

Abstract
Background/Aim: Histone methylation status is required to control gene expression. H3K27me3 is an epigenetic tri-methylation modification to histone H3 controlled by the demethylase JMJD3. JMJD3 is dysregulated in a wide range of cancers and has been shown to control the expression of a specific growth-modulatory gene signature, making it an interesting candidate to better understand prostate tumor progression in vivo. This study aimed to identify the impact of JMJD3 inhibition by its inhibitor, GSK4, on prostate tumor growth in vivo.
Materials and Methods: Prostate cancer cell lines were implanted into Balb/c nude male mice. The effects of the selective JMJD3 inhibitor GSK-J4 on tumor growth were analyzed by bioluminescence assays and H3K27me3-regulated changes in gene expression were analyzed by ChIP-qPCR and RT-qPCR.
Results: JMJD3 inhibition contributed to an increase in tumor growth in androgen-independent (AR-) xenografts and a decrease in androgen-dependent (AR+). GSK-J4 treatment modulated H3K27me3 enrichment on the gene panel in DU-145-luc xenografts while it had little effect on PC3-luc and no effect on LNCaP-luc. Effects of JMJD3 inhibition affected the panel gene expression.
Conclusion: JMJD3 has a differential effect in prostate tumor progression according to AR status. Our results suggest that JMJD3 is able to play a role independently of its demethylase function in androgen-independent prostate cancer. The effects of GSK-J4 on AR+ prostate xenografts led to a decrease in tumor growth.
Keywords: Prostate cancer, epigenetics, JMJD3, H3K27me3, epidrugs, GSK-J4, xenografts, AR status
Prostate cancer (PC) is a heterogeneous disease depending on the aggressiveness of the cancer. Annual incidence and mortality in 2020 were 1,414,259 cases and 375,304 deaths. It is the second most common cancer after lung cancer and the fourth most deadly among men (1).
PC is a multifactorial disease with risk factors that can be extrinsic or intrinsic [National Cancer Institute, Bethesda in Maryland (MD)]. An increasing number of studies highlight the link between epigenetics and tumor initiation and progression. Epigenetic processes can be defined as the modification of gene expression without changes in DNA sequence and are involved in a large number of cellular events such as cell division, differentiation, cell survival (2-4). Epigenetic modulation can be achieved through chromatin compaction by activation or inhibition of gene expression (5). The deregulation of these mechanisms can lead to the transformation of healthy cells into cancerous cells. Our study focuses on one type of epigenetic modification: post-translational modification (PTM) of histones and more specifically the tri-methylation of histone 3 at lysine 27 (H3K27me3), a repressive epigenetic mark (6). H3K27 methylation is regulated by Enhancer Zeste Homolog 2 methyltransferase (EZH2) (7-9) and jumonji domain-containing protein 3 demethylase (JMJD3), also called KDM6B, which is able to remove the di-tri methylation of H3K27 (10).
The ability of JMJD3 to demethylate H3K27me3 confers a role in activating transcription (11). JMJD3 acts in many biological processes such as neuronal differentiation (12), immunity (13), inflammation (14-16), hematopoiesis (17,18), osteogenesis (19-23) and embryonic development (24,25). Moreover, JMJD3 has a role in chromatin remodeling (26,27) and in cell reprogramming (28) independently of its demethylase activity. JMJD3 is often over or under expressed in a large variety of cancers including, but no limited to nervous system, prostate, blood, colorectal, breast, lung, liver, ovarian, gastric cancers. This makes the demethylase difficult to classify as a tumor suppressor or oncoprotein (29).
JMJD3 and EZH2 have been previously shown to be overexpressed in PC (30,31). JMJD3 expression was dependent on the aggressiveness of the cancer, as indicated by an increased Gleason score (32-34). A previous study by Daures et al. identified a gene signature on prostate biopsies, impacted by JMJD3 and EZH2, comprised of; O-methylguanine-DNA methyltransferase (MGMT), transformer 2 alpha homolog (TRA2A), 2 small nuclear RNA auxiliary factor 1 (U2AF1) and ribosomal protein S6 kinase A2 (RPS6KA2). These genes are involved in DNA damage repair, mRNA splicing and cell differentiation (34). Furthermore, a link between the androgen receptor (AR), EZH2 (35) and JMJD3 demethylase activity was demonstrated: androgen status resulted in differential accumulation of JMJD3 at candidate genes between AR+ and AR- cells (36) and this was reflected at the transcriptional level (32). JMJD3 is therefore an interesting candidate to study for a better understanding of prostate tumor progression.
Novel therapeutic strategies that target epigenetic regulatory factors, such as JMJD3, offer promising new therapeutic potential to treat aggressive prostate cancer. An increasing number of molecules inhibiting JMJD3 are currently being developed, but ethyl 3-((6-(4,5-dihydro-1H-benzo[d]azepin-3(2H)-yl)-2-(pyridin-2-yl)pyrimidin-4-yl)amino)propanoate (GSK-J4) remains the most studied in basic research. This molecule inhibits demethylase by binding to the active site in a competitive manner with α-ketoglutrate (15).
In this study, we analyzed JMJD3 involvement in tumor progression in vivo after pharmacological inhibition of JMJD3 using GSK-J4. JMJD3 being a demethylase of chromatin-bound histone 3 trimethylation, analysis of candidate gene expression as well as enrichment of these genes for trimethylation on histone 3 was performed on the collected xenografts. The study revealed that JMJD3 has a role in tumor progression and supports the hypothetical link between JMJD3 and AR. Furthermore, we identified a differential effect of the inhibitor depending on the xenograft hormonal status.
Materials and Methods
Cell culture conditions. PC3-luc (PerkinElmer, Beaconsfield, UK) and DU-145-luc (JCRB Cell Bank) cells were grown in EMEM medium (ATCC, VA, USA) and LNCaP-luc (PerkinElmer, Beaconsfield, UK) were grown in RPMI 1640 medium (Gibco, Thermo Fisher Scientific, Carlsbad, CA, USA). All cells were grown at 37˚C and 5% CO2 in a supplemented medium with 10% heat-inactivated fetal bovine serum (FBS) (Life Technologies, Carlsbad, CA, USA), 1% L-glutamine and 0,1% of gentamicin (Panpharma, Luitré, France) (DU-145-luc and LNCaP-luc) and 0,1% of puromycin (InvivoGen, Toulouse, France) (PC3-luc). All cells express firefly luciferase for in vivo bioluminescence imaging. Table I summarizes origins, AR, Phosphatase and tension homolog (PTEN) and Luciferase status of the cell lines (PC-3, DU-145, LNCaP) used to create xenografts.
Table I. Origins, AR, PTEN and luciferase status of the cell lines (PC-3, DU-145, LNCaP) used to create xenografts.

Xenografts studies. Five-week-old male athymic mice (nu/nu genotype, BALB/c background) were purchased from Janvier Laboratories (Le Genest-Saint-Isle, France) and housed under aseptic conditions. All protocols in this study were approved by the Ethics Committee (No. 2018122116517595). Mice were anesthetized by isoflurane and 3×106 cells (PC3-luc and DU-145-luc), 10×106 (LNCaP-luc) in 100 μl of cell culture media resuspended in 100 μl of matrigel (354248, Corning, NY, USA) were injected subcutaneously into the right flank of mice. To test the effects of JMJD3 inhibition by GSK-J4, mice were randomly assigned to vehicle (DMSO) and GSKJ4 treatment groups. The treatment began one day after cell injection and 50 mg/kg of GSK-J4 were administered by intraperitoneal injection every day for 10 consecutive days. Tumor growth and response to therapy were determine once a week by caliper measurement and twice a week by bioluminescent imaging. For imaging, mice were anesthetized with isoflurane and examined for tumor bioluminescence 10 min following intraperitoneal injection of XenoLight D-Luciferin, Potassium Salt at 150 mg/kg (122799, PerkinElmer, Beaconsfield, UK). Signal intensities were quantified within regions of interest, as defined by the Living Image software. Mice were sacrificed at 31, 50 and 65 days post inoculation respectively for the PC3-luc, LNCaP-luc and DU-145-luc cell lines. The protocol was ended upon reaching endpoints determined by the ethics committee. Tumors were collected and were preserved in liquid nitrogen. Tumor volumes (mm3) were measured with caliper and calculated using the formula V=L×L×S/2 where L and S are the largest and smallest diameters in millimeters. After collection, tumors were also weighed with a precision balance.
RT-qPCR. Total RNA was extracted from tumor according to the manufacter’s instruction (Macherey Nagel, Hoerdt, France). The purity of RNA was determined using the NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, Massy, France). RNase-free DNase treatment, (1 U/μl) (Thermo Scientific) was performed for 1 μg of total RNA was reverse-transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems, CA, USA). Incubation was at 25˚C for 10 min, reverse transcription was at 37˚C for 120 min, and inactivation was at 85˚C for 5 min.
All genes and control genes studied were amplified in triplicate using TaqMan Gene Expression PCR Master Mix (Applied Biosystems, Foster City, CA, USA). qPCR was performed on plates designed by Applied Biosystems (TaqMan® Array 96 well Fast Plate, Custom format 16) using ABI PRISM 7900HT sequence detection system (Applied Biosystems, Thermo Fisher Scientific). Data were collected after 50˚C during 2 min, 95˚C for 10 min, 95˚C for 15 s and 60˚C for 1 min and the two latest steps were repeated in 40 cycles. The analysis was conducted on 13 genes (Table II). Genes were considered significantly expressed and their transcript measurable if their corresponding cycle threshold (CT) value was less than 35. The relative quantification method 2−ΔΔCT was used to calculate the relative gene expression. Paired t-tests were used to compare gene expression levels. Control of the false discovery rate due to multiple testing was done according to the Holm-Sidak method.
Table II. Gene list and assay ID for TaqMan – RT-qPCR assay.

Chromatin extraction and sonication. A total of 30-40 mg of xenografts were disrupted with gentleMACS Dissociator (Miltenyie Biotec, Bergisch Gladbach, Germany) in cold PBS/PIC (Protease Inhibitor Cocktail) (Diagenode, Seraing, Belgium). Protein-DNA binding was done using 1% formaldehyde (Sigma-Aldrich, Saint-Quentin-Fallavier, France) for 8 min at room temperature. Cross-linking was stopped with 0.125 M glycine for 5 min at room temperature followed by two washes (5 min, 900 g). The cell lysis step was performed according to the manufacturer’s instructions (iDeal ChIP-seq Kit for Histones, Diagenode, Seraing, Belgium). Samples were next sonicated in shearing buffer+PIC in Bioruptor™ sonicator (Diagenode) for 10 min (10 cycles, 30 s ON/30 s OFF) at 4˚C. Lysates were clarified by centrifugation (10 min at 14,000 g, 4˚C), and supernatants were transferred to new tubes.
Chromatin immunoprecipitation assay. ChIP was performed using an iDeal ChIP-seq Kit for Histones (Diagenode) according to the manufacturer’s protocol on an SX-8G IP-Star® Compact Automated System (Diagenode). ChIP used 200 μL of sonicated chromatin and 3 μg of antibodies: anti-H3K27me3 (#C15410069, Diagenode) and anti-IgG for negative control (#C15410206, Diagenode). Antibody coating reaction with protein A-coated magnetic beads lasted 3 h, and the immunoprecipitation reaction 13 h at 4˚C. Reverse cross-linking was carried out for 4 h at 65˚C with 5 M NaCl. Immunoprecipitated DNA (IP) and total DNA (input) were purified by MicroChIP DiaPure columns (#C03040001, Diagenode) according to the manufacturer’s instructions, and analyzed by real-time PCR.
qPCR. DNA (5 μl) was analyzed by qPCR (ABI PRISM 7900HT, Applied Biosystems) in triplicate in a 25 μl reaction volume containing TaqMan Gene Expression PCR Master Mix (Applied Biosystems) with the forward and reverse primers (400 nM) and probes (250 nM) (Table III): MGMT, RPS6KA2, TRA2A, U2AF1. To calculate the relative amount of immunoprecipitated DNA compared to the INPUT DNA, the formula used was:
Table III. Primers and probes used for ChIP-qPCR assay.

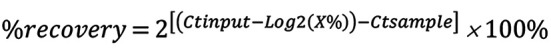
Where: log2(x) accounts for the INPUT dilution.
Ethical disclosure. For in vivo xenograft models, the authors state that they have obtained appropriate institutional review board approval (APAFIS#18214-2018122116517595 V2).
Results
Effects of JMJD3 inhibition differed depending on xenograft type. To determine the role of JMJD3 in PC tumor growth, 5-week-old male athymic mice (nu/nu genotype, BALB/c background) were subcutaneously implanted with PC3-luc, DU-145-luc and LNCaP-luc cells to form xenografts and were treated with JMJD3 inhibitor (GSK-J4). The treatment began one day after cell injection and 50 mg of GSK-J4 were administered intraperitoneally every day for 10 consecutive days. As can be seen in Figure 1a, GSK-J4 treatment induced higher xenograft growth over time for AR- cells (PC3-luc and DU-145-luc) compared to controls. The growth is reflected by the bioluminescence intensity of the xenografts. Treated AR+ xenografts (LNCaP-luc) grew less rapidly than control xenografts during the 10 days of treatment. A strong rebound in bioluminescence was noted at D16 (week after the end of treatment) in treated mice and then as early as D22 and at D50, less bioluminescence was observed in treated mice compared to control mice. These results could be matched with the measurement of tumor volume over time (Figure 1b) for the AR- xenografts (PC3-luc and DU-145-luc). Indeed, it was noted that tumor volume was larger in the treated group compared to the control group. In contrast, the difference in bioluminescence of LNCaP-luc xenografts between the control and treated groups noted in Figure 1a does not reflect what was measured in volume (mm3) for the first 37 days in Figure 1b where no difference in volume was observed between the control and treated groups. In agreement with the bioluminescence results, the xenografts showed a difference in volume from D37 until the end of the protocol. Finally, the xenografts were weighed at the end of the protocol, after mice were sacrificed. Figure 1c shows that the treated PC3-luc and DU-145-luc xenografts were heavier than the control xenografts. The treated LNCaP-luc xenografts were lighter than the control xenografts. The difference in tumor growth between the control and treated groups in the different categories of xenografts suggests that the treatment was successful and that JMJD3 and H3K27me3 are in part involved in PC tumor growth. The difference in results between AR- and AR+ xenografts suggests that AR status may also play a role, in association with JMJD3.
Figure 1. Effects of GSK-J4 treatment on tumor growth in vivo. (a) Images of measuring bioluminescence intensity of the region of interest (ROI) in p/sec/cm2/sr of PC3-luc, DU-145-luc and LNCaP-luc xenografts on the last day of the protocol (days 31, 65, 50 respectively). (b) Measurement of tumor growth by bioluminescence, tumor volume (c) and tumor weight (d) after inoculation of PC3-luc, DU-145-luc and LNCaP-luc cells in male balb/c nude mice controls or treated with GSK-J4. PC3-luc xenografts n=9, DU-145-luc xenografts n=8 and LNCaP-luc xenografts n=5. Data are expressed as means for each group, and error bars indicate the 95% confidence interval of the mean value. Data were analyzed by paired=TRUE Student’s t-test (*p<0.05; **p<0.01) for PC3-luc and DU-145-luc xenografts and data were analyzed by a non-parametric test, the Mann-Whitney test (p<0.05) for LNCaP-luc xenografts.


Three different profiles in H3K27me3 enrichment on MGMT, TRA2A, U2AF1, RPS6KA2 genes. To ascertain the impact of JMJD3 inhibition on H3K27me3 enrichment on the candidate gene promoters identified in vitro (34), we performed ChIP-qPCR on collected xenografts. ChIP undertaken on treated PC3-luc xenografts (Figure 2a) showed that H3K27me3 was found more enriched in the promoter of the control genes (Myoglobin and GAPDH) compared to the control groups. In contrast, GSK-J4 treatment on these same xenografts had no effect on the enrichment of H3K27me3 in the promoters of the four candidate genes (MGMT, TRA2A, U2AF1, RPS6KA2), even though there was a tendency to increase in the treated group. While JMJD3 demethylates H3K27me3, its inhibition by GSK-J4 in DU-145-luc xenografts (Figure 2b) showed an unexpected decrease of H3K27me3 on the promoters of the whole set of genes studied with a significant decrease for U2AF1 and RPS6KA2 genes in the treated group compared to the control group. Furthermore, GSK-J4 treatment on LNCaP-luc xenografts (Figure 2c) had no effect on H3K27me3 enrichment on the promoter of the genes since there was no difference between the control group and the treated group. These results suggest that the hormonal and PTEN status of the different xenografts could influence the treatment and again highlight a definite role for AR in relation to the treatment outcomes of LNCaP-luc xenografts. Based on these results, it appears that GSK-J4 treatment modulated H3K27me3 enrichment on the gene panel in DU-145-luc while it had little effect on PC3-luc and no effect LNCaP-luc xenografts.
Figure 2. Effects of GSK-J4 treatment on enrichment of MGMT, TRA2A, U2AF1, RPS6KA2 genes in vivo. (a) PC3-luc, (b) DU-145-luc and (c) LNCaP-luc xenografts treated with GSK-J4 (green bar) and untreated (black bar) were subjected to ChIP qPCR analysis of H3K27me3 enrichment on control genes (Myoglobin and GAPDH) and candidate genes (MGMT, TRA2A, U2AF1, RPS6KA2). Results are represented as fold enrichment over IgG. Values=mean±SD and data were analyzed by unpaired non-parametric test, the Mann-Whitney t-test (*p<0.05).

Inhibition of JMJD3 affects the expression of MGMT, TRA2A, U2AF1, RPS6KA2 and candidate oncogenes and oncosupressors in LNCaP xenografts. In order to better understand the reason of the difference in tumor growth between treated and untreated xenografts, MGMT, TRA2A, U2AF1, RPS6KA2, PTEN, JMJD3, TP53, CDKN1A, AKT1, AR, NFKB1, EZH2 and HIF1 expression analysis by RT-qPCR was conducted on PC3-luc, DU-145-luc and LNCaP-luc xenografts (Figure 3). These genes were selected after an extensive review of the literature on the roles of JMJD3 in different cancers. They have shown particular interest in other types of cancer. In agreement with gene expression profile of the different tumor types, no AR expression was found in AR- xenografts (PC-3-luc and DU-145-luc), nor PTEN expression in PC3-luc (PTEN-/-) was discovered. In contrast, PTEN expression was notable in LNCaP xenografts due to the cells harboring one deleted allele and one mutated allele (37). A slight decrease in gene expression was observed in PC3-luc and DU-145-luc xenografts compared to controls with a significant decrease in RPS6KA2, EZH2, and HIF1 expression and no difference in CDKN1A expression for DU-145-luc xenografts. In AR+ xenografts (LNCaP-luc), a significant decrease in gene expression (except JMJD3) was demonstrated in the treated group compared to the control group.
Figure 3. Effects of GSK-J4 treatment on MGMT, TRA2A, U2AF1, RPS6KA2 expression and candidate oncogenes and oncosupressor expression. mRNA level analysis of candidate genes from PC3-luc, DU-145-luc and LNCaP-luc xenografts were quantified by RT-qPCR using comparative 2(–∆∆Ct) method. Values are the average (mean±SD) from n=8 PC3-luc xenografts, n=7 DU-145-luc xenografts, n=4 LNCaP-luc xenografts and normalized to control xenografts without treatment. Data were analyzed by unpaired t-test to compare gene expression (*p<0.05).

Discussion
In this study, we demonstrated that JMJD3 and H3K27me3 have a role in prostate cancer progression.H3K27me3, whose demethylation is regulated by JMJD3, was found to be under- or over-expressed in a wide range of cancers (29). This study aims to identify the impact of JMJD3 inhibition by its inhibitor, GSK4, on prostate tumor growth in vivo. To evaluate this, we used three continuous prostate cancer lines representing different levels of aggressiveness: PC-3, DU-145 and LNCaP. These are defined as the three standard models for studying prostate cancer. The PC-3 cell line, isolated from bone metastases, is considered as the most aggressive of the three with high metastatic potential compared to the DU-145 and LNCaP cell lines that have moderate and low metastatic potential respectively. The DU-145 cell line was isolated from brain metastases and the LNCaP cell line from lymph node metastases. In contrast to the LNCaP cell line (AR+), the PC-3 and DU-145 cell lines are hormone insensitive (AR-) and do not exhibit AR at the transcriptional and protein level (38). The DU-145 cell line expresses PTEN heterozygously and exhibit a functional protein which is not the case for the LNCaP and PC-3 cell lines (37).
We first looked at in vivo the effect of JMJD3 inhibition by its chemical inhibitor, GSK-J4, on tumor growth. We demonstrated a difference in behavior between AR- and AR+ xenografts with more pronounced tumor growth for AR- treated xenografts and less for AR+ treated xenografts (Figure 1a-d). This was reflected by bioluminescence assays, tumor volume measurement and xenograft weight.
JMJD3 involvement in carcinogenesis remains unclear and the role of JMJD3 has been described in promoting cancer, but also as a tumor suppressor. A number of studies have used GSK-J4 to identify the roles of JMJD3 in carcinogenesis: In glioma, methylation maintenance by JMJD3 inhibition seems to be a new therapeutic strategy because it leads to increased survival and reduced tumor growth (39,40). In neuroblastoma, GSK-J4 decreased tumor growth in in vivo xenograft models (41). The anti-proliferative effects of GSK-J4 were also demonstrated in acute myeloid leukemia via the protein kinase A (PKA) pathway by protein down-regulation of BCL2 and caspase 3 activation (42). In breast cancer, in vivo, chemical treatment has shown a decrease in tumor growth and particularly the suppression of self-renewal and expression of stemness related markers of breast cancer stem cells (43). In contrast, in colorectal cancer, JMJD3 was identified to be positively associated with patient survival. Its inhibition by GSK-J4 showed a reduction in chemokine expression (CXCL9 and CXCL10) involved in tumor immunity (44). These differences in the effect of JMJD3 inhibition on carcinogenesis suggest that the action of JMJD3 is tissue dependent. Finally, our study shows that even in cells from the same tissue, i.e., the prostate, JMJD3 seems to have a different action depending on the properties of the cell lines studied.
The results we obtained seem to differ according to the hormonal status of the xenografts studied: AR- (PC3-luc and DU-145-luc) and AR+ (LNCaP-luc). A previous study in the laboratory had already shown that the androgen receptor status of PC-3, DU-145 (AR-) and LNCaP (AR+) cells generated opposite gene expression profiles (34). Increasingly, authors describe a demethylase action independent of their enzymatic activity, for example by interaction with transcription factors or protein, by preventing their degradation by the proteasome. This has been described in prostate cancer with KDM4B demethylase being part of the same jumonji domain-containing protein family as JMJD3. KDM4B activity is necessary to activate AR at the transcriptional level, and independently to its enzymatic activity by stabilizing AR to avoid its degradation by ubiquitination. Conversely, it has been shown that AR also regulates the activity of KDM4B. Moreover, after KDM4B inhibition by siRNA, authors demonstrated a decrease in proliferation in AR+ cell lines whereas there was no effect on AR- cell lines (45). This suggest that the effects on proliferation are mediated by AR function. This could explain the difference in cell growth after treatment between AR+ and AR- xenografts and in gene expression (Figure 3). JMJD3 could be an AR co-activator and thus there would be a differential expression of candidate genes depending on the type of xenograft. It has already been shown, in breast cancer, that JMJD3 can interact with transcription factors such as estrogen receptor (ERα). After induction of ERα by estrogen, JMJD3 is co-recruited on the BCL2 anti-apoptotic gene promoter to induce an active transcriptional state according to a genomic pathway this time (46).
Then, to demonstrate the impact of JMJD3 inhibition on H3K27me3 enrichment on four candidate genes promoters identified in vitro (34), we performed ChIP-qPCR on collected xenografts. There was no difference in H3K27me3 enrichment on the promoter of all candidate genes after treatment in LNCaP-luc (AR+) xenografts (Figure 2), whereas a difference in the expression of the same candidate genes (Figure 3) as well as an effect of treatment on tumor growth (Figure 1) were demonstrated. H3K27me3 is therefore not involved in the modulation of gene expression in the LNCaP cell line. However, this suggests that JMJD3 inhibition in this xenograft model could allow mediation of androgen-induced transcription of target genes involved in tumorigenesis or that the genes whose expression is decreased following JMJD3 inhibitory treatment could come from another type of repressive expression modification. It would be interesting to use cells transfected with shJMJD3 to form xenografts. This would allow us to examine if the physical absence of JMJD3 could have another effect than inhibition of its demethylase activity.
Surprisingly, treatment with GSK-J4 resulted in overall decrease in H3K27me3 gene enrichment in DU-145-luc xenografts, an opposite effect to what was expected. This cell line differs from PC-3 and LNCaP by its PTEN+ status, which confers an ability to negatively regulate AKT that is not shared by the other two cell lines (37). PTEN is a known tumor suppressor (47), homozygous PTEN deletion in a prostate cancer model leads to an increase in progenitor/stem cells and thus to tumor initiation (48). Although the decrease in EZH2 expression in DU-145-luc treated xenografts (Figure 3) could explain in part the loss of H3K27me3 and perhaps there is a link between JMJD3 demethylase and PTEN that could explain these results. Two previous studies in monocytic cells and renal fibrosis have made comparable observations that support this hypothesis (49,50). We noted an effect of JMJD3 inhibition on PTEN expression with a slight decrease, but there was no impact on AKT expression between control and treated mice (Figure 3). Moreover, Gong et al. found that PTEN C-terminal portion is able to maintain the heterochromatin status by interacting and stabilizing the HP1α protein (51). There may be a dynamic interaction between HP1 protein stabilization and the repressive H3K27me3 mark. Some studies have found that the balance of H3K27me3 at the genome level can be impacted by other PTMs like DNA methylation or other histone PTMs and it is well established that epigenetic modifications are complex and that there is permanent context- and cellular-dependent crosstalk between epigenetic marks (52,53). In this light, this could explain that generally, treatment resulted in a small decrease in overall gene expression (except CDKN1A) while genes were weakly enriched in H3K27me3 in DU-145-luc xenografts.
Our results on gene expression after inhibition by GSK-J4 were consistent with those previously performed in vitro on the three cell lines. By contrast, the increase in H3K27me3 gene enrichment after treatment was evident in vitro (36) while disparities have been demonstrated in vivo. It is certain that the tumor microenvironment must be taken into consideration, the tumor stroma, the infiltrating immune cells, and the blood vessels surrounding the tumor lead to molecular events that do not exist in vitro. The NF-ĸB pathway has a major role in tumor-associated chronic inflammation and HIF1 in the hypoxic environment. Activation of NF-ĸB in infiltrating leukocytes leads to secretion of TNF-α among others. This initiates and drives the regulated expression of cytokine genes responsible for cell proliferation (54). The effects of treatment on gene expression in AR+ xenografts (Figure 3) showed decreased expression of NF-ĸB as well as HIF1. The latter is widely implicated in tumor survival (55) and overexpressed in prostate cancer (56). These latter results, together with the decrease in AKT expression in treated AR+ xenografts, suggest that JMJD3 has a direct or indirect action on the expression of these genes and might contribute to the decrease in tumor growth in an AR+ prostate cancer model (Figure 1).
Conclusion
We identified a role for JMJD3 in prostate tumor progression in relation to the androgen receptor. There is a difference between AR+ and AR- xenografts in tumor growth after JMJD3 inhibition. The differential expression of the gene panel of AR- xenografts in comparison with the enrichment profile of H3K27me3 genes suggests that JMJD3 is able to play a role independently of its demethylase function in androgen-independent prostate cancer. Finally, the effects of GSK-J4 on AR+ prostate xenografts led to a decrease in tumor growth and could be used as an epidrug and a new therapeutic strategy.
Conflicts of Interest
There are no conflicts of interest to declare in relation to this study.
Authors’ Contribution
Conceptualization, A.S., L.G. and D.B.-G.; validation, D.B.-G., L.G., Y.-J.B., and F.P.-L.; experiments, A.S.; resources were collected by A.S.; writing–original draft preparation, A.S.; writing–review and editing, A.S. and D.B.-G.; supervision, D.B.-G., L.G., Y.-J.B., and F.P.-L. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors thank S.Besse for training us in the different techniques on mouse models. We also thank L.Quayle for editing the English version of the manuscript. This work was supported by the French Ligue Régionale Contre Le Cancer—Comité du Puy-de-Dôme—and the ARTP (Association pour la Recherche sur les tumeurs de la Prostate).
References
- 1.Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, Bray F. Cancer statistics for the year 2020: An overview. Int J Cancer. 2021 doi: 10.1002/ijc.33588. [DOI] [PubMed] [Google Scholar]
- 2.Ballestar E, Esteller M. Epigenetic gene regulation in cancer. Adv Genet. 2008;61:247–267. doi: 10.1016/S0065-2660(07)00009-0. [DOI] [PubMed] [Google Scholar]
- 3.Brait M, Sidransky D. Cancer epigenetics: above and beyond. Toxicol Mech Methods. 2011;21(4):275–288. doi: 10.3109/15376516.2011.562671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60(6):376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 5.Aguilera O, Fernández AF, Muñoz A, Fraga MF. Epigenetics and environment: a complex relationship. J Appl Physiol (1985) 2010;109(1):243–251. doi: 10.1152/japplphysiol.00068.2010. [DOI] [PubMed] [Google Scholar]
- 6.Wiles ET, Selker EU. H3K27 methylation: a promiscuous repressive chromatin mark. Curr Opin Genet Dev. 2017;43:31–37. doi: 10.1016/j.gde.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res. 2018;6:10. doi: 10.1186/s40364-018-0122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamagishi M, Uchimaru K. Targeting EZH2 in cancer therapy. Curr Opin Oncol. 2017;29(5):375–381. doi: 10.1097/CCO.0000000000000390. [DOI] [PubMed] [Google Scholar]
- 10.Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007;17(10):850–857. doi: 10.1038/cr.2007.83. [DOI] [PubMed] [Google Scholar]
- 11.Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol. 2015;16(8):499–513. doi: 10.1038/nrm4029. [DOI] [PubMed] [Google Scholar]
- 12.Wijayatunge R, Chen LF, Cha YM, Zannas AS, Frank CL, West AE. The histone lysine demethylase Kdm6b is required for activity-dependent preconditioning of hippocampal neuronal survival. Mol Cell Neurosci. 2014;61:187–200. doi: 10.1016/j.mcn.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manna S, Kim JK, Baugé C, Cam M, Zhao Y, Shetty J, Vacchio MS, Castro E, Tran B, Tessarollo L, Bosselut R. Histone H3 Lysine 27 demethylases Jmjd3 and Utx are required for T-cell differentiation. Nat Commun. 2015;6:8152. doi: 10.1038/ncomms9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130(6):1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488(7411):404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cribbs A, Hookway ES, Wells G, Lindow M, Obad S, Oerum H, Prinjha RK, Athanasou N, Sowman A, Philpott M, Penn H, Soderstrom K, Feldmann M, Oppermann U. Inhibition of histone H3K27 demethylases selectively modulates inflammatory phenotypes of natural killer cells. J Biol Chem. 2018;293(7):2422–2437. doi: 10.1074/jbc.RA117.000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang T, Huang K, Zhu Y, Wang T, Shan Y, Long B, Li Y, Chen Q, Wang P, Zhao S, Li D, Wu C, Kang B, Gu J, Mai Y, Wang Q, Li J, Zhang Y, Liang Z, Guo L, Wu F, Su S, Wang J, Gao M, Zhong X, Liao B, Chen J, Zhang X, Shu X, Pei D, Nie J, Pan G. Vitamin C-dependent lysine demethylase 6 (KDM6)-mediated demethylation promotes a chromatin state that supports the endothelial-to-hematopoietic transition. J Biol Chem. 2019;294(37):13657–13670. doi: 10.1074/jbc.RA119.009757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu SH, Zhu KY, Zhang F, Wang J, Yuan H, Chen Y, Jin Y, Dong M, Wang L, Jia XE, Gao L, Dong ZW, Ren CG, Chen LT, Huang QH, Deng M, Zon LI, Zhou Y, Zhu J, Xu PF, Liu TX. The histone demethylase Jmjd3 regulates zebrafish myeloid development by promoting spi1 expression. Biochim Biophys Acta Gene Regul Mech. 2018;1861(2):106–116. doi: 10.1016/j.bbagrm.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye L, Fan Z, Yu B, Chang J, Al Hezaimi K, Zhou X, Park NH, Wang CY. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11(1):50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naruse C, Shibata S, Tamura M, Kawaguchi T, Abe K, Sugihara K, Kato T, Nishiuchi T, Wakana S, Ikawa M, Asano M. New insights into the role of Jmjd3 and Utx in axial skeletal formation in mice. FASEB J. 2017;31(6):2252–2266. doi: 10.1096/fj.201600642R. [DOI] [PubMed] [Google Scholar]
- 21.Agrawal Singh S, Lerdrup M, Gomes AR, van de Werken HJ, Vilstrup Johansen J, Andersson R, Sandelin A, Helin K, Hansen K. PLZF targets developmental enhancers for activation during osteogenic differentiation of human mesenchymal stem cells. Elife. 2019;8:e40364. doi: 10.7554/eLife.40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F, Xu L, Xu L, Xu Q, Karsenty G, Chen CD. Histone demethylase JMJD3 is required for osteoblast differentiation in mice. Sci Rep. 2015;5:13418. doi: 10.1038/srep13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang F, Xu L, Xu L, Xu Q, Li D, Yang Y, Karsenty G, Chen CD. JMJD3 promotes chondrocyte proliferation and hypertrophy during endochondral bone formation in mice. J Mol Cell Biol. 2015;7(1):23–34. doi: 10.1093/jmcb/mjv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang W, Wang J, Zhang Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res. 2013;23(1):122–130. doi: 10.1038/cr.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgold T, Voituron N, Caganova M, Tripathi PP, Menuet C, Tusi BK, Spreafico F, Bévengut M, Gestreau C, Buontempo S, Simeone A, Kruidenier L, Natoli G, Casola S, Hilaire G, Testa G. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep. 2012;2(5):1244–1258. doi: 10.1016/j.celrep.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Miller SA, Mohn SE, Weinmann AS. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell. 2010;40(4):594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller SA, Weinmann AS. Molecular mechanisms by which T-bet regulates T-helper cell commitment. Immunol Rev. 2010;238(1):233–246. doi: 10.1111/j.1600-065X.2010.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao W, Li Q, Ayers S, Gu Y, Shi Z, Zhu Q, Chen Y, Wang HY, Wang RF. Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell. 2013;152(5):1037–1050. doi: 10.1016/j.cell.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez A, Houfaf Khoufaf FZ, Idrissou M, Penault-Llorca F, Bignon YJ, Guy L, Bernard-Gallon D. The functions of the demethylase JMJD3 in cancer. Int J Mol Sci. 2021;22(2):968. doi: 10.3390/ijms22020968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 31.Ngollo M, Lebert A, Dagdemir A, Judes G, Karsli-Ceppioglu S, Daures M, Kemeny JL, Penault-Llorca F, Boiteux JP, Bignon YJ, Guy L, Bernard-Gallon D. The association between histone 3 lysine 27 trimethylation (H3K27me3) and prostate cancer: relationship with clinicopathological parameters. BMC Cancer. 2014;14:994. doi: 10.1186/1471-2407-14-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daures M, Ngollo M, Judes G, Rifaï K, Kemeny JL, Penault-Llorca F, Bignon YJ, Guy L, Bernard-Gallon D. The JMJD3 histone demethylase and the EZH2 histone methyltransferase in prostate cancer. OMICS. 2016;20(2):123–125. doi: 10.1089/omi.2015.0113. [DOI] [PubMed] [Google Scholar]
- 33.Ngollo M, Lebert A, Daures M, Judes G, Rifai K, Dubois L, Kemeny JL, Penault-Llorca F, Bignon YJ, Guy L, Bernard-Gallon D. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer. 2017;17(1):261. doi: 10.1186/s12885-017-3256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daures M, Idrissou M, Judes G, Rifaï K, Penault-Llorca F, Bignon YJ, Guy L, Bernard-Gallon D. A new metabolic gene signature in prostate cancer regulated by JMJD3 and EZH2. Oncotarget. 2018;9(34):23413–23425. doi: 10.18632/oncotarget.25182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El Ouardi D, Idrissou M, Sanchez A, Penault-Llorca F, Bignon YJ, Guy L, Bernard-Gallon D. The inhibition of the histone methyltransferase EZH2 by DZNEP or SiRNA demonstrates its involvement in MGMT, TRA2A, RPS6KA2, and U2AF1 gene regulation in prostate cancer. OMICS. 2020;24(2):116–118. doi: 10.1089/omi.2019.0162. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez A, El Ouardi D, Houfaf Khoufaf FZ, Idrissou M, Boisnier T, Penault-Llorca F, Bignon YJ, Guy L, Bernard-Gallon D. Role of JMJD3 demethylase and its inhibitor GSK-J4 in regulation of MGMT, TRA2A, RPS6KA2, and U2AF1 genes in prostate cancer cell lines. OMICS. 2020;24(8):505–507. doi: 10.1089/omi.2020.0054. [DOI] [PubMed] [Google Scholar]
- 37.Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58(13):2720–2723. [PubMed] [Google Scholar]
- 38.Cunningham D, You Z. In vitro and in vivo model systems used in prostate cancer research. J Biol Methods. 2015;2(1):e17. doi: 10.14440/jbm.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, Solomon D, Mueller S, Paris PL, Zhang Z, Petritsch C, Gupta N, Waldman TA, James CD. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20(12):1394–1396. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramaswamy V, Remke M, Taylor MD. An epigenetic therapy for diffuse intrinsic pontine gliomas. Nat Med. 2014;20(12):1378–1379. doi: 10.1038/nm.3769. [DOI] [PubMed] [Google Scholar]
- 41.Lochmann TL, Powell KM, Ham J, Floros KV, Heisey DAR, Kurupi RIJ, Calbert ML, Ghotra MS, Greninger P, Dozmorov M, Gowda M, Souers AJ, Reynolds CP, Benes CH, Faber AC. Targeted inhibition of histone H3K27 demethylation is effective in high-risk neuroblastoma. Sci Transl Med. 2018;10(441):eaao4680. doi: 10.1126/scitranslmed.aao4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Illiano M, Conte M, Sapio L, Nebbioso A, Spina A, Altucci L, Naviglio S. Forskolin sensitizes human acute myeloid leukemia cells to H3K27me2/3 demethylases GSKJ4 inhibitor via protein kinase A. Front Pharmacol. 2018;9:792. doi: 10.3389/fphar.2018.00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan N, Xu L, Wu X, Zhang L, Fei X, Cao Y, Zhang F. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp Cell Res. 2017;359(2):405–414. doi: 10.1016/j.yexcr.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 44.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L, Szeliga W, Dou Y, Owens S, Marquez V, Tao K, Huang E, Wang G, Zou W. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 2016;76(2):275–282. doi: 10.1158/0008-5472.CAN-15-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coffey K, Rogerson L, Ryan-Munden C, Alkharaif D, Stockley J, Heer R, Sahadevan K, O’Neill D, Jones D, Darby S, Staller P, Mantilla A, Gaughan L, Robson CN. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013;41(8):4433–4446. doi: 10.1093/nar/gkt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svotelis A, Bianco S, Madore J, Huppé G, Nordell-Markovits A, Mes-Masson AM, Gévry N. H3K27 demethylation by JMJD3 at a poised enhancer of anti-apoptotic gene BCL2 determines ERα ligand dependency. EMBO J. 2011;30(19):3947–3961. doi: 10.1038/emboj.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen CY, Chen J, He L, Stiles BL. PTEN: Tumor suppressor and metabolic regulator. Front Endocrinol (Lausanne) 2018;9:338. doi: 10.3389/fendo.2018.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U.S.A. 2006;103(5):1480–1485. doi: 10.1073/pnas.0510652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ha SD, Cho W, Kim SO. HDAC8 prevents anthrax lethal toxin-induced cell cycle arrest through silencing PTEN in human monocytic THP-1 cells. Toxins (Basel) 2017;9(5):162. doi: 10.3390/toxins9050162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu C, Xiong C, Tang J, Hou X, Liu N, Bayliss G, Zhuang S. Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFβ and Notch signaling and preserving PTEN expression. Theranostics. 2021;11(6):2706–2721. doi: 10.7150/thno.48679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gong L, Govan JM, Evans EB, Dai H, Wang E, Lee SW, Lin HK, Lazar AJ, Mills GB, Lin SY. Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle. 2015;14(14):2323–2332. doi: 10.1080/15384101.2015.1044174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pirrotta V. The necessity of chromatin: a view in perspective. Cold Spring Harb Perspect Biol. 2016;8(1):a019547. doi: 10.1101/cshperspect.a019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. 2016;8(4):a019521. doi: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27(45):5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-inducible factors and cancer. Curr Sleep Med Rep. 2017;3(1):1–10. doi: 10.1007/s40675-017-0062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong H, Agani F, Baccala AA, Laughner E, Rioseco-Camacho N, Isaacs WB, Simons JW, Semenza GL. Increased expression of hypoxia inducible factor-1alpha in rat and human prostate cancer. Cancer Res. 1998;58(23):5280–5284. [PubMed] [Google Scholar]