

Abstract

Venezuelan and eastern equine encephalitis viruses are disease-causing, neuropathic pathogens with no approved treatment options in humans. While expanding the pharmacophoric model of antialphaviral amidines prepared via a quinazolinone rearrangement, we discovered that diamine-treated, 2-dihalomethylquinolinones unexpectedly afforded ring-expanded piperazine-fused benzodiazepinones. Notably, this new chemotype (19 examples) showed potent, submicromolar inhibition of virus-induced cell death, >7-log reduction of viral yield, and tractable structure–activity relationships across both viruses. Antiviral activity was confirmed in primary human neuronal cells. A mechanistic rationale for product formation is proposed, and key structural elements were comparatively modeled between a similarly substituted antiviral amidine and piperazinobenzodiazepinone prototypes to guide future antiviral development.
Keywords: Quinazolinone, rearrangement, alphavirus, benzodiazepinones, encephalitis
Venezuelan and eastern equine encephalitis viruses, VEEV and EEEV, respectively, are neuroinvasive RNA alphaviruses that are transmitted through the bite of infected mosquitoes. These pathogens cause significant human disease that ranges in severity from a febrile, self-resolving flu-like illness to more debilitating cases characterized by disorientation, seizure, coma, and death. While the human mortality rate associated with VEE is generally less than 1%,1 EEE affects children and the elderly population more severely2 and has an overall case fatality rate of approximately 30–50%.3 In 2019, 38 cases of EEE were confirmed in the United States by the Center for Disease Control and Prevention, resulting in 19 fatalities.4,5
Live, attenuated vaccines are available for veterinary use and at-risk lab or military personnel; however, they are considered unacceptable for broader human use due to high reactogenicity, limited seroconversion, and narrow viral strain specificity.6,7 While other therapeutic modalities8−10 also have been investigated, treatment options to date for VEEV and EEEV infection in humans are limited to supportive care. This scenario, coupled with the bioweapon potential of these viruses, along with reports that survivors of encephalitis often have neurological sequelae that results in permanent physical, behavioral, and cognitive impairment,11,12 underscores the need for safe and effective countermeasures.13,14
Following the discovery of a novel 2-chloromethylquinazolinone rearrangement15 that afforded potent, antialphaviral benzamidines 1 (Scheme 1, panel a), we have continued to explore this chemistry to elucidate the benzamidine pharmacophore and build structure–activity and structure–property relationships (SAR and SPR, respectively).16−18 Consequently, we discovered several quinazolinone-based transformations leading to differentiated, bioactive heterocyclic frameworks.19,20
Scheme 1. Structurally Divergent, Bioactive Heterocycles from the Rearrangement of 2-Substituted Quinazolinones.

Reagents and conditions: (a) NEt3, CH3CN, μW, 150 °C, 2 h; (b) K2CO3, CH3CN, 50 °C, 2 h; (c) KOAc, DMSO, μW, 60 °C, 2 h.
For instance, 2-chloroquinazolinones, when treated with 1,2-dimethylethanediamine, undergo a similar rearrangement to generate benzylguanidines 2 with improved plasma stability compared to the benzamidine series 1.19 Herein, we describe the discovery of piperazine-fused benzodiazepinones 4 that result from the ring expansion of 2-dichloromethylquinazolinones 3 when treated with N,N′-dialkylethane-1,2-diamines (Scheme 1, panel b). Further, we show that these products potently inhibit cellular VEEV and EEEV replication and provide an opportunity for differentiated scaffold development from the benzamidine template.
As part of an ongoing medicinal chemistry program aimed at developing antivirals against VEEV and EEEV, we attempted to prepare fluorobenzamidine 6 from 2-fluorochloromethylquinazolinone 5 (Scheme 2, path a). We anticipated that N,N-dimethylethane-1,2-diamine might displace the chlorine atom of a 2-dihaloalkylquinazolinone, leading to intramolecular cyclization with subsequent ring-opening to provide fluorobenzamidine 6. While none of the benzamidines were observed, we isolated and characterized a small amount (∼5%) of piperazine-fused benzodiazepinone 7a. The conversion of 2-dihaloalkylquinazolinones to benzodiazepinones bears some resemblance to the pioneering work of Dr. Leo Sternbach21 who is credited with the discovery of benzodiazepines.22 Specifically relevant is a Roche patent23 which showed that dichloromethylquinazoline-N-oxides such as 8, when treated with methylamine (15% in MeOH), afforded bis-methylaminobenzodiazepine-N-oxide 9 (Scheme 2, path b). Notably, diazepinone 7a contained benzamidine structural features known to be important for anti-VEEV activity. When 7a was evaluated in a cell-based cytopathic effect (CPE) assay employing VEEV TC83, we obtained a remarkable EC50 of 0.041 μM without observable cytotoxicity (CC50 > 30 μM). As such, we set out to improve the transformation yield and assess the antiviral SAR of the new chemotype for VEEV and EEEV.
Scheme 2. Discovery of Piperazinobenzodiazepinones and Similarity to Bis-Methylaminobenzodiazepine-N-oxide Formation.

Reagents and conditions: (a) CH3NH(CH2)2NHCH3, K2CO3, KI, DMF, μW, 150 °C, 2 h; (b) NH2CH3, CH3OH, r.t., 16 h, no yields reported; PMP = p-methoxyphenyl.
We studied the rearrangement using a 2-dichloromethylquinazolinone because the 2,2-dichloroacetyl chloride required for the quinazolinone assembly was more economical than 2-fluoro-2-chloroacetyl chloride, the same products were expected, and chloride as a leaving group was likely more favorable than the loss of fluoride, possibly improving the yield. Furthermore, we also knew from the benzamidines’ optimizations that incorporating an amido-N-3-fluoro-4-methoxyphenyl group (i.e., amide-PMP group in 6) typically provided analogs with a better overall profile, and reaction optimization could then be more easily monitored by 19F NMR. Taking these considerations into account, reaction optimization with 2-(dichloromethyl)-3-(3-fluoro-4-methoxyphenyl)-6-nitroquinolin-4(3H)-one was undertaken (Table S1 in the Supporting Information). Yield and conversion of the transformation were evaluated as a function of solvent, reaction time, and the stoichiometry and identity of the base used. When the reaction was performed in DMSO with 1.5 equiv of diamine at 60 °C for 2 h, yields of the desired product were inferior (3–15% by 19F NMR), and the most significant identifiable byproduct, 2-amino-N-(3-fluoro-4-methoxyphenyl)-5-nitro-benzamide, resulted from starting material fragmentation. However, after screening a variety of exogenous bases, we found that the desired product was maximally generated in 36% under these conditions when KOAc was added (19F NMR analysis). As a result, these conditions were used to generate a piperazinobenzodiazepinone collection that was synthesized by the protocols shown in Scheme 3.
Scheme 3. Synthesis of Piperazinobenzodiazepinones.

Reagents and conditions: (a) ArNH2, i-Pr2NEt, EDC HCl, HOBt, DMF, rt, 56–100%; (b) I2, PhNH2, EtOH, reflux, 17 h, 47%; (c) Br2, AcOH, rt, 4 h, 89%; (d) LiOH, THF, H2O, 0 °C–rt, 17 h, 82%; (e) Cl2CHCOCl, NEt3, DCM or DMF, 0 °C–rt, 17 h, then TMSCl, NEt3, rt, 17 h, 18–81%; (f) R3NH(CH2)2NHR4 or other diamine, KOAc, DMSO, μW, 60 °C, 2 h, 8–66%; (g) For 7n, morpholine, 10 mol % Pd XPhos G3, Cs2CO3, toluene, reflux, 24 h, 32% (66% brsm); (h) For 7o, 10 mol % Pd(PPh3)4, Cs2CO3, boronic acid, dioxane, H2O, 85 °C, 17 h, 69%.
Briefly, aminobenzamides 11a–j were used without purification after being prepared by one of three methods. Peptide coupling conditions were used with commercially available anthranilic acids 10a–c to afford 11a–h. Treatment of 5-fluoroisatoic anhydride 12 with iodine and aniline afforded derivative 11i, while 11j was prepared in a three-step protocol from methyl 2-amino-5-cyanobenzoate 13. Employing a modified version of our previously published procedure24 for quinazolinone core assembly, the 2-dichloromethylquinazolinones 14a–j were constructed using 2,2-dichloroacetyl chloride to form bis-amides in situ (not shown) that underwent TMSCl/NEt3-mediated ring-closure in yields ranging from 18 to 81%.
The 2-dichloromethylquinazolinone rearrangement was carried out under the optimized conditions involving treatment of 14a–j (1.0 equiv) with diamine (1.5 equiv) and KOAc (2.0 equiv) in DMSO at 60 °C for 2 h. Structural changes in each of the three essential components, i.e., anthranilic acid, aniline, and diamine, were surveyed with selection predominately driven by a known benzamidine SAR.15 For example, our benzamidine-related studies showed that a nitro group at the C5 position (see 6, Scheme 2) was critical for retaining substantial antialphavirus activity. As such, many diazepinones contain this structural feature at the analogous C8 position of the new core, but other substitutions were also examined. Improved activity and solubility resulted with benzamidines bearing an amido-N-4-methoxyphenyl ring, so analogs with this component were also made (Scheme 4).
Scheme 4. Yield of 2-Dichloroquinazolinone Ring Expansion for Various Piperazinobenzodiazepinones,

Reagents and conditions: (a) diamine, KOAc, DMSO, 60 °C, 2 h; (b) For 7n, morpholine, 10 mol % Pd XPhos G3, Cs2CO3, toluene, reflux, 24 h; (c) For 7o, 10 mol % Pd(PPh3)4, Cs2CO3, (6-methylpyridin-3-yl)boronic acid, dioxane, H2O, 85 °C, 17 h.
Isolated yields; ring expansion step performed on a 0.5 mmol scale, unless noted.
Used bis-HCl diamine salt, 2 equiv of K2CO3 added.
Scale: 0.37 mmol due to limited starting material.
Scale: 2.2 mmol (1 g) of 2-dichloromethylquinazolinone.
Parenthetical yield for aryl bromide derivatization.
Major isomer shown.
In general, piperazinobenzodiazepinones containing a nitro group and constructed with N,N′-dimethylethane-1,2-diamine were isolated from the rearrangement reaction in modest yield (8–38%), most likely due to the increased reactivity of the nitro-containing substrate (see yields for 7a–e, Scheme 4). Accordingly, moving the nitro group from the C8-position to the C7-position of the piperazinobenzodiazepinone core resulted in an 18% increase in yield, comparing 7i (38%) to 7e (20%). Notably, the rearrangement step performed appreciably better for presumably less activated substrates, including C8-cyano-substituted analogs such as 7k–7m with isolated yields in the 49–66% range. Performing the rearrangement on a 2.2 mmol (1 g) scale generated 7m in an improved 63% yield. The use of bulkier, unsymmetrical diamines on the reaction efficiency and regioselectivity was also examined. Using N,N′-dibenzylethane-1,2-diamine provided benzodiazepinone 7h in 50% yield. Interestingly, using unsymmetrical N,N′-benzylmethylethane-1,2-diamine gave benzodiazepinone 7g in 29% yield as the sole observed regioisomer. Similarly, N,N′-cyclohexylmethylethane-1,2-diamine afforded an analogous benzodiazepinone 7f as the only regioisomer, though only in 8% yield, likely due to the increased bulkiness of the cyclohexyl group. The assigned regiochemistry of both 7f and 7g was determined by 2D NOESY NMR. Additionally, each enantiomer of N,N′-dimethylcylohexyl-1,2-diamine was separately employed in the reaction, affording benzodiazepinones 7p and 7q in 30 and 34% yields, respectively, and each in 14:1 d.r., the latter of which was confirmed by NOESY NMR. Lastly, bromide 7m was derivatized by a Buchwald-Hartwig coupling to afford morpholine analog 7n (32% yield, 66% brsm), while Suzuki coupling of 7m gave pyridyl analog 7o (69% yield).
Piperazinobenzodiazepinone formation could result from several plausible mechanisms. Considering an SN2 substitution reaction between a 2-dichloromethylquinazolinone and a nucleophilic diamine, α-chloroalkylamine A may result (Scheme 5, path a), facilitated by KOAc.25−27 In this mechanism, iminium ion B formation from α-chloroalkylamine A may be trapped in an intramolecular fashion by the appended secondary amine to form aminal C. Ring expansion of the aminal through neighboring nitrogen atom assistance would then form zwitterionic spirocyclic iminium ion D, a species that is similar to those implicated for similar rearrangements. Benzodiazepinone 7 could then be formed through either a synchronous or asynchronous event involving the amide portion of the structure. Specifically, the negatively charged quinazolinone nitrogen could promote a second ring expansion via a 1,2-N-acyl migration to the iminium ion carbon or by a sequential 6-membered ring-opening to a 7-membered ring-forming process. An intramolecular 7-membered ring formation involving an NH-amide is precedented in peptidomimetic syntheses.28
Scheme 5. Mechanistic Rationale for Diazepinone Formation.

Attempts to intercept iminium ions via reductive amination were unsuccessful, but the SN2 mechanisms account for the observed regiochemical outcomes. Specifically, unsymmetrical diamines yielded products in which the bulkier group resided on the iminium ion nitrogen that was further away from the quinazolinone core in intermediate D. Alternatively, we considered a mechanism similar to the Sternbach rearrangement of 2-chloromethylquinazoline-3-oxides that, under treatment with an alkyl amine, affords aminodiazepoxides.29 In our case, nucleophilic addition of the amine to the N-acyl imine-like carbon of the quinazolinone core would form E (Scheme 5, path b). Subsequent ring-opening/ring-forming or an N-acyl shift would generate a ring-expanded chlorobenzodiazepinone F that would undergo intramolecular chloride displacement by the tethered amine. At this point, these mechanisms are not differentiated, and multiple scenarios may be relevant depending on the quinazolinone substitution and the diamine partner.
Two orthogonal antiviral, cell-based assays using VEEV and EEEV were implemented to survey the antiviral effects of these compounds (Table 1).17 Viability of infected Vero 76 cells was measured as a function of compound concentration in a CPE assay, and the reduction of viral yield was measured at a concentration of 5 μM for compounds with submicromolar CPE EC50 values. Several notable outcomes emerged from these assays. First, data between the VEEV and EEEV CPE assays were strongly congruent (<2.1-fold difference), revealing similar SAR across these two alphaviruses in this assay. Second, data showed that compounds whose viral yield reduction potential was assessed were consistently better by about 2–4-fold against EEEV compared to that of VEEV. Third, we observed that several advantageous structural modifications implemented with the amidine series were also beneficial on the piperazinobenzodiazepinones in terms of antiviral activity. For instance, an N-4-methoxyphenyl amide substituent, in combination with a nitro group present at R1, resulted in potent inhibition of CPE caused by either VEEV or EEEV (Table 1, entries 1 and 2, EC50 = 27–48 nM). Potency was eroded when the 4-methoxyphenyl substituent was exchanged for a fluorine or hydrogen atom at that para-position or if the methoxy group was migrated to the 3-position (entries 3–5), though the compounds still retained submicromolar activity. Substitution of one or more of the ring-fused piperazine nitrogen atoms with nonmethyl groups such as a benzyl or cyclohexyl moiety produced inferior results, with the N,N′-dibenzyl derivative 7h faring the worst (analogs 7f–h). Changing the piperazine ring to bicyclic, ring-fused variants (7p–q) while incorporating the N-methylpiperazine substitutions provided hit-like potencies in the range of 5 μM, suggesting that structural modifications may be permissible in this region.
Table 1. Survey of Piperazinobenzodiazepinones on VEEV and EEEV Cytopathic Effect and Viral Yield Reduction.

| VEEV
INH9813 |
EEEV
V105 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | compd | R1 | R2 | R3 | R4 | R5 | R6 | R7 | CPEa EC50 (μM) | viral yield rednb | CPEa EC50 (μM) | viral yield rednb | CC50 (μM)c |
| 1 | 7a | NO2 | H | H | CH3 | CH3 | H | OCH3 | 0.041 ± 0.9 | 5.5 ± 1.2 | 0.033 ± 0.002 | 7.9 ± 0.5 | >30 |
| 2 | 7b | NO2 | H | H | CH3 | CH3 | F | OCH3 | 0.048 ± 0.004 | 3.7 ± 1.1 | 0.027 ± 0.003 | 7.7 ± 0.8 | >30 |
| 3 | 7c | NO2 | H | H | CH3 | CH3 | H | F | 0.15 ± 0.01 | 2.9 ± 0.9 | 0.11 ± 0.004 | 6.1 ± 1.8 | >30 |
| 4 | 7d | NO2 | H | H | CH3 | CH3 | OCH3 | H | 0.53 ± 0.03 | 2.4 ± 0.4 | 0.50 ± 0.03 | 5.7 ± 1.8 | >30 |
| 5 | 7e | NO2 | H | H | CH3 | CH3 | H | H | 0.12 ± 0.02 | 2.6 ± 0.7 | 0.059 ± 0.01 | 5.7 ± 1.4 | >30 |
| 6 | 7f | NO2 | H | H | CH3 | Cy | H | OCH3 | 7.0 ± 0.8 | 6.2 ± 0.2 | >30 | ||
| 7 | 7g | NO2 | H | H | CH3 | Bn | H | OCH3 | 3.0 ± 0.1 | 3.2 ± 0.2 | >30 | ||
| 8 | 7h | NO2 | H | H | Bn | Bn | H | OCH3 | 19.8 ± 3.5 | 13.8 ± 1.5 | >30 | ||
| 9 | 7i | H | NO2 | H | CH3 | CH3 | H | H | >30 | >30 | >30 | ||
| 10 | 7j | F | H | H | CH3 | CH3 | H | H | >30 | >30 | >30 | ||
| 11 | 7k | CN | H | H | CH3 | CH3 | H | H | 7.0 ± 1.1 | 4.4 ± 0.2 | >30 | ||
| 12 | 7l | CN | H | H | CH3 | CH3 | H | OCH3 | 4.2 ± 0.4 | 2.5 ± 0.1 | >30 | ||
| 13 | 7m | CN | H | Br | CH3 | CH3 | F | OCH3 | 4.1 ± 0.1 | 3.2 ± 0.1 | >30 | ||
| 14 | 7n | CN | H | morpholine | CH3 | CH3 | F | OCH3 | 2.5 ± 0.3 | 2.5 ± 0.9 | 1.9 ± 0.1 | 4.0 ± 1.1 | >30 |
| 15 | 7o | CN | H | 6-CH3-3-pyridyl | CH3 | CH3 | F | OCH3 | 0.24 ± 0.03 | 5.2 ± 1.8 | 0.16 ± 0.003 | 6.9 ± 1.3 | 23.3 |
| 16 | ent-7od | CN | H | 6-CH3-3-pyridyl | CH3 | CH3 | F | OCH3 | 0.50 ± 0.27 | 6.1 ± 0.7 | 0.30 ± 0.06 | 5.2 ± 2.2 | >30 |
| 17 | ent-′7od | CN | H | 6-CH3-3-pyridyl | CH3 | CH3 | F | OCH3 | 0.33 ± 0.03 | 2.7 ± 0.6 | 0.26 ± 0.03 | 5.9 ± 0.8 | >30 |
| 18 | 7p | see structure at top of table (major isomer shown) | 4.7 ± 0.3 | 4.5 ± 0.3 | >30 | ||||||||
| 19 | 7q | see structure at top of table (major isomer shown) | 5.6 ± 0.8 | 3.8 ± 0.4 | >30 | ||||||||
CPE assay, V/EEEV MOI = 0.05, Vero 76 cells, assessed at 48 h; data averaged from at least 3 replicates (n ≥ 3, mean ± SD).
Viral yield reduction (Vero 76 cells) at 5 μM compound concentration calculated based on the averaged log10 transformed viral titer of the mock-treated group divided by that of the compound-treated group at 18 h postinfection (n = 4, mean ± SD).
Vero 76 cells, assessed 72 h post-treatment.
The (R)- or (S)-stereochemistry is unassigned, but each enantiomer was isolated in >99% ee by chiral HPLC.
Relocating the R1 nitro group to the R2 position (entry 9, 7i) or replacing it with a fluorine atom (entry 10, 7j) was not tolerated (EC50 > 30 μM). In fact, a limitation of the amidine series was the need for a C5-nitro group (see Scheme 2, compound 6) to achieve exceptional potency in antiviral assays, and despite a survey of many isosteres in that series, only the C5 substitution with a nitrile moiety afforded analogs with low micromolar potency. Analogously, substituting the diazepinone C8 nitro group with a nitrile functionality resulted in a 100- and 75-fold loss in antiviral activity for VEEV and EEEV, respectively (cf., 7a versus 7l); however, the retention of low micromolar activity in these assays provided hope that structural changes made elsewhere on the scaffold may remedy these losses. From a design standpoint, substitution at R3 of the diazepinone core (see general structure of 7, Table 1) seemed reasonable as a hydrogen bond acceptor in that region may mimic the tertiary amine of the cyclic amidine structures, a pharmacophoric element that improved potency for that series.
The scaffold was equipped with an aryl bromide at R3 for the purposes of coupling, and then, we explored the effect of installing a morpholine or pyridyl moiety off that position of the diazepinone core. While the morpholine modification did not appreciably improve potency, the augmentation was not deleterious, and for EEEV, it resulted in a promising 4-fold reduction of viral yield (entry 14).
The pyridyl analog 7o gratifyingly exhibited substantially improved CPE activity (EC50 = 0.16–0.24 μM) and robust reduction of viral replication, especially in the case of EEEV. Cytotoxicity was not observed for compounds tested up to 30 μM with the exception of racemic 7o which showed some liability at 23 μM. Nonetheless, the selectivity indices (CC50/EC50 = 97–146) for 7o reflect a solid margin from which to optimize.
To determine if antiviral activity was influenced by enantiomeric identity, racemic 7o was subjected to chiral separation, yielding ent-7o and ent′-7o in high enantiopurity (>99% ee). No significant difference was observed in the CPE assay between the enantiomers ent-7o and ent′-7o for VEEV or EEEV (entries 16 and 17, Table 1), and reduction in viral titer was notably different only for VEEV (entry 17). While the antiviral activity of the compounds was evaluated in a Vero 76 cell line that is well established for these viruses, antiviral activity and sensitivity to the enantiomeric identity of 7o in primary human neuronal cells were also evaluated. As such, selected compounds, 7a, 7b, 7o, and the corresponding 7o enantiomers, ent- and ent′-7o, were assayed for reduction of viral yield for both VEEV and EEEV in human brain primary neuronal cells (HNC001). Following treatment in neuronal cells, infectious viral titers were measured in a tissue culture infectious dose assay (TCID50) using VERO 76 cells (Figure 1). At a compound concentration of 5 μM, all of the compounds reduced viral titer to the limits of detection of the assay for both VEEV and EEEV compared to virus-only controls after 18 h (Figure 1A). Racemate 7o and its enantiomers were assessed in the same cell line at 1 μM concentration to see if the antiviral activity could be better differentiated; however, we observed no significant difference in activity among these compounds even at the lower concentration (Figure 1B). Importantly, these results suggest that, barring a racemization event happening during the assay assessment, the stereocenter of 7o does not significantly influence the degree of antiviral activity observed for VEEV or EEEV. Moreover, we also show that substantial reduction in VEEV and EEEV titers is achieved in a primary human neuronal cell line even at a concentration of 1 μM.
Figure 1.
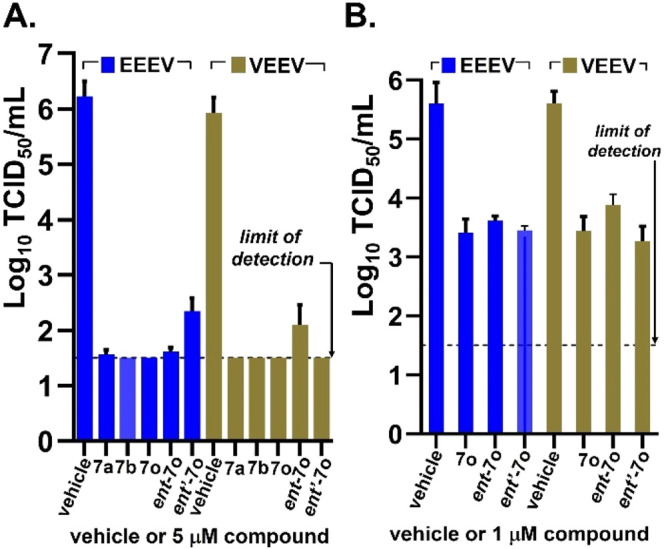
Selected 7o compounds showing reduction of EEEV and VEEV titers in a TCID50 assay using human brain primary neuronal cells at 5 μM (panel A) or 1 μM (panel B) after 18 h. Each experiment was done in triplicate, and averaged data is plotted (n = 3, mean ± SD). The graphs were generated using GraphPad Prism V.9.3.1.
Selected compounds were also profiled for kinetic aqueous solubility and stability in mouse liver microsomes (MLMs) to gauge prospects for eventual in vivo evaluation and optimization (Table 2). The nitro group containing analogs 7a,b showed limited aqueous solubility and a short MLM half-life (∼10 min), while improved solubility and stability were observed for the nitrile-containing analog 7o. Mouse plasma stability for 7o was found to be robust (T1/2 > 289 min, CD-1 strain). Nonetheless, future optimization efforts will focus on refining solubility and microsomal stability as the series is advanced.
Table 2. Solubility and Mouse Microsomal Stability Data.
Kinetic solubility in aqueous PBS buffer, pH 7.4.
MLM = CD-1 mouse liver microsomal stability.
Given the observed SAR congruency between the older benzamidine series and the piperazinobenzodiazepinone scaffold, we wanted to better visualize shared or differentiated structural features in three-dimensional space, as these pharmacophoric details may guide future optimization. Benzodiazepinone 7o, though not the most potent analog from the survey, retained significant CPE assay potency and robustly attenuated viral replication for both VEEV and EEEV in Vero 76 and human neuronal cells while also bearing an aryl nitrile moiety in place of the potential toxophoric aryl nitro group. This was significant as the benzamidine series did not reflect a similar tolerance, and notably, the presence of the pyridyl appendage of 7o was required to achieve desirable potency. A potent benchmark benzamidine, BDGR-4 (Figure 2A), was independently compared in silico to each enantiomer of benzodiazepinone 7o using the program Molecular Operating Environment (MOE).30 Interestingly, alignment of benzamidine BDGR-4 (gray-colored carbon atoms) with either isomer of 7o (shown only for the (R)-isomer of 7o as cyan-colored carbon atoms, Figure 2B) showed exceptional overlap in which the amidine portion of BDGR-4 assumed a similar spatial arrangement as the piperazine of the benzodiazepinone structure. These results, in which the best scored alignments were redundant for each enantiomer of 7o, corroborate the lack of differentiation in antiviral activity observed for ent-7o and ent′-7o in neuronal cells. Further, this model suggests that the required pyridyl appendage of 7o may engage the target with binding interactions that are differentiated from those that are possible for benzamidine BDGR-4.
Figure 2.
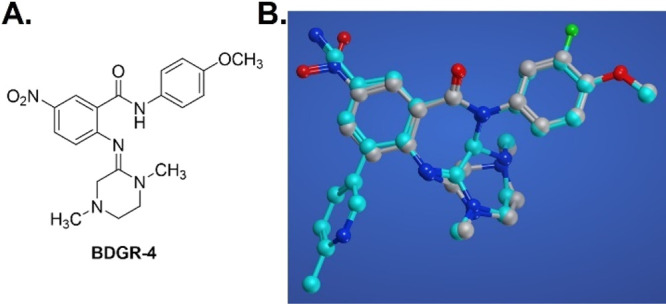
A. Structure of the benzamidine V/EEEV inhibitor, BDGR-4. B. Superimposed structures of BDGR-4 (gray structure) and (R)-7o (cyan structure) using a flexible align MOE program.
In the course of exploring a quinazolinone rearrangement that has historically afforded benzamidines or derivatives thereof, we discovered that 2-dichloroquinazolin-4-ones, when treated with N,N′-dialkylethane-1,2-diamines, undergo ring expansion to form a new class of piperazine-fused benzodiazepinones. Furthermore, the analogs generated from this effort show remarkable cellular antiviral activity in two orthogonal antiviral assays against encephalitic alphaviruses, VEEV and EEEV, and without significant cytotoxic liability. The transformation led to modest yields of analogs with nitro-group substituted cores but significantly improved to a suitable range of 49–66% yield when nitrile-group variants were generated. This yield enhancement was also notable given that submicromolar antiviral CPE inhibition and good viral yield reduction were achieved with the nitrile analogs, a milestone not met with the analogously substituted, nitrile-containing benzamidines. Additionally, we observed that several piperazinobenzodiazepinones were effective in significantly reducing both VEEV and EEEV titers in human primary neuronal cells. Preliminary solubility and mouse microsomal stability were determined to be modest, though nitrile compound 7o was comparatively better than two nitro-containing analogs. Last, we analyzed in silico the energy-minimized structures of a leading benzamidine, BDGR-4, and an optimized piperazinobenzodiazepinone 7o. This exercise revealed relatively congruent, low-energy conformations that either enantiomer of 7o can adopt a three-dimensional structural arrangement like that of benzamidine BDGR-4 and that the pyridyl appendage of this new benzodiazepinone class may offer an advantage in exploiting new target interactions. Work is ongoing to explore this aspect of the transformation. Ultimately, the discovery and development of these piperazinobenzodiazepinones represent another important milestone in expanding a sparsely stocked pipeline of small molecules that inhibit encephalitic alphaviruses.
Acknowledgments
We are grateful for support from the National Institutes of Health/NIAID (R01AI118814 and U19AI142762 to J.E.G., C.B.J., and D-H.C.) This work made use of instrumentation at the UW–Madison Medicinal Chemistry Center and the Analytical Instrumentation Center, both within the UW-Madison School of Pharmacy.
Glossary
Abbreviations
- BRSM
based on recovered starting material
- CPE
cytopathic effect
- EEEV
eastern equine encephalitis virus
- PMP
p-methoxyphenyl
- SAR
structure–activity relationships
- VEEV
Venezuelan equine encephalitis virus
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00539.
Assay and experimental details, synthetic procedures, characterization, chiral separation of 7o, including NMR spectra for all new compounds (11a-j, 14a-j, and 7a-q) reported in this work (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Griffin D. E.Alphaviruses. In Fields Virology, Knipe D. M.; Howley P. M., Eds.; Lippincott, Williams & Wilkins: 2007; pp 1023–1068. [Google Scholar]
- Silverman M. A.; Misasi J.; Smole S.; Feldman H. A.; Cohen A. B.; Santagata S.; McManus M.; Ahmed A. A. Eastern Equine Encephalitis in Children, Massachusetts and New Hampshire, USA, 1970–2010. Emerg. Infect. Dis. 2013, 19 (2), 194–201. 10.3201/eid1902.120039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar P. V.; Robich R. M.; Turell M. J.; O’Guinn M. L.; Klein T. A.; Huaman A.; Guevara C.; Rios Z.; Tesh R. B.; Watts D. M.; Olson J.; Weaver S. C. Endemic Eastern Equine Encephailitis in the Amazon Region of Peru. Am. J. Trop. Med. Hyg. 2007, 76, 293–298. 10.4269/ajtmh.2007.76.293. [DOI] [PubMed] [Google Scholar]
- Eastern Equine Encephalitis Virus Neuroinvasive Disease Cases Reported by Year, 2010–2019. ArboNET, Arboviral Disease Branch, Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases, Division of Vector-Borne Diseases: cdc.gov. https://www.cdc.gov/easternequineencephalitis/tech/epi.html#casesbyyear (accessed 2021-08-13).
- Lindsey N. P.; Martin S. W.; Staples J. E.; Fischer M. Notes from the Field: Multistate Outbreak of Eastern Equine Encephalitis Virus, US, 2019. Morb. Mortal. Wkly. Rep. 2020, 69 (2), 50–51. 10.15585/mmwr.mm6902a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg L.; Brahms A.; Hooper I.; Carey B.; Lin S.-C.; Dahal B.; Narayanan A.; Kehn-Hall K. Repurposed FDA-Approved Drug Sorafenib Reduces Replication of Venezuelan Equine Encephalitis Virus and Other Alphaviruses. Antiviral Res. 2018, 157, 57–67. 10.1016/j.antiviral.2018.07.005. [DOI] [PubMed] [Google Scholar]
- Hoke C. H. History of U.S. Military Contributions to the Study of Viral Encephalitis. Mil. Med. 2005, 170, 92–105. 10.7205/MILMED.170.4S.92. [DOI] [PubMed] [Google Scholar]
- Phelps A.; O’Brien L.; Ulaeto D. O.; Holtsberg F. W.; Liao G. C.; Douglas R.; Javad Aman M.; Glass P. J.; Moyer C. L.; Ennis J.; Zeitlin L.; Nagata L. P.; Hu W.-G. Cross-Strin Neutralizing and Protective Monoconal Antibodies against EEEV or WEEV. Viruses 2021, 13, 2231–2245. 10.3390/v13112231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L. E.; Gilliland T. Jr.; Yadav P. K.; Binshtein E.; Bombardi R.; Kose N.; Nargi R. S.; Sutton R. E.; Durie C. L.; Armstrong E.; Carnahan R. H.; Walker L. M.; Kim A. S.; Fox J. M.; Diamond M. S.; Ohi M. D.; Klimstra W. B.; Crowe J. E. Jr. Human Antibodies Protect Against Aerosolized Eastern Equine Encephalitis Virus Infection. Cell 2020, 183, 1884–1900. 10.1016/j.cell.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter G. R.; Bowen R. A.; Bluemling G. R.; DeBergh J.; Edpuganti V.; Gruddanti P. R.; Guthrie D. B.; Hagar M.; Kuiper D. L.; Lockwood M. A.; Mitchell D. G.; Natchus M. G.; Stitcher Z. M.; Kolykhalov A. A. The Prophylactic and therapeutic Activity of a Broadly Active Ribonucleoside Analog ina Murine Model of Intranasal Venezuelan Equine Encephalitis Virus Infection. Antiviral Res. 2019, 171, 104597–104607. 10.1016/j.antiviral.2019.104597. [DOI] [PubMed] [Google Scholar]
- Millet N.; Faiek S.; Gurrieri D.; Kals K.; Adams W.; Hamaty E.; Trivedi M.; Zeidwerg D. Deadly Neuroinvasive Mosquito-Borne Virus: A Case of Eastern Equine Encephalitis. Perm. J. 2021, 25 (20), 288. 10.7812/TPP/20.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronca S. E.; Dineley K. T.; Paessler S. Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2016, 7, 959. 10.3389/fmicb.2016.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens D. M.; Folkers G. K.; Fauci A. S. Eastern Equine Encephalitis Virus — Another Emergent Arbovirus in the United States. N. Engl. J. Med. 2019, 381, 1989–1992. 10.1056/NEJMp1914328. [DOI] [PubMed] [Google Scholar]
- Simpson S.; Kaufmann M. C.; Glozman V.; Chakrabarti A. Disease X: Accelerating the Development of Medical Countermeasures for the Next Pandemic. Lancet Infect. Dis. 2020, 20, e108–e115. 10.1016/S1473-3099(20)30123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder C. E.; Yao T.; Sotsky J.; Smith R. A.; Roy S.; Chu Y.-K.; Guo H.; Tower N. A.; Noah J. W.; McKellip S.; Sosa M.; Rasmussen L.; Smith L. H.; White E. L.; Aubé J.; Jonsson C. B.; Chung D.; Golden J. E. Development of (E)-2-((1,4-Dimethylpiperazin-2-ylidene)amino)-5-nitro-N-phenylbenzamide, ML336: Novel 2-Amidinophenylbenzamides as Potent Inhibitors of Venezuelan Equine Encephalitis Virus. J. Med. Chem. 2014, 57 (20), 8608–8621. 10.1021/jm501203v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder C. E.; Neuenswander S. A.; Yao T.; Aubé J.; Golden J. E. One-pot, Regiospecific Assembly of (E)-Benzamidines from δ- and γ-Amino Acids via an Intramolecular Aminoquinazolinone Rearrangement. Org. Biomol. Chem. 2016, 14 (16), 3950–3955. 10.1039/C5OB02378E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson C. B.; Cao X.; Lee J.; Gabbard J. D.; Chu Y. K.; Fitzpatrick E. A.; Julander J.; Chung D. H.; Stabenow J.; Golden J. E. Efficacy of a ML336 Derivative Against Venezuelan and Eastern Equine Encephalitis Viruses. Antiviral Res. 2019, 167, 25–34. 10.1016/j.antiviral.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffett V. A.; Nerurkar A.; Cao X.; Guzei I. A.; Golden J. E. Telescoped Synthesis of C3-Functionalized (E)-Arylamidines Using Ugi–Mumm and Regiospecific Quinazolinone Rearrangements. Org. Biomol. Chem. 2019, 17 (12), 3118–3128. 10.1039/C9OB00073A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G.; Zekarias B. L.; Li X.; Jaffett V. A.; Guzei I. A.; Golden J. E. Divergent 2-Chloroquinazolin-4(3H)-one Rearrangement: Twisted-Cyclic Guanidine Formation or Ring-Fused N-Acylguanidines via a Domino Process. Chem.—Eur. J. 2020, 26 (11), 2486–2492. 10.1002/chem.201905219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffett V. A.; Fitz-Henley J. N.; Khalifa M. M.; Guzei I. A.; Golden J. E. Diastereoselective, Multicomponent Synthesis of Pyrrolopyrazinoquinazolinones via a Tandem Quinazolinone Rearrangement/ Intramolecular Ring Closure of Tautomeric (Z)-Benzamidines. Org. Lett. 2021, 23 (15), 5799–5803. 10.1021/acs.orglett.1c01955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oransky I. Leo H Sternbach. Lancet 2005, 366 (9495), 1430. 10.1016/S0140-6736(05)67588-5. [DOI] [Google Scholar]
- Sternbach L. H. The Benzodiazepine Story. J. Med. Chem. 1979, 22 (1), 1–7. 10.1021/jm00187a001. [DOI] [PubMed] [Google Scholar]
- Stemple A.; Sternbach L. H.. Certain 2, 3-bis(substituted amino)-5-aryl-3H-1,4-benzodiazepine-4-oxide Compounds and Their Preparation. US3320239A, May 16, 1967.
- Li X.; Golden J. E. Construction of N-Boc-2-Alkylaminoquinazolin-4(3H)-Ones via a Three-Component, One-Pot Protocol Mediated by Copper(II) Chloride that Spares Enantiomeric Purity. Adv. Synth. Catal. 2021, 363 (6), 1638–1645. 10.1002/adsc.202001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordwell F. G.; Algrim D. Nitrogen acids. 1. Carboxamides and sulfonamides. J. Org. Chem. 1976, 41 (14), 2507–2508. 10.1021/jo00876a042. [DOI] [Google Scholar]
- Bordwell F. G. Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res. 1988, 21 (12), 456–463. 10.1021/ar00156a004. [DOI] [Google Scholar]
- Mucci A.; Domain R.; Benoit R. L. Solvent Effect on the Protonation of Some Alkylamines. Can. J. Chem. 1980, 58 (9), 953–958. 10.1139/v80-151. [DOI] [Google Scholar]
- Ventosa-Andrés P.; Barea Ripoll C. A.; La-Venia A.; Krchňák V. Solid-phase Synthesis of Fused 1,4-diazepanone Peptidomimetics via Tandem N-iminium Ion Cyclization–Nucleophilic Addition. Tetrahedron Lett. 2015, 56 (40), 5424–5428. 10.1016/j.tetlet.2015.08.015. [DOI] [Google Scholar]
- Stempel A.; Reeder E.; Sternbach L. H. Quinazolines and 1,4-benzodiazepines. XXVII. Mechanism of Ring Enlargement of Quinazoline 3-Oxides with Alkali to 1,4-Benzodiazepin-2-one 4-oxides. J. Org. Chem. 1965, 30 (12), 4267–71. 10.1021/jo01023a063. [DOI] [PubMed] [Google Scholar]
- Flexible alignment was done with the default Amber10 force field parameters and R-field solvation method
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.