

Abstract
Severe coronavirus disease 2019 (COVID‐19) is characterized by lung injury, cytokine storm, and increased neutrophil‐to‐lymphocyte ratio (NLR). Current therapies focus on reducing viral replication and inflammatory responses, but no specific treatment exists to prevent the development of severe COVID‐19 in infected individuals. Angiotensin‐converting enzyme‐2 (ACE2) is the receptor for SARS‐CoV‐2, the virus causing COVID‐19, but it is also critical for maintaining the correct functionality of lung epithelium and endothelium. Coronaviruses induce activation of a disintegrin and metalloprotease 17 (ADAM17) and shedding of ACE2 from the cell surface resulting in exacerbated inflammatory responses. Thus, we hypothesized that ADAM17 inhibition ameliorates COVID‐19‐related lung inflammation. We employed a preclinical mouse model using intratracheal instillation of a combination of polyinosinic:polycytidylic acid (poly(I:C)) and the receptor‐binding domain of the SARS‐CoV‐2 spike protein (RBD‐S) to mimic lung damage associated with COVID‐19. Histologic analysis of inflamed mice confirmed the expected signs of lung injury including edema, fibrosis, vascular congestion, and leukocyte infiltration. Moreover, inflamed mice also showed an increased NLR as observed in critically ill COVID‐19 patients. Administration of the ADAM17/MMP inhibitors apratastat and TMI‐1 significantly improved lung histology and prevented leukocyte infiltration. Reduced leukocyte recruitment could be explained by reduced production of proinflammatory cytokines and lower levels of the endothelial adhesion molecules ICAM‐1 and VCAM‐1. Additionally, the NLR was significantly reduced by ADAM17/MMP inhibition. Thus, we propose inhibition of ADAM17/MMP as a novel promising treatment strategy in SARS‐CoV‐2‐infected individuals to prevent the progression toward severe COVID‐19.
Keywords: acute lung injury, endothelium, IL‐10, lymphocyte, MMP, neutrophil, TACE, TNF‐α
Graphical Abstract
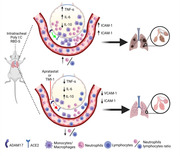
Inhibition of ADAM17 with apratastat or TMI‐1 ameliorates neutrophilia and cytokine storm in a mouse model of Covid‐19‐related lung injury induced by poly(I:C) and the receptor‐binding domain of the SARS‐CoV2 Spike protein (RBD‐S).
Abbreviations
- ACE2
Angiotensin‐converting enzyme‐2
- ADAM17
A disintegrin and metalloprotease 17
- ALI
Acute lung injury
- BALF
Bronchoalveolar lavage fluid
- COVID‐19
Coronavirus disease 2019
- ICAM‐1
Intercellular adhesion molecule 1
- MMP
Matrix metalloprotease
- NETs
Neutrophil extracellular traps
- NLR
Neutrophil‐to‐lymphocyte ratio
- Poly I:C
Polyinosinic:polycytidylic acid
- RBD‐S
Receptor‐binding domain of SARS‐CoV‐2 spike protein
- SARS‐CoV‐2
Severe acute respiratory syndrome coronavirus 2
- TACE
TNF‐α‐converting enzyme
- VCAM‐1
Vascular cell adhesion molecule 1.
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the infectious agent that causes coronavirus disease 2019 (COVID‐19), which can lead to lung failure in susceptible individuals. 1 This virus has infected 226,013,637 people and caused 4,653,053 deaths worldwide (Source: https://coronavirus.jhu.edu/map.html; November 15, 2021). SARS and severe COVID‐19 are characterized by pulmonary edema, fibrosis, activation of the coagulation cascade, hypoxia, and hypercapnia (increased levels of CO2 in the blood). 2 Moreover, severe COVID‐19 is characterized by neutrophilia (increased neutrophil numbers in the blood) and lymphopenia (reduced lymphocyte numbers in the blood) and thus increased neutrophil‐to‐lymphocyte ratio (NLR). 3 , 4 Excessive neutrophil presence and recruitment to the lung and transmigration into the alveolar luminal space contribute to lung injury as neutrophils release proteases, reactive oxygen species, and neutrophil extracellular traps (NETs), which are crucial to combat pathogens, but in large amounts are harmful to the host tissue. 5 Survival depends on the severity of lung injury, the extension of damage to other organs, comorbidities of the infected patient, and the quality of medical care. 6
SARS‐CoV‐2, via its surface protein Spike, uses angiotensin‐converting enzyme‐2 (ACE2) for host entry via airway epithelial cells. 7 ACE2 is an enzyme highly expressed on the apical side of polarized alveolar epithelial cells and lung endothelial cells that cleaves the peptide hormone angiotensin‐I into Ang1‐9 and Ang1‐7, peptides known to protect against epithelial and endothelial hyperpermeability and excessive inflammation. 8 Genetic deletion of ACE2 worsens lung inflammation, while Ang1‐7 administration improves it. 9 Coronavirus infection causes down‐regulation of ACE2 by activating enzymes known to cleave ACE2 10 including a disintegrase and metalloprotease 17 (ADAM17). 11 , 12 ADAM17 sheds the extracellular domain of ACE2 from the surface of lung epithelial cells thus reducing the protective ACE2‐dependent signaling leading to a detrimental feedback loop of exacerbated lung inflammation. Moreover, ADAM17 produces the active form of the proinflammatory cytokine TNF‐α and is critical for proinflammatory cytokine secretion thus contributing to the cytokine storm, another feature of severe COVID‐19 that triggers excessive neutrophil recruitment. 13 , 14 , 15 Although it is well known that ADAM17 gets activated after SARS‐CoV infection, 12 cleaves ACE2, and is therefore likely involved in COVID‐19 pathogenesis, 16 the role of ADAM17 in the development of severe COVID‐19 has not been experimentally analyzed.
Pharmacologic therapy of critically ill patients relies on inhibiting viral replication and inflammatory responses. 17 Although the development of vaccines has been astonishingly fast and much progress has been made with vaccinations worldwide, specific treatments to prevent the development of severe COVID‐19 in infected individuals are still desperately needed for those that do not wish to or cannot be vaccinated, or those that get infected despite being vaccinated.
We hypothesized that pharmacologic inhibition of ADAM17 protects against acute lung injury (ALI) by preventing excessive cytokine production and neutrophil recruitment. We show that intraperitoneal or intranasal administration of the ADAM17/MMP inhibitors apratastat and TMI‐1 significantly reduces proinflammatory cytokine production and leukocyte recruitment to the lungs and thus improves lung histology. Our data suggest that inhibition of ADAM17/MMP is a viable strategy to prevent the development of severe COVID‐19 and to reduce the requirement for critical care and COVID‐19 death toll.
2. METHODS
2.1. Experimental animals
Pathogen‐free 8–12 weeks old C57BL/6 male mice were obtained from the animal facility at CINVESTAV. All protocols have been approved by the institutional animal care and use committee of CINVESTAV.
2.2. Production, purification, and refolding of the RBD‐S protein
The recombinant receptor‐binding domain (RBD) of the SARS‐CoV‐2 Spike protein (RBD‐S) with amino and carboxy deletions (RDB‐NTCT) was purified from Escherichia coli SoluBL21 (DE3) (Genlantis) cells transformed with pRSET‐RDB‐NTCT. Briefly, cells were grown in 20 ml LB broth supplemented with ampicillin (100 μg/ml) at 37°C for 12 h at 150 rpm. This 20 ml of culture was added into 500 ml of 2xYT broth (tryptone16 g/L, yeast extract 10 g/L, and sodium chloride 5 g/L) and ampicillin (100 μg/ml), incubated at 37°C under constant shaking at 200 rpm until reaching an OD600 of 0.8. Expression of the protein was induced by adding 500 ml of 2xYT broth with 2 mM isopropyl β‐d‐galactopyranoside and 2 mM MgSO4 (to reach a final volume of 1 L of culture containing 1 mM IPTG and 1 mM MgSO4). The bacteria were incubated at 16°C for 16 h and harvested by centrifugation at 2500 × g for 30 min at 4°C. The bacterial pellet was resuspended in 20 ml of lysis buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM PMFS, 1 mM benzamidine, and 1 mM DTT) and sonicated in an ice bath using an Ultrasonic Homogenizer (500‐Watt Cold Palmer, USA) for 4 min. The cell lysate was centrifuged at 24,000 × g for 20 min at 4°C. The purification of the RBD‐S domain from inclusion bodies was performed as described previously 18 with the following modifications. To ensure removal of remnant endotoxins, inclusion bodies were washed 4 times with endotoxin‐free wash‐buffer (20 mM Tris–HCl pH 8, NaCl 100 mM, 2%Triton X‐100, and 1 M urea). After each wash, the samples were centrifuged and the pellet resuspended in wash buffer so that each wash step resulted in a 1000× dilution of contaminants present in the inclusion bodies after isolation from the bacteria. The detergent in the wash buffer ensured that membranal endotoxins were washed off. The insoluble fraction was homogenized using an ULTRA‐TURRAX T18 homogenizer (IKA, Germany). Inclusion bodies were solubilized in buffer A (20 mM Tris‐HCl pH 8.0, 500 mM NaCl, 5 mM imidazole, 1 mM DTT, and 8 M urea) and incubated for 16 h at 20°C with constant agitation at 150 rpm. The solubilized inclusion bodies were recovered by ultracentrifugation at 118,000 × g for 30 min at 4°C. The supernatant was filtered through a 0.45 μm membrane and applied onto a HisTrap Ni column (5 ml; Cytiva) previously equilibrated with buffer A with a FPLC (ÄKTApure 25; Cytiva). After loading the sample, the column was washed with the same buffer to eliminate unbound proteins and subsequently the RBD‐S domain was eluted through a linear gradient with buffer B (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 500 mM imidazole, 1 mM DTT, and 8 M urea). Refolding of the RBD‐S domain was performed by size‐exclusion chromatography with a Superdex 200 Increase 10/300 GL column (Cytiva). The column was equilibrated with refolding buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 5% glycerol, 0.4 oxidized glutathione, 0.2 mM reduced glutathione, 1 mM PMSF, and 100 mM arginine). For each refolding run, we applied 1 ml of sample from the previous purification step onto the column followed by a linear gradient of 8 M urea in refolding buffer. The protein was eluted with the refolding buffer. The buffer of the refolded protein was replaced with PBS using a Hi Trap desalting column (5 ml; Cytiva) for subsequent experiments.
2.3. Preclinical model of lung inflammation and ADAM17 inhibitor administration
This COVID‐19‐related preclinical model of lung inflammation is based on a recently published protocol with the exception that we here used the RBD domain of the SARS‐CoV‐2 Spike protein instead of the entire extracellular domain. 19 Mice were anesthetized by intraperitoneal injection of 200 mg/kg ketamine (Anesket, PISA, Mexico City) and 10 mg/kg xylazine (Procin, PISA, Mexico City). The trachea was surgically exposed and a sterile 31G needle was carefully inserted. A combination of 10 mg/kg poly‐I:C (Invivogen, San Diego, CA, USA) and 15 μg of the recombinant RBD‐S in 60 μl endotoxin‐free physiologic saline was instilled followed by 100 μl of air to fill the lungs. Sham‐operated animals received physiologic saline alone as control. The wounds were sutured, and the mice were left to recover in cages. The ADAM17 inhibitors apratastat and TMI‐1 were purchased from Sigma–Aldrich‐Merck (Toluca, Mexico). However, both apratastat and TMI‐1 can also inhibit other MMP; thus, we refer to them as ADAM17/MMP inhibitors. Apratastat and TMI‐1 were administered intraperitoneally at 10 mg/kg. 20 , 21 Apratastat was administered twice (4 and 16 h after surgery) and TMI‐1 was administered once (4 h after surgery) for the 24 h time point; for the 4 h time point, inhibitors were administered after 1 h; and for the 48 h time point, additionally after 32 h. DMSO was administered as vehicle control. Apratastat was also administered intranasally at 10 mg/kg; but, unfortunately, TMI‐1 was no longer available to us when we started these experiments. Four hours, 24 h, or 48 h after induction of lung inflammation, blood samples (cardiac puncture), lung tissue, and bronchoalveolar lavage fluid (BALF) were obtained for analysis of inflammation as described below.
2.4. Histology
After obtaining blood by cardiac puncture, 20 ml of PBS was transcardially perfused until the lungs turned white. The right lung was extracted and fixed in 4% formalin. Fixed lung specimens were dehydrated and embedded in paraffin. Four micrometers of cross‐sections were stained with H&E according to standard protocols. A pathologist analyzed the samples for the degree of inflammation in a blinded fashion and determined the histopathologic scores as previously published. 22
2.5. Bronchoalveolar lavage
Twenty‐four hours after induction of lung inflammation, mice were euthanized by anesthesia overdose and bronchoalveolar lavage was performed as described before. 23 Briefly, the rib cage was removed, and a plastic cannula was inserted into the trachea and lungs were flushed 3 times with 0.8 ml PBS. This procedure was repeated 4 times. Pooled BALF was analyzed by flow cytometry as described below.
2.6. Lung digestion
Left lungs were put in 2 ml microtubes with 1 ml prewarmed PBS containing 200 units of collagenase (Sigma; C9891) and 0.9 mM of calcium and magnesium. Tissue was cut into pieces using sharp scissors. A small hole was made in the cap of the microtubes to prevent hypoxia and the lung suspension was incubated for 20 min at 37°C in a water bath. Samples were vigorously vortexed and then resuspended using a pipette tip. Lung tissue suspensions were then incubated for 20 min more at 37°C. The cell suspension was then filtered through a 40 μm cell strainer (Corning) and washed with 5 ml of PBS. Cells were then centrifuged at 180 × g for 5 min and resuspended in 1 ml PBS for staining and analysis by flow cytometry.
2.7. Flow cytometry
Cells from blood, BALF, and lung suspension were blocked for 15 min with anti‐mouse TruStain FcX (Biolegend, Mexico City) and stained with anti‐mouse CD45 Pacific blue (total leukocytes), anti‐mouse Ly6G APC‐Cy7 (neutrophils), anti‐mouse F4/80 FITC (macrophages), anti‐mouse CD3 PE (T cells), and anti‐mouse PECAM‐1 and ICAM‐1 (endothelial cells) for 15 min. Total lymphocyte numbers were identified as CD45high/side scatterlow. Data were acquired using a FACS Canto II and analyzed using FlowJo V10 software.
2.8. RNA isolation, cDNA synthesis, and qRT‐PCR
Total RNA was isolated and purified from lung tissues using Trizol Reagent (Invitrogen, CA, USA). cDNA was obtained by reverse‐transcribing the purified RNA using the first stand cDNA synthesis kit (ThermoFisher, MA, USA). The following primer pairs were used: β‐actin: forw 5´‐TATCCACCTTCCAGCAGATGT‐3´; rev. 3´‐AGCTCAGTAACAGTCCGCCTA‐5´. TNF‐α: forw. 5´‐ACGGCATGGATCTCAAAGAC‐3´; rev. 3´‐AGATAGCAAATCGGCTGACG‐5´. IL‐1β: forw. 5´‐GCAACTGTTCCTGAACTCAACT‐3´; rev. 3´‐TCTTTTGGGGTCCGTCAACT‐5´. IL‐6: forw. 5´‐CCTTCCTACCCCAATTTCCAA‐3´; rev. 3´‐AGATGAATTGGATGGTCTTGGTC‐5´. IL‐10: forw. 5´‐ACTGCACCCACTTCCCAGT‐3´; rev. 3´‐TGTCCAGCTGGTCCTTTGTT‐5´. ICAM‐1: forw. 5´‐CAATTTCTCATGCCGCACAG‐3´; rev. 3´‐AGCTGGAAGATCGAAAGTCCG‐5´. VCAM‐1: forw. 5´‐TGAACCCAAACAGAGGCAGAGT‐3´; rev. 3´‐GGTATCCCATCACTTGAGCAGG‐5´. PCR was performed using Power SYBR Green PCR master mix (2×) in a final volume of 10 μl. The reaction mixture contained 5.0 μl of the master mix, 25 ng cDNA, and 0.15 μg of each primer. PCR reactions were run in a StepOne™ Real‐Time PCR System. PCR conditions were as follows: activation for 10 min at 95°C; 40 cycles of (denaturation at 95°C for 15 s and data acquisition during annealing and extension at 60°C for 60 s). The melting curve was generated afterward by heating at a rate of 0.6°C/s from 60°C to 95°C. Using β‐actin as endogenous control, expression was quantified using the 2–ΔΔCt method.
2.9. ELISA
The levels of serum TNF‐α were determined using the LEGEND MAX Mouse TNF‐α ELISA kit (Biolegend, CA, USA) according to the manufacturer´s instructions.
2.10. Statistical analysis
Data were analyzed using GraphPad Prism Version 6.0. The Shapiro–Wilk normality test was used to prove normal distribution of data. One‐way ANOVA followed by Dunnett post‐hoc test was then used for multigroup comparisons. p values < 0.05 were considered statistically significant.
3. RESULTS
3.1. Preclinical model of COVID‐19‐related lung inflammation
To investigate potential beneficial effects of ADAM17 inhibition on COVID‐19‐related lung inflammation, we employed a preclinical mouse model, in which we instilled a combination of the TLR‐3 ligand poly(I:C) and the recombinant RBD‐S into mouse lungs to mimic SARS‐CoV‐2 infection (Figure 1(A)). This preclinical COVID‐19 model is based on recently published protocols, 19 , 22 with the simplification that we used the Spike RBD‐domain instead of the whole extracellular domain. To demonstrate that our model is not just an ALI, we compared signs of inflammation such as leukocyte recruitment into lung tissue and BALF when applying poly(I:C) alone versus poly(I:C) combined with RBD‐S (Figures 1(B)–1(J)). We found that the combination of poly(I:C)/RBD‐S induced much stronger recruitment of neutrophils into lung tissue (Figure 1(B)) and BALF (Figure 1(E)) compared with poly(I:C) alone. We also observed more macrophages in both lungs (Figure 1(C)) and BALF (Figure 1(F)). However, we did not observe altered numbers of T cells in lung tissue (Figure 1(D)), whereas T cell numbers in BALF were significantly higher with poly(I:C)/RBD‐S (Figure 1(G)), suggesting that T cells recruited to lungs quickly transmigrate further into the lung lumen. We also observed a stronger neutrophilia with poly(I:C)/RBD‐S (Figure 1(H)), with unchanged numbers of T cells in peripheral blood (Figure 1(I)). Neutrophilia together with overall unchanged lymphocyte numbers resulted in a significantly higher NLR (Figure 1(J)), which is a characteristic symptom of COVID‐19 patients. Overall, these data confirm that our model yields strong lung inflammation with clinical signs resembling those of individuals suffering from severe COVID‐19. 24 , 25
FIGURE 1.
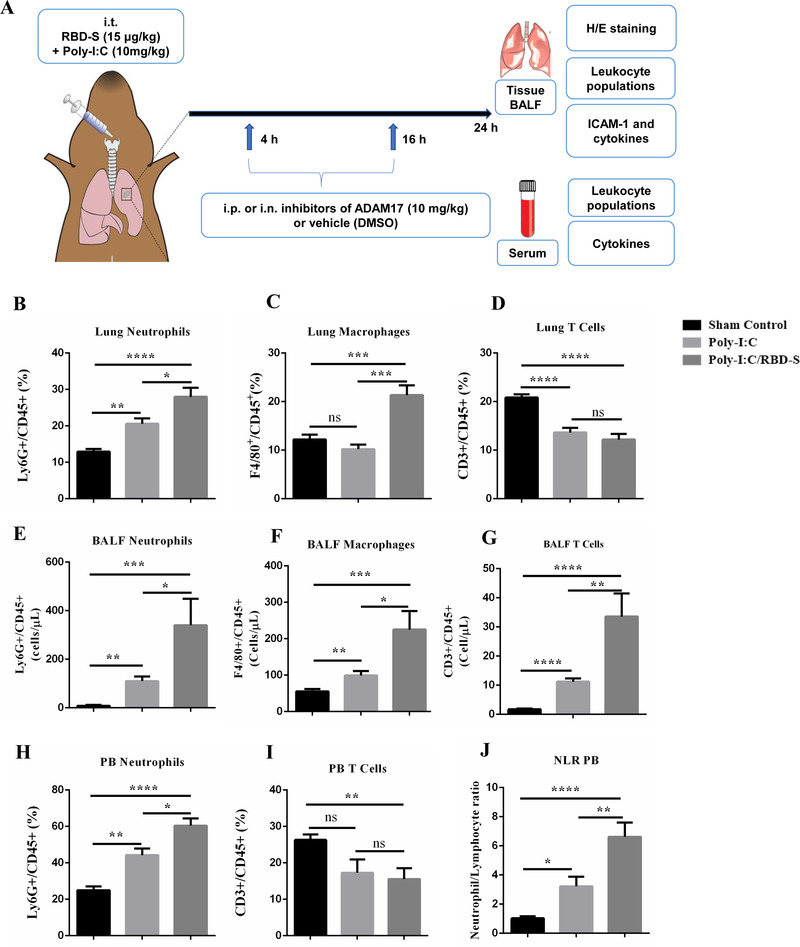
Preclinical mouse model of COVID‐19‐related lung inflammation. (A) Cartoon depicting the model consisting of intratracheal (i.t.) instillation of poly(I:C) and RBD‐S, intraperitoneal (i.p.) or intranasal (i.n.) treatment with apratastat, TMI‐1 or vehicle (DMSO), and harvesting of samples for the indicated analyses. (B–J) Comparison of lung inflammation induced by poly(I:C) alone or the combination of poly(I:C)/RBD‐S in terms of frequency of Ly6G+CD45+ neutrophils (B), F4/80+CD45+ macrophages (C), and CD3+CD45+ T cells (D) in the lungs. Cell frequencies were determined by flow cytometry 24 h after surgery. Total numbers of cells in the bronchoalveolar lavage fluids (BALF) were determined 24 h after surgery: Ly6G+ neutrophils (E), F4/80+ macrophages (F), and CD3+ T cells (G). Frequency of Ly6G+CD45+ neutrophils (H) and frequency of CD3+CD45+ T cells (I) in the peripheral blood 24 h after surgery. (J) Neutrophil–lymphocyte ratio (NLR) in the peripheral blood 24 h after surgery. Data are represented as mean ± SEM of at least 4 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
3.2. ADAM17/MMP inhibition improves lung inflammation induced by intratracheal instillation of poly(I:C) and RBD‐S
Intratracheal instillation of the combination poly‐I:C/RBD‐S induced strong signs of lung inflammation and ALI such as edema, fibrosis, alveolar thickening, closing of alveolar spaces, and leukocyte infiltration (Figure 2(A)). Lung tissues of mice that received ADAM17/MMP inhibitors showed better‐preserved lung morphology and a clear amelioration of the above‐mentioned clinical signs of lung injury (Figure 2(A)). The protective effect seemed to be more pronounced with TMI‐1 compared with apratastat. Histologic scoring confirmed these impressions. Both apratastat and TMI‐1 significantly reduced the histopathologic scores compared with the inflamed vehicle‐treated control mice, with TMI‐1 showing more pronounced protection (Figure 2(B)).
FIGURE 2.
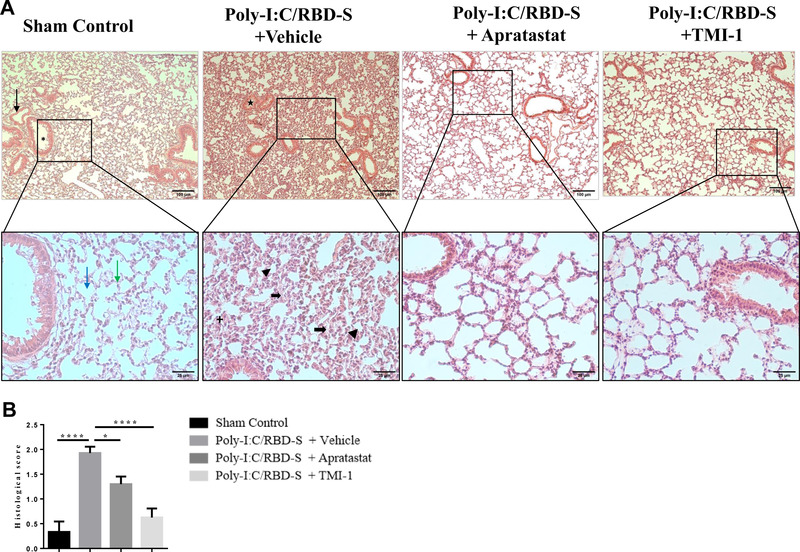
Lung injury is reduced upon ADAM17/MMP inhibition in poly(I:C)/RBD‐S‐induced lung inflammation. (A) Representative images of H&E‐stained lung tissue cross‐sections showing normal tissue morphology, and absence of edema, fibrosis, and vascular congestion in the sham control (10×, upper left panel, black arrow = blood vessel, asterisk = bronchiole; 40× magnifications of the boxed areas, lower panel, blue arrow = alveoli, green arrow = alveolar septa). The poly(I:C)/RBD‐S + vehicle group shows major inflammatory changes such as alveolar wall/septa thickening (bold black arrow) and alveolar space closing and fibrosis (arrowhead), leukocyte infiltration (+), and vascular congestion (star). The lung tissues of poly(I:C)/RBD‐S + apratastat and + TMI groups clearly show less inflammatory damage and overall a better‐preserved tissue morphology. (B) Histologic scores of the lung tissues based on the extent of inflammatory changes with 0 = absence, 1 = low, 2 = moderate, 3 = high. Data are shown as mean ± SEM of images from at least 4 independent tissue preparations. *p < 0.05, ****p < 0.0001
3.3. Neutrophil infiltration into lungs is attenuated upon ADAM17/MMP inhibition
Massive neutrophil infiltration is a key feature of COVID‐19‐associated lung injury. 26 , 27 Because the histology indicated reduced leukocyte presence in lungs after ADAM17/MMP inhibition, we quantified leukocyte populations by flow cytometry. As expected, the number of neutrophils was strongly increased 24 h after instillation of poly(I:C)/RBD‐S, and both apratastat and TMI‐1 significantly reduced the number of neutrophils in the lungs (Figure 3(A)). Also, the number of lung macrophages was much higher in inflamed lungs compared with controls, and again both apratastat and TMI‐1 strongly reduced macrophage numbers in the lungs (Figure 3(B)). By contrast, numbers of T cells were significantly reduced in inflamed lungs, and T cell numbers increased after treatment with either apratastat or TMI‐1 (Figure 3(C)).
FIGURE 3.
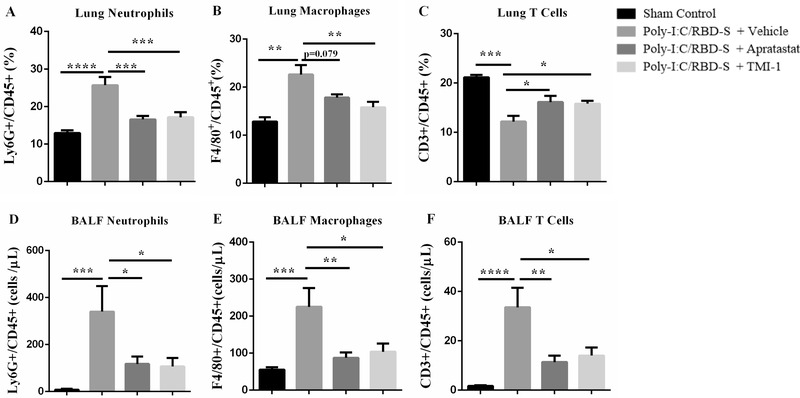
Neutrophil influx into lungs is reduced upon ADAM17/MMP inhibition. (A) Frequency of Ly6G+CD45+ cells in the lungs of the indicated groups. (B) Frequency of F4/80+CD45+ cells in the lungs. (C) Frequency of CD3+CD45+ cells in the lungs. Cell frequencies were determined by flow cytometry 24 h after surgery. Total numbers of Ly6G+ neutrophils (D), F4/80+ macrophages (E), CD3+ T cells (F) from bronchoalveolar lavage fluids (BALF) 24 h after surgery. Data are shown as mean ± SEM of at least 6 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001. ****p < 0.0001
Next, we analyzed leukocyte populations in the BALF, that is, those leukocytes that transmigrated the epithelium to reach the alveolar lumen. 28 We found similar results for neutrophils (Figure 3(D)) and macrophages (Figure 3(E)) in the BALF as in lung tissues. The strong increase in neutrophil and macrophage numbers after instillation of poly‐I:C/RBD‐S was significantly reduced by the ADAM17/MMP inhibitors apratastat and TMI‐1. By contrast, T cell numbers in the BALF were strongly increased in response to poly(I:C)/RBD‐S and were significantly reduced after ADAM17/MMP inhibition, with both apratastat and TMI‐1 showing similar effects (Figure 3(F)).
3.4. ADAM17/MMP inhibition reverses neutrophilia and lymphopenia in poly(I:C)/RBD‐S‐induced lung inflammation
Increased NLR is a well‐established risk factor for severe COVID‐19. 26 Given the changes in leukocyte numbers in the lung, we next analyzed neutrophil and T cell numbers in the blood. We found that poly(I:C)/RBD‐S‐induced lung inflammation caused neutrophilia that was clearly ameliorated by ADAM17/MMP inhibition (Figure 4(A)). Also, we observed significantly reduced T cell numbers in response to poly(I:C)/RBD‐S instillation; however, T cell numbers were not changed after ADAM17/MMP inhibition (Figure 4(B)). The reduced neutrophil numbers in combination with overall unaltered lymphocyte numbers resulted in a significant reduction of the NLR (Figure 4(C)), which likely contributes to the protective effect observed with ADAM17/MMP inhibition.
FIGURE 4.
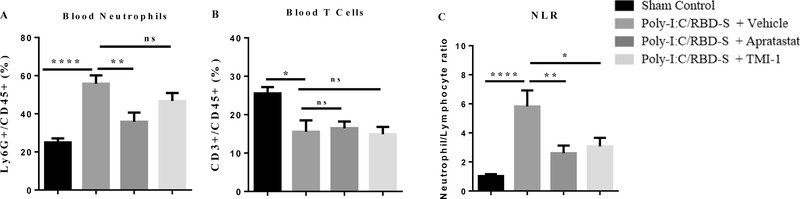
Poly‐I:C/RBD‐S‐induced neutrophilia is prevented upon inhibition of ADAM17. Frequency of Ly6G+ CD45+ neutrophils (A) and CD3+CD45+ T cells (B) in the peripheral blood 24 h after surgery. (C) Neutrophil–lymphocyte ratio (NLR) in the peripheral blood 24 h after surgery. Data are represented as mean ± SEM of at least 6 mice per group. * p < 0.05; **p < 0.01
3.5. ADAM17/MMP inhibition reduces poly(I:C)/RBD‐S‐induced overexpression of TNF‐α and endothelial adhesion molecules
Another classical feature of severe COVID‐19 is the cytokine storm, which in turn leads to endothelial activation, massive leukocyte recruitment and leukocyte hyperactivation. 27 ADAM17 is the enzyme responsible for releasing the active form of TNF‐α. 29 Thus, we expected ADAM17 inhibition to also impact cytokine expression and endothelial activation, which in consequence is the basis for excessive neutrophil recruitment. The massive overexpression of TNF‐α in response to poly(I:C)/RBD‐S was reverted to control levels by ADAM17/MMP inhibition at the mRNA level (Figure 5(A)) and the serum protein level (Figure 5(B)), indicating that not only the local but also the systemic inflammatory response is attenuated by ADAM17/MMP inhibition.
FIGURE 5.
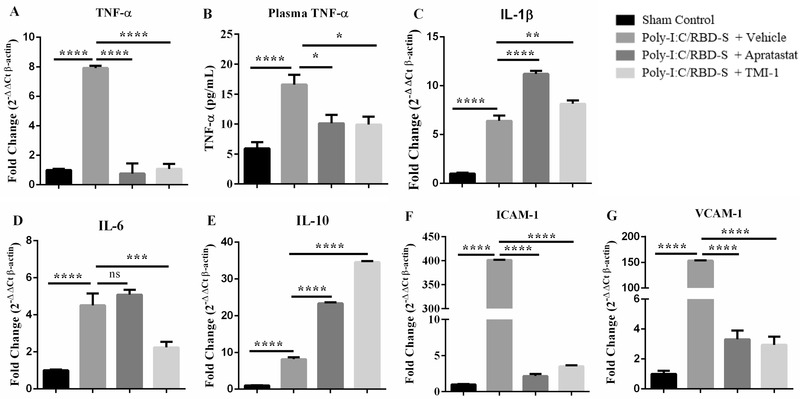
ADAM17 inhibition affects mRNA levels of cytokines and adhesion molecules. Apratastat and TMI‐1 ameliorate the poly‐I:C/RBD‐S‐induced increases in TNF‐α mRNA levels (A), and TNF‐α serum protein levels (B). (C) ADAM 17 inhibitors induce an increase in IL‐1β mRNA. (D) IL‐6 mRNA levels are significantly reduced only after TMI‐1 treatment. (E) Apratastat and TMI‐1 further increase mRNA levels of IL‐10 compared with vehicle‐treated inflamed mice. The poly‐I:C/RBD‐S‐induced increase in mRNA levels of ICAM‐1 (F) and VCAM‐1 (G) are significantly reduced by treatment with both apratastat and TMI‐1. Gene expression was analyzed using quantitative real‐time RT‐PCR with β‐actin as housekeeping gene. Data are represented as mean ± SEM of at least 4 independent cDNA preparations per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
However, ADAM17/MMP inhibitors further increased poly(I:C)/RBD‐S‐induced mRNA levels of IL‐1‐β (Figure 5(C)); and only TMI‐1 was able to reduce the levels of IL‐6 (Figure 5(D)). Of note, mRNA levels of the anti‐inflammatory cytokine IL‐10, which is known to increase during inflammation as a protective countermeasure, were significantly higher after ADAM17/MMP inhibition (Figure 5(E)), suggesting that this is another protective mechanism in the context of lung inflammation.
Proinflammatory cytokines, especially TNF‐α, are known to activate endothelial cells that respond by expressing adhesion molecules for leukocytes to enable their extravasation. 30 Accordingly, poly(I:C)/RBD‐S induced strong expression of ICAM‐1 (Figure 5(F)) and VCAM‐1 (Figure 5(G)) in lungs; and both apratastat and TMI‐1 reversed this overexpression back to control levels. This reduction in endothelial adhesion molecules likely explains the reduced leukocyte recruitment to the lungs after ADAM17/MMP inhibition.
To better understand the timing of the inflammatory response and treatments in our model, we analyzed leukocyte recruitment and cytokine production also after 4 and 48h (Figures S1 and S2). After 4 h, we only found weak increases in leukocyte recruitment in response to poly(I:C)/RBD‐S (Figure S1), although increased cytokine and adhesion molecule production was already detectable (Figure S2). However, hardly any significant effect on leukocyte recruitment was observed with the ADAM17/MMP inhibitors at this time. After 48 h, the differences in leukocyte numbers in the lung tissue were not as pronounced as after 24 h. However, we observed significantly higher leukocyte numbers in the BALF (Figure S1). Moreover, after 48 h, we only detected reduced production of TNF‐α and increased production of IL‐10, but no significant differences of other cytokines and adhesion molecules (Figure S2), suggesting that inflammation resolution has started after 48 h under these conditions. The effects of the ADAM17/MMP inhibitors on NLR was strongest after 24 h (Figure S1). Overall, these data suggest that in our model strong inflammation in response to poly(I:C)/RBD‐S and most significant protective effects of ADAM17/MMP inhibitors can be studied after 24 h.
3.6. Intranasal administration of ADAM17/MMP inhibitors also prevents excessive leukocyte recruitment to the lungs in response to poly(I:C)/RBD‐S
All the data described above were obtained after intraperitoneal administration of the ADAM17/MMP inhibitors. As this is not a desirable route of applying drugs in human patients, we wanted to know whether local application of apratastat via the intranasal route would show similar effects. Importantly, we observed similar results with respect to the leukocyte populations in lung tissue and BALF. Lung neutrophils during poly(I:C)/RBD‐S‐induced lung inflammation were reduced almost to control levels with ADAM17/MMP inhibition (Figure 6(A)); lung macrophages were significantly reduced although not to control levels (Figure 6(B)); and lung T cell numbers were increased by apratastat (Figure 6(C)). Analyzing leukocyte populations in the BALF, we found that intranasal administration of apratastat, like intraperitoneal administration, significantly reduced the numbers of neutrophils (Figure 6(D)), macrophages (Figure 6(E)), and T cells (Figure 6(F)).
FIGURE 6.
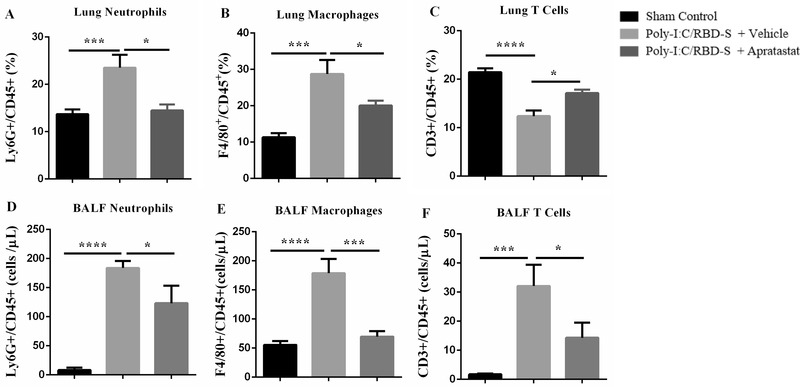
Intranasal administration of apratastat reduces neutrophil and macrophage presence in poly(I:C)/RBD‐S‐inflamed lungs. (A) Percentage of Ly6G+CD45+ neutrophils, (B) F4/80+CD45+ macrophages, and (C) CD3+CD45+ T cells in lung tissue 24 h after surgery as determined by flow cytometry. (D) Total Ly6G+ neutrophils, (E) total F4/80+ macrophages, and (F) total CD3+ T cells in the bronchoalveolar lavage fluids (BALF) as determined by flow cytometry 24 h after surgery. Results are represented as mean ± SEM of at least 5 mice per group. *p < 0.05, ***p < 0.001, and ****p < 0.0001
Moreover, surface protein expression of ICAM‐1 on endothelial cells was also significantly reduced after intranasal apratastat administration (Figures 7(A) and 7(B)). Together these data show that intranasal administration (e.g., inhalation) is also an efficient route of application of apratastat to combat COVID‐19‐associated lung inflammation.
FIGURE 7.
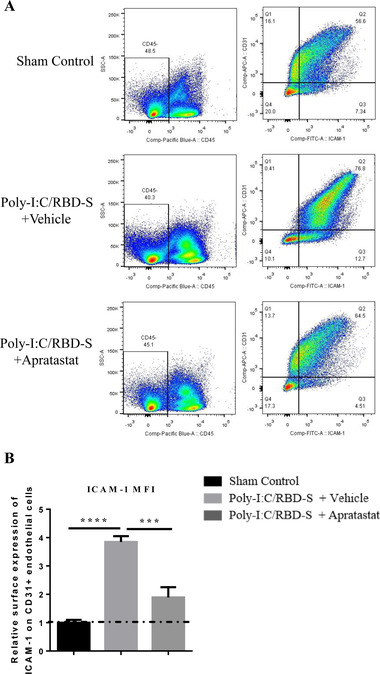
ICAM‐1 expression in lung endothelial cells is reduced after intranasal administration of apratastat. (A) Representative ICAM‐1/CD31 plots from lungs show a clear shift toward higher ICAM‐1 levels in CD31+ endothelial cells in poly‐I:C/RBD‐S‐inflamed and vehicle‐treated lungs. This shift is reverted in apratastat‐treated inflamed mice. (B) Quantification of the MFI of the ICAM‐1 signal from CD31+ICAM1+CD45– cells. Results are represented as mean ± SEM of at least 5 mice per group. i.n.: intranasal. ***p < 0.001 and ****p < 0.0001
4. DISCUSSION
Albeit herculean research efforts worldwide to understand COVID‐19 pathogenesis, it is still not clear under which circumstances a SARS‐CoV‐2‐infected individual develops severe COVID‐19. Risk factors include age, comorbidities such as diabetes and obesity, and smoking, but sometimes even healthy young adults develop severe COVID‐19 and die. Thus, specific and efficient treatments are desperately needed. One obstacle for research is that working with the virus itself requires high biosafety level infrastructure that is not available at many research facilities. Another obstacle is that mice, the most common animal in preclinical research, are not very susceptible to SARS‐CoV‐2 infection due to lower affinity of the spike protein for mouse ACE2 caused by sequence differences (His 353). 31 Thus, mouse models that can be used to study the molecular mechanisms related to COVID‐19 without using the actual virus have been developed. 19 , 31 , 32 , 33 Here, we used a simple mouse model based on the use of the TLR3 ligand poly(I:C) 19 , 22 and the recombinant RBD of the SARS‐CoV‐2 spike protein in C57Bl/6 mice. We showed that these mice developed severe but not lethal lung injury with features characteristic for COVID‐19‐associated lung injury such as edema, fibrosis, leukocyte infiltration, and increased NLR. Importantly, the combination of poly(I:C) with RBD‐S led to a stronger lung inflammation compared with poly(I:C) alone and to a significantly higher NLR, an important symptom of severe COVID‐19. These findings show that this simplified mouse model is convenient to study molecular mechanisms of COVID‐19 pathogenesis. However, given that this model is not based on live virus infection and that ADAM17/MMP inhibitors can affect SARS‐CoV‐2 viral load due to potential suppression of innate immune responses, it will be critical to investigate in future studies the effects of inhibiting ADAM17/MMP on COVID‐19 pathogenesis using a model of live virus infection. Moreover, we only used relatively young mice (8–10 weeks). It will be interesting to see whether older or obese animals will develop a more severe disease, because, if so, this model will also be appropriate to study the molecular differences in high‐risk groups.
ACE2 is not just the receptor for SARS‐CoV and SARS‐CoV‐2, 7 , 34 but it is also a critical enzyme regulating the production of angiotensinogenic peptides, inflammation, and vascular and epithelial permeability. 8 Thus, correct regulation of its enzymatic functions is of utmost importance for correct lung functionality, and loss of ACE2 by enzymatic cleavage is detrimental for lung functions. 29 Two proteases have been shown to cleave ACE2, that is, TMPRSS2 and ADAM17. 7 , 12 , 34 While ACE2 cleavage by TMPRSS2 is critical for SARS‐CoV‐2 entry into lung epithelial cells, the relevance of ADAM17‐mediated ACE2 shedding is less clear. Here, we show that ADAM17 has a critical role in mediating COVID‐19‐related lung inflammation. Our data clearly show that ADAM17 inhibition ameliorates lung inflammation by attenuating the cytokine storm and neutrophil infiltration into the lung (compare graphical abstract). ADAM17 is a well‐known metalloproteinase expressed on the surface of many cells including lung epithelial cells and neutrophils that gets activated in response to many stimuli including coronavirus infections. 35 ADAM17, also known as TNF‐α‐converting enzyme (TACE), is responsible for producing the active form of TNF‐α. 11 We showed that TNF‐α production is indeed significantly reduced after ADAM17 inhibition in our COVID‐19 model. Reduced TNF‐α levels in turn may lead to reduced synthesis of other proinflammatory cytokines due to less NF‐κB activation. This is likely an important mechanism responsible for the protective effect of ADAM17 inhibition against COVID‐19‐associated lung injury. Whether ADAM17 directly affects the production of other proinflammatory cytokines and chemokines or whether this an indirect effect of altered TNF‐α production needs to be investigated in the future.
ADAM17 is also expressed in neutrophils, where it gets activated in response to integrin activation leading to cleavage of l‐selectin, enhanced neutrophil effector functions and bacterial clearance in the lung. 36 It will be important to analyze whether such mechanisms, at least to some degree, also play a role in viral clearance in mild to moderate COVID‐19. However, in the context of severe COVID‐19, it is rather important to dampen neutrophil hyperactivation in response to the cytokine storm because excessive uncontrolled neutrophil effector functions including release of proteases, reactive oxygen species, and NETs contribute to tissue damage as those substances do not distinguish between host and pathogens. 5 On the other hand, lack of ADAM17 in neutrophils 37 , 38 or ADAM17 inhibition in the context of sepsis 15 has been shown to accelerate neutrophil recruitment. We observed here significantly reduced neutrophil presence in lung tissue and BALF and consequently an improved NLR. However, it is still unclear whether this inhibitory effect is due to ADAM17 inhibition in neutrophils, endothelial cells, epithelial cells, or all cell types. It is also important to consider that neutrophil recruitment into the lung follows molecular steps different from those in other tissues 39 and that recruitment to different lung compartments in different COVID‐19 stages is also governed by distinct mechanisms. 40 Thus, more research is required to unravel the mechanisms underlying the altered neutrophil recruitment patterns in our model of COVID‐19‐related lung inflammation.
Apratastat (TMI‐005) and TMI‐1 are structurally related orally bioavailable ADAM17/MMP inhibitors of the thiomorpholine sulfonamide hydroxymate family. 21 These compounds are specific for ADAM17 and barely inhibit the related ADAM10. 41 However, they also inhibit other MMP to a certain extent. Thus, we cannot exclude that some of the observed effects are a result of partial MMP inhibition. Especially TMI‐1 has been reported to inhibit MMP‐1, −2, and −13 at concentrations similar to those needed to inhibit ADAM17. 21 Thus, it is tempting to speculate that, in the cases where we observed stronger effects of TMI‐1 compared with apratastat, for example, in the histologic score (Figure 2(B)), the reduction of IL‐1 and IL‐6 (Figures 5(C) and 5(D)), the differences are caused by additional inhibition of MMPs by TMI‐1. It will be important to prove this idea in future studies.
Both apratastat and TMI‐1 have been shown to inhibit TNF‐α secretion in preclinical models of rheumatoid arthritis (RA); and apratastat (but not TMI‐1) has been tested in clinical trials to treat RA patients. 21 , 42 However, although effective in reducing TNF‐α levels, apratastat did not improve the clinical manifestations in RA patients so that the study was terminated. 43 Despite this disappointing clinical result, apratastat and TMI‐1 could still be useful for the treatment of COVID‐19 patients because in this context we observed not only a protective effect on TNF‐α production, but also on neutrophil recruitment and NLR, all critical features of severe COVID‐19 pathogenesis. Moreover, pharmacodynamics and pharmacokinetics data from clinical studies showed that at least apratastat is well tolerated and bioavailable in humans 44 so that it will be feasible to test apratastat in clinical trials for efficacy in inhibiting severe COVID‐19 development in SARS‐CoV‐2‐infected patients. Unfortunately, pharmacodynamics and pharmacokinetics data for TMI‐1 in humans are not available.
In summary, we provide experimental evidence that ADAM17/MMP inhibition by apratastat and TMI‐1 protects against lung inflammation in a novel, simplified mouse model of COVID‐19‐associated lung injury. ADAM17/MMP inhibitors reduced the cytokine storm and excessive neutrophil recruitment to the lung while improving NLR. Thus, we propose to use these drugs in clinical trials to test their efficacy in COVID‐19 patients.
AUTHORSHIP
N. L. L. and S. V. R. contributed equally to this work. NLL and SVR performed research and statistical analysis, analyzed and interpreted data and wrote the manuscript; HVR, KEJC, IMGF, RMC, AMG, JGC, JGFV, DRV, and DIZV performed research and analyzed data; LCB, and PN analyzed and interpreted data; EMR analyzed and interpreted data and provided essential reagents; CSL performed pathological analysis of tissue samples; EV analyzed and interpreted data and contributed to the study design and manuscript writing; MS conceived and supervised the study, analyzed and interpreted data and wrote the manuscript. All authors reviewed the manuscript.
DISCLOSURE
N. L. L., S. V. R., H. V. R., E. V., and M. S. declare that they have submitted a patent application for the use of ADAM17 inhibitors as COVID‐19 treatment (Mx/a2020/012431). J. G. F. V., D. R. V., D. I. Z. V., and E. M. R. declare that they are in the process of filing a patent application including the methodology of purification and refolding of the RBD‐S protein.
Supporting information
FIGURE S1. Time course of leukocyte numbers in response to intratracheal instillation of poly(I:C)/RBD‐S and ADAM17 inhibition.
Figure S2. Time course of cytokine and adhesion molecule expression in response to intratracheal instillation of poly(I:C)/RBD‐S and ADAM17 inhibition.
ACKNOWLEDGMENTS
This work was supported by funds from the Agencia Mexicana de Cooperación Internacional para el Desarrollo (AMEXCID) of the Secretaría de Relaciones Exteriores (SRE) (Projects AMEXCID_2020‐4 to M. S. and AMEXCID_2020‐3 to E. M. R.). M. S. acknowledges funding from Consejo Nacional de Ciencia y Tecnología (CONACYT; CB‐2016‐284292; and PRONAII Leukemia grant 302978 of the National Strategic Health Program, PRONACE). E. M. R. acknowledges funding from CONACYT (CB2017‐2018: A1‐S‐10743; Apoyo para proyectos de investigación científica, desarrollo tecnológico e innovación en salud ante la contingencia por COVID‐19: 311835), SEP‐CINVESTAV (2018:1), and Gobierno del Estado de Hidalgo (Proyectos Sincrotrón 20201120 and 20201039). S. V. R. received a postdoctoral fellowship from CONACYT. N. L. L., K. E. J. C., I. M. G. F., R. M. C., A. M. G., J. G. F. V., D. R. V., and D. I. Z. V. received a predoctoral fellowship from CONACYT.
Lartey NL, Valle‐Reyes S, Vargas‐Robles H, et al. ADAM17/MMP inhibition prevents neutrophilia and lung injury in a mouse model of COVID‐19. J Leukoc Biol. 2022;111:1147–1158. 10.1002/JLB.3COVA0421-195RR
REFERENCES
- 1. Team CDC COVID‐19 Response . Severe outcomes among patients with coronavirus disease 2019 (COVID‐19) ‐ United States, February 12‐March 16, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:343‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tatum D, Taghavi S, Houghton A, Stover J, Toraih E, Duchesne J. Neutrophil‐to‐lymphocyte ratio and outcomes in Louisiana COVID‐19 patients. Shock. 2020;54:652‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vadillo E, Taniguchi‐Ponciano K, Lopez‐Macias C, et al. A shift towards an immature myeloid profile in peripheral blood of critically Ill COVID‐19 patients. Arch Med Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Narasaraju T, Tang BM, Herrmann M, Muller S, Chow VTK, Radic M. Neutrophilia and NETopathy as key pathologic drivers of progressive lung impairment in patients with COVID‐19. Front Pharmacol. 2020;11:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matthay MA, Wick KD. Corticosteroids, COVID‐19 pneumonia, and acute respiratory distress syndrome. J Clin Invest. 2020;130:6218‐6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271‐280 e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin‐converting enzyme 2: sARS‐CoV‐2 receptor and regulator of the renin‐angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126:1456‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cao Y, Liu Y, Shang J et al. Ang‐(1‐7) treatment attenuates lipopolysaccharide‐induced early pulmonary fibrosis. Lab Invest. 2019;99:1770‐1783. [DOI] [PubMed] [Google Scholar]
- 10. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nat Med. 2005;11:875‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zunke F, Rose‐John S. The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res. 2017;1864:2059‐2070. [DOI] [PubMed] [Google Scholar]
- 12. Patel VB, Clarke N, Wang Z, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM‐17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167‐176. [DOI] [PubMed] [Google Scholar]
- 13. Cui SN, Tan HY, Fan GC. Immunopathological roles of neutrophils in virus infection and COVID‐19. Shock. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borges L, Pithon‐Curi TC, Curi R, Hatanaka E. COVID‐19 and neutrophils: the relationship between hyperinflammation and neutrophil extracellular traps. Mediators Inflamm. 2020;2020:8829674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mishra HK, Ma J, Walcheck B. Ectodomain shedding by ADAM17: its role in neutrophil recruitment and the impairment of this process during sepsis. Front Cell Infect Microbiol. 2017;7:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palau V, Riera M, Soler MJ. ADAM17 inhibition may exert a protective effect on COVID‐19. Nephrol Dial Transplant. 2020;35:1071‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kupferschmidt K, Cohen J. Race to find COVID‐19 treatments accelerates. Science. 2020;367:1412‐1413. [DOI] [PubMed] [Google Scholar]
- 18. de la Cruz JJ, Villanueva‐Lizama L, Dzul‐Huchim V, et al. Production of recombinant TSA‐1 and evaluation of its potential for the immuno‐therapeutic control of Trypanosoma cruzi infection in mice. Hum Vaccin Immunother. 2019;15:210‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gu T, Zhao S, Jin G, et al. Cytokine signature induced by SARS‐CoV‐2 spike protein in a mouse model. Front Immunol, 2020;11:621441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ford BM, Eid AA, Gooz M, et al. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am J Physiol Renal Physiol. 2013;305:F323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Xu J, Levin J, et al. Identification and characterization of 4‐[[4‐(2‐butynyloxy)phenyl]sulfonyl]‐N‐hydroxy‐2,2‐dimethyl‐(3S)thiomorpholinecar boxamide (TMI‐1), a novel dual tumor necrosis factor‐alpha‐converting enzyme/matrix metalloprotease inhibitor for the treatment of rheumatoid arthritis. J Pharmacol Exp Ther. 2004;309:348‐355. [DOI] [PubMed] [Google Scholar]
- 22. Gan T, Yang Y, Hu F, et al. TLR3 regulated Poly(I:c)‐induced neutrophil extracellular traps and acute lung injury partly through p38 MAP kinase. Front Microbiol. 2018;9:3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun F, Xiao G, Qu Z. Murine bronchoalveolar lavage. Bio Protoc. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klopfleisch R. Multiparametric and semiquantitative scoring systems for the evaluation of mouse model histopathology–a systematic review. BMC Vet Res. 2013;9:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schaller T, Hirschbuhl K, Burkhardt K et al. Postmortem examination of patients with COVID‐19. JAMA. 2020;323:2518‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li X, Liu C, Mao Z et al. Predictive values of neutrophil‐to‐lymphocyte ratio on disease severity and mortality in COVID‐19 patients: a systematic review and meta‐analysis. Crit Care. 2020;24:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hussman JP Cellular and molecular pathways of COVID‐19 and potential points of therapeutic intervention. Front Pharmacol. 2020;11:1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID‐19 patients. Emerg Microbes Infect. 2020;9:761‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zipeto D, Palmeira JDF, Arganaraz GA, Arganaraz ER. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID‐19. Front Immunol. 2020;11:576745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692‐704. [DOI] [PubMed] [Google Scholar]
- 31. Ren W, Zhu Y, Wang Y, et al. Comparative analysis reveals the species‐specific genetic determinants of ACE2 required for SARS‐CoV‐2 entry. PLoS Pathog. 2021;17:e1009392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dinnon KH 3rd, Leist SR, Schafer A, et al. A mouse‐adapted model of SARS‐CoV‐2 to test COVID‐19 countermeasures. Nature. 2020;586:560‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCray PB Jr., Pewe L, Wohlford‐Lenane C, et al. Lethal infection of K18‐hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. 2007;81:813‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heurich A, Hofmann‐Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88:1293‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haga S, Yamamoto N, Nakai‐Murakami C, et al. Modulation of TNF‐alpha‐converting enzyme by the spike protein of SARS‐CoV and ACE2 induces TNF‐alpha production and facilitates viral entry. Proc Natl Acad Sci USA. 2008;105:7809‐7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cappenberg A, Margraf A, Thomas K, et al. L‐selectin shedding affects bacterial clearance in the lung: a new regulatory pathway for integrin outside‐in signaling. Blood. 2019;134:1445‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long C, Wang Y, Herrera AH, Horiuchi K, Walcheck B. In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli‐mediated peritonitis. J Leukoc Biol. 2010;87:1097‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang J, Zarbock A, Gomez I, et al. Adam17‐dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood. 2011;118:786‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Margraf A, Ley K, Zarbock A. Neutrophil recruitment: from model systems to tissue‐specific patterns. Trends Immunol. 2019;40:613‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alon R, Sportiello M, Kozlovski S, et al. Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID‐19. Nat Rev Immunol. 2021;21:49‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ludwig A, Hundhausen C, Lambert MH, et al. Metalloproteinase inhibitors for the disintegrin‐like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester‐inducible shedding of cell surface molecules. Comb Chem High Throughput Screen. 2005;8:161‐171. [DOI] [PubMed] [Google Scholar]
- 42. Moss ML, Sklair‐Tavron L, Nudelman R. Drug insight: tumor necrosis factor‐converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2008;4:300‐309. [DOI] [PubMed] [Google Scholar]
- 43. Thabet MM, Huizinga TW. Drug evaluation: apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr Opin Investig Drugs. 2006;7:1014‐1019. [PubMed] [Google Scholar]
- 44. Shu C, Zhou H, Afsharvand M et al. Pharmacokinetic‐pharmacodynamic modeling of apratastat: a population‐based approach. J Clin Pharmacol. 2011;51:472‐481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Time course of leukocyte numbers in response to intratracheal instillation of poly(I:C)/RBD‐S and ADAM17 inhibition.
Figure S2. Time course of cytokine and adhesion molecule expression in response to intratracheal instillation of poly(I:C)/RBD‐S and ADAM17 inhibition.