

INTRODUCTION
The vascular endothelium is composed of a monolayer of specialized cells (endothelial cells), which form the interface between the underlying smooth muscle cells from the vascular lumen. Endothelial cells may exhibit significant plasticity in function depending on the milieu in which they exist; for example, the endothelium comprising the blood–brain barrier has significantly different functional characteristics than that lining the aorta. However, all endothelial cells share a common set of functions, including the regulation of hemostasis, maintenance of vascular permeability, mediation of both acute and chronic immune responses to various types of injury, and control of vascular tone.1 There are several molecular mechanisms that govern these critical processes, but none are as critical as the nitric oxide (NO) signaling pathway.2 NO is a small, soluble gas with strong vasodilatory, anti-inflammatory, and antioxidant properties that plays a central role in the maintenance of vascular homeostasis.3 Highlighting this, the concept of endothelial dysfunction (ED) is centrally linked to decreased NO production and sensitivity, which ultimately results in an imbalance in vascular homeostasis leading to a prothrombotic, proinflammatory, and less compliant blood vessel wall.2,4 In the present review, we highlight the mechanisms underpinning the regulatory effects of NO on ED and link these to currently understood clinical phenomena.
NITRIC OXIDE IN NORMAL ENDOTHELIAL FUNCTION
Nitric Oxide Synthesis and Nitric Oxide Synthase Enzyme Isoforms
NO is a highly reactive, readily diffusible gaseous free radical with strong intrinsic oxidant properties. It is synthesized by 3 distinct subtypes of the NO synthase (NOS) enzyme, each with unique expression patterns and functional properties: neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3).5,6 Broadly, these proteins catalyze the production of NO and l-citrulline from l-arginine and O2, using electrons donated from dihydronicotinamide-adenine dinucleotide phosphate (NADPH). This process is tightly regulated requiring several key protein–protein interactions and multiple prosthetic groups and cofactors. In monomeric form, the NOS subtypes are incapable of binding to l-arginine, and subsequently can function primarily as weak NADPH oxidases resulting in the production of harmful superoxide radical anion (O2•−). When bound by the calcium signaling protein calmodulin (CaM), the transfer of electrons through a flavin adenine mononucleotide and flavin adenine dinucleotide domain is enhanced. Finally, in the presence of heme and tetrahydrobiopterin (BH4), NOS monomers form homodimers capable of using the donated NADPH electrons to catalyze the 2-step oxidation of l-arginine to l-citrulline and NO. In the first step, NOS promotes the hydroxylation of l-arginine to Nω-hydroxy-l-arginine, which remains bound by the enzyme. In the second step, NOS catalyzes the oxidation of Nω-hydroxy-l-arginine to l-citrulline, thereby releasing NO (Fig. 1).
Fig. 1.
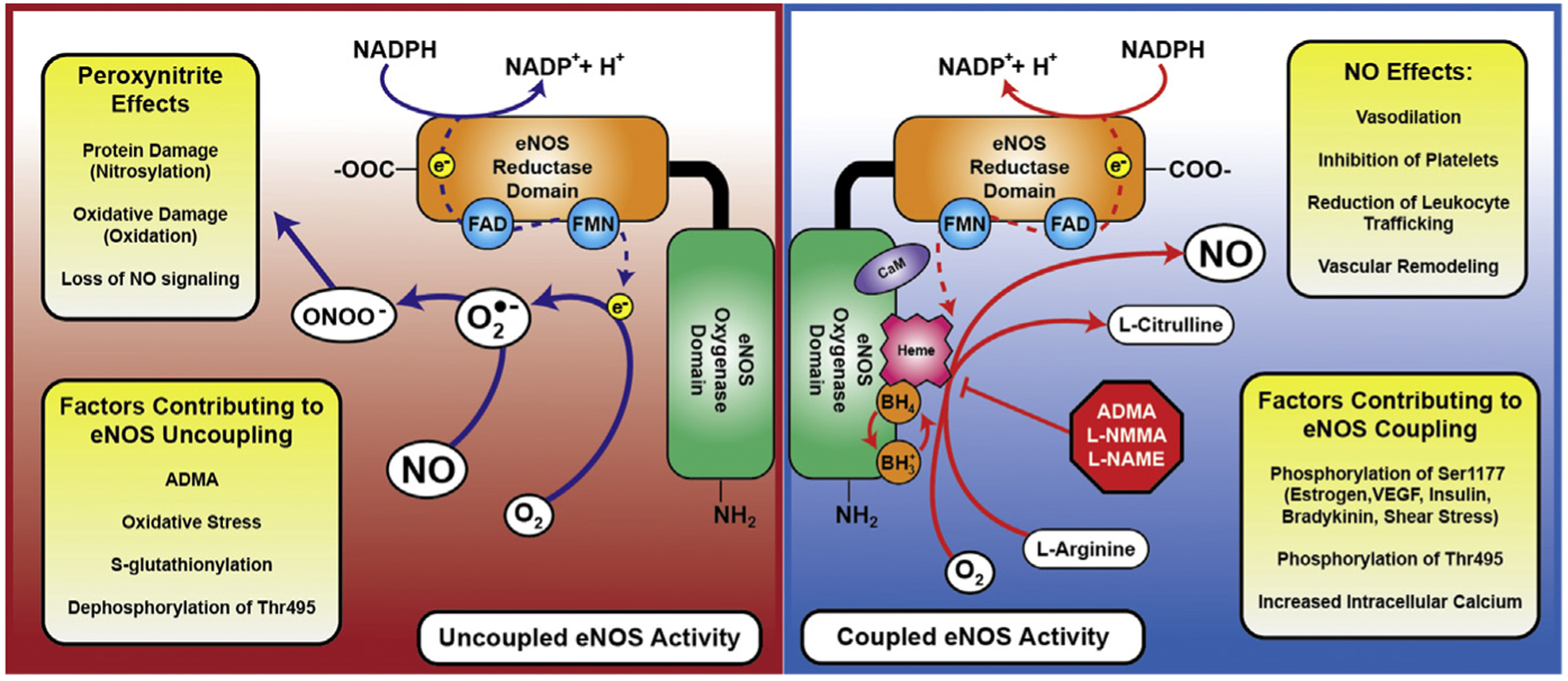
eNOS regulation, coupling, and uncoupling. During coupled eNOS activity (blue side), in the presence of heme and tetrahydrobiopterin, electrons from NADPH are passed through a core of flavin adenine dinucleotide–flavin adenine mononucleotide in the reductase domain to the Heme prosthetic group on the oxygenase domain. Here, l-arginine and O2 are consumed to create l-citrulline and NO. In the uncoupled state (red side), electrons are passed directly from the flavin adenine dinucleotide (FAD)–flavin adenine mononucleotide (FMN) core of the reductase domain to O2, generating superoxide (O2•−), which can ultimately combine with locally produced NO to make peroxynitrite (ONOO−). Several of the effects of ONOO− and NO are listed here, as well as factors contributing to both coupling and uncoupling of eNOS activity. These are explained in greater detail in the body of the text. VEGF, vascular endothelial growth factor.
NOS isoforms are differentially regulated through post-translational modifications as well as interactions with the associated scaffolding proteins. nNOS and eNOS, for example, are highly dependent on Ca2+-activated CaM for homodimerization and activity, whereas iNOS is minimally dependent on calcium concentration.6 These nuances have critical functional effects. In neurons, NO produced by nNOS signaling functions to modulate longer term synaptic transmission, leading to critical involvement in learning, memory, and neurogenesis.7 Additionally, some data suggest that nNOS may play a significant role in the maintenance of vascular tone through effects of nNOS-expressing end neurons that innervate smooth muscle in microvasculature.8 In contrast, iNOS is strongly induced in activated macrophages, in which it creates an NO burst representing the sentinel event in the acute inflammatory cascade. This often leads to significant collateral damage to bystander healthy tissue through the generation of downstream reactive oxygen species (ROS) and reactive nitrogen species.9 Herein, we focus on the regulation and function of eNOS, the native NOS of vascular endothelial cells.
Endothelial Nitric Oxide Synthase Regulation and Function
eNOS is predominantly expressed in vascular endothelial cells, although its expression has been detected in other specialized groups of cells with important circulatory roles. These include cardiac myocytes, platelets, certain neurons in the brain, placental cells, and kidney tubular epithelium.10 Nominally, NO production by eNOS increases substantially with increasing calcium concentrations secondary to its dependence on CaM. However, there are several alternative mechanisms for eNOS activation that lessen the importance of sustained increases in intracellular calcium for enzyme activity. The most well-studied of these is phosphorylation of the Ser1177 residue in the eNOS reductase domain, which leads to a higher flux of electrons and increased calcium sensitivity.6,11,12 This modification is the end result of multiple signaling cascades associated with a variety of different protein kinase activation pathways. Signals include estrogen and vascular endothelial growth factor, which activate protein kinase B (Akt); insulin, which is thought to function through both Akt and AMP-activated protein kinase; bradykinin, which signals via Ca2+/CaM-dependent protein kinase II; and mechanical shear stress, which activates protein kinase A as well as Akt.6,11,13 Another important residue is Thr495, which is phosphorylated by protein kinase C under resting conditions in endothelial cells. Phosphorylation of Thr495 limits CaM binding to eNOS, slowing electron transfer, and studies have demonstrated that Thr495 becomes dephosphorylated under stimuli that promote increases in intracellular Ca2+.11 However, this may be deleterious under certain circumstances, as dephosphorylated Thr495 also seems to favor uncoupling of eNOS activity.14 Several other sites of post-translational modification have been identified, and are reviewed elsewhere.15
eNOS function is critical for maintenance of appropriate vascular homeostasis. NO signaling directly leads to blood vessel dilation by stimulating soluble guanylyl cyclase, leading to an increase in cyclic guanosine monophosphate and subsequent relaxation of vascular smooth muscle.16–18 However, NO has multiple other distinct roles in vascular physiology. NO exerts antiplatelet effects through inhibition of platelet aggregation and adhesion; furthermore, eNOS-derived NO from platelets likely has both autocrine and paracrine inhibitory effects in a developing thrombus to limit pathologic clot formation.5 NO decreases the expression of macrophage chemoattractant protein-1, which limits leukocyte trafficking to endothelium.19 NO additionally alters the functionality of CD11/CD18 proteins on leukocytes, further altering their ability to adhere to the endothelial wall.20 With regard to vascular remodeling, eNOS-derived NO plays a critical role in angiogenesis, and NO production is one of the final products of angiogenic signaling cascades. Studies have shown roles for eNOS in early neonatal lung capillary development, as well as for the appropriate development of collateral circulation and neovascularization following ischemic insult.21 Broadly, this may be associated with the mobilization of appropriate progenitor cells from the bone marrow, as demonstrated by murine studies in eNOS-deficient mice.22
Endothelial Nitric Oxide Synthase in Vascular Pathophysiology
NO and its signaling functions, particularly in thrombosis and vascular tone, are central to the maintenance of vascular homeostasis. Highlighting this, ED is defined biochemically as a decreased amount of bioavailable NO in the vasculature. Broadly, the mechanisms underpinning ED can be placed into 2 basic categories: consumptive processes that transform bioavailable NO into other species and deficiencies in production of NO in the endothelium (Fig. 2).
Fig. 2.
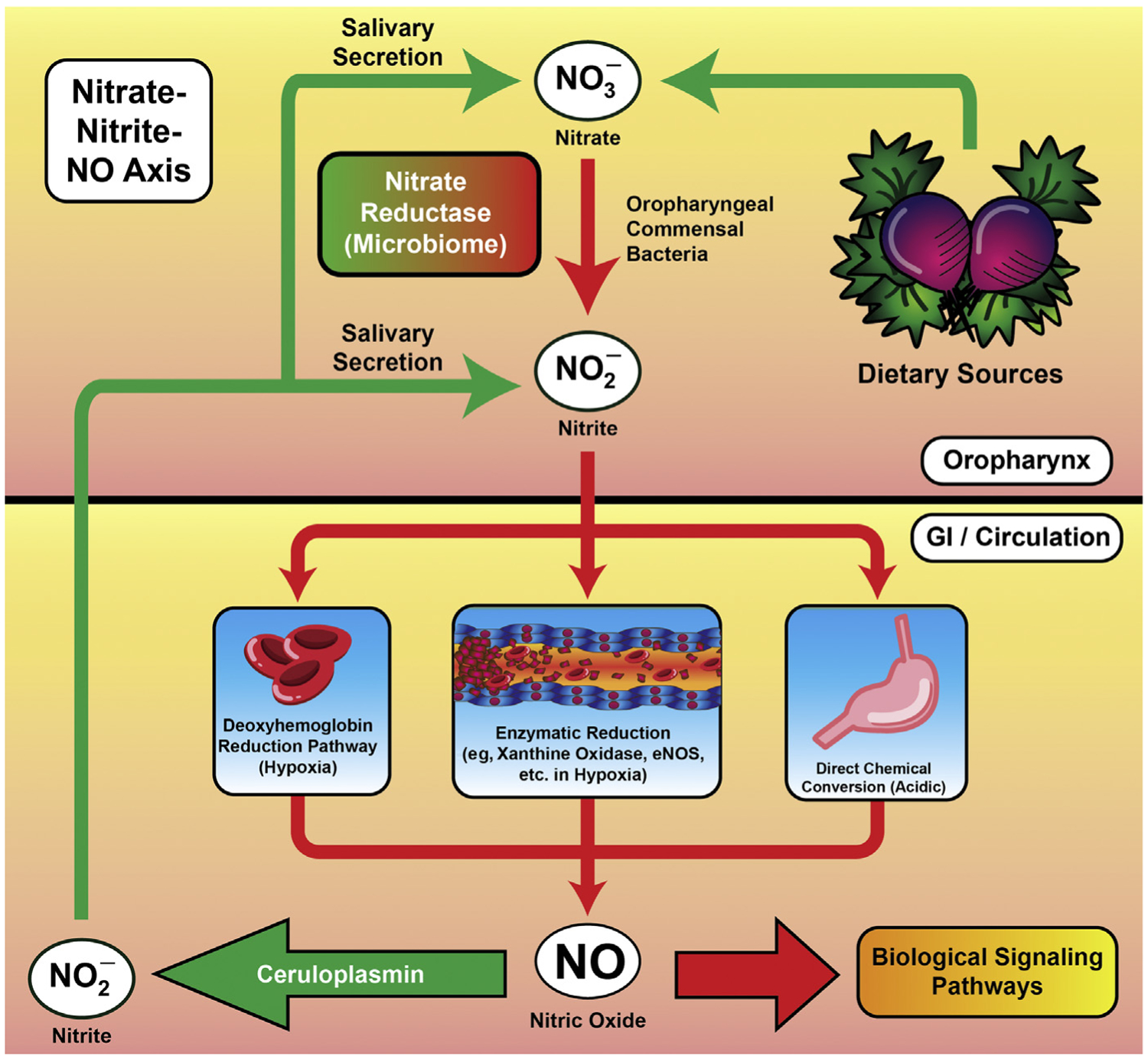
The nitrate–nitrite–NO pathway. Increasingly, research is demonstrating that dietary nitrates and nitrites serve as an endogenous reservoir for noncanonically produced NO, particularly in hypoxic conditions when NOS may be less functional or predisposed to being uncoupled. A detailed explanation of this pathway can be found in the body of the text.
Because NO is a highly diffusible and reactive species with an unpaired electron, there are a variety of chemical fates that prevent appropriate signaling. A primary driver of this deficiency is ROS, and O2•− in particular. O2•− reacts readily with NO, forming peroxynitrite (ONOO−), a deleterious reactive nitrogen species that itself reacts readily with biological molecules both as a potent oxidant as well as a nitrating agent. Myriad acute and chronic pathologic states can potentiate the overproduction of ROS and subsequent ONOO− formation; a full discussion of these reactions is beyond the scope of this review.
In addition to the consumption of bioavailable NO as a mechanism to reduce NO signaling in ED, modifications to eNOS itself can also alter the production of NO at the source. As mentioned elsewhere in this article, eNOS requires dimerization in the presence of heme and BH4 for effective electron movement to l-arginine. If this relationship is disrupted, the end result is that eNOS functions as a weak NADPH oxidase, producing O2•− instead of NO—a situation referred to as eNOS uncoupling. Multiple mechanisms contribute to eNOS uncoupling, which enhances local oxidative stress in addition to eliminating the vasoprotective effects of NO signaling. One such pathway involves ONOO− produced by initial oxidative stressors and functional eNOS. ONOO− both disrupts a key zinc-thiolate cluster in eNOS and oxidizes BH4 to BH3•, which is biologically inactive and leads to eNOS uncoupling—creating a vicious cycle in which more ROS are produced instead of NO.23,24
Another potential mechanism for eNOS uncoupling involves the bioavailability of l-arginine or its inhibitor, asymmetric dimethyl-l-arginine (ADMA). l-Arginine supplementation has been shown to partially alleviate ED in various animal and human subject models.25–28 This does not seem to be associated with substantially altered global l-arginine levels, but instead with relative bioavailability at the endothelium. Endothelial cells and acute inflammatory cells such as macrophages can express arginases that locally decrease l-arginine pools and effectively starve eNOS of substrate.29–31 ADMA, in contrast, is an endogenous inhibitor of eNOS and its production is strongly governed by redox status. Both the production of ADMA by protein arginine N-methyltransferase type 1 and its subsequent degradation by dimethylarginine dimethylaminohydrolase are altered by oxidative stress. Protein arginine N-methyltransferase type 1 is more active under oxidative conditions and dimethylarginine dimethylaminohydrolase is less active, leading to increased steady-state concentrations of ADMA and eNOS inhibition and uncoupling.32
Post-translational modifications of eNOS may also contribute to pathologic uncoupling and reduction of NO production. As mentioned elsewhere in this article, dephosphorylation of Thr495 may lead to uncoupling and production of deleterious ROS. Another increasingly well-characterized modification is S-glutathionylation, which is involved in signaling under conditions of oxidative stress and may serve to protect redox-sensitive cysteine residues in the eNOS protein. S-glutathionylation, which is reversible under reducing conditions, is associated with reduced eNOS activity and enhanced production of O2•−.33,34
ALTERNATIVE NITRIC OXIDE NO PRODUCTION: NITRATE–NITRITE–NITRIC OXIDE PATHWAY
NO produced enzymatically from l-arginine by NOS isoforms may not always be sufficient to maintain appropriate endothelial homeostasis, particularly under hypoxic conditions. Recently, more research has focused on the contributions of alternative routes to NO production—namely, from the reduction of dietary nitrates and nitrites to NO.35–38 Nitrate (NO3−) and nitrite (NO2−) were previously thought to be inert metabolites of NO, and their detection was considered a surrogate for NO production.38 However, more recent studies suggest that NO3−, NO2− and NO all exist in a complex equilibrium mediated by microenvironmental stimuli, the microbiome, and dietary nitrate/nitrite consumption. A full discussion of this pathway is beyond the scope of the present review, but a summary of the relevant material can be seen in Fig. 2. Briefly, dietary NO3− is reduced by oral nitrate reductase-expressing commensal bacteria to NO2−. NO2− then has multiple fates that result in NO production, both via enzymatic and nonenzymatic means. For example, in the acidic environment of the stomach, NO2− is nonenzymatically converted to NO, which then exerts local effects. NO2− may also enter the bloodstream via gastrointestinal absorption. Here, under relative hypoxia, NO2− can be reduced by deoxyhemoglobin, releasing NO in local microcirculation. Xanthine oxidase and eNOS itself also likely reduce NO2− to NO under acidic or hypoxic conditions, such as tissue ischemia. Critically, circulating NO2− also may be oxidized to NO3−, approximately 25% of which is recirculated into the saliva by the salivary glands to maintain the signaling axis.36 The potential health benefits associated with modulating dietary NO2− and NO3− are currently under active investigation, and recent studies demonstrate that NO3− supplementation is beneficial in ischemia–reperfusion injury,39,40 pulmonary hypertension,41–43 and hypertension,44–46 among other conditions.
CLINICAL IMPLICATIONS OF ENDOTHELIAL NITRIC OXIDE AND NITRIC OXIDE REGULATION
NO signaling pathways, involving the regulation of NO production as well as downstream activation of second messengers, are heavily associated with a variety of pathologies. Here, we discuss broadly how the NO axis plays a role in both the multisystem organ dysfunction of critical illness and sepsis, as well as provide brief summaries of some of the data regarding NO signaling in specific organ pathology. This process is also summarized in Fig. 3.
Fig. 3.
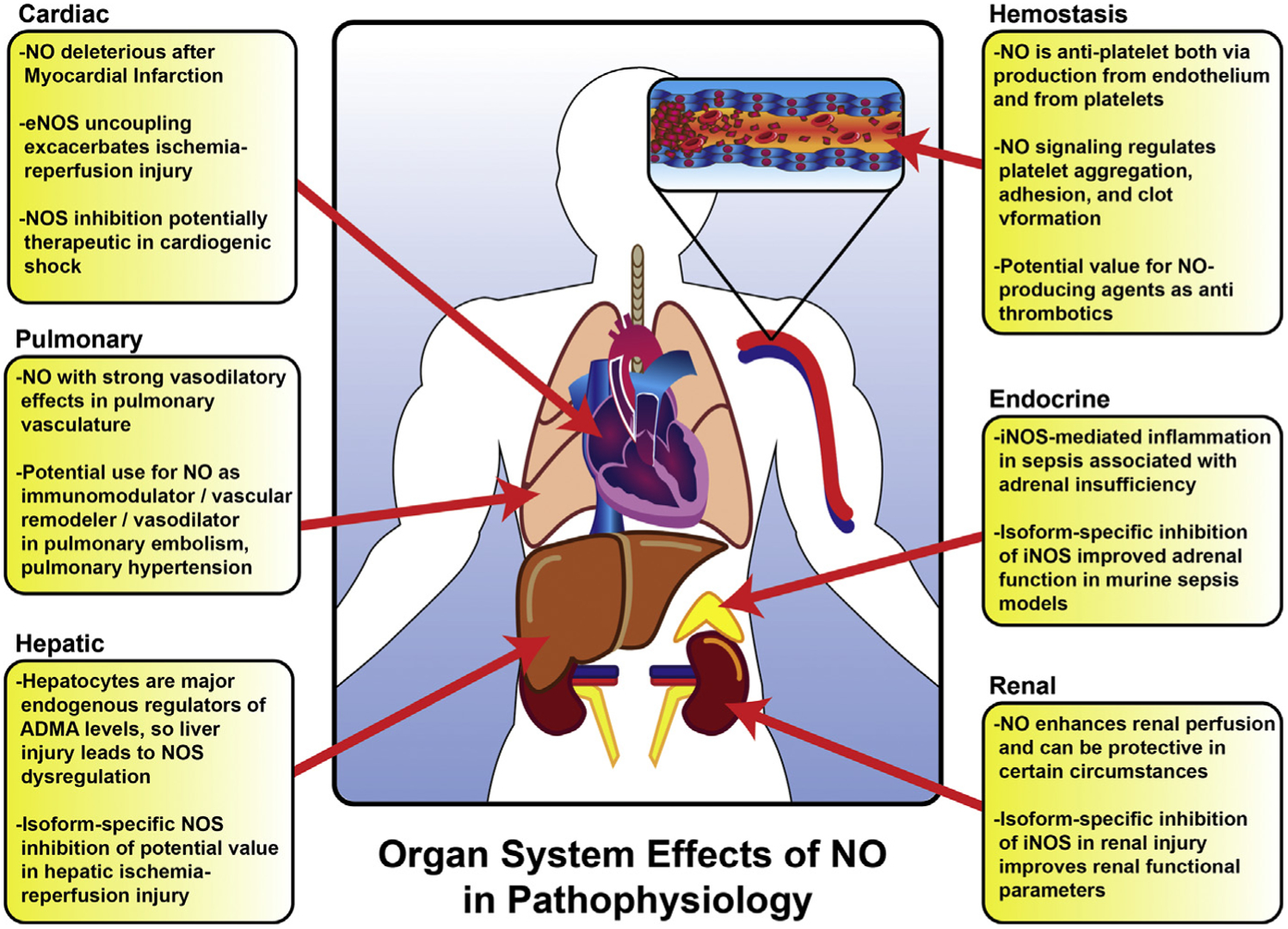
A brief overview of NO signaling in specific organ system pathophysiology. For more specific details and references, please refer to the body of the text.
Sepsis and Infectious Disease
Sepsis is characterized by a dysregulated immune response to an initial infectious insult, and has gained significant attention owing to its emergence as a leading cause of in-hospital mortality.47 ED is a key feature of sepsis and septic shock, resulting in microcirculatory dysfunction and end-organ hypoperfusion. Subsequently, NO has been studied extensively as a potential therapeutic target in treating sepsis.
NO mediates the inflammatory response to host pathogens through various mechanisms. Endogenous NO from iNOS expressed during the acute inflammatory response has been identified as a regulator of the NLRP3 inflammasome via stabilization of mitochondria.48 Using either genetic knockout (iNOS−/−) or pharmacologic inhibition of iNOS in a murine endotoxemia model, inflammatory cytokine production was significantly enhanced and associated with increased mortality.48 In another study, supplementation with l-arginine decreased neutrophil adhesion and increased rolling velocity, which was reversed with a nonselective NOS inhibitor but not a selective iNOS inhibitor.49 This result suggests that eNOS mediates these effects, although it is unclear what the relative contributions of endothelium-derived eNOS versus platelet-derived eNOS are in the regulation of the inflammatory response. Nevertheless, selective regulation of eNOS in relation to the deleterious impact of iNOS may attenuate the negative effects on microvascular tone.49
Despite its vasodilatory properties, administration of inhaled NO (iNO) has not been demonstrated to cause systemic hypotension. However, this treatment does increase nitrite levels, which might supply NO and promote beneficial vasodilatation in targeted microcirculatory beds. Because local shunting and imbalance in regional flow may contribute to sepsis-induced organ dysfunction, this paradigm has remained a therapeutic target. A randomized controlled trial of iNO compared with a sham treatment in severe sepsis was designed to address this issue. However, on evaluation the authors found no differences in microcirculatory flow, lactate clearance, or organ dysfunction, suggesting little value for NO in the later stages of sepsis.50
Methylene blue has also been studied for its potential therapeutic value in NO-mediated vasodilatation. In addition to being a powerful reducing agent, methylene blue inhibits guanylate cyclase and has been demonstrated to increase systemic vascular resistance in patients with shock. Its widespread use is currently limited owing to associations with renal failure and hyperbilirubinemia, in addition to broader questions about its efficacy.51
Although multisystem organ dysfunction is a deadly consequence of sepsis, isolated organ dysfunction can occur secondary to a wide array of noninfectious critical illnesses. Global tissue hypoperfusion results in hypoxia, accumulation of toxic metabolites, and subsequent ischemia–reperfusion injury, all of which may result in permanent organ damage or compound preexisting injuries to other organ systems. Regulation of microvascular flow mediated by NO in these circumstances may thus determine outcomes in critically ill patients. Several specific studied roles for NO and ED in the in the pathophysiology of various organ systems are discussed in the subsequent sections.
Cardiovascular System
NO has demonstrable direct inotropic effects in addition to its role in peripheral vasodilatation. Cardiomyocytes express both nNOS and eNOS constitutively, whereas iNOS expression can be induced by inflammatory mediators. Abnormally high amounts of NO produced in response to acute inflammation following myocardial infarction may have negative inotropic effects, compounding circulatory dysfunction after myocardial infarction. Moreover, NOS uncoupling leads to additional ROS formation (as detailed elsewhere in this article), which further exacerbates cardiac injury after ischemic insult.52
Owing to these effects, NOS inhibition has been studied as a potential therapeutic option for cardiogenic shock.53 In a randomized trial in patients with refractory cardiogenic shock, N-γ-nitro-l-arginine methyl ester (l-NAME), a nonspecific NOS inhibitor, was associated with significantly improved mortality at 30 days (27% vs 67%).53 Additionally, l-NAME therapy was associated with increased mean arterial blood pressure, increased urine output, and decreased requirements for intra-aortic blood pump support and need for mechanical ventilation. Similar results were seen in a small exploratory trial with the nonspecific NOS inhibitor NG-monomethyl-l-arginine (L-NMMA).54 However, a larger randomized controlled trial evaluating L-NMMA in patients with myocardial infarction and refractory cardiogenic shock (TRIUMPH) was terminated early owing to a lack of efficacy after demonstrating no difference in 30-day mortality.55 Despite these conflicting findings, NOS inhibition-associated increases in blood pressure were present in all treated patients suggesting some biological effect. It is also important to note that these drugs carry a relatively safe risk profile.55 One question that remains is whether isoform-specific NOS inhibition may provide a more robust outcome, given the confluence of NO signaling from all 3 isoforms in cardiomyocytes.
Pulmonary Function and Nitric Oxide
iNO has theoretic benefit in the lungs owing to its ability induce selective pulmonary vasodilatation and improve ventilation–perfusion mismatch.56 Studies have demonstrated that iNO exerts a vasodilatory effect only in conditions of elevated pulmonary vascular tone and has little effect in normal pulmonary vasculature.57 The US Food and Drug Administration has approved iNO administration for neonatal hypoxic respiratory failure; at present, there is no US Food and Drug Administration approval for treatment in adults. However, there are several lung pathologies for which iNO is presently being investigated as a potential therapy.
The theoretic benefit of iNO in adult acute respiratory distress syndrome is 2-fold: immune modulation in the setting of exuberant inflammation, as well as alleviation pulmonary vasoconstriction. However, a Cochrane review from 2010 found no benefit to iNO administration in acute respiratory distress syndrome, despite the theoretic benefit of direct pulmonary administration.56 Indeed, iNO administration in acute respiratory distress syndrome was associated with harmful effects, most notably renal impairment.
In pulmonary arterial hypertension (PAH) with associated right ventricular dysfunction, localized ED of the pulmonary vasculature is a known entity. In PAH, pulmonary arterial eNOS is frequently uncoupled, favoring O2•− generation and leading to less production of NO.57 The NO pathway is 1 of 3 well-characterized molecular pathways in PAH, alongside prostacyclin and endothelin-1 signaling.58 Several therapies in current clinical use target downstream elements of the NO signaling pathway, including phosphodiesterase type 5 inhibitors (sildenafil) as well as direct soluble guanylate cyclase stimulators (riociguat).59 Collectively, these agents enhance cyclic guanosine monophosphate-based signaling, which comprises the second message portion of the NO signaling cascade. Other approaches have examined the production of NO itself: the PHACeT trial was designed as a phase I clinical trial to deliver eNOS-overexpressing endothelial progenitor cells to patients with refractory PAH.60 Endothelial cells were delivered via pulmonary artery catheter to 5 patients resulting in a transient improvement in pulmonary resistance with a sustained increase in 6-minute walk distance even at 6 months.60 This promising approach requires further study to examine efficacy.
Given the vasodilatory effect of iNO on the pulmonary vasculature and potential to alter shunt fraction, iNO therapy has theoretic benefit in patients with acute pulmonary embolism. In the recently published iNOPE trial, patients with submassive pulmonary embolism and right ventricular dysfunction were randomized to receive either iNO or O2. The authors demonstrated that iNO failed to normalize troponin or echocardiogram endpoints, but patients receiving iNO were more likely to display improvements in right ventricular hypokinesis and dilation on echocardiography.61 This area is another that demands additional study for the delineation of exact benefits of iNO on pulmonary physiology.
Hepatic Function and Nitric Oxide
NO has been implicated in the hyperdynamic circulation seen in acute liver failure, subsequent to presumed overproduction of NO in splanchnic circulation.62 In a devas cularized porcine model of acute liver failure, Sharma and colleagues62 measured plasma levels of l-arginine, citrulline, ornithine, NO, and ADMA, as well as plasma arginase activities in both sham and injured animals. Plasma arginine was significantly depleted 6 hours after injury, concomitant with an increase in plasma ADMA as well as enhanced plasma arginase activity. Despite this, systemic levels of NO were not significantly decreased, suggesting that in this model of acute liver failure NO production was not limited by plasma substrate or inhibitor bioavailability. However, other data suggest that hepatic clearance of ADMA may play a central role in the propagation of multiorgan failure in shock states. Hepatocytes are the primary expressors of dimethylarginine dimethylaminohydrolase, which metabolize ADMA, and liver dysfunction through a variety of means leads to increasing plasma ADMA levels. Increasing ADMA concentration, in turn, is an independent risk factor for multiorgan failure in critically ill patients, highlighting the importance of this NOS regulatory capacity.63
Hepatic ischemia–reperfusion injury studies have similarly provided a wealth of data that could be extrapolated to whole organism shock states and global hypoperfusion. These studies have been recently reviewed elsewhere by Zhang and colleagues.64 In summary, NO has multiple complex functions in hepatic ischemia–reperfusion injury, depending on the source of production and depending on the downstream pathways favored. For example, eNOS expression in hepatic vasculature is protective against hepatic ischemia–reperfusion injury, but overexpression can be detrimental. In contrast, inhibition of iNOS induced during the inflammatory cascade after reperfusion is also protective, suggesting that overproduction of NO leads to detrimental downstream effects. Clearly, NOS regulation involves a delicate homeostasis, and extreme overproduction or underproduction of NO after liver injury can lead to harmful downstream effects.
Renal Function and Nitric Oxide
NO broadly regulates renal hemodynamics and function, which has been extensively reviewed elsewhere.65 In shock states, as modeled by ischemic models of acute renal failure, targeting the NO pathway seems to be beneficial.66 In addition to its direct vasodilatory effects, NO also suppresses production of endothelin-1, a potent vasoconstrictor. In a rat model of renal ischemia-reperfusion injury, a key pathologic driver of acute kidney injury, pretreatment with the NO donor FK409 attenuated renal dysfunction, histologic damage, and endothelin-1 overproduction. Conversely, pretreatment with the NOS inhibitor l-NAME resulted in increased endothelin-1 production, demonstrating that the inhibition of NO production led to deleterious effects.66 The effects of NOS inhibition seems to be isoform specific, similar to what is seen in the liver. In a similar model system, selective iNOS inhibition attenuated the decrease in renal oxygen delivery and microvascular oxygen pressure seen with reperfusion injury; it also improved renal oxygen extraction and consumption.67 Taken together, these findings suggest that iNOS may be contributing to harmful pathways in ischemia–reperfusion events and that inhibition of iNOS in selected scenarios may be of clinical value.
Nitric Oxide and Coagulation
Studies in human patients have demonstrated activation of coagulation factors as well as platelets in severe sepsis and septic shock.68 This may represent a protective mechanism to contain pathogen spread that is exhausted in severe sepsis and septic shock. However, overexuberant coagulation with platelet activation leads to the consumptive pathology of disseminated intravascular coagulation, a devastating condition that is associated with significantly increased mortality.69 NO signaling plays a major role in regulating platelet aggregation, adhesion, and clot formation overall.5 NO produced by healthy endothelium nominally serves a local antiplatelet role by limiting platelet adhesion and activation through activation of the NO-soluble guanylate cyclase axis in platelets themselves. There is additionally a substantial amount of eNOS expressed by platelets, suggesting that both autocrine and paracrine NO signaling occurs within developing clots, thereby limiting progression. Indeed, there is some consideration that NO-enhancing drugs may provide both cardiovascular and antiplatelet benefit, and this area of research is constantly evolving.5
Endocrine Signaling and Nitric Oxide
Sepsis is marked by low cortisol levels and blunted response to adrenocorticotrophic hormone stimulation, which together lead to adrenal insufficiency. Because NO has been linked to organ dysfunction in various other tissues, studies have focused on NO-mediated effects in the adrenal glands. In an experimental endotoxemia model, both iNOS expression and NO production were increased in the adrenal glands of mice.70 This was associated with the development of an adrenal insufficiency phenotype in these animals, as well as enhanced mitochondrial superoxide production in adrenal cortical cells. This effect was mitigated by iNOS inhibition, but was exacerbated by treatment with a systemic NO donor. Notably, increased expression of iNOS was localized to the endothelium and resident macrophages rather than adrenocortical cells, suggesting that some degree of ED contributes to adrenal insufficiency in sepsis.70
SUMMARY
NO is a critically important gaseous transmitter with a large number of context-dependent functions, and its production and consumption are tightly regulated to limit potential damage. The endothelium is a major source of NO, as it constitutively expresses eNOS and modifies NO production profiles in response to a variety of endogenous and exogenous stressors. ED occurs when this signaling axis is perturbed, limiting the ability of the vasculature and local structures either to produce NO itself or to respond appropriately to its presence. This is a central pathologic process in a number of different acute and chronic conditions, and NO and its downstream signaling partners are thus a prime therapeutic target. Although enthusiasm regarding iNO as a therapy is partially hindered by its exorbitant cost, numerous pharmacologic approaches for NO delivery have been researched and more are certainly in development.
Underscoring this, there are currently 101 registered clinically trials in the United States involving NO that are actively recruiting subjects. These trials span a wide breadth of disease processes, echoing the diverse effects and widespread therapeutic potential of this gaseous mediator. With the increasing recognition of the roles of specific NOS isoforms in certain tissues and a focus on optimizing NO delivery given its intrinsic properties, future work stands poised to optimize this pharmacologic target and provide a helpful toolbox to mitigate ED associated with critical illness.
KEY POINTS.
Nitric oxide is a small highly reactive signaling molecule that is a primary determinant of vascular homeostasis.
Nitric oxide production is tightly regulated through transcriptional and post-translational control of 3 main nitric oxide synthase isoforms.
Endothelial nitric oxide synthase is constitutively expressed in platelets and endothelial cells and is associated with maintenance of appropriate vascular tone, regulation of angiogenesis, and regulation of platelet function.
In disease states nitric oxide production can be deleterious through production of reactive oxygen and nitrogen species, and nitric oxide signaling plays a major role in the pathophysiology of numerous systemic illnesses including sepsis.
Nitric oxide and its effect on endothelial function continues to be a promising target for pharmacologic manipulation in a diverse number of critical illnesses.
Footnotes
DISCLOSURE
The authors have nothing to disclose.
REFERENCES
- 1.Sturtzel C Endothelial cells. In: Sattler S, Kennedy-Lydon T, editors. The Immunology of Cardiovascular Homeostasis and Pathology. Advances in Experimental Medicine and Biology, vol. 1003. Cham: Springer; 2017. [Google Scholar]
- 2.Incalza MA, D’Oria R, Natalicchio A, et al. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol 2018;100(June 2017):1–19. [DOI] [PubMed] [Google Scholar]
- 3.Tousoulis D, Kampoli A-M, Tentolouris Nikolaos Papageorgiou C, et al. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol 2012;10(1):4–18. [DOI] [PubMed] [Google Scholar]
- 4.Vallet B Bench-to-bedside review: endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Crit Care 2003;7(2):130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gresele P, Momi S, Guglielmini G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem Pharmacol 2019;166(June):300–12. [DOI] [PubMed] [Google Scholar]
- 6.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J 2012;33(7):829–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou L, Zhu D-Y. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009;20(4):223–30. [DOI] [PubMed] [Google Scholar]
- 8.Melikian N, Seddon MD, Casadei B, et al. Neuronal nitric oxide synthase and human vascular regulation. Trends Cardiovasc Med 2009;19(8):256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lind M, Hayes A, Caprnda M, et al. Inducible nitric oxide synthase: good or bad? Biomed Pharmacother 2017;93:370–5. [DOI] [PubMed] [Google Scholar]
- 10.Förstermann U, Closs EI, Pollock JS, et al. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994; 23(6_pt_2):1121–31. [DOI] [PubMed] [Google Scholar]
- 11.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Integr Comp Physiol 2003;284(1): R1–12. [DOI] [PubMed] [Google Scholar]
- 12.Fulton D, Gratton J-P, McCabe TJ, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999;399(6736):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixit M, Loot AE, Mohamed A, et al. Gab1, SHP2, and protein kinase a are crucial for the activation of the endothelial NO synthase by fluid shear stress. Circ Res 2005;97(12):1236–44. [DOI] [PubMed] [Google Scholar]
- 14.Lin MI, Fulton D, Babbitt R, et al. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem 2003;278(45):44719–26. [DOI] [PubMed] [Google Scholar]
- 15.Fleming I Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 2010;459(6):793–806. [DOI] [PubMed] [Google Scholar]
- 16.Ignarro LJ, Harbison RG, Wood KS, et al. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther 1986;237(3):893–900. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2872327. [PubMed] [Google Scholar]
- 17.Rapoport RM, Draznin MB, Murad F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 1983;306(5939):174–6. [DOI] [PubMed] [Google Scholar]
- 18.Förstermann U, Mülsch A, Böhme E, et al. Stimulation of soluble guanylate cyclase by an acetylcholine-induced endothelium-derived factor from rabbit and canine arteries. Circ Res 1986;58(4):531–8. [DOI] [PubMed] [Google Scholar]
- 19.Zeiher AM, Fisslthaler B, Schray-Utz B, et al. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res 1995;76(6):980–6. [DOI] [PubMed] [Google Scholar]
- 20.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A 1991;88(11):4651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murohara T, Asahara T, Silver M, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 1998;101(11):2567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aicher A, Heeschen C, Mildner-Rihm C, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med 2003; 9(11):1370–6. [DOI] [PubMed] [Google Scholar]
- 23.Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003;111(8):1201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuzkaya N, Weissmann N, Harrison DG, et al. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem 2003;278(25):22546–54. [DOI] [PubMed] [Google Scholar]
- 25.Hishikawa K, Nakaki T, Suzuki H, et al. Role of L-arginine-nitric oxide pathway in hypertension. J Hypertens 1993;11(6):639–45. [DOI] [PubMed] [Google Scholar]
- 26.Ishikawa K, Calzavacca P, Bellomo R, et al. Effect of selective inhibition of renal inducible nitric oxide synthase on renal blood flow and function in experimental hyperdynamic sepsis. Crit Care Med 2012;40(8):2368–75. [DOI] [PubMed] [Google Scholar]
- 27.Drexler H, Zeiher AM, Meinzer K, et al. Correction of endothelial dysfunction in coronary microcirculation of hypercholesterolaemic patients by L-arginine. Lancet 1991;338(8782–8783):1546–50. [DOI] [PubMed] [Google Scholar]
- 28.Rossitch E, Alexander E, Black PM, et al. L-arginine normalizes endothelial function in cerebral vessels from hypercholesterolemic rabbits. J Clin Invest 1991; 87(4):1295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu W, Kaneko FT, Zheng S, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 2004;18(14):1746–8. [DOI] [PubMed] [Google Scholar]
- 30.Ming X-F, Barandier C, Viswambharan H, et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation 2004;110(24):3708–14. [DOI] [PubMed] [Google Scholar]
- 31.Berkowitz DE, White R, Li D, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003;108(16):2000–6. [DOI] [PubMed] [Google Scholar]
- 32.Karbach S, Wenzel P, Waisman A, et al. eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr Pharm Des 2014;20(22): 3579–94. [DOI] [PubMed] [Google Scholar]
- 33.Chen C-A, Wang T-Y, Varadharaj S, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010;468(7327):1115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zweier JL, Chen C-A, Druhan LJ. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid Redox Signal 2011;14(10):1769–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qu XM, Wu ZF, Pang BX, et al. From nitrate to nitric oxide: the role of salivary glands and oral bacteria. J Dent Res 2016;95(13):1452–6. [DOI] [PubMed] [Google Scholar]
- 36.Ma L, Hu L, Feng X, et al. Nitrate and nitrite in health and disease. Aging Dis 2018;9(5):938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeMartino AW, Kim-Shapiro DB, Patel RP, et al. Nitrite and nitrate chemical biology and signalling. Br J Pharmacol 2019;176(2):228–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlstrom M, Montenegro MF. Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J Intern Med 2019;285(1):2–18. [DOI] [PubMed] [Google Scholar]
- 39.Yang T, Zhang X-M, Tarnawski L, et al. Dietary nitrate attenuates renal ischemia-reperfusion injuries by modulation of immune responses and reduction of oxidative stress. Redox Biol 2017;13:320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeddi S, Khalifi S, Ghanbari M, et al. Effects of nitrate intake on myocardial ischemia-reperfusion injury in diabetic rats. Arq Bras Cardiol 2016;107(4): 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malikova E, Carlström M, Kmecova Z, et al. Effects of inorganic nitrate in a rat model of monocrotaline-induced pulmonary arterial hypertension. Basic Clin Pharmacol Toxicol 2019. 10.1111/bcpt.13309. [DOI] [PubMed] [Google Scholar]
- 42.Cortés-Puch I, Sun J, Schechter AN, et al. Inhaled nebulized nitrite and nitrate therapy in a canine model of hypoxia-induced pulmonary hypertension. Nitric Oxide 2019;91:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch CD, Gladwin MT, Freeman BA, et al. Enterosalivary nitrate metabolism and the microbiome: intersection of microbial metabolism, nitric oxide and diet in cardiac and pulmonary vascular health. Free Radic Biol Med 2017;105:48–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lara J, Ashor AW, Oggioni C, et al. Effects of inorganic nitrate and beetroot supplementation on endothelial function: a systematic review and meta-analysis. Eur J Nutr 2016;55(2):451–9. [DOI] [PubMed] [Google Scholar]
- 45.Ashor AW, Lara J, Siervo M. Medium-term effects of dietary nitrate supplementation on systolic and diastolic blood pressure in adults: a systematic review and meta-analysis. J Hypertens 2017;35(7):1353–9. [DOI] [PubMed] [Google Scholar]
- 46.Kapil V, Khambata RS, Robertson A, et al. Dietary nitrate provides sustained blood pressure lowering in hypertensive patients: a randomized, phase 2, double-blind, placebo-controlled study. Hypertension 2015;65(2):320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall MJ, Levant S, Defrances CJ. Trends in inpatient hospital deaths: national hospital discharge survey, 2000–2010. NCHS Data Brief 2013;(118):1–8. [PubMed] [Google Scholar]
- 48.Mao K, Chen S, Chen M, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 2013;23(2): 201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan R, Kirschenbaum LA, Larow C, et al. Augmentation of platelet and endothelial cell enos activity decreases sepsis-related neutrophil-endothelial cell interactions. Shock 2010;33(3):242–6. [DOI] [PubMed] [Google Scholar]
- 50.Trzeciak S, Glaspey LJ, Dellinger RP, et al. Randomized controlled trial of inhaled nitric oxide for the treatment of microcirculatory dysfunction in patients with sepsis. Crit Care Med 2014;42(12):2482–92. [DOI] [PubMed] [Google Scholar]
- 51.Weiner MM, Lin H-M, Danforth D, et al. Methylene blue is associated with poor outcomes in vasoplegic shock. J Cardiothorac Vasc Anesth 2013;27(6):1233–8. [DOI] [PubMed] [Google Scholar]
- 52.Umar S, Van Der Laarse A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem 2010;333(1–2):191–201. [DOI] [PubMed] [Google Scholar]
- 53.Cotter G, Kaluski E, Milo O, et al. LINCS: L-NAME (a NO synthase inhibitor) in the treatment of refractory cardiogenic shock: a prospective randomized study. Eur Heart J 2003;24(14):1287–95. [DOI] [PubMed] [Google Scholar]
- 54.Cotter G, Kaluski E, Blatt A, et al. L-NMMA (a nitric oxide synthase inhibitor) is effective in the treatment of cardiogenic shock. Circulation 2000;101(12): 1358–61. Available at: http://ovidsp.ovid.com/ovidweb.cgi?T5JS&PAGE5=reference&D5emed5&NEWS5N&AN52000114347. [DOI] [PubMed] [Google Scholar]
- 55.The TRIUMPH Investigators. Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock. JAMA 2007;297(15):1657. [DOI] [PubMed] [Google Scholar]
- 56.Afshari A, Brok J, Møller AM, et al. Inhaled nitric oxide for acute respiratory distress syndrome and acute lung injury in adults and children. Cochrane Database Syst Rev 2010;7. 10.1097/sa.0b013e318242c0bc. [DOI] [PubMed] [Google Scholar]
- 57.Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;188(6):639–46. [DOI] [PubMed] [Google Scholar]
- 58.Huertas A, Tu L, Guignabert C. New targets for pulmonary arterial hypertension: going beyond the currently targeted three pathways. Curr Opin Pulm Med 2017; 23(5):377–85. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe H Treatment selection in pulmonary arterial hypertension: phosphodiesterase type 5 inhibitors versus soluble guanylate cyclase stimulator. Eur Cardiol 2018;13(1):35–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Granton J, Langleben D, Kutryk MB, et al. Endothelial NO-synthase gene-enhanced progenitor cell therapy for pulmonary arterial hypertension: the PHACeT Trial. Circ Res 2015;117(7):645–54. [DOI] [PubMed] [Google Scholar]
- 61.Kline JA, Puskarich MA, Jones AE, et al. Inhaled nitric oxide to treat intermediate risk pulmonary embolism: a multicenter randomized controlled trial. Nitric Oxide 2019;84:60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma V, Ten Have GAM, Ytrebo L, et al. Nitric oxide and l -arginine metabolism in a devascularized porcine model of acute liver failure. Am J Physiol Gastrointest Liver Physiol 2012;303(3):G435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferrigno A, Di Pasqua LG, Berardo C, et al. Liver plays a central role in asymmetric dimethylarginine-mediated organ injury. World J Gastroenterol 2015; 21(17):5131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y-Q, Ding N, Zeng Y-F, et al. New progress in roles of nitric oxide during hepatic ischemia reperfusion injury. World J Gastroenterol 2017;23(14):2505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnan SM, Kraehling JR, Eitner F, et al. The impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) signaling cascade on kidney health and disease: a preclinical perspective. Int J Mol Sci 2018;19(6). 10.3390/ijms19061712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurata H, Takaoka M, Kubo Y, et al. Protective effect of nitric oxide on ischemia/reperfusion-induced renal injury and endothelin-1 overproduction. Eur J Pharmacol 2005;517(3):232–9. [DOI] [PubMed] [Google Scholar]
- 67.Legrand M, Almac E, Mik EG, et al. l -NIL prevents renal microvascular hypoxia and increase of renal oxygen consumption after ischemia-reperfusion in rats. Am J Physiol Renal Physiol 2009;296(5):F1109–17. [DOI] [PubMed] [Google Scholar]
- 68.Mavrommatis AC, Roussos C, Zakynthinos S, et al. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit Care Med 2000;28(2): 451–7. [DOI] [PubMed] [Google Scholar]
- 69.Iba T, Levi M, Levy JH. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Semin Thromb Hemost 2019. 10.1055/s-0039-1694995. [DOI] [PubMed] [Google Scholar]
- 70.Wang CN, Duan GL, Liu YJ, et al. Overproduction of nitric oxide by endothelial cells and macrophages contributes to mitochondrial oxidative stress in adrenocortical cells and adrenal insufficiency during endotoxemia. Free Radic Biol Med 2015;83:31–40. [DOI] [PubMed] [Google Scholar]