

Abstract
Neonatal hypoxic-ischemic encephalopathy (HIE) causes lifelong neurologic disability. Despite the use of therapeutic hypothermia, memory deficits and executive functions remain severely affected. Cholinergic neurotransmission from the basal forebrain to neocortex and hippocampus is central to higher cortical functions. We examined the basal forebrain by light microscopy and report loss of choline acetyltransferase-positive (ChAT)+ neurons, at postnatal day (P) 40, in the ipsilateral medial septal nucleus (MSN) after neonatal hypoxia-ischemia (HI) in mice. There was no loss of ChAT+ neurons in the ipsilateral nucleus basalis of Meynert (nbM) and striatum. Ipsilateral striatal and nbM ChAT+ neurons were abnormal with altered immunoreactivity for ChAT, shrunken and crenated somas, and dysmorphic appearing dendrites. Using confocal images with 3D reconstruction, nbM ChAT+ dendrites in HI mice were shorter than sham (p=0.0001). Loss of ChAT+ neurons in the MSN directly correlated with loss of ipsilateral hippocampal area. In the nbM and striatum, percentage of abnormal ChAT+ neurons correlated with loss of ipsilateral cerebral cortical and striatal area, respectively. Acetylcholinesterase (AChE) activity increased in adjacent ipsilateral cerebral cortex and hippocampus and the increase was linearly related to loss of cortical and hippocampal area. Numbers and size of cathepsin D+ lysosomes increased in large neurons in the ipsilateral nbM. After neonatal HI, abnormalities were found throughout the major cholinergic systems in relationship to amount of forebrain area loss. There was also an upregulation of cathepsin D+ particles within the nbM. Cholinergic neuropathology may underlie the permanent dysfunction in learning, memory, and executive function after neonatal brain injury.
Keywords: Choline acetyltransferase, cathepsin D, somato-dendritic neuronal attrition, nucleus basalis of Meynert, medial septal nucleus, target deprivation, learning, memory, executive function
Graphical Abstract text:
Mouse cholinergic systems: Poor executive function and decreases in memory and learning are seen after neonatal HIE. We examined the cholinergic systems to determine if there is pathology there that could contribute to these functional outcomes. In contrast to normal, after neonatal HI on P10, loss of permanent attrition of ChAT neurons in the MSN, nbM and striatum is found. ChAT+ neurons are crenated with shorter dystrophic dendrites. Increased and larger lysosomes are also present in the nbM. Within the injured ipsilateral cortex, AChE activity is abnormally increased. We hypothesize that permanent injury to the cholinergic systems after neonatal HI is responsible for these poor neurologic outcomes.

1. Introduction
Complications from hypoxic-ischemic (HI) brain injury contribute to one-quarter of neonatal deaths worldwide and cause significant long-term neurological morbidity. (“Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy,” 2014; Lawn, Cousens, Zupan, & Lancet Neonatal Survival Steering, 2005; Lee et al., 2013; Shankaran et al., 2012). Although, there is a decrease in both mortality and motor handicap seen in the large randomized clinical trials of therapeutic hypothermia (TH) (Azzopardi et al., 2014; Shankaran et al., 2012), survivors and their families are left to struggle with deficits in learning, cognition, memory, and behavior (Azzopardi et al., 2014; Shankaran et al., 2012). The Total Body Hypothermia trial for neonatal encephalopathy found hypothermic-mediated neuroprotection in attention and executive function at 6–7 years of age. No improvement was seen in another ten neuro-behavioral measures tested, including learning and memory, and visuospatial processing (Azzopardi et al., 2014). The National Institute of Child Health and Development (NICHD) TH trial failed to find TH mediated protection against a low intelligence quotient (<70), poor attention, poor executive function, or a visuospatial score <70 (Shankaran et al., 2012). The neuropathology causing these deficits is not clearly defined. Injury to the hippocampus, cerebral cortex, basal ganglia, and thalamus (L. Martin, Brambrink, Koehler, & Traystman, 1997; Roland, Poskitt, Rodriguez, Lupton, & Hill, 1998; Salas et al., 2019; Wood et al., 2016) play a role, but other brain regions and connectome defects are likely to contribute to this complex pathology.
The cholinergic systems of the forebrain, including the medial septal nucleus (MSN) and the nucleus basalis of Meynert (nbM) with its peripallidal (PP) neurons, and interneurons in the striatum, are essential for normal executive function, cognitive flexibility, learning and memory, arousal, decision-making, modulation of cortical activity and the default mode network (Alves et al., 2019; Deiana, Platt, & Riedel, 2011; Schmitz & Duncan, 2018; Zaborszky, Pol, & Gyengesi, 2012). The effects of disruption in the brain’s cholinergic system are well described and extensively studied in human adults with neurodegenerative diseases, particularly Alzheimer’s Disease (AD) (M. Grothe, Heinsen, & Teipel, 2012; Mesulam, 2013). Striatal cholinergic activity has been thought to be transiently effected by HI, returning to normal by adult age (Johnston, 1983). Recently, loss of volume in the basal forebrain region was linked to neonatal complications in preterm infants and deficits in functional IQ in young adulthood (M. J. Grothe et al., 2017). These data suggest that there may be injury to the cholinergic basal forebrain resulting in permanent deficits in cholinergic neurotransmission after neonatal HI.
Mice show significant deficits in cognitive flexibility after neonatal HI (Maxwell et al., 2020; van der Kooij et al., 2010). We thus tested the hypothesis that there is basal forebrain cholinergic pathology in mice after neonatal HI. Following neonatal HI at postnatal (P) day 10 (Ditelberg, Sheldon, Epstein, & Ferriero, 1996; Graham et al., 2004), we examined the MSN, the nbM, and the striatum at P40 for pathology in choline acetyltransferase (ChAT+) containing neurons, queried the relationship of abnormal ChAT+ neuronal phenotype to hippocampal, cerebral cortical and striatal area loss, measured dendrite length, and explored abnormalities in cerebral cortical and hippocampal cholinergic innervation by acetylcholinesterase (AChE) activity. Finally, we probed for evidence of autophagy in basal forebrain neurons.
2. Material and methods
2.1. Animals
C57BL6 mice from Charles River Laboratories (Wilmington, MA) were used for all experiments with approval from the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine. All animal studies were carried out with standards of care and housing following the National Institutes of Health Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services 85–23, 2011.
2.2. Neonatal HI brain injury model
We used the method originally described by Rice-Vannucci using rat (Rice, Vannucci, & Brierley, 1981) to induce HI in C57BL6 mice at P10 (Ditelberg et al., 1996; Graham et al., 2004). Sham mice were exposed to anesthesia but not to surgery. Mice were killed at P40 via exposure to 20% (v/v) mixture of isoflurane in propylene glycol via one drop method (Markovic & Murasko, 1993). Following exsanguination with intracardiac cold 0.1 M phosphate-buffered saline (PBS, pH 7.4, Amresco, Solon, OH), mice were perfused with 4% paraformaldehyde in 0.1 M PBS for 15 minutes (min) at 4 ml/min. Brains from sham (n=11) and HI (n=12) mice were used for these experiments. The specific n for each experiment is provided.
2.3. Immunocytochemistry, immunofluorescence, antibody characterization and AChE enzyme histochemistry
Whole brains were cryoprotected with graded immersion in 15% and 30% sucrose in PBS until they sank, frozen at −30oC in isopentane and stored at −80oC. Brains were sectioned serially in the coronal plane at 50 μm on a freezing microtome. A subset of sections (1 in 12) was mounted on glass microscope slides for cresyl violet (CV) (Sigma-Aldrich Company Ltd, Dorset, UK) staining.
Floating immunohistochemistry (IHC) was performed on every 11th and 10th brain sections after incubating in cold methanol/H2O2 (1%) (30 min), permeabilizing in 0.04% triton X-100 (TX), and blocking in 10% normal donkey serum (NDS) for 60 min. Details on antibodies and selected reagents are included in Table 1. Sections were incubated overnight at 4°C in goat anti-ChAT antibody (Millipore). Incubation in donkey anti-goat IgG H&L secondary followed for 60 min at room temperature. Sections were then incubated in goat anti-horseradish peroxidase at 1:400 for 60 min. Immunoreactivity (IR) was detected using 3,3’diaminobenzidine (DAB) (Sigma-Aldrich Company Ltd, Dorset, UK) (0.5mg/mL, 15 min incubation). Slides were dehydrated in ethanol, cleared in xylene, and cover slipped using Permount. For cathepsin D IHC, the same procedure was followed with incubation overnight in rabbit anti-cathepsin D antibody (Abcam) at (4°C), and then in goat anti-rabbit IgG F(ab’)2 secondary for 60 min and rabbit peroxidase anti-peroxidase for 60 min. Slides were counterstained with CV.
Table 1.
Antibody and immunohistology resources
| Antibody | Use | Company | Catalogue # | RRID | Dilution |
|---|---|---|---|---|---|
| Goat anti-ChAT | 1° | Millipore | AB 144P | 2079751 | 1:800 IHC 1:400 IF |
| Rabbit anti-cathepsin D | 1° | Abcam | AB 75852 | 1523267 | 1:1000 IHC 1:500 IF |
| Donkey anti- IgG H&L | 2° | Abcam | AB 6880 | 955987 | 1:200 |
| Goat anti-rabbit IgG F(ab’)2 | 2° | ThermoFisher Scientific | 31234 | 228343 | 1:200 |
| Donkey anti-goat IgG (H+L) Alexa Fluor Plus 488 | 2° | Thermo Fisher Scientific | A32814TR | 2866497 | 1:400 |
| Donkey anti-rabbit IgG (H+L) Alexa Fluor 594 | 2° | Thermo Fisher Scientific | A32754 | 2762828 | 1:800 |
| Normal donkey serum | Blocking | EMD Millipore | S30 | 10% | |
| Normal goat serum | Blocking | Jackson ImmunoResearch Laboratories, Inc | 005-000-001 | 10% | |
| Goat anti- horseradish peroxidase | PAP | Jackson ImmunoResearch Laboratories, Inc | 123-005-021 | 2338952 | 1:400 |
| Rabbit peroxidase anti-peroxidase | PAP | Jackson ImmunoResearch Laboratories, Inc | 323-005-024 | 2337254 | 1:400 |
10-primary antibody, 20-secondary antibody, PAP -used in the peroxidase anti-peroxidase step of IHC
For immunofluorescent (IF) staining, brain sections, (one in every 9th), were washed in tris-buffered saline (TBS, pH 7.6) and underwent antigen retrieval in sodium citrate buffer (pH 6.0) at 80°C (90 min), permeabilized with 0.06% TX, blocked in normal goat serum (NGS) for 60 min and incubated overnight at 4°C in anti-cathepsin D antibody in TBS-T and NDS (4%). After washing, tissue was incubated overnight at 4°C in anti-ChAT antibody in TBS-T and 4% NGS. Tissue was then incubated in donkey anti-goat IgG (H+L) Alexa Fluor Plus 488 and donkey anti-rabbit IgG (H+L) Alexa Fluor 594 with NDS (4%) and TBS-T for120 min, washed, mounted, cover slipped and stored at 4°C.
Antibody characterization:
The anti-choline acetyltransferase antibody was a purified goat polyclonal raised against choline acetyltransferase in human placenta. As a negative control, incubation of sections with non-immune goat immunoglobulin at the same concentration as the primary antibody showed no staining of ChAT+ neurons, thus confirming the specificity of this antibody for choline acetyltransferase found in cholinergic neurons in the central nervous system as shown previously by us using IHC (Chang & Martin, 2009; Fayzullina & Martin, 2014) and also shown to detect mono-specifically a single ~68 kDa protein in mouse CNS by western blotting (L. J. Martin & Wong, 2020). Anti-cathepsin D antibody is a rabbit monoclonal antibody produced against a proprietary recombinant synthetic peptide including human cathepsin D aa 350 to the C-terminus. Incubation with non-immune rabbit immunoglobulin at the same concentration as the cathepsin IgG gave no similar staining. This antibody recognizes cathepsin D + lysosomes in the human and mouse developing brain and neurons undergoing autophagy and cell death (Aoto et al., 2021).
Assay for AChE activity was performed using the Hedreen and Bacon modified Karnovsky Roots method (Hedreen, Bacon, & Price, 1985) on one in every 8th section from sham (n=7) and HI (n=9) mice. Staining was intensified with in 0.1% sodium nitrate (1 min) and then 3, 5-minute exposures in 100 mM sodium nitrate solution. Tissue was mounted, dehydrated in ethanol, cleared in xylene, and cover slipped.
2.4. Assessment of ChAT+ neuronal counts and morphology
Counts of ChAT+ neurons, determined by cell profile counting, and their morphology were assessed in different cholinergic cell groups in sham and the ipsilateral HI mouse brains. The MSN was counted at the level of the nucleus accumbens corresponding to bregma 0.74 mm (Paxinos & Franklin, 2003). The nbM was counted at two levels corresponding to the area of the fourth cholinergic cell group (Ch4) in the mouse (Paxinos & Franklin, 2003) with the counting level being immediately ventral to the anterior commissure and the body of the striatum (bregma 0.26 mm), and the second counting level 600 μm posterior to the more anterior level and ventral to the globus pallidus (bregma −0.34). The nbM was also counted at two additional levels to capture the PP neurons. These levels were at bregma −0.34 and 600 μm more posterior at bregma −0.94. ChAT+ interneurons in the striatum were assessed in the body of the striatum on the same mouse brains at bregma 0.26 mm and −0.34 mm. ChAT+ neurons were counted in the brainstem in the pedunculopontine nucleus (PPN) and in the trochlear nucleus of cranial nerve IV (CNIV) ipsilateral and contralateral to the forebrain injury between bregma −4.26 and −4.36. Based on the surrounding neuroanatomy, this nucleus is the anterior trochlear nucleus. In the sections counted, the surrounding posterior telencephalon has only subiculum and the overlying occipital-visual cortex and there is the mesencephalic nucleus of CNV within the periaqueductal gray. Ventrally on the section is the emergent pedunculopontine tegmental nucleus. Therefore, the overall neuroanatomy is consistent with this being the paratrochlear-trochlear nucleus in brainstem.
Counts were made in one section per level per region per mouse. Care was taken to avoid treatment group bias in sample selection and counting by having independent individuals (FJN, LJM) identify and count precisely anatomical matched forebrain levels and by independent and collective screening and verification of counts.
If the number of ChAT+ neurons did not differ between sham and HI brains in a region, we then analyzed the morphology of the ChAT+ neurons in those regions. Normal ChAT+ neurons were large, strongly IR multipolar neurons whose processes extended into the surrounding neuropil. Features considered abnormal were i) altered immunoreactivity for ChAT+ compared to surrounding or contralateral ChAT+ neurons ii) loss or truncation of dendrites iii) displacement of the nucleus from its normal central position to the periphery of the cell soma, and iv) a crenated appearance of the cell or shrinkage of the cell soma. If the ChAT+ neurons displayed one or more of these abnormalities, we classified them as attritional.
2.5. ChAT neuronal dendritic length in nbM
Fifty μm-thick slices containing the nbM were imaged unbiasedly using a Zeiss LSM700 confocal microscope. All multichannel IF-IHC experiments were run under the same conditions and antibody concentrations, images were set medial to the region of interest (ROI) and stitched in 2 × 2 high magnification (63X x 1.4) fields to generate a single z-stack for 3D-reconstruction using slicing set at 0.5 air unit (0.4 μm) to the maximal wavelength. All imaging was performed under similar gain and laser intensity settings, using the reuse function of the Zeiss ZEN software. Images were stored in CZI format and transferred to the IMARIS software (Oxford Instruments, Zurich, Switzerland) workstation for processing.
An automatic IMARIS creation algorithm was used for processing. Following definition of channel sourcing and level of surface detailing, the surface renderings of ChAT+ neurons were detected, adjusted automatically with background subtraction, and masked. Split touching objects function, with the seed point diameter set to the average diameter of ChAT + neurons was used to create ChAT somas. The filament feature was used to trace visible dendrites originating from ChAT+ neuronal somas. Computer generated path analysis using starting and seed points were compared to manual tracing and if disagreement was noted, tracing was stopped at the most distal point of agreement. Quantifications of total dendrite length, length of unbranched and branched dendrites and number of dendrites per soma were determined within the section.
2.6. Measurement of regional area and optical densitometry (OD)
Corresponding CV and AChE stain sections were scanned at high resolution using TissueScope™ LE Slide Scanner, HURON Digital Pathology. Sections with the anterior hippocampi (bregma −1.70) and somatosensory cortices (bregma 0.26 mm) were used for area and AChE OD measures. Regional margins were determined using an atlas of mouse brain (Paxinos & Franklin, 2003). Using the measurement tool in Objective View (Objective Pathology Services. 2018, Objective View™. Version 1.48), a scale bar was drawn for the section, a screenshot taken with the view set to 50% zoom and opened in Fiji (Schindelin et al., 2012) at 50% zoom. Scale bars were used to calibrate the scale in Fiji for each photomicrograph. Area of the somatosensory cortex from the midline to the border of the retrosplenial cortex, the anterior hippocampus, and the body of the striatum was determined. For AChE staining, images were imported to Fiji in grayscale, and OD calibration performed per NIH. normally distributed OD Calibration https://imagej.nih.gov/ij/docs/examples/calibration. OD was measured in the hippocampus and somatosensory cortex of each hemisphere. Area measures were reported as percent difference in contralateral/ipsilateral area with greater percent differences reflecting greater ipsilateral area loss. OD was reported as a ratio of ipsilateral to contralateral OD corrected for background.
2.7. Cathepsin D+ lysosomal burden in nbM neurons
Sections stained for cathepsin D and CV were analyzed for cathepsin D + lysosomes within large multipolar cells of the nbM, at the levels previously described. Cells were photographed on a Nikon Eclipse E400 (Nikon, USA) at 1000x and analyzed using NIH ImageJ and CaseViewer Image Browser 2.4 (3DHISTECH Ltd. Budapest, Hungary). Although, multiple neural cell types express cathepsin, neurons are easily differentiated from other cell types within the nbM at 1000x magnification. Images were processed using the ImageJ “Cell Counter” plugin and cathepsin+ lysosomes counted manually within each neuron and the area of each neuron measured in one focal plane within the z-axis. The cathepsin D+ lysosomes were classified as large (diameter ~1μm), medium (diameter ~0.5μm) and small (diameter ~0.2 μm) (Araujo, Liebscher, Hess, & Huber, 2020; Guyton, 1981).
2.7. Statistics
All measurements were done without knowledge of the treatment group. Nonparametric statistics were applied including Mann-Whitney U test for independent groups, Wilcoxon test for non-independent groups, and Spearman correlation to evaluate relationships. Best fit was calculated with 95% confidence, resulting in a quadratic relationship. P < 0.05 was considered significant in all analyses. Scale bars were shown in each figure as appropriate.
2. Results
2.4. Regions of basal magnocellular cholinergic system studied
In P40 HI and sham mice, ChAT+ neurons were studied in the MSN at the level of the nucleus accumbens (Figure 1a), in the nbM within the substantia innominata beneath the striatum and the globus pallidus (Figure 1b and c), in the striatum at the levels shown in Figure 1a and b, and in PP neurons (Figure 1c) and one additional section as described in methods. The sections in Figure 1a, b and c were separated by 600 μm in the coronal plane.
Figure 1. Distribution and area of study of cholinergic neurons.

ChAT+ neurons were studied in the MSN (a)- the nbM (b)- and the striatum and in PP neurons (c). Sections were separated by 600μ in the coronal plane.
3.2. ChAT+ neurons in the MSN were lost or attritional at P40 after neonatal HI
ChAT+ neurons are vertically distributed through the MSN in a cone shaped arrangement (Figure 2a, and b, arrows). They are isodendritic with a large polygonal or triangular cell body and multiple dendrites. After HI, MSN ChAT+ neurons were the most uniformly and severely affected of the basal forebrain cholinergic cell groups with phenotypic alterations including loss of immunoreactivity, truncated and dystrophic neurites, shrunken soma, and prominent axonal pathology (Figure 2c and d, neurons closed arrows, axonal beading (open arrows)). In the ipsilateral MSN there was a decrease in percent remaining ipsilateral ChAT+ neurons compared to contralateral ChAT+ MSN neurons in HI brains (n=6, median 65% (IQR 45.5–75%)) vs sham brains (n=5, median 92% (IQR 89–100%) (p< 0.01)) (Figure 2e)). Percent remaining ipsilateral MSN ChAT+ neurons directly correlated with the % residual ipsilateral hippocampal area after HI (R2= 0.5894, p = 0.0066) (Figure 2f).
Figure 2. ChAT+ neurons were injured and lost after HI (hypoxia-ischemia) in MSN.
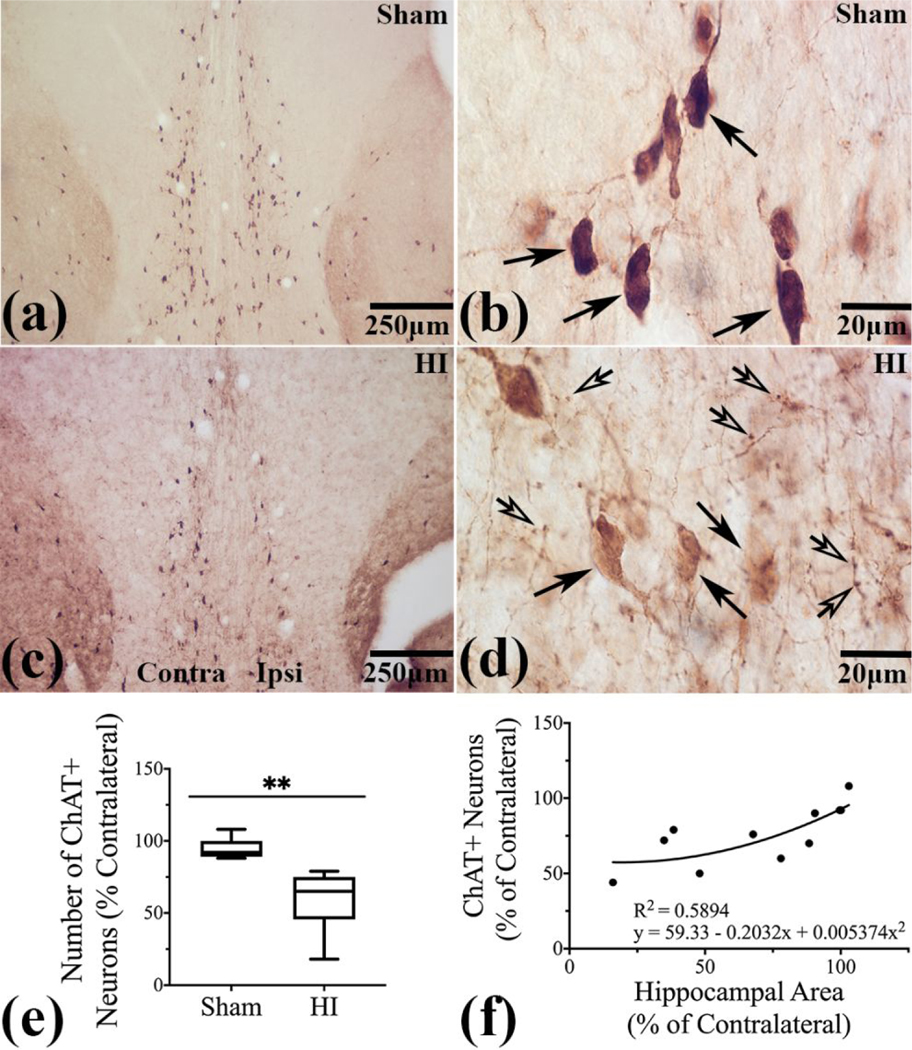
(a and b)-Normal distribution and morphology of ChAT+ neurons in MSN in sham (arrows). (c)- Loss of ChAT+ neurons in ipsilateral MSN. (d)- After HI, MSN ChAT+ neurons with loss of IR and with truncated, dystrophic neurites and shrunken soma (closed arrows) and prominent axonal beading in the neuropil (open arrows). (e)- Percent loss of ChAT+ neurons ipsilateral to the injury (p<0.01) (f)-Residual ipsilateral hippocampal area correlates with percent remaining ChAT+ neurons in ipsilateral MSN (R2= 0.5894, p = 0.0066). N=5 sham and n=6 HI brains
3.3. nbM and PP ChAT+ neurons were attritional at P40 after neonatal HI
ChAT+ neurons populate the ventral forebrain substantia innominata without forming a discretely bordered nucleus (Figure 3a, brown immunoreactivity). The Ch4 regions of nbM and PP ChAT + neurons were diffusely distributed, but consistent in general pattern localization, and constitute a single population of neurons. The location of PP ChAT+ neurons surrounding the GP was distinct in the mouse forebrain (Figure 3a). nbM ChAT+ neurons in the medial aspect of the substantia innominata and ventral to the GP were counted as nbM neurons (Figure 3a). Sham brains had normal ChAT+ neurons, being large, deeply stained with multiple processes (arrows) (Figure 3b and d, PP) (Figure 3f and h, nbM) extending into the neuropil and (Figure 3d, PP) (Figure 3h, nbM) (open arrows). HI injury eventuated in ChAT+ neurons with attenuated processes and altered ChAT IR (arrows) (Figure 3c, PP), (Figure 3g, nbM). Shrunken soma and truncated dendrites were present after HI in nbM ChAT+ neurons (Figure 3e, arrow indicates crenated soma and open arrows short dendrites, some with retraction bulbs) and in PP neurons (Figure 3i). Individual neurons with decreased immunoreactivity and eccentrically placed nucleus and shortened processes flanked cells with crenated cell bodies (Figure 3i, open arrow). Eccentric misplacement of the nucleus within HI ChAT+ neurons in the nbM was evident with CV counterstaining (Figure 3k) (arrows outline the medial aspect of the nucleus), in contrast to normal central location of the nucleus in nbM sham ChAT+ neurons (Figure 3j). Withering of processes and loss of ChAT IR occurred in concert with eccentric misplacement of the nucleus in HI nbM neurons (Figure 3k), but not in sham ChAT+ neurons, (Figure 3j).
Figure 3. ChAT+ Neurons in the ipsilateral nbM and PP region were injured after neonatal HI.

(a)- IR for ChAT was distributed throughout the basal forebrain, around the GP and in the striatum. PP ChAT+ neurons (b and d)- with normal cholinergic neuronal morphology (b and d, arrows) (d, neurites, open arrows) in sham brains. (c)- ChAT IR was lost after HI in PP neurons (arrows). (e)- Crenated soma (closed arrow) and truncated dendrites (open arrows), in PP ChAT+ neurons after HI. (f and g)- ChAT+ neurons (arrows) in sham (f) and HI (g) nbM. ChAT+ neurons (larger arrows) in sham (h) and HI (i) nbM (dendrites, small arrows, h, and i). (j and k)- Nuclear placement in ChAT+ neuron in sham (j) and HI (k) nbM neuron. (l) Total ChAT + neurons at 2 levels of nbM in sham (n=7) and HI (n=9). (m)- Percentage of abnormal ChAT+ neurons was higher in HI nbM (p<0.001). (n)- Area loss in ipsilateral cortex correlates with percent abnormal ChAT+ neurons, (Sham n=5, HI n=9) (R2= .814, p <0.001).
At P40, total number of ChAT+ neurons in the nbM was equal in sham and HI brains (Figure 3l, Sham (n=7): median 116.0, IQR 100.0 – 126.0, HI (n=9): median 113.0, IQR 92.50 – 119.0, p=NS). At P40 following neonatal HI (n=9), 66.06% (IQR 54.22–75.05%) of the ChAT+ neurons in the ipsilateral nbM displayed at least one abnormality compared to 5.00% of shams (n=7) (IQR 3.914–6.275%, p <0.001) (Figure 3m). Percent abnormal ChAT+ neurons in the nbM directly correlated (quadratic) with percent loss of ipsilateral cortical area after HI (Sham n=5, HI n=9) R2= 0.814, p < 0.001) (Figure 3n).
Length of dendrites on ChAT+ neurons in the nbM was altered at P40 after HI. ChAT+ neurons were easily identified in both sham and HI nbM (Figure 4 A and B) and with surface rendering of the soma and dendrite tracing (Figures 4c and d). In n=22 neurons from 6 sham mice and n=33 neurons from 7 HI mice, nbM ChAT+ dendrites were longer in sham mice compared to HI (Figure 4e, (Sham: median 167.6 μm, IQR 89.8–235, HI: median 60.1 μm, IQR 29.8–78.8, p<0.0001). Number of dendrites per ChAT+ neuron did not differ between groups (data not shown). To determine whether neurons with more extensive dendritic branching have greater susceptibility to HI than those with less extensive branching, individual dendrites were identified in IMARIS as unbranched or branched and compared in length. Both unbranched and branched dendrites of HI nbM ChAT+ neurons were shorter than those in sham neurons (Figure 4f, (unbranched dendrites, sham n=31, median 19.7 μm, IQR 87.7–31.2, HI n=104, median 7.3 μm, IQR 4.3–13.2, p<0.0001), (branched dendrites, sham n=31, median 51.7 μm, IQR 36.7–73.4, HI n=35: median 29.4 μm, IQR 18.2–41.2, p=0.0001)). Median distance between ChAT+ neurons did not differ in shams and HI (data not shown), thus overall neuropil compaction is not the cause of the shorter dendrite length.
Figure 4. Dendrites of nbM cholinergic neurons were shorter at P40 after HI.

ChAT+ neurons and their dendrites (green IF) from (a)-sham and (b)-HI nbM neurons. (c) and (d)- Surface rendering of the soma and dendrite tracing. (e)- Dendritic lengths in Sham (n=22) and HI (n=33) nbM ChAT+ neurons, p<0.0001. (f)-Length of unbranched (sham n=90), (HI n=104) and branched (sham n=31), (HI n=35) dendrites in HI nbM ChAT+ neurons (p<0.0001and p=0.0001). (Sham n=6, HI n=7 mice).
3.4. ChAT+ neurons in the striatum at P40 were attritional after neonatal HI
Although striatal ChAT+ neurons are interneurons (Bolam, Wainer, & Smith, 1984; Takagi, Somogyi, & Smith, 1984) lacking cortical projections, they too, were damaged ipsilateral to HI injury. ChAT+ interneurons were widely dispersed throughout the striatum (Figure 5a). Phenotypically normal neurons had full soma and multiple processes (Figure 5b and d, arrows). After HI, their IR was altered (Figure 5c, arrows) and they were attritional (Figure 5e, closed arrows -shrunken, rounded soma, open arrows- interrupted and simplified dendrites) compared to uniformly and evenly stained ChAT+ neurons with healthy dendrites in shams (Figure 5d, closed arrow-soma, open arrow-dendrites). Sham (Figure 5f) and HI (Figure 5g, ipsilateral on the right) striatum is outlined. At P40, total ChAT + neurons in the striatum did not differ between sham (n=5) and HI (n=8); (sham: median 141.0, (IQR 126.0 – 156.0), HI: median 128.0, (IQR 106.0 – 158.5, p=NS) (Figure 5h). Attritional ChAT+ neurons in P40 mice made up 62.4% (IQR 46.7–76.2%) of the total ChAT+ interneuron population in the ipsilateral striatum after neonatal HI compared to 3.8% in shams (IQR 1.6–5.3%, p< 0.001, Figure 5i). The percentage of abnormal ChAT+ neurons in the striatum increased in concert with increase loss of ipsilateral striatal area after HI (sham n=5, HI n=8) (R2= 0.871, p<0.001, Figure 5j).
Figure 5. Striatal interneurons were injured after neonatal HI.

(a)- ChAT+ interneurons were widely dispersed throughout the striatum (b and d)-Normal ChAT+ neurons in shams. Soma (closed arrow) and processes (open arrows) in (d). (c)- Decreased ChAT IR (arrows) and (e)- shrunken, contracted soma (closed arrows) and interrupted and simplified dendrites (open arrows) after HI. (f and g)- Region of striatal area measurements in sham and HI. (h)-Total ChAT + neurons in sham and HI striatum at P40 (p=NS). (i)- 62.4% of ChAT+ HI interneurons were abnormal (p< 0.001 vs. sham). (j)-Residual ipsilateral striatal area correlates with percent abnormal ChAT+ neurons after HI (R2= 0.871, p<0.001) (Sham n=5, HI n=8).
3.5. ChAT+ neurons in the brainstem at P40 appeared normal after neonatal HI
No abnormalities of ChAT+ neurons were detected in the midbrain and pontine tegmentum after neonatal HI (Figure 6). Number of ChAT+ neurons in the ipsilateral and contralateral PPN did not differ (Figure 6a, and g, n=6 sham, n=7 HI) nor did number of ChAT+ neurons in CNIV (Figure 6d and g, n=6 sham, n=7 HI). ChAT+ soma and processes were normal in the contralateral and ipsilateral PPN (Figure 6b and 6c) and in contralateral and ipsilateral CNIV (Figure 6e and 6f).
Figure 6. ChAT+ neurons in the midbrain and pontine tegmentum after neonatal HI.

Number of ChAT+ neurons in the ipsilateral and contralateral PPN were equivalent (a) and (g) as were number of ChAT+ neurons in CNIV (d and g), (Sham n=6, HI n=7). (b and c)- Normal ChAT+ neurons in contralateral and ipsilateral PPN. (e and f)- Normal ChAT+ neurons in contralateral and ipsilateral CNIV.
3.6. Ipsilateral cerebral cortex and hippocampus had loss of area but AChE was sustained at P40 after neonatal HI
OD of AChE staining and area measurements (Figure 7) were made in bilateral sensory-motor cortex (black outline) and anterior hippocampus (red outline) in sham (Figure 7d, n=5) and HI (Figure 7e, n=5) brains. OD was measured in AChE-stained brains and residual area in adjacent CV sections. Relative ipsilateral/ contralateral OD increased in both the injured cortex (sham, median OD= 0.9950, (IQR 0.9810–1.058) HI median OD= 1.317, (IQR1.213–1.383), p <0.0025) and hippocampus (sham, median OD= 0.9973, (IQR 0.9400–1.023)) (HI, median OD=1.407, (IQR 1.219–2.017), p <0.0025)) (Figure 7a). Increase of AChE OD in the HI brain regions was linearly related to the % difference in area of the ipsilateral and contralateral cortex (Figure 7b, R2= 0.707, p= 0.0438) and hippocampus (Figure 7c, R2=0.777, p=0.0083).
Figure 7. AChE activity preserved in penumbral hippocampus and cortex after HI.

(a)- OD of AChE increased in ipsilateral somatosensory cortex and anterior hippocampus after HI (p<0.01vs Sham). (b and c)- Increased OD was linearly related to increased area loss (% Ipsilateral /Contralateral difference) (Cortex- R2 =0.71, p< 0.05, Hippocampus- R2 =0.77, p< 0.01). (d, e and f)- OD (f) and area measurements were made in outlined areas of bilateral sensory-motor cortex (black outline) and anterior hippocampus (red outline) in sham (d) and HI (e) brains. (Sham n=5, HI n=5)
3.7. Large cells in nbM accumulated cathepsin D at P40 after neonatal HI
Large cells, most likely neurons throughout the nbM, had many cathepsin D+ lysosomes (Figure 8a and b, arrows). Cathepsin D+ lysosomes were present in smaller cells as well (Figure 8b, open arrow). Cells (sham, n=28, HI, n= 37) analyzed in both sham (n=3) and HI (n=5) brains were of equal surface area (Figure 8c, p=NS) but cathepsin D+ lysosomes were more numerous (Figure 8b and d, sham median 36.5, (IQR 23.0–48.75), HI median 53.5, (IQR 44.25–71.0) p<0.0001)) and there were more large cathepsin D+ lysosomes per cell in HI brains (Figure 8b and e, sham median 4.0, (IQR 2.0–5.0), HI median 9.0, (IQR 7.0–13.5), p<0.0001). Double labeling IF demonstrated cathepsin D+ lysosomes (red IF) in both sham and HI in ChAT+ neurons (green IF), (Figure 8f and 8g, respectively).
Figure 8. Lysosomal burden increased in magnocellular neurons of the ipsilateral nbM after HI.

(a)- Large cells presumably neurons, throughout the nbM have cathepsin D+ lysosomes (arrows), (b) -larger and increased density of cathepsin D+ lysosomes in HI neurons (arrows), cathepsin D+ lysosomes are present in smaller cells (open arrow). (c)- Neurons in sham and HI brains were of equal area (p=NS). (d)- Cathepsin D+ lysosomes were more numerous (p<0.0001)) and (e)- larger in HI brains (p<0.0001 vs sham), (Neurons analyzed, Sham n=28, HI n=37) (f and g) Both sham and HI ChAT+ nbM neurons (green IF) have cathepsin + lysosomes (red IF).
4. Discussion
Cholinergic systems were not thought to be permanently disturbed by neonatal HI heretofore, but we now find evidence of permanent injury to forebrain cholinergic systems after neonatal HI. At P40 after neonatal HI, most of the ipsilateral ChAT+ neurons in the mouse nbM, MSN, and striatum have altered localization ChAT, were attritional, and had dendritic pathology. These findings coincided with loss of area in the brain regions to which these ChAT+ neurons project; cortex for nbM, hippocampus for MSN, and striatum, for striatal interneurons. ChAT+ neurons in the MSN were most severely affected. We were also able to quantitate the dendritic shortening previously reported in other disorders of cholinergic basal forebrain function. Combined with maintenance of AChE and an increase in cathepsin D+ lysosomal burden, the present results suggest that learning, memory, and executive function deficits after neonatal HI may have a hitherto unexplored neuroanatomical basis manifested by perturbations of the forebrain cholinergic systems.
Arousal, attention, decision making, motivation, memory, modulation of cortical activity, the default-mode network, and cortical blood flow are all controlled in part by the basal forebrain cholinergic system (Alves et al., 2019; Deiana et al., 2011; Hotta, Uchida, Kagitani, & Maruyama, 2011; Mesulam, 2013; Raver & Lin, 2015; Zaborszky et al., 2012). All ChAT+ neurons that project to the cortex and hippocampus are found in the basal forebrain (Mesulam, 2013): the nbM, including PP neurons (Mesulam, 2013; Whitehouse, Struble, Hedreen, Clark, & Price, 1985), being the largest of the areas of the basal forebrain magnocellular complex. The basal cholinergic forebrain, with its widespread corticopedal projections that rapidly release acetylcholine (Ach) in a distributed fashion, may serve as the biochemical basis for attention (Mesulam, 2013; Schmitz & Duncan, 2018). The cholinergic system is an important subcortical node in the default mode network (Alves et al., 2019; Nair et al., 2018), and cholinergic input exerts control of the default-mode network (Shah et al., 2015). ChAT+ neurons in the nbM and ChAT+ interneurons in the striatum are critical for cognitive flexibility (Prado, Janickova, Al-Onaizi, & Prado, 2017; Ragozzino, Mohler, Prior, Palencia, & Rozman, 2009). Dorsomedial striatal ChAT+ neurons are essential for establishment of a new strategy in reversal learning while ChAT+ neurons in the nbM modulate initial inhibition of a previously learned strategy (Prado et al., 2017). This dependance of large domains of cognition and brain function on cholinergic function, suggests early life injury to ChAT+ neurons may be responsible for abnormalities in cognition, learning, memory, executive function, sleep, and attention.
Deficits in learning, memory, executive function, and attention are widely reported following neonatal HI especially in preterm infants (Edgin et al., 2007; M. J. Grothe et al., 2017; Marlow, Rose, Rands, & Draper, 2005; Smith, Alexander, Rosenkrantz, Sadek, & Fitch, 2014). These deficits have previously been correlated with white matter injury (Edgin et al., 2007; Tymofiyeva et al., 2018), however, scant literature exists on the role of injury of the cholinergic system in neonatal life and subsequent deficits. A transient decrease in ChAT+ activity and in 3H-choline uptake occurs in the ipsilateral striatum at P21 after neonatal HI with recovery by adult age (Johnston, 1983). Our morphologic findings suggest permanent changes to ChAT+ forebrain neurons after neonatal HI.
Most literature on the responses of basal forebrain ChAT+ neurons to neocortical and hippocampal damage follows from findings that nbM neurons are highly attritional and lost in AD (Whitehouse et al., 1982);(Vogels et al., 1990). Humans and macaque monkeys with large cortical lesions, leucotomy, or infarctions have attrition of nbM neurons (Liberini, Pioro, Maysinger, & Cuello, 1994; Pearson, Gatter, & Powell, 1983). Cortical ablations through P14 and removal of the pia induce phenotypic changes to ChAT+ neurons identical to those described here (Sophou, Dori, Antonopoulos, Parnavelas, & Dinopoulos, 2006) (Sofroniew, Pearson, Eckenstein, Cuello, & Powell, 1983). The present results indicate that basal forebrain ChAT+ neurons respond to injury with a common attritional phenotype consistently across the lifespan, regardless of the type of insult or species. Within the boundary of the tissue imaged we found a 65% decrease in dendritic length in HI ChAT+ nbM neurons at P40. It is unclear how this percent decrease translates to overall loss of dendritic length within the intact brain. We used loss of IF for ChAT along the path of the dendrite to signal end of the process leaving open the possibility that it is loss of staining rather than actual truncation of the dendrite. Finding retraction bulbs and beaded dendrites with IHC (Figure 3(e) and (i) support our conclusion that the dendrites are truncated rather than just phenotypically losing their ChAT signature. This report is the first of its kind. The branching morphology of the dendrite was not determinant of vulnerability to HI injury, but shortened and injured dendrites would impair the vital connection of basal forebrain ChAT+ neurons to the cortex and hippocampus. The forebrain pathology following neonatal HI in mice is well described and presents in the distribution of the middle cerebral artery (Burnsed et al., 2015; Sheldon, Sedik, & Ferriero, 1998). Continued progression of cortical and hippocampal injury has been less well studied but does occur (Geddes, Vannucci, & Vannucci, 2001; Stone et al., 2008). Among the possible mechanisms for this late progression of injury, loss of cholinergic input from the basal forebrain should be considered.
Quantifiable loss of ChAT neurons differentiates the response of the MSN from the nbM to neonatal HI. Septal neurons are more vulnerable and degenerate after hippocampal injury early in life (Sofroniew, Galletly, Isacson, & Svendsen, 1990). Transection of the fimbria fornix or removal of the hippocampus causes attrition and loss of MSN neurons (Daitz & Powell, 1954; Sofroniew, Pearson, Cuello, Tagari, & Stephens, 1986). MSN ChAT+ neurons that remain are pyknotic and lightly stained (Daitz & Powell, 1954; Ginsberg & Martin, 1998). Loss of ChAT+ neurons and abundant axonal beading in the MSN 4 weeks after neonatal HI, suggests that axonal injury and target deprivation could have prominent roles in cholinergic neurodegeneration after neonatal HI. The difference in response of MSN versus nbM ChAT+ neurons has been attributed to the more restricted and reciprocal connections of the MSN to the hippocampus compared to the widely distributed connections of the nbM to the cortex (Sophou et al., 2006). Additionally, the hippocampus is the more severely injured region in this model of neonatal HI (Burnsed et al., 2015).
Severe direct excitotoxic lesion of nbM cholinergic neurons causes loss of AChE, the enzyme necessary to terminate ChAT+ signaling at synapses, (Zaborszky et al., 2012) in the cortex (Johnston, McKinney, & Coyle, 1981). AChE histochemical staining did not decrease in the ipsilateral cortex and hippocampus of our mice. Injuring distal cholinergic axons indirectly may allow preserved axons to sprout, perhaps aberrantly. Preservation of AChE activity after neonatal HI, combined with a decrease of cholinergic input from the MSN and the nbM may augment a cholinergic deficit in cerebral cortex and hippocampus and be an aberrant and functionally harmful tissue plasticity response. Traditional inhibitors of AChE (Hiremathad, Chaves, & Keri, 2021) have not been tested after neonatal HI but clearly there was AChE enzymatic activity present at P40 in the injured cortex and hippocampus that might be inhibitable.
No injury to ChAT+ neurons in the ipsilateral PPN or nucleus of CN IV was detected, but their axonal arborizations were not assessed. Whether abnormalities of cholinergic neurotransmission from the PPN affects cognition is unclear (Janickova et al., 2019) (Dautan et al., 2020). The anatomical complexity of the PPN and large variability in ChAT+ neuron counts in the PPN may obscure an effect of HI, nevertheless, impairments in cognitive flexibility after neonatal HI (Maxwell et al., 2020) can be explained by the observed injury to striatal ChAT+ interneurons.
Cathepsin D, a lysosomal marker of autophagy was increased in large neurons in the ipsilateral nbM after HI. Acute ischemia induces neuronal autophagy including increased expression of cathepsin D (Carloni, Buonocore, & Balduini, 2008; Liu et al., 2018; Xiao et al., 2015). The lentiform nuclei in human infants that die after severe HIE, have increased number and size of cathepsin D+ lysosomes (Xie et al., 2016). Our findings were consistent with nbM neurons upregulating lysosomal-related autophagy but do not determine which type of nbM neurons were responsible. ChAT+ neurons clearly have cathepsin D+ lysosomes, however analysis failed to show either an increase in number or size of the cathepsin D+ lysosomes in nbM ChAT+ neurons (data not shown). Other neurons in the basal forebrain may be the source of the cathepsin D increase seen here.
This work did not address the effects of HI on non-ChAT+ basal forebrain neurons and other systems within the substantia innominata, including the substantia bed nucleus-amygdala continuum (L. J. Martin, Powers, Dellovade, & Price, 1991). Injury to these neurons could also contribute to deficits in learning, memory, and behavioral flexibility seen after HI in this model (Maxwell et al., 2020). GABAergic interneurons intermingle with ChAT+ neurons in the basal forebrain as well as neurons with other neuropeptide neurotransmitters, so damage to these neurons, may affect other neurotransmitter systems (Prado et al., 2017).
Despite these limitations, this study highlights a previously unrecognized vulnerability of the neonatal brain to HI. New data on ChAT+ dendritic attrition after HI might underlie a deficit in afferent input to the ChAT+ basal forebrain neurons which could then cascade to abnormal cholinergic modulation of the cerebral cortex. Because of the extensive abnormalities found in important ChAT+ neuronal populations after neonatal HI, therapeutics being investigated as treatments for the loss of cholinergic signaling and dementia in Trisomy 21(Granholm et al., 2002) and AD (Hiremathad et al., 2021) may be useful as therapies for memory, learning, cognitive flexibility, and attention deficits suffered by survivors of HIE, after proper animal testing. Finally, because of the newly recognized importance of the nbM in the default-mode network (Alves et al., 2019), these data contribute to an overarching hypothesis for behavioral, cognitive, learning, and executive dysfunctions seen in survivors of neonatal HIE.
Funding and Acknowledgements
FJN is supported by AG061643, HD086058, NS103882, and HD074593, LJM by NS079348 HD074593, AG061643, and the Johns Hopkins University Alzheimer’s Disease Research Center (AG005146), RCV by NS096115 and Johns Hopkins University Clinician Scientist Award and Sutland Pakula Endowment for Neonatal Research. PK is supported by the K12HD001399 (NIH/NICHD) Child Health Research Career Development Award (CHRCDA) and by the Children’s National Board of Visitors. The authors declare they have no competing interests.
Abbreviations:
- Ach
Acetylcholine
- AChE
Acetylcholinesterase
- AD
Alzheimer’s disease
- ChAT
Choline acetyltransferase
- Ch4
Cholinergic cell group 4
- CV
Cresyl violet
- GP
Globus Pallidus
- HIE
Hypoxic-ischemic encephalopathy
- HI
Hypoxic ischemic
- IHC
Immunohistochemistry
- IF
Immunofluorescent
- IR
Immunoreactivity
- IQR
Interquartile range
- IQ
Intelligence quotient
- MSN
Medial Septal Nucleus
- min
Minute
- NICHD
National Institute of Child Health and Development
- NDS
Normal donkey serum
- NGS
Normal goat serum
- nbM
Nucleus basalis of Meynert
- OD
Optical density
- PPN
Pedunculopontine nucleus
- PP
Peri-pallidal (neurons)
- P
Post-natal
- ROI
Region of interest
- TH
Therapeutic hypothermia
- TX
Triton X-100
- CNIV
Trochlear cranial nerve IV nucleus
Footnotes
Dedication: The authors dedicate this paper to our friend and mentor, Dr. Michael V. Johnston.
Data Availability Statement: The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Alves PN, Foulon C, Karolis V, Bzdok D, Margulies DS, Volle E, & Schotten M. T. d. (2019). An improved neuroanatomical model of the default-mode network reconciles previous neuroimaging and neuropathological findings. Communications biology, 2(1), 370. doi: 10.1038/s42003-019-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto K, Kato M, Akita T, Nakashima M, Mutoh H, Akasaka N, . . . Saitsu H. (2021). ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice. Nature Communications, 12(1), 2107. doi: 10.1038/s41467-021-22389-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo MEG, Liebscher G, Hess MW, & Huber LA (2020). Lysosomal size matters. Traffic, 21(1), 60–75. doi: 10.1111/tra.12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzopardi D, Strohm B, Marlow N, Brocklehurst P, Deierl A, Eddama O, . . . Group, T. (2014). Effects of hypothermia for perinatal asphyxia on childhood outcomes. The New England journal of medicine, 371(2), 140–149. doi: 10.1056/nejmoa1315788 [DOI] [PubMed] [Google Scholar]
- Bolam JP, Wainer BH, & Smith AD (1984). Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience, 12(3), 711–718. doi: 10.1016/0306-4522(84)90165-9 [DOI] [PubMed] [Google Scholar]
- Burnsed JC, Chavez-Valdez R, Hossain MS, Kesavan K, Martin LJ, Zhang J, & Northington FJ (2015). Hypoxia-ischemia and therapeutic hypothermia in the neonatal mouse brain--a longitudinal study. PLoS One, 10(3), e0118889. doi: 10.1371/journal.pone.0118889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni S, Buonocore G, & Balduini W. (2008). Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis, 32(3), 329–339. [DOI] [PubMed] [Google Scholar]
- Chang Q, & Martin LJ (2009). Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am J Pathol, 174(2), 574–585. doi: 10.2353/ajpath.2009.080557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daitz HM, & Powell TP (1954). Studies of the connexions of the fornix system. J Neurol Neurosurg Psychiatry, 17(1), 75–82. doi: 10.1136/jnnp.17.1.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautan D, Huerta-Ocampo I, Gut NK, Valencia M, Kondabolu K, Kim Y, . . . Mena-Segovia J. (2020). Cholinergic midbrain afferents modulate striatal circuits and shape encoding of action strategies. Nature Communications, 11(1), 1739. doi: 10.1038/s41467020-15514-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiana S, Platt B, & Riedel G. (2011). The cholinergic system and spatial learning. Behavioural Brain Research, 221(2), 389–411. doi: 10.1016/j.bbr.2010.11.036 [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, & Ferriero DM (1996). Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res, 39(2), 204–208. Retrieved from [DOI] [PubMed] [Google Scholar]
- Edgin JO, Inder TE, Anderson PJ, Hood KM, Clark CAC, & Woodward LJ (2007). Executive functioning in preschool children born very preterm: Relationship with early white matter pathology. Journal of the International Neuropsychological Society, 14(1), 90–101. doi: 10.1017/s1355617708080053 [DOI] [PubMed] [Google Scholar]
- Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. (2014). Obstet Gynecol, 123(4), 896–901. doi: 10.1097/01.AOG.0000445580.65983.d2 [DOI] [PubMed] [Google Scholar]
- Fayzullina S, & Martin LJ (2014). Skeletal Muscle DNA Damage Precedes Spinal Motor Neuron DNA Damage in a Mouse Model of Spinal Muscular Atrophy (SMA). PLoS One, 9(3), e93329. doi: 10.1371/journal.pone.0093329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes R, Vannucci RC, & Vannucci SJ (2001). Delayed cerebral atrophy following moderate hypoxia-ischemia in the immature rat. Dev Neurosci, 23(3), 180–185. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, & Martin LJ (1998). Ultrastructural analysis of the progression of neurodegeneration in the septum following fimbria-fornix transection. Neuroscience, 86(4), 1259–1272. [DOI] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O’Riordan DP, & Northington FJ (2004). Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis, 17(1), 89–98. [DOI] [PubMed] [Google Scholar]
- Granholm A-CE, Ford KA, Hyde LA, Bimonte HA, Hunter CL, Nelson M, . . . Crnic LS (2002). Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiology & Behavior, 77(2–3), 371–385. doi: 10.1016/s00319384(02)00884-3 [DOI] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, & Teipel SJ (2012). Atrophy of the Cholinergic Basal Forebrain Over the Adult Age Range and in Early Stages of Alzheimer’s Disease. Biological Psychiatry, 71(9), 805–813. doi: 10.1016/j.biopsych.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Scheef L, Bauml J, Meng C, Daamen M, Baumann N, . . . Sorg C. (2017). Reduced Cholinergic Basal Forebrain Integrity Links Neonatal Complications and Adult Cognitive Deficits After Premature Birth. Biol Psychiatry, 82(2), 119–126. doi: 10.1016/j.biopsych.2016.12.008 [DOI] [PubMed] [Google Scholar]
- Guyton AC (1981). Textbook of Medical Physiology (6th ed.). Phliladelphia: W. B. Saunders Company. [Google Scholar]
- Hedreen JC, Bacon SJ, & Price DL (1985). A modified histochemical technique to visualize acetylcholinesterase-containing axons. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society, 33(2), 134–140. doi: 10.1177/33.2.2578498 [DOI] [PubMed] [Google Scholar]
- Hiremathad A, Chaves S, & Keri RS (2021). Multi-Targeting Tacrine Conjugates with Cholinesterase and Amyloid-Beta Inhibitory Activities: New Anti-Alzheimer’s Agents. Chemistry & Biodiversity, 18(2). doi: 10.1002/cbdv.202000083 [DOI] [PubMed] [Google Scholar]
- Hotta H, Uchida S, Kagitani F, & Maruyama N. (2011). Control of cerebral cortical blood flow by stimulation of basal forebrain cholinergic areas in mice. The Journal of Physiological Sciences, 61(3), 201–209. doi: 10.1007/s12576-011-0139-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janickova H, Kljakic O, Rosborough K, Raulic S, Matovic S, Gros R, . . . Prado MAM (2019). Selective decrease of cholinergic signaling from pedunculopontine and laterodorsal tegmental nuclei has little impact on cognition but markedly increases susceptibility to stress. The FASEB Journal, 33(6), 7018–7036. doi: 10.1096/fj.201802108r [DOI] [PubMed] [Google Scholar]
- Johnston MV (1983). Neurotransmitter alterations in a model of perinatal hypoxic-ischemic brain injury. Ann Neurol, 13(5), 511–518. [DOI] [PubMed] [Google Scholar]
- Johnston MV, McKinney M, & Coyle JT (1981). Neocortical cholinergic innervation: A description of extrinsic and intrinsic components in the rat. Experimental Brain Research, 43(2), 159–172. doi: 10.1007/bf00237760 [DOI] [PubMed] [Google Scholar]
- Lawn JE, Cousens S, Zupan J, & Lancet Neonatal Survival Steering, T. (2005). 4 million neonatal deaths: when? Where? Why? Lancet, 365(9462), 891–900. doi: 10.1016/S01406736(05)71048-5 [DOI] [PubMed] [Google Scholar]
- Lee AC, Kozuki N, Blencowe H, Vos T, Bahalim A, Darmstadt GL, . . . Lawn JE (2013). Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res, 74 Suppl 1, 50–72. doi: 10.1038/pr.2013.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberini P, Pioro EP, Maysinger D, & Cuello AC (1994). Neocortical infarction in subhuman primates leads to restricted morphological damage of the cholinergic neurons in the nucleus basalis of Meynert. Brain Res, 648(1), 1–8. doi: 10.1016/00068993(94)91897-x [DOI] [PubMed] [Google Scholar]
- Liu Y, Xue X, Zhang H, Che X, Luo J, Wang P, . . . Yang J. (2018). Neuronal-targeted TFEB rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy, 15(3), 493–509. doi: 10.1080/15548627.2018.1531196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic SN, & Murasko DM (1993). Anesthesia inhibits interferon-induced natural killer cell cytotoxicity via induction of CD8+ suppressor cells. Cell Immunol, 151(2), 474–480. doi:S0008–8749(83)71256–6 [pii] 10.1006/cimm.1993.1256 [DOI] [PubMed] [Google Scholar]
- Marlow N, Rose AS, Rands CE, & Draper ES (2005). Neuropsychological and educational problems at school age associated with neonatal encephalopathy. Archives of Disease in Childhood - Fetal and Neonatal Edition, 90(5), F380–F387. doi: 10.1136/adc.2004.067520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Brambrink A, Koehler R, & Traystman R. (1997). Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. The Journal of comparative neurology, 377(2), 262–285. doi: 10.1002/(sici)1096-9861(19970113)377:2<262::aid-cne8>3.0.co;2-1 [DOI] [PubMed] [Google Scholar]
- Martin LJ, Powers RE, Dellovade TL, & Price DL (1991). The bed nucleus-amygdala continuum in human and monkey. J Comp Neurol, 309(4), 445–485. doi: 10.1002/cne.903090404 [DOI] [PubMed] [Google Scholar]
- Martin LJ, & Wong M. (2020). Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front Neurol, 11, 592851. doi: 10.3389/fneur.2020.592851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JR, Zimmerman AJ, Pavlik N, Newville JC, Carlin K, Robinson S, . . . Jantzie LL (2020). Neonatal Hypoxic-Ischemic Encephalopathy Yields Permanent Deficits in Learning Acquisition: A Preclinical Touchscreen Assessment. Front Pediatr, 8, 289. doi: 10.3389/fped.2020.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM (2013). Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. Journal of Comparative Neurology, 521(18), 4124–4144. doi: 10.1002/cne.23415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair J, Klaassen A-L, Arato J, Vyssotski AL, Harvey M, & Rainer G. (2018). Basal forebrain contributes to default mode network regulation. Proceedings of the National Academy of Sciences, 115(6), 1352–1357. doi: 10.1073/pnas.1712431115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, & Franklin KBJ (2003). The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press. [Google Scholar]
- Pearson RC, Gatter KC, & Powell TP (1983). Retrograde cell degeneration in the basal nucleus in monkey and man. Brain Res, 261(2), 321–326. doi: 10.1016/00068993(83)90637-6 [DOI] [PubMed] [Google Scholar]
- Prado VF, Janickova H, Al-Onaizi MA, & Prado MA (2017). Cholinergic circuits in cognitive flexibility. Neuroscience, 345, 130–141. doi: 10.1016/j.neuroscience.2016.09.013 [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Mohler EG, Prior M, Palencia CA, & Rozman S. (2009). Acetylcholine activity in selective striatal regions supports behavioral flexibility. Neurobiology of Learning and Memory, 91(1), 13–22. doi: 10.1016/j.nlm.2008.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raver SM, & Lin S-C (2015). Basal forebrain motivational salience signal enhances cortical processing and decision speed. Frontiers in behavioral neuroscience, 9, 277. doi: 10.3389/fnbeh.2015.00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JE, Vannucci RC, & Brierley JB (1981). The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol, 9(2), 131–141. [DOI] [PubMed] [Google Scholar]
- Roland EH, Poskitt K, Rodriguez E, Lupton BA, & Hill A. (1998). Perinatal hypoxic-ischemic thalamic injury: clinical features and neuroimaging [see comments]. Ann Neurol, 44(2), 161–166. [DOI] [PubMed] [Google Scholar]
- Salas J, Reddy N, Orru E, Carson KA, Chavez-Valdez R, Burton VJ, . . . Huisman T. (2019). The Role of Diffusion Tensor Imaging in Detecting Hippocampal Injury Following Neonatal Hypoxic-Ischemic Encephalopathy. J Neuroimaging, 29(2), 252259. doi: 10.1111/jon.12572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, . . . Cardona A. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods, 9(7), 676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz TW, & Duncan J. (2018). Normalization and the Cholinergic Microcircuit: A Unified Basis for Attention. Trends in cognitive sciences, 22(5), 422–437. doi: 10.1016/j.tics.2018.02.011 [DOI] [PubMed] [Google Scholar]
- Shah D, Blockx I, Keliris GA, Kara F, Jonckers E, Verhoye M, & Linden A. V. d. (2015). Cholinergic and serotonergic modulations differentially affect large-scale functional networks in the mouse brain. Brain Structure and Function, 221(6), 3067–3079. doi: 10.1007/s00429-015-1087-7 [DOI] [PubMed] [Google Scholar]
- Shankaran S, Pappas A, McDonald S, Vohr B, Hintz S, Yolton K, . . . Eunice Kennedy Shriver NNRN (2012). Childhood outcomes after hypothermia for neonatal encephalopathy. The New England journal of medicine, 366(22), 2085–2092. doi: 10.1056/NEJMoa1112066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Sedik C, & Ferriero DM (1998). Strain-related brain injury in neonatal mice subjected to hypoxia- ischemia. Brain Res, 810(1–2), 114–122. [DOI] [PubMed] [Google Scholar]
- Smith AL, Alexander M, Rosenkrantz TS, Sadek ML, & Fitch RH (2014). Sex differences in behavioral outcome following neonatal hypoxia ischemia: Insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic brain injury. Experimental neurology, 254, 54–67. doi: 10.1016/j.expneurol.2014.01.003 [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Galletly NP, Isacson O, & Svendsen CN (1990). Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science, 247(4940), 338–342. doi: 10.1126/science.1688664 [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Pearson RC, Eckenstein F, Cuello AC, & Powell TP (1983). Retrograde changes in cholinergic neurons in the basal forebrain of the rat following cortical damage. Brain Res, 289(1–2), 370–374. doi: 10.1016/0006-8993(83)90045-8 [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Pearson RCA, Cuello AC, Tagari PC, & Stephens PH (1986). Parenterally administered GM1 ganglioside prevents retrograde degeneration of cholinergic cells of the rat basal forebrain. Brain Research, 398(2), 393–396. doi: 10.1016/0006-8993(86)91503-9 [DOI] [PubMed] [Google Scholar]
- Sophou S, Dori I, Antonopoulos J, Parnavelas JG, & Dinopoulos A. (2006). Apoptosis in the rat basal forebrain during development and following lesions of connections. Eur J Neurosci, 24(2), 573–585. doi: 10.1111/j.1460-9568.2006.04929.x [DOI] [PubMed] [Google Scholar]
- Stone BS, Zhang J, Mack DW, Mori S, Martin LJ, & Northington FJ (2008). Delayed neural network degeneration after neonatal hypoxia-ischemia. Ann Neurol, 64(5), 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Somogyi P, & Smith AD (1984). Aspiny neurons and their local axons in the neostriatum of the rat: a correlated light and electron microscopic study of Golgi-impregnated material. J Neurocytol, 13(2), 239–265. doi: 10.1007/BF01148118 [DOI] [PubMed] [Google Scholar]
- Tymofiyeva O, Gano D, Trevino RJ, Glass HC, Flynn T, Lundy SM, . . . Xu D. (2018). Aberrant Structural Brain Connectivity in Adolescents with Attentional Problems Who Were Born Prematurely. American Journal of Neuroradiology, 39(11), 2140–2147. doi: 10.3174/ajnr.a5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kooij MA, Ohl F, Arndt SS, Kavelaars A, van Bel F, & Heijnen CJ (2010). Mild neonatal hypoxia-ischemia induces long-term motor- and cognitive impairments in mice. Brain Behav Immun, 24(5), 850–856. doi: 10.1016/j.bbi.2009.09.003 [DOI] [PubMed] [Google Scholar]
- Vogels OJM, Broere CAJ, Laak THJ, Donkelaar THJ, Nieuwenhuys R, & Schulte BPM (1990). Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiology of aging, 11(1), 3–13. doi: 10.1016/01974580(90)90056-6 [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, & Delon MR (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science, 215(4537), 1237–1239. doi: 10.1126/science.7058341 [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Struble RG, Hedreen JC, Clark AW, & Price DL (1985). Alzheimer’s disease and related dementias: selective involvement of specific neuronal systems. CRC Crit Rev Clin Neurobiol, 1(4), 319–339. [PubMed] [Google Scholar]
- Wood T, Osredkar D, Puchades M, Maes E, Falck M, Flatebo T, . . . Thoresen (2016). Treatment temperature and insult severity influence the neuroprotective effects of therapeutic hypothermia. Sci Rep, 6, 23430. doi: 10.1038/srep23430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, . . . Lee J-M (2015). Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Aβ Generation and Amyloid Plaque Pathogenesis. The Journal of Neuroscience, 35(35), 12137–12151. doi: 10.1523/jneurosci.0705-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li T, . . . Zhu C. (2016). Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy, 12(2), 410–423. doi: 10.1080/15548627.2015.1132134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Pol A. v. d., & Gyengesi E. (2012). The Mouse Nervous System. Section D: Behavioral and Emotional States, 684–718. doi: 10.1016/b978-0-12-369497-3.10028-7 [DOI] [Google Scholar]