

Abstract
Background:
Cytoplasmic inclusions of α-synuclein in brainstem neurons are characteristic of idiopathic Parkinson’ disease. Parkinson’s disease also entails α-synuclein buildup in sympathetic nerves. Among genetic forms of Parkinson’s disease the relative extents of sympathetic intra-neuronal accumulation of α-synuclein have not been reported.
Objective:
This cross-sectional observational study compared magnitudes of intra-neuronal deposition of α-synuclein in common and rare genetic forms of Parkinson’s disease.
Methods:
α-Synuclein deposition was quantified by the α-synuclein-tyrosine hydroxylase colocalization index in C2 cervical skin biopsies from 65 subjects. These included 30 subjects with pathogenic mutations in SNCA (N=3), PRKN (biallelic [N=7] and monoallelic [N=3]), LRRK2 (N=7), GBA (N=7), or PARK7/DJ1 (biallelic [N=1] and monoallelic [N=2]). 25 of the mutation carriers had Parkinson’s disease and 5 did not. Data were also analyzed from 19 idiopathic Parkinson’s disease patients and 16 control participants.
Results:
α-Synuclein deposition varied as a function of genotype (F=16.7, p<0.0001). It was above the control range in 100% of subjects with SNCA mutations, 100% with LRRK2 mutations, 95% with idiopathic Parkinson’s disease, 83% with GBA mutations, and 0% with biallelic PRKN mutations. α-Synuclein deposition in the biallelic PRKN group was significantly higher than in the control group. Additionally, patients with biallelic PRKN mutations had higher α-synuclein deposition than did their unaffected siblings.
Conclusions:
Individuals with SNCA, DJ-1, LRRK2, or GBA mutations have substantial intra-neuronal α-syn deposition in sympathetic noradrenergic nerves in skin biopsies, whereas those with biallelic PRKN mutations do not. Biallelic PRKN patients may have mildly increased α-synuclein deposition compared to controls.
Keywords: Parkinson’s disease, alpha-synuclein, skin biopsy, sympathetic, Parkin
INTRODUCTION
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, with a lifetime risk of about 7% in individuals 45 years old or older.1 An important source of PD risk is genetic. Monogenic forms of PD (mutations in a single gene) account for 5–10% of PD cases, with mutations most commonly found in the genes LRRK2, PRKN (encoding the protein Parkin), and SNCA (encoding the protein α-synuclein (α-syn)).2–5 More frequent but lower risk polygenic variants are responsible for about a quarter of the risk in idiopathic PD (iPD).6 Between these extremes, heterozygous mutations in GBA are considered high-risk alleles and increase PD risk by 2–10 fold.7 Biallelic GBA mutations cause Gaucher disease and also increase PD risk. Understanding these genetic risk factors has been a major research focus over the last 20 years and has induced new concepts about PD pathogenesis, identified novel therapeutic targets, and aided diagnosis in patients with genetic PD.
A dominant mutation in SNCA was the first identified monogenic cause of PD.2 Shortly after this discovery in 1997, aggregated α-syn was found to be a prominent component of Lewy bodies (LBs), a neuropathological hallmark of iPD.8 Additionally, common variants in SNCA have been shown to increase the risk of PD, suggesting that intra-neuronal α-syn deposition may not only be a biomarker but also a pathogenetic factor in iPD.9, 10 Several ongoing therapeutic trials target α-syn.11–13
Mutations in LRRK2 are another common genetic cause of PD. Although brainstem α-syn deposits are characteristic for iPD and observed in most SNCA cases, such deposits have been reported in only about half of LRRK2 cases and a quarter of PRKN cases.14 Even within a single LRRK2 family, members can have different pathologies, with α-syn deposits observed in some affected carriers and tau or no deposits observed in others.4 Autopsy studies have rarely directly compared multiple genotypes within a study or reported the magnitude of α-syn deposition in affected neurons.
Until recently, identifying intra-neuronal α-syn deposition required brain biopsy or post-mortem analyses; however, extra-cranial α-syn deposition occurs in sympathetic noradrenergic neurons in PD patients, both in autonomic ganglia and in post-ganglionic nerve fibers.16 In idiopathic PD, α-syn deposition also occurs in cutaneous sympathetic nerve fibers,15 introducing the possibility that biopsying skin, a procedure that involves low risk and relative ease, may provide pathophysiologically relevant biomarkers of synucleinopathy. Additionally, across genetic forms of PD, the relative extents of sympathetic intra-neuronal α-syn deposition have been unknown. Filling this knowledge gap is important, because the disease mechanisms and potential treatments may depend on whether the particular forms involve intra-neuronal α-syn deposition. Similarly, approaches that allow systematic assessments of the extent of intra-neuronal synucleinopathy in skin biopsies may provide insights into the relationships between synucleinopathy and PD genes.
Synucleinopathy in skin biopsies typically has been assessed semi-quantitatively. We introduced quantitative immunofluorescence confocal microscopic methodology that enables measurement of the burden of intra-neuronal α-syn in sympathetic noradrenergic nerve fibers.16 In this approach a colocalization index is calculated based on the signal intensities of immunoreactive α-syn and of immunoreactive tyrosine hydroxylase (TH), which is a marker of catecholaminergic neurons. We previously validated our quantitative method of colocalization indices of total α-syn with TH by analyses of post-mortem sympathetic ganglion tissues from patients with autopsy-proven Lewy body diseases and control subjects.16 We previously applied this method to detect increased α-syn-TH colocalization in a PD patient with biallelic DJ1 (PARK7) mutations, a cause of autosomal recessive early-onset PD (EOPD).17 No previous study has quantitatively compared the extents of sympathetic α-syn deposition in skin biopsies from patients with different genetic forms of PD. In particular, no data are available from LRRK2 pathogenic mutation carriers.
In this study, we measured α-syn deposition in sympathetic noradrenergic nerve fibers in skin biopsies from individuals with SNCA, PRKN, LRRK2, GBA, or DJ1 mutations and control subjects. Our approach enabled direct, quantitative comparisons of peripheral intra-neuronal α-syn deposition among these forms of genetic PD.
METHODS
Participants were recruited to the Parkinson’s Disease Clinic of the National Institute of Neurological Disorders and Stroke (NINDS) and the National Institutes of Health Clinical Center. All the subjects in this cross-sectional observational study gave written informed consent to protocols approved by the Institutional Review Board of the NINDS before undergoing any research procedures.
Subject groups
All subjects seen in the NIH PD clinic and identified to have a genetic cause of PD were invited for inclusion in the study. When available, first degree family members who were not affected were also included in the study. Additionally, a patient with Gaucher disease who did not have PD was identified from a natural history study of Gaucher disease. We also analyzed skin biopsies from patients with iPD; patients with unrelated diseases (UD) such as multiple system atrophy, autoimmune autonomic ganglionopathy, or autoimmunity-associated autonomic failure with sympathetic denervation; and healthy volunteers (HVs).
Biopsies in patients with EOPD or genetic forms of PD, as well as a subset of late-onset PD (LOPD) patients without a mutation, were performed between March, 2018 and February, 2020. For all genetic PD patients, a clinical examination was conducted, along with olfactory testing using the University of Pennsylvania Smell Identification Test (UPSIT) and cognitive testing using the Montreal Cognitive Assessment (MoCA) during the same visit as the biopsy. An asymptomatic carrier of a SNCA duplication also received formal neuropsychological testing 17 months after his biopsy, after developing new cognitive symptoms. Biopsies in the other groups were performed between March, 2011 and September, 2018. Data from participants with DJ1 mutations, HVs, and a subset of LOPD patients have been published previously.17 Autopsy findings from one patient with a GBA mutation, who had a skin biopsy while living, have also been reported.18
Genotyping
Initial genotyping in patients without a known mutation was performed using a genome-wide coverage array (NeuroX or Neuro Consortium Array, Illumina, Inc., San Diego, CA),19, 20 whole-genome sequencing, Sanger sequencing, or commercial next-generation sequencing of a panel of PD-related genes (GeneDx Pakinson Disease Panel; Gaithersburg, MD). All genotypes were confirmed with CLIA-certified testing performed by GeneDx (Gaithersburg, MD), with the exception of one patient with a GBA mutation and an unaffected Gaucher patient, both of whom were genotyped by Sanger sequencing in the laboratory of Dr. Ellen Sidransky at NHGRI (Bethesda, MD).
Skin biopsies
The location of the skin biopsies was the C2 region of the nape of the neck, where hair is relatively abundant. This ensured that the biopsies would include arrector pili muscles, which receive prominent sympathetic noradrenergic innervation.21
After cleansing the skin and intradermal and subcutaneous local anethesia using lidocaine without adrenaline, 3-mm full-thickness punches were obtained. The samples were placed in Zamboni fixative solution and kept at 4 °C for 18–20 hours, washed with Sorenson’s phosphate buffer (133 mM, pH 7.6), and placed in 20% glycerol for cryoprotection. Samples were then embedded in optimum cutting temperature compound, frozen, and sliced into 8–10 μm thick sections (Histoserv, Germantown, MD).
Alpha-smooth muscle actin (SMA)-containing and sympathetic noradrenergic fibers are arranged in parallel in arrector pili muscles. This characteristic feature enabled identification of even small pieces of arrector pili in the relatively thin sections.
Immunofluorescence microscopy
The immunofluorescence staining procedures for visualizing α-syn and TH followed our previously published methodology.16 The primary antibody to TH was rabbit anti-TH (1:1000; Pel-Freez Biologicals, Rogers, AR), to α-syn mouse IgG1 monoclonal anti-AS (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), and to SMA mouse IgG2a monoclonal anti-SMA (1:400; Santa Cruz Biotechnology).
The primary immunoreactions were visualized using the following secondary antibodies—Alexa 488-conjugated anti-mouse IgG1, Alexa 555-conjugated anti-rabbit, and Alexa 647-conjugated anti-mouse IgG2a (Thermo Scientific, Inc, Rockford, IL). Coverslips were mounted on slides with ProLong Gold antifade reagent (Thermo Scientific). The slides were examined by immunofluorescence confocal microscopy using a Zeiss LSM 880 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany). Image sizes were 1024×1024 pixels. For each tissue, images were obtained using the same microscopic settings. Each slide had positive and negative control slices.
Avoidance of biases
The slide staining, imaging, and image analyses were conducted at the NIH by personnel strictly blinded as to the diagnostic group until the data were tabulated. The clinical team was also blinded as to the imaging data until the data were tabulated.
Data analysis and statistics
α-Syn-TH colocalization indices in entire images were measured using Fiji software, with background subtraction as described previously.16 Briefly, the automated method of Jaskolski et al. was used.22 α-Syn-TH colocalization indices in the entire image were calculated in the following steps: (1) normalized mean deviation product (nMDP) values from −1.0 to +1.0 were tabulated; (2) counts corresponding to nMDP values from 0.3 to 1.0 were summed; (3) 0.1 was added, so that the sum of the counts was greater than zero; and (4) the log of the number from step (3) was calculated.
As an index of nerve density, we calculated the TH signal intensities in regions of interest in arrector pili muscles, as described previously.16
Mean α-syn-TH colocalization indices and TH intensities across subject groups were compared by factorial analyses of variance using GraphPad Prism (www.graphpad.com). Dunnett’s multiple comparisons post-hoc test was used to compare the patient groups to the control group. Across PD patients, Pearson correlation coefficients were calculated relating individual α-syn-TH colocalization indices to age at the time of skin biopsy and age at motor onset of PD.
RESULTS
A total of 65 individuals participated in this study. There were 30 participants with pathogenic PD mutations in SNCA (N=3), PRKN (biallelic (N=7); heterozygous (N=3)), LRRK2 (N=5), GBA (heterozygous (N=6), homozygous (N=1)), LRRK2/GBA digenic (N=2), or DJ1 (biallelic (N=1); heterozygous (N=2)); and 19 patients with iPD (4 EOPD, 15 LOPD). 25 of the mutation carriers had PD and 5 did not. Demographics, clinical status, group assignment, and genotype are summarized in Table 1. The colocalization index data in the 12 patients with diseases unrelated to PD did not differ from those in the 4 HVs who were not family members of any of the patients. Therefore, for statistical testing the data for the 2 comparison groups were combined in a single control (CTRL) group. The mean age of the CTRL group was 57.0±12.8 years old, and the mean age of the affected patients was 59.5±13.9 years old. The CTRL group (N=16) consisted of 10 males and 6 females, the patient group (N=44) consisted of 21 males and 23 females, and mutation carriers without PD (N=5) consisted of 3 males and 2 females.
Table 1:
Demographic, clinical, and genotypic characteristics of study participants
| GROUP | Family | Sex | PD Status | Age | AAO | Disease Duration | Genotype | Coloc. Index |
|---|---|---|---|---|---|---|---|---|
| DJ1 | DJ1 | M | A | 35 | 20 | 15 | DJ1, p.A35Cfs*12; DJ1, delE3 | 1.998 |
| GBA | F | A | 51 | 47 | 4 | GBA, p.N409S | 2.059 | |
| GBA | M | A | 68 | 42 | 26 | GBA, p.N409S | 2.361 | |
| GBA | F | A | 66 | 52 | 14 | GBA, p.N409S | 2.952 | |
| GBA | F | A | 41 | 35 | 6 | GBA, p.L483P | 1.831 | |
| GBA | F | A | 36 | 32 | 4 | GBA, p.L483P | 0.536 | |
| GBA | F | A | 83 | 61 | 22 | GBA, p.N409S | 1.578 | |
| PRKN | PRKN 1 | F | A | 50 | 35 | 15 | PRKN p.V56E; PRKN delE7 | 1.061 |
| PRKN | PRKN 1 | F | A | 39 | 32 | 7 | PRKN p.V56E; PRKN delE7 | 0.830 |
| PRKN | M | A | 67 | 19 | 48 | PRKN, p.R275W; PRKN, delE3–4 | 1.083 | |
| PRKN | M | A | 72 | 37 | 35 | PRKN, p.R275W; PRKN, delE3–4 | 1.296 | |
| PRKN | PRKN 2 | F | A | 47 | 27 | 20 | PRKN, c.7+5G>T; PRKN, delE3–4 | 1.262 |
| PRKN | M | A | 58 | 37 | 21 | PRKN, E3; PRKN, E3–4 | 1.140 | |
| PRKN | F | A | 67 | 30 | 37 | PRKN, p.Asn52fs; PRKN , p.Asn52fs | 1.117 | |
| PRKN Het | M | A | 55 | 37 | 18 | PRKN, delE2 | 1.207 | |
| PRKN Het | M | A | 45 | 35 | 10 | PRKN, p.Gln34Argfs | 1.545 | |
| LRRK2 | F | A | 45 | 45 | 0 | LRRK2, p.G2019S; LRRK2, p.R1441G | 2.848 | |
| LRRK2 | M | A | 48 | 47 | 1 | LRRK2, p.G2019S | 2.471 | |
| LRRK2 | M | A | 66 | 46 | 20 | LRRK2, p.G2019S | 2.105 | |
| LRRK2 | M | A | 70 | 55 | 15 | LRRK2, p.G2019S | 2.708 | |
| LRRK2 | LRRK2 | M | A | 76 | 61 | 15 | LRRK2, p.G2019S | 1.994 |
| LRRK2, GBA | F | A | 68 | 58 | 10 | LRRK2, p.G2019S; GBA, p.N409S | 2.075 | |
| LRRK2, GBA | LRRK2 | M | A | 73 | 66 | 7 | LRRK2, p.G2019S; LRRK2, p.G2019S; GBA, p.N409S | 1.802 |
| SNCA | M | A | 42 | 37 | 5 | SNCA, p.A53T | 2.790 | |
| SNCA | F | A | 52 | 49 | 3 | SNCA, Duplication | 2.499 | |
| EOPD | M | A | 40 | 32 | 8 | N/A | 1.058 | |
| EOPD | M | A | 46 | 41 | 5 | N/A | 3.238 | |
| EOPD | F | A | 40 | 36 | 4 | N/A | 2.151 | |
| EOPD | F | A | 44 | 41 | 3 | N/A | 1.659 | |
| EOPD | F | A | 62 | 56 | 6 | N/A | 1.880 | |
| LOPD | F | A | 75 | 73 | 2 | N/A | 3.536 | |
| LOPD | F | A | 61 | Missing | N/A | 2.062 | ||
| LOPD | M | A | 78 | 61 | 17 | N/A | 1.891 | |
| LOPD | M | A | 76 | 68 | 8 | N/A | 2.361 | |
| LOPD | F | A | 69 | 64 | 5 | N/A | 2.355 | |
| LOPD | M | A | 67 | 56 | 11 | N/A | 2.457 | |
| LOPD | M | A | 77 | 67 | 10 | N/A | 2.208 | |
| LOPD | F | A | 62 | 58 | 4 | N/A | 2.576 | |
| LOPD | M | A | 74 | 70 | 4 | N/A | 2.146 | |
| LOPD | F | A | 75 | 69 | 6 | N/A | 3.071 | |
| LOPD | F | A | 66 | 57 | 9 | N/A | 3.240 | |
| LOPD | F | A | 72 | 67 | 5 | N/A | 1.726 | |
| LOPD | F | A | 42 | 42 | 0 | N/A | 2.941 | |
| LOPD | M | A | 71 | Missing | N/A | 2.172 | ||
| Unaffcted SNCA* | M | U | 68 | N/A | SNCA, Duplication | 2.175 | ||
| GBA homo | M | U | 72 | N/A | GBA, p.N409S; GBA, p.N409S |
1.004 | ||
| Unaffected Parkin Het | PRKN 2 | F | U | 43 | N/A | PRKN, delE3–4 | 0.663 | |
| Unaffected DJ1 Carrier | DJ1 | F | U | 59 | N/A | DJ1, delE3 | 1.277 | |
| Unaffected DJ1 Carrier | DJ1 | M | U | 64 | N/A | DJ1, p.A35Cfs*12 | 0.184 | |
| HV | PRKN 1 | M | U | 35 | N/A | N/A | 0.259 | |
| HV | F | U | 59 | N/A | N/A | −1.000 | ||
| HV | F | U | 56 | N/A | N/A | −0.456 | ||
| HV | M | U | 65 | N/A | N/A | 1.279 | ||
| Unrelated Disease Group | F | U | 73 | N/A | N/A | 1.402 | ||
| Unrelated Disease Group | M | U | 63 | N/A | N/A | 1.305 | ||
| Unrelated Disease Group | M | U | 71 | N/A | N/A | 0.602 | ||
| Unrelated Disease Group | M | U | 72 | N/A | N/A | 0.477 | ||
| Unrelated Disease Group | F | U | 62 | N/A | N/A | −1.000 | ||
| Unrelated Disease Group | F | U | 31 | N/A | N/A | −1.000 | ||
| Unrelated Disease Group | M | U | 43 | N/A | N/A | 1.052 | ||
| Unrelated Disease Group | M | U | 49 | N/A | N/A | 0.827 | ||
| Unrelated Disease Group | M | U | 64 | N/A | N/A | −1.000 | ||
| Unrelated Disease Group | M | U | 63 | N/A | N/A | −1.000 | ||
| Unrelated Disease Group | M | U | 62 | N/A | N/A | −0.648 | ||
| Unrelated Disease Group | F | U | 46 | N/A | N/A | 1.571 |
Abbreviations: A=Affected PD; AAO=Age at onset; Coloc. Index = Colocalization Index; EOPD=early onset PD; F=female; Het= heterozygous; Homo=homozygous; HV=healthy volunteer; LOPD=late onset PD; M=Male; N/A=not applicable; PD=Parkinson disease; U=unaffected PD
Participant did not have symptoms at the time of the biopsy but subsequently developed mild cognitive impairment, which could reflect incipient dementia with Lewy bodies.
α-Synuclein (α-syn) deposition in genetic forms of PD
Figure 1A shows individual values for α-syn-TH colocalization indices in genetic PD patients grouped by genotype and in the iPD and CTRL groups. Only mutation carriers with PD were included Figure 1A. Each group had data for at least 2 subjects.
Figure 1: Individual values for α-synuclein (α-syn)-tyrosine hydroxylase (TH) colocalization indices and representative confocal microscopic images.
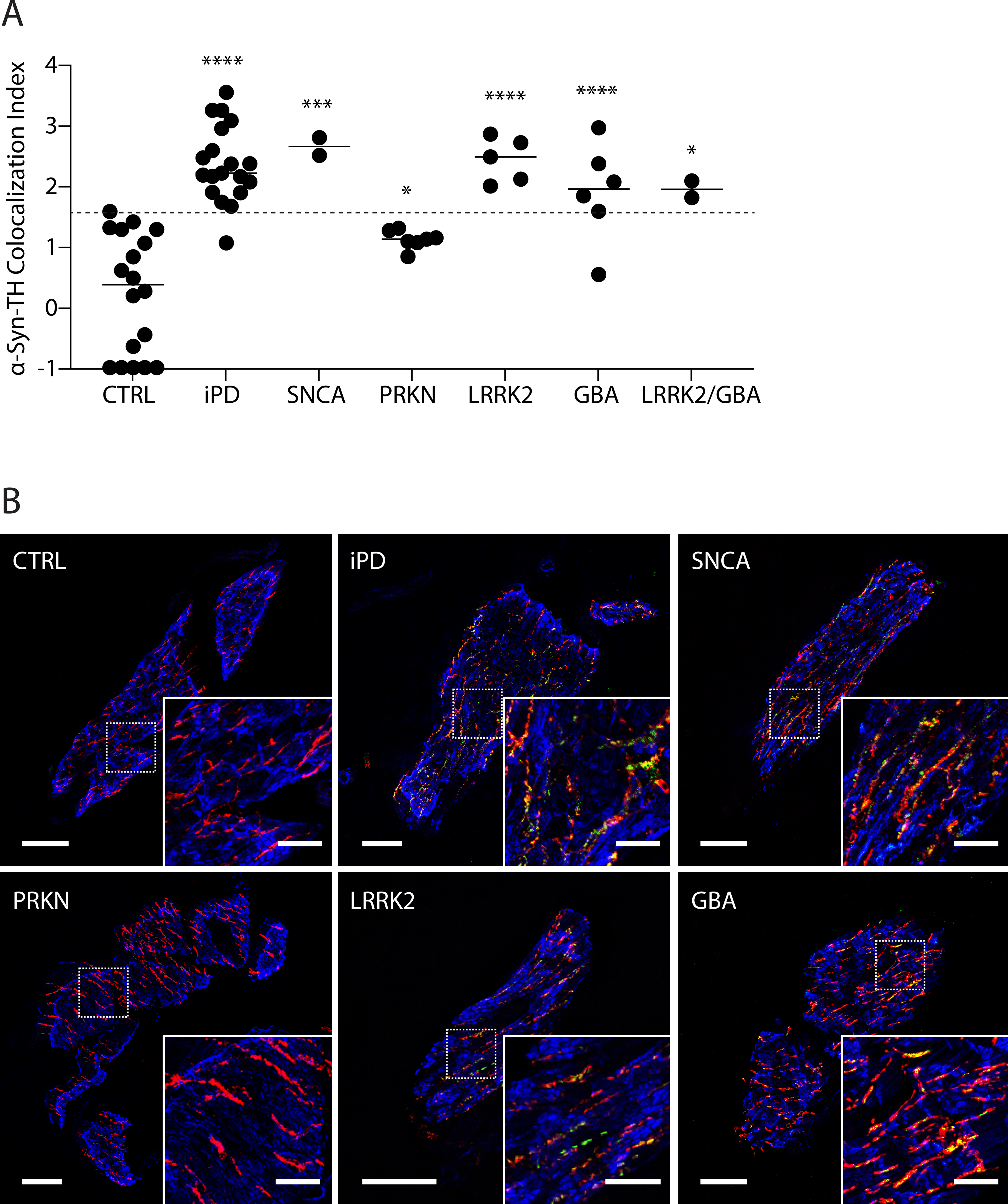
(A) individual data for control subjects (CTRL) and patients with idiopathic Parkinson disease (iPD) or mutations in SNCA, PRKN, LRRK2, GBA, or digenic LRRK2/GBA. Dashed line shows the upper limit of CTRLs. Horizontal lines indicate median values in each group. Abbreviations: ns=not significant. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, and (****) p < 0.0001 vs. CTRL. (B) Representative confocal microscopic images of immunoreactive α-syn (green), tyrosine hydroxylase (TH, red), smooth muscle actin (blue), and α-syn colocalized with TH (yellow) in arrector pili muscles from skin biopsies. Scale bars = 50 μm for main image, 20 μm inserts. Note that the iPD, SNCA, LRRK2, and GBA groups have elevated colocalization indices compared to CTRL.
Values for α-syn-TH colocalization indices varied highly significantly as a function of subject group (F=16.7, p<0.0001). Mean α-syn-TH colocalization indices were higher in the iPD (p<0.0001), SNCA (p=0.0002), biallelic PRKN (p=0.045), LRRK2 (p<0.0001), GBA (p<0.0001), and LRRK2/GBA digenic (p=0.014) PD groups than in the CTRL group. All the colocalization indices in the biallelic PRKN patients were between the mean values in the CTRL and iPD groups. Mean TH signal intensities in the arrector pili muscles in the iPD (13.3±1.6), SNCA (11.1±1.1), biallelic PRKN (16.4±2.7), LRRK2 (10.1±2.7), GBA (13.3±1.0) and LRRK2/GBA digenic (7.9±12.5) groups did not differ from those in the CTRL group (9.4±1.5).
Based on the present data about colocalization indices in iPD and control groups, the sensitivity for identifying iPD is 95% and the specificity for excluding iPD is 100%, with a colocalization index cutoff of 1.58. Frequencies of colocalization indices above the upper limit of CTRL (horizontal dashed line in Figure 1A) were 100% for SNCA, 100% for LRRK2, 100% for LRRK2/GBA digenic, 95% for iPD, 83% for GBA, and 0% for biallelic PRKN patients with PD.
Within the biallelic PRKN PD group, colocalization indices were similar across specific genotypes (Table 1). Two biallelic PRKN PD patients carried a p.R275W point mutation, with colocalization indices of 1.083 and 1.296, within the CTRL range. There were 2 PD patients who were heterozygous for PRKN mutations, delE2 and p.Gln34Argfs, with colocalization indices of 1.207 and 1.545, the latter at about the upper limit of the CTRL range (1.571) (Table 1).
Two PD patients in the study were LRRK2/GBA digenic. One had a p.G2019S mutation in the LRRK2 gene and a p.N409S (N370S) mutation in the GBA gene. The other was homozygous for the p.G2019S mutations in the LRRK2 gene and carried a p.N409S mutation in the GBA gene. The colocalization indices for these LRRK2/GBA digenic PD patients were similar to those in PD patients with a single LRRK2 p.G2019S mutation (Figure 1A).
Figure 1B shows representative images demonstrating minimal α-syn-TH colocalization (yellow) in a CTRL subject and in a PRKN biallelic PD patient, in contrast with substantial colocalization in iPD and in PD patients carrying SNCA, LRRK2, or GBA mutations.
α-Syn deposition among affected and unaffected family members
Three patients with biallelic PRKN mutations and the DJ1 patient had unaffected first-degree relatives who were biopsied at the same time as the probands (Figure 2 top). Two of the PRKN patients were sisters and had an unaffected brother who did not carry PRKN mutations (PRKN Family 1 in Figure 2; black). Another PRKN patient had an unaffected sister who carried one PRKN mutation (PRKN Family 2 in Figure 2; red). Each of the unaffected parents of the DJ1 patient carried a single DJ1 mutation (DJ1 Family 1 in Figure 2; blue).
Figure 2: Pedigrees and individual values for α-synuclein (α-syn)-tyrosine hydroxylase (TH) colocalization indices in 3 families with mutations of PRKN or DJ1.
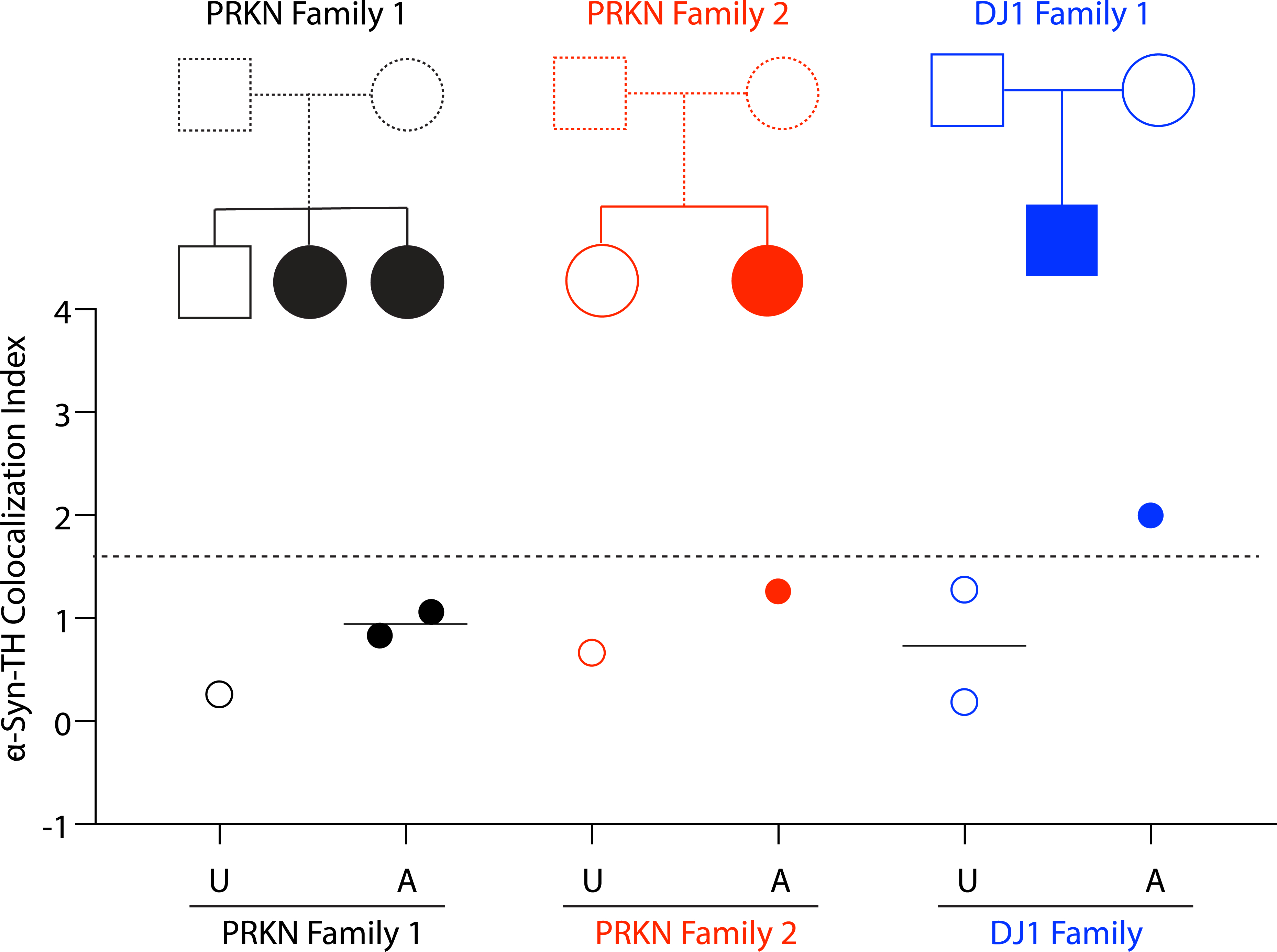
The unaffected member of PRKN family 1 (black open circle) has no mutation, and both affected members have PRKN biallelic mutations (PRKN p.V56E; PRKN delE7). The unaffected member of PRKN family 2 (red open circle) is a PRKN heterozygote (PRKN, delE3–4), and the affected member in this family is biallelic. In the DJ1 family (blue) both of the unaffected family members (black open circles) have single DJ1 mutations, and the affected member (black closed circles) has biallelic DJ1 mutations (DJ1, p.A35Cfs*12; DJ1, delE3). Dashed line shows the upper limit of CTRL. Horizontal lines indicate median values in each group. Abbreviations: A=affected, CTRL=control group, iPD=idiopathic Parkinson’s disease, U=unaffected.
In the two PRKN families and in the DJ1 family, α-syn-TH colocalization indices were higher in the affected individuals than in their unaffected first-degree relatives (Figure 2 bottom).
α-Syn-TH colocalization indices as a function of age or disease duration
Across all patients individual values for α-syn-TH colocalization indices were unrelated to subject age at the time of skin biopsy (Figure 3A). α-Syn-TH colocalization indices were positively correlated with age at motor onset (Figure 3B; r=0.52, p=0.0009); the relationship was no longer significant when the data from PD patients with biallelic PRKN mutations were excluded (r=0.33, p=0.068).
Figure 3: Individual values for α-synuclein (α-syn)-tyrosine hydroxylase (TH) colocalization indices as functions of age at the time of biopsy and age at the time of symptom onset.
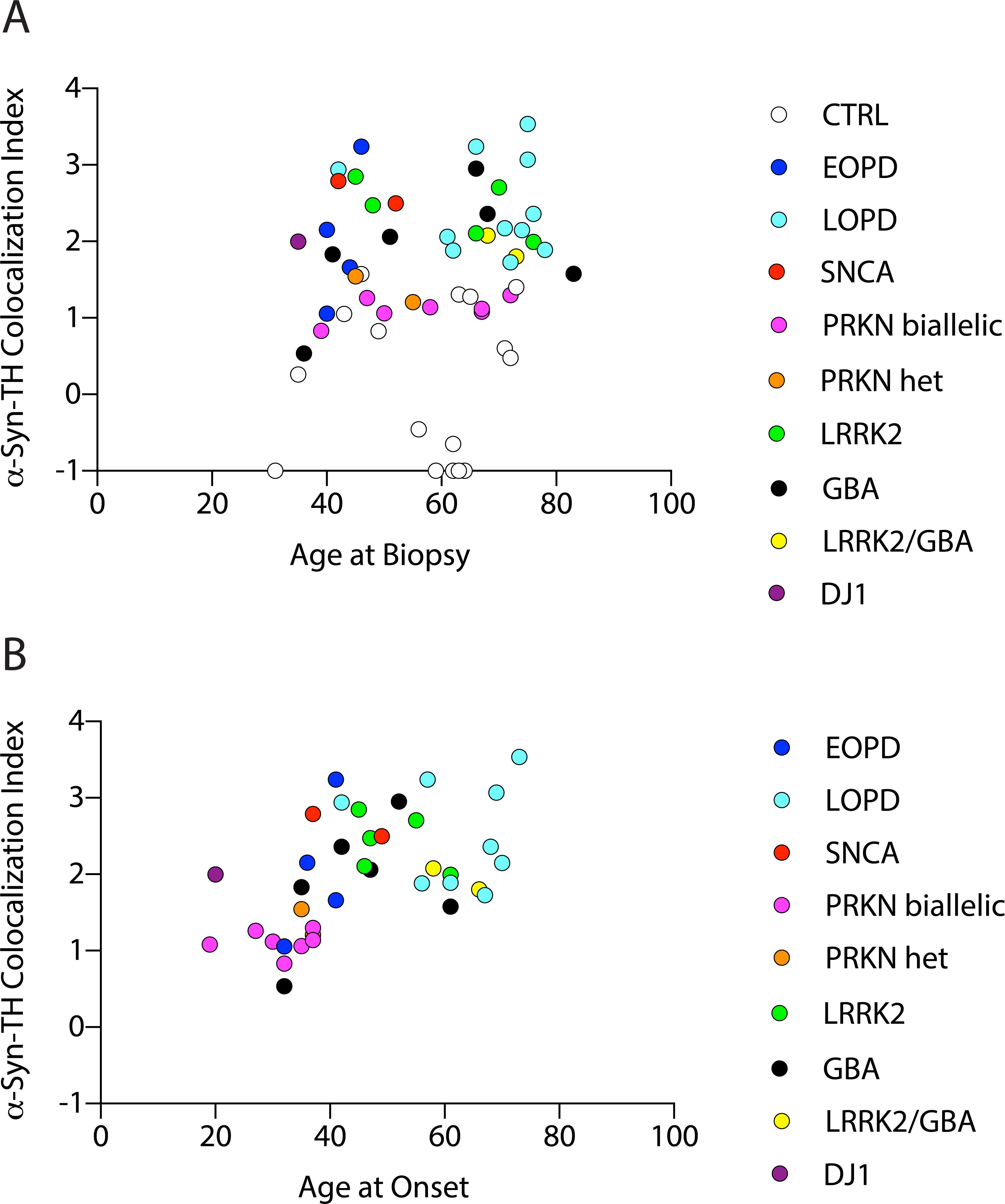
Subject groups are controls (CTRL), early-onset PD (EOPD), late-onset PD (LOPD), SNCA mutation, PRKN biallelic mutation (PRKN), PRKN heterozygous mutation (PRKN Het), LRRK2 mutation, GBA heterozygous mutation, LRRK2/GBA digenic (LRRK2/GBA), and biallelic DJ1 mutations. Across patients α-syn-TH colocalization indices are unrelated to age at biopsy (A) but positively related to age at motor onset of PD (B).
Among the 5 affected GBA mutation carriers with colocalization indices above the CTRL range, the mean disease duration was 14 years (range 4–26, SD=9.6), while in the single affected GBA mutation carrier with a colocalization index within the CTRL range, the disease duration was 4 years.
α-Syn deposition in unaffected carriers of pathogenic mutations
Our study included one SNCA mutation carrier without parkinsonism and one homozygous GBA mutation carrier without parkinsonism but with longstanding Gaucher disease (these individuals are referred to as unaffected (U) in Figure 4). The unaffected SNCA duplication carrier was initially identified as an obligate carrier in a family with autosomal dominantly transmitted PD and was confirmed to carry a SNCA duplication on genetic testing (spanning ~6 Mb) (Figure 4A and B). At the time of the biopsy he did not have parkinsonism or non-motor symptoms. Seventeen months later he complained of a new onset of memory difficulties. An isolated memory deficit was identified on formal neuropsychological testing, indicating mild cognitive impairment, and he scored 26/30 on the Montreal Cognitive Assessment. He did not have parkinsonism at that time either. He had moderate microsmia (27/40 on the University of Pennsylvania Smell Identification Test, SNCA Dup 1 in Figure 4C). The 3 affected SNCA patients in this study (including his son) had severe microsmia or anosmia.
Figure 4: α-Synuclein (α-syn)-tyrosine hydroxylase (TH) colocalization indices in affected or unaffected carriers of SNCA or GBA mutations.
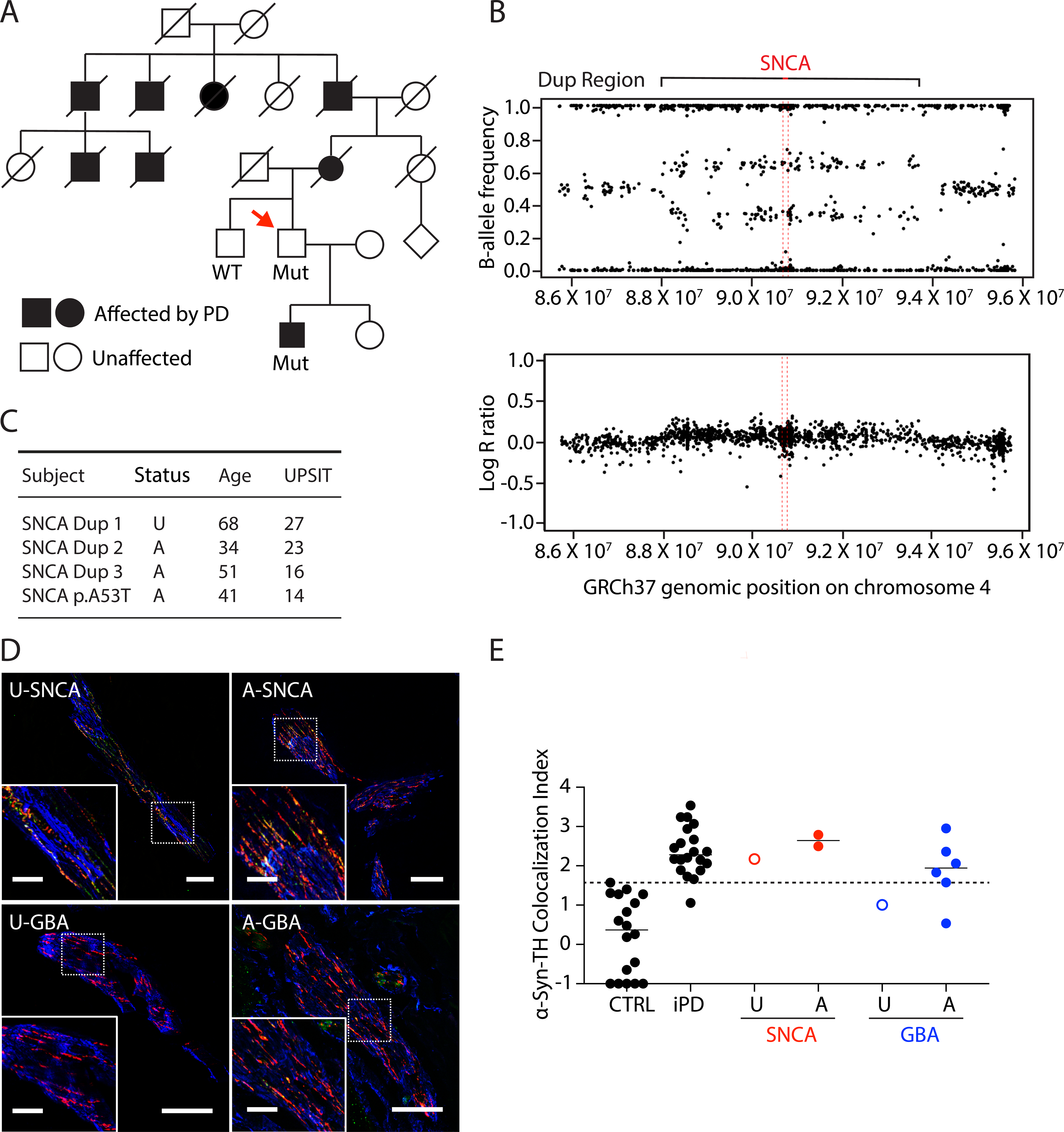
(A) Pedigree of an unaffected SNCA duplication carrier. Red arrow shows the patient. Filled in symbol indicates affected individual. (B) Genome sequencing results of the unaffected SNCA duplication carrier. (C) Clinical characteristics of the unaffected SNCA duplication carrier and 3 affected SNCA duplication carrier. SNCA Dup 2 is the first-degree relative of the unaffected SNCA duplcation carrier, in the filled square in (A). These patients had available University of Pennsylvania Smell Identification Test (UPSIT) data. (D) Confocal microscopic images of immunoreactive α-syn (green), tyrosine hydroxylase (TH, red), smooth muscle actin (blue), and α-syn colocalized with TH (yellow) in arrector pili muscles from skin biopsies. The images are from the patients who are unaffected (U) or affected (A) carriers of SNCA or GBA mutations. Scale bars = 50 μm for main image, 20 μm inserts. (E) Individual data for control subjects (CTRL), patients with idiopathic Parkinson’s Disease (iPD), and unaffected (U) or affected (A) with SNCA or GBA genotypic abnormalities. Dashed line shows the upper limit of the CTRL. Horizontal lines indicate median values in each group. Note that SNCA abnormalities are associated with increased α-syn-TH colocalization indices even if the individual is unaffected.
Of the 4 participants with SNCA mutations, 3 had skin biopsies (2 affected, 1 unaffected) and each had elevated α-syn-TH colocalization indices (Figure 4D, 4E). The patient with Gaucher disease did not have signs of parkinsonism at the time of the biopsy, and the α-syn-TH colocalization index was within the CTRL range (Figure 4D, 4E).
DISCUSSION
In this cross-sectional observational study we surveyed genetic forms of PD to ascertain the extent of α-syn deposition in sympathetic noradrenergic nerves in skin biopsies. Increased values for α-syn-TH colocalization indices were found in most individuals with mutations of SNCA, LRRK2, GBA, LRRK2/GBA, or DJ1, in contrast to patients with biallelic PRKN mutations, in whom the colocalization indices all were within the control range.
A major strength of the study was the application of quantitative methodology for measuring intra-neuronal α-syn. We used a validated assay based on colocalization of α-syn with TH.16 Further validation came from the data in this study from carriers of SNCA mutations, all of whom had increased α-syn-TH colocalization indices. To date our quantitative methodology has been applied in Lewy body forms of neurogenic orthostatic hypotension. No previous study has reported on this methodology as applied to groups with genetic forms of PD. Another major strength of the study is direct comparison α-syn deposition in the four most common forms of genetic PD—LRRK2, GBA, SNCA, and PRKN. Prior reports have compared at most two forms. In particular, we provide the first assessment of skin biopsies for α-syn deposition from patients with pathogenic LRRK2 mutations, all of whom had elevated α-syn-TH colocalization indices.
Yang et al. recently reported that patients carrying the LRRK2 risk variant G2385R have phosphorylated α-syn deposition in the skin similar to patients without the variant.23 This variant increases the risk of PD by about 2-fold and is present in about 2% of the East Asian population, according to the Genome Aggregation Database (gnomAD) v.2.1.1.24–26 In contrast, we report here on two rare mutations, p.G2019S and p.R1441G, which cause monogenic forms of PD. Most of the disease risk is conferred by the mutations, establishing a direct link between the phenotype and the mutation. Our findings provide evidence that pathogenic LRRK2 mutations drive intra-neuronal α-syn deposition in sympathetic nerve fibers.
In our study, we detected peripheral synucleinopahty in an unaffected SNCA duplication carrier, who subsequently developed mild cognitive impairment at follow-up, which could indicate incipient dementia with Lewy bodies (DLB). Analogously, Carmona-Abellan et al.27 detected peripheral synucleinopathy in an unaffected SNCA p.E46K mutation carrier, suggesting that peripheral synucleinopathy may be present in SNCA mutation carriers before the onset of parkinsonism and other non-motor symptoms; however, no follow-up studies have been performed to test whether SNCA mutation carriers with peripheral synucleinopathy subsequently develop PD or DLB.
Based on the post-mortem literature (Supplemental Tables 1 and 3) only about 50% of LRRK2 patients have brainstem synucleinopathy. Resolution of the possible discrepancy between peripheral and central synucleinopathy in LRRK2 may come from a larger cohort of LRRK2 patients in whom α-syn-TH colocalization indexes are calculated. Alternatively, some LRRK2 patients with peripheral synucleinopathy may not have central synucleinopathy. Autopsy studies of LRRK2 patients finding synucleinopathy in the skin and not in the brainstem would provide additional support for this interpretation.
We observed increased α-syn-TH colocalization indices in most but not all affected GBA carriers. Similarly, Doppler et al. reported phosphorylated α-syn deposition in 6 of 10 affected GBA carriers.28 In their study, the affected GBA carriers without increased peripheral α-syn deposition had a shorter disease duration than did those with increased peripheral α-syn deposition. In the present study, the sole affected GBA mutation carrier with a colocalization index within the control range had a relatively short disease duration. Similarly, the 72-year old Gaucher disease patient with homozygous GBA p.N409S mutations had neither parkinsonism nor increased α-syn-TH colocalization in his skin biopsy. These findings are consistent with the view that in GBA patients peripheral synucleinopathy might be delayed relative to the onset of parkinsonism. A longitudinal study would be required to test this idea formally.
All the PRKN cases in our study had α-syn-TH colocalization indices within the control range. This finding is consistent with three prior studies that found α-syn was not increased in skin nerve fibers in PRKN cases.27, 29, 30 A recent study of submandibular gland tissue also found no evidence of α-syn buildup in carriers of PRKN mutations.31 Similar to these reports based on biopsy tissues, sympathetic neuroimaging studies have shown that most PRKN cases do not have evidence for a cardiac noradrenergic lesion.32–36 In particular, Orimo et al. reported normal myocardial 123I-metaiodobenzylguanidine- (123I-MIBG)-derived radioactivity in 3 PRKN patients, 2 of whom came to autopsy and had normal immunoreactive TH in epicardial nerves.33
As with the LRRK2 carriers, PRKN carriers had uniformity in their peripheral α-syn findings. This was unexpected given the heterogeneity in brainstem synucleinopathy. From the available literature (Supplemental Tables 1–3), 28% of published post-mortem PRKN cases have brainstem synucleinopathy. The presence of the p.R275W mutation, which has been suggested to be a determinant of brainstem synucleinopathy,37, 38 was not associated with the extent of peripheral synucleinopathy in our study.
While we found that α-syn-TH colocalization indices were lower in PRKN patients than in the other genetic subtypes, the mean colocalization index was significantly higher than that in the CTRL group; and within PRKN families three affected participants had higher α-syn-TH colocalization indices than their unaffected siblings. These findings raise the possibility that some patients with PRKN mutations might have a small amount of intra-neuronal α-syn deposition. As our methodology detects total α-syn, it is not clear if this elevation reflects a low level of the misfolded, pathological form of α-syn or an elevation of correctly folded α-syn in the PRKN patients. Studies using methods such as protein misfolding cyclic amplification (PMCA)39 and real-time quaking-induced conversion (RT-QuIC),40 might help clarify if low but detectable levels of α-syn are present in peripheral nerve fibers of PRKN patients.
Study Limitations
A potential limitation of the present study is that data about α-syn deposition in sympathetic nerves in vivo skin biopsies might not directly correlate with α-syn deposition in the nigrostriatal system. Simultaneously measuring α-syn seeds in skin and CSF by PMCA in the same subjects may help clarify the extent to which peripheral and central synucleinopathy correlate. This may be particularly useful in genetic forms like PRKN and LRRK2, where studies of peripheral synucleinopathy suggest uniformity but autopsy studies of central synucleinopathy suggest variability.
The main advantages of the colocalization index methodology are that it is quantitative, it is validated, and it gives an estimate of the intra-neuronal burden of α-syn. As the conformation of α-syn changes dynamically in PD pathogenesis, it is possible that antibodies against total, phosphorylated, or oligomeric α-syn might yield different results in preclinical vs. established PD or monogenic vs. iPD. Another potential limitation is that low levels of α-syn are normally present in skin nerve fibers. An expanded study employing a panel of antibodies for different α-syn species may give insight into the evolution of α-syn pathology in peripheral nerve fibers.
The CTRL group and patient groups had similar mean ages but were not sex-matched. As the sample sizes were small, only limited analyses were done. Consequently, age and sex were not explicitly addressed as potential correlates of α-syn-TH colocalization indexes. The number of healthy controls in this study was small. Greater normative data from healthy controls are needed in future studies employing this assay, particularly if it is to be used as a surrogate biomarker in clinical trials. While measures were taken to reduce bias, such as blinding the investigator performing immunostaining and analysis of sections, other participants such as those performing clinical evaluations and performing the biopsies were not blinded. This is a potential source of bias. Finally, while we were interested to explore how other genes like PINK1 and VPS35 compare to the groups included in this report, there were no PINK1 or VPS35 mutation carriers in our study.
Conclusions and Perspective
In conclusion, we report that, based on calculation of α-syn-TH colocalization indices in skin biopsies from living patients with genetic forms of PD, most individuals with mutations in SNCA, DJ1, LRRK2, GBA, or LRRK2/GBA have substantial α-syn deposition in sympathetic noradrenergic nerves, whereas most individuals with PRKN mutations do not. PRKN patients may have mildly increased α-Syn deposition compared to controls. Targeting intra-neuronal α-syn deposition would be rational in individual patients with demonstrable α-syn deposition in skin catecholaminergic nerve fibers; and tracking α-syn-TH colocalization indices might provide a surrogate biomarker in experimental therapeutic trials.
Supplementary Material
Supplemental Table 1: Comparison of peripheral synucleinopathy in skin and in brainstem Lewy bodies in genetic PD from this study and published literature.
Supplemental Table 3: Results of literature review of original studies describing α-syn deposition in brainstem from autopsy studies. We consulted the systematic 2017 review by Schneider and Alcalay14 and the internally referenced articles to identify studies describing the presence of α-syn or typical Lewy pathology in the brainstem in carriers of pathogenic mutations in SNCA, PRKN, DJ1, LRRK2, or GBA. The 2017 review was updated by searching PubMed for articles that were published since 2016, using the search terms “Parkinson and (lrrk2 or gba or PRKN or snca or synuclein or park1 or park2 or park4 or park7 or dj1 or park8) and autopsy” (date of literature review: 9/23/2020).
Supplemental Table 2: Results of literature review of original studies describing α-syn deposition in skin nerve fibers. To identify published articles about α-syn in dermal nerve fibers, PubMed was searched for articles using the search terms “skin and (synuclein or snca)” (date of literature review: 9/13/2020). The abstracts were reviewed, and 32 studies were identified that described assessment of α-syn in cutaneous nerve fibers.
ACKNOWLEDGEMENT
This work was supported by the Intramural Research Program of the NINDS, National Institutes of Health. This work was supported in part by the Intramural Research Programs of the National Institute on Aging (NIA). This work was supported in part by the Intramural Research Programs of the National Human Genome Research Institute (NHGRI).
Financial support: The research reported here was supported by the Division of Intramural Research, NINDS, NIH
Financial Disclosures of all authors
Dr. Gonzalez-Alegre has received consulting fees from Spark Therapeutics, Eisai Therapeutics and NeuExcell Therapeutics, licensing fees from Spark Therapeutics via the University of Iowa, research funding from the NIH/NINDS, is the Principal Investigator of the clinical trial NeuroSEQ (sponsored by Illumina) and is a site-investigator for clinical studies or trials sponsored by CHDI and BioHaven. Dr. Ehrlich receives grants for research in dystonia from Medtronic, Inc. The other authors declare no competing interests.
Footnotes
Conflicts of interest: The authors do not have a conflict of interest to report.
REFERENCES
- 1.Wanneveich M, Moisan F, Jacqmin-Gadda H, Elbaz A, Joly P. Projections of prevalence, lifetime risk, and life expectancy of Parkinson’s disease (2010–2030) in France. Mov Disord 2018;33(9):1449–1455. [DOI] [PubMed] [Google Scholar]
- 2.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392(6676):605–608. [DOI] [PubMed] [Google Scholar]
- 4.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44(4):601–607. [DOI] [PubMed] [Google Scholar]
- 5.Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44(4):595–600. [DOI] [PubMed] [Google Scholar]
- 6.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18(12):1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- 9.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009;41(12):1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 2009;41:1303–1307. [DOI] [PubMed] [Google Scholar]
- 11.Volc D, Poewe W, Kutzelnigg A, et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol 2020;19(7):591–600. [DOI] [PubMed] [Google Scholar]
- 12.Brys M, Fanning L, Hung S, et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord 2019;34(8):1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jankovic J, Goodman I, Safirstein B, et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol 2018;75(10):1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 2017;32(11):1504–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsukita K, Sakamaki-Tsukita H, Tanaka K, Suenaga T, Takahashi R. Value of in vivo alpha-synuclein deposits in Parkinson’s disease: A systematic review and meta-analysis. Mov Disord 2019;34(10):1452–1463. [DOI] [PubMed] [Google Scholar]
- 16.Isonaka R, Rosenberg AZ, Sullivan P, et al. Alpha-Synuclein deposition within sympathetic noradrenergic neurons Is associated with myocardial noradrenergic deficiency in neurogenic orthostatic hypotension. Hypertension 2019;73(4):910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narendra DP, Isonaka R, Nguyen D, et al. Peripheral synucleinopathy in a DJ1 patient with Parkinson disease, cataracts, and hearing loss. Neurology 2019;92(23):1113–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isonaka R, Sullivan P, Jinsmaa Y, Corrales A, Goldstein DS. Spectrum of abnormalities of sympathetic tyrosine hydroxylase and alpha-synuclein in chronic autonomic failure. Clin Auton Res 2018;28(2):223–230. [DOI] [PubMed] [Google Scholar]
- 19.Nalls MA, Bras J, Hernandez DG, et al. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 2015;36(3):1605 e1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blauwendraat C, Faghri F, Pihlstrom L, et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol Aging 2017;57:247 e249–247 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donadio V, Incensi A, Vacchiano V, Infante R, Magnani M, Liguori R. The autonomic innervation of hairy skin in humans: an in vivo confocal study. Sci Rep 2019;9(1):16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaskolski F, Mulle C, Manzoni OJ. An automated method to quantify and visualize colocalized fluorescent signals. J Neurosci Methods 2005;146(1):42–49. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Wang H, Yuan Y, et al. Peripheral synucleinopathy in Parkinson disease with LRRK2 G2385R variants. Ann Clin Transl Neurol 2021;8(3):592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mata IF, Kachergus JM, Taylor JP, et al. Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 2005;6(4):171–177. [DOI] [PubMed] [Google Scholar]
- 25.Di Fonzo A, Wu-Chou YH, Lu CS, et al. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics 2006;7(3):133–138. [DOI] [PubMed] [Google Scholar]
- 26.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carmona-Abellan M, Gabilondo I, Murueta-Goyena A, et al. Small fiber neuropathy and phosphorylated alpha-synuclein in the skin of E46K-SNCA mutation carriers. Parkinsonism Relat Disord 2019;65:139–145. [DOI] [PubMed] [Google Scholar]
- 28.Doppler K, Brockmann K, Sedghi A, et al. Dermal phospho-alpha-synuclein deposition in patients with Parkinson’s disease and mutation of the glucocerebrosidase gene. Front Neurol 2018;9:1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fadda L, Lombardi R, Soliveri P, Lauria G, Giovanni D, Tagliavini F. Skin nerve alpha-synuclein deposits in a parkinsonian patient with heterozygous parkin mutation. Parkinsonism Relat Disord 2019;60:182–183. [DOI] [PubMed] [Google Scholar]
- 30.Donadio V, Incensi A, Leta V, et al. Skin nerve alpha-synuclein deposits: a biomarker for idiopathic Parkinson disease. Neurology 2014;82(15):1362–1369. [DOI] [PubMed] [Google Scholar]
- 31.Shin JH, Park SH, Shin C, et al. Negative alpha-synuclein pathology in the submandibular gland of patients carrying PRKN pathogenic variants. Parkinsonism Relat, Disord 2020;81:179–182. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki M, Hattori N, Orimo S, et al. Preserved myocardial [123I]metaiodobenzylguanidine uptake in autosomal recessive juvenile parkinsonism: first case report. Mov Disord 2005;20(5):634–636. [DOI] [PubMed] [Google Scholar]
- 33.Orimo S, Amino T, Yokochi M, et al. Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord 2005;20:1350–1353. [DOI] [PubMed] [Google Scholar]
- 34.Quattrone A, Bagnato A, Annesi G, et al. Myocardial 123metaiodobenzylguanidine uptake in genetic Parkinson’s disease. Mov Disord 2008;23(1):21–27. [DOI] [PubMed] [Google Scholar]
- 35.Kim YD, Song IU, Kim JS, Chung SW, Lee KS. Cardiac (123)I-metaiodobenzylguanidine Scintigraphy in a Patient with Familial Parkinsonism with Parkin Gene Mutation. J Mov Disord 2010;3(2):42–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Rosa A, Pellegrino T, Pappata S, et al. Myocardial (123)I-metaiodobenzylguanidine scintigraphy in patients with homozygous and heterozygous parkin mutations. J Nucl Cardiol 2017;24(1):103–107. [DOI] [PubMed] [Google Scholar]
- 37.Farrer M, Maraganore DM, Lockhart P, et al. alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet 2001;10(17):1847–1851. [DOI] [PubMed] [Google Scholar]
- 38.Miyakawa S, Ogino M, Funabe S, et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov Disord 2013;28(3):388–391. [DOI] [PubMed] [Google Scholar]
- 39.Manne S, Kondru N, Jin H, et al. Blinded RT-QuIC Analysis of alpha-Synuclein Biomarker in Skin Tissue From Parkinson’s Disease Patients. Mov Disord 2020;35(12):2230–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Becker K, Donadio V, et al. Skin alpha-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Comparison of peripheral synucleinopathy in skin and in brainstem Lewy bodies in genetic PD from this study and published literature.
Supplemental Table 3: Results of literature review of original studies describing α-syn deposition in brainstem from autopsy studies. We consulted the systematic 2017 review by Schneider and Alcalay14 and the internally referenced articles to identify studies describing the presence of α-syn or typical Lewy pathology in the brainstem in carriers of pathogenic mutations in SNCA, PRKN, DJ1, LRRK2, or GBA. The 2017 review was updated by searching PubMed for articles that were published since 2016, using the search terms “Parkinson and (lrrk2 or gba or PRKN or snca or synuclein or park1 or park2 or park4 or park7 or dj1 or park8) and autopsy” (date of literature review: 9/23/2020).
Supplemental Table 2: Results of literature review of original studies describing α-syn deposition in skin nerve fibers. To identify published articles about α-syn in dermal nerve fibers, PubMed was searched for articles using the search terms “skin and (synuclein or snca)” (date of literature review: 9/13/2020). The abstracts were reviewed, and 32 studies were identified that described assessment of α-syn in cutaneous nerve fibers.