inhibitors in heavily pretreated phase-I patients looks encouraging: With BGJ398, a similar overall DCR of 37% was achieved in patients with FGFR alterations and 7 PRs (1 unconfirmed) were reported in patients with FGFR1-amplified sqNSCLC and FGFR3-mutant bladder/urothelial cancer (20). In 23 patients with FGFR alterations receiving JNJ-42756493, there were 5 PRs (1 unconfirmed) after 6–8 weeks of treatment and 8 patients had stable disease for >3 months (21). With LY2874455, the majority of patients had stable disease, except 1 PR in a gastric cancer patient (28).
For patient selection, tumor type seems less informative than the kind of underlying FGFR alteration. In our study, we observed some clustering of responses in patients with FGFR fusions, the depth of response appearing to increase over time (Supplement 8). However, the targeted approach of testing for FGFR1–3 alterations beforehand is not without problems as demonstrated by the discordance between local pre-screen and central post-hoc results. This might have been due to intratumor heterogeneity and sampling errors, the dynamics of acquired somatic mutations and genomic evolution or technical discrepancies between local pre-screening and central confirmatory assays. The latter could be due to (i) evolving analysis algorithms during the study, to (ii) differences in annotating structurally complex fusions, or to (iii) the more accurate reading of amplifications using FISH as compared to NGS. Those discrepancies exemplify in our opinion the current diagnostics landscape and the issues that may be encountered when using such complex technologies.
Our findings for serial DUSP6 mRNA are of interest for future study, although presented data are still limited: In preclinical models its decrease had previously been reported as potential marker of FGFR inhibition by Debio 1347 (26) or AZD4547 (29). In patient-derived xenograft models, a 50% decrease after Debio 1347 was interpreted as clear evidence of on-target effect (30). To our knowledge this study is the first to report on DUSP6 mRNA assessment in the clinical setting. In keeping with preclinical findings, we saw decreases in DUSP6 levels after initiation of Debio 1347. The finding that some of those patients with the deepest decrease in DUSP6 levels were able to achieve PRs (Figure 4) is notable and deserves further investigation in future studies of FGFR inhibitors.
Based on the findings presented here, which demonstrate manageable toxicity and encouraging efficacy in adequately selected patients further clinical development of Debio 1347 is warranted and will be pursued at continuous once-daily doses of 80 mg. Our study findings support development across diseases in a molecularly defined patient population, which will be continued in the next steps of clinical development for this new agent.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Fibroblast growth factor receptors (FGFR), in their function as upstream tyrosine kinases activating the MAPK and the PI3K signaling pathways can regulate cellular growth and have established roles in human tumorigenesis. In that setting, increased kinase signaling can be brought about by a variety of genetic alterations which can be detected in patient tumor samples, identifying those patients most likely to benefit from FGFR-directed therapy. Debio 1347 is a highly selective, orally available FGFR 1–3 inhibitor. In the present phase I trial, its safety and efficacy was explored in a target population of patients screened positive for harboring activating FGFR1–3 alterations in their tumors. This approach proved to be feasible with promising tolerability and efficacy results.
ACKNOWLEDGMENTS
We thank Uwe Totzke (TDS, Basel) for medical writing support and Marie-Claude Roubaudi-Fraschini (Debiopharm International) for the bioanalytical work.
Financial support:
This study was sponsored by Debiopharm International SA.
Conflict of interest disclosure statement:
M. H. Voss: Research grants from Bristol-Myers Squibb and Genentech/Roche; honoraria from Novartis; travel/accommodation from Novartis and Takeda; consultant/advisory board member for Alexion, Bayer, Calithera Biosciences, Corvus, Exelixis, Eisai, GlaxoSmithKline, Natera, Novartis, and Pfizer. J. M. Cleary: Research funding from Merck and Tesaro, travel funding from Bristol Myers Squibb, Agios, and Roche; consultant for Bristol-Myers Squibb. F. Meric-Bernstam: Research grants from Novartis, AstraZeneca, Taiho, Genentech, Calithera, Debiopharm International, Bayer, PUMA, Aileron, Jounce, CytoMx, Effector, Zymeworks, Curis, and Pfizer; consultant/advisory board member for Dialecta, Sumitomo Dainippon, Pieris Pharmaceuticals, Darwin Health, Samsung Bioepis, Aduro, Spectrum, OrigiMed, Debiopharm International, Inflection Biosciences, Xencor, and Genentech. J. Tabernero: Scientific consultant for Bayer, Boehringer Ingelheim, Chugai, Genentech, Ipsen, Lilly, MSD, Merck Serono, Merrimack, Merus, Novartis, Peptomyc, Pfizer, Rafael Pharmaceuticals, Roche, Sanofi, Symphogen, and Taiho. L. Gandhi: Consultant/advisory board member for Merck, Genentech/Roche, Astra-Zeneca, Ignyta and Syndax at the time of the study; currently employee of Lilly Research Laboratories. A. J. Iafrate: Stock/ownership interest in Archer DX; consultant/advisory board member for Roche, Chugai Pharmaceutical and Debiopharm International. N. Ishii: employee of Chugai Pharmaceutical. Y. Hu, Y. Kirpicheva, V. Nicolas-Metral, A. Pokorska-Bocci, A. Vaslin, C. Zanna: Employees of Debiopharm International. K. Flaherty: Honoraria and travel/accommodation from Debiopharm International; consultant/advisory board member for Debiopharm International. J. Baselga: Board of Directors for Varian Medical Systems, Bristol-Myers Squibb and Foghorn, and past board member of Grail, Aura Biosciences and Infinity Pharmaceuticals; consultant for Grail, PMV Pharma, ApoGen, Juno, Roche, Lilly, Novartis and Northern Biologics. Stock or other ownership interests in PMV Pharma, Grail, Juno, Varian, Foghorn, Aura, Infinity, ApoGen, Tango and Venthera, of which he is a co-founder; honoraria or travel expenses from Roche, Novartis, and Lilly. No potential conflicts of interest were disclosed by the other authors.
Abbreviations:
- AE
adverse event
- AUC
area under the curve
- BOR
best overall response
- CLIA
Clinical Laboratory Improvement Amendments
- CR
complete response
- CTC
common toxicity criteria
- DCR
disease control rate
- DLT
dose limiting toxicities
- DUSP6
dual-specificity phosphatase 6
- ECOG
Eastern Cooperative Oncology Group
- FGF(R)
fibroblast growth factor (receptor)
- FISH
fluorescence in situ hybridization
- LC
liquid chromatography
- MAPK
mitogen-activated protein kinase
- MS
mass spectrometry
- MTD
maximum tolerated dose
- NGS
next generation sequencing
- PD
pharmacodynamics
- PI3K
phosphoinositide 3 kinase
- PK
pharmacokinetics
- PR
partial response
- RP2D
recommended phase 2 dose
- TEAE
treatment-emergent AE
- TKI
tyrosine kinase inhibitor
- ULN
upper limit of normal
REFERENCES
- 1.Hierro C, Rodon J, Tabernero J. Fibroblast Growth Factor (FGF) Receptor/FGF Inhibitors: Novel Targets and Strategies for Optimization of Response of Solid Tumors. Semin Oncol. 2015;42:801–19. [DOI] [PubMed] [Google Scholar]
- 2.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–29. [DOI] [PubMed] [Google Scholar]
- 3.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katoh M FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int J Mol Med. 2016;38:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007;9:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plowright EE, Li Z, Bergsagel PL, Chesi M, Barber DL, Branch DR, et al. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood. 2000;95:992–8. [PubMed] [Google Scholar]
- 7.Knowles MA. Role of FGFR3 in urothelial cell carcinoma: biomarker and potential therapeutic target. World J Urol. 2007;25:581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emoto N, Isozaki O, Ohmura E, Ito F, Tsushima T, Shizume K, et al. Basic fibroblast growth factor (FGF-2) in renal cell carcinoma, which is indistinguishable from that in normal kidney, is involved in renal cell carcinoma growth. J Urol. 1994;152:1626–31. [DOI] [PubMed] [Google Scholar]
- 10.Rand V, Huang J, Stockwell T, Ferriera S, Buzko O, Levy S, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A. 2005;102:14344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macdonald D, Reiter A, Cross NC. The 8p11 myeloproliferative syndrome: a distinct clinical entity caused by constitutive activation of FGFR1. Acta Haematol. 2002;107:101–7. [DOI] [PubMed] [Google Scholar]
- 12.Peng DF, Sugihara H, Mukaisho K, Tsubosa Y, Hattori T. Alterations of chromosomal copy number during progression of diffuse-type gastric carcinomas: metaphase- and array-based comparative genomic hybridization analyses of multiple samples from individual tumours. J Pathol. 2003;201:439–50. [DOI] [PubMed] [Google Scholar]
- 13.Byron SA, Gartside MG, Wellens CL, Mallon MA, Keenan JB, Powell MA, et al. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res. 2008;68:6902–7. [DOI] [PubMed] [Google Scholar]
- 14.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. [DOI] [PubMed] [Google Scholar]
- 15.Knights V, Cook SJ. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol Ther. 2010;125:105–17. [DOI] [PubMed] [Google Scholar]
- 16.Jeffers M, LaRochelle WJ, Lichenstein HS. Fibroblast growth factors in cancer: therapeutic possibilities. Expert Opin Ther Targets. 2002;6:469–82. [DOI] [PubMed] [Google Scholar]
- 17.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–49. [DOI] [PubMed] [Google Scholar]
- 18.Basilico C, Moscatelli D. The FGF family of growth factors and oncogenes. Adv Cancer Res. 1992;59:115–65. [DOI] [PubMed] [Google Scholar]
- 19.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–97. [DOI] [PubMed] [Google Scholar]
- 20.Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J Clin Oncol. 2017;35:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol. 2015;33:3401–8. [DOI] [PubMed] [Google Scholar]
- 22.Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3:264–79. [DOI] [PubMed] [Google Scholar]
- 23.Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Sakata K, Sase H, et al. The Fibroblast Growth Factor Receptor Genetic Status as a Potential Predictor of the Sensitivity to CH5183284/Debio 1347, a Novel Selective FGFR Inhibitor. Molecular Cancer Therapeutics. 2014;13:2547–58. [DOI] [PubMed] [Google Scholar]
- 24.Schram AM, Voss MH, Hyman DM. Genome-Driven Paradigm for the Development of Selective Fibroblast Growth Factor Receptor Inhibitors. J Clin Oncol. 2017;35:131–4. [DOI] [PubMed] [Google Scholar]
- 25.Nicolas-Metral V, Vaslin A, Supko JG, Nakai K, Ishii N, Menetrey A, et al. Abstract CT228: Formulation switch and pharmacokinetics/pharmacodynamics of Debio 1347 (CH5183284), a novel FGFR inhibitor, in a first-in-human dose escalation trial in solid tumors patients. Cancer Res. 2015;75:CT228–CT. [Google Scholar]
- 26.Nakanishi Y, Mizuno H, Sase H, Fujii T, Sakata K, Akiyama N, et al. ERK Signal Suppression and Sensitivity to CH5183284/Debio 1347, a Selective FGFR Inhibitor. Mol Cancer Ther. 2015;14:2831–9. [DOI] [PubMed] [Google Scholar]
- 27.van der Noll R, Leijen S, Neuteboom GH, Beijnen JH, Schellens JH. Effect of inhibition of the FGFR-MAPK signaling pathway on the development of ocular toxicities. Cancer Treat Rev. 2013;39:664–72. [DOI] [PubMed] [Google Scholar]
- 28.Michael M, Bang YJ, Park YS, Kang YK, Kim TM, Hamid O, et al. A Phase 1 Study of LY2874455, an Oral Selective pan-FGFR Inhibitor, in Patients with Advanced Cancer. Target Oncol. 2017;12:463–74. [DOI] [PubMed] [Google Scholar]
- 29.Delpuech O, Rooney C, Mooney L, Baker D, Shaw R, Dymond M, et al. Identification of Pharmacodynamic Transcript Biomarkers in Response to FGFR Inhibition by AZD4547. Mol Cancer Ther. 2016;15:2802–13. [DOI] [PubMed] [Google Scholar]
- 30.Brichory F, Pokorska-Bocci A, Nuciforo P, Rigotti S, Lembrez N, Vuagniaux G, et al. Abstract 2088: The activity of the FGFR selective inhibitor Debio 1347 is correlated with high mRNA expression. Cancer Res. 2017;77:2088-. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.