

Abstract
Focused ultrasound can deliver energy safely and non-invasively into tissues at depths of centimetres. Here we show that the genetics and cellular functions of chimeric antigen receptor T cells (CAR-T cells) within tumours can be reversibly controlled by the heat generated by short pulses of focused ultrasound via a CAR cassette under the control of a promoter for the heat-shock protein. In mice with subcutaneous tumours, locally injected T cells with the inducible CAR and activated via focused ultrasound guided by magnetic resonance imaging mitigated on-target off-tumour activity and enhanced the suppression of tumour growth, compared with the performance of non-inducible CAR-T cells. Acoustogenetic control of the activation of engineered T cells may facilitate the design of safer cell therapies.
Optogenetics enables the control of specific molecular events and cellular functions in living systems with high spatiotemporal resolutions. However, optogenetics cannot reach deep tissues, with the penetration depth of light typically limited at micrometre to millimetre scales1. Ultrasound can be focused to deliver mechanical energy safely and non-invasively into small volumes of tissue deep inside the body up to tens of centimetres1. The rapidly oscillating pressure of focused ultrasound (FUS) waves and the resultant cycles of mechanical loading/unloading can lead to local heat generation in biological tissues. Aided by magnetic resonance imaging (MRI) thermometry, FUS has been widely applied clinically to ablate tumours and to control drug delivery, vasodilation, neuromodulation2 and transgene expression3–7. Transcription factors and genetic circuits have also been engineered to convert the FUS-generated heat into genetic regulation to control microbial systems in vivo8. However, there is a lack of general methods using FUS to control mammalian cell functions in vivo for therapeutic applications.
Chimeric antigen receptor (CAR) T-cell therapy, where T cells are genetically programmed with redirected specificity against malignant cells, is becoming a paradigm-shifting approach for cancer treatment, especially for blood cancers9. However, major challenges remain for solid tumours before CAR-based immunotherapy can be widely adopted. For instance, the non-specific targeting of the CAR-T cells against normal tissues (on-target off-tumour toxicities) can be life-threatening: off-tumour toxicities against the lung, the brain and the heart have caused multiple cases of deaths9–12. Immunosuppressive corticosteroid therapy and suicide-gene engineering are relatively effective in suppressing off-tumour toxicities and related cytokine release syndrome, but they fail to discriminate between beneficial T-cell functions and toxic side effects13–15. Synthetic biology and elegant genetic circuits have been used to enhance specificity and reduce off-tumour toxicity by creating chemically inducible dimerization of split CARs, inhibitory CARs and synthetic Notch (SynNotch) to control CAR activation10,16–20. However, given the extensive overlaps of antigens between solid tumours and normal tissues, especially those under conditions of tissue injury/inflammation21, it remains very difficult to identify ideal antigens and their combinations to differentiate tumours from normal tissues. There is thus an urgent need for high-precision control of CAR-T cells to confine the activation at local sites of solid tumours. Recently, we demonstrated that ultrasound signals can be amplified by microbubbles coupled to cells engineered with the mechanosensor Piezo1 to precisely control CAR- T-cell activation22. However, the presence of microbubbles as cofactors limits the application of this system in vivo. Here we have engineered a class of inducible CAR-T cells that can be acoustogenetically and directly controlled by FUS without any exogenous cofactor. We show that short-pulsed FUS stimulation can activate the engineered T cells at the desired time and location to suppress tumour growth in vivo with greater safety than standard CAR-T cells.
Results
Heat-induced reporter gene activation.
We propose to genetically engineer T cells with inducible CAR cassettes that can be remotely and directly activated without any exogenous cofactor, by MRI-guided FUS at local tumour sites for recognizing and eradicating tumour cells (Fig. 1a).
Fig. 1 |. Heat-inducible gene activation.
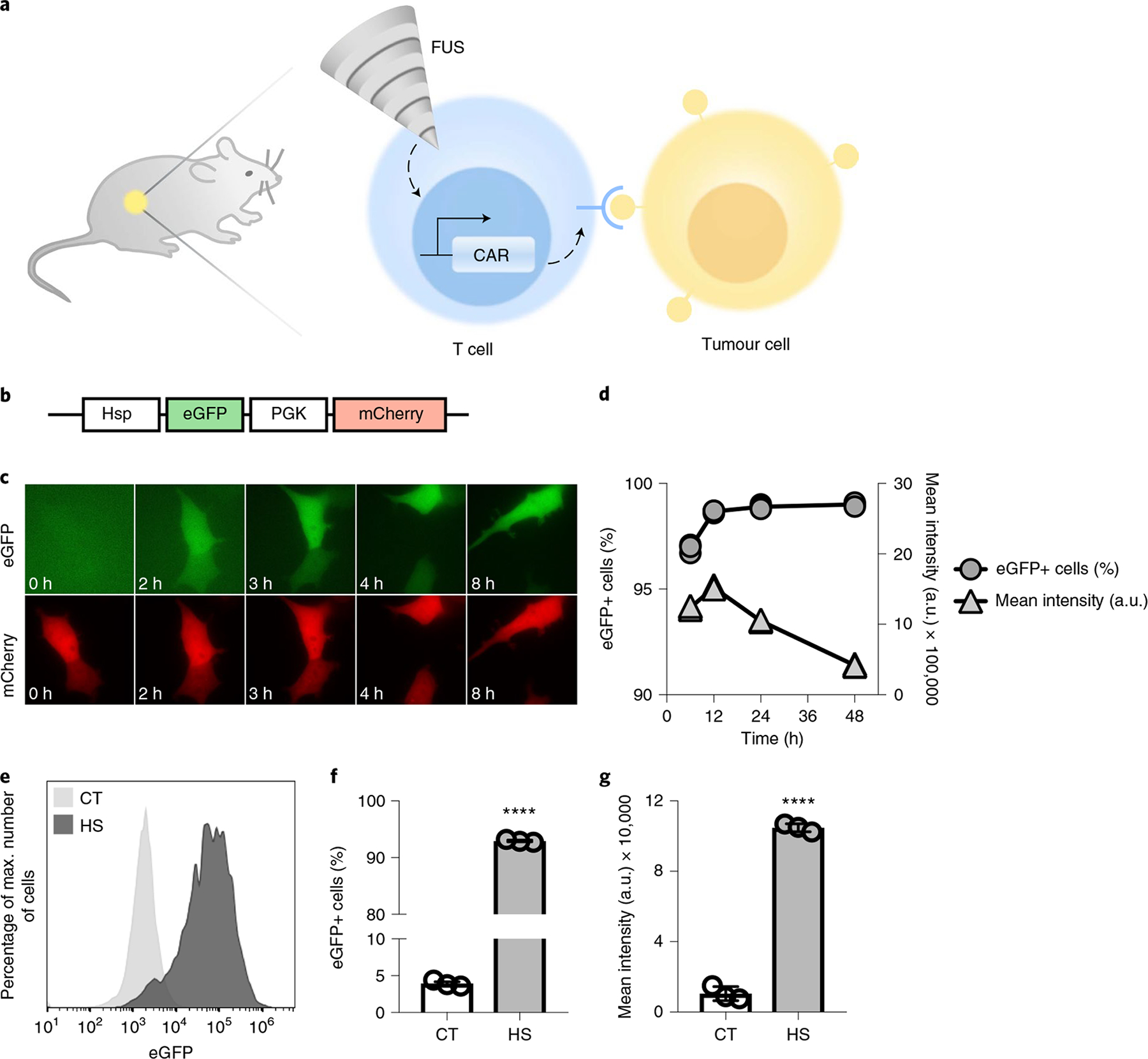
a, Design of the FUS-CAR-T-cell therapy technology. MRI-guided FUS induces localized activation of FUS-CAR-T cells to recognize and eradicate target tumour cells. b, Schematics of the dual-promoter eGFP reporter. c,d, HEK293 T cells containing the dual-promoter reporter received a 15 min, 43 °C HS at t = 0 h. Shown are fluorescent images of inducible eGFP and constitutive mCherry (c) and the percentages of eGFP+ cells and their mean fluorescence intensities (d). e–g, Gene induction in primary human T cells with the dual-promoter eGFP reporter. Shown are representative flow cytometry profiles of eGFP expression (e), the percentages of eGFP+ cells (****P = 5.5 × 10−14) (f) and their mean fluorescence intensities (****P = 5.0 × 10−10) (g). Plots in f and g are parts of complete bar graphs shown in Supplementary Fig. 3d,e, with one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. Bar heights and error bars represent means of three biological replicates ± s.e.m. mCherry+ cells were gated for eGFP expression analysis.
We first tested the inducible activation of a reporter eGFP under the control of the heat-shock-protein promoter (Hsp). We assembled a dual-promoter reporter construct containing the Hsp-driven eGFP and a constitutive mouse phosphoglycerate kinase 1 promoter (PGK)-driven mCherry (Fig. 1b). HEK293 T cells infected with the reporter lentivirus (Supplementary Fig. 1a) were heated at 43 °C for 15 min. Real-time fluorescence imaging revealed that the heat-induced eGFP expression started as early as 2 h after heat shock (HS) and persisted throughout the course of observation (Fig. 1c and Supplementary Video 1). Quantitative tracking of the dynamics of heat-induced eGFP expression by flow cytometry showed that an average of 97% of the cells expressed eGFP at 6 h post HS, and the percentage increased to 99% at 12 h and remained stable for 2 d, while the mean fluorescence intensity peaked at 12 h followed by a steady decrease (Fig. 1d). We then investigated the inducible effect of HS in primary human T cells hosting the dual-promoter eGFP reporter (Supplementary Fig. 1b). A 15 min HS induced an eGFP expression in an average of 93% of the engineered T cells, in contrast to a background of 4% in control cells without HS (Fig. 1e,f). The mean fluorescence intensity of the eGFP+ cells in the HS group was tenfold of that in the control group without HS (Fig. 1g).
Heat-induced CAR expression and its functionality in Jurkat and primary human T cells.
To convert the transient heat stimulation to sustained gene activation and cellular functions for therapeutic actions, we integrated the Cre–lox gene switch into the inducible system. The design is composed of two constructs, one containing the Hsp-driven Cre recombinase and the PGK-driven membrane c-Myc tag for cell sorting (‘inducible Cre’, Fig. 2a), and the other containing a lox-flanked ZsGreen-STOP sequence between PGK and an anti-CD19 CAR (‘lox-stop CAR reporter’, Fig. 2a). As such, the excision of the STOP cassette mediated by the transiently expressed heat-induced Cre can cause a switch from ZsGreen expression to a sustained CD19CAR production.
Fig. 2 |. Heat-inducible CD19CAR expression and functionality in Jurkat and primary T cells.
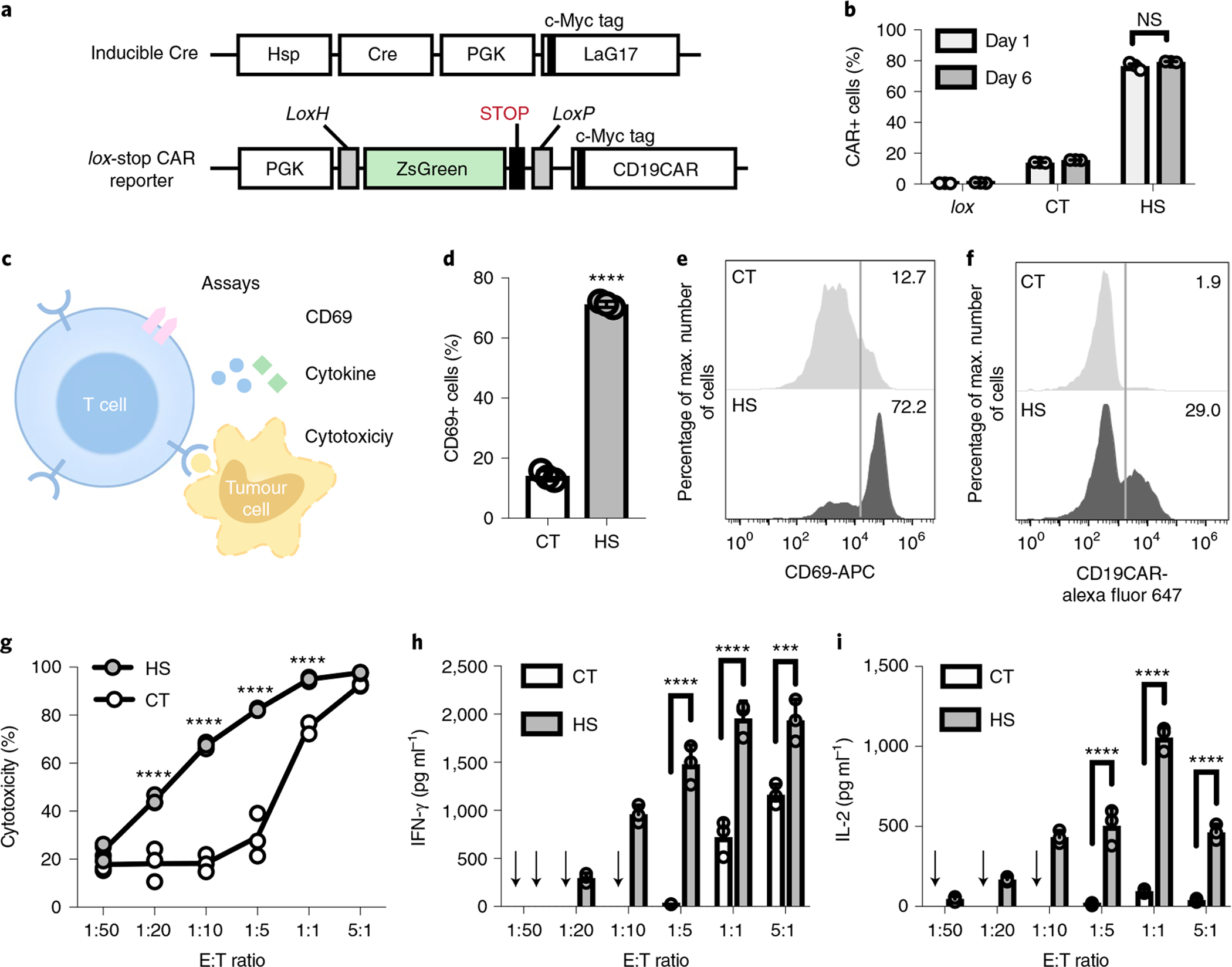
a, Schematics of transgenes: inducible Cre and lox-stop CAR reporter. b, Inducible CAR expression in Jurkat cells hosting the lox-stop CAR reporter alone (lox), or both transgenes in a with HS or without HS (CT); NS, no significant difference, P = 0.086. c, Schematics of assays accessing the functionality of the heat-induced CAR-T cells, including CD69 expression, cytotoxicity and cytokine release. d, The percentage of CD69+ cells in Jurkat with both transgenes in a. ****P = 6.1 × 10−6, two-tailed Student’s t-test. e, Representative flow cytometry histograms of CD69 expression in d. f, Representative flow cytometry histograms of the percentage of CD19CAR-expressing cells in primary T cells with both transgenes in a. g–i, T cells from f were co-cultured with Nalm-6 tumour cells at various E:T ratios for 24 h. Shown are quantified cytotoxicities of the T cells (****P = 8.3 × 10−8 at 1:20, ****P = 3.1 × 10−13 at 1:10, ****P = 5.9 × 10−14 at 1:5, ****P = 1.2 × 10−5 at 1:1) (g), and the associated release of cytokines IFN-γ (****P = 5.2 × 10−6 at 1:5, ****P = 8.7 × 10−6 at 1:1, ****P = 6.1 × 10−4 at 5:1) (h) and IL-2 (****P = 1.0 × 10−6 at 1:5, ****P = 4.9 × 10−10 at 1:1, ****P = 4.8 × 10−6 at 5:1) (i). Arrows in h and i indicate that cytokine level was not detectable. Two-way ANOVA followed by Sidak’s multiple comparisons test was used in b, g–i. Bar heights and error bars represent means of three biological replicates ± s.e.m.
We first tested this system in Jurkat T-cell lines (Supplementary Fig. 2a). A 15 min HS induced CAR expression in an average of 77% of the cells when measured 24 h after HS (day 1), in contrast to an average basal level of 14% in control cells without HS and a minimal leakage of 0.6% in cells infected with the lox-stop CAR reporter alone (Fig. 2b and Supplementary Fig. 2b). The heat-induced CAR expression remained stable when measured 6 d after HS (day 6, Fig. 2b and Supplementary Fig. 2b). We further examined the functionality of the induced CD19CAR in the engineered cells (Fig. 2c). Engineered Jurkat cells with HS or without (CT, control) HS were co-cultured with CD19-expressing Nalm-6 tumour cells for 24 h. Quantification of the expression level of CD69 (an early T-cell activation marker) revealed an average of 71% CD69+ cell population in the engineered Jurkat cells in the HS group, in contrast to an average of 14% in the CT group (Fig. 2d,e). These results indicate that the HS-induced CD19CAR is efficient for functional changes in engineered Jurkat T cells.
We then examined our system in primary human T cells (Fig. 2a,c and Supplementary Fig. 2c). Antibody staining showed that a 15 min HS induced CAR expression in 29% of the T cells, in contrast to 1.9% in control cells without HS (Fig. 2f). The heat-inducible CAR-T cells were then co-cultured with firefly luciferase (Fluc)-expressing Nalm-6 cells at different effector-to-target (E:T) ratios for cytotoxicity assays. The luminescence of the remaining Nalm-6 cells was quantified after a 24 h co-culture. The heat-stimulated T cells (HS) demonstrated increased cytotoxicity with increased E:T ratio, with the largest contrast in cytotoxicity being observed between the HS and the control (CT) T cells at E:T = 1:5, eliminating 82.9% and 29.3% of the target tumour cells, respectively (Fig. 2g). The heat-stimulated CAR-T cells also secreted substantially higher concentrations of cytokines interferon-γ (IFN-γ) and interleukin-2 (IL-2) than the control cells (Fig. 2h,i), verifying the functional capability gained by the HS-induced CAR-T cells.
While the continuous 15 min HS led to strong gene inductions (Figs. 1 and 2), it may cause toxicity to cells23. We therefore investigated the effect of different HS patterns in primary human T cells (Supplementary Fig. 3). Our results showed that longer HS resulted in more cell death; however, pulsed HS was able to alleviate this toxicity while achieving induction levels comparable to that in response to continuous HS with the same total heating time (Supplementary Fig. 3). In particular, a pulsed HS with 50% duty cycle and a total heating time of 15 min (Supplementary Fig. 3a, pattern 2) caused a strong induction of eGFP expression in 91.4% of the engineered T cells with minimal toxicity (92.2% cell viability) measured 24 h after HS (Supplementary Fig. 3b–d). Therefore, we applied this HS pattern (Supplementary Fig. 3a, pattern 2) for in vivo therapeutic studies.
MRI-guided FUS-induced gene activation in phantom and in vivo.
MRI-guided FUS enables the delivery of thermal energy in vivo at confined local regions with high spatiotemporal resolutions3,4. We integrated a FUS system with a 7T MRI as described in Methods. An annular array transducer was placed above the target region of the object (phantoms or small animals) in the MRI bore. MR images were acquired and transferred to Thermoguide software to calculate the temperature of the target region in real time; this temperature was then fed back to the proportional-integral-derivative controller to automatically regulate the output power of the FUS generator, maintaining the temperature of the target region at the desired level (Fig. 3a and Supplementary Fig. 4).
Fig. 3 |. MRI-guided FUS-inducible gene activation in phantom and in vivo.
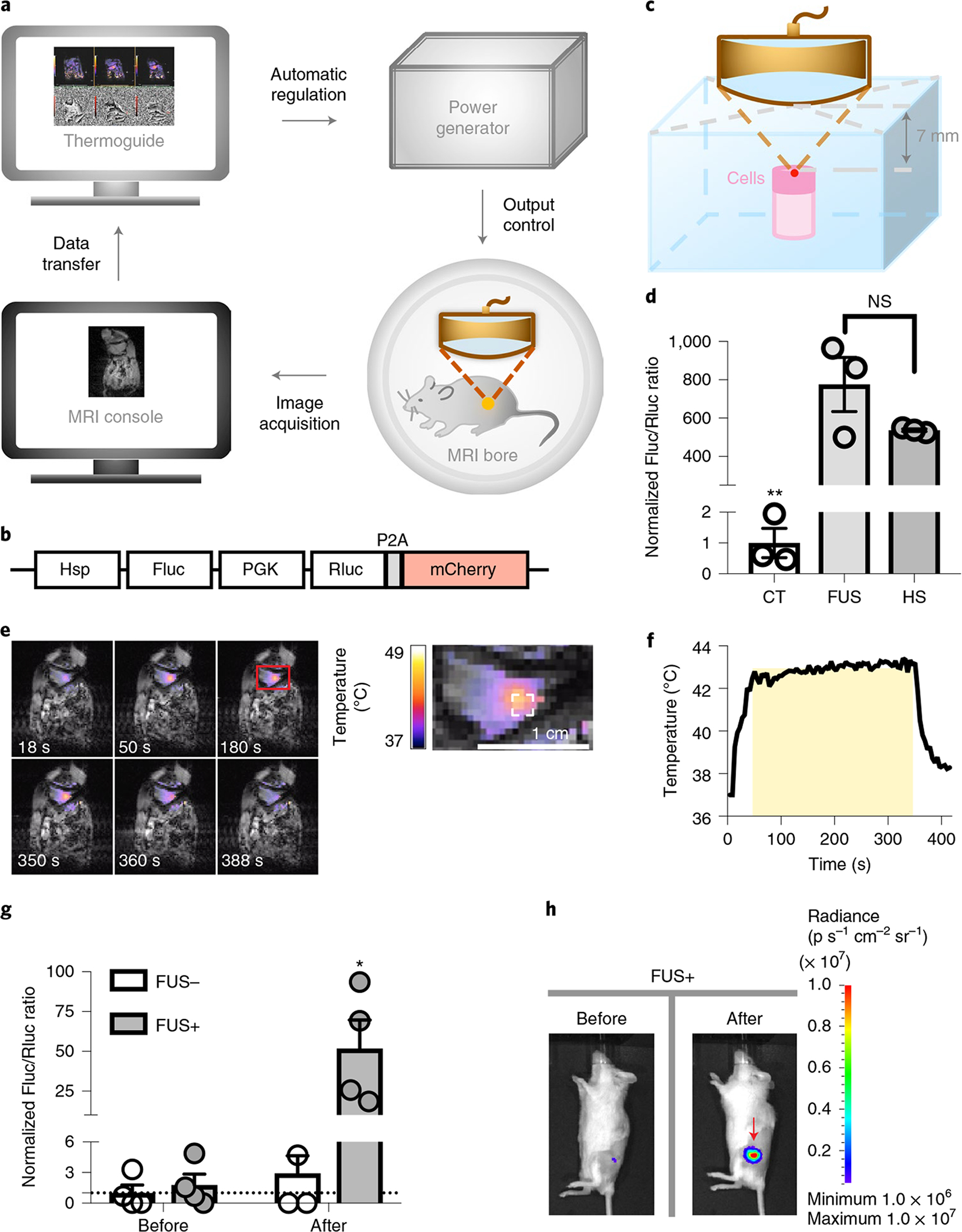
a, Schematics of the MRI-guided FUS system. b, Schematics of the dual-luciferase reporter. P2A, 2A self-cleaving peptide derived from porcine teschovirus-1. c, Experimental setup of FUS stimulation on cells in a tofu phantom. d, Gene induction level in Nalm-6 cells containing the dual-luciferase reporter with three pulses of 5 min heating by MRI-guided FUS in tofu phantom (FUS) or by thermal cycler (HS). Gene induction level was quantified by the Fluc/Rluc ratio and normalized to CT. **P = 0.0013 between CT and FUS; **P = 8.4 × 10−3 between CT and HS; NS, no significant difference, P = 0.1767. One-way ANOVA followed by Tukey’s multiple comparisons test. N = 3 biological replicates. e, Left: colour-coded temperature map superimposed on MRI images at different time points during a 5 min FUS stimulation at 43 °C on the hindlimb of an anaesthetised mouse. Right: close-up of the red rectangle region on the left. The dashed white square outlines the region of interest for temperature regulation. f, The average temperature of the region of interest during FUS stimulation in e. The yellow shadow represents the predefined target temperature (43 °C) and duration (300 s) of FUS stimulation. g, Gene induction in vivo by MRI-guided FUS on Nalm-6 cells with the dual-luciferase reporter. FUS+ or FUS−, with or without two pulses of 5 min FUS stimulation at 43 °C. Gene induction was quantified by the in vivo Fluc/Rluc ratio and normalized to the ‘FUS−, Before’ group, as indicated by the dotted line (y = 1). *P = 1.09 × 10−2 between ‘FUS+, After’ and ‘FUS−, After’; **P = 9.3 × 10−3 between ‘FUS+, After’ and ‘FUS+, Before’. Two-way ANOVA followed by Sidak’s multiple comparisons test. N = 4 mice. h, Representative BLI images of Fluc expression before and after FUS stimulation in g. Bar heights and error bars represent mean ± s.e.m.
We transduced Nalm-6 cells with a lentiviral dual-luciferase reporter containing inducible Fluc and constitutive Rluc (Hsp–Fluc–PGK–Rluc–mCherry; Rluc, Renilla luciferase; Fig. 3b) and embedded the cells in a tofu phantom (Fig. 3c and Methods). We then focused the ultrasound on the embedded cells by changing the focal distance in the z direction. Three pulses of 5 min FUS stimulations caused a significant induction of gene expression as quantified by the Fluc/Rluc ratio of the cells assayed 8 h later (Fig. 3d and Methods). The induction level was comparable to that of the positive control using a thermal cycler with the same heating pattern (Fig. 3d), suggesting that this approach can acoustogenetically control gene activation in engineered cells with high efficiency.
We then used MRI-guided FUS to control local temperature in vivo (Fig. 3e,f and Supplementary Video 2) and tested the FUS-induced gene activation using Nalm-6 cells with the dual-luciferase reporter in vivo in mice. Significant gene induction was observed in the injected cells with only two pulses of 5 min FUS stimulation (FUS+, after), compared with the basal level (FUS+, before) and the control groups (FUS−, before and after) (Fig. 3g,h).
FUS-inducible cytotoxicity of the engineered CAR-T cells in vivo.
To test the cytotoxicity of the FUS-inducible CAR (FUS-CAR) T cells in vivo, we subcutaneously injected Fluc+ Nalm-6 cells on both sides of NOD scid gamma (NSG) mice to generate matched bilateral tumours (Fig. 4a). Four days later, engineered CD19CAR T cells were injected subcutaneously at both tumour sites locally, followed by three pulses of 5 min FUS stimulation at 43 °C on the left but not on the right tumour (Fig. 4a). Our results showed that the growth of the left tumour was significantly suppressed compared with that of the right tumour, demonstrating the in vivo cytotoxicity of the FUS-CAR-T cells (Fig. 4b,c). We included a control group where bilateral-tumour-bearing mice received FUS stimulation on one side, with neither site subjected to the injection of T cells (Supplementary Fig. 5a). The tumours on both sides exhibited similar growth profiles, indicating that FUS itself (with the chosen pattern) had negligible impact on tumour growth (Supplementary Fig. 5b,c). An additional control where bilateral-tumour-bearing mice received local injections of naive T cells on both tumours followed by FUS stimulation on the left tumour also showed similar growth profiles of both tumours (Supplementary Fig. 6). Therefore, our results demonstrated that FUS can be used to precisely control the cytotoxicity of FUS-CAR-T cells in vivo against target tumour cells.
Fig. 4 |. Tumour suppression by FUS-CAR-T cells in vivo.
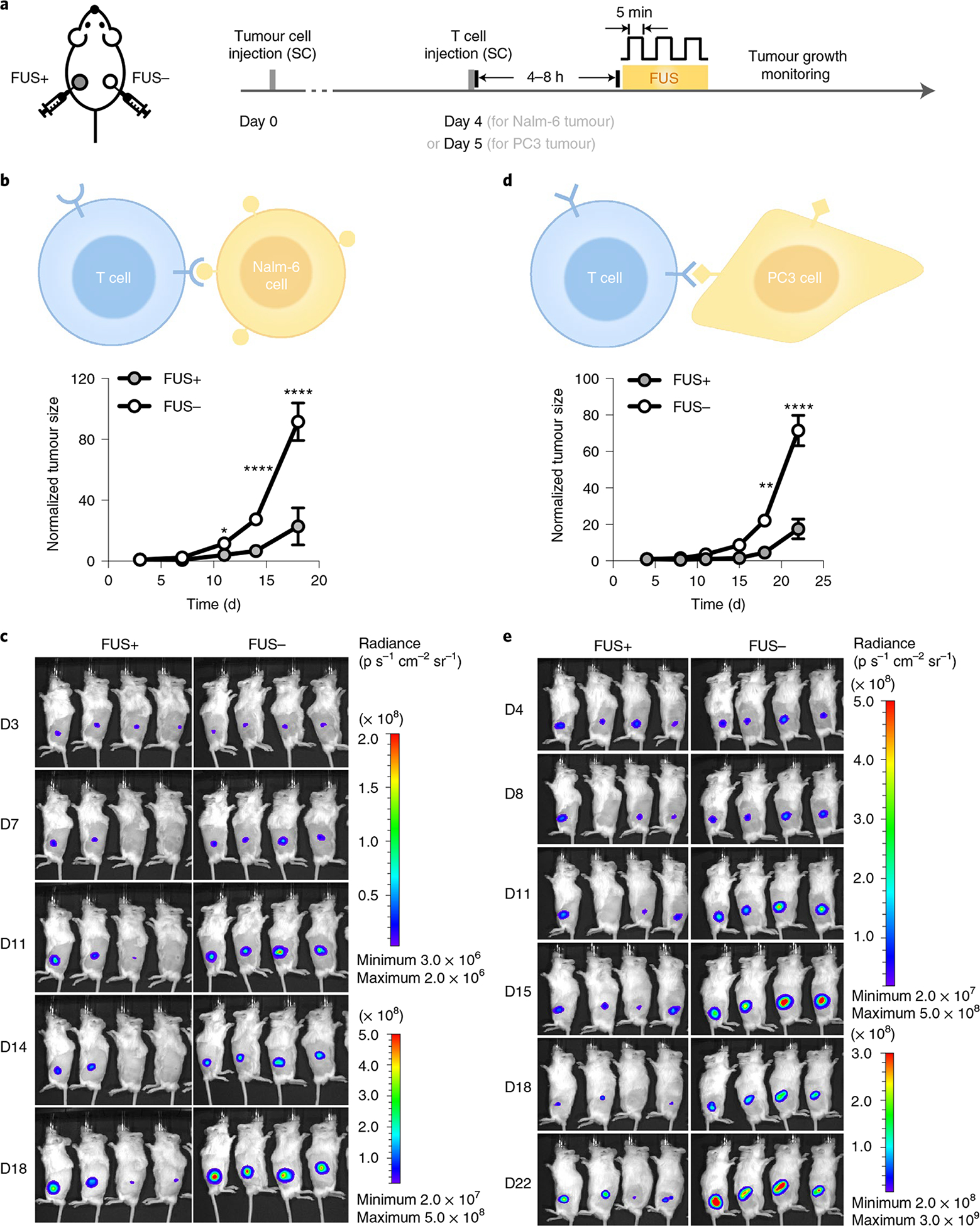
a, Timeline of in vivo experiment using NSG mice bearing matched bilateral tumours. Both tumours received local subcutaneous (SC) injection of engineered CAR-T cells. The tumour on the left received FUS stimulation (FUS+) and the one on the right received no FUS (FUS−). b–e, Normalized tumour sizes (b,d) and representative BLI images (c,e) of Nalm-6 tumours (b,c) and PC3 tumours (d,e). Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. In b, *P = 2.7 × 10−2 at day 11 (D11), ****P = 4.52 × 10−6 at D14, ****P = 5.12 × 10−12 at D18. In d, **P = 2.6 × 10−3 at D18, ****P = 3.5 × 10−9 at D22. Two-way ANOVA followed by Sidak’s multiple comparisons test. Data points and error bars represent means of four mice ± s.e.m.
To further validate the effectiveness of our technology, we examined the cytotoxicity of the FUS-CAR-T cells against more established tumours. We subcutaneously injected Fluc+ Nalm-6 cells on the left side of NSG mice to generate a single tumour model. Ten days after tumour challenge, FUS-CAR-T cells were locally injected at the tumour site and activated by FUS stimulation. The control group received an injection of naive T cells followed by the same FUS stimulation (Extended Data Fig. 1a). FUS-CAR-T cells significantly suppressed tumour growth compared with the control, showing the efficacy of FUS-CAR-T-cell therapy in treating larger tumours (Extended Data Fig. 1b–d).
Moreover, we tested this acoustogenetic technology in controlling inducible CAR-T cells against other types of tumours, particularly solid tumours. We engineered human prostate cancer PC3 cells to express the prostate-specific membrane antigen (PSMA) and Fluc, and engineered primary human T cells with the Cre–lox mediated FUS-inducible anti-PSMA CAR (PSMACAR; Extended Data Fig. 2a,b). We verified the functionality of the FUS-inducible PSMACAR T cells through in vitro co-culture cytotoxicity assays and the associated cytokine assays (Extended Data Fig. 2c,e). We then generated matched bilateral subcutaneous PC3 tumours (PSMA+, Fluc+) in NSG mice; five days later we subcutaneously injected FUS-inducible PSMACAR T cells locally at both tumour sites. The tumour region on the left side was treated with three pulses of 5 min FUS, while that on the right remained unstimulated. Consistently, the tumours with FUS stimulation showed significantly inhibited growth compared with the controls (Fig. 4d,e). We further collected the tumour tissues on the last day of monitoring (day 22, 17 d after FUS stimulation; Methods) and quantified the related messenger RNA amount. CD3 mRNA in the FUS-treated tumours averaged threefold of that in the untreated ones, indicating more T-cell infiltration and/or expansion in the FUS-treated solid prostate tumours (Supplementary Fig. 7a). Moreover, the amount of Cre-mediated recombined CAR mRNA in the FUS-treated tumours was ninefold of that in the untreated controls, verifying the FUS-induced DNA recombination and subsequent CAR expression in the engineered T cells at the tumour sites (Supplementary Fig. 7 and Methods). Additionally, in a mock control where the bilateral PC3 tumour-bearing mice received local injections of naive T cells on both tumours followed by FUS stimulation only on the left tumour, no significant difference in growth between the left and right tumours was observed (Supplementary Fig. 8). These results demonstrate the efficacy of FUS-based acoustogenetics in the control of CAR-T cells for treating different types of tumours in vivo, including solid tumours of prostate cancer.
Safety of FUS-CAR-T-cell therapy.
We hypothesized that FUS-CAR-T cells have less on-target off-tumour toxicity than standard CAR-T cells with constitutive CAR expression. To test this, we adopted the above-mentioned bilateral Nalm-6 tumour model and treated the tumour on one side (the proximal tumour) with a local injection of the same number of standard CD19CAR T (Supplementary Fig. 9 and Methods) or FUS-inducible CD19CAR T cells. The distal tumour was used to mimic normal tissues expressing the target antigen and received no treatment (Supplementary Fig. 10a,b). Standard CAR-T cells not only eliminated the proximal tumour but also attacked the normal-tissue-mimicking distal tumour (Fig. 5a,b and Supplementary Fig. 10c). In contrast, FUS-CAR-T cells, after FUS activation, dramatically inhibited the growth of the proximal tumour, but spared the distal tumour (Fig. 5c,d and Supplementary Fig. 10d). Using tumour growth in control mice (with bilateral tumours, without T-cell treatment; Extended Data Fig. 3) as reference, we quantified the in vivo cytotoxicity of the standard CAR and FUS-CAR-T cells on the last day of monitoring (Methods). Standard CAR-T cells demonstrated 100% and 99.8% cytotoxicity against proximal and distal tumours, respectively; FUS-CAR-T cells showed comparable effectiveness of 95.7% against proximal tumours, but merely 9.8% effectiveness against distal tumours (Fig. 5e). These results suggest that FUS-CAR-T-cell therapy is safer, with significantly less on-target off-tumour toxicity than the standard CAR-T-cell therapy.
Fig. 5 |. Comparison of on-target off-tumour side effect between standard and FUS-CAR-T-cell therapy.
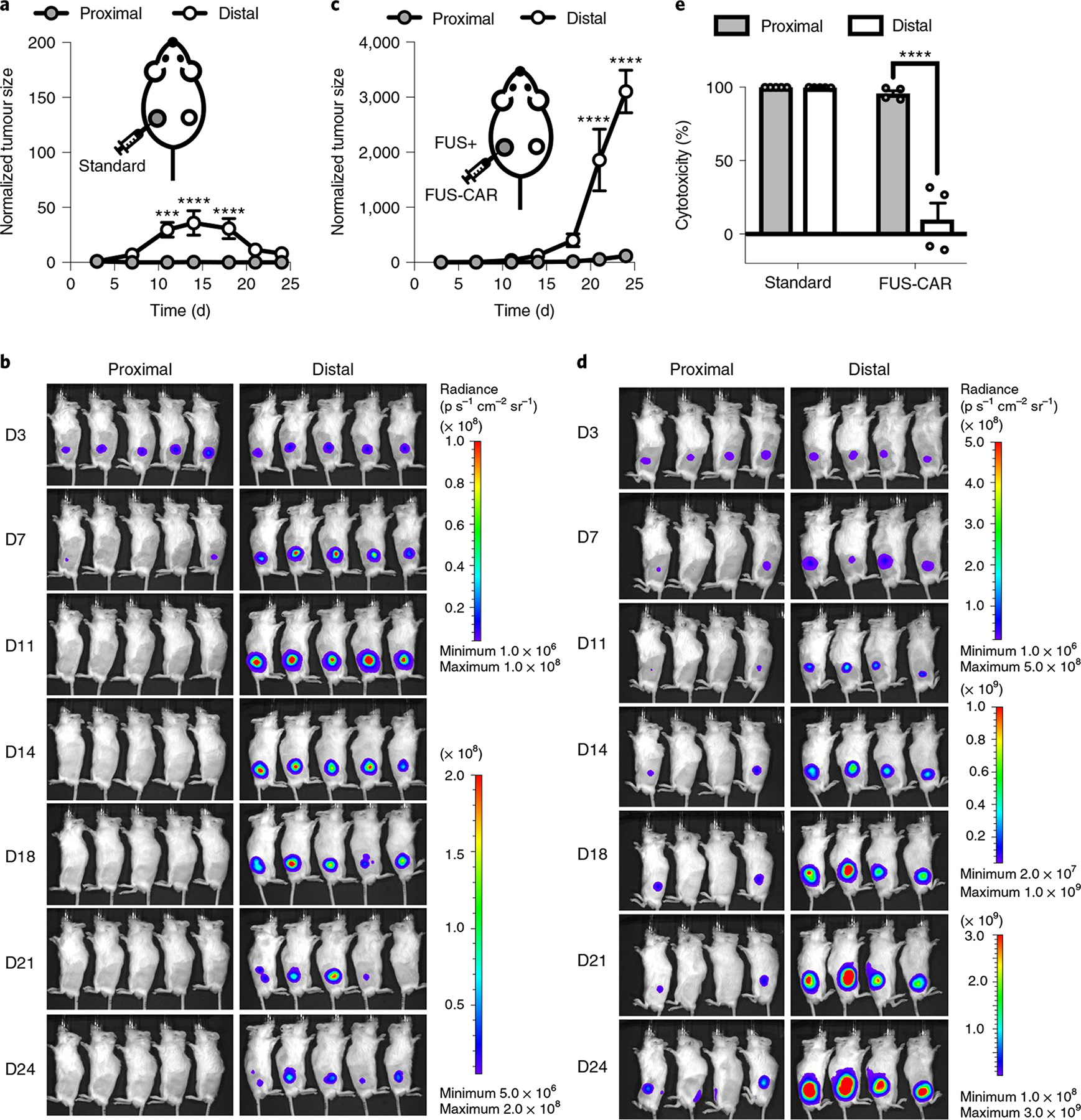
a–d, Tumour growth (a,c) and BLI images (b,d) of the proximal and distal tumours in mice receiving standard CD19CAR T cells (a,b) or FUS-inducible CD19CAR (FUS-CAR) T cells (c,d). Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. In a, ***P = 1.6 × 10−4 at D11, ****P = 5.5 × 10−6 at D14, ****P = 9.0 × 10−5 at D18. In c, ****P = 3.7 × 10−6 at D21, ****P = 1.7 × 10−9 at D24. e, Quantified in vivo cytotoxicities of standard and FUS-CAR-T cells against proximal and distal tumours on day 24 after tumour challenge. ****P = 3.2 × 10−8 between proximal and distal tumours in the FUS-CAR group. Two-way ANOVA followed by Sidak’s multiple comparisons test. N = 5 and 4 mice for standard and FUS-CAR groups, respectively. Data points in a and c and bar heights in e represent means. Error bars represent s.e.m.
Developing a clinically compatible and reversible FUS-CAR-T cell.
To minimize the usage of exogenous components in our technology for higher clinical compatibility, we further characterized a FUS-CAR-T cell where CAR expression was directly driven by Hsp (Hsp-CAR) without the Cre–lox switch, with PGK-driven mCherry as an indicator of gene delivery efficiency (Fig. 6a and Supplementary Fig. 11a). The FUS-CAR-T cell containing Hsp-CAR represents a reversible system: CAR expression reached an average of 43.9% in primary Hsp-CAR-T cells 6 h after the first HS (43 °C, 15 min), which dwindled down to 10.5% at 12 h and returned to a basal level comparable to that of the untreated control by 24 h (Fig. 6b,c). When treated with a second HS (43 °C, 15 min), we observed CAR expression in an average of 44.2% of cells, followed by the same degradation dynamics (Fig. 6c), indicating the reversible nature of the Hsp-CAR system. We examined the functionality of the reversible FUS-CAR-T cells using the cytotoxicity assay as described above, and observed stronger cytotoxicity of the FUS-CAR cells with HS activation than those without (Fig. 6d). The largest contrast was seen at an E:T ratio of 1:5, with the activated reversible FUS-CAR-T cells eliminating 83% of tumour cells compared with the 36% elimination by the control T cells (Fig. 6d). The specific killing was verified by cytokine secretions (Fig. 6e).
Fig. 6 |. Reversible FUS-CAR-T cells.
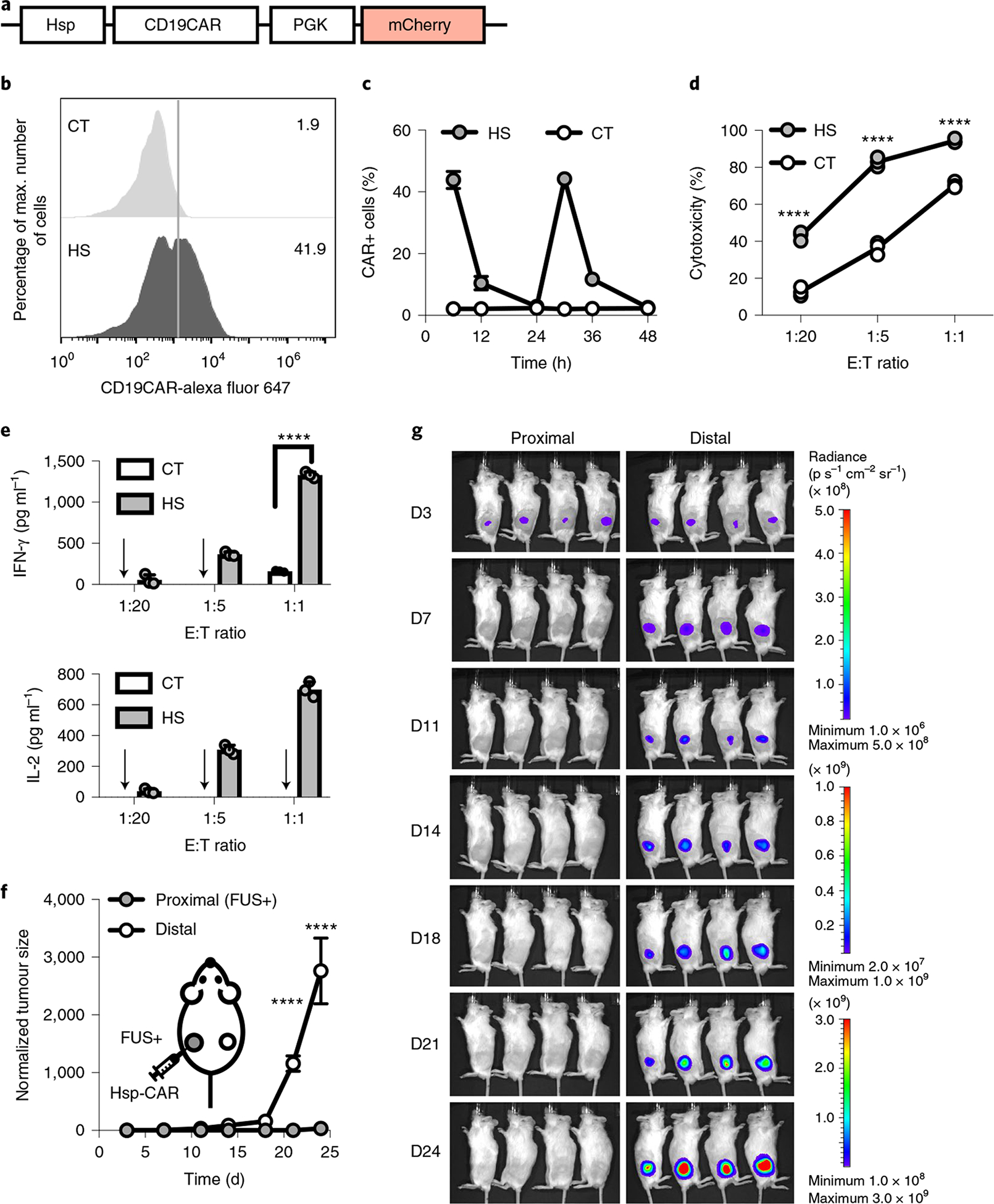
a, Schematics of the Hsp-CD19CAR transgene. b, Representative flow cytometry data of CAR expression profile 6 h after HS. c, Dynamics of CAR expression in cells with repeated HS stimulation at 0 h and 24 h. d,e, Cytotoxicities of the engineered T cells against Nalm-6 cells at different E:T ratios (left to right: ****P = 7.9 × 10−9, ****P = 5.2 × 10−11, ****P = 1.1 × 10−7) (d) and the associated IFN-γ (****P = 2.8 × 10−14) and IL-2 cytokine secretion (e). Arrow, cytokine level not detectable. N = 3 biological replicates. f,g, Normalized tumour size (f) and BLI images (g) of bilateral tumour-bearing mice with local administration of reversible FUS-CAR-T cells followed by FUS stimulation at the proximal tumour on D4 and D7. Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. In f, ****P = 3.6 × 10−5 at D21, ****P = 9.0 × 10−15 at D24. N = 4 mice. Two-way ANOVA followed by Sidak’s multiple comparisons test. Data points in c and f and bar heights in e represent means. Error bars represent s.e.m.
To investigate the performance of the reversible FUS-CAR-T cells in vivo, we generated bilateral Fluc+ Nalm-6 tumours in NSG mice. The tumour on one side (proximal tumour) received a local injection and FUS activation of the reversible FUS-CAR-T cells; this treatment was repeated on day 4 and day 7 (Supplementary Fig. 11b). The contralateral distal tumour received no treatment. Our results showed that the growth of the proximal tumour was drastically inhibited compared with that of the distal tumour (Fig. 6f,g), demonstrating sufficient efficacy and minimal side effect of the reversible FUS-CAR-T cells. In a control group where the proximal tumour was repeatedly stimulated by FUS on day 4 and day 7 but without T-cell administration, the proximal tumour demonstrated similar aggressiveness as the distal tumour, suggesting that the repeated FUS stimulation itself had no direct effect on tumour growth (Extended Data Fig. 4). We also included a control group where the proximal tumour received local injections of the reversible FUS-CAR-T cells on day 4 and day 7 but without FUS stimulation (Extended Data Fig. 5a). While the growth of the proximal tumour slightly slowed down, probably due to basal CAR expressions and allogeneic responses of the T cells, it still demonstrated an average of 1,318-fold growth by day 24 (Extended Data Fig. 5b,c). Taken together, our results demonstrate the reversibility, efficacy and safety of the reversible FUS-CAR-T cells that minimized the use of clinically untested exogenous components.
Discussion
We developed a FUS-based approach to acoustogenetically control the engineered T cells to transduce ultrasound signals into genetic and cellular activations for therapeutic applications in vivo. This acoustogenetics technology enables the activation of CAR-T cells at confined tissue regions, allowing the targeting of less ideal antigens without causing non-specific off-tumour toxicity. The short-pulsed patterns of FUS stimulation should also minimize potential detrimental effects of hyperthermia and induce transient expression of synthetic protein regulators to circumvent severe immune responses. This acoustogenetic approach is highly modular, with the target CAR genes switchable to aim at different cancer types.
We first employed the Cre-mediated gene switch to convert transient FUS inputs into sustained outputs of genetic and cellular activities for sufficient therapeutic efficiency. The nature of local activation limited the number of activated cells off the tumour site and the potential non-specific toxicity against normal tissues, making the FUS-CAR-T-cell therapy safer than the standard one (Fig. 5). It is of note that our model utilizing contralaterally localized tumours as a readout should only be considered as a substitute method to investigate side effects on normal tissues, and further validation is therefore needed.
We further characterized a reversible FUS-CAR-T cell without the Cre–lox gene switch. In this design, Hsp directly drives the production of CAR under FUS stimulation (Supplementary Note). Upon the withdrawal of FUS stimulation, heat shock factors gradually dissociate from Hsp, returning Hsp and its downstream transcriptional activities to the resting state. This recovery process is relatively fast, within 45 min after HS for Drosophila Hsp70 and approximately 60 min after HS for human Hsp7024,25, allowing repeated activation of CAR by multiple FUS stimulations. Such a reversible FUS-CAR-T-cell system with multiple repeats of short-pulsed stimulations can further allow a high-efficiency therapy and prevent on-target off-tumour toxicity of standard CAR-T-cell therapy, as the T cells leaving the tumour site will no longer receive FUS stimulation and gradually lose CAR expression. The tunable FUS stimulation pattern and reversible CAR-T activation should also allow optimal efficacy and controllable exhaustion in the future.
We utilized local injection at the tumour site to deliver T cells in vivo. Local administration of CAR-T cells has been tested in animals and patients to overcome the obstacle of T-cell homing associated with intravenous delivery, and has achieved promising therapeutic effects17,26,27. Local delivery of CAR-T cells was applied in orthotopic mouse models for solid tumour types, including glioblastoma28, breast cancer brain metastases29, metastatic medulloblastoma and ependymoma30, as well as atypical teratoid/rhabdoid tumours31. Regarding the prostate tumour model in our study, the prostate is positioned near critical organ structures including the urethra and neurovascular bundles. Surgery or radiation therapy targeting the whole prostate gland to treat prevalent locally progressed prostate cancer32 may cause adverse effects that would substantially impact quality of life33,34. Local delivery and activation of FUS-CAR-T cells using clinically available MRI-guided FUS systems should allow a high degree of precision and safety in eradicating tumour cells in patients harbouring locally progressed prostate cancer34. Further work is needed before the current FUS-CAR-T cells can be used in applications requiring intravenous delivery, possibly by equipping them with additional antigen binders and/or chemokine receptors to promote trafficking, infiltration and the enrichment of these engineered cells at the tumour site before FUS activation35,36.
The short-pulsed stimulation and the biocompatible Hsp capable of inducing transient expressions of different synthetic protein regulators can potentially enhance the safety of gene therapy, circumventing detrimental host immune response. For instance, CRISPR–Cas9 proteins have been a powerful tool for research in genetic and epigenetic engineering, but can evoke adaptive immune responses and tissue damage in vivo, and are therefore potentially pathogenic if applied to correct inherited genetic defects to treat diseases37. Protein engineering to remove immunogenic epitopes and humanize these synthetic proteins to circumvent this issue can be difficult owing to the high diversity of the human leukocyte antigen (HLA) loci38. Using our acoustogenetic approach, the transiently induced Hsp-driven synthetic regulators (for example, Cas9) can be cleared in a timely manner to mitigate or evade the adaptive immune response, hence offering another option for gene editing and therapy.
Each component of this FUS-based acoustogenetics, that is, ultrasound devices, molecular thermosensors and genetic/epigenetic transducing modules, is highly modular and will continue to evolve for greater precision and reduced immunogenicity. In fact, stretchable electronic circuits are being developed to fabricate wearable patches of ultrasound transducers39. The leverage of technological advancements in different fields into FUS-based acoustogenetics should be able to drive the development of these fields to open up various frontiers. We envision that the current state of acoustogenetics is analogous to optogenetics at its infancy. Before the functional demonstration of channelrhodopsin in neuronal cells40, it was challenging to manipulate molecular activities in live cells at high spatiotemporal resolutions. With the technological integration and convergence of optics, genetic circuits and light-sensitive proteins, optogenetics is rapidly reaching its full potential. Based on this analogy, acoustogenetics may undergo a similar trajectory to provide a broadly applicable method and usher in an era of applying ultrasound for the direct and non-invasive control of genetically engineered cells for therapeutics.
Methods
Cloning.
Plasmids used in this paper are listed in Supplementary Table 1. Cloning strategies include Gibson Assembly (NEB, E2611L) and T4 ligation (NEB, M0202L). PCR was performed using synthesized primers (Integrated DNA Technologies) and Q5 DNA polymerase (NEB, M0491). The sequences of the constructed plasmids were verified by Sanger sequencing (Genewiz).
General cell culture.
Human embryonic kidney HEK293 T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, 11995115) with 10% foetal bovine serum (FBS) (Gibco, 10438026) and 1% penicillin–streptomycin (P/S) (Gibco, 15140122). Jurkat, Nalm-6 and PC3 cells were cultured in Roswell Park Memorial Institute Medium (RPMI 1640) (Gibco, 22400105) with 10% FBS and 1% P/S. Primary human T cells were cultured in complete RPMI 1640 supplemented with 100 U ml−1 recombinant human IL-2 (PeproTech, 200-02). Cells were cultured at 37 °C in a humidified 5% CO2 incubator.
Staining and flow cytometry.
Staining of cell surface markers (for example, c-Myc, CD69, and so on) for flow cytometry was performed using fluorophore-conjugated antibodies according to manufacturers’ protocols. In general, cells were washed and resuspended in 100 μl staining buffer (PBS + 0.5% BSA) containing the suggested amounts of antibodies, incubated in the dark at room temperature for suggested durations, and washed three times before flow cytometry analysis (BD Accuri C6 or SONY SH800). Gating was based on non-engineered cells with the same staining. Flow cytometry data were analysed using FlowJo software (FlowJo).
In vitro heat shock.
For Fig. 1c and Supplementary Video 1, cells were seeded in a glass bottom dish and heated at 43 °C for 15 min using a heating stage (Instec) integrated with a Nikon Eclipse Ti inverted microscope. Images were acquired in real time to obtain the kinetics of the induced fluorescent protein. For the remainder of the in vitro heat shock (HS) experiment, unless otherwise specified, cells were resuspended in a cell culture medium at a concentration of 2 × 106 cells per ml, aliquoted into 8-strip PCR tubes with 50 μl per tube, and received 43 °C HS in a thermal cycler (Bio-Rad, 1851148) with various patterns as indicated (Supplementary Table 2). Cells were returned to standard culture condition after HS. The gene induction levels were quantified by flow cytometry 12 h after HS in Fig. 1f,g and Supplementary Fig. 3d,e.
Engineered cells.
The engineered cells (excluding primary human T cells) used in this work are listed in Supplementary Table 3. Lentiviruses were used to deliver transgenes into the cells. Fluorescence-activated cell sorting (FACS), when needed, was performed at the University of California, San Diego (UCSD) Human Embryonic Stem Cell Core Facility or with the in-house SONY SH800 following standard protocols.
Quantification of CAR expression in Jurkat cells.
Jurkat cells were either transduced with a lentiviral cocktail (inducible Cre and lox-stop CAR reporter, Fig. 2a) followed by the indicated HS (Fig. 2b), or transduced with the lox-stop CAR reporter lentivirus alone without HS. CAR expression was quantified by flow cytometry of CAR antibody-stained cells (anti-mouse IgG, F(ab’)2 fragment specific antibody; Jackson ImmunoResearch, 115-606-072) 24 h after HS. Non-engineered Jurkat cells were stained with the same antibody to generate the CAR+ gate.
Quantification of CD69 expression in Jurkat cells.
Jurkat cells transduced with a lentiviral cocktail (inducible Cre and lox-stop CAR reporter, Fig. 2a) were treated with or without HS at 43 °C for 15 min, and co-cultured with target tumour cells for 24 h. The cells were then stained by an antigen-presenting cell (APC) anti-human CD69 antibody (BioLegend, 310910) and analysed by flow cytometry. ZsGreen+ cells (representing the engineered Jurkat cells) were gated for analysis of CD69 expression. Non-engineered Jurkat cells co-cultured with target tumour cells were stained with the same antibody to generate the CD69+ (APC+) gate.
Isolation, culture, transduction and MACS of primary human T cells.
Human peripheral blood mononuclear cells were isolated from buffy coats (San Diego Blood Bank) using lymphocyte separation medium (Corning, 25-072-CV) following the manufacturer’s instructions. Primary human T cells were isolated from peripheral blood mononuclear cells using Pan T Cell Isolation Kit (Miltenyi, 130-096-535) and activated with Dynabeads Human T-Expander CD3/CD28 (Gibco, 11141D) (day 0). On day 3, lentivirus concentrated using PEG-it (SBI, LV825A-1) was added to the T cells at multiplicity of infection equals 10, followed by spinoculation in a 24-well plate coated with Retronectin (Takara, T100B). T cells were further expanded and Dynabeads were removed on day 6. T cells were used for downstream applications (for example, magnetic-activated cell sorting (MACS), HS, killing assays, and so on) on days 7–10. When needed, T cells were restimulated by Dynabeads following the manufacturer’s instructions.
For in vitro and in vivo cytotoxicity studies, T cells were transduced with a lentiviral cocktail of inducible Cre and lox-stop CAR reporter (Fig. 2a). MACS was performed using Anti-c-Myc-Biotin antibodies and Anti-Biotin microbeads (Miltenyi, 130-092-471 and 130-097-046) following the manufacturer’s instructions to enrich c-Myc+ cells. A representative double positive efficiency after MACS is 69%: 95% for the c-Myc+ and 71.4% for the ZsGreen+ cells (Supplementary Fig. 2c). CAR expression in the engineered inducible CAR-T cells with or without HS was quantified using the CAR antibody as described above.
Luciferase-based cytotoxicity assay.
A constant number of 5 × 104 Fluc+ Nalm-6 cells were mixed with engineered primary human T cells with or without HS (pre-washed and resuspended with complete RPMI without IL-2) at E:T ratios of 1:50, 1:20, 1:10, 1:5, 1:1, 5:1 or no T cells (‘target cell only’). The mixtures were then cultured in round-bottom 96-well plates for 24 h, centrifuged to remove the supernatant (which was collected for quantification of cytokine production), and assayed with the Bright-Glo Luciferase Assay System (Promega, E2610) following the manufacturer’s instructions to quantify the luminescence of each sample. The cytotoxicity (%) of sample X was calculated as (1 – luminescence of X / luminescence of ‘target cell only’) × 100%.
For cytotoxicity assay using PC3 cells as the target, 1 × 104 PSMA + Fluc+ PC3 cells were seeded onto tissue-culture-treated flat-bottom 96-well plates (Corning, 3603). Except for ‘target cell only’ wells, engineered primary human T cells with or without HS (washed and resuspended with complete RPMI without IL-2) were added 6 h later at E:T ratios of 1:10, 1:5, 1:1, 5:1, 10:1 and 20:1. Luminescence was quantified 24 h after co-culture as described above.
Quantification of cytokine production.
The supernatant of effector-target cell co-culture was collected. The concentrations of cytokines IL-2 and IFN-γ were quantified using the corresponding ELISA kits (BD, 555190 and 555142). Samples with negative values were denoted as ‘cytokine level not detectable’ in figure captions and excluded from statistical analysis.
T-cell viability assay.
Non-engineered primary human T cells received HS as described above and were returned to normal culture condition. Cell viability was assessed 24 h later using the FITC Annexin V Apoptosis Detection Kit I (BD, 556547) following the manufacturer’s instructions. The cells that stained negative for both Annexin V and propidium iodide were counted as live cells.
Animals.
Animal experiments were performed following Protocol S15285 approved by the UCSD Institutional Animal Care and Use Committee (IACUC). All researchers involved in animal experiments complied with relevant animal-use guidelines and ethical regulations during this study. NSG mice (6–8 weeks old, male) purchased from Jackson Laboratory (JAX) or UCSD Animal Care Program (ACP) were used in all animal experiments.
MRI-guided FUS in animals.
The MRI-guided FUS system is composed of a 1.5 MHz 8-element annular array transducer, a 16-channel broadband radiofrequency generator, a piezo motor-based X–Y positioning stage, and a degassing and water circulation system (Image Guided Therapy). MR images acquired using a Bruker 7 T MRI system were transferred to Thermoguide software (Image Guided Therapy) to generate phase images and real-time temperature maps. Using a proportional-integral-derivative controller, the software automatically regulates the output power of the generator to maintain the temperature at the focal spot at a desired value as described elsewhere8,41.
NSG mice were shaved before FUS stimulation. Anaesthesia was induced using a 2% isoflurane–oxygen mixture and maintained with 1.5% isoflurane–oxygen mixture during FUS stimulation. The mouse was laid on its side on an MR bed containing an agarose gel pad and a surface coil. A pressure pad was placed under the mouse to monitor its respiration rate, and a rectal thermal probe was used to provide feedback for the delivery of warm air into the bore to maintain the mouse’s core temperature at approximately 37 °C. The ultrasound transducer was positioned right above the targeted region on the mouse’s hindlimb. Thin layers of SCAN ultrasound gel (Parker labs) were applied at the skin–transducer and skin–bed interfaces.
The Thermoguide software regulates the temperature in a 3 × 3 pixel square (3–4 mm2) centred at the ultrasound focus (Fig. 3e). A proportional-integral-derivative controller was used to maintain the average temperature of the target region at 6 °C above reference by controlling the output power of the FUS generator, with the reference temperature being 37 °C as measured by the rectal thermal probe. As such, the MRI-guided FUS enabled temperature elevation to 43 °C locally at the focal area in the hindlimb of an anaesthetised mouse.
FUS stimulation in tofu phantom.
For FUS stimulation of cells in the tofu phantom, Nalm-6 cells were lentivirally transduced with the dual-luciferase reporter (Fig. 3b, Hsp–Fluc–PGK–Rluc–mCherry) and FACS-sorted. The cells were resuspended in culture medium and mixed with matrigel (Corning, 354262) at 1:1 volume ratio on ice. Extra-firm tofu was cut into a 15-mm-thick pad, and an 8-mm-deep hole of 8 mm diameter was drilled from the top. A microcentrifuge tube of 7.5 mm diameter (Fisherbrand, 05-408-120) was cut to 8 mm length by removing the lid and the conical bottom, and inserted into the hole in the tofu phantom. A 150 μl cell–matrigel mixture was added into the hole (~3 mm thick) and allowed to gel at room temperature. The rest of the hole and the gap between the tube and the tofu phantom were filled up with matrigel. After gelation, the assembly was inverted and positioned onto the MR bed containing the surface coil. The ultrasound transducer was positioned above the tofu phantom with its centre aligned with that of the tube. Thin layers of ultrasound gel were applied at the tofu–transducer and tofu–bed interfaces. A thermal probe was inserted into the distal end of the tofu phantom to provide reference temperature readings.
MR images of the assembly were acquired and transferred to Thermoguide to calculate the theoretical ultrasound focal position. Test FUS shots were delivered to determine the actual focal position. Steering was applied to focus the ultrasound at the region immediately above the cells. Three pulses of 5 min FUS stimulations at 43 °C were applied. The cell–matrigel mixture was then recovered from the tube, placed in cell culture medium, and returned to a standard 37 °C cell culture incubator. After 6 h, the culture was centrifuged to remove the supernatant, and the cell–matrigel pellet was incubated in a Cell Recovery Solution (Corning, 354253) at 4 °C for 1 h to retrieve the Nalm-6 cells from the matrigel. The Fluc and Rluc luminescence of the cells was quantified using the Dual-Luciferase Reporter Assay System (Promega, E1910) following the manufacturer’s instructions.
In vivo bioluminescence imaging.
In vivo bioluminescence imaging (BLI)was performed using an In vivo Imaging System (IVIS) Lumina LT Series III (PerkinElmer). For Fluc imaging, 150 mg kg−1 d-luciferin (GoldBio, LUCK) was administered intraperitoneally. BLI started 10 min after substrate injection until peak signal was acquired. For Rluc imaging, 200 μl 0.295 mM ViviRen (Promega, P1232)42 was administered intraperitoneally. BLI started 15 min after substrate injection until peak signal was acquired. BLI of Fluc and Rluc in the same mouse, when needed, was performed 4 h apart. Images were analysed using Living Image software (PerkinElmer). For tumour aggressiveness monitoring, the integrated Fluc luminescence intensity of the tumour region was quantified and normalized to that of the same tumour on the first measurement to obtain the normalized tumour size.
FUS-inducible gene activation in vivo.
NSG mice were subcutaneously injected with 2 × 106 dual-luciferase reporter Nalm-6 cells at the hindlimb. One week later, the experimental mice received two pulses of 5 min FUS stimulation at 43 °C targeted at the implanted cells, while the control mice remained unstimulated. The in vivo Fluc and Rluc luminescence was quantified 4 h before and 12 h after FUS stimulation, as described above.
In vivo cytotoxicity of FUS-CAR-T cells.
NSG mice were subcutaneously injected with 2 × 105 Fluc+ Nalm-6 cells (or 2 × 105 PSMA + Fluc+ PC3 cells, for PC3 tumours) on both hindlimbs to generate matched bilateral tumours. Four days later (or five days later, for PC3 tumours), 1 × 106 inducible primary human CAR-T cells prepared as described above were injected subcutaneously and locally at tumour regions. Within 4–8 h after T-cell injection, three pulses of 5 min FUS stimulation targeted at 43 °C were applied on the left tumour region as described above, while the tumour on the right hindlimb received no FUS stimulation to serve as control. Tumour aggressiveness was monitored by BLI twice a week as described above.
Quantification of mRNA expression in tumour tissue.
PC3 tumours (Fig. 4d,e) were collected 22 d after tumour implantation (17 d after T-cell injection and FUS stimulation). The tumours were disrupted and homogenized, and the same amount of lysate from each tumour was used to extract total RNA with the RNeasy Mini Kit (Qiagen, 74104) followed by reverse transcription using the same amount of template RNA. Quantitative PCR (qPCR) was performed using iTaq Universal SYBRRTM Green Supermix (Bio-Rad, 1725121), the same amount of template complementary DNA and the specific primers described below. The mRNA levels were normalized to β-actin.
The first pair of specific primers were designed on human CD3γ chain to detect the presence of human T cells. The second pair of specific primers were designed based on the lox-stop PSMACAR reporter sequence to reflect CAR expression after FUS-induced Cre recombination (Supplementary Fig. 7a,b). In the second pair, the forward primer anneals from −60 bp of the mouse PGK promoter, downstream of the transcription starting site, and the reverse primer anneals from +20 bp of the PSMACAR gene43. With the presence of FUS-induced Cre recombinase, the sequence from the second half of LoxH to the first half of LoxP will be excised, resulting in a 200-bp qPCR product. Without Cre-mediated recombination, this pair of primers will theoretically generate a 984-bp fragment. We adopted a two-step qPCR protocol with combined annealing/extension at 60 °C for only 15 s to ensure the specific amplification of the 200-bp fragment, but not the 984-bp fragment, as confirmed by gel electrophoresis and Sanger sequencing of the qPCR product (Supplementary Fig. 7c,d; sequence alignment performed in Serial Cloner). Therefore, the second pair of specific primers can detect the successfully recombined CAR mRNA amount.
Single tumour model.
NSG mice were subcutaneously injected with 2 × 105 Fluc+ Nalm-6 cells on the left flank. Ten days later, 2 × 106 FUS-CAR-T cells or naive T cells were injected subcutaneously and locally at tumour regions. Both groups received three pulses of 5 min FUS stimulation targeted at 43 °C after T-cell transfer. Tumour growth was monitored by BLI and caliper measurement (volume = length × width2/2).
Preparation of standard CAR-T cells.
Primary human T cells were transduced with eGFP-tagged CD19CAR driven by the constitutive PGK promoter (Supplementary Fig. 9a). The multiplicity of infection was adjusted to achieve ~30% CAR+ percentage (indicated by eGFP+ %) efficiency to be comparable with that in the FUS-CAR-T cells after induction (Supplementary Fig. 9b and Fig. 2f). The functionality of the standard constitutive CD19CAR T cells was verified via in vitro cytotoxicity assay and cytokine secretion measurements (Supplementary Fig. 9c–e).
On-target off-tumour side effect test in vivo.
NSG mice were subcutaneously injected with 2 × 105 Fluc+ Nalm-6 cells on both hindlimbs to generate bilateral tumours. Four days later, 1 × 106 standard or FUS-CAR-T cells were injected subcutaneously and locally at the tumour on the left flank (denoted as the proximal tumour). The tumour on the right flank (denoted as the distal tumour) was used to mimic normal tissues expressing the same target antigen and received no T-cell injection. The FUS-CAR-T-group mice received FUS stimulation after T-cell injection. A ‘tumour only’ control group was also included where the mice bearing bilateral tumours received no treatment on either tumour (Extended Data Fig. 3), serving as the reference for cytotoxicity quantification. Tumour growth was monitored by BLI and caliper measurement. The in vivo cytotoxicity of experimental CAR-T cells at a given time was quantified as (1–BLI reading of experimental tumour/BLI reading of control tumour) × 100%.
For staining of tumour re-isolates, tumours were collected on day 24 after implantation, minced using razor blade, and digested with 0.5 mg ml−1 collagenase P (MilliporeSigma, 11213857001) and 1 mg ml−1 DNase I (MilliporeSigma, 10104159001) in RPMI at 37 °C with constant shaking for 30 min36. The digested tumours were passed through cell strainer (Fisher Scientific, 352235), treated with red blood cell lysis buffer (Biolegend, 420301), and stained with FITC anti-human HLA-A,B,C antibody (Biolegend, 311404) and Alexa Fluor 647 anti-human CD3 antibody (Biolegend, 300322) according to the manufactures’ manuals. The samples were then subjected to flow cytometry (Sony SH800) to analyse tumour infiltrating T cells.
Statistical analyses.
Statistical analyses were performed using Prism software (GraphPad). Detailed statistical information for each figure is provided in the corresponding figure captions.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Effectiveness of FUS-CAR T cells in the single-tumour model.
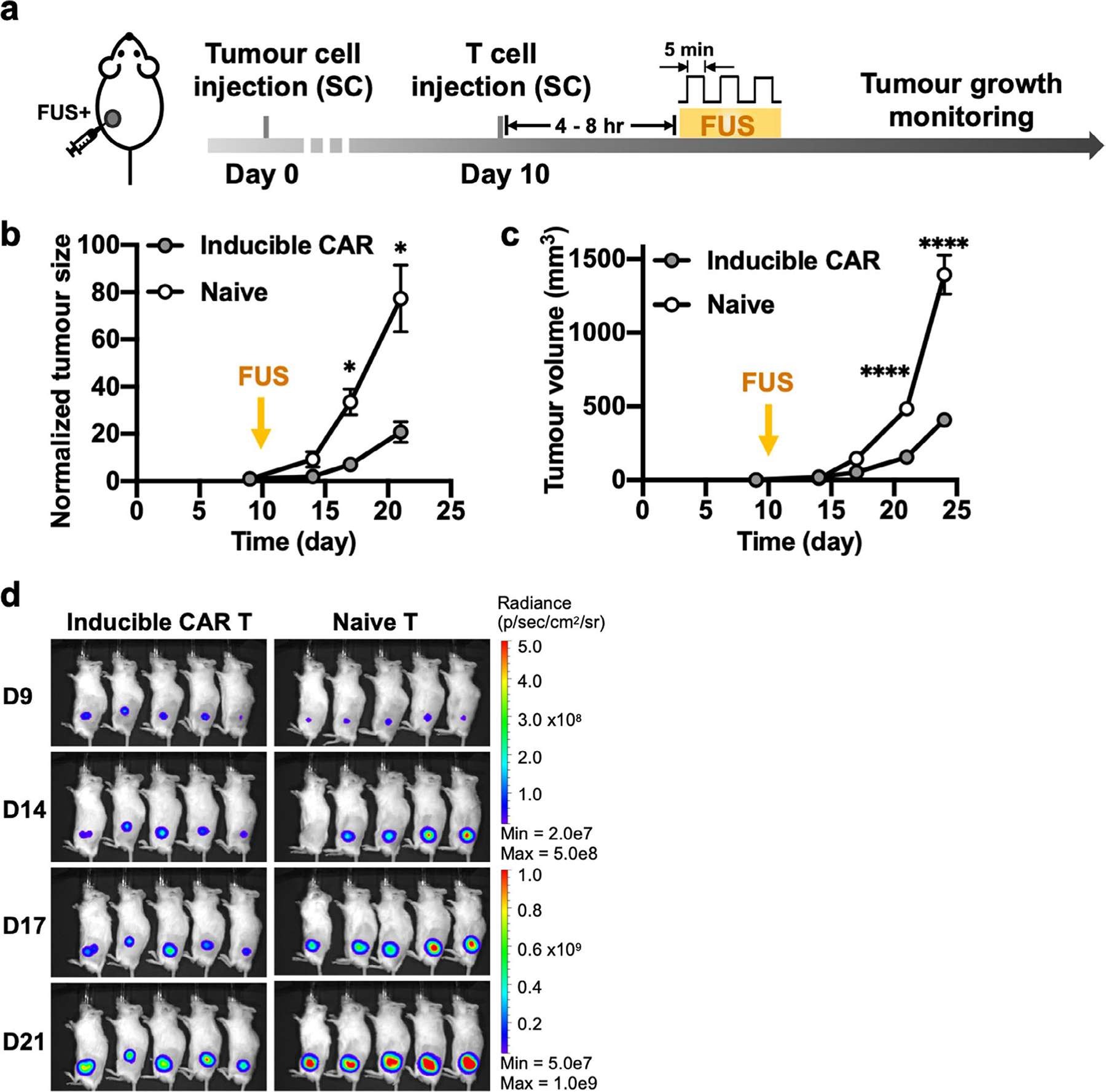
a, Timeline of the experiment. Fluc+ Nalm-6 tumour cells were injected subcutaneously into NSG mice to generate single tumour model. Ten days after tumour challenge, FUS-inducible CAR T or naive T cells were locally injected at the tumour site, followed by FUS stimulation. b-d, Normalized tumour size (*P = 0.027 at D17, *P = 0.021 at D21) (b), tumour volume (****P = 2.4 × 10−5, ****P < 1.0 × 10−15) (c) and BLI images (d). Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. Tumour volume was calculated based on caliper measurement as described in Methods. Two-way ANOVA followed by Sidak’s multiple comparisons test. Data points and error bars represent means of 5 mice ± SEM.
Extended Data Fig. 2 |. Functionality of FUS-inducible PSMACAR T cells in vitro.
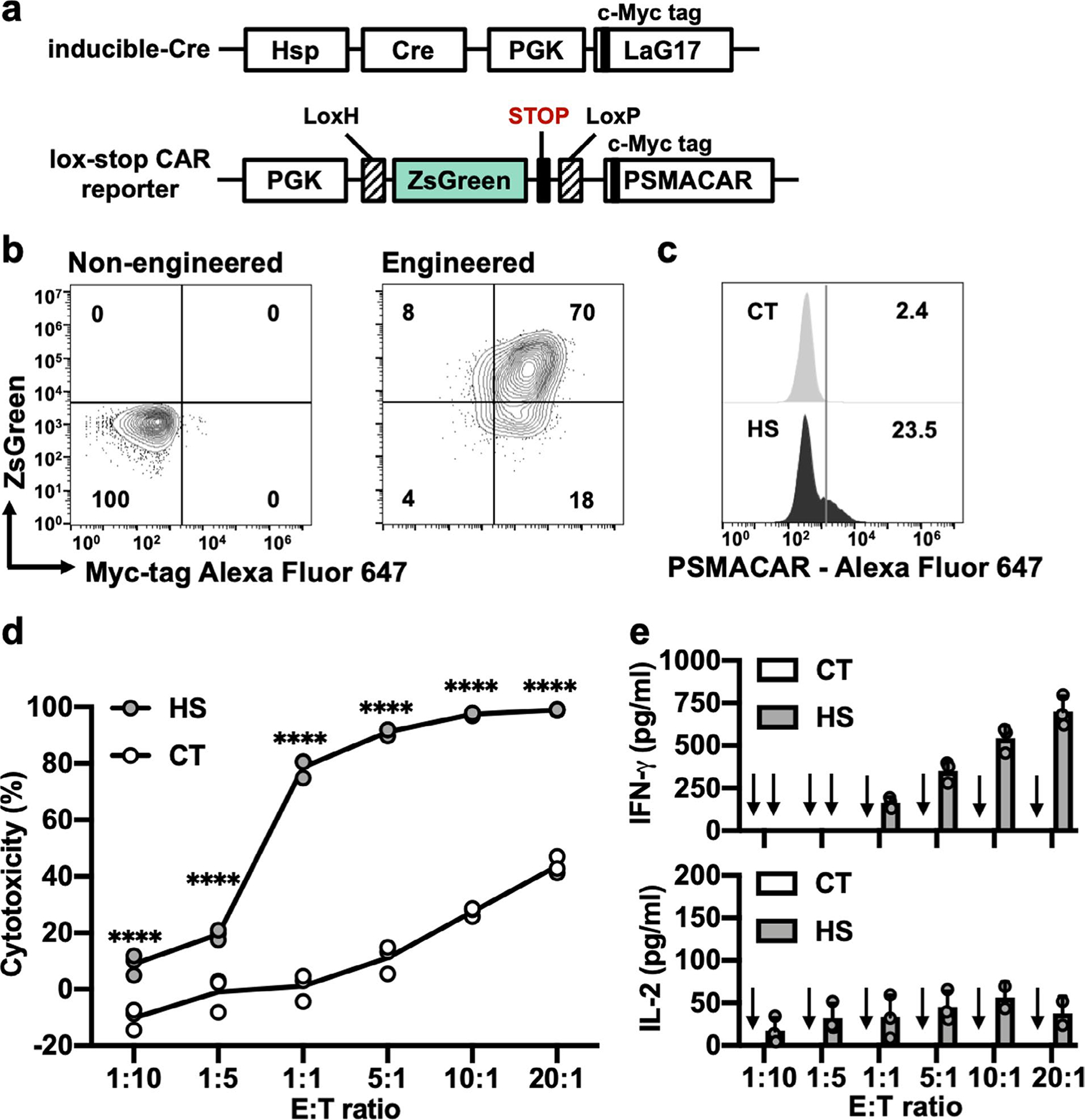
a, Schematics of transgenes: heat-inducible Cre and lox-stop PSMACAR reporter. b, Representative flow cytometry data of the percentage of double positive T cells after MACS. c, Representative PSMACAR induction by HS. d, Cytotoxicities of the T cells engineered with the transgenes in a against Fluc+ PSMA + PC3 tumour cells at various E:T ratios. From left to right: ****P = 3.09 × 10−6, P = 1.11 × 10−6, P < 1 × 10−15, P < 1 × 10−15, P < 1 × 10−15, P = 3 × 10−15. e, Quantification of IFN-γ and IL-2 cytokine release associated with d. Arrow: cytokine level not detectable. In c to e, CT: without HS. HS: with a continuous 15-min HS. Two-way ANOVA followed by Sidak’s multiple comparisons test. Bar heights and error bars represent means of 3 biological replicate ± SEM.
Extended Data Fig. 3 |. ‘Tumour only’ in vivo control experiment associated with Fig. 5.
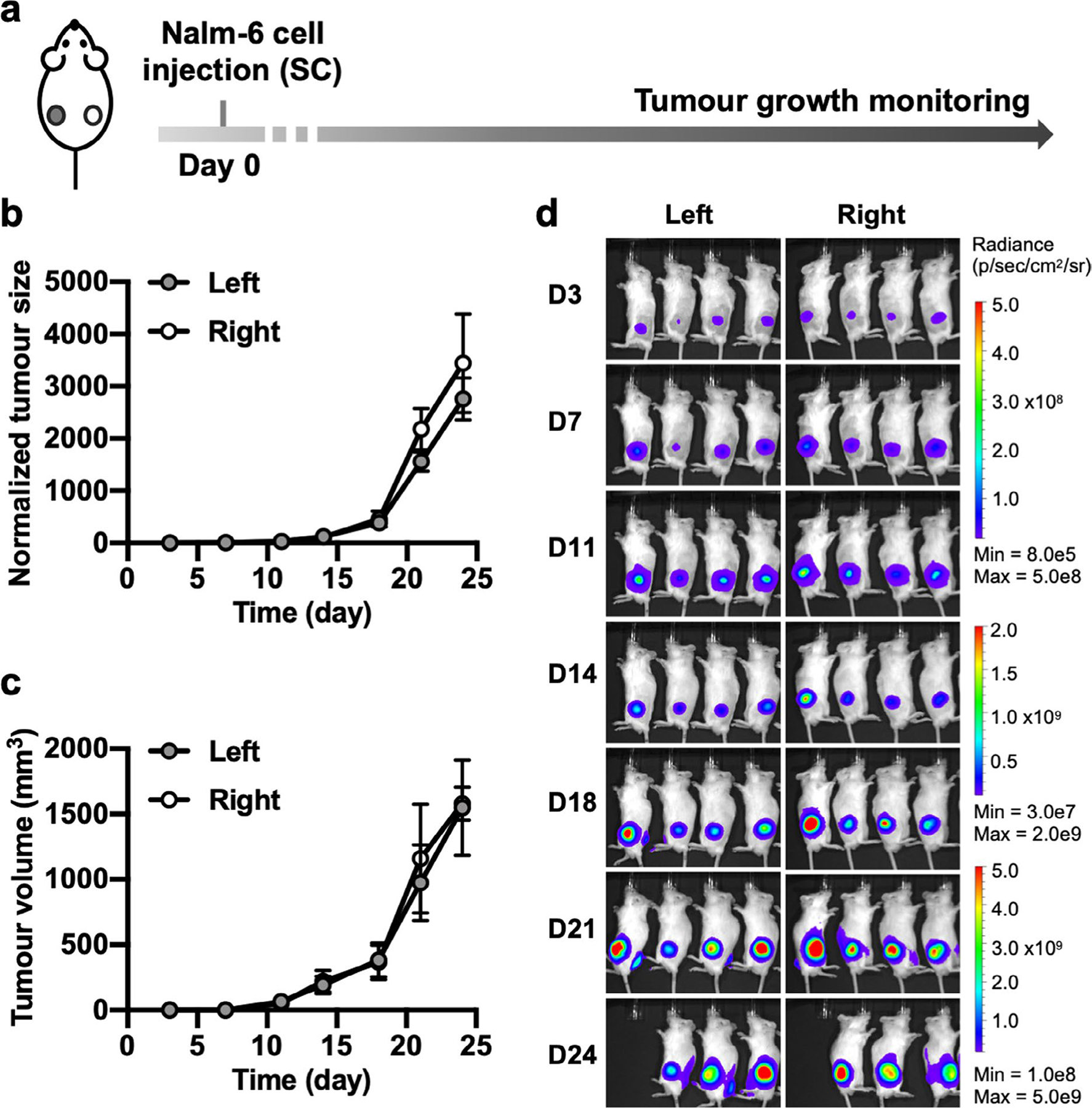
a, Timeline of the ‘tumour only’ control group used as the reference for calculating in vivo cytotoxicity in Fig. 5e. NSG mice were subcutaneously injected with Nalm-6 tumours on both sides and received no other treatment. b–d, Monitoring of tumour aggressiveness: normalized tumour size (b), tumour volume (c) and BLI images (d). Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. Tumour volume was calculated based on caliper measurement as described in Methods. No significant difference was detected by mixed-effects analysis followed by Sidak’s multiple comparisons test. Data points and error bars represent means of 4 mice ± SEM. One mouse was sacrificed on D24 according to euthanasia criteria.
Extended Data Fig. 4 |. Repeated FUS stimulation in vivo control experiment associated with Fig. 6.
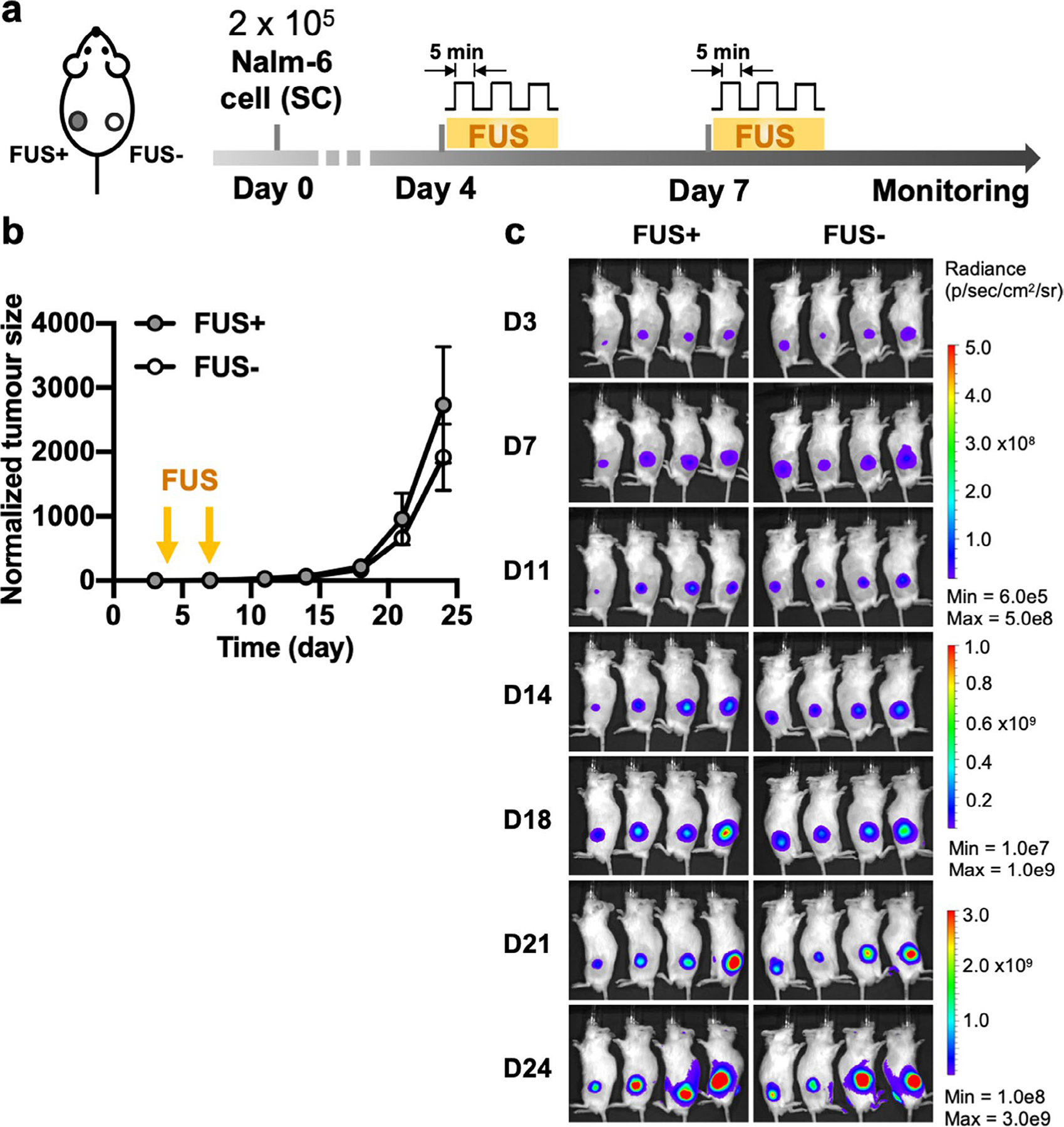
a, Timeline of the experiment. Bilateral tumour-bearing mice received local FUS stimulation without T cell injection at the left tumour on Day 4 and Day 7. The right tumour received no treatment. b,c, Normalized tumour size (b) and BLI images of the tumours on both sides (c). Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. Data points and error bars represent means of 4 mice ± SEM.
Extended Data Fig. 5 |. Unstimulated reversible FUS-CAR T in vivo control experiment associated with Fig. 6.
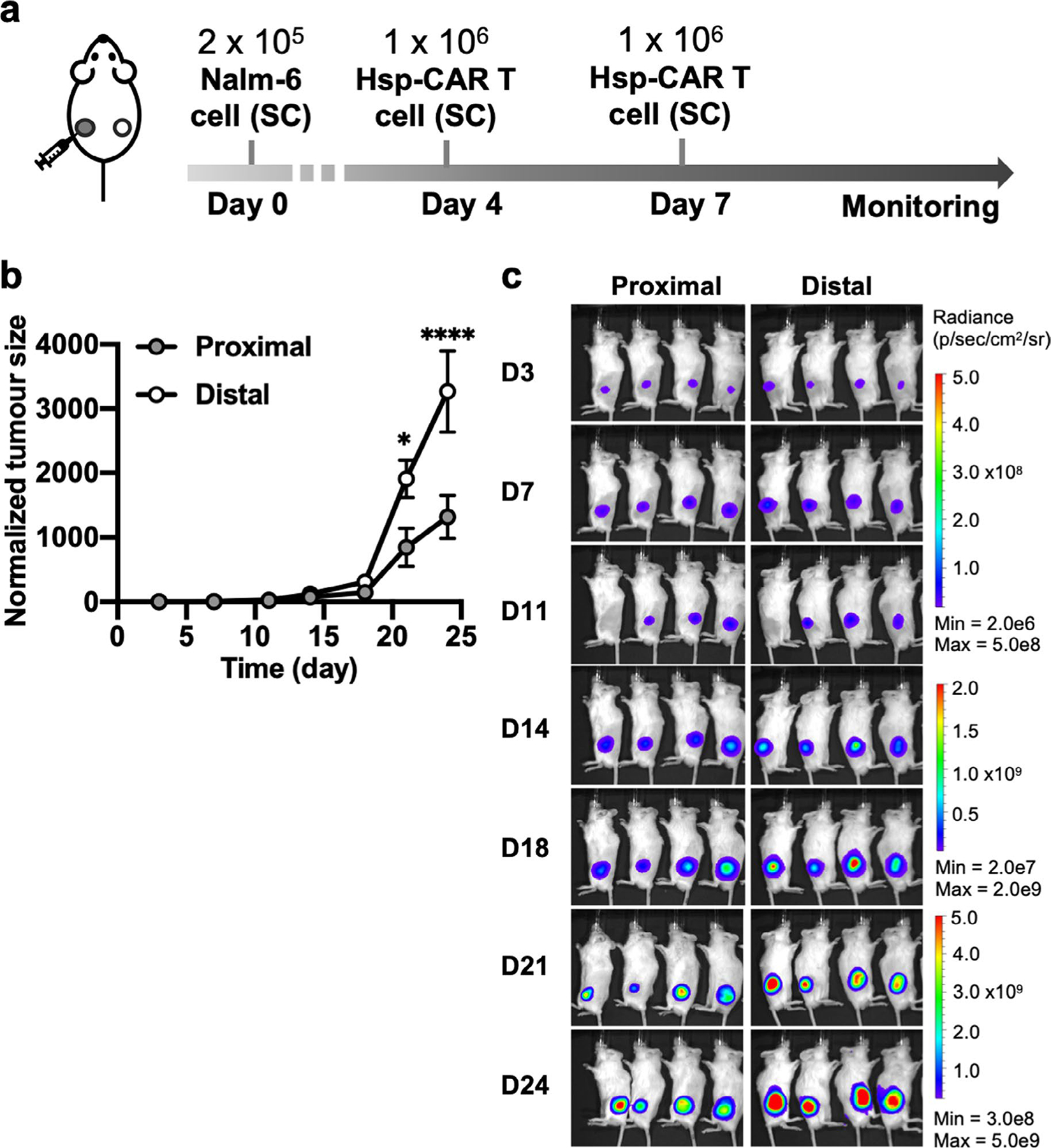
a, Timeline of the experiment. Bilateral tumour-bearing mice received local injections of the reversible FUS-CAR (Hsp-CAR) T cells without FUS stimulation at the left tumour (proximal) on Day 4 and Day 7. The right tumour (distal) received no treatment. b,c, Normalized tumour size (*P = 0.011, ****P = 1.4 × 10−5, Two-way ANOVA followed by Sidak’s multiple comparisons test) (b) and BLI images of the proximal and distal tumours (c). Tumour size was quantified using the integrated Fluc luminescence intensity of the tumour region and normalized to that of the same tumour on the first measurement. Data points and error bars represent means of 4 mice ± SEM.
Supplementary Material
Acknowledgements
This work was supported in part by grants from NIH HL121365, GM125379, GM126016, CA204704 and CA209629 (Y. Wang). We thank F. Couillaud (University of Bordeaux, France) for providing the Hsp template; M. Sadelain (Sloan Kettering Institute, USA) for the PSMA scFv and PSMA constructs and the Nalm-6 cells; E. Dumont and S. Hoarau-Recco (Image Guided Therapy, France) for their most valuable help on the FUS system; and P. Mali for the insightful discussion.
Footnotes
Competing interests
Y. Wang is scientific co-founder of Cell E&G Inc. and Acoustic Cell Therapy Inc. These financial interests do not affect the design, conduct or reporting of this research.
Extended data is available for this paper at https://doi.org/10.1038/s41551-021-00779-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-021-00779-w.
Data availability
The main data supporting the results of this study are available within the paper and its Supplementary Information. Source data for tumour growth curves are provided with this paper. Other raw data generated during this study are available from the corresponding authors on reasonable request.
References
- 1.Tsien RY Imagining imaging’s future. Nat. Rev. Mol. Cell Biol 4 (Suppl.), SS16–SS21 (2003). [PubMed] [Google Scholar]
- 2.Thanou M & Gedroyc W MRI-guided focused ultrasound as a new method of drug delivery. J. Drug Deliv 2013, 616197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deckers R et al. Image-guided, non-invasive, spatiotemporal control of gene expression. Proc. Natl Acad. Sci. USA 106, 1175–1180 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guilhon E et al. Image-guided control of transgene expression based on local hyperthermia. Mol. Imaging 2, 11–17 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Wang S, Zderic V & Frenkel V Extracorporeal, low-energy focused ultrasound for noninvasive and nondestructive targeted hyperthermia. Future Oncol. 6, 1497–1511 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Smith RC, Machluf M, Bromley P, Atala A & Walsh K Spatial and temporal control of transgene expression through ultrasound-mediated induction of the heat shock protein 70B promoter in vivo. Hum. Gene Ther 13, 697–706 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Madio DP et al. On the feasibility of MRI-guided focused ultrasound for local induction of gene expression. J. Magn. Reson. Imaging 8, 101–104 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Piraner DI, Abedi MH, Moser BA, Lee-Gosselin A & Shapiro MG Tunable thermal bioswitches for in vivo control of microbial therapeutics. Nat. Chem. Biol 13, 75–80 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Davila ML et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med 6, 224ra225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakravarti D & Wong WW Synthetic biology in cell-based cancer immunotherapy. Trends Biotechnol. 33, 449–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maus MV, Grupp SA, Porter DL & June CH Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123, 2625–2635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akpek G, Lee SM, Anders V & Vogelsang GB A high-dose pulse steroid regimen for controlling active chronic graft-versus-host disease. Biol. Blood Marrow Transpl 7, 495–502 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Di Stasi A et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med 365, 1673–1683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Themeli M & Sadelain M Combinatorial antigen targeting: ideal T-cell sensing and anti-tumor response. Trends Mol. Med 22, 271–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho JH, Collins JJ & Wong WW Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426–1438 e1411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedorov VD, Themeli M & Sadelain M PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med 5, 215ra172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roybal KT et al. Precision tumor recognition by t cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu CY, Roybal KT, Puchner EM, Onuffer J & Lim WA Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L & Alimandi M CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 9, 282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grivennikov SI, Greten FR & Karin M Immunity, inflammation, and cancer. Cell 140, 883–899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan Y et al. Mechanogenetics for the remote and non-invasive control of cancer immunotherapy. Proc. Natl Acad. Sci. USA 115, 992–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller IC, Gamboa Castro M, Maenza J, Weis JP & Kwong GA Remote control of mammalian cells with heat-triggered gene switches and photothermal pulse trains. ACS Synth. Biol 7, 1167–1173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abravaya K, Phillips B & Morimoto RI Attenuation of the heat shock response in HeLa cells is mediated by the release of bound heat shock transcription factor and is modulated by changes in growth and in heat shock temperatures. Genes Dev. 5, 2117–2127 (1991). [DOI] [PubMed] [Google Scholar]
- 25.Ghosh SK, Missra A & Gilmour DS Negative elongation factor accelerates the rate at which heat shock genes are shut off by facilitating dissociation of heat shock factor. Mol. Cell. Biol 31, 4232–4243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez M & Moon EK CAR-T cells for solid tumours: new strategies for finding, infiltrating and surviving in the tumour microenvironment. Front. Immunol 10, 128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sridhar P & Petrocca F Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers 9, 92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown CE et al. Optimization of IL13R alpha 2-targeted chimeric antigen receptor T cells for improved anti-tumour efficacy against glioblastoma. Mol. Ther 26, 31–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Priceman SJ et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2 thorn breast cancer metastasis to the brain. Clin. Cancer Res 24, 95–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donovan LK et al. Locoregional delivery of CAR-T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med 26, 720–731 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Theruvath J et al. Locoregionally administered B7-H3-targeted CAR-T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med 26, 712–719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahmood U et al. Current clinical presentation and treatment of localized prostate cancer in the United States. J. Urol 192, 1650–1656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musunuru HB et al. Active surveillance for intermediate risk prostate cancer: survival outcomes in the sunnybrook experience. J. Urol 196, 1651–1657 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Rastinehad AR et al. Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study. Proc. Natl Acad. Sci. USA 116, 18590–18596 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boice M et al. Loss of the HVEM tumour suppressor in lymphoma and restoration by modified CAR-T Cells. Cell 167, 405–418. e413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roybal KT et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167, 419–432. e416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chew WL et al. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 13, 868–874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moreno AM et al. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng 3, 806–816 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang CH et al. Monitoring of the central blood pressure waveform via a conformal ultrasonic device. Nat. Biomed. Eng 2, 687–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyden ES, Zhang F, Bamberg E, Nagel G & Deisseroth K Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci 8, 1263–1268 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Fite BZ et al. Magnetic resonance thermometry at 7T for real-time monitoring and correction of ultrasound-induced mild hyperthermia. PLoS ONE 7, e35509 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otto-Duessel M et al. In vivo testing of Renilla luciferase substrate analogs in an orthotopic murine model of human glioblastoma. Mol. Imaging 5, 57–64 (2006). [PubMed] [Google Scholar]
- 43.Mcburney MW et al. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 19, 5755–5761 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results of this study are available within the paper and its Supplementary Information. Source data for tumour growth curves are provided with this paper. Other raw data generated during this study are available from the corresponding authors on reasonable request.