

Abstract
Atherosclerosis is the major cause of ischemic heart disease and stroke, the leading causes of mortality worldwide. The central pathological features of atherosclerosis include macrophage infiltration and foam cell formation. However, the detailed mechanisms regulating these two processes remain unclear. Here we show that oxidative stress-activated Ca2+-permeable transient receptor potential melastatin 2 (TRPM2) plays a critical role in atherogenesis. Both global and macrophage-specific Trpm2 deletion protect Apoe−/− mice against atherosclerosis. Trpm2 deficiency reduces oxidized low-density lipoprotein (oxLDL) uptake by macrophages, thereby minimizing macrophage infiltration, foam cell formation and inflammatory responses. Activation of the oxLDL receptor CD36 induces TRPM2 activity, and vice versa. In cultured macrophages, TRPM2 is activated by CD36 ligands oxLDL and thrombospondin-1 (TSP1), and deleting Trpm2 or inhibiting TRPM2 activity suppresses the activation of CD36 signaling cascade induced by oxLDL and TSP1. Our findings establish the TRPM2-CD36 axis as a molecular mechanism underlying atherogenesis, and suggest TRPM2 as a potential therapeutic target for atherosclerosis.
Keywords: Atherosclerosis, Ca2+ signaling, TRPM2, macrophages, CD36, oxLDL, TSP1
Introduction
Atherosclerosis and its complications, myocardial infarction and stroke, are the leading causes of death worldwide1. Atherosclerosis is a chronic inflammatory disease. The initial and central pathological feature of atherosclerosis is the formation of foam cells after infiltrated macrophages uptake oxidized low density lipoprotein (oxLDL) and become overloaded with cholesterol2. These lipid-laden macrophages are the culprit for the progression of atherosclerotic lesions by secreting pro-inflammatory cytokines and matrix-degrading proteases, which cause profound inflammatory responses and tissue damage in the vessel walls3. Therefore, inhibiting foam cell formation and inflammatory cytokine production could be a promising strategy for developing more effective therapies for atherosclerosis1.
The uptake of oxLDL by macrophages is mediated by several scavenger receptors. CD36 is the predominant receptor for oxLDL as it is responsible for over 70% of oxLDL uptake4. The binding of oxLDL to CD36 not only triggers internalization of oxLDL, but also elicits downstream signaling cascades, including Fyn, JNK and p38, which further induces oxidative stress and expression of pro-inflammatory genes5. Moreover, binding of oxLDL to CD36 promotes the activation of NLRP3 inflammasome by interacting with Toll like receptor 4 and 6 (TLR4/6) heterodimer, driving the differentiation of macrophages toward a pro-inflammatory phenotype6. Thus, CD36 plays a critical role in macrophage activation and foam cell formation in atherosclerotic lesions. However, the underlying mechanisms regarding how oxLDL binding triggers CD36 signaling activation remain unclear4,7.
TRPM2 is a nonselective cation channel activated by heat, reactive oxygen species (ROS), intracellular Ca2+, and ADP-ribose (ADPR)8–10, which are substantially generated in inflammatory responses11. TRPM2 is widely expressed in myeloid cells, and TRPM2-mediated Ca2+ signaling is important for macrophage activation and phagocytic functions12,13. Knockout of Trpm2 was found to reduce ROS production in macrophages and mitigate tissue damage in a lung injury mouse model14. However, whether TRPM2 is involved in foam cell formation and atherogenesis is unknown. Considering atherosclerosis is also an inflammatory disease and TRPM2 is activated by oxidative stress under inflammatory conditions, we proposed that TRPM2 in macrophages plays a key role in atherogenesis by integrating extracellular stimuli and intracellular signaling cascade.
In this study, we demonstrate that both global Trpm2 deletion and macrophage-specific Trpm2 deletion protect Apoe−/− mice against HFD-induced atherosclerosis, characterized by reduced atherosclerotic lesions, decreased macrophage burden, and suppressed inflammasome activation in the vessel walls. We find that deletion of Trpm2 or inhibiting the activation of TRPM2 in macrophages reduces oxLDL uptake, inhibits macrophage infiltration, and improves the impaired macrophage emigration. We reveal the mechanism by which Trpm2 deletion inhibits macrophage uptake of oxLDL is through impairing CD36 activation. Moreover, we demonstrate that TRPM2 can be activated by CD36 ligands in a CD36-dependent manner. Our studies establish an important, mutually regulating, and positive feedback mechanism between CD36 and TRPM2 in atherogenesis. Targeting TRPM2 inhibits both TRPM2-dependent and CD36-dependent inflammatory responses thereby producing potent protective effects against atherosclerosis.
Results
Global Trpm2 deletion protects mice against atherosclerosis
To investigate whether TRPM2 plays a role in atherosclerosis, global Trpm2−/− mice were crossed with Apoe/− mice. Successful Trpm2 deletion was confirmed by PCR (Extended Data Fig.1a), Western blot (Extended Data Fig.1b) and whole-cell current recordings (Extended Data Fig.1c–e). After mice were fed with a high-fat food (HFD) for 4 months, Trpm2+/+ mice developed severe atherosclerosis, with a lesion ratio of 0.36±0.17 quantified from en-face aortas stained with Oil Red O (ORO) (Fig. 1a, b). In contrast, Trpm2−/− mice exhibited significantly reduced atherosclerotic plaque lesion ratio (0.13±0.02) (Fig. 1a, b). Similarly, significant reduced lesion areas by Trpm2 deletion were observed in the ORO-stained cross-sections of aorta roots (0.24±0.01 vs 0.17±0.01 mm2; Fig. 1c, d). Atherosclerosis is a chronic inflammatory disease and is associated with systemic inflammation15,16. HFD treatment induced a marked increase of total cholesterol level, and this increase is not influenced by Trpm2 deletion (Fig. 1e), which is consistent with other studies showing unaltered cholesterol level by anti-atherosclerosis manipulation17. In Trpm2+/+ mice, there was a significant increase of interleukin-1β (IL-1β) level in serum induced by HFD (430.70±73.69 pg/mL), whereas this increase was significantly attenuated in Trpm2−/− mice (138.03±27.10 pg/mL) (Fig. 1f), suggesting global Trpm2 deletion attenuates systemic inflammation caused by HFD treatment. These results indicate that Trpm2 deletion protects Apoe−/− mice against HFD induced atherogenesis.
Fig. 1: Global Trpm2 deletion protects mice against atherosclerosis.
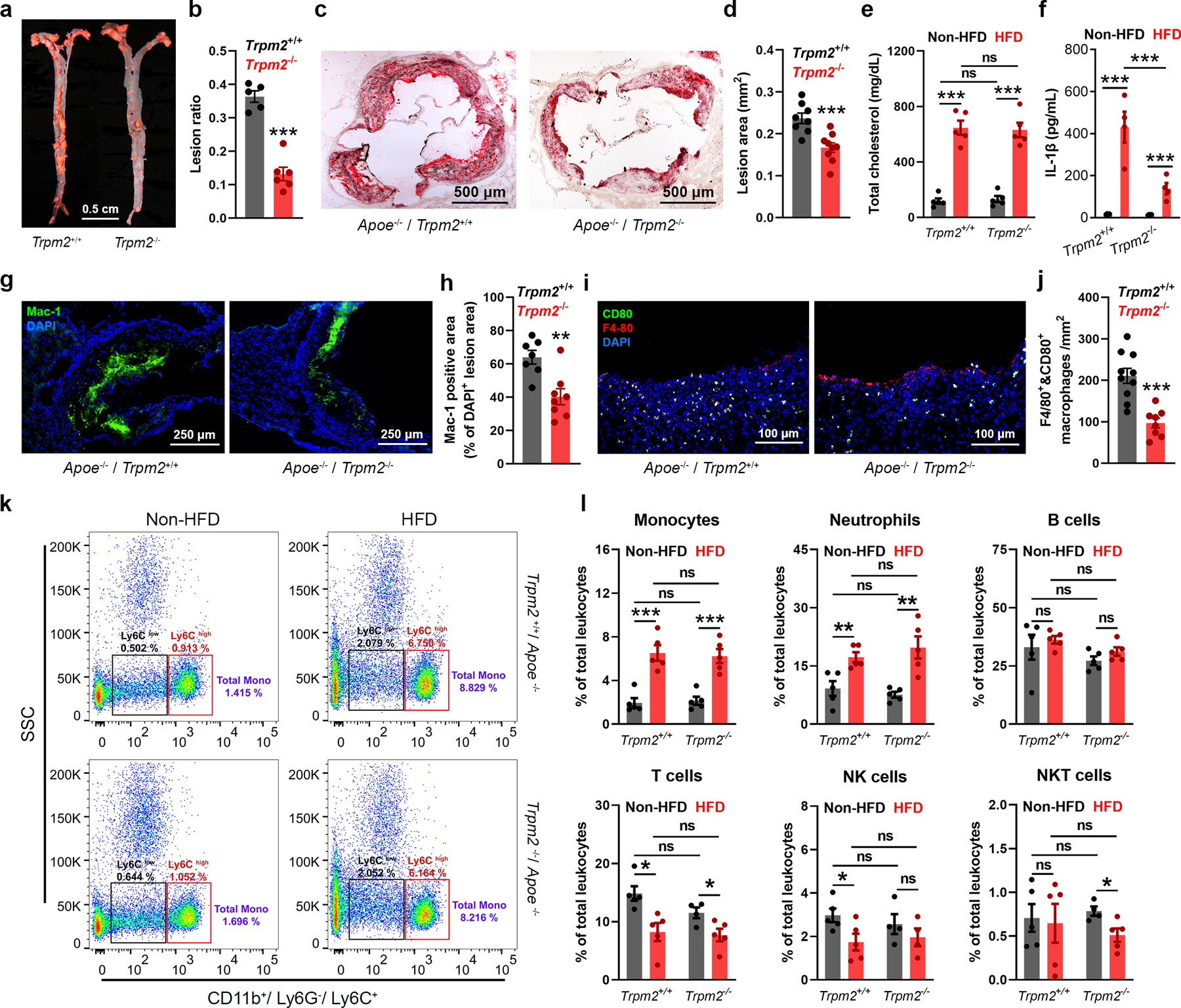
(a,b) Global Trpm2 deletion (Trpm2−/−) inhibited atherosclerotic plaque formation. a, Representative images of Oil Red O (ORO) staining of full-length aorta (red areas represents plaque). b, Mean atherosclerotic lesion ratio based on ORO staining from Trpm2+/+ (n=5) and Trpm2−/− mice (n=6), P < 0.0001. (c, d) Representative images and quantification of ORO staining of the aortic root sections from Trpm2+/+ (n=8) and Trpm2−/− mice (n=9), P = 0.0009. (e) Trpm2−/− did not influence the total cholesterol level in serum (n=5/group) (HFD: high-fat diet). (f) Trpm2−/− inhibited systemic inflammation evaluated by measuring IL-1β level in serum (n=4/group). (g-j) Trpm2−/− reduced macrophage burden in atherosclerotic plaque. g, h Representative merged images and quantification of Mac-1 staining of aortic root sections from Trpm2+/+ (n=7) and Trpm2−/− mice (n=8), P = 0.0027. (i, j) Representative merged images and quantification of F4/80 and CD80 staining of aorta cross-sections using the plaque areas as shown in Extended Data Fig. 1f (Red: F4/80; Blue: DAPI; Green: CD80) from Trpm2+/+ (n=9) and Trpm2−/− mice (n=8), P = 0.0001. (k, l) Trpm2−/− did not influence the leukocyte population in the peripheral blood. k, Representative monocyte population identified using flow cytometry. Ly6C+ monocyte population was identified from CD11b+ and Ly6G− leukocytes. l, Quantification of monocyte, neutrophil, B cell, T cell, NK cell and NKT cell population in the peripheral blood from Trpm2+/+ (n=5) and Trpm2−/− mice (n=5) with or without HFD treatment. (ns: no statistical significance; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Macrophage infiltration plays a critical role in the initiation and progression of atherosclerosis3. Using a recently reported method18, we found that macrophage content in the plaque represented by the ratio of Mac-1 positive versus DAPI positive areas is significantly reduced in Trpm2−/− compared with Trpm2+/+ mice (64±4.11 vs 40.27±4.79 %, Fig. 1g, h). Moreover, the number of F4/80 and CD80 positive macrophages in atherosclerotic plaque was decreased from 210.70±18.12 /mm2 in Trpm2+/+ mice to 96.90±11.82 /mm2 in Trpm2−/− mice (Extended Data Fig.1f, Fig. 1i, j). Importantly, Trpm2 deletion did not influence the leukocyte populations, including monocytes, in the peripheral blood (Fig. 1k, l). Interestingly, we observed that TRPM2 current amplitude recorded in peritoneal macrophages isolated from HFD fed Apoe−/− mice was significantly larger than that from regular chow fed mice (Fig. 2a–c). The increase of TRPM2 currents resulted from the upregulated TRPM2 expression (Extended Data Fig.2a–d) could be caused by systemic inflammation such as enhanced Il-1β as previously reported19 20. Consistent with the immunofluorescence staining, HFD-induced increase of CD11b and CD80 expression in the aortas was inhibited in Trpm2−/− mice (Fig. 2d, g). These data suggest that Trpm2 deletion reduces macrophage infiltration in the plaques.
Fig. 2: Trpm2 deletion attenuates inflammation in the aortas.

(a-c) a, Time-dependent activation of TRPM2 currents (Green: outward current at +100 mV; Purple: inward current at −100 mV) recorded from isolated peritoneal macrophages. N-Methyl-D-glucamine (NMDG) blocks inward current indicating the tightness of seal. b, Representative TRPM2 currents recorded with ramp protocol. c, Quantification of current amplitude (HFD: high-fat diet). (d, g) Representative western blot (WB) analysis and quantification of the expression of CD11b and CD80 in aorta (n=6/group). (e, h) Representative WB analysis and quantification of the expression of MCP1 and MIF in aorta (n=6/group). (f, i) Representative WB analysis and quantification of iNOS expression in aortas (n=6/group). (j, k) Representative WB analysis and quantification of the expression of NLRP3, ASC, cleaved caspase-1 (cCas1), and cleaved IL-1β (cIL-1β) expression in aortas (n=6/group). (l, m) In vitro macrophage migration assay using the methods shown in Extended Data Fig. 3a. l, F4/80 and CD80 co-staining of macrophages in the lower chamber (Red: F4/80; Blue: DAPI; Green: CD80). c, Quantification of the number of infiltrated macrophages within a x 10 field. Six dishes from each group were chosen for quantification. (n, o) In vitro macrophage emigration assay using the methods shown in Extended Data Fig. 3b. Note that macrophages were pre-treated with oxLDL prior this assay. n, F4/80 and CD80 co-staining of macrophages in the lower chamber (Red: F4/80; Blue: DAPI; Green: CD80). o, Quantification of the number of emigrated macrophages within a x 10 field. Six dishes from each group were chosen for quantification. (p, q) Scratch assay of cultured macrophages. p, images of macrophages under bright field taken right after (0 h), 24 h, 48 h and 72 h after the scratch. q, Quantification of the percentage of wound healing. Six dishes from each group were chosen for quantification. (ns: no statistical significance; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Macrophage infiltration in atherosclerosis is strongly influenced by two chemokines, monocyte chemoattractant protein-1 (MCP1) and macrophage migration inhibitory factor (MIF)21. We found that Trpm2 deletion drastically inhibited the HFD-induced increase of both MCP1 and MIF expression in the aortas (Fig. 2e, h), suggesting that TRPM2 may influence the recruitment of macrophages into the plaques. Macrophages that infiltrate into the atheroprone site quickly become the center of inflammatory cues. Inducible nitric oxide synthase (iNOS) shifts the production of nitric oxide by NOS toward the production of ROS22, and was previously found to be abundantly expressed in human atherosclerotic lesions23. We found that Trpm2 deletion significantly inhibited the increase of iNOS expression in aortas after HFD treatment (Fig. 2f, i), implicating that Trpm2 deletion may reduce ROS production in atherosclerotic plaques. Activation of NLRP3 inflammasome in macrophages has been shown to be required for atherogenesis24,25. We found that in Trpm2+/+ mice fed with HFD, there was a significant increase of NLRP3, ASC, cleaved Caspase-1 and cleaved IL-1β expression in the, whereas this increase was inhibited in Trpm2−/− mice (Fig. 2j, k), indicating Trpm2 deletion reduces in vivo inflammasome activation.
Deletion of Trpm2 inhibits macrophage activation
To investigate the mechanisms underlying the reduced macrophage contents in plaques (Fig. 1g–j), we established an in vitro assay to examine whether TRPM2 influences the infiltration ability of macrophages (Detailed in Extended Data Fig. 3a). Different from previous reported infiltration assays26, we plated aortic endothelial cells isolated from wild-type (WT) mice onto the upper surface of transwell inserts to better mimic the pathophysiological conditions during atherosclerosis. We found that MCP1 induced a significant increase of macrophage infiltration into the lower chamber in WT group, but this increase was inhibited in M2KO group (Fig. 2l, m). Moreover, MIF-induced macrophage infiltration was also inhibited by Trpm2 deletion (Extended Data Fig. 3c, d). These results indicate that Trpm2 deletion in macrophages reduces their infiltration ability.
Macrophage emigration refers to the returning of macrophages from atherosclerotic lesion sites back into circulation, which is important for atherosclerosis plaque regression2,27. However, uptake of oxLDL markedly impairs the migration ability of macrophages, resulting in macrophages being trapped in atherosclerotic areas thereby contributing to sustained inflammatory responses in the vessel walls26. We designed an in vitro assay to examine the emigration ability of macrophages (Detailed in Extended Data Fig.3b). We found that preloading with oxLDL dramatically inhibited the migration of WT macrophages, whereas this inhibition was not observed in M2KO macrophages (Fig. 2n, o). We also found that the non-directed migration ability of macrophages evaluated by scratch assay was also significantly inhibited by Trpm2 deletion (Fig. 2p, q). Taken together, the above data suggest that Trpm2 deletion inhibits macrophage migration, and alleviates impaired emigration caused by oxLDL overload.
Macrophage-specific Trpm2 deletion inhibits atherogenesis
To understand the mechanisms by which Trpm2 deletion protects mice against atherosclerosis as shown in Fig. 1, we generated cell type specific deletion of Trpm2 by breeding Apoe−/−Trpm2fl/fl mice28 with Cd11b-cre29 mice. Cd11b-cre induces gene deletion predominantly in macrophages and granulocytes, which provides a powerful tool to investigate the function of a specific gene in macrophages29,30. Macrophage deletion of Trpm2 in Apoe−/− mice was confirmed by Western blot and TRPM2 current recording (Extended Data Fig.1g–i). Littermates of Cd11b-cre- mice were used as control for Trpm2fl/fl Cd11b-cre+ (or Cd11b-cre+ Trpm2 KO) mice. HFD treatment induced significant atherosclerotic lesions in aortas and aorta roots in Cd11b-cre- mice, which was inhibited in Cd11b-cre+ Trpm2 deletion mice (Fig. 3a–d). Similar to global Trpm2−/−, Cd11b-cre+ Trpm2 deletion did not influence serum cholesterol level (Fig. 3e). The macrophage contents evaluated by Mac-1 staining of aorta root sections and F4/40 and CD80 double staining of aorta sections was significantly reduced by Cd11b-cre+ Trpm2 deletion (Fig. 3 f–i), suggesting that Trpm2 deletion in macrophages inhibit their infiltration ability. The peripheral leukocyte populations were not influenced by Cd11b-cre+ Trpm2 deletion (Fig. 3j, k).
Fig. 3: Trpm2 deletion in macrophages protects mice against atherosclerosis.

(a,b) Trpm2 deletion in Cd11b expressing cells (Trpm2fl/flCd11b-cre+) inhibited atherosclerotic plaque formation. a, Representative images of Oil Red O (ORO) staining of full-length aorta (red areas represents plaque). b, Mean atherosclerotic lesion ratio based on ORO staining from Trpm2fl/flCd11b-cre− (n=5) and Trpm2fl/flCd11b-cre+ mice (n=8), P < 0.0009. (c, d) Representative images and quantification of ORO staining of the aortic root sections from Trpm2fl/flCd11b-cre− (n=9) and Trpm2fl/flCd11b-cre+ mice (n=10), P = 0.0016. (e) Trpm2fl/flCd11b-cre+ did not influence the total cholesterol level in serum (n=5/group) (HFD: high-fat diet). (f-i) Trpm2fl/flCd11b-cre+ reduced macrophage burden in atherosclerotic plaque. f, g Representative merged images (Blue:DAPI; Green: Mac-1) and quantification of Mac-1 staining of aortic root sections from Trpm2fl/flCd11b-cre− (n=8) and Trpm2fl/flCd11b-cre+ mice (n=7), P = 0.0071. (h, i) Representative merged images and quantification of F4/80 and CD80 staining of aorta cross-sections using the plaques areas as shown in Extended Data Fig. 1f (Red: F4/80; Blue: DAPI; Green: CD80) from Trpm2fl/flCd11b-cre− (n=9) and Trpm2fl/flCd11b-cre+ mice (n=10), P = 0.0005. (j, k) Trpm2fl/flCd11b-cre+ did not influence the leukocyte population in the peripheral blood. j, Representative monocyte population identified using flow cytometry. Ly6C+ monocyte population was identified from CD11b+ and Ly6G− leukocytes. k, Quantification of monocyte, neutrophil, B cell, T cell, NK cell and NKT cell population in the peripheral blood from Trpm2fl/flCd11b-cre− (n=3) and Trpm2fl/flCd11b-cre+ mice (n=4) with or without HFD treatment. (l, o) Representative WB analysis and quantification of the CD80 expression in aorta (n= 6 mice/group). (m, p) Representative WB analysis and quantification of the expression of MCP1 and MIF in aorta (n= 6 mice/group). (n, q) Representative WB analysis and quantification of iNOS expression in aortas (n= 6 mice/group). (r, s) Representative WB analysis and quantification of the expression of NLRP3, ASC, cleaved caspase-1 (cCas1), and cleaved IL-1β (cIL-1β) expression in aortas (n= 6 mice/group). (ns: no statistical significance; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Consistent with the increased macrophage contents in the atherosclerotic plaques, the expression of macrophage marker CD80 was much higher in Cd11b-cre- mice compared to that in Cd11b-cre+ mice (Fig. 3l, o). During atherosclerosis, macrophages are the predominant cell type in producing inflammatory cytokines such as MCP1, MIF, and Il-1β21,31,32. Cd11b-cre+ Trpm2 deletion markedly reduced MCP1, MIF, iNOS and Il-1β production (Fig. 3m–q), and significantly inhibited inflammasome activation (Fig. 3r, s). These results indicate that Cd11b-cre+ Trpm2 deletion produced similar anti-atherosclerosis effects as those of global Trpm2 deletion (Fig. 1; Fig. 2), underscoring the essential role of TRPM2 in macrophages in promoting atherosclerosis.
Trpm2 deletion inhibits foam cell formation
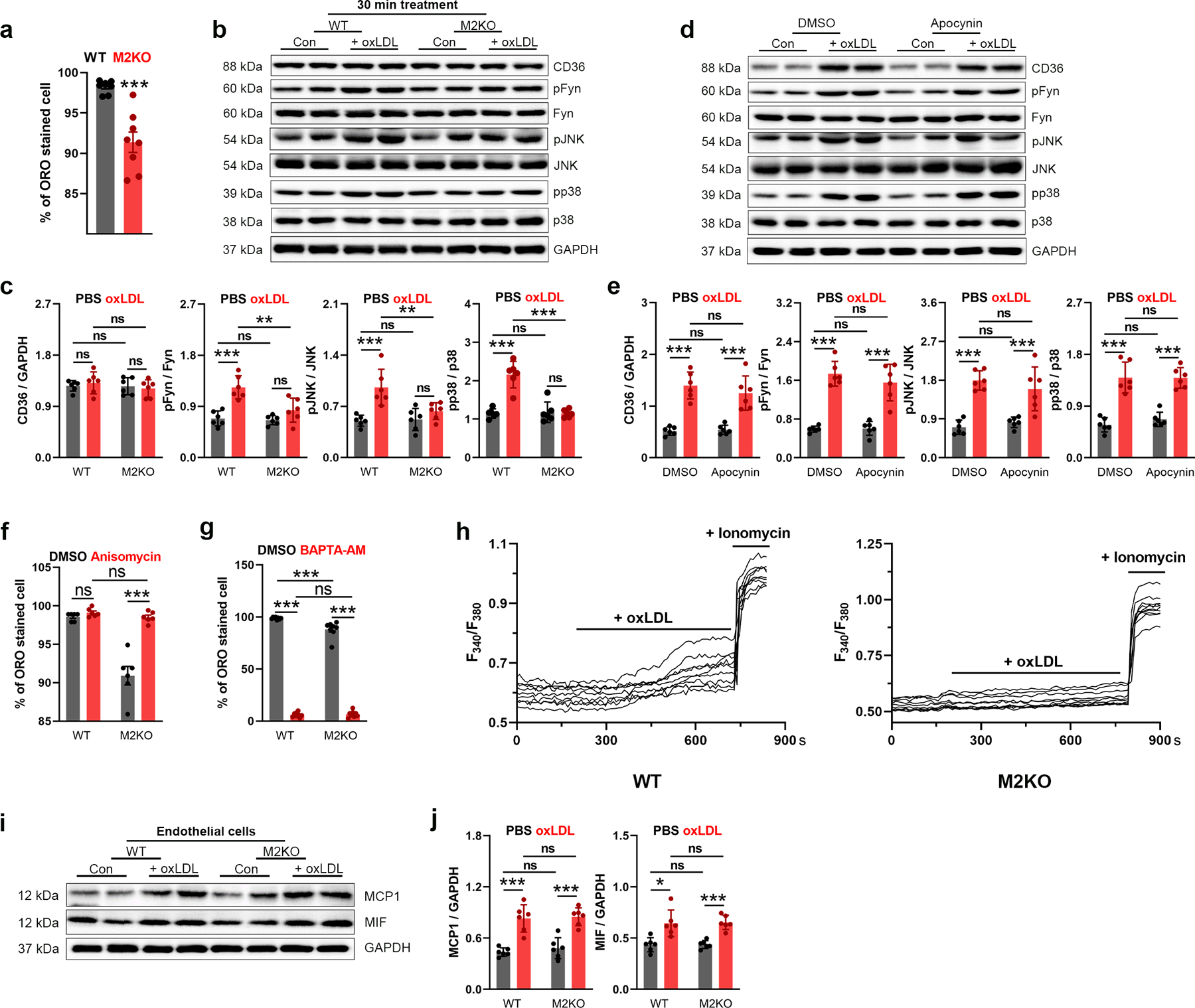
Over-uptake of oxLDL transforms macrophages into highly pro-inflammatory foam cells, which produces inflammatory cytokines thereby promoting atherosclerosis progression2. We found that deletion of Trpm2 markedly inhibited oxLDL uptake in macrophages, as shown by ORO staining (Fig. 4a, b; Extended Data Fig. 4a). The reduced oxLDL uptake in Trpm2 KO macrophages may explain the improved macrophage emigration in vitro (Fig. 2n, o) as well as reduced macrophage burden in vivo (Fig. 1 g–j; Fig. 3f–i).
Fig. 4: Deletion of Trpm2 inhibits foam cell formation.

(a, b) Representative images and quantification of ORO staining in macrophages from wild-type (WT) or Trpm2 knockout (M2KO) mice after 24h oxLDL treatment (n=6/group). (c, d) Representative WB analysis (WB) of CD36, pFyn, pJNK and pp38 expression in macrophages (n=6/group). (e, f) Representative images and quantification of ORO staining of macrophages (n=8/group) treated with and without anisomycin (10 μM), a p38 and pJNK activator. (g-h) Representative WB of CD36, pFyn, pJNK and pp38 expression in macrophages. BAPTA-AM (1 μM), an intracellular Ca2+ chelator. (i, j) Representative images and quantification of ORO staining of macrophages treated with BAPTA-AM (n=3/group). (k, l) k, Representative images of Rhodamine-123 (R123) real-time imaging before and 5 min after oxLDL treatment in macrophages. Control group (PBS treatment) was used to show the rapid photo bleaching of R123. l, Quantification of R123 fluorescence changes 5 min after oxLDL treatment (n=30,40,40,43, respectively). (m, n) Quantification and representative WB of iNOS expression in macrophages (n=6/group). (o, p) o, Representative real-time ratio Ca2+ imaging traces during oxLDL treatment. The averaged traces were from 10 macrophages randomly chosen from a representative culture dish for each group. p, Quantification of Fura-2 fluorescence changes 5 min after oxLDL treatment (n=20/group). (q, r) q, Representative images of R123 imaging before and 5 min after oxLDL treatment in macrophages with DMSO or BAPTA-AM (1 μM) preloading for 30 min. r, Quantification of R123 fluorescence changes 5 min after oxLDL treatment (n=20/group). (s) Measurement of IL-1β level in culture medium of isolated macrophages after oxLDL treatment using ELISA (n=4/group). (t, v) Representative WB of MCP1 and MIF expression in macrophages (n=6/group). (u, w) Representative WB of pp65 expression in macrophages (n=6/group). (oxLDL (50 μg/ml) was treated for 24 h for all the ORO staining and WB analysis; ns: no statistical significance; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
CD36 is the major receptor mediating oxLDL uptake in macrophages5, and activation of signaling cascades downstream of CD36, such as Fyn, JNK, and p38, has been shown to promote macrophage activation, and inhibit the emigration of macrophages from atherosclerotic plaques5,26. We found that Trpm2 deletion did not influence the basal expression of CD36, but largely abolished the oxLDL induced up-regulation of CD36 (Fig. 4c, d). Also, Trpm2 deletion drastically inhibited oxLDL-mediated phosphorylation of Fyn, JNK and p38. To understand whether this reduced phosphorylation results from the reduced CD36 expression in Trpm2 KO macrophages, we treated macrophages with oxLDL for a shorter time (30 min) so that the CD36 was activated but CD36 expression was not upregulated (Extended Data Fig.4b, c). We found that activation of CD36 downstream signaling by a 30-min oxLDL treatment in WT macrophages was almost completely abolished by Trpm2 KO. These data indicate that TRPM2 is critical for CD36 signaling activation in macrophages elicited by oxLDL.
Activation of p38 MAP kinase and JNK2 are required for foam cell formation33,34, and CD36-mediated JNK2 activation is required for oxLDL uptake by macrophages35. Anisomycin is a potent activator for both JNK2 and p38. As shown in Fig. 4e, anisomycin significantly increased lipid uptake by 29.85% in WT macrophages (Fig. 4f; Extended Data Fig.4f), and this increase is more pronounced in M2KO macrophages (299.28%, Fig. 4f; Extended Data Fig.4f), suggesting that Trpm2 deletion reduces oxLDL uptake by inhibiting CD36 signaling activation. To understand the mechanisms by which TRPM2 regulates CD36 activation, we applied BAPTA-AM to chelate intracellular Ca2+ including the TRPM2-mediated Ca2+ influx, and found that oxLDL-induced CD36 activation was abolished (Fig. 4g, h). This result suggests that TRPM2-mediated Ca2+ influx may play an important role in regulating CD36-mediated oxLDL uptake. Indeed, BAPTA-AM also eliminated the oxLDL uptake in both WT and Trpm2 KO macrophages (Fig. 4i, j; Extended Data Fig. 4g).
Reactive oxygen species (ROS) production is a hallmark of macrophage activation during atherosclerosis22,36. Rhodamine-123 (R123) imaging is a commonly used method for monitoring mitochondrial oxidative stress and ROS production37,38. We found that oxLDL induced a marked and rapid increase of R123 signal in WT macrophages within 5 min of oxLDL exposure, but this increase was largely inhibited by Trpm2 deletion (Fig. 4k, l). Besides mitochondria, ROS can be produced by NADPH oxidase in the cell, which cannot be detected by measuring R123 signal. However, we found that NADPH oxidase inhibitor apocynin did not directly influence CD36 activation in response to oxLDL treatment (Extended Data Fig. 4d, e). To further understand how TRPM2 influences ROS production, we evaluated the expression of iNOS, a known driver for ROS production in macrophages22. As shown in Fig. 4m, n, Trpm2 deletion significantly inhibited the increase of iNOS expression in macrophages treated by oxLDL.
Increased intracellular Ca2+ is crucial for macrophage activation12,13,22,36,39. We found that oxLDL induced a robust increase of intracellular Ca2+ in WT macrophages, but this increase was significantly inhibited in M2KO macrophages (Fig. 4o, p; Extended Data Fig. 4h), suggesting oxLDL-induced intracellular Ca2+ changes depends on TRPM2-mediated Ca2+ influx. Moreover, increase of intracellular Ca2+ is required for oxLDL-induced increase of R123 signal, as BAPTA-AM abolished R123 signal changes induced by oxLDL (Fig. 4q, r). NLRP3 inflammasome activation induced by oxLDL uptake is critical for macrophage activation during atherogenesis6. We found that Trpm2 deletion inhibited IL-1β secretion induced by oxLDL, suggesting that activation of NLRP3 inflammasome was suppressed by Trpm2 deletion (Fig. 4s). These results suggest that Trpm2 deletion inhibits oxLDL-mediated macrophage activation.
In atherosclerotic plaque, MCP1 and MIF produced by foam cells lead to the recruitment of more macrophages into the lesion sites, thereby resulting in a positive feed-back vicious cycle accelerating atherosclerosis progression3. We found that MCP1 and MIF expression were significantly increased after oxLDL treatment in WT macrophages, whereas this increase was inhibited in M2KO macrophages treated with oxLDL (Fig. 4t, v). Interestingly, Trpm2 deletion did not change MCP1 and MIF production induced by oxLDL in endothelial cells (Extended Data Fig.4i, j), which is consistent with previous reports that macrophages are the predominant cells for MCP1 and MIF production21,31,32, and also supports our hypothesis that TRPM2 in macrophages plays the key role in promoting atherosclerosis. As expression of MCP140 and MIF32 are regulated by NFκB pathway, we examined whether TRPM2 influences NFκB signaling in macrophages exposed to oxLDL by detecting p65 phosphorylation. We found that WT macrophages exhibited significantly enhanced p65 phosphorylation after oxLDL treatment, which was largely inhibited by Trpm2 KO (Fig. 4u, w), suggesting that TRPM2 regulates oxLDL induced MCP1 and MIF production through activating NFκB pathway41.
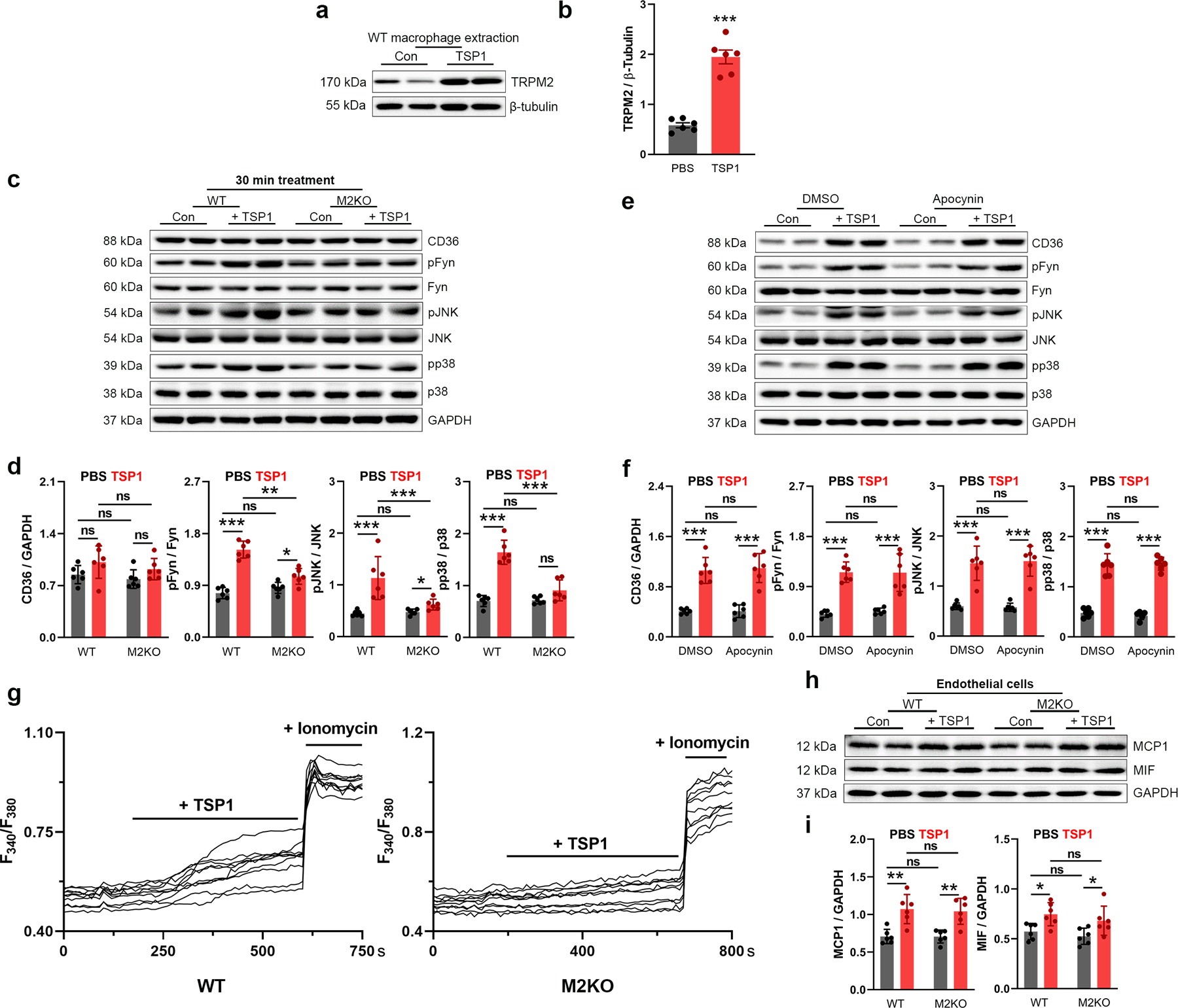
To exclude any potential non-specific effect caused by oxLDL and confirm the TRPM2-dependent activation of CD36, we used another CD36 ligand, thrombospondin-1 (TSP1). TSP1 is an extracellular glycoprotein secreted by macrophages and other types of cells, and is known to promote inflammation42,43. It was recently demonstrated that global deletion of TSP1 protects mice against leptin induced atherosclerosis by inhibiting the abnormal activation of smooth muscle cells44. However, whether TSP1 is involved in HFD induced atherogenesis and in macrophage activation in the plaques remains unknown. We found that similar to the up-regulated TRPM2 expression in macrophages induced by oxLDL (Extended Data Fig.2c, d), a 24-h TSP1 treatment significantly increased the expression of TRPM2 in macrophages (Extended Data Fig.5a, b). Moreover, TSP1 also elicited activation of CD36 signaling, whereas these changes were largely abolished in M2KO macrophages (Fig 5a, b; Extended Data Fig. 5c, d).
Fig. 5: Trpm2 deletion inhibits the activation of CD36 signaling by TSP1.

(a, b) Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in macrophages. (c, d) c, Representative R123 imaging before and 5 min after TSP1 treatment in macrophages. Control group (Con: PBS treatment) was used to show the rapid photo bleaching of R123. d, Quantification of changes of R123 fluorescence 5 min after TSP1 treatment. WT (n=45 for TSP1 treatment, n=47 for control) and M2KO (n=43 for TSP1 treatment, n=47 for control) macrophages were from 4 dishes of cultured macrophages isolated from 4 mice/group. (e, f) Representative WB of iNOS expression in isolated macrophages treated with TSP1 (10 μg/ml). (g), Representative real-time ratio Ca2+ imaging traces during TSP1 treatment. The averaged traces were from 10 macrophages randomly chosen from a representative culture dish of each group. (h), Quantification of Fura-2 fluorescence changes 5 min after TSP1 treatment. WT (n=20 for TSP1 treatment, n=20 for control) and M2KO (n=20 for TSP1 treatment, n=20 for control) macrophages were from 3 dishes of cultured cells isolated from 3 mice in each group. (i, j) Representative WB of CD36, pFyn, pJNK and pp38 expression in macrophages after treatment with TSP1 and DMSO or BAPTA-AM (1 μM), an intracellular Ca2+ chelator (n=3/group). (k, l) k, Representative images of R123 imaging in macrophages before and 5 min after oxLDL treatment with DMSO or BAPTA-AM (1 μM) preloading for 30 min. l, Quantification of R123 fluorescence changes 5 min after TSP1 treatment (n=20/group). (m) Measurement of IL-1β level in culture medium after TSP1 treatment using ELISA. (n, o) Representative WB of MCP1 and MIF expression in macrophages (n=6/group). (p, q) Representative WB analysis and quantification of pp65 expression in macrophages (n=6/group). (TSP1 (10 μg/ml) was treated for 24 h for all the WB analysis; ns: no statistical significance; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Similar to oxLDL, TSP1 treatment induced an increase of R123 fluorescence in WT macrophages, but this increase was markedly inhibited in M2KO macrophages (Fig. 5c, d), suggesting that ROS production induced by TSP1 is inhibited by Trpm2 deletion. Also, NADPH oxidase inhibitor apocynin did not influence CD36 activation in response to TSP1 (Extended Data Fig. 5e, f). Moreover, the increased expression of iNOS induced by TSP1 was inhibited in M2KO macrophages (Fig. 5e, f). Interestingly, TSP1 at a lower concentration induced a larger increase of intracellular Ca2+ concentration than that induced by oxLDL (TSP1 at 10 μg/ml vs oxLDL at 50 μg/ml) in macrophages from WT group, whereas Trpm2 deletion inhibited this increase (Fig. 5g, h; Extended Data Fig.5g). Importantly, intracellular Ca2+ chelator BAPTA-AM abolished TSP1 induced activation of CD36 signaling (Fig. 5i, j) and increase of ROS production (Fig. 5k, l). These results indicate that TSP1-induced macrophage activation is inhibited by Trpm2 deletion.
Next, we sought to determine the effects of TRPM2 on TSP1-induced cytokine production. As shown in Fig. 5m–o, TSP1 induced an increase in Il-1β, MCP1, and MIF expression in WT macrophages, which was markedly inhibited in M2KO macrophages. However, Trpm2 deletion did not influence the increased expression of MCP1 and MIF in endothelial cells treated with TSP1 (Extended Data Fig. 5h, i). Also, TSP1 increased p65 phosphorylation in WT macrophages, and Trpm2 KO significantly inhibited this increase (Fig. 5p, q), suggesting that TRPM2 contributes to TSP1 induced MCP1 and MIF expression through activating NFκB pathway. As the expression of TSP1 was shown to be significantly increased in atherosclerotic aortas45, our data suggest that TSP1 induced activation of CD36 signaling in macrophages may play an important role in macrophage activation during atherogenesis, and that the in vivo protective effects of TRPM2 deletion may partially be mediated by inhibiting TSP1 induced CD36 activation.
In summary, our results suggest a hypothetical working model: the mutual activating relationship of TRPM2 and CD36 forms a positive feedback loop to increase oxLDL uptake and promote foam cells formation, and enhance the production of inflammatory cytokines (MCP1 and MIF). The increased cytokine production (Fig. 2e & Fig. 3m; Fig. 4t & Fig. 5n) further attracts macrophage infiltration to atherosclerotic areas, forming a vicious cycle perpetuating atherosclerosis progression.
TRPM2 is required for CD36 signaling activation
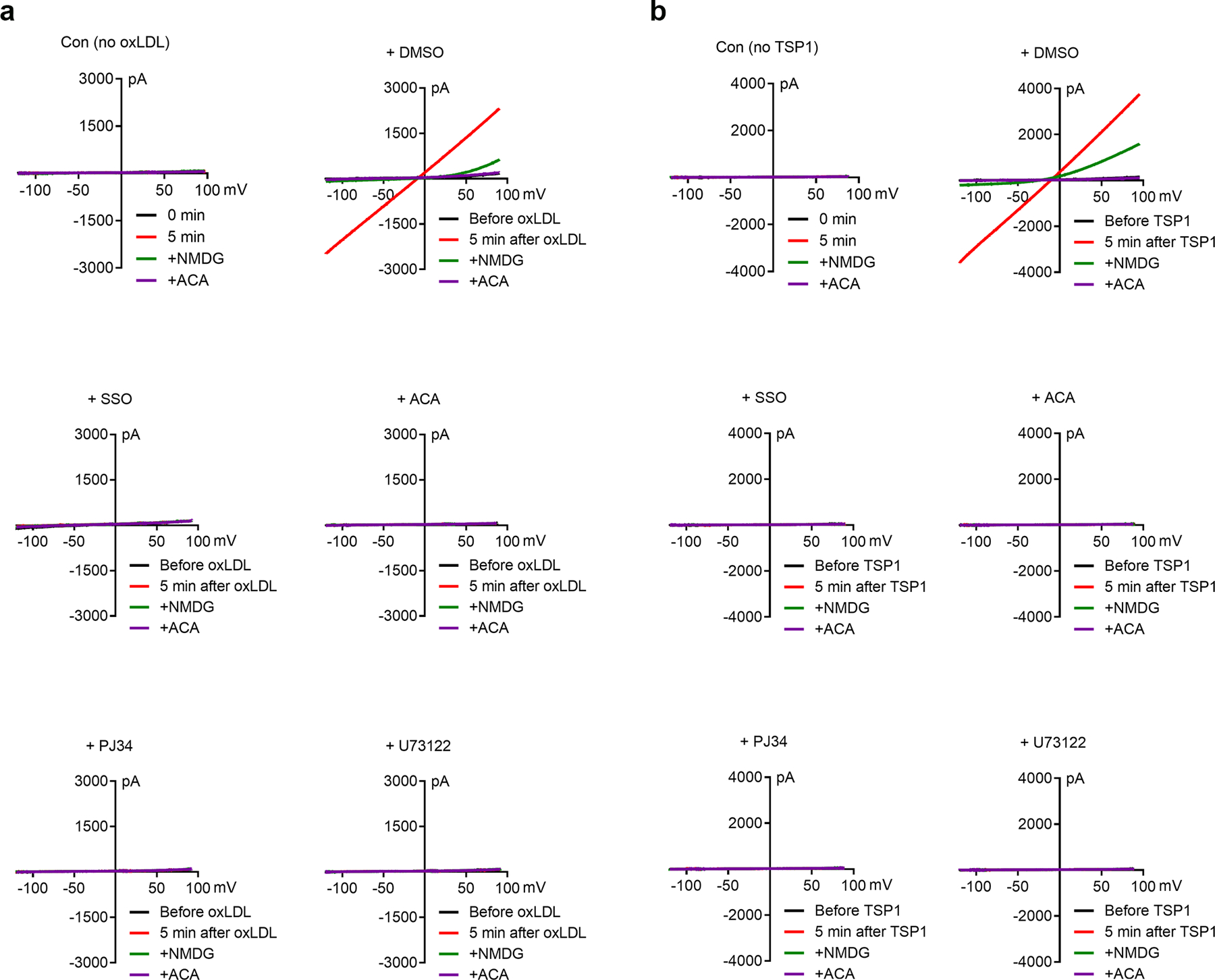
The requirement of TRPM2 for CD36 activation by oxLDL and TSP1 is an unexpected discovery. To understand the underlying mechanisms, we first determined whether TRPM2 is indeed activated during oxLDL and TSP1 treatment. We found that oxLDL substantially activated TRPM2 currents in HEK293T cells co-transfected with both TRPM2 and CD36 in 5 min (Fig. 6a, upper). In contrast, there was no TRPM2 current activation in cells transfected with TRPM2 only, even after perfusion with oxLDL for 10 min (Fig. 6a, lower). The currents activated by oxLDL display typical TRPM2 characteristics such as linear I-V relation (Extended Data Fig.6a) and can be blocked by 30 μM N-(p-amylcinnamoyl) anthranilic acid (ACA), a TRPM2 blocker. Moreover, preincubation with sulfosuccinimidyl oleate (SSO), a CD36 specific inhibitor, effectively abolished the activation of TRPM2 by oxLDL (2156.00 ± 342.00 pA vs 50.02 ± 10.65 pA) (Fig. 6b, Extended Data Fig. 6a). To further confirm that oxLDL-elicited TRPM2 activation was mediated by influencing intracellular signaling pathways, we performed perforated-patch for TRPM2 current recording so that the intracellular content is not disturbed by pipette solution. We found that oxLDL induced a smaller but similar TRPM2 activation (Fig. 6c) as that obtained by regular whole-cell current recording in Fig. 6a.
Fig. 6: TRPM2 mediates the activation of CD36 signaling.

(a, b) Whole-cell recording of TRPM2 currents in response to oxLDL (50 μg/ml). a, Representative TRPM2 current traces (Green: outward current at +100 mV; Purple: inward current at −100 mV) in HEK293T cells transfected with both CD36 and TRPM2 (upper) during oxLDL treatment. N-Methyl-D-glucamine (NMDG) blocks inward current indicating the tightness of seal. ACA is a TRPM2 blocker. Representative recording traces in HEK293T cells transfected with only TRPM2 (lower) during oxLDL treatment. b, Quantification of TRPM2 current amplitude (n=5,9,6,6,6,6, respectively). (c) TRPM2 current recorded under perforated patch in HEK293T cells transfected with both CD36 and TRPM2 (upper) or transfected with only TRPM2 (lower) during oxLDL treatment. (d, e) Inhibiting TRPM2 activation impairs the activation of CD36 signaling cascade induced by oxLDL (50 μg/ml) in macrophages. Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in isolated macrophages from WT (n=3 in each group) and M2KO mice (n=3 in each group). e, Quantification of WB bands. 3 dishes of macrophages were used for protein extraction in each group. (f, g) Whole-cell recording of TRPM2 current in response to TSP1 (10 μg/ml) (n=5,6,6,6,6,6, respectively). f, Representative TRPM2 current traces (Green: outward current at +100 mV; Purple: inward current at -+100 mV) in HEK293T cells transfected with both CD36 and TRPM2 (upper) during TSP1 treatment. Representative recording traces in HEK293T cells transfected with only TRPM2 (lower) during TSP1 treatment. g, Quantification of TRPM2 current amplitude. (h) TRPM2 current recorded under perforated patch in HEK293T cells transfected with both CD36 and TRPM2 (upper) or transfected with only TRPM2 (lower) during TSP1 treatment. (i, j) Inhibiting TRPM2 activation impairs the activation of CD36 signaling cascade induced by TSP1 (10 μg/ml) in macrophages. Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in isolated macrophages from WT (n=3 in each group) and eM2KO mice (n=3/group). (*: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Since the activation of TRPM2 needs intracellular Ca2+ and ADP ribose (ADPR)46, we reasoned that oxLDL may influence intracellular Ca2+ or ADPR thereby activating TRPM2. We found that PJ34, a specific inhibitor for poly ADP-ribose polymerase to inhibit ADPR production, effectively abolished the activation of TRPM2 by oxLDL. Similarly, U73122, a potent PLC inhibitor to suppress Ca2+ release from endoplasmic reticulum, also eliminated the activation of TRPM2 by oxLDL (Fig. 6b, Extended Data Fig. 6a). These results indicate that oxLDL activates TRPM2 by increasing intracellular ADPR and Ca2+ levels. To exclude the non-specific effect caused by oxLDL, we applied CD36 inhibitor SSO, and found that the activation of TRPM2 induced by oxLDL was abolished (Fig. 6c). This rapid activation of TRPM2 induced by CD36 (by ligand binding) also explains the observed oxLDL-induced Ca2+ entry in macrophages (Fig. 4o).
To understand how TRPM2-mediated Ca2+ influences CD36 activation, we inhibited TRPM2 activation using ACA, PJ34, and U73122, and evaluated activation of CD36 signaling cascade in WT and TRPM2-KO macrophages (Fig. 6d). In WT macrophages, inhibition of TRPM2 by ACA, PJ34 and U72122 effectively inhibited CD36 activation (pFyn, pJNK and pp38) induced by oxLDL (Fig. 6d, e). As a Ca2+-permeable non-selective cation channel47, TRPM2-mediated Ca2+ signaling has been found to be important for various cellular functions47. Given that Trpm2 deletion or inhibiting TRPM2 activation produced similar inhibitory effects on CD36 signaling as those produced by chelating internal Ca2+ with BAPTA-AM (Fig. 4g, h), our results suggest that TRPM2-mediated Ca2+ signaling is essential for oxLDL activated CD36 signaling cascades in macrophages.
We further evaluated CD36-dependent TRPM2 activation by TSP1. Interestingly, compared to oxLDL, TSP1 treatment induced a more robust activation (4117.00 ± 454.90 pA by TSP1 versus 2156.00 ± 342.00 pA by oxLDL) of TRPM2 currents in HEK293T cells transfected with both TRPM2 and CD36 (Fig. 6f, upper), but not in cells transfected with TRPM2 alone (Fig. 6f, lower). This observation was further supported by the perforated-patch results showing TRPM2 was activated by TSP1 in TRPM2 and CD36 co-expressed HEK293 cells, but not in TRPM2 single-transfected cells (Fig. 6h). Moreover, CD36 inhibitor SSO completely inhibited the activation of TRPM2 by TSP1 (Fig. 6g, Extended Data Fig. 6b), suggesting CD36-dependent activation of TRPM2 induced by TSP1. Similar to oxLDL induced activation of TRPM2, TRPM2 activation by TSP1 disappeared when transfected cells were treated with ACA, PJ34 and U73122 (Fig. 6g, Extended Data Fig. 6b). Together with CD36-dependent activation of TRPM2 by oxLDL (Fig. 6a–c), these results indicate that ligand engagement to CD36 induces TRPM2 activation by increasing intracellular Ca2+ and ADPR. Moreover, CD36 signaling activation in WT macrophages by TSP1 was inhibited by ACA, PJ34, and U73122 treatment (Fig. 6i, j), presumably through inhibiting TRPM2 activation. Indeed, SSO, ACA, PJ34 and U73122 treatments did not produce any additional effect on activating CD36 signaling cascades in TRPM2-KO macrophages subjected to oxLDL or TSP1 (Extended Data Fig. 7a–d), indicating these inhibitors did not affect CD36 activation in the absence of TRPM2. The above data suggest that during oxLDL and TSP1 treatment, CD36 signaling activates TRPM2 by increasing the production of ADPR and intracellular Ca2+ concentration, and TRPM2-mediated Ca2+ signaling is also critical for the activation of CD36 signaling cascades.
TRPM2 mediates macrophage activation by oxLDL or TSP1
We then sought to understand whether inhibiting TRPM2 influences the activation of macrophages by oxLDL or TSP1. As a negative control, inhibition of CD36 by preincubating macrophages with SSO significantly mitigated the increase of R123 signal in macrophages induced by either oxLDL or TSP1 treatment (Fig. 7a–d). Moreover, ACA, PJ34 and U73122 preincubation markedly inhibited the increase of R123 signal in macrophages induced by oxLDL or TSP1 treatment (Fig. 7a– d). Furthermore, TRPM2 inhibitor ACA and inhibiting the activation of TRPM2 by PJ34 or U73122 (with low Ca2+ extracellular medium) inhibited the expression iNOS in macrophages exposed to oxLDL and TSP1 treatment (Fig. 7e– g). Noticeably, SSO, ACA, PJ34 and U73122 treatments did not cause additional inhibition on the expression of iNOS in M2KO macrophages subjected to oxLDL or TSP1 (Extended Data Fig.7e–h). Similarly, SSO, ACA, PJ34 and U73122 preincubation markedly inhibited the increase of intracellular Ca2+ concentration in macrophages induced by either oxLDL (Fig. 7h, i) or TSP1 treatment (Fig. 7j, k). These data indicate that inhibiting TRPM2 activation suppressed production of ROS and elevation of intracellular Ca2+ in macrophages during oxLDL or TSP1 treatment. Since ROS and Ca2+ signaling are critical for the activation of pro-inflammatory pathways in macrophages12,36, we examined whether these inhibitors affect the activation of NLRP3 inflammasome by measuring the concentration of IL-1β in culture medium. Our data showed that by inhibiting the activation of TRPM2 using ACA, PJ34 and U73122, the secretion of IL-1β by macrophages induced by oxLDL or TSP1 was significantly inhibited (Fig. 7l, m). Given the crucial role of ROS and Ca2+ signaling in the activation of macrophages, the above results suggest that inhibition of TRPM2 activation significantly suppresses macrophage activation induced by oxLDL and TSP1.
Fig. 7: TRPM2 mediates macrophage activation induced by oxLDL or TSP1.

(a-d) Representative picture of Rhodamine-123 real-time imaging of macrophages before and 5 min after oxLDL treatment (50 μg/ml) as in a, and 5 min after TSP1 treatment (10 μg/ml) as in c in isolated macrophages. Quantification of changes of R123 fluorescence 5 min after oxLDL treatment as in b, and 5 min after TSP1 treatment as in d. For oxLDL treatment, WT (n=40 for PBS, n=38 for DMSO, n=35 for SSO, n=38 for ACA, n=39 for PJ34, n=44 for U73122) and M2KO (n=38 for PBS, n=35 for DMSO) macrophages were from 4 dishes of cultured cells isolated from 3 mice in each group. For TSP1 treatment, WT (n=48 for PBS, n=50 for DMSO, n=53 for SSO, n=51 for ACA, n=57 for PJ34, n=56 for U73122) and M2KO (n=48 for PBS, n=52 for DMSO) macrophages were from 4 dishes of cultured cells isolated from 3 mice in each group. (e-g) Representative WB analysis of iNOS expression in isolated macrophages. 3 dishes of cells from 3 mice from each group were chosen for quantification. (h-k) Representative real-time Fura-2 Ca2+ imaging traces during oxLDL (50 μg/ml) as in h, and during TSP1 treatment (10 μg/ml) as in j. The averaged traces were from 10 macrophages randomly chosen from a representative culture dish of each group. Quantification of fluorescence changes 5 min after oxLDL treatment as in i, and 5 min after TSP1 treatment as in k. For oxLDL treatment and TSP1 treatment, 20 macrophages in each group from 3 dishes isolated from 3 mice were chosen for quantification. (l, m) Measurement of IL-1β level in culture medium of isolated macrophages after the treatment of oxLDL (50 μg/ml) or TSP1 (10 μg/ml) for 24 h using ELISA. 3 dishes of cells from 3 mice from each group were chosen for quantification. (*: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM)
Inhibiting TRPM2 suppresses foam cell formation
After confirming that inhibiting TRPM2 activation could suppress macrophage activation, we examined whether these inhibitors affect the phenotypic changes of macrophages induced by oxLDL and TSP1. CD36 inhibitor SSO significantly inhibited the uptake of oxLDL in macrophages derived from bone marrow (Fig. 8a, b), and TRPM2 inhibitor ACA, as well as PJ34 and U73122 also suppressed oxLDL uptake in macrophages (Fig. 8a, b). Moreover, the increased expression of MCP1 and MIF induced by oxLDL or TSP1 was inhibited by ACA, PJ34 and U73122 (Fig. 8c–f) in WT macrophages, whereas these inhibitors did not produce any suppression of MCP1 and MIF expression in M2KO macrophages subjected to oxLDL or TSP1 treatment (Extended Data Fig.7i–l). Furthermore, preincubation with ACA, PJ34 and U73122 inhibited in vitro macrophage infiltration induced by MCP1 (Fig. 8g, h) and prevented the impairment of emigration ability caused by oxLDL preloading (Fig. 8i, j). These results recapitulate the reduced oxLDL uptake by macrophages, inhibited macrophage infiltration, and preserved macrophage emigration by deleting Trpm2 in vitro (Fig.2, Extended Data Fig. 3a, b).
Fig. 8: Inhibiting TRPM2 activation suppresses foam cell formation.

(a, b) Representative images and quantification of Oil Red O (ORO) staining of cultured macrophages after the treatment with oxLDL (50 μg/ml) for 24 h. 3 dishes of cells from 3 mice from each group were chosen for quantification. (c-f) Representative WB analysis and quantification of the expression of MCP1 and MIF in cultured macrophages treated with oxLDL (50 μg/ml) or TSP1 (10 μg/ml) for 24 h (n=3/group). (g, h) Inhibiting the activation of TRPM2 suppressed macrophage infiltration. in vitro macrophage infiltration test was performed as graphic illustration in Extended Data Fig. 3a (Red: F4/80; Blue: DAPI; Green: CD80). h, Quantification of the number of infiltrated macrophages within a x 10 field (n=6/group). (i, j) Inhibiting TRPM2 activation prevented the loss of emigration ability in oxLDL-pre-loaded macrophages. in vitro macrophage emigration test was performed as graphic illustration in Extended Data Fig. 3b. Macrophage emigration across endothelial cells induced by MCP1. Aorta-derived endothelial cells were plated on the transwell inserts (pore size: 12 μm) for 2–3 days. Macrophages preloaded with oxLDL for 24 h were added into the upper chamber after endothelial cells completely covered the upper surface of transwells. After 24 h, F4/80 and CD80 staining of macrophages in lower chamber was performed as in i (Red: F4/80; Blue: DAPI; Green: CD80). j, Quantification of the number of infiltrated macrophages within a x 10 field (n=6/group). (ns: no statistical significance; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Discussion
Atherosclerosis is a chronic inflammatory disease with the central pathological features of macrophage infiltration and foam cell formation1,2. Mitigating atherosclerosis is essential for minimizing its complications such as myocardial infarction and ischemic stroke, which are the leading causes of mortality and morbidity1. Current available therapies which controls dyslipidermia have proven effective only to some extents1, mainly due to the poor patient compliance to lifelong lifestyle modification48. Therapeutic strategies which directly target the culprit, macrophages, in the pathogenesis of atherosclerosis have been lacking due to the incomplete understanding of atherogenic mechanisms. In this study, we revealed that TRPM2 plays a key role in promoting the activation of macrophages during atherogenesis, and established that targeting TRPM2 in macrophages could be a promising therapeutic strategy for atherosclerosis.
With the unique feature of being activated by oxidative stress during inflammation, TRPM2 has been implicated in several pathological conditions including Alzheimer’s disease, ischemic stroke, inflammatory bowel disease, and inflammatory lung injury20. However, whether TRPM2 is involved in atherogenesis was unknown. Here, we demonstrate that both Trpm2 deletion globally and Trpm2 deletion in macrophages markedly attenuates atherosclerosis in Apoe−/− mice fed with HFD. Moreover, we discovered an unknown link between TRPM2 and atherosclerosis initiators, the CD36 ligands oxLDL and TSP1. We found that inhibiting TRPM2 markedly suppressed the pro-inflammatory activation of macrophages induced by oxLDL and TSP1, both of which have been implicated as potent atherogenic activators44,45,49.
One of the intriguing discoveries in this study is the TRPM2 activation mediated by CD36 ligands oxLDL and TSP1, which can be blocked by CD36 inhibitor SSO. The activation of TRPM2 by TSP1 may provide new mechanistic insights about the important role of TSP1 in atherosclerosis shown in a recent study44. By utilizing R123 real-time cell imaging, we demonstrated previously unknown phenomenon that oxLDL and TSP1 induced a rapid increase of ROS production and mitochondrial oxidative stress in cultured macrophages. The increased ROS and oxidative stress promote ADPR production. As CD36 activation also leads to PLCγ activation which enhances intracellular Ca2+ 50, the enhanced ADPR and Ca2+ will further activate TRPM2, resulting in a mutually activating feedback loop between TRPM2 and CD36, which perpetuates the inflammatory response in atherosclerosis.
The TRPM2-CD36 axis constitutes a strong self-promoting mechanism in the initiation and progression of atherosclerosis. We observed a marked TRPM2-mediated intracellular Ca2+ increase in macrophages induced by TSP1 and oxLDL. Inhibiting this Ca2+ signaling by deleting or inhibiting TRPM2 reduces production of ROS, MCP1 and MIF, inhibits oxLDL uptake and suppresses macrophage migration. Moreover, we reveal that TRPM2-CD36 mediated Ca2+ signaling also promotes NLRP3 inflammasome activation. Consistent with our results, previous studies have implicated that Ca2+ is involved in NLRP3 induced inflammasome activation in cultured macrophages24,51. Thus, the TRPM2-CD36 activation loop triggers multiple factors and signaling pathways in promoting atherosclerosis. Controlling this atherogenic TRPM2-CD36 axis by targeting TRPM2 provides a promising anti-atherosclerosis strategy.
One major limitation of our study is lack of flow cytometry analysis of immune cell populations inside the aorta, which is generally considered as the golden standard for quantifying immune cell infiltration in atherosclerotic aortas52. Another limitation is our heavy reliance on in vitro assays for examining the migration ability of macrophages. In the future, fate mapping experiments53 will be needed to further confirm our observation in in vitro experiments that Trpm2 deletion inhibits the migration of macrophages while preserves their emigration ability.
In conclusion (Extended Data Fig. 8), we found that at the animal level, Trpm2 deletion globally or specifically in macrophages protected Apoe−/− mice against HFD induced atherosclerosis, which was characterized by reduced plaque burden in the aorta. At the tissue level, Trpm2 deletion resulted in decreased macrophage burden and suppressed inflammasome activation. At the cellular level, deletion of Trpm2 or inhibiting TRPM2 activation in macrophages suppressed macrophage infiltration, decreased oxLDL uptake, thereby improving the impaired macrophage emigration. At the molecular level, oxLDL and TSP1 activated TRPM2 through CD36 activation, and TRPM2 was required for the activation of CD36 signaling cascades in macrophages induced by oxLDL or TSP1. Taken together, our studies reveal an important mechanism for understanding the development and progression of atherosclerosis, and establish a therapeutic strategy that targets TRPM2, a key player in atherogenesis, for attenuating atherosclerosis.
Methods
Animals Care
All the experimental mice bred and hosted in the animal facility building of University of Connecticut School (UCONN Health) were fed with standard chow diet or high-fat diet (HFD) (Harlan, TD.88137), and water ad libitum. Standard housing conditions were maintained at a controlled temperature (18–23°C) and humidity (40–60%) with a 12-h light/dark cycle. Male mice for atherosclerosis study were fed with HFD for 16 weeks. All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Connecticut School of Medicine (animal protocol: AP-200135–0723), and were conducted in accordance with the U.S. National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Knockout of TRPM2 (TRPM2-KO)
The global TRPM2 knockout (Trpm2−/−) mice were generated by Dr. Yasuo Mori’s lab at Kyoto University, Japan. The deletion of Trpm2 was developed in C57B6J mice by replacing the third exon (S5–S6 linker in the pore domain) with a neomycin coding region. The knockout mice exhibited no differences in behavior or impairment in breeding, compared to wild type (WT) C57BL/6 mice. Trpm2−/− mice were back-crossed to C57BL/6 mice for ≥10 generations before being used for experiments. Trpm2−/− mice were crossed with Apoe−/− mice (JAX laboratory, 002052) to generate Trpm2−/− and Apoe−/− mice. Knockout was confirmed by genotyping. The mice were backcrossed with C57BL/6 mice for ≥10 generations before being used for experiments.
Trpm2fl/fl mice were generated by Dr. Barbara Miller (Penn State University, Pennsylvania). The exons 21 and 22 encoding transmembrane domain 5 and 6 and pore loop were flanked by loxp recombination sites and will be deleted by Cre recombinase 54. Trpm2fl/fl mice were first bred with Apoe−/− mice (JAX laboratory, 002052) to generate Trpm2fl/fl and Apoe−/− mice, and then bred with Cd11b-Cre mice generated by Dr. Vacher’s lab at Institut de Recherches Cliniques de Montreal, Canada29. All mice were on the C57BL/6 background for more than 10 generations before breeding. Specific deletion of Trpm2 in Cd11b expressing cells was achieved by crossing Trpm2fl/fl mice with Cd11b-cre mice with C57BL/6 background. Knockout was confirmed by genotyping.
Male mice (8~12 weeks) were used for all the experiments.
Flow cytometry of leukocytes population in the peripheral blood
Peripheral blood (~ 200 μL) was harvested from mouse tail based on approved animal protocol, and incubated with 1 mL of red blood cell (RBC) lysis buffer at room temperature for 15 min. Antibody cocktail was prepared by mixing CD11b (eFluor 450), NK1.1 (PE-Cyanine7), Ly-6C (Alexa Fluor® 700), Ly-6G (FITC), CD3ε (PE) and anti-mouse/human CD45R/B220 (PerCP/Cyanine5.5) in washing buffer at a dilution at 1:100. 100 μL antibody cocktail was used for incubation at room temperature for 15 min. Flow cytometry was performed using ZE5 Cell Analyzer (BIO-RAD). Collected data were analyzed using FlowJo (9.9.6). Firstly, T cell, NK cell and NKT cell populations were identified using the T cell marker CD3ε and NK cell marker NK1.1. Secondly, B cell population was identified using the B cell maker B220 from the non-T/NK/NKT population. Then neutrophil population was identified using the neutrophil maker Ly6G from the non-B/T/NK/NKT population. Lastly, monocyte population was identified using CD11b and Ly6C from the non-neutrophil/B/T/NK/NKT population.
Measurement of total cholesterol in serum
Cholesterol E kit was ordered from Fujifilm (999–02601). 10 μL of serum was added into each well followed by 250 μL of working solution. After incubation at 37 °C for 5 min, absorbance was measured at 600 nm. The concentration of total cholesterol was determined by comparing the absorbance of sample to the absorbance of standard solution.
Oil Red O (ORO) staining
Oil Red O (Sigma-Aldrich, O0625) was dissolved to a 0.5% stock solution. 30 min prior to use, ORO stock solution was diluted with water at a 6 : 4 ratio, and filtered to make the working solution.
For en-face analysis of atherosclerotic areas, mice were euthanized based on our animal protocol, and the full-length aorta was carefully dissected out. Aortas were washed, and fixed in 10% formaldehyde for 30 min at room temperature. Then aortas were washed, and stained by ORO working solution for 10 min at room temperature. After washing 3 times using PBS, the aorta was ready for imaging.
For aortic root staining, aortic roots were cut into slices at a thickness of 6 μm (based on a well-established protocol55). Prior to staining, slides were left at room temperature for enough dehydration. Slices were fixed in 10% formaldehyde for 15 min, and stained by ORO working solution for 5 min at room temperature. After washing 3 times using PBS, the aorta was ready for mounting and imaging.
For in vitro cultured macrophages staining, mature bone-marrow derived macrophages were plated on 25 mm square coverslips, and treated with oxLDL at a concentration of 50 μg/ml for 24 h. Macrophages were fixed in 10% formaldehyde, and stained by ORO working solution for 30 s at room temperature, following a wash using 60% isopropanol for 60 s. After washing 3 times using PBS, the coverslip was mounted using Prolong® Gold antifade reagent with DAPI.
Antibodies, chemicals and reagents
Rabbit polyclonal antibodies to TRPM2 (Novus, NB110–81601, 1:500 in 5% BSA); Rabbit polyclonal antibodies to Mac-1 (Cell Signaling Technology, 49420, 1:250 in 10% goat serum and 5% BSA for immunofluorescence (IF)); Rabbit polyclonal antibodies to F4/80 (Santa Cruz Biotechnology, sc-377009–594, 1:100 in 10% goat serum and 5% BSA for immunofluorescence (IF)); Rabbit polyclonal antibodies to CD80 (Santa Cruz Biotechnology, sc-376012 AF488, 1:1000 in 5% BSA for western blot (WB), 1:100 in 10% goat serum and 5% BSA for IF); Prolong® Gold antifade reagent with DAPI (Life technologies, P36935); Rabbit polyclonal antibodies to MCP-1 (E8Y7P) (Cell Signaling Technology, 81559, 1:1000 in 5% BSA); Rabbit polyclonal antibodies to MIF (E7T1W) (Cell Signaling Technology, 87501, 1:1000 in 5% BSA); Rabbit polyclonal antibodies to CD36 (D8L9T) (Cell Signaling Technology, 14347S, 1:1000 in 5% BSA); Rabbit polyclonal antibodies to Fyn (Cell Signaling Technology, 4023S, 1:2500 in 5% BSA); Rabbit polyclonal antibodies to Phospho-Src Family (Tyr416) (E6G4R) (Cell Signaling Technology, 59548S, 1:2500 in 5% BSA); Rabbit polyclonal antibodies to SAPK/JNK (Cell Signaling Technology, 9252S, 1:2500 in 5% BSA for WB); Rabbit polyclonal antibodies to Phospho-SAPK/JNK (Thr183/Tyr185) (81E11) (Cell Signaling Technology, 4668S, 1:2500 in 5% BSA); Rabbit polyclonal antibodies to p38 MAPK (Cell Signaling Technology, 9212S, 1:2500 in 5% BSA for WB); Rabbit polyclonal antibodies to Phospho-p38 MAPK (Thr180/Tyr182) (Cell Signaling Technology, 9211S, 1:2500 in 5% BSA for WB); Rabbit polyclonal antibodies to iNOS (Santa Cruz Biotechnology, sc-7271, 1:1000 in 5% BSA for WB); Rabbit polyclonal antibodies to GAPDH (Cell Signaling Technology, 7074S, 1:5000 in 5% BSA for WB); HRP-linked anti-rabbit IgG (Cell Signaling Technology, 7074S); Rabbit polyclonal antibodies to Phospho-NF-κB p65 (Ser536) (93H1) (Cell Signaling Technology, 3033, 1:1000 in 5% BSA); Rabbit polyclonal antibodies to NF-κB p65 (D14E12) (Cell Signaling Technology, 8242, 1:1000 in 5% BSA); CD11b Monoclonal Antibody (M1/70), eFluor 450, eBioscience™ (Thermal Fisher Scientific, 48–0112-80); NK1.1 Monoclonal Antibody (PK136), PE-Cyanine7, eBioscience™ (Thermal Fisher Scientific, 25–5941-81); Alexa Fluor® 700 Rat Anti-Mouse Ly-6C (BD Biosciences, 561237); FITC anti-mouse Ly-6G Antibody (BioLegend, 127605); PE anti-mouse CD3ε Antibody (BioLegend, 100308); PerCP/Cyanine5.5 anti-mouse/human CD45R/B220 Antibody (BioLegend, 103236). NP40 (Thermal Fisher Scientific, 28324), Triton™ X-100 (T-9284), Bovine Serum Albumin (Sigma-Aldrich, 9048–46-8), Goat Serum (Thermal Fisher Scientific, 16210–064). Sulfosuccinimidyl Oleate (sodium salt) (SSO) (Cayman chemical, 11211), N-(p-amylcinnamoyl) Anthranilic Acid (ACA) (Cayman chemical, 14531), PJ-34 (hydrochloride) (Cayman chemical, 14440), U73122 (Cayman chemical, 70740), BAPTA-AM (Cayman chemical, 15551), Apocynin (Cayman chemical, 11976). Recombinant Human Thrombospondin-1 Protein, CF (TSP1) (R&D systems, 3074-TH-050), Recombinant Human CCL2/MCP-1 Protein, (MCP-1) (R&D systems, 279-MC-050/CF), Recombinant Human MIF Protein (R&D systems, 289-MF-002/CF). All chemicals for making Tyrode solution and recording solution (see U73122ow) were purchased from Sigma-Aldrich.
Plasmids and enzymes
CD36 (Addgene, 52025). The pcDNA4/TO-FLAG-hTRPM2 construct was a kind gift from Dr. A.M. Sharenberg(University of Washington, Seattle).
Cell culture and transfection
HEK293T cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) (Thermal Fisher Scientific, 12100–038) supplemented with 10% Bovine Growth Serum (BGS) (HyClone, SH30541.03) and 0.5% penicillin/streptomycin (Thermal Fisher Scientific, 15140–122) at 37 °C and 5% CO2. Cells were transfected when at a confluence about 80–90% using Lipofectamine® 3000 Transfection Kit (Thermal Fisher Scientific, 2232162) based on manufacturer’s instruction.
Isolation and culture of aorta-derived endothelial cell
Endothelial culture medium was made prior to isolation: DMEM: Nutrient Mixture F-12 (DMEM/F12) (Thermal Fisher Scientific, 11330) was supplemented with 100 μg/ml Endothelial cell growth supplement from bovine neural tissue (Sigma, E2759–15MG), 10% Fetal Bovine Serum (FBS) (Thermal Fisher Scientific, A4766) and 0.5% penicillin/streptomycin (Thermal Fisher Scientific, 15140–122).
Wild-type mice were euthanized based on IACUC-approved protocols. Thoracic aorta was quickly dissected out, and the lumen was filled with collagenase II (Worthington, 4177) in DMEM/F12 at a concentration of 1 mg/ml with 2 ends ligated, and digested at 37 °C for 30 min. Then the ligations were released, and homogenate in the aorta was centrifuged at 1000 g for 10 min at 4 °C. The supernatant was carefully removed, and the cell pellet was re-suspended using 20% BSA in DMEM/F12 and centrifuged at 1000 g for 20 min at 4 °C. Then the cell pellet was re-suspended with prewarmed endothelial cells culture medium, and cells were plated onto 35 mm culture dishes precoated with Corning® Collagen I, Rat Tail (Corning, 354236). Culture medium was changed every 2 days. Puromycin (Sigma, P8833–25MG) at a concentration 2 μg/ml was be added in the 1st week to inhibit the growth of other non-endothelial cells. After 1 week, immunofluorescence staining of CD31 was performed to confirm the purity of isolated endothelial cells. Then endothelial cells were plated onto transwell inserts with 12 μm pore size (Costar, 3403) at a density of ~5 × 106 cells/ml.
Isolation and culture of bone marrow derived macrophages
Mice were euthanized and femurs were quickly removed. Two ends for femurs were cut using a scissor, and bone marrow was washed out using PBS. Collected bone marrow was thoroughly resuspended with DMEM: Nutrient Mixture F-12 (DMEM/F12) (Thermal Fisher Scientific, 11330) supplemented with 25 ng/ml Macrophage Colony-Stimulating Factor from mouse (Sigma, M9170–10UG), 10% BGS (HyClone, SH30541.03) and 0.5% penicillin/streptomycin (Thermal Fisher Scientific, 15140–122). Culture medium was changed every 3 days. After culturing for 7 ~ 9 days, macrophages were usable for experiments.
Isolation of peritoneal macrophages
1 ml of thioglycolate medium (sodium thioglycolate, 0.5 g/L; yeast extract, 5 g/L; glucose, 5.5 g/L; sodium chloride, 2.5 g/L; L-cystine, 0.5 g/L) was injected intraperitoneally. After 4 days, mice were euthanized, and 8 ml of sterilized ice-cold PBS was injected into the peritoneal cavity to collect peritoneal macrophages. Collected peritoneal exudate cells in PBS were centrifuged at 800 g for 10 min at 4°C. Then cells were resuspended and plated on culture dishes at a density of ~2.5 × 106 cells/ml. At 3rd day of isolation, macrophages were subjected to whole-cell TRPM2 current recording.
Treatment of macrophages
SSO was used to specifically inhibit the activation of CD36 by oxLDL or TSP1. ACA, PJ34 and U73122 were used to inhibit TRPM2 activation by oxLDL or TSP1. SSO, PJ34 and U73122 were all diluted to a working concentration of 1 μM. U73122 was used in combination with low Ca2+ medium and solution. For cell culture, U73122 was diluted in HBSS medium supplemented with BGS and penicillin/streptomycin. For current recording, Fura-2 and Rhodamine-123 imaging, U73122 was diluted in Ca2+ free Tyrode solution.
In vitro macrophage infiltration and emigration test
As shown in the graphic illustrations in Extended Data Fig. 3, macrophage infiltration and emigration across cultured aorta-derived endothelial cells were examined. Transwell inserts were plated with endothelial cells as described above. For fluid permeation test, ~100000 isolated bone marrow derived macrophages were added into the upper chamber, and recombinant human MCP1 or MIF was added into the lower chamber at a concentration of 50 nM to promote macrophage infiltration. For the macrophage infiltration test, macrophages were treated with oxLDL at a concentration of 50 μM for 24h. Then ~100000 oxLDL preloaded macrophages were added into the upper chamber, and recombinant human MCP1 was added into the lower chamber at a concentration of 50 nM to promote macrophage emigration.\
Scratch assay of isolated macrophages56
Macrophage migration ability was further examined by scratch assay. 14-days old bone marrow derived macrophages were plated at appropriate density on 60 mm dish. On 2nd day after plating, a 200 μL sterile micropipette tip was used to scratch the cell layer. The migration ability was quantified by analyzing the proportion of “healing” area to the scratched area using imageJ56.
Real-time monitoring of mitochondrial function
Mitochondria function was evaluated using Rhodamine-123 dye (Rh123, Thermal Fisher Scientific, R302) quenching as previously reported. Pre-warmed DMEM/F12 medium was used to dilute Rhodamine-123 to a 20 μg/ml working concentration. Cells were incubated with Rh123 at 37 °C for 15 min. Fluorescence intensities at 509 nm with excitation at 488nm were collected every 15 s for 30 min using CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon).
Ratio calcium imaging experiments
Changes of intracellular Ca2+ were measured using ratio Ca2+ imaging as we describe previously. Pre-warmed DMEM/F12 medium was used to dilute Fura-2 AM (Thermal Fisher Scientific, F1221) to a working concentration at 2.5 μM, and 0.02% Pluronic™ F-127 (Thermal Fisher Scientific, P3000MP) was added to facilitate loading of Fura-2 AM. Ca2+ influx was measured by perfusing the cells with Tyrode’s solution under different treatments. Ionomycin (Iono) at 1 μM was applied at the end of the experiment as an internal control. Fluorescence intensities at 510 nm with 340 nm and 380 nm excitation were collected at a rate of 1 Hz using CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon). Ca2+ influx induced by oxLDL or TSP1 was normalized to the maximal response caused by ionomycin as previously performed 57.
Western blotting
NP-40/Triton lysis buffer (10% NP40, 1% Triton™ X-100, 150 mM NaCl, 1 mM EDTA, 50 mM Tris, pH=8.0) containing proteinase inhibitors and phosphatase inhibitors was used to lyse both cultured cells and frozen aorta tissue. For cultured macrophages, proteins were extracted 8 h after either oxLDL treatment. For tissue, full-length aortas were collected 4 months after HFD treatment. Protein concentration was measured using Pierce™ Rapid Gold BCA Protein Assay Kit. 50–80 μg of total protein per lane was loaded and separated proteins were transferred to nitrocellulose membranes. Membranes were blocked with 5% BSA and 2.5% goat serum in Tris buffered saline (TBS, pH=7.4), and incubated with primary antibodies in TBS with 0.05% Tween (TBS-T). Then membranes were incubated with secondary antibodies. Blots were developed with ImageQuant LAS 4000 imaging system. Band intensity was quantified using ImageJ software and normalized with appropriate loading controls.
Electrophysiology
Whole cell currents were recorded using an Axopatch 200B amplifier. Data were digitized at 10 or 20 kHz and digitally filtered offline at 1 kHz. Patch electrodes were pulled from borosilicate glass and fire-polished to a resistance of ∼3 MΩ when filled with internal solutions. Series resistance (Rs) was compensated up to 90% to reduce series resistance errors to <5 mV. Cells in which Rs was >10 MΩ were discarded 46. For recordings using SSO, ACA, PJ34 and U73122, these inhibitors were added into the extracellular recording solution at the same concentration as used during pre-incubation.
Normal Tyrode solution contained (mM): 145 NaCl, 5 KCl, 2 CaCl2, 10 HEPES, 10 glucose, osmolarity=290–320 mOsm/Kg, and pH=7.4 was adjusted with NaOH. NMDG-Cl solution contained (mM): 150 NMDG-Cl, 10 HEPES, 10 glucose, osmolarity=290–320 mOsm/Kg, and pH=7.4 was adjusted with NMDG. The internal pipette solution for whole cell current recordings of TRPM2 contained: 135 mM Cs-methanesulfonate (CsSO3CH3), 8 mM NaCl, 500 nM CaCl2, 5 μM EGTA, and 10 mM HEPES, with pH adjusted to 7.2 with CsOH. Free [Ca2+]i buffered by EGTA was about 500 nM calculated using Max chelator46. ADPR 1 μM was included in the pipette solution for all experiments.
Perforated-patch recordings were conducted to maintain intact intracellular contents as we previously reported 46. In brief, freshly prepared nystatin (Sigma, N6261) was added into the internal pipette solution (135 mM Cs-methanesulfonate (CsSO3CH3), 8 mM NaCl, 3 mM MgCl2 and 10 mM HEPES, with pH adjusted to 7.2 with CsOH) to make the nystatin working concentration at 100 μg/mL. There was no added Ca2+ or EGTA in the pipette solution. Patch electrodes were pulled from borosilicate glass and fire-polished to a resistance of 3~5 MΩ when filled with internal pipette solutions. After forming tight seal, cells were kept in the cell-attached mode at a holding potential of 0 mV until the series resistance was <20 MΩ (typically within 5 to 10 min), and then the ramp protocol for current recording (see above) was applied. Normally, 10 min was allowed for achieving perforated-patch prior to current recording.
Immunofluorescence staining
Full length aortas or the heart with aortic valve harvested from mice were mounted in Fisher Healthcare™ Tissue-Plus™ O.C.T. Compound (Thermal Fisher Scientific, 23–730-571) prior to cutting. Aortas were cut into slices at a thickness of 6 μm. Prior to staining, slides were left at room temperature for enough dehydration. Slices were fixed in 10% formaldehyde for 15 min, and incubated in blocking solution containing 5% BSA, 15% goat serum and 1% Triton X-100 at room temperature for 2 h. Primary antibodies were diluted as described previously in TBS-T containing 15% goat serum. Slices were incubated with primary antibodies for at least 12 h at 4 °C, and incubated with secondary antibodies at room temperature for 2 h. Then slices were washed 3 times using PBS, mounted using Prolong® Gold antifade reagent with DAPI.
Software
NIS Elements AR4 (Nikon), pClamp 9.2 (Molecular Devices) and ZE5 Cell Analyzer (BIO-RAD) were used for data collection. FlowJo (9.9.6) and ImageJ (US NIH) were used for quantification. GraphPad Prism 6.0 (GraphPad Software) was used for statistical analysis. Biorender was used for drawing graphic illustrations. Adobe Illustrator 6.0 (Adobe) was used for preparing figures.
Data analysis
The investigators were blinded to group allocation during data collection and/or analysis. All data are expressed as mean ± SEM. For two groups’ comparison, statistical significance was determined using two-tailed Student’s t-test. For multiple groups data analysis, statistical significance was determined using one-way or two-way analysis of variance (ANOVA), followed by Bonferroni post-test. P<0.05 with confidence interval of 95% was considered as significant.
Statistics and Reproducibility
All the experiments were independently replicated or reproduced for at least three times. The exact number of replications is described in the result of each experiment.
Extended Data
Extended Data Fig. 1. Knockout of Trpm2 in Apoe−/− mice.
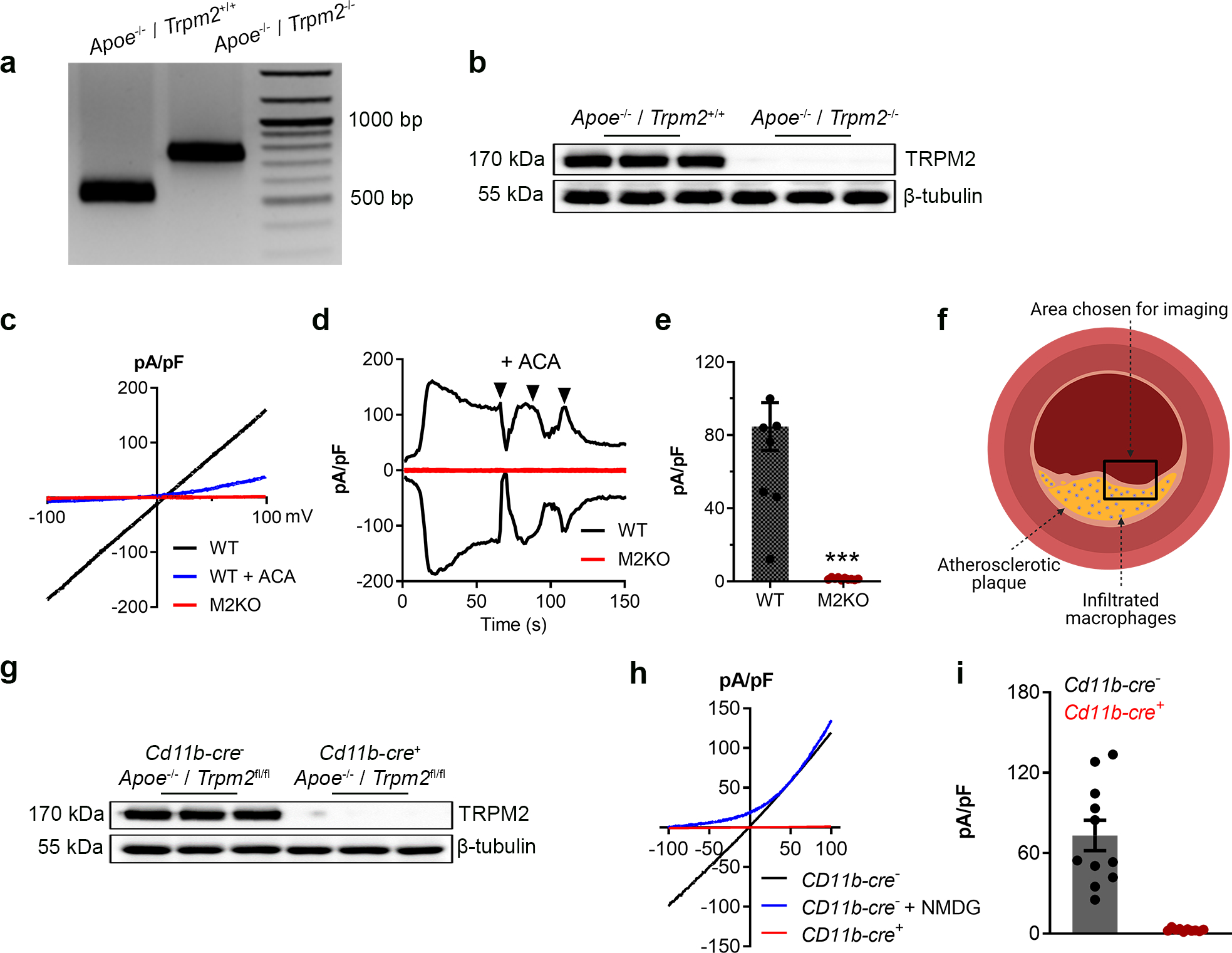
(a) Representative PCR genotyping results showing a 514bp and 740 bp products for WT and M2KO mice. (b) Representative WB analysis of TRPM2 expression in macrophages isolated from Apoe single knockout (WT (n=3)) and Apoe / Trpm2 double knockout (M2KO (n=3)) mice (c-e) Representative recording (c, I-V curve; d, time-current trace) and quantification of TRPM2 current in macrophages isolated from Apoe single knockout (WT) and Apoe / Trpm2 double knockout (M2KO) mice. ACA is a TRPM2 blocker. (***: p < 0.001; unpaired t test; mean ± SEM) (f) Graphic illustration showing the atherosclerotic area chosen for taking images of F4/80&CD80 staining in Fig 1i and Fig 3h. (g) Representative WB analysis of TRPM2 expression in macrophages isolated from Trpm2fl/flCd11b-cre− (n=3) and Trpm2fl/flCd11b-cre+ mice (n=3) with Apoe knockout. (h, i) Representative recording and quantification of TRPM2 current in macrophages isolated from Trpm2fl/flCd11b-cre− and Trpm2fl/flCd11b-cre+ mice with Apoe knockout.
Extended Data Fig. 2. Expression of TRPM2 is increased during atherogenesis.
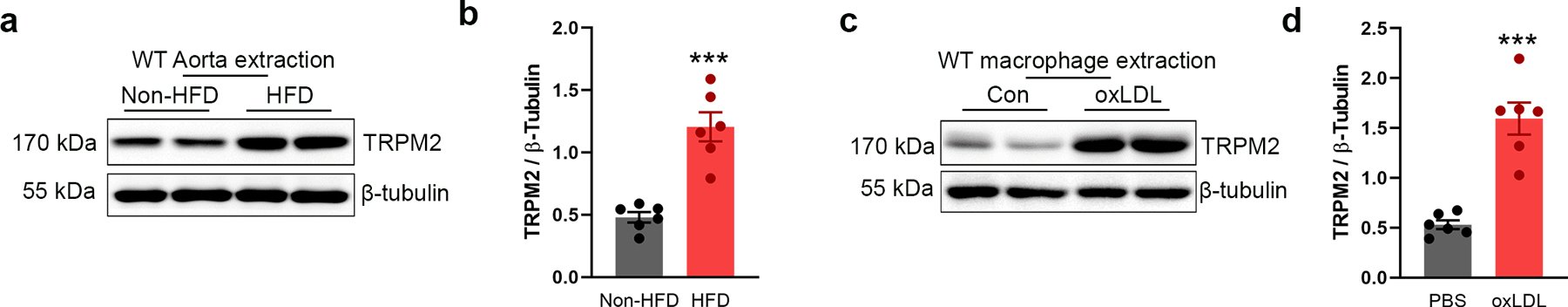
(a, b) Representative WB analysis of TRPM2 expression in aorta from wild-type mice (WT) with or without the treatment of high-fat diet (HFD) for 4 months (n=6/group). (c, d) Representative WB analysis of TRPM2 expression in macrophages isolated from WT mice with or without the treatment of oxLDL (50 μg/ml) for 24 h (n=6/group). (***: p < 0.001; unpaired t test, two-tailed; mean ± SEM)
Extended Data Fig. 3. In vitro macrophage migration and emigration assay.
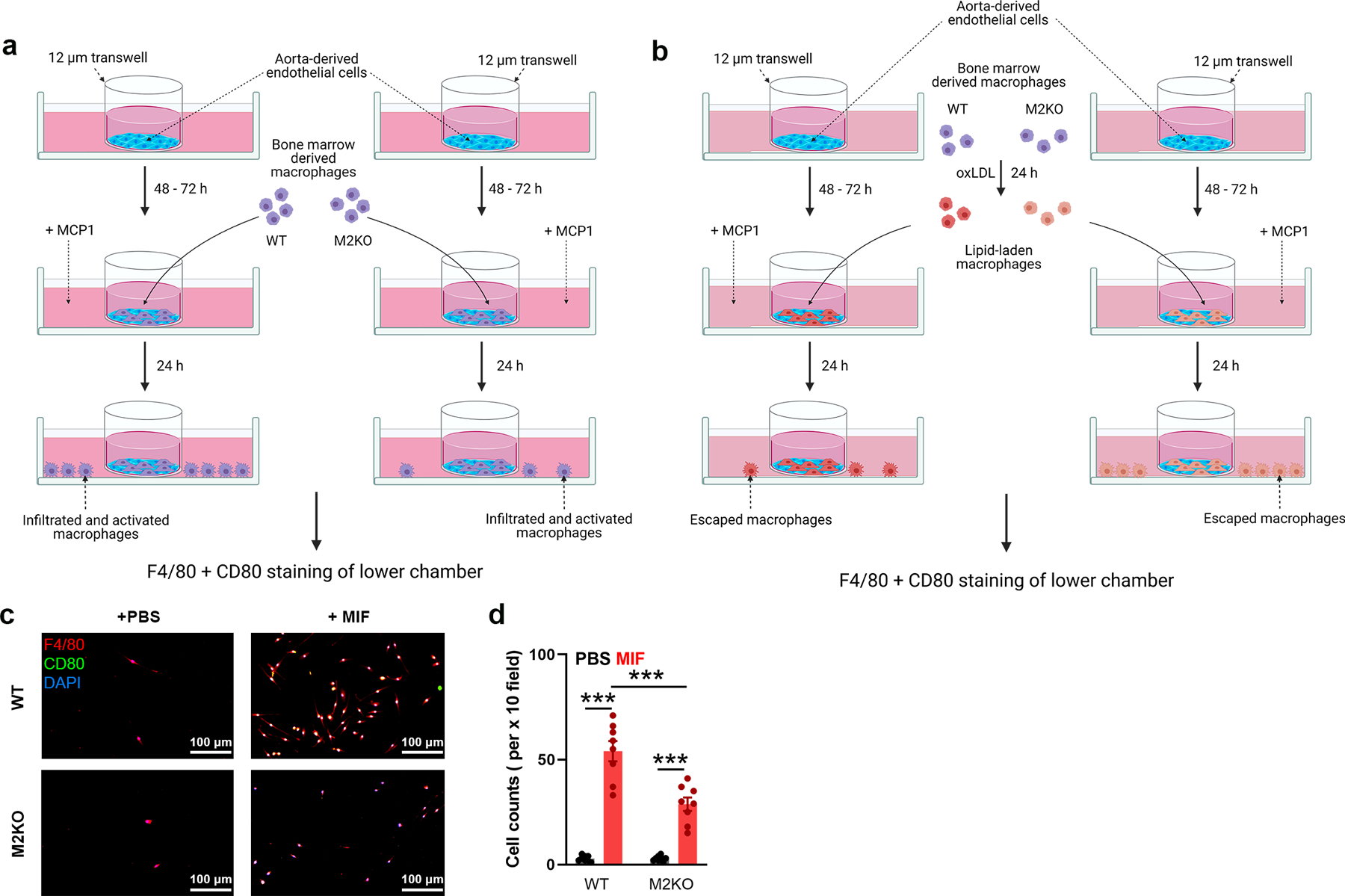
(a) Graphic illustration of in vitro examination of macrophage infiltration across endothelial cells induced by MCP1. Aorta-derived endothelial cells were plated on the transwell inserts (pore size: 12 μm) for 3–5 days. Bone marrow derived macrophages were added into the upper chamber after endothelial cells completely covered the upper surface of transwells. After 4 h, F4/80 and CD80 staining of macrophages in the lower chamber was performed as in Fig 2l. (b) Graphic illustration of in vitro examination of macrophage emigration across endothelial cells induced by MCP1. Aorta-derived endothelial cells were plated on the transwell inserts (pore size: 12 μm) for 3–5 days. Bone marrow derived macrophages preloaded with oxLDL for 24 h were added into the upper chamber after endothelial cells completely covered the upper surface of transwells. After 24 h, F4/80 and CD80 staining of macrophages in lower chamber was performed as in Fig 2n. (c, d) In vitro macrophage migration assay induced by MIF instead of MCP1 as shown in Extended Data Fig. 3a. l, F4/80 and CD80 staining of macrophages in the lower chamber (Red: F4/80; Blue: DAPI; Green: CD80). c, Quantification of the number of infiltrated macrophages within a x 10 field. 6 dishes from each group were chosen for quantification. (***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Extended Data Fig. 4. TRPM2 is required for CD36 activation in macrophages induced by oxLDL.
(a) Quantification of Fig 4a by counting percentage of Oil red O staining macrophages (n=8/group). (b, c) 30-min oxLDL treatment (50 μg/ml) induce the activation of CD36 signaling without upregulating CD36 expression. Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in macrophages after oxLDL treatment for 30 min (n=6/group). (d, e) NADPH oxidase inhibitor apocynin does not inhibit CD36 activation induced by 24-h oxLDL treatment (50 μg/ml) in macrophages isolated from wild-type (WT) mice. Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in macrophages after oxLDL treatment for 24 h (n=6/group). (f) Quantification of Fig 4k by counting percentage of Oil red O staining macrophages (n=6/group). (g) Quantification of Fig 4o by counting percentage of Oil red O staining macrophages (n=8/group). (h) A set of original Fura-2 real time recording traces without normalization during oxLDL treatment as in Fig 4o. (i, j) Trpm2 deletion does not influence the production of MCP1/MIF in endothelial cells isolated from aorta in response to 24-h oxLDL treatment (50 μg/ml). Representative WB analysis of MCP1 and MIF expression in endothelial cells after oxLDL treatment for 24 h (n=6/group). (ns: no statistical significance; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Extended Data Fig. 5. TRPM2 is required for CD36 activation in macrophages induced by TSP1.
(a, b) Representative WB analysis of TRPM2 expression in macrophages isolated from WT mice with or without the treatment of TSP1 (10 μg/ml) for 24 h (n=6/group). (c, d) 30-min TSP1 (10 μg/ml) treatment induce the activation of CD36 signaling without upregulating CD36 expression. Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in macrophages after TSP1 treatment for 30 min (n=6/group). (e, f) NADPH oxidase inhibitor apocynin does not inhibit CD36 activation induced by 24-h TSP1 (10 μg/ml) treatment in macrophages isolated from wild-type (WT) mice. Representative WB analysis of CD36, pFyn, pJNK and pp38 expression in macrophages after TSP1 treatment for 24 h. (g) A set of original Fura-2 real time recording traces without normalization during TSP1 treatment as in Fig 5g. (h, i) Trpm2 deletion does not influence the production of MCP1/MIF in endothelial cells isolated from aorta in response to 24-h TSP1 (10 μg/ml) treatment. Representative WB analysis of MCP1 and MIF expression in endothelial cells after TSP1 treatment for 24 h (n=6/group). (ns: no statistical significance; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Extended Data Fig. 6. Different inhibitors suppressed the activation of TRPM2 by oxLDL or TSP1 treatment.
(a, b) Representative recording of TRPM2 current in HEK293T cells transfected with CD36 and TRPM2 during oxLDL treatment (50 μg/ml) as in a, and during TSP1 treatment (10 μg/ml) as in b. Transfected cells were treated with different inhibitors as indicated before current recording.
Extended Data Fig. 7. Inhibition of CD36 or TRPM2 did not produce additional inhibitory effect on M2KO macrophages.

(a) Representative WB analysis of the expression of CD36, pFyn, Fyn, pJNK, JNK, pp38 and p38 in isolated macrophages from M2KO mice after oxLDL treatment (50 μg/ml). Macrophages were treated with different inhibitors as indicated before protein extraction. (b) Quantification of WB bands (n=3/group). (c) Representative WB analysis of the expression of CD36, pFyn, Fyn, pJNK, JNK, pp38 and p38 in isolated macrophages from M2KO mice after TSP1 treatment (10 μg/ml). Macrophages were treated with different inhibitors as indicated before protein extraction. (d) Quantification of WB bands (n=3/group). (e) Representative WB analysis of the expression of iNOS in isolated macrophages from M2KO mice after oxLDL treatment (50 μg/ml). Macrophages were treated with different inhibitors as indicated before protein extraction. (f) Quantification of WB bands (n=3/group). (e) Representative WB analysis of the expression of iNOS in isolated macrophages from M2KO mice after TSP1 treatment (10 μg/ml). Macrophages were treated with different inhibitors as indicated before protein extraction. (f) Quantification of WB bands (n=3/group). (i) Representative WB analysis of the expression of MCP1 and MIF in isolated macrophages from M2KO mice after oxLDL treatment (50 μg/ml). Macrophages were treated with different inhibitors as indicated before protein extraction. (j) Quantification of WB bands (n=3/group). (k) Representative WB analysis of the expression of MCP1 and MIF in isolated macrophages from M2KO mice after TSP1 treatment (10 μg/ml). Macrophages were treated with different inhibitors as indicated before protein extraction. (l) Quantification of WB bands (n=3/group). (ns: no statistical significance; ANOVA, two-tailed, Bonferroni’s test; mean ± SEM).
Extended Data Fig. 8. Graphic illustration, the activation of CD36 and TRPM2 form a positive feedback loop in atherogenesis.
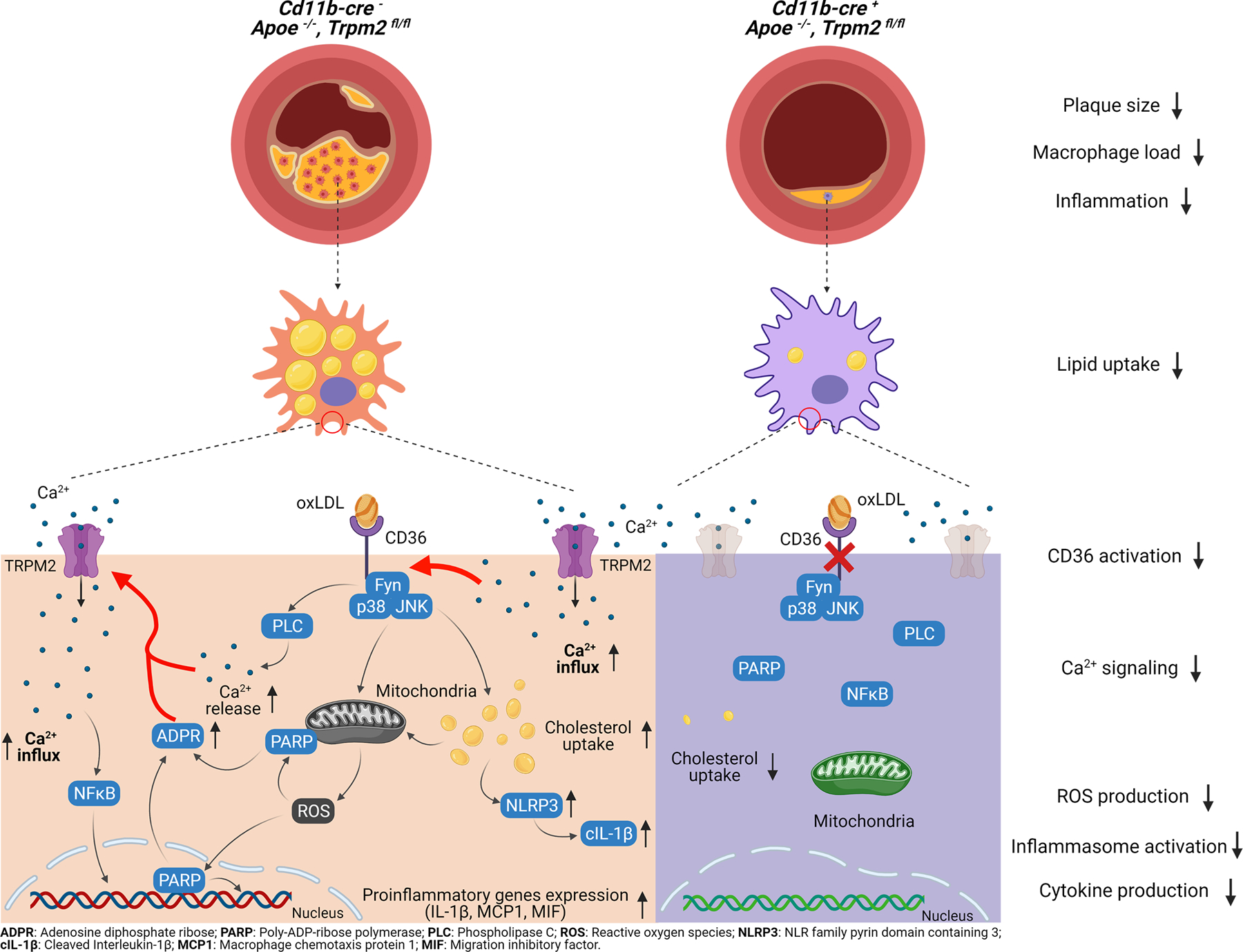
In summary, we found that: (1) Global Trpm2 deletion and macrophage-specific Trpm2 deletion protect against atherosclerosis in Apoe−/− mice fed with a high-fat diet (HFD). (2) Trpm2 deficiency in macrophages inhibits atherogenesis by inhibiting macrophage infiltration and minimizing foam cell formation. (3) TRPM2 activation is required for CD36-induced oxLDL uptake and subsequent inflammatory responses in macrophages. (4) The ligands of CD36, oxLDL and TSP1, activate TRPM2, thereby perpetuating TRPM2-CD36 inflammatory cycle in atherogenesis cascade. (5) Our data establish TRPM2-CD36 axis in macrophages as an important atherogenesis mechanism and TRPM2 as a promising therapeutic target for atherosclerosis
Supplementary Material
Acknowledgements
We thank Dr. Andrew M. Scharenberg (University of Washington) for kindly providing TRPM2 plasmid. CD36-bio-His was a gift from Gavin Wright (Addgene plasmid #52025; http://n2t.net/addgene:52025; RRID:Addgene_52025)58. We thank Qiaoshan Lin (University of Connecticut, Storrs) for preparing artistic graphic works for our study.
This work was partially supported by the National Institute of Health (R01-HL143750) and American Heart Association (19TPA34890022) to L.Y., and Canadian Institutes of Health Research (MOP 86655) to J.V. .
Footnotes
Competing Interests
All authors declare no competing interests.
Data availability
All data generated or analyzed are included in the main article and associated files. Source data are provided with this paper.
References
- 1.Libby P et al. Atherosclerosis. Nat Rev Dis Primers 5, 56, doi: 10.1038/s41572-019-0106-z (2019). [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Sheedy FJ & Fisher EA Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13, 709–721, doi: 10.1038/nri3520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore KJ & Tabas I Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355, doi: 10.1016/j.cell.2011.04.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverstein RL, Li W, Park YM & Rahaman SO Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Trans Am Clin Climatol Assoc 121, 206–220 (2010). [PMC free article] [PubMed] [Google Scholar]
- 5.Moore KJ & Freeman MW Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol 26, 1702–1711, doi: 10.1161/01.ATV.0000229218.97976.43 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Baldrighi M, Mallat Z & Li X NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 267, 127–138, doi: 10.1016/j.atherosclerosis.2017.10.027 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Cho S CD36 as a therapeutic target for endothelial dysfunction in stroke. Current pharmaceutical design 18, 3721–3730, doi: 10.2174/138161212802002760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hara Y et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Molecular cell 9, 163–173. (2002). [DOI] [PubMed] [Google Scholar]
- 9.Perraud AL et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411, 595–599. (2001). [DOI] [PubMed] [Google Scholar]
- 10.Sano Y et al. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293, 1327–1330 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Takahashi N, Kozai D, Kobayashi R, Ebert M & Mori Y Roles of TRPM2 in oxidative stress. Cell calcium 50, 279–287, doi: 10.1016/j.ceca.2011.04.006 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Desai BN & Leitinger N Purinergic and calcium signaling in macrophage function and plasticity. Front Immunol 5, 580, doi: 10.3389/fimmu.2014.00580 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syed Mortadza SA, Wang L, Li D & Jiang LH TRPM2 Channel-Mediated ROS-Sensitive Ca(2+) Signaling Mechanisms in Immune Cells. Frontiers in immunology 6, 407, doi: 10.3389/fimmu.2015.00407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di A et al. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol 13, 29–34, doi: 10.1038/ni.2171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolf D & Ley K Immunity and Inflammation in Atherosclerosis. Circ Res 124, 315–327, doi: 10.1161/CIRCRESAHA.118.313591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arida A, Protogerou AD, Kitas GD & Sfikakis PP Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int J Mol Sci 19, doi: 10.3390/ijms19071890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheedy FJ et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nature immunology 14, 812–820, doi: 10.1038/ni.2639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsiantoulas D et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature 597, 92–96, doi: 10.1038/s41586-021-03818-3 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Fonfria E et al. TRPM2 is elevated in the tMCAO stroke model, transcriptionally regulated, and functionally expressed in C13 microglia. Journal of receptor and signal transduction research 26, 179–198, doi: 10.1080/10799890600637522 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Belrose JC & Jackson MF TRPM2: a candidate therapeutic target for treating neurological diseases. Acta pharmacologica Sinica 39, 722–732, doi: 10.1038/aps.2018.31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deshmane SL, Kremlev S, Amini S & Sawaya BE Monocyte chemoattractant protein-1 (MCP-1): an overview. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 29, 313–326, doi: 10.1089/jir.2008.0027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nowak WN, Deng J, Ruan XZ & Xu Q Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler Thromb Vasc Biol 37, e41–e52, doi: 10.1161/ATVBAHA.117.309228 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Buttery LD et al. Inducible nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab Invest 75, 77–85 (1996). [PubMed] [Google Scholar]
- 24.Zhong Z et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nature communications 4, 1611, doi: 10.1038/ncomms2608 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duewell P et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361, doi: 10.1038/nature08938 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park YM, Febbraio M & Silverstein RL CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. The Journal of clinical investigation 119, 136–145, doi: 10.1172/JCI35535 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett TJ Macrophages in Atherosclerosis Regression. Arterioscler Thromb Vasc Biol 40, 20–33, doi: 10.1161/ATVBAHA.119.312802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller BA et al. TRPM2 channels protect against cardiac ischemia-reperfusion injury: role of mitochondria. The Journal of biological chemistry 289, 7615–7629, doi: 10.1074/jbc.M113.533851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferron M & Vacher J Targeted expression of Cre recombinase in macrophages and osteoclasts in transgenic mice. Genesis 41, 138–145, doi: 10.1002/gene.20108 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Beckers CML et al. Cre/lox Studies Identify Resident Macrophages as the Major Source of Circulating Coagulation Factor XIII-A. Arteriosclerosis, thrombosis, and vascular biology 37, 1494–1502, doi: 10.1161/ATVBAHA.117.309271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshimura T, Robinson EA, Tanaka S, Appella E & Leonard EJ Purification and amino acid analysis of two human monocyte chemoattractants produced by phytohemagglutinin-stimulated human blood mononuclear leukocytes. Journal of immunology 142, 1956–1962 (1989). [PubMed] [Google Scholar]
- 32.Harris J, VanPatten S, Deen NS, Al-Abed Y & Morand EF Rediscovering MIF: New Tricks for an Old Cytokine. Trends in immunology 40, 447–462, doi: 10.1016/j.it.2019.03.002 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Zhao M et al. Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 110, 458–468, doi: 10.1034/j.1600-0463.2002.100604.x (2002). [DOI] [PubMed] [Google Scholar]
- 34.Ricci R et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science 306, 1558–1561, doi: 10.1126/science.1101909 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Rahaman SO et al. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell metabolism 4, 211–221, doi: 10.1016/j.cmet.2006.06.007 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Wang GZ, Rabinovitch PS & Tabas I Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res 114, 421–433, doi: 10.1161/CIRCRESAHA.114.302153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan J, Bengtson CP, Buchthal B, Hagenston AM & Bading H Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 370, doi: 10.1126/science.aay3302 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Dugan LL et al. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci 15, 6377–6388 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK & Kettenmann H Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci 23, 4410–4419 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshimura T The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cellular & molecular immunology 15, 335–345, doi: 10.1038/cmi.2017.135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto S et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature medicine 14, 738–747, doi: 10.1038/nm1758 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu LY, Ramakrishnan DP & Silverstein RL Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 122, 1822–1832, doi: 10.1182/blood-2013-01-482315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Dee Z, Pidcock K & Gutierrez LS Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm 2011, 296069, doi: 10.1155/2011/296069 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganguly R et al. TSP-1 (Thrombospondin-1) Deficiency Protects ApoE(−/−) Mice Against Leptin-Induced Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 41, e112–e127, doi: 10.1161/ATVBAHA.120.314962 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ganguly R et al. Oral chromium picolinate impedes hyperglycemia-induced atherosclerosis and inhibits proatherogenic protein TSP-1 expression in STZ-induced type 1 diabetic ApoE(−/−) mice. Scientific reports 7, 45279, doi: 10.1038/srep45279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du J, Xie J & Yue L Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci U S A 106, 7239–7244, doi: 10.1073/pnas.0811725106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sumoza-Toledo A & Penner R TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 589, 1515–1525, doi: 10.1113/jphysiol.2010.201855 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leung AWY, Chan RSM, Sea MMM & Woo J An Overview of Factors Associated with Adherence to Lifestyle Modification Programs for Weight Management in Adults. Int J Environ Res Public Health 14, doi: 10.3390/ijerph14080922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeurissen MLJ et al. Prevention of oxLDL uptake leads to decreased atherosclerosis in hematopoietic NPC1-deficient Ldlr(−/−) mice. Atherosclerosis 255, 59–65, doi: 10.1016/j.atherosclerosis.2016.10.038 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Rahaman SO, Zhou G & Silverstein RL Vav protein guanine nucleotide exchange factor regulates CD36 protein-mediated macrophage foam cell formation via calcium and dynamin-dependent processes. The Journal of biological chemistry 286, 36011–36019, doi: 10.1074/jbc.M111.265082 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murakami T et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences of the United States of America 109, 11282–11287, doi: 10.1073/pnas.1117765109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez DM & Giannarelli C Immune cell profiling in atherosclerosis: role in research and precision medicine. Nat Rev Cardiol 19, 43–58, doi: 10.1038/s41569-021-00589-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin JD et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 4, doi: 10.1172/jci.insight.124574 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller BA et al. The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. American journal of physiology. Heart and circulatory physiology 304, H1010–1022, doi: 10.1152/ajpheart.00906.2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paigen B, Morrow A, Holmes PA, Mitchell D & Williams RA Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 68, 231–240, doi: 10.1016/0021-9150(87)90202-4 (1987). [DOI] [PubMed] [Google Scholar]
- 56.Amini-Nik S et al. beta-Catenin-regulated myeloid cell adhesion and migration determine wound healing. The Journal of clinical investigation 124, 2599–2610, doi: 10.1172/JCI62059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Du J et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circulation research 106, 992–1003, doi:CIRCRESAHA.109.206771 [pii] 10.1161/CIRCRESAHA.109.206771 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun Y et al. A Human Platelet Receptor Protein Microarray Identifies the High Affinity Immunoglobulin E Receptor Subunit alpha (FcepsilonR1alpha) as an Activating Platelet Endothelium Aggregation Receptor 1 (PEAR1) Ligand. Mol Cell Proteomics 14, 1265–1274, doi: 10.1074/mcp.M114.046946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed are included in the main article and associated files. Source data are provided with this paper.