

Abstract
In late 2019, an outbreak of coronavirus disease 2019 (COVID‐19) arose, caused by severe acute respiratory syndrome coronavirus type 2 (SARS‐CoV‐2). This disease rapidly became a public health event of international concern. In addition to the most typical symptoms of dyspnea, numerous patients with COVID‐19 exhibited systemic symptoms, such as cardiovascular disease, liver and kidney failure, and disorders in coagulation. At present, clinical data indicates that numerous patients who are critically ill die from multiple organ dysfunction syndromes (MODS). Moreover, the entry of SARS‐CoV‐2 into cells causing severe pathology and progressive organ failure is precisely mediated by the human angiotensin‐converting enzyme 2 protein. This plays a role in maintaining both fluid and electrolyte homeostasis, ensuring the stability of the internal environment. Therefore, the present review aimed to investigate the pathogenesis of MODS caused by SARS‐CoV‐2 infection based on the current clinical data and previous studies.
Keywords: COVID‐19, multiple organ dysfunction syndrome, SARS‐CoV‐2
Highlights
Inflammatory factor storm, oxidative stress, and disseminated intravascular coagulation cause multiple organ dysfunction syndromes (MODS) in coronavirus disease 2019 patients.
Angiotensin‐converting enzyme 2 (ACE2) protein, closely related to viral infection, mediates organ damage and causes MODS.
Aging, underlying disease, and obesity downregulate ACE2 and may exacerbate MODS.
1. INTRODUCTION
The coronavirus disease 2019 (COVID‐19) caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has become a public health emergency of international concern since its outbreak in late December 2019, causing mass transmission worldwide. As the mutation of the virus increases, more and more people are at risk of infection. By November 2021, there were over 2.5 billion cumulative confirmed cases of COVID‐19 worldwide, including more than 5.16 million deaths.
All humans are susceptible to developing the disease associated with SARS‐CoV‐2, but the conditions following infection differed between patients. 1 Although there have been a considerable number of asymptomatic infections, numerous patients with COVID‐19 exhibited symptoms of fever, dry cough, fatigue, and myalgia, and severely ill patients in particular certain specific populations presented with dyspnea and hypoxemia, which may result in acute respiratory distress syndrome (ARDS). 2 In addition to severe respiratory damage, cardiovascular digestive system, urinary system, and immune system are also greatly affected, affecting the prognosis. Researchers 3 analyzed the clinical characteristics of 41 COVID‐19 patients and found that five patients had combined acute myocardial injury, three patients had combined acute kidney injury, and most of them were severe patients. In a statistical study by Chen 4 on 99 COVID‐19 patients, 11% exhibited short term deterioration and later died of multiple organ dysfunction syndromes (MODS), including heart failure, acute renal injury, and shock.
MODS is a clinical syndrome in which two or more systemic or organ dysfunctions occur simultaneously or sequentially following acute damage, such as infection, poisoning, severe trauma, and extensive burns. Previous studies have demonstrated that patients with COVID‐19 who also exhibited comorbid MODS have extremely high rates of severe illness and mortality. Therefore, futher understanding the mechanisms underlying SARS‐CoV‐2 in MODS development is critical to reduce the impact of COVID‐19 (Figure 1).
Figure 1.
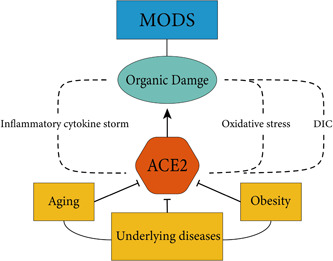
The factors contributing to multiple organ dysfunction syndromes (MODS). Factors including aging, obesity, and underlying diseases can degrade angiotensin‐converting enzyme 2 (ACE2) and exacerbate downregulation of ACE2 induced by virus. In addition, the infection of severe acute respiratory syndrome coronavirus type 2 and the downregulation of ACE2 further cause inflammatory cytokine storms, oxidative stress and disseminated intravascular coagulation (DIC). These events lead to organ damage, eventually causing MODS
2. MECHANISMS OF INFECTED WITH COVID‐19 LEADING TO MODS
2.1. Inflammatory cytokine storm causes MODS
An inflammatory cytokine storm occurs when an organism is infected by microorganisms that cause the release of a large number of inflammatory factors, which not only kills the virus, bacteria but also causes permanent damage to normal cells, resulting in damage to various organs. 5 The study 6 found that plasma cytokine levels were higher in COVID‐19 patients compared to healthy subjects, while plasma IL‐2, IL‐7, IL‐10, granulocyte colony‐stimulating factor and recombinant human interferon‐inducible protein‐10, MCP‐1, macrophage inflammatory protein‐1A, and TNF‐α levels were incrsased in critically ill patients, compared with non critically ill patients. Moreover, results of a further studiey demonstrated that IL‐6 played a unique role in the development of a hyper‐inflammatory response following SARS‐CoV‐2 infection, and this could be used as a marker for understanding the severity of the disease and predicting patient prognosis. 7 , 8 When SARS‐CoV‐2 directly infects endothelial cells that highly express angiotensin‐converting enzyme 2 (ACE2), thus causing inflammation, IL‐6 is released to increase vascular permeability and promote the release of pro‐inflammatory factors in the endothelial cells themselves, thereby enhancing the release of cytokine. 9 The release of a large number of inflammatory factors eventually evolves into an uncontrolled systemic inflammatory cytokine storm that damages various organs leading to MODS.
2.2. Oxidative stress causes MODS
Oxidative stress is a phenomenon caused by the loss of regulation of the production and accumulation of reactive oxygen species (ROS) in cells and tissues and the inhibition of the activity of endogenous antioxidant system. 10 ROS include superoxide radicals (•O2 −), hydrogen peroxide (H2O2), hydroxyl radicals (•OH), and singlet oxygen (1O2), which are closely associated with multiple physiological activities, such as protein phosphorylation, activation of several transcription factors, apoptosis, immunity, and differentiation. 11 The increase of ROS will irreversibly destroy proteins, lipids, and nucleic acids, inhibit the enzyme complex of the respiratory chain and cause mitochondrial dysfunction, leading to cell damage, and further organ dysfunction. 12 Oxidative stress may be a contributing factor in the pathogenesis of MODS due to the association of elevated plasma levels of ROS, NO, and ONOO− with MODS. 13 By investigating the clinical characteristics of 198 patients with COVID‐19, results of a previous study demonstrated that glutathione (GSH) reductase (GR) levels were significantly increased in 40.2% patients with COVID‐19. 14 GR is known to participate in an oxidative defense system required for effective immune responses against bacterial infection. From an accumulation of literature data, an endogenous deficiency in GSH may underly the serious manifestations and death resulting from COVID‐19 infection. 15 The receptor‐binding domain of the viral spike proteins and ACE2 has several cysteine residues. Molecular dynamic simulations showed that the binding affinity was significantly impaired when all the disulfide bonds of both ACE2 and SARS‐CoV/CoV‐2 spike proteins were reduced to thiol groups. 16 GSH‐peroxidase is a key antioxidant enzyme that converts peroxides and hydroxyl radicals into nontoxic forms by the oxidation of reduced GSH into GSH disulfide, which is then reduced to GSH by GR. 17 Under oxidative stress, the lack of a reducing environment would significantly favor viral protein binding to cell surface ACE2. Therefore, it is speculated that SARS‐CoV‐2 may cause ACE2‐expressing cells with GSH depletion, thus leading to oxidative stress. This may mean that the histocyte is more susceptible to SARS‐CoV‐2 infection and ultimately causing systemic multiple organ damage.
2.3. Disseminated intravascular coagulation (DIC) causes MODS
d‐Dimer (DD) levels are commonly used as an indicator of fibrinolytic system function, and elevated DD levels indicate fibrinolytic system activity. Clinical reports demonstrated that patients infected with SARS‐CoV‐2 often present with thrombocytopenia and increased DD, and patients with severe infection exhibit an increased risk of DIC. 4 DIC, a secondary syndrome of intravascular coagulation due to local damage caused by different etiologies, is a manifestation of coagulation failure and an intermediate link in the development of multiorgan failure. 18 Abnormal coagulation in patients with COVID‐19 is characterized by elevated fibrinogen and DD in parallel with increased inflammatory markers. 4 At the same time, prothrombin time and partial thrombin activation time were prolonged, as well as platelet reduction, suggesting that an excessive inflammatory response may activate the clotting pathway, leading to excessive consumption of clotting factors and platelets. In parallel with the systemic organ inflammation, the microvascular system is damaged, leading to abnormal activation of the coagulation system, which manifests itself pathologically as systemic microangiitis and extensive microthrombosis, eventually leading to MODS.
2.4. Distribution of ACE2 in the body
ACE2, the first homolog of the human angiotensin‐converting enzyme (ACE), is a key members of the renin−angiotensin−aldosterone system (RAAS). 19 The RAAS is an important component in regulating the function of numerous tissues and organs, including the cardiovascular system, kidneys, lungs, and liver, and especially by maintaining a balance of blood pressure, electrolyte balance, and inflammatory response. 20 In the classical RAAS system, renin secreted by the paranephric cells of the kidney acts on angiotensinogen in the blood and converts it into angiotensinI (AngI). AngI forms angiotensinII (AngII) and AngIII under the action of ACE. AngII can increase blood pressure by contracting the smooth muscle of arteries, promoting the release of vasopressin and oxytocin from the pituitary gland, stimulating the secretion of aldosterone by the adrenal cortex, and promoting the reabsorption of water and sodium by the renal tubules. In contrast, angiotensin (1−7) (Ang (1−7)) exhibit a number of biological effects, including vasodilation, regulation of blood pressure, inhibition of cell proliferation, diuretic, and natriuretic properties, and theses closely associated with slow peptide interactions. 20 , 21 ACE2 converts AngI to angiotensin (1−9) and further to Ang (1−7). 22 In addition, ACE2 can also directly degrades AngII to Ang (1−7). 23 Using single‐cell RNA sequencing, results of previous study demonstrated that ACE2 is predominantly expressed in type II alveolar cells in the lung, proximal tubule cells in the nephron, cardiomyocytes, epithelial cells of the ileum and esophagus, and urothelial cells of the bladder in humans. 24 ACE2 could mediate both SARS‐CoV and SARS‐CoV‐2 S‐mediated entry into cells, but the affinity of the S protein of SARS‐CoV‐2 to the ACE2 receptor is nearly 20 times that of SARS. 25 Thus, although SARS‐CoV‐2 exhibits a lower lethality than SARS, the high affinity for ACE2 may explain its greater susceptibility to human infection and transmission. The expression and distribution of ACE2 in the human body may act as a target of SARS‐CoV‐2 infection. 21
2.4.1. Lung injury mediated by ACE2
A recent study of patients with COVID‐19 by Wang's team 26 demonstrated that the pathology caused by SARS‐CoV‐2 was characterized by diffuse alveolar damage with pulmonary edema and formation of lung hyaline membranes. Fibrous mucus‐like exudates were also observed in the lung tissue. ARDS is a syndrome with bilateral pulmonary effusion as the clinical manifestation without heart failure, and the mortality rate of patients with ARDS is as high as 30%. 27 ACE2 may play a key role in the progression of ARDS. 28 It has previously been demonstrated that in a lipopolysaccharide‐induced ARDS rat model, bronchoalveolar lavage fluid increased ACE activity and AngII expression levels, while ACE2 activity and Ang (1−7) expression levels were decreased. 29 , 30 Moreover, ACE2 is abundantly expressed in alveolar type II pneumocytes and associated with endothelial cells, making these cells major targets for viral entry and replication. 31 It is thus speculated that following infection of alveolar epithelial cells by SARS‐CoV‐2, the corresponding S protein acts on the ACE2 receptor to allow the virus to enter the cells via endocytosis. This activates A disintegrin and metalloproteinase‐17 (ADAM17) to cleave the ACE2 N‐terminal, resulting in the downregulation of ACE2 expression. 32 Decreased expression of ACE2 increases vascular permeability, thus leading a cycle of the alveolar wall in a vicious cycle of ongoing destruction and repair, ultimately resulting in diffuse alveolar damage.
2.4.2. Cardiac injury mediated by ACE2
Results of previous studies have suggested that high activity of the RAAS system in the heart and the production of cardiomyocyte AngII accelerate the progression of heart failure. Yamamoto 33 demonstrated that ACE2 knockout mice were more likely to develop severe cardiac dysfunction under stress overload, while myocardial AngII levels were significantly increased. Moreover, Burrell 34 demonstrated that cardiac ACE2 expression was increased in the infarcted and peripheral ischemic zone in experimental myocardial infarction. Results by, Donoghue 35 indicated that ACE2 induced vascular diastole in the heart and maintained myocardial perfusion during acute ischemia. Furthermore, researchers 36 measured ACE2 mRNA levels in the left ventricular myocardium of patients with normal donor (9 cases), primary dilated cardiomyopathy (11 cases), and ischemic cardiomyopathy (12 cases). The results of this study indicated that ACE2 mRNA levels were found to be locally upregulated in the hearts of patients with ischemic cardiomyopathy and idiopathic dilated cardiomyopathy. Previous studies have demonstrated that upregulation of ACE2 levels is a compensatory response to heart failure, 37 , 38 , 39 as increased levels of Ang (1−7) can counteract the effects of AngII to some extent. These findings may imply that when SARS‐CoV‐2 is infected with ACE2, levels in the cardiac system may consequently decrease, and symptoms such as heart failure may occur in critically ill patients as well.
2.4.3. Liver injury mediated by ACE2
Previous research indicated that patients with COVID‐19 exhibit a 14.8%−53% probability of developing concurrent liver injury, with abnormal serum ALT/AST levels and mildly elevated bilirubin levels. 4 , 40 In severe cases, the albumin level decreased to 26.3−30.9 g/L, and the proportion of liver injury was significantly higher in patients with severe disease than in those with mild disease. 40 Liver injury caused by SARS‐CoV‐2 through multiple pathways mainly includes moderate steatosis, lobular and portal inflammation, apoptosis, and bile duct proliferation. 41 The expression of ACE2 in hepatic duct cells is much higher than that in hepatocytes, so the liver damage caused by SARS‐CoV‐2 infection in hepatic duct cells with immune and protective effects may be more serious. On the one hand, SARS‐CoV‐2 infection attenuated bile duct barrier function leading to liver injury by reducing cholangiocyte tight junction protein and mRNA expression. On the other hand, it impairs bile acid signaling and causes biliary changes by negatively regulating hepatobiliary transporters. 41 Moreover, infected hepatocytes also upregulated the expression of inflammatory pathways. 42 Other factors such as hypoxia and inflammatory factor storms, also cause liver injury by affecting the expression of ACE2. 41
2.4.4. Kidney injury mediated by ACE2
In the kidney, ACE2 is predominantly found in the brush‐like proximal margin of the tubules, the endothelium of renal vessels, and smooth muscle cells, and in parietal epithelial cells. 43 , 44 , 45 Results of previous studies highlighted the role of renal ACE in renal damage. Notably, Tikellis 46 demonstrated a two fold increase in circulating ACE2 activity in diabetic mice, which may be a potential cause of impaired reactivity to AngII. Oudit 47 demonstrated that ACE2 deficiency is associated with the formation of lipid peroxidation products in the kidney, and the increased activation of mitogen‐activated protein kinase and extracellular signal‐regulated kinases 1 and 2 in the glomeruli. It has previously been reported that SARS‐CoV‐2 can be directly mediated through ACE2, causing acute renal failure, tubular necrosis, Bowman's capsule protein leakage, glomerular collapse, and mitochondrial damage. 48 A cause of acute kidney injury in patients with COVID‐19 may be that SARS‐CoV‐2 reduced ACE2 expression after entering the cells to inhibit its anti‐inflammatory protective effect. The disrupted balance between intrarenal ACE and ACE2 may elevate the levels of Ang2 concentrations in the injured tubules, thus leading to further kidney damage.
3. SARS‐CoV‐2 INFECTION IN THE ELDERLY CAUSES MODS
Previous studies have demobnstrated that the prevalance of COVID‐19 is increased in patients with underlying diseases such as chronic obstructive pulmonary disease, coronary heart disease, liver and kidney failure, hypertension, diabetes, and cerebrovascular disease, and so forth. 49 Investigating fatal cases of COVID‐19 in China demonstrated that the majority of the non‐survivors died of multiple organ failure, and most of these cases were in male over 50 years old with noncommunicable chronic diseases. 50 Apparently, there is a link between basic diseases and COVID‐19 infection. Clinical reports have indicated that ACE2 deficiency in patients with COVID‐19 with underlying disease, exacerbated the damage caused by ACE2 downregulation during viral infections. 48 As numerous underlying diseases afflicting the elderly may be more complex and severe, this age group may be affected the most. Studies reported “inflame‐aging,” a chronic mild inflammation in aging that is associated with elevated systemic levels of pro‐inflammatory cytokines which may promote underlying diseases. 51 It is speculated that “inflame‐aging” caused a cytokine storm in elderly patients with COVID‐19, by altering the expression of ACE2 receptor, producing excessive ROS, aging adipocyte activity, altering the levels of autophagy and mitochondrial autophagy, immune aging, and reducing vitamin D (VD). 52 This is precisely verified by the age‐related ACE2 dysregulation. 53 This may also explain why elderly people with these underlying diseases exhibited a high rate of COVID‐19 infection (31.2%). 54 Most of the critically ill patients in the ICU are also elderly, 55 and acutely ill patients progress to ARDS, sepsis, shock, and multi‐organ dysfunction, and even failure within a week. Moreover, previous that described the pathology and the molecular changes in patients with COVID‐19 suggested that immunosenescence is also a major driver of high mortality in elderly patients, which hampers pathogen recognition, alert signaling, and clearance. 56 The main manifestations of immune aging are thymus atrophy resulting in reduced T cell output, decreased natural killer cell activity, ineffective pathogen recognition, and macrophage activation. 57 These age‐related changes are thought to be due to pathogenic, genetic, and lifestyle factors that affect the cells' epigenetic status and the diversity of immune cells. 58 These factors indicated that both aging and numerous diseases may exacerbate the impact of SARS‐COV‐2 in the elderly and may eventually lead to MODS.
4. SARS‐CoV‐2 INFECTION IN THE OBESE SUBJECTS CAUSES MODS
Results of previous studies have demonstrated that COVID‐19 and its severity is associated with overweight and obesity. Notably, obese patients frequently suffer from cardiovascular dysfunction, hypertension, type 2 diabetes, and other fundamental diseases. 59 Therefore, obesity may be an important risk factor for the development of severe COVID‐19. In patients who are overweight or obese, macronutrient excess in the adipose tissues stimulates adipocytes to release TNF‐α, IL‐6, and other pro‐inflammatory mediators and to reduce the production of the anti‐inflammatory adiponectin, thus predisposing to a pro‐inflammatory state and oxidative stress. 60 The lack of cardiovascular and anti‐inflammatory protective factors of the ACE2/ang(1−7)/Masr axis triggered a cytokine storm due to the physiological conditions of the adipose tissue and viral induction. Results of aprevious study demonstrated that both SARS‐CoV‐2 infection and obesity appear to share some common metabolic and inflammatory response pathways. 61 In Germany, patients with ARDS were more commonly overweight or obese (83%) versus those with normal body mass index (BMI) (42%). 62 In China, patients with overweight/obesity were more likely to be hospitalized longer than those with normal BMI. 63 A high BMI not only reflects the risk of infection and complications but also tends to indicate the severity of the SARS‐CoV‐2 infection. Other complications of severe obesity, such as heart failure and renal failure, 64 , 65 may exacerbate the occurrence of multiple organ dysfunction.
5. CONCLUSION AND PROSPECT
In conclusion, the severity of COVID‐19 may be impacted by multisystem failure, which includes underlying disease and ACE2 expression in humans (Figure 2). We also need to consider that the current rate of SARS‐CoV‐2 variation is rapid, with implications for both infection and lethality, and that this is the reason why COVID‐19 was not controlled despite widespread vaccination in China. In the face of the possible long‐term coexistence of COVID‐19 with the human body, we should more adhere to standardized prevention and control, cutting the transmission chain of the virus and blocking it to a minimum. ACE2 has been identified as the cell receptor of SARS‐CoV‐2, 21 so we can start from the targeted combination of SARS‐CoV‐2 S protein and ACE2 receptor to find treatment schemes or develop related drugs. At present, therapeutic method involves treatment with soluble recombinant human ACE2, in order to disrupt viral entry via the spike protein‐ACE2 interaction and reduce viral load in a dose‐dependent manner. 1 In response to the effects of the underlying disease and age factors, we are aiming for therapeutic intervention with pro‐inflammatory factors and chemokines, while also actively attempting to reduce mortality by means of activating the body's defenses against aging. In the future, the management of COVID‐19 patients with possible MODS should take into account individual differences such as age, weight, different and complex underlying diseases and infection levels, and adopt a more flexible and personalized treatment approach.
Figure 2.
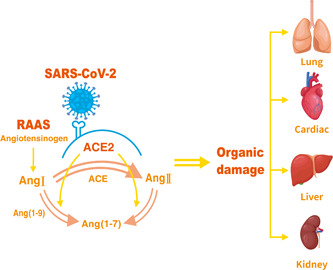
Angiotensin converting enzyme 2 (ACE2)‐mediated injury of different organs. The spike protein of severe acute respiratory syndrome coronavirus type 2 (SARS‐CoV‐2) can bind to the ACE2 receptor, mediating virus entry into host cells. The balance of angiotensin‐converting enzyme (ACE)/ACE2 is disrupted by the action of the virus, promoting the development of inflammation and fibrosis. These intensify the attack of the virus, leading to organ damage including the lungs, heart, liver, and kidneys. AngI, angiotensinI; AngII, angiotensinII; Ang (1−7), angiotensin (1−7); Ang (1−9), angiotensin (1−9); RAAS, renin−angiotensin−aldosterone system
CONFLICT OF INTERESTS
The author declares that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Jian Xu and Hanmeng Li designed the study and prepared the outline. Hanmeng Li performed literature review research and prepared the first draft. Wenbin Zhao and Hanmeng Li cooperated in drafting and revising the sentences. All authors contributed to revising the manuscript. All authors read and signed the paper manuscript.
Zhao W, Li H, Li J, Xu B, Xu J. The mechanism of multiple organ dysfunction syndrome in patients with COVID‐19. J Med Virol. 2022;94:1886‐1892. 10.1002/jmv.27627
DATA AVAILABILITY STATEMENT
This is a review article; data openly available in a public repository that issues datasets with DOIs. The data that support the findings of this study are openly available in PubMed at doi:10.1152/physiolgenomics.00089.2020; doi:10.1136/postgradmedj-2020-138234; doi:10.1007/s11606-020-05762-w; doi:10.1016/j.ijid.2020.03.053; doi:10.1007/s00281-017-0639-8; doi:10.1111/bjh.16659; doi:10.1038/s41591-020-1051-9; doi:10.3389/fimmu.2021.613422; doi:10.1038/s41581-020-00357-4; doi:10.1155/2017/8416763; doi:10.1007/s00101-009-1537-9; doi:10.1080/13510002.2021.1891808; doi:10.1101/2020.03.04.20030395; doi:10.1021/acsinfecdis.0c00288; doi:10.1096/fj.202001807; doi:10.2147/CIA.S158513; doi:10.1016/S0140-6736(20)30211-7; doi:10.1111/jth.14820; doi:10.1253/circj.cj-10-0045; doi:10.1111/jvim.15454; doi:10.1016/j.ejim.2020.04.037; doi:10.1186/s40249-020-00662-x; doi:10.1007/s11684-020-0754-0; doi:10.3390/cells9122638; doi:10.1016/j.jsbmb.2020.105719; doi:10.1055/s-0039-1683996; doi:10.1038/srep27911; doi:10.1038/nm1267; doi:10.1002/path.5471; doi:10.1136/openhrt-2020-001424; doi:10.1161/01.HYP.0000205833.89478.5b; doi:10.1093/eurheartj/ehi114; doi:10.3389/fendo.2014.00016; doi:10.1038/nature00786; doi:10.3760/cma.j.issn.1001-0939.2020.03.013; doi:10.3760/cma.j.issn.1001-0939.2020.03.016; doi:10.1007/s00134-020-05985-9; doi:10.1016/S0140-6736(20)30183-5; doi:10.1016/S0140-6736(20)30211-7; doi:10.1111/liv.14730; doi:10.1016/j.stem.2020.06.015; doi:10.1016/S2213-2600(20)30076-X; doi:10.1016/j.jhep.2020.06.006; doi:10.1093/ndt/gft320; doi:10.1002/path.1670; doi:10.1681/ASN.2006050423; doi:10.1002/rmv.2176; doi:10.1164/rccm.202003-0543OC; doi:10.1007/s00011-020-01372-8; doi:10.1016/S1473-3099(20)30235-8; doi:10.1001/jama.2020.1585; doi:10.18632/aging.103344; doi:10.5114/aoms.2016.58928; doi:10.1016/j.dsx.2020.04.033; doi:10.3238/arztebl.2020.0271; doi:10.1038/s41366-020-0634-3
REFERENCES
- 1. Pollard CA, Morran MP, Nestor‐Kalinoski AL. The COVID‐19 pandemic: a global health crisis. Physiol Genomics. 2020;52(11):549‐557. 10.1152/physiolgenomics.00089.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Umakanthan S, Sahu P, Ranade AV, et al. Origin, transmission, diagnosis and management of coronavirus disease 2019 (COVID‐19). Postgrad Med J. 2020;96(1142):753‐758. 10.1136/postgradmedj-2020-138234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiang F, Deng L, Zhang L, Cai Y, Cheung CW, Xia Z. Review of the clinical characteristics of coronavirus disease 2019 (COVID‐19). J Gen Intern Med. 2020;35(5):1545‐1549. 10.1007/s11606-020-05762-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen N, zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507‐513. 10.1016/j.ijid.2020.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517‐528. 10.1007/s00281-017-0639-8 [DOI] [PubMed] [Google Scholar]
- 6. Wan S, Yi Q, Fan S, et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID‐19) infected patients. Br J Haematol. 2020;189(3):428‐437. 10.1111/bjh.16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Del Valle DM, Kim‐Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26(10):1636‐1643. 10.1038/s41591-020-1051-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santa Cruz A, Mendes‐Frias A, Oliveira AI, et al. Interleukin‐6 is a biomarker for the development of fatal severe acute respiratory syndrome coronavirus 2 pneumonia. Front Immunol. 2021;12:613422. 10.3389/fimmu.2021.613422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perico L, Benigni A, Casiraghi F, Ng L, Renia L, Remuzzi G. Immunity, endothelial injury and complement‐induced coagulopathy in COVID‐19. Nat Rev Nephrol. 2021;17(1):46‐64. 10.1038/s41581-020-00357-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pizzino G, Irrera N, Cucinotta M, et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. 10.1155/2017/8416763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kudryavtseva AV, Krasnov GS, Dmitriev AA, et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016;7(29):44879‐44905. 10.18632/oncotarget.9821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wendel M, Heller AR, Koch T. Pathomechanismen des Organversagens. Zelluläre Sauerstoffverwertungsstörung im Rahmen der Sepsis [Pathomechanisms of organ failure. Mitochondrial dysfunction in sepsis]. Anaesthesist. 2009;58(4):343‐352. 10.1007/s00101-009-1537-9 [DOI] [PubMed] [Google Scholar]
- 13. Toro‐Pérez J, Rodrigo R. Contribution of oxidative stress in the mechanisms of postoperative complications and multiple organ dysfunction syndrome. Redox Rep. 2021;26(1):35‐44. 10.1080/13510002.2021.1891808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao M, Zhang D, Wang Y, et al. Clinical features of patients infected with the 2019 novel coronavirus (COVID‐19) in Shanghai, China. medRxiv. 2020. 10.1101/2020.03.04.20030395 [DOI] [Google Scholar]
- 15. Polonikov A. Endogenous deficiency of glutathione as the most likely cause of serious manifestations and death in COVID‐19 patients. ACS Infect Dis. 2020;6(7):1558‐1562. 10.1021/acsinfecdis.0c00288 [DOI] [PubMed] [Google Scholar]
- 16. De Flora S, Balansky R, La Maestra S. Rationale for the use of N‐acetylcysteine in both prevention and adjuvant therapy of COVID‐19. FASEB J. 2020;34(10):13185‐13193. 10.1096/fj.202001807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liguori I, Russo G, Curcio F, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757‐772. 10.2147/CIA.S158513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arachchillage DRJ, Laffan M. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(5):1233‐1234. 10.1111/jth.14820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imai Y, Kuba K, Ohto‐Nakanishi T, Penninger JM. Angiotensin‐converting enzyme 2 (ACE2) in disease pathogenesis. Circ J. 2010;74(3):405‐410. 10.1253/circj.cj-10-0045 [DOI] [PubMed] [Google Scholar]
- 20. Ames MK, Atkins CE, Pitt B. The renin‐angiotensin‐aldosterone system and its suppression. J Vet Intern Med. 2019;33(2):363‐382. 10.1111/jvim.15454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexandre J, Cracowski JL, Richard V, Bouhanick B, ‘Drugs, COVID‐19’ Working Group of the French Society of Pharmacology, Therapeutics . Renin‐angiotensin‐aldosterone system and COVID‐19 infection. Ann Endocrinol. 2020;81(2−3):63‐67. 10.1016/j.ando.2020.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS‐CoV‐2 infection. Eur J Intern Med. 2020;76:14‐20. 10.1016/j.ejim.2020.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS‐CoV‐2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9(1):45. 10.1186/s40249-020-00662-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med. 2020;14(2):185‐192. 10.1007/s11684-020-0754-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hatmal MM, Alshaer W, Al‐Hatamleh MAI, et al. Comprehensive structural and molecular comparison of spike proteins of SARS‐CoV‐2, SARS‐CoV and MERS‐CoV, and their interactions with ACE2. Cells. 2020;9(12):2638. 10.3390/cells9122638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quesada‐Gomez JM, Entrenas‐Castillo M, Bouillon R. Vitamin D receptor stimulation to reduce acute respiratory distress syndrome (ARDS) in patients with coronavirus SARS‐CoV‐2 infections: revised Ms SBMB 2020_166. J Steroid Biochem Mol Biol. 2020;202:105719. 10.1016/j.jsbmb.2020.105719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. 2019;40(1):31‐39. 10.1055/s-0039-1683996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wösten‐van Asperen RM, Bos AP, Bem RA, et al. Imbalance between pulmonary angiotensin‐converting enzyme and angiotensin‐converting enzyme 2 activity in acute respiratory distress syndrome. Pediatr Crit Care Med. 2013;14(9):e438‐e441. 10.1097/PCC.0b013e3182a55735 [DOI] [PubMed] [Google Scholar]
- 29. Li Y, Zeng Z, Cao Y, et al. Angiotensin‐converting enzyme 2 prevents lipopolysaccharide‐induced rat acute lung injury via suppressing the ERK1/2 and NF‐κB signaling pathways. Sci Rep. 2016;6:27911. 10.1038/srep27911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nat Med. 2005;11(8):875‐879. 10.1038/nm1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bourgonje AR, Abdulle AE, Timens W, et al. Angiotensin‐converting enzyme 2 (ACE2), SARS‐CoV‐2 and the pathophysiology of coronavirus disease 2019 (COVID‐19). J Pathol. 2020;251(3):228‐248. 10.1002/path.5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaudhry F, Lavandero S, Xie X, et al. Manipulation of ACE2 expression in COVID‐19. Open Heart. 2020;7(2):e001424. 10.1136/openhrt-2020-001424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamamoto K, Ohishi M, Katsuya T, et al. Deletion of angiotensin‐converting enzyme 2 accelerates pressure overload‐induced cardiac dysfunction by increasing local angiotensin II. Hypertension. 2006;47(4):718‐726. 10.1161/01.HYP.0000205833.89478.5b [DOI] [PubMed] [Google Scholar]
- 34. Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26(4):369‐324. 10.1093/eurheartj/ehi114 [DOI] [PubMed] [Google Scholar]
- 35. Ruiz‐Ortega M, Rupérez M, Esteban V, et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant. 2006;21(1):16‐20. 10.1093/ndt/gfi265 [DOI] [PubMed] [Google Scholar]
- 36. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822‐828. 10.1038/nature00786 [DOI] [PubMed] [Google Scholar]
- 37. Chen L, Liu HG, Liu W, et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43(3):203‐208. 10.3760/cma.j.issn.1001-0939.2020.03.013 [DOI] [PubMed] [Google Scholar]
- 38. Sun ML, Yang JM, Sun YP, Su GH. Inhibitors of RAS might be a good choice for the therapy of COVID‐19 pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43(3):219‐222. 10.3760/cma.j.issn.1001-0939.2020.03.016 [DOI] [PubMed] [Google Scholar]
- 39. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin‐converting enzyme 2 (ACE2) as a SARS‐CoV‐2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586‐590. 10.1007/s00134-020-05985-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nardo AD, Schneeweiss‐Gleixner M, Bakail M, Dixon ED, Lax SF, Trauner M. Pathophysiological mechanisms of liver injury in COVID‐19. Liver Int. 2021;41(1):20‐32. 10.1111/liv.14730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang L, Han Y, Nilsson‐Payant BE, et al. A human pluripotent stem cell‐based platform to study SARS‐CoV‐2 tropism and model virus infection in human cells and organoids. Cell Stem Cell. 2020;27(1):125‐136e7. 10.1016/j.stem.2020.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420‐422. 10.1016/S2213-2600(20)30076-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M. COVID‐19 and the liver. J Hepatol. 2020;73(5):1231‐1240. 10.1016/j.jhep.2020.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soler MJ, Wysocki J, Batlle D. ACE2 alterations in kidney disease. Nephrol Dial Transplant. 2013;28(11):2687‐2697. 10.1093/ndt/gft320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tikellis C, Bialkowski K, Pete J, et al. ACE2 Deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57(4):1018‐1025. 10.2337/db07-1212 [DOI] [PubMed] [Google Scholar]
- 47. Oudit GY, Herzenberg AM, Kassiri Z, et al. Loss of angiotensin‐converting enzyme‐2 leads to the late development of angiotensin II‐dependent glomerulosclerosis. Am J Pathol. 2006;168(6):1808‐1820. 10.2353/ajpath.2006.051091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahmadian E, Hosseiniyan Khatibi SM, Razi Soofiyani S, et al. Covid‐19 and kidney injury: pathophysiology and molecular mechanisms. Rev Med Virol. 2021;31(3):e2176. 10.1002/rmv.2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS‐CoV‐2: a systematic review and meta‐analysis. Int J Infect Dis. 2020;94:91‐95. 10.1016/j.ijid.2020.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Du Y, Tu L, Zhu P, et al. Clinical features of 85 fatal cases of COVID‐19 from Wuhan. A retrospective observational study. Am J Respir Crit Care Med. 2020;201(11):1372‐1379. 10.1164/rccm.202003-0543OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia S, Zhang X, Zheng S, et al. An update on Inflamm‐aging: mechanisms, prevention, and treatment. J Immunol Res. 2016;2016:8426874. 10.1155/2016/8426874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meftahi GH, Jangravi Z, Sahraei H, Bahari Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID‐19 infection: the contribution of “inflame‐aging”. Inflamm Res. 2020;69(9):825‐839. 10.1007/s00011-020-01372-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Y, Li L. SARS‐CoV‐2: virus dynamics and host response. Lancet Infect Dis. 2020;20(5):515‐516. 10.1016/S1473-3099(20)30235-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. The Novel Coronavirus Pneumonia Emergency Response Epidemiology Team . The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID‐19) ‐ China, 2020. China CDC Wkly. 2020;2(8):113‐122. [PMC free article] [PubMed] [Google Scholar]
- 55. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061‐1069. 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Perrotta F, Corbi G, Mazzeo G, et al. COVID‐19 and the elderly: insights into pathogenesis and clinical decision‐making.Aging Clin Exp Res. 2020;32(8):1599‐1608. 10.1007/s40520-020-01631-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol. 2020;13(1):151. 10.1186/s13045-020-00986-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mueller AL, McNamara MS, Sinclair DA. Why does COVID‐19 disproportionately affect older people? Aging. 2020;12(10):9959‐9981. 10.18632/aging.103344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barazzoni R, Gortan Cappellari G, Ragni M, Nisoli E. Insulin resistance in obesity: an overview of fundamental alterations. Eat Weight Disord. 2018;23(2):149‐157. 10.1007/s40519-018-0481-6 [DOI] [PubMed] [Google Scholar]
- 60. Ellulu MS, Patimah I, Khaza'ai H, Rahmat A, Abed Y. Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci. 2017;13(4):851‐863. 10.5114/aoms.2016.58928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Michalakis K, Ilias I. SARS‐CoV‐2 infection and obesity: common inflammatory and metabolic aspects. Diabetes Metab Syndr. 2020;14(4):469‐471. 10.1016/j.dsx.2020.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dreher M, Kersten A, Bickenbach J, et al. The characteristics of 50 hospitalized COVID‐19 patients with and without ARDS. Dtsch Arztebl Int. 2020;117(16):271‐278. 10.3238/arztebl.2020.0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hu X, Pan X, Zhou W, et al. Clinical epidemiological analyses of overweight/obesity and abnormal liver function contributing to prolonged hospitalization in patients infected with COVID‐19. Int J Obes. 2020;44(8):1784‐1789. 10.1038/s41366-020-0634-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med. 2002;347(5):305‐313. 10.1056/NEJMoa020245 [DOI] [PubMed] [Google Scholar]
- 65. Silva Junior GB, Bentes AC, Daher EF, Matos SM. Obesity and kidney disease. J Bras Nefrol. 2017;39(1):65‐69. 10.5935/0101-2800.20170011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is a review article; data openly available in a public repository that issues datasets with DOIs. The data that support the findings of this study are openly available in PubMed at doi:10.1152/physiolgenomics.00089.2020; doi:10.1136/postgradmedj-2020-138234; doi:10.1007/s11606-020-05762-w; doi:10.1016/j.ijid.2020.03.053; doi:10.1007/s00281-017-0639-8; doi:10.1111/bjh.16659; doi:10.1038/s41591-020-1051-9; doi:10.3389/fimmu.2021.613422; doi:10.1038/s41581-020-00357-4; doi:10.1155/2017/8416763; doi:10.1007/s00101-009-1537-9; doi:10.1080/13510002.2021.1891808; doi:10.1101/2020.03.04.20030395; doi:10.1021/acsinfecdis.0c00288; doi:10.1096/fj.202001807; doi:10.2147/CIA.S158513; doi:10.1016/S0140-6736(20)30211-7; doi:10.1111/jth.14820; doi:10.1253/circj.cj-10-0045; doi:10.1111/jvim.15454; doi:10.1016/j.ejim.2020.04.037; doi:10.1186/s40249-020-00662-x; doi:10.1007/s11684-020-0754-0; doi:10.3390/cells9122638; doi:10.1016/j.jsbmb.2020.105719; doi:10.1055/s-0039-1683996; doi:10.1038/srep27911; doi:10.1038/nm1267; doi:10.1002/path.5471; doi:10.1136/openhrt-2020-001424; doi:10.1161/01.HYP.0000205833.89478.5b; doi:10.1093/eurheartj/ehi114; doi:10.3389/fendo.2014.00016; doi:10.1038/nature00786; doi:10.3760/cma.j.issn.1001-0939.2020.03.013; doi:10.3760/cma.j.issn.1001-0939.2020.03.016; doi:10.1007/s00134-020-05985-9; doi:10.1016/S0140-6736(20)30183-5; doi:10.1016/S0140-6736(20)30211-7; doi:10.1111/liv.14730; doi:10.1016/j.stem.2020.06.015; doi:10.1016/S2213-2600(20)30076-X; doi:10.1016/j.jhep.2020.06.006; doi:10.1093/ndt/gft320; doi:10.1002/path.1670; doi:10.1681/ASN.2006050423; doi:10.1002/rmv.2176; doi:10.1164/rccm.202003-0543OC; doi:10.1007/s00011-020-01372-8; doi:10.1016/S1473-3099(20)30235-8; doi:10.1001/jama.2020.1585; doi:10.18632/aging.103344; doi:10.5114/aoms.2016.58928; doi:10.1016/j.dsx.2020.04.033; doi:10.3238/arztebl.2020.0271; doi:10.1038/s41366-020-0634-3