

Abstract
Cotton (Gossypium spp.) is an important natural fiber plant. Lint percentage (LP) is one of the most important determinants of cotton yield and is a typical quantitative trait with high variation and heritability. Many cotton LP genetic linkages and association maps have been reported. This work summarizes the inheritance, quantitative trait loci (QTLs), and candidate genes of LP to facilitate LP genetic study and molecular breeding. More than 1439 QTLs controlling LP have been reported. Excluding replicate QTLs, 417 unique QTLs have been identified on 26 chromosomes, including 243 QTLs identified at LOD >3. More than 60 are stable, major effective QTLs that can be used in marker-assisted selection (MAS). More than 90 candidate genes for LP have been reported. These genes encode MYB, HOX, NET, and other proteins, and most are preferentially expressed during fiber initiation and elongation. A putative molecular regulatory model of LP was constructed and provides the foundation for the genetic study and molecular breeding of LP.
Keywords: upland cotton (Gossypium hirsutum L.), lint percentage, inheritance, quantitative trait locus, gene
Introduction
Cotton (Gossypium spp.) is an important natural fiber plant. The genus Gossypium consists of 52 wild and cultivated species, including 6 allotetraploid species and 46 diploids (Wendel and Cronn, 2003; Chen et al., 2016; Wang et al., 2021). Cultivated species include two diploid species (2n = 2x = 26), Gossypium herbaceum L. (A1) and Gossypium arboreum L. (A2); and two tetraploid species (2n = 4x = 52), upland cotton [Gossypium hirsutum L. (AD1)] and sea island cotton [Gossypium barbadense L. (AD2)]. Among the cultivated species, upland cotton is the most widely cultivated, accounting for approximately 95% of worldwide cotton production (Chen et al., 2007; Imran et al., 2012). Cotton yield is typically influenced by multiple complex quantitative traits, including boll number per plant (BN), single boll weight (BW), lint percentage (LP), seed index (SI), and lint index (LI) (Qin et al., 2008; Yu et al., 2016). Of these, LP is a key contributor to lint yield and a critical economic index for cotton cultivars, and measurement of LP is easy during cotton breeding (Tang and Xiao, 2013; Su et al., 2016; Song et al., 2019).
Cotton breeding is performed to achieve simultaneous improvement of cotton yield and fiber quality (Wang et al., 2011). However, LP is positively or negatively correlated with other yield and fiber quality components (Kong et al., 2000; Ulloa and Meredith, 2000; Wang et al., 2007; Imran et al., 2012; Tang and Xiao, 2013; Wang et al., 2014; Carvalho et al., 2015; Chen et al., 2019; Zhu et al., 2021). Selecting for LP alone to increase lint yield reduced boll and seed mass, but also fiber length and fiber strength (Wang et al., 2011). The negative correlation between these traits and the reduction in a cotton cultivar due to the introduction of deleterious genes along with the beneficial ones make it challenging to synchronously improve fiber quality and lint yield. Thus, effective cotton breeding requires characterization of the inheritance, quantitative trait loci (QTLs), candidate genes, molecular regulatory networks of LP, and other yield and fiber quality components. By using big segregating populations and whole genome selection techniques, it may be possible to break tight linkages of QTLs with the opposite genetic effects.
Genetic study, germplasm resource enhancement, and breeding of upland cotton are very important for efforts to improve cotton production. Abundant cotton germplasm resources with high genetic diversity can be used to improve upland cotton, and LP can be used as an indicator of genetic diversity. There have been recent advances in the molecular and genetic basis of cotton LP, however, the genetic basis of cotton LP remains incompletely understood. To provide detailed genetic information related to cotton LP for future cotton lint yield studies and breeding, we comprehensively summarized the inheritance, QTLs, and related candidate genes of cotton LP. We reanalyzed available data and mined the QTLs and candidate LP genes, screened the stable and most effective QTLs for use in marker-assisted selection (MAS), identified the key biological pathways regulating LP, and outlined a molecular regulatory network for cotton LP development. These results can guide cotton yield improvement.
Inheritance of Cotton Lint Percentage
LP is a Typical Quantitative Trait Controlled by Major Genes Plus Polygenes
Cotton yield per unit area includes four major components, plant number per unit area, boll number per plant, single boll weight, and LP (Yu et al., 2016). LP (also known as ginning out-turn) is the ratio of lint weight to seed cotton weight [or lint index/(lint index + seed index)] as a percentage. LP is determined by fiber number, fiber length, fiber diameter, and the weight of seed on which the fibers are grown. LP is a critical criterion for cotton cultivars, and it influences the purchase price of the seed cotton. Historic data of Huang-Huai Regional Trials in China from 1973 to 1996 reported an increase in yield from 1200 kg·hm−2–1425 kg·hm−2 that was explained by improvements of LP, which increased 5.5% (Jiang et al., 2000). Obviously, LP is a key selection trait for cotton yield breeding.
The phenotypes of complex traits are often determined by the combined actions of multiple genes and environmental factors (Mackay et al., 2009). Cotton yield and its components, BN, BW, LP, SI, and LI, are inherited quantitatively and are genetically related to each other (Wang et al., 2007; Qin et al., 2009). Of these components, the heritability of LP is the highest (49%; Badigannavar and Myers, 2015), and LP correlates with seed cotton yield, lint yield and other yield components at different strengths (Wang et al., 2014; Chen et al., 2019; Zhu et al., 2021). LP is a typical quantitative trait of cotton cultivars, except in fuzz-less and lint-less mutants where it is considered a qualitative trait (An et al., 2010).
We can choose appropriate breeding methods to improve cotton yield when there are good genetic models of yield-related traits. Studies of the inheritance of cotton LP began in the 1970s in China. First, simple crosses were performed between high and low LP cultivars, and then the LP of the offspring was evaluated. Overall, the inheritance of LP was not clear. In the 1990s, the model of major genes plus polygenes became more widely studied (Mo, 1993), allowing the later establishment of a mixed genetic model of major genes plus polygenes. The model assumed that the studied traits were diploid inherited, and lacked female effects, interaction, and linkage among major genes and polygenes (Gai et al., 2003). A genetic model to simultaneously analyze genetic effects of nuclear, cytoplasm, and nuclear-cytoplasmic interaction (NCI) and genotype by environment (GE) interactions for quantitative traits was subsequently developed (Han et al., 2007). These methods have been applied in genetic studies on cotton yield related traits, including LP (Du et al., 1999; Yuan et al., 2002; Zeng and Wu, 2012; Xia et al., 2014; Li B et al., 2016; Gong et al., 2019).
Lint yield and yield components are mainly controlled by genetic effects, i.e., additive and dominant variance account for ≥66% of the total phenotypic variance (Zeng and Wu, 2012). LP has the highest additive variance as a proportion of the total phenotypic variance (Zeng and Wu, 2012). LP is controlled mainly by genetic effects and is less affected by GE interaction effects (Yuan et al., 2002; Han et al., 2007). The variances of additive effect (VA), cytoplasmic effect (VC), and the dominant nuclear-cytoplasmic interaction effect (DC) interaction variance (VDC) are significant for LP (Han et al., 2007). LP shows great genetic variability. Other quality parameters have high heritability values (up to 90%), while LP exhibits moderate heritability (49%) (Badigannavar and Myers, 2015).
Segregation analysis of the mixed genetic model of major genes plus polygenes was used to identify the major genes for cotton yield-related traits using six generations of P1, P2, F1, B1, B2, and F2 generated from a cross of Baimian1 × TM-1 (TexasMarker-1) (Jia et al., 2014). One major gene of LP was detected, with positive complete dominance, and an additive effect value equal to the dominant effect value (0.512) (Xia et al., 2014). A major QTL, qLP-3b(F2)/qLP-3(F2:3), of LP was detected in both F2 and F2:3 using QTL mapping, with additive effect values (1.86 and 1.49, respectively) slightly smaller than the dominant effect values (1.95 and 1.51, respectively), thus displaying a dominant effect (Xia et al., 2014).
We studied LP using a female parent sGK Zhong156, male parent 901-001, and their 250 introgression lines (ILs) and found that LP is controlled by two to four pairs of major genes or two pairs of major genes plus polygenes. The heritability of major genes ranged from 1.26 to 83.13%, the heritability of polygenes ranged from 27.35 to 90.83%, and the heritability of major genes plus polygenes ranged from 92.00 to 99.35% (Gong et al., 2019).
Overall, the contributions of VG and VGE to total genetic variances of LP are about 73.62 and 26.38% (Han et al., 2007). LP is determined by additive effects (65.4%) (Yuan et al., 2002; Saha et al., 2006, 2010; Liu et al., 2012; Li C et al., 2016), non-additive effects (Liang et al., 2012), dominant effects (16.8%) (Yuan et al., 2002; Saha et al., 2006; Xia et al., 2014), cytoplasmic effects, and dominant nuclear-cytoplasmic interaction effect variance (Saha et al., 2006; Han et al., 2007; Liu et al., 2012; Li et al., 2017). The results indicated that LP is mainly controlled by major genes plus polygenes with additive and dominant effects (Cheng et al., 1998; Du et al., 1999; Xia et al., 2014; Gong et al., 2019).
LP Correlates With Other Components of Yield and Fiber Quality
Many agronomic traits are correlated due to linkage, pleiotropy, or the correlated traits are components of a more complex variable (Ulloa et al., 2005). LP is positively or negatively correlated with other components. Generally, LP has significantly negative correlation with seeds per boll, seed index, seed cotton yield, fiber length (FL), and fiber strength; and is positively correlated with boll number per plant, lint per seed, lint index, and lint yield (Kong et al., 2000; Ulloa and Meredith, 2000; Wang et al., 2007; Imran et al., 2012; Tang and Xiao, 2013; Carvalho et al., 2015; Zhu et al., 2021).
Germplasm Resources and Enhancement of High LP Cotton
Excellent germplasm resources are required for the successful breeding of cotton cultivars (Zeng and Wu, 2012). Based on introducing foreign cotton cultivars, three cultivar substitutions happened in China from the 1920s, first, Jinzimian, Tuozimian, and Longzimian from the United States substituted some Asiatic cotton cultivars planted in north China in 1920s; second, Sizimian4, Dezimian531, and Daizimian14 substituted the Asiatic cotton cultivars planted in a large area of China in the 1930s–1940s; third, Daizimian15, Sizimian2B, and Sizimian5A substituted the Asiatic cotton cultivars planted for a long period, and the degenerated foreign upland cultivars in China in the 1950s (Yin et al., 2002).
Chinese researchers traveled to Mexico, the United States, France, Australia, and other countries to explore and collect cotton germplasm resources from the 1970s to 1980s, with more than 4800 cultivars, lines, wild species, semi-wild species, and special lines with genetic markers collected by 1984 (Gao et al., 2021). A total of 8868 accessions were collected and preserved by 2020, including 7362 G. hirsutum, 350 G. hirsutum wild races, 633 G. barbadence, 433 G. arbareum, 18 G. herbaceous, and 32 wild species, making the cotton collection in China the fourth largest in the world (Du et al., 2012). Many of these cotton resources have been characterized. For example, 28 cotton lines with high LP (43–47%) were screened by Li and Wang (1992), and 8 high LP (> 42%) lines were screened from former Soviet Union germplasm resources by Liu et al. (1998). Cultivars introduced from the former Soviet Union (108Ф, C1470, 611B, and KK1543), the United States, and Africa (DPL15, STV2B, and UGDM) were applied for modern cultivar improvement in Xinjiang, China (Han et al., 2020). Using foreign germplasm resources, many high LP (> 43%) cultivars/lines have been bred in China.
In addition to traditional methods of plant cultivar improvement, such as intraspecific crosses and physical and chemical mutagenesis (Gai, 1997; Hulse-Kemp et al., 2015), the introduction of elite alien genes or chromosome fragments by interspecific crosses is a main approach for cotton germplasm enhancement, especially for LP. Upland cotton has a high lint yield but undesirable fiber quality, sea island cotton has superior fiber quality but lower lint yield. Therefore, significant efforts have been made worldwide to combine the desirable lint yield of G. hirsutum with the superior fiber quality of G. barbadense by interspecific hybridization. Many germplasm resources with improved fiber quality containing chromosomal introgressions from G. barbadense have been constructed (Saha et al., 2006; Saha et al., 2010; Zhu et al., 2010; Wang et al., 2011; Zhu et al., 2017; Chen et al., 2018; Li et al., 2019) (Supplementary Table S1). Compared to G. barbadense, the superior introgression lines (IL) have fiber quality more similar to that of G. barbadense and are usually more convenient to use for upland cotton improvement (Wang et al., 2011).
Artificial selection efforts have reduced genetic diversity but significantly increased LP. A cotton variation map of an intact breeding pedigree including 7 elite and 19 backbone parents was constructed (Ma et al., 2019). The 26 pedigree accessions were subjected to strong artificial selection during domestication and have reduced genetic diversity but stronger linkage disequilibrium and higher extents of selective sweeps. The elite parents acquired significantly improved agronomic traits, with an especially pronounced increase in LP. Although there is a negative correlation between LP and FL in long fiber materials (Carvalho et al., 2015), two cultivars and one advanced line have the highest LP and FL as well as high productivity: FM 993, FM 910, and CNPA MT 04 2088, suggesting these materials can be used to increase LP and FL values (Santos et al., 2017).
A set of chromosome segment introgression lines (CSILs) were developed by crossing the recipient parent TM-1 and the donor parent Hai7124. TM-1 is a genetic G. hirsutum standard accession developed in the United States, and Hai7124 is an early, high-yielding G. barbadense accession developed in China (Song and Zhang, 2009). This set of CSILs has been used to explore the genetic basis of heterosis for interspecific hybrids (Guo et al., 2013). A set of 14 chromosome substitutions (CS-B) with specific chromosomes or chromosome arms from G. barbadense substituted into G. hirsutum and chromosome-specific F2 families has been reported, and additive and dominant effects are significant for LP (Saha et al., 2006). Another set of 17 CS-B lines (2n = 52) were reported that contain G. barbadense doubled-haploid line “3-79” germplasm systematically introgressed into “TM-1”. TM-1 yield is much greater than 3-79, but cotton from 3-79 has superior fiber properties. CS-B14sh, 17, 22Lo, and 25 produce positive homozygous dominant effects on lint yield (Saha et al., 2010). A CSSL population was developed via crossing and backcrossing of G. hirsutum and G. barbadense by PCR-based MAS (Zhu D et al., 2020). By whole genome re-sequencing, 11,653,661 high-quality SNPs were identified with 1211 recombination chromosome introgression segments from G. barbadense. Six QTLs for LP were identified that have negative alleles from G. hirsutum background.
Extensive efforts have generated successful intraspecific crosses of G. hirsutum, and interspecific crosses between G. hirsutum and G. barbadense. Many interspecific ILs or CSILs have been developed and studied (Shi et al., 2015; Li et al., 2019), and some ILs have been identified with positive effects that increase LP, such as the IL008 lines (Zhu et al., 2017). Many advanced new cotton germplasm resources and cultivars have been bred, such as C24 (LP = 48–50%; Wang and Xiao, 1996), IL-10-1 (LP > 37%; Ma et al., 2012), S26-1 (LP > 46%), Malan (LP > 46%), Xingtai6871 (LP > 46%), Shan815 (LP > 45%), Kang35 (LP > 45%) (Li and Wang, 1992), Lumianyan22 (Chen et al., 2018), and Sumian16 and Simian3 (both LP > 43%; Zhang Z et al., 2005). These enhanced cotton germplasm resources can be used for LP breeding in upland cotton.
The Heterosis of LP
Utilization of heterosis has greatly improved the productivity of many crops worldwide, including cotton. Hybrid cotton varieties exhibit strong heterosis that confers high fiber yields (Ma et al., 2019). Understanding the potential molecular mechanism underlying how hybridization produces superior yield in upland cotton is critical for effective breeding (Shahzad et al., 2020). Although the role of loci with over-dominant effects continues to be discussed by biologists, the genetic basis of heterosis has not yet been determined. Genetic models are important, as heterosis is a nonlinear effect from multiple heterozygous gene combinations (Gai et al., 2007). Trait phenotype and heterosis may be controlled by different sets of loci and many genetic factors likely function together to yield heterotic output (Shahzad et al., 2020). All three forms of genetic effects, additive, over-dominant, and epistasis, and environmental interaction contribute to the heterosis of yield and its components in upland cotton, with over-dominance and epistasis the most important (Guo et al., 2013; Li et al., 2018).
A study with 286 upland cotton cultivars found that female parent groups based on genetic distance (GD) and F1 performance significantly differ in LP. The correlations between GD and F1 performance, mid-parent heterosis (MPH), and best parent heterosis (BPH) are significant for LP (Geng et al., 2021). Fourteen putative candidate genes were identified as associated with heterosis of LP (Sarfraz et al., 2021). A study with CP-15/2, NIAB Krishma, CIM-482, MS-39, and S-12 found greater specific combining ability (SCA) variance than general combining ability (GCA) variance for LP (0.470), indicating the predominance of non-additive genes (Imran et al., 2012). Dominance plays an important role in the genetic basis of heterosis in Xiangzamian2, and non-additive inheritance is also an important genetic mode for LP (Liu et al., 2012).
Studies on the interspecific heterosis of G. hirsutum × G. barbadense were performed starting in the 1960s in China (Diao and Huang, 1961; Hua et al., 1963). Although many traits exhibit heterosis such as boll setting, fiber quality, photosynthesis, and drought and disease resistance, some hybrid cultivars suffer from severe defects that preclude planting in many areas, with characteristics such as small bolls, low LP, and low yield (Chen et al., 2002). Breeding chromosome segment substitution lines (CSSLs) is a main strategy to utilize excellent genes from G. barbadense and partial interspecific heterosis in upland cotton.
Studies with CSSLs and their 50 F1 hybrids found that yield and yield components are controlled by combined additive and dominant effects, and LP is mainly controlled by additive effects. Significant positive MPD was detected for all yield traits, and significant positive BPH was detected for all yield traits except LP (Shen et al., 2006). A proposed model was studied in G. hirsutum introgression lines (ILs) harboring a segment of G. barbadense. These ILs contained 396 QTLs for 11 yield and other traits. The yield group has significantly higher over-dominant QTL ratios, with 16 over-dominant QTLs for 5 yield-related traits consistently detected during 5 cotton growing seasons, one of which is LP. Overdominance is the major genetic basis of lint yield heterosis in interspecific hybrids between G. barbadense and G. hirsutum (Tian et al., 2019).
Over-dominance is a major factor for yield heterotic output, so genetic diversity is especially important. Genitors with short GD share many alleles, and there are fewer complementary genes in heterozygotes. The genetic diversity among 16 cotton cultivars was studied. Variables with low magnitude, redundancy, and non-stability are not ideal for genetic diversity study, but LP is the trait that most contributes to genetic diversity at 31.3% (Santos et al., 2017).
Although the genetic basis of heterosis has not been resolved in cotton, the available data indicate that LP is a significant contributor to genetic diversity (Santos et al., 2017). SCA variance is greater than GCA variance for LP, showing the predominant contribution of non-additive genes (Imran et al., 2012). Overdominance is the major genetic basis of lint yield heterosis in interspecific hybrids of G. barbadense and G. hirsutum, including LP (Tian et al., 2019), and LP has significant negative heterosis value (Yuan et al., 2002; Shen et al., 2007). Additive and over-dominant effects have been found for QTLs of LP (Liu et al., 2011; 2012). At the single-locus level, the genetic bases of heterosis vary in different populations. In conclusion, additive effects, over-dominant effects, epistasis, and environmental interactions all contribute to the heterosis of yield and its components in upland cotton, with over-dominance and epistasis the most important (Li et al., 2018).
QTLs of Cotton Lint Percentage
High-Density Genetic Maps of Cotton LP
A QTL is a “gene” controlling a certain quantitative trait. Although where it is and what it encodes may be unknown, the term QTL theoretically links a gene and a trait. QTLs can be detected and located by genetic linkage analysis and QTL mapping relying on a similar set of genetic assumptions. Genetic linkage analysis can detect both major gene and polygene effects, does not need a linkage map and provides no information about the location of the QTL. QTL mapping can detect and locate a possible QTL if a linkage map is available (Gai et al., 2007). Significant advances in QTL mapping occurred in the 1990s, enabling the study of individual QTLs and interaction between QTLs for heterosis, providing a powerful tool to identify the genetic bases of yield and yield components (Liu et al., 2012; Li et al., 2018). QTL mapping using populations segregating for mutants can increase the sensitivity of this method due to the enlarged quantitative variation scale by the expression of qualitative mutant genes (An et al., 2010). In the discovery of major genes or major QTLs, segregation analysis and QTL mapping are mutually complementary methods that can verify results.
The first cotton linkage map was reported in 1985, with 11 linked groups located on chromosomes, and mapping of some mutant genes, including two LP genes (Endrizzi et al., 1985). Currently, more than 31 genetic linkage maps have been reported in upland cotton (Supplementary Table S1). Crosses including intra-species of G. hirsutum, inter-species of G. hirsutum and G. barbadense, populations of immortalized F2 and F2:3, RIL, backcross inbred lines (BIL), and immortalized backcross populations (DHBCF1s and JMBCF1s). Various molecular markers such as simple sequence repeats (SSR), expressed sequence tag-SSR (EST-SSR), fragment length polymorphism (RFLP), random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), chip-SNP, and whole genome re-sequencing have been employed to make genetic linkage maps of cotton. The density of the genetic maps has increased, and the marker span has reached 5115.16 cM, with an average marker interval that is less than 1 cM 0.92, 0.72, and 0.5 cM (Blenda et al., 2012; Shi et al., 2015; Diouf et al., 2018; Gu et al., 2020). We have constructed a consensus genetic linkage map covering the whole genome with 8295 markers (458 SSR, 5521 SLAF-SNP, and 2316 chip-SNP), spanning 5197.17 cM, with 2384.94 cM for the At subgenome and 2812.23 cM for the Dt subgenome. The average interval between adjacent markers is 0.88 cM (Zhang K et al., 2019).
With the development of techniques related to genetic mapping, the coverage and accuracy of the maps are constantly increasing, enabling discovery of more QTLs. High density maps make it possible to clone candidate genes of major effective QTLs, including LP. As more QTLs are discovered, the number of high-quality QTLs for LP with closely linked markers has increased, leading to the discovery of more effective candidate genes. Thus, these improved techniques have greatly facilitated molecular breeding and cotton LP improvement efforts. Some genetic and association maps reported in cotton, including the numbers of the identified QTLs for LP, are listed in Supplementary Table S1.
High-Density Genetic Maps of G. hirsutum
More than 17 crosses or populations of upland cotton have been used to construct genetic maps (Supplementary Table S1), such as crosses of Yumian1 × T586 (Zhang P et al., 2005; Wan et al., 2007; Liu D et al., 2015), Yumian1 × Zhongmiansuo35 (Chen et al., 2008), NC05AZ06 × NC11-2091 (Zhang Z et al., 2019), DH962 × Jimian5 (Lin et al., 2009a, b; Wang et al., 2016), Zhongmiansuo12 (ZMS12) × 8891 (Wang et al., 2007), (Simian3 × Sumian12) × (Zhong4133 × 8891) (Qin et al., 2015), Baimian1 × TM-1 (Wang et al., 2014; Xia et al., 2014), Xiangzamian2 (Liu et al., 2011; Liu et al., 2012), HS46 × MARCABUCAG8US-1-88 (Wu et al., 2009; Li C et al., 2016), and CCRI35 × Nan Dan Ba Di Da Hua (NH) (Diouf et al., 2018). With more advanced studies, the densities of the genetic maps have increased significantly. For example, a high-density genetic map was drawn using 588 F7 RILs derived from the cross of Nongdamian13 × Nongda601 by a whole genome re-sequencing strategy. This high-density bin linkage map contains 6187 bin markers spanning 4478.98 cM with an average distance of 0.72 cM. Fifty-eight individual QTLs and 25 QTL clusters harboring 94 QTLs, and 119 previously undescribed QTLs controlling 13 fiber quality and yield traits across 8 environments were identified, including 17 QTLs for LP (Gu et al., 2020).
High-Density Genetic Maps of G. hirsutum × G. barbadense
Cotton genetic maps have been constructed with interspecific crosses of G. hirsutum × G. barbadense (Supplementary Table S1) (Li et al., 2009; Blenda et al., 2012; Yu et al., 2013; Shi et al., 2015). For example, a high-density SSR genetic linkage map has been drawn using a BC1F1 population developed from a cross of G. hirsutum × G. barbadense. The map comprises 2292 loci and covers 5115.16 cM of the cotton AD genome, with an average marker interval of 2.23 cM. For 1577 common loci on this map, 90.36% agree well with the marker order on the D genome sequence genetic map. Twenty-six QTLs for LP were identified on 9 chromosomes, and 50% of the QTLs are from G. barbadense and increase LP by 1.07–2.41% (Shi et al., 2015).
High-Density Genetic Maps of QTLs From G. barbadense
Upland cotton CSILs with chromosome fragments from G. barbadense are ideal materials to identify new agronomic genes from sea island cotton, which can be identified using genetic maps (Supplementary Table S1) (Zhu et al., 2010; Wang et al., 2011; Guo et al., 2013; Said et al., 2015; Zhai et al., 2016; Zhu et al., 2017; Chen et al., 2018; Kong et al., 2018). For example, F2 and F2:3 populations derived from the cross of CSILs-IL-15-5 × IL-15-5-1 were used to map QTLs for LP and seed index with SSR markers. The SI and LP characteristics of 774 F2 individuals and F2:3 families were analyzed by composite interval mapping (CIM), and two QTLs were detected for LP: qLP-15-1 in the F2 and F2:3 populations located between NAU3040 and JESPR152, with confident genetic distances of 5.40 and 3.20 cM, respectively, and qLP-15-2, mapped between NAU5302 and NAU2901 with confident genetic distance of 0.08 cM (Zhu et al., 2010). In this way, more than 107 QTLs for LP have been reported (Said et al., 2015) (Supplementary Table S1).
QTLs Controlling Cotton LP
LP is controlled by many QTLs. Study of the F2:3 population derived from the cross of HS46 × MARCABUCAG8US-1-88 identified a single QTL that explains 15.45% of phenotypic variance of LP, with other QTLs that explain 10.03–15.46% (Diouf et al., 2018). Mapping where these genes reside on the chromosomes will facilitate breeding, especially if easily measured molecular markers are closely linked with the specific QTLs so they can be used in MAS (Shappley et al., 1998; Liu et al., 2014). The classical genetic method of studying quantitative traits is based on a linkage analysis of both parents, enabling the mapping of multiple QTLs responsible for LP using segregating populations from different parents (Dong et al., 2018). To date, many QTLs controlling LP have been reported (Table 1; Supplementary Table S2).
TABLE 1.
The unique, tightly linked (LOD >3) and major effective QTLs for LP.
| Chromosome No | Chr01 | Chr02 | Chr03 | Chr04 | Chr05 | Chr06 | Chr07 | Chr08 | Chr09 |
|---|---|---|---|---|---|---|---|---|---|
| Unique QTLs | 9 | 8 | 27 | 16 | 15 | 15 | 24 | 8 | 16 |
| Tightly linked QTLs | 3 | 5 | 19 | 10 | 8 | 12 | 14 | 5 | 6 |
| Major QTLs | 0 | 2 | 10 | 4 | 2 | 2 | 3 | 1 | 0 |
| Chromosome No | Chr10 | Chr11 | Chr12 | Chr13 | Chr14 | Chr15 | Chr16 | Chr17 | Chr18 |
| Unique QTLs | 14 | 10 | 15 | 29 | 18 | 16 | 11 | 28 | 15 |
| Tightly linked QTLs | 7 | 6 | 8 | 20 | 12 | 10 | 8 | 19 | 4 |
| Major QTLs | 1 | 2 | 1 | 3 | 4 | 4 | 3 | 2 | 0 |
| Chromosome No | Chr19 | Chr20 | Chr21 | Chr22 | Chr23 | Chr24 | Chr25 | Chr26 | Total |
| Unique QTLs | 9 | 18 | 16 | 15 | 10 | 21 | 28 | 6 | 417 |
| Tightly linked QTLs | 2 | 10 | 11 | 10 | 7 | 11 | 13 | 3 | 243 |
| Major QTLs | 0 | 2 | 3 | 0 | 2 | 2 | 4 | 3 | 60 |
QTLs for LP in Upland Cotton
To identify QTLs for LP, mapping populations have been constructed using a high LP variety, Baimian1, and a low LP variety, TM-1. Six major effective QTLs (M-QTL) for LP were detected on chromosomes 3, 5, 19, and 26. Three of these, qLP-3(2010), qLP-3(2011), and qLP-19(2010), are significant M-QTLs (Wang et al., 2014). Four common QTLs for LP were discovered from the cross of Baimian1 × TM-1: qLP-3b(F2)/qLP-3(F2:3) and qLP-19b(F2)/qLP-19(F2:3) explaining 23.47 and 29.55% of phenotypic variation (Xia et al., 2014). Similarly, using a cross of Yumian1 × T586, four QTLs for LP were detected (Zhang P et al., 2005). Using 219 F2 individuals developed from a cross of TM-1 × T586, three LP QTLs were tagged and mapped on the A03 linkage group and chromosome 6 (Guo et al., 2006). The allele(s) originating from T586 of QTLs controlling LP can increase the trait phenotypic value (Wan et al., 2007). A recombinant inbred line (RIL) population including 188 RILs developed from 94 F2-derived families and their two parental lines, “HS46” and “MARCABUCAG8US-1-88”, were evaluated, and five QTLs responsible for 38.1% of the phenotypic variance for LP were mapped to chromosomes 3, 4, 9, 12, and 26. Chromosomes 3, 5, 12, 13, 14, 16, 20, and 26 harbor important QTLs for both yield and fiber quality. Chromosomes 3 and 26 are associated with QTLs for LP (Wu et al., 2009). Seventeen SSRs out of 304 markers tested from MGHES (EST-SSR), JESPR (genomic SSR), and TMB (BAC-derived SSR) collections show significant linkage associations with LP QTLs in a set of RILs segregating for LP (Abdurakhmonov et al., 2007). One dominant QTL for LP, qLP43 from DH962, is near marker MGHES-75 (Lin et al., 2009a; b). Two QTLs for LP, qLP-C13-1 and qLP-C25-1 were detected from the high LP parent Lumianyan22, and explain 5.57–8.87% of phenotypic variation (Chen et al., 2018). The detailed information of QTLs for LP in upland cotton is summarized in Table 1 and Supplementary Tables S1, S2, S3, and S4.
QTLs for LP in Upland Cotton With Lintless Mutants
Using mapping populations developed from a G. barbadense line (fuzzy-linted) and a single fuzzless locus mutant (G. hirsutum, N1 or n2), considerable quantitative variation in lint fiber production superimposed on the discrete effects of fuzz mutants was observed and both loci mapped within the intervals of QTL for LP and lint index (Rong et al., 2005). These results were confirmed by QTL mapping for LP and fiber quality traits (micronaire, uniformity, and fiber elongation) using an n2 mutant derived population (Rong et al., 2007). Two F2 populations were studied from crosses of MD17, a fuzzless-lintless line containing three fuzzless loci, N1, n2 and a postulated n3, with line 181, fuzzless-linted and with FM966, a fuzzy-linted cultivar. QTLs for LP and LI explain a high percentage of the phenotypic variation (62.8–87.1%) and are probably on the same location on chromosome 26 with the common marker BNL3482-138 in both populations. The MD17 BNL3482-138 allele at this locus decreases both LP and LI (An et al., 2010) (Supplementary Table S1).
QTLs for LP From G. barbadense
Many QTLs for LP from G. barbadense have been identified. Eight QTLs for LP were discovered in an RIL population of G. barbadense (Fan et al., 2018). The F2 populations from two CSSLs MBI7455 and MBI7358 were quantified using SSR, and five LP QTLs were identified (Guo et al., 2015). Four CSILs, MBI7115, MBI7412, MBI7153, and MBI7346, were obtained by advanced backcrossing and continuous inbreeding of upland cotton variety CCRI45 and sea island cotton variety Hai1 and were used to construct double-cross segregating populations F1 and F1:2 through the following crosses [(MBI7115 × MBI7412) × (MBI7153 × MBI7346)]. The analysis revealed that three QTLs control LP (Kong et al., 2018). Four BC1F1 populations, E(E3), E(3E), (E3)E, and (3E)E, were developed by crossing sea island cotton 3-79 (“3”) and upland cotton “Emian22” (“E”), using “Emian22” as the recurrent parent. Using composite interval mapping, two LP QTLs were detected on a single chromosome (Dai et al., 2018). We previously developed a set of CSSLs using CCRI8 of G. hirsutum as the recipient parent and Pima90-53 of G. barbadense as the donor parent. We genotyped the BC3F5 generation of CSSLs with SSR markers, constructed a QTL map for fiber quality and yield traits, and identified the stable QTLs in three different environments (Baoding, Qingxian, and Luntai). Seven QTLs for LP were detected, and each QTL explained 2.03–19.38% of the phenotypic variation (Li et al., 2018).
Stable QTLs for LP Can Be Used in MAS
DNA markers linked to agronomic traits increase the efficiency of breeding and significantly decrease the cost, time, and risk of subjective phenotypic assays. MAS can help by pyramiding favorable genes or alleles into a cultivar and breaking the negative relation between yield and quality (Zhang et al., 2011; Zhu et al., 2017). The application of MAS for the improvement of traits of interest depends on the availability of QTL information (QTL positions and effects) and flanking marker information (flanking marker positions and allele sizes) (Wu et al., 2009).
Most reported QTLs controlling LP have closely linked markers, enabling their use in MAS (Shappley et al., 1998). Some of this kind of QTLs have been reported, such as the stable qLP-A10-1 from the cross of Zhongmiansuo12 (ZMS12) × 8891 (Wang et al., 2007); qLP-15-1 and qLP-15-2 in CSILs-IL-15 (Zhu et al., 2010); qLP-2-1, qLP-2-2 and qLP-4-2 from RILs derived from the cross of 0-153 × SGK9708 (Jia et al., 2011); qLP-3b(F 2 )/qLP-3(F 2:3 ), detected from the cross of Baimian1 × TM-1 (Xia et al., 2014); six main-effect QTLs (M-QTL) on chromosomes 3, 5, 19, and 26; qLP-3 and qLP-19 (Wang et al., 2014); major QTL qLP-3 from G. mustelinum (Chen et al., 2019); and three major QTLs, qLP-CH24-D3-1, qLP-CH25-D12-1, and qLP-17-CH14-A8-1, derived from NC05AZ06 (Zhang Z et al., 2019). Additionally, 26 QTLs on 9 chromosomes were reported from a cross of G. hirsutum × G. barbadense, including 9 stable or common QTLs that can be used in MAS (Shi et al., 2015). Currently, more than 60 stable major QTLs have been reported for LP and can be used in MAS breeding (Table 1 and 2; Supplementary Table S4).
TABLE 2.
Some stable, major effective QTLs for LP which can be used in MAS.
| QTL | Chromosome | Genetic position (cM) | LOD | R 2 (%) |
|---|---|---|---|---|
| qLP-3-1 | Chr03 | 26.54 | 3.20 | 20.10 |
| qLP-A3-1 | Chr03 | 14.01 | 4.59 | 11.14 |
| qLP-A3-1 | Chr03 | 22.90 | 7.37 | 17.48 |
| qLP-A3-2 | Chr03 | 46.16 | 3.89 | 10.90 |
| qLP06.1 | Chr06 | 63.20 | 4.70 | 8.60 |
| qLP07.1 | Chr07 | 7.80 | 20.90 | 36.70 |
| qLP-A11-1 | Chr11 | 41.22 | 3.18 | 6.27 |
| qLP12.1 | Chr12 | 70.00 | 58.20 | 63.40 |
| qLP-15-1 | Chr15 | 24.41 | 7.35 | 19.86 |
| qLP-15-1 | Chr15 | 9.21 | 9.61 | 6.90 |
| qLP-15-2 | Chr15 | 33.25 | 6.35 | 18.47 |
| qLP-D2-1 | Chr15 | 0.01 | 4.15 | 8.69 |
| qLP-C16-6 | Chr16 | 177.70 | 4.39 | 10.11 |
| qLP-C16-7 | Chr16 | 189.30 | 5.79 | 11.60 |
| qLP-C17-1 | Chr17 | 91.30 | 10.81 | 27.51 |
| qLP-C17-2 | Chr17 | 94.90 | 11.70 | 26.40 |
| qLP21.1 | Chr21 | 165.60 | 15.60 | 24.30 |
| qLP-23-1 | Chr23 | 3.00 | 3.71 | 6.99 |
| qLP-D6-1 | Chr25 | 146.96 | 4.52 | 8.23 |
| qLP26.1 | Chr26 | 49.50 | 3.90 | 8.00 |
High-Density Association Maps and QTNs of Cotton LP
A QTN is a marker associated with a QTL/gene and is likely in a candidate gene of the associated QTL if it is in a functional gene region. A QTN links a QTL with a nucleotide mutation. Different genome-wide association study (GWAS), single locus-GWAS (SL-GWAS), multi-locus GWAS (ML-GWAS), and restricted two-stage, multi-locus, multi-allele GWAS (RTM-GWAS) approaches have been used to search QTNs for LP in a large number of cotton accessions (Supplementary Table S1). For example, using RTM-GWAS, 86 single-nucleotide polymorphism linkage disequilibrium block (SNPLDB) loci for LP were identified from 315 cotton accessions (Su J et al., 2020). A total of 719 upland cotton accessions were screened by GWAS combined with cottonSNP63K array. High density SNP maps have been constructed (Sun et al., 2018), including more than 16 association maps (Supplementary Table S1). Many candidate genes for agronomic traits have also been reported (Fang et al., 2017; Huang et al., 2017; Sun et al., 2018; Shen et al., 2019; Su et al., 2019; Su J et al., 2020; Zhu G et al., 2020; Yu et al., 2021).
Using GWAS, six significant SNPs associated with LP were identified on chromosomes A02, A04, D03, and D12, together explaining approximately 43.41% of the total phenotypic variation. Three loci, i03389Gh, i33922Gh, and i03391Gh, are near each other on chromosome D03 (Sun et al., 2018). A comprehensive genomic assessment of modern improved upland cotton was performed based on the genome-wide re-sequencing of 318 landraces and modern improved cultivars or lines. Two major haplotypes with two nonsynonymous SNPs are associated with higher LP and more bolls per plant (GhLYI-A02 HLB ), and lower LP and fewer bolls per plant (GhLYI-A02 LLB ) (Fang et al., 2017). A diversity panel consisting of 355 upland cotton accessions was subjected to specific-locus amplified fragment sequencing (SLAF-seq). Twelve SNPs associated with LP were detected via GWAS, with five SNP loci distributed on chromosomes At3 (A02) and At4 (A08) that contain two major effective QTLs that were detected in best linear unbiased predictions (BLUPs) and in more than three environments simultaneously (Su et al., 2016). To date, more than 1396 QTNs for LP have been reported (Supplementary Table S1).
A Consensus Physical Map of QTLs for LP in Upland Cotton
QTLs of LP represent genes controlling the phenotype of LP. QTNs are markers of the QTLs. Referring to the cotton genome sequences of G. hirsutum and G. barbadense (Li et al., 2015; Liu X et al., 2015; Yuan et al., 2015; Zhang et al., 2015; Huang et al., 2020), the QTLs of LP can be mapped using the locations of the associated QTNs or markers. In this way, more than 1439 QTLs for LP have been reported via genetic linkage maps. Excluding the replicate QTLs, there are more than 417 unique QTLs for LP. QTLs (183) with available tightly linked marker sequences were mapped to a consensus physical map of upland cotton (Figure 1). Obviously, many QTLs are clustered.
FIGURE 1.

A consensus physical map of QTLs for LP in upland cotton. The QTLs were mapped to G. hirsutum genome by the locations of their linked markers. Left: the marker unit is Mb.
Candidate Genes and Regulatory Network of Lint Percentage
Candidate Genes of Cotton LP QTLs
Genes control phenotypes and isolation of the genes regulating LP is essential for functional gene marker development for MAS and gene engineering breeding. Fine mapping of the major effective QTLs for LP makes it possible to clone the candidate genes by map-based cloning. The availability of whole-genome sequences also paves a way to identify and clone functional genes, e.g., those relating to fiber development and increased LP (Pan et al., 2020; Sarfraz et al., 2021).
A diversity panel of 355 upland cotton accessions was subjected to specific-locus amplified fragment sequencing (SLAF-seq). Twelve SNPs associated with LP were detected via GWAS, in which five SNP loci distribute on chromosomes At3 (A02) and At4 (A08). G. hirsutum_A02G1268 may determine LP (Su et al., 2016). A GWAS uncovered 23 polymorphic SNPs and 15 QTLs significantly associated with LP and identified two candidate genes, G. hirsutum_D05G0313 and G. hirsutum_D05G1124, as promising regulators of LP (Song et al., 2019). Two ethylene-pathway-related genes are associated with increasing lint yield in improved cultivars, with two QTLs for LP, qLP-5-17.8 and Cs9_PF_21, located within the two association loci, respectively. G. hirsutum_A02G1392 is a homolog of the AP2/ethylene response factor (ERF)-type transcription-factor-encoding gene AINTEGUMENTA-like 6 (AIL6) in Arabidopsis thaliana (Fang et al., 2017). Another candidate gene, G. hirsutum_D08G2376, may control LP (Huang et al., 2017). Two genes, G. hirsutum_D03G1064 and G. hirsutum_D12G2354, increase lint yield and with G. hirsutum_D03G1067 are candidate genes of LP (Sun et al., 2018). Cotton PROTODERMAL FACTOR1 (GbPDF1) gene is related with LP (Deng et al., 2012). A non-synonymous SNP (A-to-G) site in a gene encoding the cell wall-associated receptor-like kinase 3 (GhWAKL3) protein is highly correlated with increased LP (Ma et al., 2019). A class I TCP transcription factor (designated GbTCP), from G. barbadense affects LP (Hao et al., 2012). A cotton GhH 2 A 12 gene encoding a typical SPKK motif in the carboxyterminal and a plant-unique peptide-binding A/T-rich DNA region is involved in fiber differentiation (Hao et al., 2014). Candidate gene Ghir_A03G020290 of a stable QTL qLPA03-1 for LP encodes a thioredoxin domain-containing protein 9 homolog (Gu et al., 2020). G. hirsutum_D03G0919 (GhCOBL4), G. hirsutum_D09G1659 (GhMYB4), and G. hirsutum_D09G1690 (GhMYB85) of LP QTLs were identified from RILs with G. barbadense introgressions (Wang et al., 2020). A MYB gene (GhMYB103), with two SNP variations in cis-regulatory and coding regions, is significantly correlated with LP, implying a crucial role in lint yield (Zhu G et al., 2020). A gene orthologous to Arabidopsis receptor-like protein kinase HERK 1 (GB_A07G1034) is predicated as a candidate gene for LP improvement (Yu et al., 2021). Two NET genes, GH_A08G0716 and GH_A08G0783, in novel QTL hotspots (qtl24 and qtl25), are mainly expressed during early fiber development stages and exhibit significant correlation with LP (Zhu et al., 2021). More than 90 candidate genes of QTLs have been reported (Supplementary Table S5).
None of the genes of the major QTLs for LP have been isolated via map-based cloning, however, high density genetic linkage maps and multi-genome sequence data identified more than 90 candidate genes of QTLs for LP (Supplementary Table S5). According to the putative protein functions, most candidate genes of QTLs for LP are transcription factor (TF) and transcription related genes, with many candidate genes belonging to the MYB family of TF. According to the predicted gene functions of the candidate genes, transcription; signaling pathways including hormone, calcium (Ca2+), and MAPK; energy metabolism; substance transport; cell division; and cell wall development are important biological processes for LP formation (Figure 2; Table 3; Supplementary Table S5).
FIGURE 2.
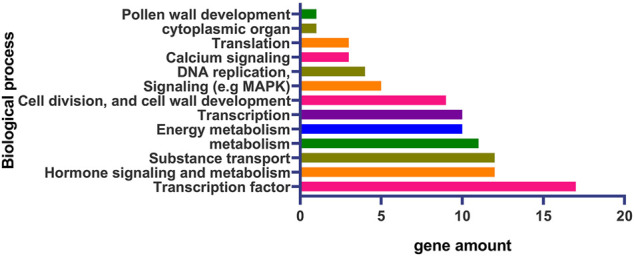
The major biological processes of the candidate genes of the QTLs for LP.
TABLE 3.
The major biological processes related to the QTLs for LP.
| Biological process | The candidate genes (homologs) of the QTLs for LP |
|---|---|
| Hormone signaling and metabolism | ET: AIL6 (AP2), EIN3; BR: BZR1, LUT2; JA: TIFY10A; IAA: T85; CTK: MPK4; ABA: MYB52, KEG, PAA1, NET1D, NET3A |
| Calcium signaling | CPK17, CRCK2, CIPK6 |
| Signaling (e.g., MAPK) | ASK8, PPC4-2, At1g12280, HERK1, Gpr107 |
| Transcription factor | ZHD1, GhMYB4, GhMYB25, GhMYB25l, MYB52, GhMYB85, MYB86, GhMYB103, GbTCP, HDG11, GhHD-1, GhHD1, GhHOX1, VRN1, MYB308, ODO1, TTG1 |
| DNA replication | PAA1, GhAATF1, bsdc1, GhmTERF1 |
| Transcription | FRI, Bicc1, rpc11, At5g12190, SPP, SEN1, GhAATF1, GhmTERF1, GhH2A12, H2A |
| Translation | RPS10A, asnS, RPS1 |
| Cell division and cell wall development | MPK4, FLA12, NCD80, At3g43860, CAP1, GhWAKL3, GbPDF1, GhCOBL4, NDC80 |
| Substance transport | PHO1, TSC10B, COPT5, PRA1B1, UDP-GALT2, PYRAB13050, Gpr107, xpr1, bro1, G. hirsutum_D13G0342, TMN1, PLT5 |
| Energy metabolism | APL2, COX5B-2, CRR21, WAXY, At3g43860, G. hirsutum_D03G1407, Acot9, GhGRAM31, GRAM35, GRAM5 |
| Pollen wall development | TKPR1 |
| Metabolism | INO1, RHA2A, CYP78A7, bsdc1, DTX33, YLS8, ROD1, UGT80A2, RAB71, TXND9, GEML5 |
| Cytoplasmic organ | ATJ20 |
Note: ET, ethylene; BR, brassinosteroid; JA, jasmonate acid; IAA, auxin; CTK, cytokinesis; ABA, abscisic acid. The detailed information of the reported genes is listed in Supplementary Table S5.
To identify the major metabolic pathways of the candidate genes, pathway enrichment analysis was carried out using the Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.kegg.jp/kegg/) database. KEGG annotation analysis indicated that the 64 candidate genes of the QTLs for LP belonged to 44 KEGG pathways in G. hirsutum (Supplementary Tables S6 and S7). Among them, metabolic pathways, biosynthesis of secondary metabolites, MAPK signaling pathway, plant hormone signal transduction, plant-pathogen interaction, starch and sucrose metabolism, spliceosome, and two-component systems were most enriched (Table 4; Figure 3). The above results indicated that most of the candidate genes that affect LP in upland cotton do so through various metabolic pathways.
TABLE 4.
Major metabolic pathways related to LP in cotton.
| KEGG pathway | Ko Id | Candidate gene number |
|---|---|---|
| Metabolic pathways | Ko 01100 | 8 |
| Biosynthesis of secondary metabolites | ko 01110 | 5 |
| MAPK signaling pathway—plant | ko 04016 | 3 |
| Plant hormone signal transduction | ko 04075 | 3 |
| Plant-pathogen interaction | ko 04626 | 3 |
| Starch and sucrose metabolism | ko 00500 | 2 |
| Spliceosome | ko 03040 | 2 |
| Two-component system | ko 02020 | 2 |
FIGURE 3.

Candidate genes (gene number) that regulate LP are involved in major metabolic pathways.
Molecular Regulatory Network of Cotton Fiber Development
The coat of normal cottonseed is covered with lint and fuzz. The spinnable long fibers are called lint (25–35 mm) and initiate around flowering and then elongate rapidly afterward; the short fibers are called fuzz (∼5 mm), which develops at a later stage (Stewart, 1975; Du et al., 2001). Anatomically, cotton fibers are single-celled trichomes that differentiate from the outermost cell layer (protoderm) of the ovule. Cotton fiber is a highly elongated single cell, and its development synchronizes with and depends on seed development (Liu D et al., 2015). Cotton fiber development is a highly programmed and regulated process that can be divided into four discrete, yet overlapping stages on the basis of morphological characteristics during a 50-d period: initiation (0-3 days post anthesis, DPA), elongation (3-20 DPA), secondary cell wall thickening (16-40 DPA), and maturation (40-50 DPA) (Rong et al., 2005; Liu D et al., 2015; Qin et al., 2019). LP is a representative phenotypic trait of fiber development and the candidate genes of QTLs for LP (Table 3; Supplementary Tables S5 and S6) suggest relationships between LP and fiber development.
Many candidate genes of QTLs for LP participate in the regulation of fiber cell development. One candidate gene of QTLs for LP, G. hirsutum_A02G1268 (MIPS), is highly expressed during the early fiber development stage and may determine LP by regulating seed and fiber development (Su et al., 2016). Two candidate genes of QTLs for LP, G. hirsutum_D05G0313 and G. hirsutum_D05G1124, are highly expressed during ovule and fiber development stages (Song et al., 2019). A core genomic segment has a non-synonymous SNP (A-to-G) site in a gene encoding the cell wall-associated receptor-like kinase 3 (GhWAKL3), which may be involved in signaling and fiber cell wall-development to increase LP (Ma et al., 2019). Analysis of CSILs of SL15 (low-LP) and LMY22 (high-LP) indicate that delayed secondary-cell-wall thickening results in altered LP (Gai et al., 2007). In selective sweeps, an apoptosis-antagonizing transcription factor gene (GhAATF1) and mitochondrial transcription termination factor family protein gene (GhmTERF1) were found to be highly involved in the determination of LP (Han et al., 2020). Most of these genes are preferentially expressed at the initiation of cotton fiber and early elongation stages (Table 3; Supplementary Table S5).
Transgenic and functional gene assays have demonstrated that some genes that regulate fiber development can increase LP. GbPDF1 is expressed preferentially during fiber initiation and early elongation. GbPDF1 plays a critical role together with interaction partners in hydrogen peroxide homeostasis and steady biosynthesis of ethylene and pectin during fiber development via the core cis-element HDZIP2ATATHB2 (Deng et al., 2012). GbTCP is preferentially expressed in the elongating cotton fiber from 5 to 15 DPA and GbTCP positively regulates the level of jasmonic acid (JA) to activate down-stream genes necessary for fiber elongation (reactive oxygen species, calcium signaling, ethylene biosynthesis and response, and several NAC [for NAM, ATAF1/2, and CUC2] and WRKY transcription factors) (Hao et al., 2012). GhH 2 A 12 is preferentially expressed during cotton fiber initiation and early elongation stages, suggesting a role in fiber differentiation (Hao et al., 2014). GhFSN1 is a cotton NAC transcription factor that acts as a positive regulator to control secondary cell wall (SCW) formation of cotton fibers by activating downstream SCW-related genes, including GhDUF231L1, GhKNL1, GhMYBL1, GhGUT1, and GhIRX12 (Zhang et al., 2018). Overexpression of GhCIPK6a enhances expression levels of co-expressed genes induced by salt stress that scavenge reactive oxygen species (ROS), are involved in MAPK signaling pathways, and increase LP under salt stress (Su Y et al., 2020). MiR319, GhTCP4, and GhHOX3 dynamically coordinate the regulation of fiber elongation (Cao et al., 2020). The glucosyltransferases, Rab-like GTPase activators, and myotubularins (GRAM) domain gene GhGRAM31 (Ghir_D02G018120) regulate fiber elongation and affect LP. GhGRAM31 directly interacts with GhGRAM5 and GhGRAM35. GhGRAM5 also interacts with the transcription factor GhTTG1, while GhGRAM35 interacts with the transcription factors GhHOX1 and GhHD1 (Ye et al., 2021). GhMYB109 is required for cotton fiber development (Pu et al., 2008). Overexpression of GhMYB3 in cotton leads to a slight increase in LP (Jiang et al., 2021). Over-expressing TF genes, GhHD-1, GhMYB25, and GhMYB25-like, in cotton can increase the density of fiber and fiber yield under field conditions. There are positive relationships between lint yield and LP and lint yield and fiber density in transgenic lines (Liu et al., 2020). Currently, more than 50 genes related to fiber development have been reported, including the miR319 gene (Supplementary Table S8). The MYB-bHLH-WD40 (including MYB-DEL-TTG; CPC-MYC-TTG) (Liu D et al., 2015; Shangguan et al., 2021) and TCP-HOX-HD (Cao et al., 2020; Ye et al., 2021) regulatory complexes play key roles in cotton fiber development. The NAC-MYB-CESAs module is a central pathway of SCW cellulose biosynthesis (Cao et al., 2020). Phytohormone balance, Ca2+ signaling, and ROS also play key roles in the regulation of fiber development to affect cotton LP (Tang et al., 2014; Cheng et al., 2016) (Figure 4).
FIGURE 4.

A molecular regulatory network of cotton fiber development. In the schematic, arrows indicate positive regulation; symbols of ‘T’ indicate negative regulation; lines indicate receptor or interaction; three tightly connected rectangles indicate tri-molecular complexes; red letters in clouds indicate phytohormones. Et, ethylene; ABA, abscisic acid; GA, gibberellin; JA, jasmonic acid; ROS, reactive oxygen species. “Gh” before gene symbols was omitted for space. The detailed information of the genes and their functions in cotton fiber development is listed in Supplementary Table S8.
Transcriptomics studies have discovered many candidate genes and noncoding RNAs regulating LP. Cotton fibers initiate from the ovule epidermis on the day of anthesis. After fiber initiation, fiber cells expand rapidly as a result of increased intracellular swelling pressure and cell wall relaxation (Li et al., 2012; Qin et al., 2019). Comparative transcriptomics studies exploring differences in gene expression between long- and short-fiber cotton lines at five developmental stages identified 22 consistently down-regulated differentially expressed genes (DEGs) involved in fiber initiation and 31 consistently up-regulated genes involved in fiber elongation. These may be good candidate genes for improving LP. Of the candidate genes, ERF1 is involved in ethylene metabolism and is expressed at a higher level in long-fiber cotton lines at fiber initiation; TUA2 and TUB1 are involved in microtubule synthesis and expressed at a higher level in long-fiber cotton lines at fiber elongation; and PER64 encodes a peroxidase (POD) and is expressed at a higher level in short-fiber cotton lines (Qin et al., 2019). Three lines with different LP were developed by crossing Xu142 with its fiberless mutant, Xu142 fl, and three long noncoding RNAs (lncRNAs) were identified as involved in fiber development. Silencing XLOC_545639 and XLOC_039050 in Xu142 fl increases the number of initiating fibers on the ovules, but silencing XLOC_079089 in Xu142 results in a short fiber phenotype (Hu et al., 2018). This demonstrated that lncRNAs manipulated the initiation of lint and fuzz fibers and affected LP. There is good evidence for the involvement of at least 62 genes, 45 miRANs, and 3 lncRNAs, which has been reported (Supplementary Table S9). Because the numbers of involved genes and potentially involved genes are exceptionally large, further work is required to determine the exact functions of these genes or miRNAs and the importance for LP development.
Molecular Regulatory Network of Cotton LP
Although there have been many studies about cotton LP, the molecular mechanism of LP development remains largely unknown. The number of initial fibers determines the LP. The isolated candidate genes of QTLs for LP and the functionally analyzed genes suggest that key genes involved in fiber initiation and early elongation are the more important regulators, particularly those involved in hormone signaling, transcription, cell wall development, and signal transduction, as described above (Figure 4; Tables 3 and 4; Supplementary Tables S5, S6, S7, and S8).
Based on the above summarized data, we proposed a molecular regulatory network for LP (Figure 5). Calcium signaling regulates hormone signaling, such as ABA signaling in leaf guard cells, and regulates gene expression, energy, and substance transport (McAinsh et al., 1997). TFs modulate transcription and hormone signaling and can affect LP. For example, MYB52 is involved in ABA signaling (Su J et al., 2020) and AIL6, a AP2-like ethylene-responsive TF, involved in ethylene (ET) signaling (Liu et al., 2014).
FIGURE 5.

Proposed molecular regulatory network for LP. The gray rectangle indicates the four overlapped fiber developmental stages. The arrows indicate the regulatory relationships. The yellow rectangles indicate biological processes. The circles indicate different regulators. All genes and favorable alleles are preferentially and highly expressed during the initiation and early elongation stages; however, BR and JA pathways are repressed. The red rectangle indicates the active biological processes that help increase LP.
Most candidate genes of QTLs for LP (27/90) are TFs or otherwise related to transcription, suggesting that gene expression is more active during fiber initiation and elongation. The second most genes (12/90) are related to hormone signaling, indicating key roles in regulating fiber development (Table 3). The biological processes of signaling, cell division, cell wall development, energy metabolism, and substance transport are important for LP formation (Deng et al., 2012; Ma et al., 2019; Qin et al., 2019). In addition, some lncRNAs are involved in fiber development, and increase LP (Hu et al., 2018). According to the currently available data, longer fiber initiation and elongation stages should result in higher LP.
Conclusion and Perspective
The current understanding of inheritance, QTLs, and candidate genes of LP is systemically summarized here. A large amount of genetic research results has been reported, including more than 417 unique QTLs for LP, 60 major effective QTLs, and more than 90 candidate genes of the QTLs. Additionally, more than 50 genes related to fiber development and microRNAs and long noncoding RNAs affecting LP have been reported. Most LP QTL candidate genes and genes related to fiber development affecting LP are preferentially expressed during fiber initiation and elongation. The biological processes of signaling, transcription, cell division, cell wall development, energy metabolism, and substance transport are important for LP development. By summarizing and analyzing the available data, this work reports the current state of the field and reveals what is known about cotton LP formation.
Although great genetic achievements have been made in cotton LP studies, it is difficult to improve LP without altering other yield components. The main reasons are as follows: (1) the inheritance of LP is complex; (2) LP is significantly negatively correlated with other key yield and quality components; (3) most QTLs for LP are unstable in different populations and environments, and may explain too little phenotype variance to trace with MAS; (4) LP has a significant negative heterosis value; (5) most genes of QTLs for LP are unknown and the molecular regulatory mechanism of LP is unclear. To address the above problems, future studies should: (1) Fully characterize the orchestrated inheritance model of LP and the relationship of LP with other yield and quality components; (2) identify more stable and major effective QTLs for LP; (3) improve elite germplasm resources of LP by breaking the linkage between LP and other deleterious yield and quality component QTLs/alleles; (4) study the molecular mechanisms of fiber development and increasing LP using omics and gene functional validation; and (5) develop effective techniques to pyramid favorable genes or alleles for high yield and high LP cotton breeding.
Author Contributions
HN searched and analyzed the data and drafted the manuscript. QG helped with analysis of the data. HS and YY designed the whole study, revised the manuscript, and gave the final approval to the version of the manuscript that is being sent for consideration for publication.
Funding
This work was funded by the Natural Science Foundation of China (No. 31621005), Central Public-interest Scientific Institution Basal Research Fund (No. 1610162019010101), the State Key Laboratory of Cotton Biology Open Fund (CB 2017A05), and the Agricultural Science and Technology Innovation Program for CAAS (CAAS-ASTIP-ICRCAAS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.855574/full#supplementary-material
References
- Abdurakhmonov I. Y., Buriev Z. T., Saha S., Pepper A. E., Musaev J. A., Almatov A., et al. (2007). Microsatellite Markers Associated with Lint Percentage Trait in Cotton, Gossypium hirsutum . Euphytica 156, 141–156. 10.1007/s10681-007-9361-2 [DOI] [Google Scholar]
- An C., Jenkins J. N., Wu J., Guo Y., McCarty J. C. (2010). Use of Fiber and Fuzz Mutants to Detect QTL for Yield Components, Seed, and Fiber Traits of upland Cotton. Euphytica 172, 21–34. 10.1007/s10681-009-0009-2 [DOI] [Google Scholar]
- Badigannavar A., Myers G. O. (2015). Genetic Diversity, Population Structure and Marker Trait Associations for Seed Quality Traits in Cotton (Gossypium hirsutum). J. Genet. 94 (1), 87–94. 10.1007/s12041-015-0489-x [DOI] [PubMed] [Google Scholar]
- Blenda A., Fang D. D., Rami J.-F., Garsmeur O., Luo F., Lacape J.-M. (2012). A High Density Consensus Genetic Map of Tetraploid Cotton that Integrates Multiple Component Maps through Molecular Marker Redundancy Check. PLoS ONE 7 (9), e45739. 10.1371/journal.pone.0045739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.-F., Zhao B., Huang C.-C., Chen Z.-W., Zhao T., Liu H.-R., et al. (2020). The miR319-Targeted GhTCP4 Promotes the Transition from Cell Elongation to wall Thickening in Cotton Fiber. Mol. Plant 13, 1063–1077. 10.1016/j.molp.2020.05.006 [DOI] [PubMed] [Google Scholar]
- Carvalho L. P., Farias F. J. C., Rodrigues J. I. S. (2015). Selection for Increased Fiber Length in Cotton Progenies from Acala and Non-acala Types. Crop Sci. 55 (5), 985–991. 10.2135/cropsci2014.08.0547 [DOI] [Google Scholar]
- Chen L., Zhang Z. S., Hu M. C., Wang W., Zheng F. M., Ma J., et al. (2008). Genetic Linkage Map Construction and QTL Mapping for Yield and Fi-Ber Quality in Upland Cotton (Gossypium hirsutum L.). A a S 34 (7), 1199–1205. 10.3724/sp.j.1006.2008.01199 [DOI] [Google Scholar]
- Chen Q., Zhou S. J., Sun K. T., Liu J. J., Wang B. H., Zhuang Z. M., et al. (2019). QTL Mapping of Lint Percentage in Gossypium mustelinum Introgression Lines. Southwest. China J. Agric. Sci. 32 (8), 1735–1739. 10.16213/j.cnki.scjas.2019.8.008 [DOI] [Google Scholar]
- Chen X. S., Di J. C., Xu N. Y., Liu J. H., Xiao S. G. (2002). Research Situation and Develop Tendency of Sea-Island and Land Hybrid Cotton. Jiangxi Cottons 24 (4), 3–6. [Google Scholar]
- Chen Y., Liu G., Ma H., Song Z., Wang F., Zhang J., et al. (2018). Identification of Introgressed Alleles Conferring High Fiber Quality Derived from Gossypium barbadense L. In Secondary Mapping Populations of G. Hirsutum L. Fron. Plant Sci. 9, 1023. 10.3389/fpls.2018.01023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Feng K., Grover C. E., Li P., Liu F., Wang Y., et al. (2016). Chloroplast DNA Structural Variation, Phylogeny, and Age of Divergence Among Diploid Cotton Species. PLoS ONE 11 (6), e0157183. 10.1371/journal.pone.0157183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. J., Scheffler B. E., Dennis E., Triplett B. A., Zhang T., Guo W., et al. (2007). Toward Sequencing Cotton (Gossypium) Genomes: Figure 1. Plant Physiol. 145, 1303–1310. 10.1104/pp.107.107672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H., He X., Pan G., Zhang M., Leng S. (1998). Analysis of Quantitative Traits in Cotton Breeding for Lint Yield. Acta Goss. Sin. 10 (6), 285–291. [Google Scholar]
- Cheng Y., Lu L., Yang Z., Wu Z., Qin W., Yu D., et al. (2016). GhCaM7-like , a Calcium Sensor Gene, Influences Cotton Fiber Elongation and Biomass Production. Plant Physiol. Biochem. 109, 128–136. 10.1016/j.plaphy.2016.09.009 [DOI] [PubMed] [Google Scholar]
- Dai B., Guo H., You C., Zhang X., Lin Z. (2018). QTL Mapping of Important Agronomic Traits on Chromosomes Showing Marker Segregation Distortion in Interspecific Backcross Cotton Populations. Cotton Sci. 30 (6), 435–447. 10.11963/1002-7807.dbslzx.20181119 [DOI] [Google Scholar]
- Deng F., Tu L., Tan J., Li Y., Nie Y., Zhang X. (2012). GbPDF1 Is Involved in Cotton Fiber Initiation via the Core Cis-Element HDZIP2ATATHB2. Plant Physiol. 158, 890–904. 10.1104/pp.111.186742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao G. Z., Huang Z. K. (1961). Utilization of the Heterosis between Gossypium hirsutum and Gossypium barbadense . Sci. Agric. Sin. 1 (7), 49–50. [Google Scholar]
- Diouf L., Magwanga R., Gong W., He S., Pan Z., Jia Y., et al. (2018). QTL Mapping of Fiber Quality and Yield-Related Traits in an Intra-specific upland Cotton Using Genotype by Sequencing (GBS). Ijms 19, 441. 10.3390/ijms19020441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Wang J., Chen Q., Yu Y., Li B. (2018). Detection of Favorable Alleles for Yield and Yield Components by Association Mapping in upland Cotton. Genes Genom 40, 725–734. 10.1007/s13258-018-0678-0 [DOI] [PubMed] [Google Scholar]
- Du X. M., Pan J. J., Wang R. H., Zhang T. Z., Shi Y. Z. (2001). Genetic Analysis of Presence and Absence of Lint and Fuzz in Cotton. Plant Breed. 120, 519–522. 10.1046/j.1439-0523.2001.00643.x [DOI] [Google Scholar]
- Du X. M., Sun J. L., Zhou Z. L., Jia Y. H., Pang B. Y., Wang L. R., et al. (2012). Current Situation and the Future in Collection, Preservation, Evaluation and Utilization of Cotton Germplasm in China. Plant Genet. Resour. 13 (2), 163–168. 10.13430/j.cnki.jpgr.2012.02.003 [DOI] [Google Scholar]
- Du X. M., Wang R. H., Liu G. Q., Fu H. Q., Pan J. J., Zhang T. Z. (1999). Inheritance Analysis of the Characters Related with Fiber by Using Mixed Major Gene and Polygene Model. Acta Goss. Sin. 11 (2), 73–78. [Google Scholar]
- Endrizzi J. E., Turcotte E. L., Kohel R. J. (1985). Genetics, Cytology, and Evolution of Gossypium . Adv. Genet. 23, 271–375. 10.1016/S0065-2660(08)60515-5 [DOI] [Google Scholar]
- Fan L., Wang L., Wang X., Zhang H., Zhu Y., Guo J., et al. (2018). A High-Density Genetic Map of Extra-long Staple Cotton (Gossypium barbadense) Constructed Using Genotyping-By-Sequencing Based Single Nucleotide Polymorphic Markers and Identification of Fiber Traits-Related QTL in a Recombinant Inbred Line Population. BMC Genomics 19, 489. 10.1186/s12864-018-4890-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L., Wang Q., Hu Y., Jia Y., Chen J., Liu B., et al. (2017). Genomic Analyses in Cotton Identify Signatures of Selection and Loci Associated with Fiber Quality and Yield Traits. Nat. Genet. 49 (7), 1089–1098. 10.1038/ng.3887 [DOI] [PubMed] [Google Scholar]
- Gai J., Liu Y., Lv H., Xing H., Zhao T., Yu D., et al. (2007). Identification, Inheritance and QTL Mapping of Root Traits Related to Tolerance to Rhizo-Spheric Stresses in Soybean (G. max (L.) Merr.). Front. Agric. China 1 (2), 119–128. 10.1007/s11703-007-0022-y [DOI] [Google Scholar]
- Gai J. Y. (1997). Crop Breeding for Various Species. China, Beijing: Agriculture Press, pp181. [Google Scholar]
- Gai Y. J., Zhang Y. M., Wang J. K. (2003). Genetic System of Quantitative Traits in Plants. China, Beijing: Science Press. [Google Scholar]
- Gao Y., Chen Y., Song Z., Zhang J., Zhang J., Zhang T., et al. (2021). Comparative Dynamic Transcriptome Reveals the Delayed Secondary-Cell-wall Thickening Results in Altered Lint Percentage and Fiber Elongation in a Chromosomal Segment Substitution Line of Cotton (Gossypium hirsutum L.). Front. Plant Sci. 12, 756434. 10.3389/fpls.2021.756434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X., Qu Y., Jia Y., He S., Pan Z., Wang L., et al. (2021). Assessment of Heterosis Based on Parental Genetic Distance Estimated with SSR and SNP Markers in upland Cotton (Gossypium hirsutum L.). BMC Genomics 22 (1), 123. 10.1186/s12864-021-07431-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Liu A., Li J., Jiang X., Geng H., Yuan Y., et al. (2019). Major Gene Plus Polygene Genetic Analysis of Lint Percent in upland Cotton. Cotton Sci. 31 (3), 192–200. 10.11963/1002-7807.gjwyyl.20190416 [DOI] [Google Scholar]
- Gu Q., Ke H., Liu Z., Lv X., Sun Z., Zhang M., et al. (2020). A High-Density Genetic Map and Multiple Environmental Tests Reveal Novel Quantitative Trait Loci and Candidate Genes for Fibre Quality and Yield in Cotton. Theor. Appl. Genet. 133, 3395–3408. 10.1007/s00122-020-03676-z [DOI] [PubMed] [Google Scholar]
- Guo L., Shi Y., Li J., Gong J., Sun J., Yuan Y., et al. (2015). Mapping QTL of Fiber Yield and Quality Traits in F2 Populations of Chromosome Segment Substitution Lines from Gossypium hirsutum × Gossypium barbadense . Cotton Sci. 27 (6), 550–560. 10.11963/issn.1002-7807.201506007 [DOI] [Google Scholar]
- Guo W.-Z., Ma G.-J., Zhu Y.-C., Yi C.-X., Zhang T.-Z. (2006). Molecular Tagging and Mapping of Quantitative Trait Loci for Lint Percentage and Morphological Marker Genes in upland Cotton. J. Integr. Plant Biol. 48 (3), 320–326. 10.1111/j.1744-7909.2006.00174.x [DOI] [Google Scholar]
- Guo X., Guo Y., Ma J., Wang F., Sun M., Gui L., et al. (2013). Mapping Heterotic Loci for Yield and Agronomic Traits Using Chromosome Segment Introgression Lines in Cotton. J. Integr. Plant Biol. 55 (8), 759–774. 10.1111/jipb.12054 [DOI] [PubMed] [Google Scholar]
- Han L., Yang J., Zhu J. (2007). Analysis of Genetic Effects of Nuclear-Cytoplasmic Interaction on Quantitative Traits: Genetic Model for Diploid Plants. J. Genet. Genomics 34 (6), 562–568. 10.1016/s1673-8527(07)60062-9 [DOI] [PubMed] [Google Scholar]
- Han Z., Hu Y., Tian Q., Cao Y., Si A., Si Z., et al. (2020). Genomic Signatures and Candidate Genes of Lint Yield and Fibre Quality Improvement in Upland Cotton in Xinjiang. Plant Biotechnol. J. 18, 2002–2014. 10.1111/pbi.13356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J., Tu L., Hu H., Tan J., Deng F., Tang W., et al. (2012). GbTCP, a Cotton TCP Transcription Factor, Confers Fibre Elongation and Root Hair Development by a Complex Regulating System. J. Exp. Bot. 63 (2), 6267–6281. 10.1093/jxb/ers278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J., Chen S., Tu L., Hu H., Zhang X. (2014). GhH2A12, a Replication-dependent Histone H2A Gene from Gossypium hirsutum, Is Negatively Involved in the Development of Cotton Fiber Cells. Plant Cel Rep 33, 1711–1721. 10.1007/s00299-014-1649-9 [DOI] [PubMed] [Google Scholar]
- Hu H., Wang M., Ding Y., Zhu S., Zhao G., Tu L., et al. (2018). Transcriptomic Repertoires Depict the Initiation of Lint and Fuzz Fibres in Cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 16, 1002–1012. 10.1111/pbi.12844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X. D., Zhou X., Huang J. Q., Zhu S. L., Zhang L. S., Liu X. M., et al. (1963). Study on the Utilization of the F1 Heterosis between Gossypium hirsutum and Gossypium barbadense . Zuo Wu Xue Bao 2 (1), 1–28. [Google Scholar]
- Huang C., Nie X., Shen C., You C., Li W., Zhao W., et al. (2017). Population Structure and Genetic Basis of the Agronomic Traits of upland Cotton in China Revealed by a Genome-wide Association Study Using High-Density SNPs. Plant Biotechnol. J. 15 (11), 1374–1386. 10.1111/pbi.12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Wu Z., Percy R. G., Bai M., Li Y., Frelichowski J. E., et al. (2020). Genome Sequence of Gossypium herbaceum and Genome Updates of Gossypium arboreum and Gossypium hirsutum Provide Insights into Cotton A-Genome Evolution. Nat. Genet. 52, 516–524. 10.1038/s41588-020-0607-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse-Kemp A. M., Lemm J., Plieske J., Ashrafi H., Buyyarapu R., Fang D. D., et al. (2015). Development of a 63K SNP Array for Cotton and High-Density Mapping of Intraspecific and Interspecific Populations of Gossypium spp. G3-genes Genom. Genet. 5, 1187–1209. 10.1534/g3.115.018416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imran M., Shakeel A., Azhar F. M., Farooq J., Saleem M. F., Saeed A., et al. (2012). Combining Ability Analysis for Within-Boll Yield Components in upland Cotton (Gossypium hirsutum L.). Genet. Mol. Res. 11 (3), 2790–2800. 10.4238/2012.august.24.4 [DOI] [PubMed] [Google Scholar]
- Jia F., Sun F., Li J., Liu A., Liu Z., Yuan Y., et al. (2011). Identification of QTL for Boll Weight and Lint Percentage of Upland Cotton (Gossypium hirsutum L.) RIL Population in Multiple Environments. Mol. Plant Breed. 9 (3), 318–326. 10.3969/mpb.009.000318 [DOI] [Google Scholar]
- Jia Y., Sun X., Sun J., Pan Z., Wang X., He S., et al. (2014). Association Mapping for Epistasis and Environmental Interaction of Yield Traits in 323 Cotton Cultivars under 9 Different Environments. PLoS ONE 9 (5), e95882. 10.1371/journal.pone.0095882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B. G., Kong F. L., Zhang Q. Y., Jiang R. Q., He J. X., Zhang X. X. (2000). Effects of Improvements of Cotton Yield Components. Acta Goss. Sin. 12 (5), 258–260. [Google Scholar]
- Jiang X., Fan L., Li P., Zou X., Zhang Z., Fan S., et al. (2021). Co-expression Network and Comparative Transcriptome Analysis for Fiber Initiation and Elongation Reveal Genetic Differences in Two Lines from upland Cotton CCRI70 RIL Population. PeerJ 9, e11812. 10.7717/peerj.11812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F. L., Jiang B. G., Zhang Q. Y., Yang F. X. (2000). Genetic Improvements of Cotton Varieties in Huang-Huai Region in China since 1950s. I. Improvements on Yield and Yield Components. Acta Agron. Sin. 26 (2), 148–156. 10.3321/j.issn:0496-3490.2000.02.004 [DOI] [PubMed] [Google Scholar]
- Kong L., Shi Y., Li S., Li B., Song W., Yuan Y., et al. (2018). QTL Mapping for Yield and Fiber Quality Traits in Double-Cross Population of Chromosome Segment Introgression Lines from Gossypium hirsutum × Gossypium barbadense . Cotton Sci. 30 (2), 119–127. 10.11963/1002-7807.kllyyl.20180321 [DOI] [Google Scholar]
- Li B., Shi Y., Gong J., Li J., Liu A., Shang H., et al. (2016). Genetic Effects and Heterosis of Yield and Yield Component Traits Based on Gossypium barbadense Chromosome Segment Substitution Lines in Two Gossypium hirsutum Backgrounds. PLoS ONE 11 (6), e0157978. 10.1371/journal.pone.0157978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Dong Y., Zhao T., Li L., Chen J., Zhu S., et al. (2016). Genome-wide SNP Linkage Mapping and QTL Analysis for Fiber Quality and Yield Traits in the upland Cotton Recombinant Inbred Lines Population. Front. Plant Sci. 7, 1356. 10.3389/fpls.2016.01356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Zhao T., Yu H., Li C., Deng X., Dong Y., et al. (2018). Genetic Basis of Heterosis for Yield and Yield Components Explored by QTL Mapping across Four Genetic Populations in upland Cotton. BMC Genomics 19, 910. 10.1186/s12864-018-5289-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Fan G., Lu C., Xiao G., Zou C., Kohel R. J., et al. (2015). Genome Sequence of Cultivated Upland Cotton (Gossypium hirsutum TM-1) Provides Insights into Genome Evolution. Nat. Biotechnol. 33 (5), 524–530. 10.1038/nbt.3208 [DOI] [PubMed] [Google Scholar]
- Li J., Yang Z., Li J., Shi Y., Wang T., Yuan Y., et al. (2009). Using G. Hirsutum × G. barbadense BC1 Populations Construction Cotton Genetic Linkage Map and Primary QTL Analysis of Yield Related Traits. Chin. Agric. Sci. Bull. 25 (09), 11–18. [Google Scholar]
- Li M., Wang Z. Z. (1992). Characterization, Screening and Enhancement of Cotton Germplasm. Zuo Wu Pin Zhong Zi Yuan 1 (3), 11–12. 10.19462/j.cnki.1671-895x.1992.03.009 [DOI] [Google Scholar]
- Li P.-t., Rashid M. H. o., Chen T.-t., Lu Q.-w., Ge Q., Gong W.-k., et al. (2019). Transcriptomic and Biochemical Analysis of upland Cotton (Gossypium hirsutum) and a Chromosome Segment Substitution Line from G. Hirsutum × G. barbadense in Response to Verticillium dahliae Infection. BMC Plant Biol. 19 (1), 19. 10.1186/s12870-018-1619-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.-t., Wang M., Lu Q.-w., Ge Q., Rashid M. H. o., Liu A.-y., et al. (2017). Comparative Transcriptome Analysis of Cotton Fiber Development of Upland Cotton (Gossypium hirsutum) and Chromosome Segment Substitution Lines from G. Hirsutum × G. barbadense. BMC Genomics 18 (1), 705. 10.1186/s12864-017-4077-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Jin X., Zhu Y.-X. (2012). Identification and Analyses of miRNA Genes in Allotetraploid Gossypium hirsutum Fiber Cells Based on the Sequenced Diploid G. Raimondii Genome. J. Genet. Genomics 39, 351–360. 10.1016/j.jgg.2012.04.008 [DOI] [PubMed] [Google Scholar]
- Liang Z., Lv Y., Cai C., Tong X., Guo X., Guo W., et al. (2012). Toward Allotetraploid Cotton Genome Assembly: Integration of a High-Density Molecular Genetic Linkage Map with DNA Sequence Information. BMC Genomics 13, 539. 10.1186/1471-2164-13-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z. X., Feng C. H., Guo X. P., Zhang X. L. (2009a). Genetic Analysis of Major QTLs and Epistasis Interaction for Yield and Fiber Quality in upland Cotton. Sci. Agric. Sin. 42 (9), 3036–3047. 10.3864/j.issn.0578-1752.2009.09.004 [DOI] [Google Scholar]
- Lin Z., Zhang Y., Zhang X., Guo X. (2009b). A High-Density Integrative Linkage Map for Gossypium hirsutum . Euphytica 166, 35–45. 10.1007/s10681-008-9822-2 [DOI] [Google Scholar]
- Liu C., Zhai X., Wang G., Li J., Li Z., Geng J. (1998). Studies on Main Agronomic and Economic Characters of Germplasm Resources of upland Cotton Introduced from Former Soviet Union. Acta Goss. Sin. 10 (2), 68–73. [Google Scholar]
- Liu D., Liu F., Shan X., Zhang J., Tang S., Fang X., et al. (2015). Construction of a High-Density Genetic Map and Lint Percentage and Cottonseed Nutrient Trait QTL Identification in upland Cotton (Gossypium hirsutum L.). Mol. Genet. Genomics 290, 1683–1700. 10.1007/s00438-015-1027-5 [DOI] [PubMed] [Google Scholar]
- Liu R., Ai N., Zhu X., Liu F., Guo W., Zhang T. (2014). Genetic Analysis of Plant Height Using Two Immortalized Populations of "CRI12 × J8891" in Gossypium hirsutum L. Euphytica 196, 51–61. 10.1007/s10681-013-1013-0 [DOI] [Google Scholar]
- Liu R., Wang B., Guo W., Qin Y., Wang L., Zhang Y., et al. (2012). Quantitative Trait Loci Mapping for Yield and its Components by Using Two Immortalized Populations of a Heterotic Hybrid in Gossypium hirsutum L. Mol. Breed. 29, 297–311. 10.1007/s11032-011-9547-0 [DOI] [Google Scholar]
- Liu R., Wang B., Guo W., Wang L., Zhang T. (2011). Differential Gene Expression and Associated QTL Mapping for Cotton Yield Based on a cDNA-AFLP Transcriptome Map in an Immortalized F2. Theor. Appl. Genet. 123, 439–454. 10.1007/s00122-011-1597-5 [DOI] [PubMed] [Google Scholar]
- Liu S. M., Koebernick J. C., Walford S. A., Constable G. A., Stiller W. N., Llewellyn D. J. (2020). Improved Lint Yield under Field Conditions in Cotton Over-expressing Transcription Factors Regulating Fibre Initiation. Transgenic Res. 29 (5-6), 529–550. 10.1007/s11248-020-00214-x [DOI] [PubMed] [Google Scholar]
- Liu X., Zhao B., Zheng H.-J., Hu Y., Lu G., Yang C.-Q., et al. (2015). Gossypium barbadense Genome Sequence Provides Insight into the Evolution of Extra-long Staple Fiber and Specialized Metabolites. Sci. Rep. 5, 14139. 10.1038/srep14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Wang F., Guo Y. P., Guo X., Sun X. Z., Song X. L., et al. (2012). Study on Lint Percentage Formation Mechanism of Chromosome Segment Introgression Line IL-10-1 with High Lint Percentage. Shandong Agri. Sci. 44 (10), 25–29. 10.14083/j.issn.1001-4942.2012.10.014 [DOI] [Google Scholar]
- Ma X., Wang Z., Li W., Zhang Y., Zhou X., Liu Y., et al. (2019). Resequencing Core Accessions of a Pedigree Identifies Derivation of Genomic Segments and Key Agronomic Trait Loci during Cotton Improvement. Plant Biotechnol. J. 17 (4), 762–775. 10.1111/pbi.13013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay T. F. C., Stone E. A., Ayroles J. F. (2009). The Genetics of Quantitative Traits: Challenges and Prospects. Nat. Rev. Genet. 10, 565–577. 10.1038/nrg2612 [DOI] [PubMed] [Google Scholar]
- McAinsh M. R., Brownlee C., Hetherington A. M. (1997). Calcium Ions as Second Messengers in Guard Cell Signal Transduction. Physiol. Plant 100, 16–29. 10.1111/j.1399-3054.1997.tb03451.x [DOI] [Google Scholar]
- Mo H. D. (1993). Genetic Analysis for Qualitative-Quantitative Traits I. The Genetic Constitution of Generation Populations and Identification of Major Gene Genotypes. Acta Agron. Sin. 19 (1), 1–6. [Google Scholar]
- Pan Y., Meng F., Wang X. (2020). Sequencing Multiple Cotton Genomes Reveals Complex Structures and Lays Foundation for Breeding. Front. Plant Sci. 11, 560096. 10.3389/fpls.2020.560096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L., Li Q., Fan X., Yang W., Xue Y. (2008). The R2R3 MYB Transcription Factor GhMYB109 Is Required for Cotton Fiber Development. Genetics 180 (2), 811–820. 10.1534/genetics.108.093070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Chen M., Yi X., Bie S., Zhang C., Zhang Y., et al. (2015). Identification of Associated SSR Markers for Yield Component and Fiber Quality Traits Based on Frame Map and upland Cotton Collections. PLoS ONE 10 (1), e0118073. 10.1371/journal.pone.0118073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Guo W., Zhang Y.-M., Zhang T. (2008). QTL Mapping of Yield and Fiber Traits Based on a Four-Way Cross Population in Gossypium hirsutum L. Theor. Appl. Genet. 117, 883–894. 10.1007/s00122-008-0828-x [DOI] [PubMed] [Google Scholar]
- Qin Y.-S., Liu R.-Z., Mei H.-X., Zhang T.-Z., Guo W.-Z. (2009). QTL Mapping for Yield Traits in upland Cotton (Gossypium hirsutum L.). A a S 35 (10), 1812–1821. 10.3724/sp.j.1006.2009.01812 [DOI] [Google Scholar]
- Qin Y., Sun H., Hao P., Wang H., Wang C., Ma L., et al. (2019). Transcriptome Analysis Reveals Differences in the Mechanisms of Fiber Initiation and Elongation between Long- and Short-Fiber Cotton (Gossypium hirsutum L.) Lines. BMC Genomics 20 (1), 633. 10.1186/s12864-019-5986-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong J., Feltus F. A., Waghmare V. N., Pierce G. J., Chee P. W., Draye X., et al. (2007). Meta-analysis of Polyploid Cotton QTL Shows Unequal Contributions of Subgenomes to a Complex Network of Genes and Gene Clusters Implicated in Lint Fiber Development. Genetics 176, 2577–2588. 10.1534/genetics.107.074518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong J., Pierce G. J., Waghmare V. N., Rogers C. J., Desai A., Chee P. W., et al. (2005). Genetic Mapping and Comparative Analysis of Seven Mutants Related to Seed Fiber Development in Cotton. Theor. Appl. Genet. 111 (6), 1137–1146. 10.1007/s00122-005-0041-0 [DOI] [PubMed] [Google Scholar]
- Saha S., Jenkins J. N., Wu J., McCarty J. C., Gutiérrez O. A., Percy R. G., et al. (2006). Effects of Chromosome-specific Introgression in upland Cotton on Fiber and Agronomic Traits. Genetics 172 (3), 1927–1938. 10.1534/genetics.105.053371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Wu J., Jenkins J. N., McCarty J. C., Hayes R., Stelly D. M. (2010). Genetic Dissection of Chromosome Substitution Lines of Cotton to Discover Novel Gossypium barbadense L. Alleles for Improvement of Agronomic Traits. Theor. Appl. Genet. 120 (6), 1193–1205. 10.1007/s00122-009-1247-3 [DOI] [PubMed] [Google Scholar]
- Said J. I., Song M., Wang H., Lin Z., Zhang X., Fang D. D., et al. (2015). A Comparative Meta-Analysis of QTL between Intraspecific Gossypium hirsutum and Interspecific G. hirsutum × G. barbadense Populations. Mol. Genet. Genomics 290 (3), 1003–1025. 10.1007/s00438-014-0963-9 [DOI] [PubMed] [Google Scholar]
- Santos I. G., Teodoro P. E., Farias F. C., Farias F. J., Carvalho L. P., Rodrigues J. I., et al. (2017). Genetic Diversity Among Cotton Cultivars in Two Environments in the State of Mato Grosso. Genet. Mol. Res. 16 (2), gmr16029628. 10.4238/gmr16029628 [DOI] [PubMed] [Google Scholar]
- Sarfraz Z., Iqbal M. S., Geng X., Iqbal M. S., Nazir M. F., Ahmed H., et al. (2021). GWAS Mediated Elucidation of Heterosis for Metric Traits in Cotton (Gossypium hirsutum L.) across Multiple Environments. Front. Plant Sci. 12, 565552. 10.3389/fpls.2021.565552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad K., Zhang X., Guo L., Qi T., Bao L., Zhang M., et al. (2020). Comparative Transcriptome Analysis between Inbred and Hybrids Reveals Molecular Insights into Yield Heterosis of upland Cotton. BMC Plant Biol. 20, 239. 10.1186/s12870-020-02442-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan X., Yang Q., Wu X., Cao J. (2021). Function Analysis of a Cotton R2R3 MYB Transcription Factor GhMYB3 in Regulating Plant Trichome Development. Plant Biol. J. 23, 1118–1127. 10.1111/plb.13299 [DOI] [PubMed] [Google Scholar]
- Shappley Z. W., Jenkins J. N., Zhu J., McCarty J. C., Jr. (1998). Cotton Improvement, Quantitative Trait Loci Associated with Agronomic and Fiber Traits of upland Cotton. J. Cotton Sci. 2, 153–163. [Google Scholar]
- Shen C., Wang N., Huang C., Wang M., Zhang X., Lin Z. (2019). Population Genomics Reveals a fine‐scale Recombination Landscape for Genetic Improvement of Cotton. Plant J. 99 (3), 494–505. 10.1111/tpj.14339 [DOI] [PubMed] [Google Scholar]
- Shen X., Guo W., Lu Q., Zhu X., Yuan Y., Zhang T. (2007). Genetic Mapping of Quantitative Trait Loci for Fiber Quality and Yield Trait by RIL Approach in Upland Cotton. Euphytica 155, 371–380. 10.1007/s10681-006-9338-6 [DOI] [Google Scholar]
- Shen X., Zhang T., Guo W., Zhu X., Zhang X. (2006). Mapping Fiber and Yield QTLs with Main, Epistatic, and QTL × Environment Interaction Effects in Recombinant Inbred Lines of Upland Cotton. Crop Sci. 46, 61–66. 10.2135/cropsci2005.0056 [DOI] [Google Scholar]
- Shi Y., Li W., Li A., Ge R., Zhang B., Li J., et al. (2015). Constructing a High-Density Linkage Map for Gossypium hirsutum × Gossypium barbadense and Identifying QTLs for Lint Percentage. J. Integr. Plant Biol. 57 (5), 450–467. 10.1111/jipb.12288 [DOI] [PubMed] [Google Scholar]
- Song C., Li W., Pei X., Liu Y., Ren Z., He K., et al. (2019). Dissection of the Genetic Variation and Candidate Genes of Lint Percentage by a Genome-wide Association Study in upland Cotton. Theor. Appl. Genet. 132 (7), 1991–2002. 10.1007/s00122-019-03333-0 [DOI] [PubMed] [Google Scholar]
- Song X., Zhang T. (2009). Quantitative Trait Loci Controlling Plant Architectural Traits in Cotton. Plant Sci. 177, 317–323. 10.1016/j.plantsci.2009.05.015 [DOI] [Google Scholar]
- Stewart J. M. (1975). Fiber Initiation on the Cotton Ovule (Gossypium hirsutum). Am. J. Bot. 62 (7), 723–730. 10.1002/j.1537-2197.1975.tb14105.x [DOI] [Google Scholar]
- Su J., Fan S., Li L., Wei H., Wang C., Wang H., et al. (2016). Detection of Favorable QTL Alleles and Candidate Genes for Lint Percentage by GWAS in Chinese Upland Cotton. Front. Plant Sci. 7, 1576. 10.3389/fpls.2016.01576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J., Wang C., Hao F., Ma Q., Wang J., Li J., et al. (2019). Genetic Detection of Lint Percentage Applying Single-Locus and Multi-Locus Genome-wide Association Studies in Chinese Early-Maturity upland Cotton. Front. Plant Sci. 10, 964. 10.3389/fpls.2019.00964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J., Wang C., Ma Q., Zhang A., Shi C., Liu J., et al. (2020). An RTM-GWAS Procedure Reveals the QTL Alleles and Candidate Genes for Three Yield-Related Traits in upland Cotton. BMC Plant Biol. 20 (1), 416. 10.1186/s12870-020-02613-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Guo A., Huang Y., Wang Y., Hua J. (2020). GhCIPK6a Increases Salt Tolerance in Transgenic upland Cotton by Involving in ROS Scavenging and MAPK Signaling Pathways. BMC Plant Biol. 20 (1), 421. 10.1186/s12870-020-02548-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Wang X., Liu Z., Gu Q., Zhang Y., Li Z., et al. (2018). A Genome-wide Association Study Uncovers Novel Genomic Regions and Candidate Genes of Yield-Related Traits in upland Cotton. Theor. Appl. Genet. 131 (11), 2413–2425. 10.1007/s00122-018-3162-y [DOI] [PubMed] [Google Scholar]
- Tang F., Xiao W. (2013). Genetic Effects and Heterosis of Within-Boll Yield Components in upland Cotton (Gossypium hirsutum L.). Euphytica 194, 45–51. 10.1007/s10681-013-0958-3 [DOI] [Google Scholar]
- Tang W., Tu L., Yang X., Tan J., Deng F., Hao J., et al. (2014). The Calcium Sensor GhCaM7 Promotes Cotton Fiber Elongation by Modulating Reactive Oxygen Species ( ROS ) Production. New Phytol. 202 (2), 509–520. 10.1111/nph.12676 [DOI] [PubMed] [Google Scholar]
- Tian S., Xu X., Zhu X., Wang F., Song X., Zhang T. (2019). Overdominance Is the Major Genetic Basis of Lint Yield Heterosis in Interspecific Hybrids between G. Hirsutum and G. barbadense . Heredity 123 (3), 384–394. 10.1038/s41437-019-0211-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa M., Meredith W. R., Jr. (2000). Genetic Linkage Map and QTL Analysis of Agronomic and Fiber Quality Traits in an Intraspecific Population. J. Cotton Sci. 4, 161–170. [Google Scholar]
- Ulloa M., Saha S., Jenkins J. N., Meredith W. R., jr., McCarty J. C., jr., Stelly D. M. (2005). Chromosomal Assignment of RFLP Linkage Groups Harboring Important QTLs on an Intraspecific Cotton (Gossypium hirsutum L.) Joinmap. J. Hered. 96 (2), 132–144. 10.1093/jhered/esi020 [DOI] [PubMed] [Google Scholar]
- Wan Q., Zhang Z., Hu M., Chen L., Liu D., Chen X., et al. (2007). T1 Locus in Cotton Is the Candidate Gene Affecting Lint Percentage, Fiber Quality and Spiny Bollworm (Earias spp.) Resistance. Euphytica 158, 241–247. 10.1007/s10681-007-9446-y [DOI] [Google Scholar]
- Wang B., Guo W., Zhu X., Wu Y., Huang N., Zhang T. (2007). QTL Mapping of Yield and Yield Components for Elite Hybrid Derived-RILs in upland Cotton. J. Genet. Genomics 34 (1), 35–45. 10.1016/s1673-8527(07)60005-8 [DOI] [PubMed] [Google Scholar]
- Wang F., Gong Y., Zhang C., Liu G., Wang L., Xu Z., et al. (2011). Genetic Effects of Introgression Genomic Components from Sea Island Cotton (Gossypium barbadense L.) on Fiber Related Traits in upland Cotton (G. Hirsutum L.). Euphytica 181, 41–53. 10.1007/s10681-011-0378-1 [DOI] [Google Scholar]
- Wang F., Zhang J., Chen Y., Zhang C., Gong J., Song Z., et al. (2020). Identification of Candidate Genes for Key Fibre‐related QTL S and Derivation of Favourable Alleles in Gossypium hirsutum Recombinant Inbred Lines with G. barbadense Introgressions. Plant Biotechnol. J. 18, 707–720. 10.1111/pbi.13237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Huang C., Zhao W., Dai B., Shen C., Zhang B., et al. (2016). Identification of QTL for Fiber Quality and Yield Traits Using Two Immortalized Backcross Populations in upland Cotton. PLoS ONE 11 (12), e0166970. 10.1371/journal.pone.0166970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Li C., Wang Q. (2014). Quantitative Trait Loci Mapping and Genetic Dissection for Lint Percentage in upland Cotton (Gossypium hirsutum). J. Genet. 93, 371–378. 10.1007/s12041-014-0385-9 [DOI] [PubMed] [Google Scholar]
- Wang M., Li J., Wang P., Liu F., Liu Z., Zhao G., et al. (2021). Comparative Genome Analyses Highlight Transposon-Mediated Genome Expansion and the Evolutionary Architecture of 3D Genomic Folding in Cotton. Mol. Biol. Evol. 38 (9), 3621–3636. 10.1093/molbev/msab128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. P., Xiao S. D. (1996). Breeding advance of the New Cotton Line C24 with High Lint Percentage. Chin. Agric. Sci. Bull. 12 (6), 27–28. [Google Scholar]
- Wendel J. F., Cronn R. C. (2003). Polyploidy and the Evolutionary History of Cotton. Adv. Agron. 78, 139–186. 10.1016/s0065-2113(02)78004-8 [DOI] [Google Scholar]
- Wu J., Gutierrez O. A., Jenkins J. N., McCarty J. C., Zhu J. (2009). Quantitative Analysis and QTL Mapping for Agronomic and Fiber Traits in an RI Population of upland Cotton. Euphytica 165, 231–245. 10.1007/s10681-008-9748-8 [DOI] [Google Scholar]
- Xia Z., Zhang X., Liu Y.-y., Jia Z.-f., Zhao H.-h., Li C.-q., et al. (2014). Major Gene Identification and Quantitative Trait Locus Mapping for Yield-Related Traits in upland Cotton (Gossypium hirsutum L.). J. Integr. Agric. 13 (2), 299–309. 10.1016/S2095-3119(13)60508-0 [DOI] [Google Scholar]
- Ye Z., Qiao L., Luo X., Chen X., Zhang X., Tu L. (2021). Genome-wide Identification of Cotton GRAM Family Proteins Reveals that GRAM31 Regulates Fiber Length. J. Exp. Bot. 72 (7), 2477–2490. 10.1093/jxb/eraa597 [DOI] [PubMed] [Google Scholar]
- Yin J. M., Wu Y. T., Zhang J., Zhang T. Z., Guo W. Z., Zhu X. F. (2002). [Tagging and Mapping of QTLs Controlling Lint Yield and Yield Components in upland Cotton (Gossypium hirsutum L.) Using SSR and RAPD Markers]. Sheng Wu Gong Cheng Xue Bao 18 (2), 162–166. 10.13345/j.cjb.2002.02.031 [DOI] [PubMed] [Google Scholar]
- Yu J., Hui Y., Chen J., Yu H., Zhu S., Zhao T., et al. (2021). Whole-genome Resequencing of 240 Gossypium barbadense Accessions Reveals Genetic Variation and Genes Associated with Fiber Strength and Lint Percentage. Theor. Appl. Genet. 134, 3249–3261. 10.1007/s00122-021-03889-w [DOI] [PubMed] [Google Scholar]
- Yu J., Zhang K., Li S., Yu S., Zhai H., Wu M., et al. (2013). Mapping Quantitative Trait Loci for Lint Yield and Fiber Quality across Environments in a Gossypium hirsutum × Gossypium barbadense Backcross Inbred Line Population. Theor. Appl. Genet. 126, 275–287. 10.1007/s00122-012-1980-x [DOI] [PubMed] [Google Scholar]
- Yu S. X., Fan S. L., Wang H. T., Wei H. L., Pang C. Y. (2016). Progresses in Research on Cotton High Yield Breeding in China. Sci. Agric. Sin. 49 (18), 3465–3476. 10.3864/j.issn.0578-1752.2016.18.001 [DOI] [Google Scholar]
- Yuan D. J., Tang Z. H., Wang M. J., Gao W. H., Ruan Y., Zhang X., et al. (2015). The Genome Sequence of Sea-Island Cotton (Gossypium barbadense) Provides Insights into the Allopolyploidization and Development of superior Spinnable Fibres. Sci. Rep-uk 5, 17662. 10.1038/srep17662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y. L., Zhang T. Z., Guo W. Z., Pan J. J., Kohel R. J. (2002). Heterosis and Gene Action of Boll Weight and Lint Percentage in High Quality Fiber Property Varieties in Upland Cotton. Acta Agron. Sin. 28 (2), 196–202. [Google Scholar]
- Zeng L., Wu J. (2012). Germplasm for Genetic Improvement of Lint Yield in Upland Cotton: Genetic Analysis of Lint Yield with Yield Components. Euphytica 187, 247–261. 10.1007/s10681-012-0708-y [DOI] [Google Scholar]
- Zhai H., Gong W., Tan Y., Liu A., Shi Y., Yuan Y., et al. (2016). Identification of Chromosome Segment Substitution Lines of Gossypium barbadense Introgressed in G. Hirsutum and Quantitative Trait Locus Mapping for Fiber Quality and Yield Traits. PLoS ONE 11 (9), e0159101. 10.1371/journal.pone.0159101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Huang G.-Q., Zou D., Yan J.-Q., Li Y., Hu S., et al. (2018). The Cotton (Gossypium hirsutum) NAC Transcription Factor (FSN1) as a Positive Regulator Participates in Controlling Secondary Cell wall Biosynthesis and Modification of Fibers. New Phytol. 217, 625–640. 10.1111/nph.14864 [DOI] [PubMed] [Google Scholar]
- Zhang K., Kuraparthy V., Fang H., Zhu L., Sood S., Jones D. C. (2019). High-density Linkage Map Construction and QTL Analyses for Fiber Quality, Yield and Morphological Traits Using CottonSNP63K Array in upland Cotton (Gossypium hirsutum L.). BMC Genomics 20, 889. 10.1186/s12864-019-6214-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P. T., Zhu X. F., Guo W. Z., Zhang T. Z. (2005). Genetic Analysis and QTLs Tagging of Lint Percentage and its Closely Related Yield Compnents in upland Cotton. Jiang Su J. Agric. Sci. 21 (4), 264–271. [Google Scholar]
- Zhang T., Hu Y., Jiang W., Fang L., Guan X., Chen J., et al. (2015). Sequencing of Allotetraploid Cotton (Gossypium hirsutum L. Acc. TM-1) Provides a Resource for Fiber Improvement. Nat. Biotechnol. 33 (5), 531–537. 10.1038/nbt.3207 [DOI] [PubMed] [Google Scholar]
- Zhang W., Liu F., Li S.-H., Wang W., Wang C.-Y., Zhang X.-D., et al. (2011). QTL Analysis on Yield and its Components in Upland Cotton RIL. Acta Agron. Sin. 37 (3), 433–442. 10.3724/sp.j.1006.2011.00433 [DOI] [Google Scholar]
- Zhang Z.-S., Xiao Y.-H., Luo M., Li X.-B., Luo X.-Y., Hou L., et al. (2005). Construction of a Genetic Linkage Map and QTL Analysis of Fiber-Related Traits in upland Cotton (Gossypium hirsutum L.). Euphytica 144, 91–99. 10.1007/s10681-005-4629-x [DOI] [Google Scholar]
- Zhang Z., Li J., Jamshed M., Shi Y., Liu A., Gong J., et al. (2019). Genome‐wide Quantitative Trait Loci Reveal the Genetic Basis of Cotton Fibre Quality and Yield‐related Traits in a Gossypium hirsutum Recombinant Inbred Line Population. Plant Biotechnol. J. 18 (1), 239–253. 10.1111/pbi.13191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D., Li X., Wang Z., You C., Nie X., Sun J., et al. (2020). Genetic Dissection of an Allotetraploid Interspecific CSSLs Guides Interspecific Genetics and Breeding in Cotton. BMC Genomics 21, 431. 10.1186/s12864-020-06800-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G., Gao W., Song X., Sun F., Hou S., Liu N., et al. (2020). Genome-wide Association Reveals Genetic Variation of Lint Yield Components under Salty Field Conditions in Cotton (Gossypium hirsutum L.). BMC Plant Biol. 20, 23. 10.1186/s12870-019-2187-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G., Hou S., Song X., Wang X., Wang W., Chen Q., et al. (2021). Genome-wide Association Analysis Reveals Quantitative Trait Loci and Candidate Genes Involved in Yield Components under Multiple Field Environments in Cotton (Gossypium hirsutum). BMC Plant Biol. 21, 250. 10.1186/s12870-021-03009-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.-F., Wang P., Si Z.-F., Zhang T.-Z. (2017). QTL Mapping for Yield Components in Gossypium barbadense Chromosome Segment Introgression Lines Based on Gossypium hirsutum Background. Acta Agronomica Sinica 43 (12), 1784–1790. 10.3724/sp.j.1006.2017.01784 [DOI] [Google Scholar]
- Zhu Y.-J., Wang P., Guo W.-Z., Zhang T.-Z. (2010). Mapping QTLs for Lint Percentage and Seed Index Using Gossypium Bar-Badense Chromosome Segment Introgression Lines. A a S 36 (8), 1318–1323. 10.3724/sp.j.1006.2010.01318 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.