

Abstract
Hairy cell leukemia (HCL) and HCL-like disorders such as hairy cell leukemia variant (HCL-V) and splenic diffuse red pulp lymphoma (SDRPL) are rare indolent B-cell malignancies. Purine analogs (PNAs), alone or in association with rituximab (R), are the standard of care for HCL in the first-line setting. However, PNAs are toxic and patients may become resistant to these drugs. Therefore, new therapeutic strategies are needed. Several recent in vitro studies highlighted the importance of the interactions between HCL cells and their microenvironment, in particular with bone marrow stromal cells, endothelial cells, and the extracellular matrix. In these interactions, chemokine receptors and adhesion molecules play a major role. Moreover, the importance of signaling pathways, like BRAF, BCR, and CXCR4 has been underlined. Bruton’s tyrosine kinase (BTK) is a fundamental signal transmitter of BCR and CXCR4 in HCL. Preclinical and recent clinical data showed an efficacy of ibrutinib, a BTK inhibitor (BTKi), in HCL and HCL-V. These promising results joined those of other emerging drugs like BRAF or MEK inhibitors and anti-CD22 immunotoxins.
Plain Language Summary
Bruton’s tyrosine kinase (BTK) inhibitors (BTKi) in hairy cell leukemia (HCL) and variant-type HCL
The treatment of hairy cell leukemia (HCL) has changed significantly in recent years. In the first-line settings, treatment with purine analogs (PNAs) with or without anti-CD20 monoclonal antibodies remains the gold standard in 2022. In relapsed/refractory HCL, other drugs are needed: BRAF inhibitors: vemurafenib monotherapy with or without rituximab or dabrafenib in combination with trametinib, an MEK inhibitor (MEKi), as well as the anti-CD22 antibody drug conjugate moxetumomab pasudotox.
There are arguments for the use of Bruton’s tyrosine kinase inhibitors (BTKi). Ibrutinib was recently tested in a multisite phase 2 study in 37 patients with either HCL (28 patients: 76%) or HCL-V (nine patients: 24%) including two who were previously untreated. Patients received single-agent ibrutinib at 420 mg daily (24 patients) or 840 mg daily (13 patients) until disease progression or unacceptable toxicity. The overall response rate (ORR) at 32 weeks was 24%, increasing to 36% at 48 weeks and reaching 54% at any time since starting ibrutinib. Seven patients achieved a complete response (CR) as the best response at any time on study, while 13 patients had a partial response (PR) and 10 patients had stable disease (SD). Interestingly, the response rate was not statistically different between HCL and HCL-V patients, suggesting that ibrutinib could be an option in both entities. The estimated 36-month progression-free survival (PFS) was 73% and the estimated 36-month overall survival (OS) was 85%, with no differences between HCL and HCL-V. The frequency of cardiovascular grade 1–2 adverse events (AEs) was 16% for atrial fibrillation; 3% for atrial flutter; 32% for hypertension; and 0%, 3%, and 11%, respectively, for grade ⩾ 3 AEs. Unlike in chronic lymphocytic leukemia (CLL), where the mechanism of action of ibrutinib is well known, the mechanism of action of ibrutinib in HCL appears to be unclear. No mutations were identified in patients with progressive disease, suggesting that the mechanisms of resistance could be different between HCL and CLL. The BTKi that are not yet approved are challenged by the new other targeted treatments.
Keywords: BCR, BTK, hairy cell leukemia, hairy cell leukemia variant, ibrutinib
Introduction
Classic hairy cell leukemia (HCL) and HCL-like disorders, including the hairy cell leukemia variant (HCL-V) and splenic diffuse red pulp lymphoma (SDRPL), are a very heterogeneous group of mature lymphoid B-cell disorders characterized by the identification of hairy cells, a specific immunophenotypic and genetic profile, a different clinical course and the need for appropriate treatment.
Initially described in 1958, 1 HCL is a well-defined and distinct entity in the fourth revised 2017 classification of the World Health Organization (WHO) of hematopoietic and lymphoid tumors. 2 In 2016, the number of new HCL cases is estimated at 1100 in the United States. 3 It is four to five times more frequent in men than in women. The median age of patients at diagnosis is 63 years in men and 59 years in women. 4 HCL patients present with splenomegaly, pancytopenia, or infections. HCL diagnosis is based on morphological evidence of circulating leukemic cells, usually in low numbers in the blood, (Figure 1(a)), an HCL immunologic score of 3 or 4 based on the CD11c, CD103, CD123, and CD25 expression, 5 a variable degree of medullary infiltration and the presence of a BRAF V600E mutation in the B-raf proto-oncogene (BRAF gene) (7q34). 6 The BRAFV600E mutation results in a constitutive activation of the Ras-Raf-MEK-ERK pathway. Ex vivo and human studies have demonstrated that HCL cells present high levels of mitogen-activated extracellular signal-regulated protein kinase (MEK) and extracellular signal-regulated kinase (ERK) phosphorylation the levels of which are reduced by inhibitors of BRAF (BRAFi) (vemurafenib and dabrafenib). 7 Suggesting an involvement of B-cell receptor (BCR) signaling in HCL pathogenesis, Weston-Bell et al. 8 reported that BCRs of HCL cells respond to antibody-mediated cross-linking with an increase in cellular calcium levels, ERK phosphorylation, and apoptosis. Conversely, the ability of BCR cross-linking to protect primary HCL cells from undergoing spontaneous apoptosis was reported in vitro. Importantly, pretreatment with the Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib, completely abrogated these effects, suggesting a therapeutic relevance of the BCR pathway in HCL. 9
Figure 1.
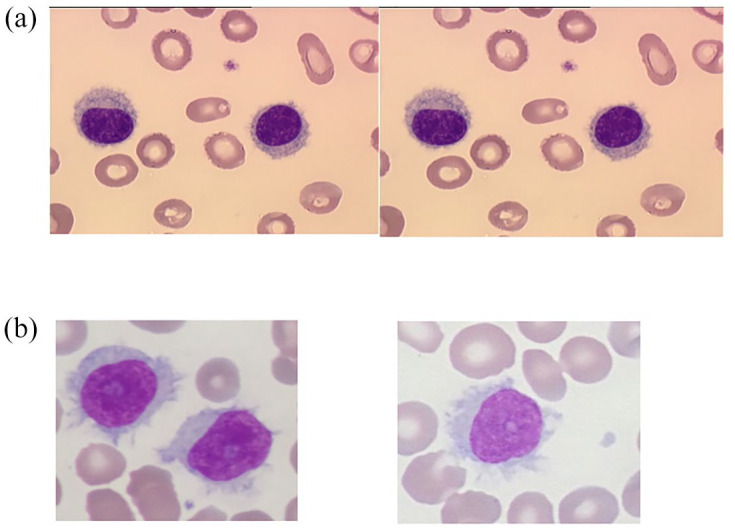
Hairy cells. (a) Typical HCL and (b) variant-type of HCL (HCL-V). From the Laboratoire d’Hématologie, CHU de Caen Normandie, France.
HCL is sometimes confused with other HCL-like disorders, especially splenic B-cell lymphoma/leukemia, unclassifiable, including HCL-V, SDRPL, and splenic marginal zone lymphoma (SMZL).
HCL-V is a provisional entity representing 810 new incident cases in the United States in 2016. 3 The circulating abnormal lymphoid cells have a morphology intermediate between prolymphocytes and hairy cells (Figure 1(b)). The HCL immunological score is low (0 or 1): there is no expression of CD25 and CD200. CD123 expression is inconstant and weak.10–12 A high prevalence of activating mutations in the mitogen-activated protein kinase 1 (MAP2 K1) gene (15q22.1-q22.3) was identified with an overall frequency of 48%.13,14 MAP2 K1 mutations are predominantly detected in HCL patients expressing immunoglobulin (Ig) heavy variable 4-34 gene (IGHV4-34) and in HCL-V whatever the IGHV rearrangements. TP53 aberrations, accounting for 30–40% of patients, are associated with a significant risk for chemoresistance.15–18
SDRPL is a provisional entity, very close if not identical to HCL-V. 19 SDRPL could be the first step before the occurrence of HCL-V and is characterized by the presence of a large proportion (median: 60%) of small to medium-sized villous lymphoid cells in peripheral blood. The abnormal lymphoid cells have a polar distribution of the villi and the nucleolus is small or not visible. The monoclonal B cells express CD11c (97%), inconsistently CD103 (38%), and rarely CD123 (16%) or CD25 (3%). 20 The CD200/CD180 median fluorescence intensity (MFI) ratio may be helpful to distinguishing HCL from SDRPL, with a ratio of 0.5 or less in favor of SDRPL. The BRAFV600E mutation has not been described in this entity. CCND3 mutations involving the regulatory PEST domain, leading to cyclin D3 overexpression were observed in less than 25% of SDRPL patients. 21
SMZL is characterized by the presence of abnormal lymphoid cells with round nuclei, condensed chromatin, and basophilic cytoplasm with polar short villi (also known as ‘villous lymphocytes’) in the peripheral blood. Heterogeneity in blood morphology is common, ranging from small lymphoid cells without specific features, to various degrees of monocytoid and plasmacytoid differentiation. A scoring system based on CD11c, CD22, CD76, CD38, and CD27 expression has been designed to differentiate SDRPL from SMZL. 20 Unlike HCL and HCL-V, SMZL develops in the white pulp of the spleen with a biphasic picture; lymphoma cells may involve the red pulp in patchy or diffuse fashion, with subsequent spread to the sinuses.
HCL patients should be treated only if they are symptomatic.22,23 Chemotherapy with risk adapted therapy purine analogs (PNAs) are indicated in first-line HCL patients. The use of chemoimmunotherapy combining PNAs and rituximab (R) represents an increasingly used therapeutic approach. 24 Long remissions are typically achieved, but most cases relapse and require further therapy. 25 In relapsed/refractory (R/R) HCL patients, novel therapeutic approaches are needed in utilizing less-immunosuppressive drugs than PNAs. 26 In this review, we will focus on the interest of BTK inhibitors (BTKi) as a strategy in HCL and HCL-V.
Purine analogs: a standard option in symptomatic and early HCL without active infection
PNAs are the mainstay of HCL for physically fit and symptomatic HCL patients, conferring in most cases a long overall survival (OS) in first line. 27 Either cladribine (CDA) ± R or pentostatin (DCF) ± R are the recommended standard first-line treatments. 28 In large retrospective series, including both agents, 76–92% of patients receiving chemotherapy achieved a complete response (CR) with an overall response rate (ORR) of 95–100%, with no significant difference in CR or ORR observed between both agents.25,29–31 Therapy with PNAs increases the risk of myelosuppression and infection with common pathogens and opportunistic infections (OIs). PNA therapy is effective but associated with significant toxicities that increase cost.
Recent data suggest that adding R to CDA has therapeutic potential and could improve the duration of CR. The treatment schedule varies between studies: simultaneous versus sequential schedule, number of R infusions, frequency of R infusions, R only for minimal residual disease (MRD). Chemoimmunotherapy associating CDA followed by R 1 month later, scheduled weekly for 8 weeks was introduced in early HCL to achieve undetectable MRD (MRDu) and get durable CR in untreated patients. 32 In a phase II clinical trial enrolling 80 patients (59 untreated patients), CR rate was 100% demonstrating a high efficacy of chemoimmunotherapy in frontline therapy. Five-year failure-free survival (FFS) and OS were 94.8% and 96.8%, respectively. The regimen was well tolerated, with no severe or unexpected toxicity. In a randomized study, 68 patients received CDA with eight weekly doses of R. 33 The first group received R on day 1 (CDAR) and the second started R later at 6 months after CDA. At 6 months, the CR rate was 100% for CDAR versus 88% for CDA. The CR rate with undetectable MRD was significantly different: 97% and 24%, respectively. In addition, at a median follow-up of 96 months, the MRD-free rate was 94% for CDAR and only 12% for CDA, suggesting that CDAR could be an option in first-line therapy.
Chemoimmunotherapy is currently the standard option in HCL-V as frontline treatment. 18 Twenty patients with HCL-V, including eight previously untreated, received CDAR. Patients received a second course of eight weekly doses of R at least 6 months after the first course if MRD was detected in the peripheral blood. Fourteen patients achieved CR at 4 weeks, with 9/18 evaluable patients achieving negative MRD. At 6 months after CDAR, 18/20 (90%) patients sustained CR with 16 (80%) patients MRDu by immunohistochemistry and flow cytometry (blood and bone marrow). The 5-year progression-free survival (PFS) was 63.3% and the 5-year OS was 74%. Eleven patients received delayed R between 6 and 82 months (median: 345.8 months) and two additional patients received chemoimmunotherapy because of rapid progression of the disease. Patients who achieved MRDu CR at 6 months had longer PFS (not reached versus 17.4 months) and OS (not reached versus 38.2 months) compared with those who did not. Five of 19 evaluable patients (26%) had TP53 mutations: these patients had significantly more MRD positivity at 6 months compared with those without mutation.
One of the most challenging clinical situations involves the patient with symptomatic HCL and febrile infection. Attempts to control the infection should be pursued prior to instituting the PNA. If it is not possible to control the infection or in the event of a health crisis (Sars-Cov-2), the use of interferon alpha (IFNα) or vemurafenib as bridging therapy could be required transiently. 34 Note that the use of IFN, a possible alternative in pregnant women, becomes more and more difficult due to a production stop by laboratories.
Toxicities of repeated treatments with PNA and the loss of sensitivity to PNA in R/R HCL patients have led to a need for emerging and less toxic therapeutic strategies in HCL. The study of the tumor microenvironment and the signaling pathways allowed the development of new molecules.
Focus on the tumor microenvironment in HCL
Chemokine receptors and adhesion molecules are involved in the trafficking, homing, and retention of normal and malignant B cells, allowing access to different microenvironmental niches and also retention in different sites, such as the spleen, liver, lymph nodes, and/or bone marrow. The hairy cells accumulate in the red pulp of the spleen (the white pulp is typically atrophic) and in the sinusoids of the liver and tends to spare the lymph nodes. The marrow microenvironment is made up of a complex network of extracellular matrix (ECM) (proteins, soluble growth factors, and cytokines) and cellular niches. The niches are divided into two compartments. Osteoblasts and osteoclasts, bone marrow mesenchymal stroma cells (BMSC), T regulatory cells (Treg), and macrophages form the endosteal (osteoblastic) niche. The vascular niche includes endothelial cells and marrow stromal reticular cells (CAR cells). The hematopoietic stem cells (HSCs) are located into endosteal niche: they are maintained in a quiescent state and if needed can migrate to the vascular niche where they can actively cycle. 35 The hairy cells can access different niches that are normally restricted to HSC. 36
Figure 2 presents the interactions existing between HCL cells and the various components of the tumor microenvironment. HCL cells express on their surface high levels of the chemokine receptor CXCR4 (CD184):37–39 CXCR4 is the receptor for the chemokine (C-X-C motif) ligand 12, which is secreted by BMSC and the CXCR4/CXCL12 cross-talk directs the abnormal lymphoid cells from the circulation into the marrow. HCL cells also express various adhesion molecules that cooperate with chemokine receptors for retention of HCL cells in the marrow. HCL cells express high level of integrins, such as α4β1 or CD49d [also called very late activation antigen-4 (VLA-4)] binding to its ligand CD106 [also called vascular cell adhesion molecule 1 (VCAM-1)] in bone marrow, hepatic, and splenic sinusoids accounting for the preferential homing of HCL cells to the sinusoids.40,41 The expression of CXCR4 and VLA-4 by HCL cells and their respective ligands by BMSC (CXCL12 and VCAM-1) induces adhesion of HCL cells to BMSC, which favors the retention of HCL cells in the BM and its protective milieu, and displaces normal hematopoietic progenitor cells. Moreover, in vitro experiments with coculture of HCL cells with BMSC showed pseudoemperipolesis, a phenomenon of migration of HCL cells underneath the layer of BMSC. This adhesion and migration of HCL cells can be reduced after blockade of CXCR4 and CD49d receptors, highlighting the role of chemokine receptors and adhesion molecules in HCL cells interactions with the BM environment. 36
Figure 2.

Interactions between HCL and its microenvironment.
BMSC, bone marrow stromal cell; ECM, extracellular matrix.
Localization in the vitronectin-rich splenic red pulp could be related to HCL cell expression of the vitronectin receptor CD51 (αvβ3 integrin). CD51 also interacts with CD31 [platelet/endothelial cell adhesion molecule 1 (PECAM-1)] and plays a role in HCL cell motility. 42 Moreover, interactions between laminin expressed at the surface of HCL cells and the basement membrane could mediate replacement of endothelial cells by HCL cells, which could be responsible for hepatic hemangiomatous lesions and splenic pseudosinuses.43,44 The lack of lymph nodes in HCL could be explained by the downregulation of several chemokine receptors, such as CD62 L (L-selectin), CXCR5, and CCR7.37,38
Chemokine receptors and adhesion molecules allow HCL cells to interact with cellular components and the ECM of the marrow environment. These interactions offer stimuli for HCL cell survival, proliferation, and a protective milieu against certain treatments. For example, HCL cells have a high affinity for BMSC. 45 This interaction activates signaling pathways into HCL cells, like NF-κB and MAP kinase, favoring their survival. When HCL cells are co-cultured with BMSC, there is an inhibition of apoptosis induced by BRAF inhibitors. 7 Moreover, HCL cells express CD40 and are activated by CD40 crosslinking. CD40 ligand (CD154) is expressed at the surface of T cells. Thus, T cells of the tumor microenvironment could play a role in HCL progression. 46 Interactions of CD49d (VLA-4; α4β1) and CD49e (VLA-5; α5β1) expressed by HCL cells with FN prevent the apoptosis induced by tumor necrosis factor-alpha (TNFα) secreted by HCL cells as a response to IFNα. 47 Importantly, HCL cells highly express CD44, which binds to hyaluronan (HA) expressed by BMSC and hepatic portal tracts. This interaction not only favors the localization of HCL cells in BM and liver, but also induces the autocrine production of fibronectin (FN) and basic fibroblast growth factor (bFGF/FGF-2) by HCL cells which, in addition to the synthesis of transforming growth factor beta 1 (TGF-β1), is responsible for the typical BM fibrosis found in HCL.48–51
Focus on the B-cell receptor pathway and BTK in HCL
Figure 3 presents signaling pathways of importance in HCL pathophysiology. The BCR complex of each B-cell comprises Ig with uniform non-covalently binding activity associated with CD79a and CD79b. The cytoplasmic tails of CD79a/CD79b contain an immunotyrosine-based activation motif (ITAM) that serves as a docking site for kinases and adapter proteins that participate in BCR signaling. B-cell activation following BCR stimulation is mediated through signaling cascades that involve activation of membrane-proximal kinases, such as spleen tyrosine kinase (SYK), BTK, and phosphoinositide 3-kinase δ (PI3Kδ). The three kinases (SYK, BTK, and PI3Kδ) have become the target of kinase inhibitors, which have emerged over the past few years in patients with chronic lymphocytic leukemia (CLL), 52 mantle cell lymphoma (MCL), 53 Waldenström macroglobulinemia (WM), 54 and diffuse large B-cell lymphoma (DLBCL) of the activated B-cell-like (ABC) 55 subtype and more recently in HCL. 56 Active BTK signals through further phosphorylation and activation of phospholipase Cγ2 (PLCG2) accompanied by calcium mobilization. Stimulation of this pathway ultimately leads to activation of NF-κB and MAP kinase pathways, which in turn results in increased proliferation, survival, and migration of B cells. Moreover, activation of the BCR pathway induces secretion of chemokines like CCL3 and CCL4 by normal or malignant B cells, which in turn attracts monocytes and T cells. 57
Figure 3.

Signaling pathways and their inhibitors in HCL.
BCR signaling has emerged as a key pathway for the expansion of neoplastic B-cell clones in several B-cell malignancies. The mechanisms that activate BCR signaling differ among subtypes of B-cell lymphoma and leukemia. These include BCR stimulation by foreign or self-antigens, or the acquisition of mutations in components of the BCR pathway that result in autonomous or enhanced antigen-induced BCR signaling (tonic BCR signaling).
The most solid evidence for the involvement of the BCR and BCR signaling in HCL pathogenesis arises from structural analyses of HCL BCR molecules. These reveal a biased Ig heavy (IGH) and Ig light (IGL) variable gene segment repertoire, as well as the existence of mutated and unmutated HCL cases. Analysis of Ig heavy chain variable (IGHV) region gene mutation status allows to identify a mutated profile in nearly 80% of HCL patients (M-HCL). The M-HCL profile is defined by less than 98% sequence homology with the corresponding consensus germline sequence. Conversely, 20% of HCL patients are unmutated (U-HCL), with 98% or more sequence homology: patients with U-HCL define a minor subgroup of patients with more aggressive disease and shorter survival, whereas M-HCL patients typically have slower disease progression and longer survival.15,58 U-HCL and the usage of the IGHV4-34 gene segment are considered poor prognostic markers. 59 The pattern of IGHV mutations is identical in HCL-V and SMZL, with 22% and 15% of cases that show no evidence of somatic hypermutation in the IGHV genes. 60 Interestingly, IGHV4-34 is over-represented in both HCL-V and SMZL and is preferentially used in unmutated cases.
Suggesting an involvement of BCR signaling in HCL pathogenesis, Weston-Bell et al. 8 reported that BCR of HCL cells respond to antibody-mediated cross-linking with an increase in cellular calcium levels, ERK phosphorylation, and apoptosis. On the contrary, we recently reported on the ability of BCR cross-linking to protect primary HCL cells from undergoing spontaneous apoptosis in vitro. Similarly, Sivina et al. 9 demonstrated that BCR triggering with anti IgA/IgG/IgM increases HCL cells viability. They also demonstrated that BTK is expressed in HCL cells and that BCR stimulation increases phosphorylated (p)BTK, pERK, and pAKT levels, and increases the secretion of CCL3 and CCL4 by HCL cells. 9 Altogether, these data suggest that activation of the BCR in HCL cells induces BTK phosphorylation, calcium mobilization, activation of MAP kinase, and Akt pathways, favoring survival of HCL cells, and secretion of CCL3 and CCL4.
In addition to its role in the BCR pathway, BTK is a signal transmitter for CXCR4 in normal 61 and in CLL B cells.62,63 Thus, BTK plays a major role in B-cell homing. Preclinical data on ibrutinib in HCL are in favor of a role of BTK in the CXCR4 pathway in HCL cells as well. 9
BRAF is a mediator of the BCR pathway and activation of the MAP kinases. 64 Interestingly, the BRAF V600E mutation does not alter the response of B cells to BCR activation.9,65 Finally, HCL cells express different isotypes of the BCR. Therefore, there are various functional responses to BCR activation in HCL cells. 65
Thus, the importance of BTK in the BCR and CXCR4 pathways in HCL has led to the use of BTKi in HCL and HCL-V.
Therapeutic advances in HCL and HCL-V: focus on ibrutinib
Ibrutinib (PCI-32765) is a first-in class, oral, once daily, and irreversible BTKi which covalently binds to a cysteine residue (Cys-481) in the BTK kinase domain. Ibrutinib inhibits survival and proliferation of CLL cells in vitro. 63 In addition, ibrutinib antagonizes the migration of CLL cells toward the chemokines CXCL12 and CXCL13 and decreases adhesion to FN and VCAM. 63 Ibrutinib has been recently proven to have therapeutic activity in R/R HCL. Currently, ibrutinib is not yet approved by the FDA in HCL and HCL-V.
Preclinical data in experiments using HCL cell lines and primary HCL cells showed promising efficacy of ibrutinib in HCL. 9 Indeed, pre-incubation of these cells with ibrutinib inhibits BTK phosphorylation, downstream kinases phosphorylation, BCR-induced secretion of CCL3 and CCL4, and reduces HCL cell survival after BCR stimulation with anti IgA/IgG/IgM. Interestingly, this study showed that low ibrutinib concentrations induce full BTK target occupancy in HCL cells. Finally, pre-incubation of these cells with ibrutinib reduces pERK levels after CXCR4 stimulation with CXCL12. 9 Thus, ibrutinib, inhibiting the BCR and CXCR4 pathways, both playing a major role in HCL cells proliferation, survival, and migration, could be a good alternative therapeutic option for HCL patients.
Clinically, the efficacy of ibrutinib in HCL was noted in some case reports. Visentin et al. 66 reported two R/R HCL-V patients treated with ibrutinib. The first one received ibrutinib 560 mg/day in third line (first line: splenectomy and second line: R bendamustine) and achieved an unconfirmed partial response (PR) at 6 months, with a duration of response (DOR) of 16.5 months before relapsing. The relapse was unsuccessfully treated with venetoclax. The second one received ibrutinib 560 mg/day, but it was stopped early because of gastrointestinal toxicities and easy bruising. The authors observed a spleen size reduction after 3 months of treatment. Bohn et al. reported the rapid effectiveness of ibrutinib 420 mg/day in a patient treated for R/R HCL-V, with resolution of constitutional symptoms, decrease of spleen size and lymphocytosis, and increase of hemoglobin level. Despite the patient not meeting criteria for PR due to persistent thrombocytopenia, he was kept on ibrutinib after 16 months of follow-up, with no major toxicity. 67
Ibrutinib was recently tested in a multisite phase 2 study in 37 patients with either R/R HCL (28 patients: 76%) or HCL-V (nine patients: 24%) including two untreated. 56 The median number of prior treatments was 4 (range: 0–12) in HCL patients, with all of them having previously received PNAs. Patients received single-agent ibrutinib at 420 mg daily (24 patients) or 840 mg daily (13 patients) until disease progression or unacceptable toxicity. The primary endpoint was ORR, including CR and PR at 32 weeks and the best response at any time since starting ibrutinib. The ORR at 32 weeks was 24%, increasing to 36% at 48 weeks and reaching 54% at any time starting ibrutinib. One patient had a CR at 32 weeks, eight patients a PR, 11 patients a stable disease (SD) and three patients a progressive disease (PD). Four patients were not evaluable for response. Seven patients achieved a CR as the best response at any time on study while 13 patients had PR and 10 patients SD. Interestingly, the response rate was not statistically different between HCL and HCL-V patients, suggesting that ibrutinib could be effective in both entities. However, the trial was not designed to detect a significant difference between both entities: 6 (21%) of 28 HCL patients versus 1 (11%) of 9 HCL-V patients achieved CR, which certainly does not mean a higher CR rate in HCL versus HCL-V. The estimated 36-month PFS was 73% and the estimated 36-month OS was 85%, with no significant differences between HCL and HCL-V. These data suggest ibrutinib could be an option in some cases of HCL and HCL-V. The results are clearly inferior to those obtained with BRAFi, even if there are no comparative studies between both treatments. Adverse events (AEs) included diarrhea (59%), myalgia (54%), nausea (51%), and bruising (43%). The frequency of cardiovascular grade 1–2 AEs was 16% for atrial fibrillation, 3% for atrial flutter; 32% for hypertension; and 0%, 3%, and 11%, respectively, for grade > or = 3 AEs. Unlike in CLL where the mechanism of effect of ibrutinib is well known, it is unclear in HCL. The mobilization of leukemic cells in the peripheral blood is not observed in the majority of patients, the persistence of pERK in HCL cells is not associated with a shorter PFS and pERK cannot be used as a marker of response. Several patients with presence of pERK after 32 and 48 weeks of treatment have a durable benefit from ibrutinib. Resistance to ibrutinib is related to mutations in BTK or PLCG2 in CLL patients. Contrary to CLL, no mutation was identified in four patients (two HCL and two HCL-V) with progressive disease, suggesting a difference in the mechanisms of resistance between HCL and CLL, despite the number of HCL patients being insufficient to establish any conclusion.
New BTKi, acalabrutinib (ACP-196), tirabrutinib (ONO/GS-4059), zanubrutinib (BGB-3111), and pirtobrutinib (LOXO-305) are being widely tested in CLL patients. Acalabrutinib, tirabrutinib, and zanubrutinib are irreversible BTKi and bind irreversibly to cysteine 481 of BTK. On the contrary, pirtobrutinib is a highly selective, reversible BTKi, and could be effective even in the presence of the C481 S mutation of BTK. 68 However, ibrutinib remains the most well studied and predictable BTKi at present. The place of these new drugs in the treatment of HCL and HCL-V remains to be specified.
The isoform-selective phosphatidylinositol 3-kinase (PI3 K) inhibitors also disrupt the BCR signaling. Idelalisib in combination with R has been approved in R/R CLL and those with TP53 disruption. However, toxic side effects, such as colitis, infections, and hepatitis were mentioned. 69 Other PI3 K inhibitors like duvelisib, copanlisib, acalisib, parsaclisib, umbralisib, and buparlisib, have been developed in CLL and non-Hodgkin lymphoma (NHL), with less toxicity than idelalisib for some of them. 70 To our knowledge, no data are available in HCL and HCL-V patients.
Other targeted therapies in HCL and HCL-V
Other emerging drugs can be used for R/R HCL patients (Table 1). However, the optimal sequence of the different treatments remains to be determined.
Table 1.
New drugs in HCL.
| New drugs in HCL | N | ORR (%) | CR (%) | MRD negative | PFS | OS | Relapses | Follow-up | Study |
|---|---|---|---|---|---|---|---|---|---|
| Vemurafenib | 50 | 96–100 | 35–42 | – | 73% at 1 year a | 91% at 1 year a | 27% at 1 year a | 11.7 months a | Tiacci et al. 71 |
| Rituximab + vemurafenib | 30 | 87 | 87 | 65% of CR pts | 78% | – | 15% | 37 months | Tiacci et al. 74 |
| Dabrafenib | 10 | 80 | 30 | 0% | 10% | 90% | 88% | 64 months | Tiacci et al. 73 |
| Dabrafenib + trametinib | 43 | 78 | 49 | 30% of CR pts | 98% at 1 year | 98% at 1 year | 2% | – | Kreitman et al. 75 |
| Moxetumomab pasudotox | 80 | 75 | 41 | 82% of CR pts | median 71.7 months | – | – | 24.6 months | Kreitman et al. 78 |
| Ibrutinib | 37 | 54 b | 19 b | – | 73% at 36 months | 85% at 36 months | – | 3.5 years | Rogers et al. 56 |
CR, complete response; HCL, hairy cell leukemia; MRD, minimal residual disease; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
In the US trial.
As best response at any time.
Vemurafenib monotherapy has demonstrated significant activity in HCL. 7 When using high dose (960 mg twice daily) for 16–18 weeks, the CR rates were 35% and 42% in two clinical trials, respectively. 71 Vemurafenib is also effective at lower doses. Indeed, in a study, including 21 heavily pretreated HCL patients, vemurafenib was given at doses from 240 to 1920 mg/day. 72 Responses were observed independently of the dose and duration of treatment. In another study, low-dose vemurafenib (480–960 mg/day) was given in six HCL patients with an active infection. 34 All patients achieved a response and recovered from the infectious disease. The drug-related AEs include arthralgia or arthritis, rash or erythema, photosensitivity reaction, increased pancreatic enzymes, secondary cutaneous tumors (squamous-cell carcinoma and cutaneous superficial melanoma), and ocular toxicity, including central retinopathy and retinal vein occlusion. Results are similar with dabrafenib (150 mg twice daily), an another BRAFi for 8–12 weeks. 73 The association of vemurafenib with R has recently been proven to have a drastic therapeutic activity. 74 In a phase II trial, a CR was achieved in 87% evaluable patients, a rate twice as high as that obtained with vemurafenib monotherapy. PFS was 78% at a median follow-up of 37 months.
MEK inhibitors (MEKi) are also increasingly used in R/R HCL. In a phase II study, including 43 heavily pretreated HCL patients, dabrafenib, an oral BRAFi, was given at 150 mg twice daily in combination with trametinib, an oral MEKi, at 2 mg once daily, continuously until disease progression or unacceptable toxicity. 75 The efficacy was impressive, with an ORR of 78% and a CR of 49% (6/41 evaluable patients achieved MRD negativity). The 12-month PFS and OS were 98%. However, toxicities were important with 49% of patients experiencing grade 3–4 AEs and 42% of patients having a dose reduction.
Another major therapeutic advance for R/R HCL is moxetumomab pasudotox (Moxe), the variable region of an anti-CD22 monoclonal antibody conjugated to the PE38 exotoxin of Pseudomonas aeruginosa. 76 In the pivotal study, Moxe was given at 40 µg/kg/day iv at d1, d3, and d5, for a maximum of six 28-day cycles. 77 The ORR, CR rate, and durable (>180 days) CR rate were 75%, 41%, and 36%, respectively. Among patients achieving a CR, 82% were MRD negative, which can explain the rate of durable responses. 78 With the exception of some patients experiencing low-grade capillary leak syndromes or hemolytic uremic syndromes, the tolerance was good. Strikingly, the three HCL-V patients did not achieve a CR. Moxe is now Food and Drug Administration (FDA) and European Medicines Agency (EMA)-approved for HCL patients after at least two previous lines of therapy (including a PNA). Note that the drug should be difficult to obtain, due to a development stop in Europe.
Bcl2-inhibitors (Bcl-2i) can play a major role in the future. In a recent preclinical work, venetoclax, a Bcl-2i, showed its ability to induce apoptosis of HCL cells in in vitro experiments. However, stimuli from the microenvironment can protect HCL cells from venetoclax-induced cell death. 79 In a case report with a patient having both IGHV-4-34 positive HCL-V with complex karyotype and TP53 mutation and a small CLL clone in the BM, ibrutinib was given at 420 mg/day alone for 3 months, then in combination with venetoclax. There was a clear improvement in symptoms and spleen size, even if there was no response on BM assessment. 80
Conclusion
With the advent of targeted strategies, treatment choices have expanded particularly in R/R patients and patients are living longer. Careful consideration is needed regarding treatment durations and optimal sequences, how best to respect patients’ needs, balancing toxicities while improving long-term survival and maximizing depth of response. In R/R patients, BTKi are promising in HCL and HCL-V but are challenged by other therapeutic advances, in particular Moxe, BRAFi and MEKi. As observed in CLL, new drug combinations could emerge and prove to be effective in R/R patients: ibrutinib plus R, ibrutinib plus obinutuzumab or fixed duration treatments, such as ibrutinib plus venetoclax, a Bcl-2i that can play a major role in the future.
We use PNA ± R in the frontline setting. The association with R is encouraged in cases of HCL-V, TP53 mutation, unmutated IGHV, and/or IGHV-4-34 use, because these situations are associated with refractoriness or short-lasting response to PNA alone. In first relapse and if the CR duration is > 2 years, we use again PNA ± R, because patients may have long-lasting response after re-treatment with PNA. However, in case of CR < 2 years, primary refractory disease, or beyond the second relapse, we use targeted therapies like vemurafenib ± R, Moxe or ibrutinib. Because the results of clinical trials (even if not comparative) seem to show deeper and faster responses with vemurafenib ± R or Moxe than with ibrutinib, we preferentially use the first two options. Moreover, we propose these targeted therapies in the frontline setting for patients ineligible for standard chemotherapy because of age, comorbidities, or active infection.
Footnotes
Author contributions: Jérôme Paillassa: Conceptualization; Formal analysis; Methodology; Validation; Writing – original draft; Writing – review & editing.
Firas Safa: Formal analysis; Writing – review & editing.
Xavier Troussard: Conceptualization; Formal analysis; Methodology; Supervision; Validation; Writing – original draft; Writing – review & editing.
Conflict of interest statement: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Xavier Troussard: Consultant for Innate Pharma, Astra Zeneca, Abbvie, Beigene.
Jérôme Paillassa and Firas Safa: The authors declare that there is no conflict of interest.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Permission to reproduce material from other sources: Personal pictures: no formatting required
ORCID iD: Xavier Troussard  https://orcid.org/0000-0001-6863-9992
https://orcid.org/0000-0001-6863-9992
Contributor Information
Jérôme Paillassa, Service des Maladies du Sang, CHU d’Angers, Angers, France.
Firas Safa, Service des Maladies du Sang, CHU d’Angers, Angers, France.
Xavier Troussard, Laboratoire Hématologie, CHU de Caen Normandie, avenue de Côte de Nacre, 14033 Caen Cedex, France.
References
- 1. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958; 13: 609–630. [DOI] [PubMed] [Google Scholar]
- 2. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127: 2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016; 66: 443–459. [DOI] [PubMed] [Google Scholar]
- 4. Le Guyader-Peyrou S, Defossez G, Dantony E, et al. Estimations nationales de l’incidence et de la mortalité par cancer en France métropolitaine entre 1990 et 2018. Etude à partir des registres des cancers du réseau Francim. Volume 2: Hémopathies malignes. Saint-Maurice: Santé publique France, 2019. [Google Scholar]
- 5. Matutes E, Morilla R, Owusu-Ankomah K, et al. The immunophenotype of hairy cell leukemia (HCL). Proposal for a scoring system to distinguish HCL from B-cell disorders with hairy or villous lymphocytes. Leuk Lymphoma 1994; 14(Suppl. 1): 57–61. [PubMed] [Google Scholar]
- 6. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 364: 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pettirossi V, Santi A, Imperi E, et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood 2015; 125: 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weston-Bell NJ, Hendriks D, Sugiyarto G, et al. Hairy cell leukemia cell lines expressing annexin A1 and displaying B-cell receptor signals characteristic of primary tumor cells lack the signature BRAF mutation to reveal unrepresentative origins. Leukemia 2013; 27: 241–245. [DOI] [PubMed] [Google Scholar]
- 9. Sivina M, Kreitman RJ, Arons E, et al. The Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014; 166: 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Angelova EA, Medeiros LJ, Wang W, et al. Clinicopathologic and molecular features in hairy cell leukemia-variant: single institutional experience. Mod Pathol 2018; 31: 1717–1732. [DOI] [PubMed] [Google Scholar]
- 11. Pillai V, Pozdnyakova O, Charest K, et al. CD200 flow cytometric assessment and semiquantitative immunohistochemical staining distinguishes hairy cell leukemia from hairy cell leukemia-variant and other B-cell lymphoproliferative disorders. Am J Clin Pathol 2013; 140: 536–543. [DOI] [PubMed] [Google Scholar]
- 12. Favre R, Manzoni D, Traverse-Glehen A, et al. Usefulness of CD200 in the differential diagnosis of SDRPL, SMZL, and HCL. Int J Lab Hematol 2018; 40: e59–e62. [DOI] [PubMed] [Google Scholar]
- 13. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014; 46: 8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mason EF, Brown RD, Szeto DP, et al. Detection of activating MAP2K1 mutations in atypical hairy cell leukemia and hairy cell leukemia variant. Leuk Lymphoma 2017; 58: 233–236. [DOI] [PubMed] [Google Scholar]
- 15. Forconi F, Sozzi E, Cencini E, et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 2009; 114: 4696–4702. [DOI] [PubMed] [Google Scholar]
- 16. Hockley SL, Else M, Morilla A, et al. The prognostic impact of clinical and molecular features in hairy cell leukaemia variant and splenic marginal zone lymphoma. Br J Haematol 2012; 158: 347–354. [DOI] [PubMed] [Google Scholar]
- 17. Durham BH, Getta B, Dietrich S, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood 2017; 130: 1644–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chihara D, Arons E, Stetler-Stevenson M, et al. Long term follow-up of a phase II study of cladribine with concurrent rituximab with hairy cell leukemia variant. Blood Adv 2021; 5: 4807–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Traverse-Glehen A, Baseggio L, Salles G, et al. Splenic diffuse red pulp small-B cell lymphoma: toward the emergence of a new lymphoma entity. Discov Med 2012; 13: 253–265. [PubMed] [Google Scholar]
- 20. Baseggio L, Traverse-Glehen A, Callet-Bauchu E, et al. Relevance of a scoring system including CD11c expression in the identification of splenic diffuse red pulp small B-cell lymphoma (SRPL). Hematol Oncol 2011; 29: 47–51. [DOI] [PubMed] [Google Scholar]
- 21. Curiel-Olmo S, Mondéjar R, Almaraz C, et al. Splenic diffuse red pulp small B-cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood 2017; 129: 1042–1045. [DOI] [PubMed] [Google Scholar]
- 22. Cornet E, Delmer A, Feugier P, et al. Recommendations of the SFH (French Society of Haematology) for the diagnosis, treatment and follow-up of hairy cell leukaemia. Ann Hematol 2014; 93: 1977–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017; 129: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paillassa J, Troussard X. Biology and treatment of hairy cell leukemia. Curr Treat Options Oncol 2020; 21: 44. [DOI] [PubMed] [Google Scholar]
- 25. Paillassa J, Cornet E, Noel S, et al. Analysis of a cohort of 279 patients with hairy-cell leukemia (HCL): 10 years of follow-up. Blood Cancer J 2020; 10: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paillassa J, Troussard X. Patients with relapsed/refractory hairy-cell leukemia. Cancer Rep. Epub ahead of print 12 July 2021. DOI: 10.1002/cnr2.1495. [DOI] [Google Scholar]
- 27. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014; 166: 390–400. [DOI] [PubMed] [Google Scholar]
- 28. Sundar H, Fletcher CD, Gaballa S, et al. NCCN Guidelines version 1.2022 hairy cell leukemia continue NCCN Guidelines Panel Disclosures, 2021, https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1481
- 29. Zinzani PL, Pellegrini C, Stefoni V, et al. Hairy cell leukemia: evaluation of the long-term outcome in 121 patients. Cancer 2010; 116: 4788–4792. [DOI] [PubMed] [Google Scholar]
- 30. Else M, Dearden CE, Catovsky D. Long-term follow-up after purine analogue therapy in hairy cell leukaemia. Best Pract Res Clin Haematol 2015; 28: 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. López Rubio M, Da Silva C, Loscertales J, et al. Hairy cell leukemia treated initially with purine analogs: a retrospective study of 107 patients from the Spanish Cooperative Group on Chronic Lymphocytic Leukemia (GELLC). Leuk Lymphoma 2014; 55: 1007–1012. [DOI] [PubMed] [Google Scholar]
- 32. Chihara D, Kantarjian H, O’Brien S, et al. Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia: update of a phase II trial. Br J Haematol 2016; 174: 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chihara D, Arons E, Stetler-Stevenson M, et al. Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. J Clin Oncol 2020; 38: 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bohn J-P, Pircher A, Wanner D, et al. Low-dose vemurafenib in hairy cell leukemia patients with active infection. Am J Hematol 2019; 94: E180–E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 2008; 132: 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sivina M, Burger JA. The importance of the tissue microenvironment in hairy cell leukemia. Best Pract Res Clin Haematol 2015; 28: 208–216. [DOI] [PubMed] [Google Scholar]
- 37. Wong SWJ, Fulcher DA. Chemokine receptor expression in B-cell lymphoproliferative disorders. Leuk Lymphoma 2004; 45: 2491–2496. [DOI] [PubMed] [Google Scholar]
- 38. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004; 199: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dürig J, Schmücker U, Dührsen U. Differential expression of chemokine receptors in B cell malignancies. Leukemia 2001; 15: 752–756. [DOI] [PubMed] [Google Scholar]
- 40. Vincent AM, Burthem J, Brew R, et al. Endothelial interactions of hairy cells: the importance of α4β1 in the unusual tissue distribution of the disorder. Blood 1996; 88: 3945–3952. [PubMed] [Google Scholar]
- 41. Burthem J, Baker PK, Hunt JA, et al. Hairy cell interactions with extracellular matrix: expression of specific integrin receptors and their role in the cell’s response to specific adhesive proteins. Blood 1994; 84: 873–882. [PubMed] [Google Scholar]
- 42. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014; 15: 187–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nanba K, Soban EJ, Bowling MC, et al. Splenic pseudosinuses and hepatic angiomatous lesions. Distinctive features of hairy cell leukemia. Am J Clin Pathol 1977; 67: 415–426. [DOI] [PubMed] [Google Scholar]
- 44. Karttunen T, Apaja-Sarkkinen M, Alavaikko M, et al. Altered basement membrane structure of the spleen in hairy cell leukaemia. Demonstration of laminin in hairy cells. Pathol Res Pract 1987; 182: 233–239. [DOI] [PubMed] [Google Scholar]
- 45. Sivina M, Kreitman RJ, Peled A, et al. Adhesion of hairy cells leukemia (HCL) cells to stromal cells can be inhibited by blocking VLA-4 integrins and CXCR4 chemokine receptors. Blood 2011; 118: 1760–1760. [Google Scholar]
- 46. Kluin-Nelemans HC, Beverstock GC, Mollevanger P, et al. Proliferation and cytogenetic analysis of hairy cell leukemia upon stimulation via the CD40 antigen. Blood 1994; 84: 3134–3141. [PubMed] [Google Scholar]
- 47. Baker PK, Pettitt AR, Slupsky JR, et al. Response of hairy cells to IFN-alpha involves induction of apoptosis through autocrine TNF-alpha and protection by adhesion. Blood 2002; 100: 647–653. [DOI] [PubMed] [Google Scholar]
- 48. Burthem J, Cawley J. The bone marrow fibrosis of hairy-cell leukemia is caused by the synthesis and assembly of a fibronectin matrix by the hairy cells. Blood 1994; 83: 497–504. [PubMed] [Google Scholar]
- 49. Aziz KA, Till KJ, Chen H, et al. The role of autocrine FGF-2 in the distinctive bone marrow fibrosis of hairy-cell leukemia (HCL). Blood 2003; 102: 1051–1056. [DOI] [PubMed] [Google Scholar]
- 50. Aziz KA, Till KJ, Zuzel M, et al. Involvement of CD44-hyaluronan interaction in malignant cell homing and fibronectin synthesis in hairy cell leukemia. Blood 2000; 96: 3161–3167. [PubMed] [Google Scholar]
- 51. Shehata M, Schwarzmeier JD, Hilgarth M, et al. TGF-beta1 induces bone marrow reticulin fibrosis in hairy cell leukemia. J Clin Invest 2004; 113: 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol 2014; 15: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013; 369: 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med 2015; 372: 1430–1440. [DOI] [PubMed] [Google Scholar]
- 55. Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015; 21: 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rogers KA, Andritsos LA, Wei L, et al. Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood 2021; 137: 3473–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Burger JA, Quiroga MP, Hartmann E, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood 2009; 113: 3050–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Forconi F. Hairy cell leukaemia: biological and clinical overview from immunogenetic insights. Hematol Oncol 2011; 29: 55–66. [DOI] [PubMed] [Google Scholar]
- 59. Arons E, Suntum T, Stetler-Stevenson M, et al. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood 2009; 114: 4687–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hockley SL, Giannouli S, Morilla A, et al. Insight into the molecular pathogenesis of hairy cell leukaemia, hairy cell leukaemia variant and splenic marginal zone lymphoma, provided by the analysis of their IGH rearrangements and somatic hypermutation patterns. Br J Haematol 2010; 148: 666–669. [DOI] [PubMed] [Google Scholar]
- 61. de Gorter DJJ, Beuling EA, Kersseboom R, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 2007; 26: 93–104. [DOI] [PubMed] [Google Scholar]
- 62. De Rooij MFM, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012; 119: 2590–2594. [DOI] [PubMed] [Google Scholar]
- 63. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012; 119: 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brummer T, Shaw PE, Reth M, et al. Inducible gene deletion reveals different roles for B-Raf and Raf-1 in B-cell antigen receptor signalling. EMBO J 2002; 21: 5611–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Weston-Bell NJ, Forconi F, Kluin-Nelemans HC, et al. Variant B cell receptor isotype functions differ in hairy cell leukemia with mutated BRAF and IGHV genes. PLoS ONE 2014; 9: e86556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Visentin A, Imbergamo S, Trimarco V, et al. Ibrutinib in relapsed hairy cell leukemia variant: a case report and review of the literature. Hematol Oncol 2020; 38: 823–826. [DOI] [PubMed] [Google Scholar]
- 67. Bohn JP, Wanner D, Steurer M. Ibrutinib for relapsed refractory hairy cell leukemia variant. Leuk Lymphoma 2017; 58: 1224–1226. [DOI] [PubMed] [Google Scholar]
- 68. Tambaro FP, De Novellis D, Wierda WG. The role of BTK inhibition in the treatment of chronic lymphocytic leukemia: a clinical view. J Exp Pharmacol 2021; 13: 923–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014; 370: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kienle DL, Stilgenbauer S. Approved and emerging PI3K inhibitors for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma. Expert Opin Pharmacother 2020; 21: 917–929. [DOI] [PubMed] [Google Scholar]
- 71. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015; 373: 1733–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dietrich S, Pircher A, Endris V, et al. BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood 2016; 127: 2847–2855. [DOI] [PubMed] [Google Scholar]
- 73. Tiacci E, De Carolis L, Simonetti E, et al. Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: a pilot phase-2 clinical trial. Leukemia 2021; 35: 3314–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tiacci E, De Carolis L, Simonetti E, et al. Vemurafenib plus rituximab in refractory or relapsed hairy-cell leukemia. N Engl J Med 2021; 384: 1810–1823. [DOI] [PubMed] [Google Scholar]
- 75. Kreitman RJ, Moreau P, Hutchings M, et al. Treatment with combination of dabrafenib and trametinib in patients with recurrent/refractory BRAF V600E-mutated hairy cell leukemia (HCL). Blood 2018; 132(Suppl. 1): 391. [Google Scholar]
- 76. Lin AY, Dinner SN. Moxetumomab pasudotox for hairy cell leukemia: preclinical development to FDA approval. Blood Adv 2019; 3: 2905–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kreitman RJ, Dearden C, Zinzani PL, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018; 32: 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kreitman RJ, Dearden C, Zinzani PL, et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol 2020; 14: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vereertbrugghen A, Colado A, Gargiulo E, et al. In vitro sensitivity to venetoclax and microenvironment protection in hairy cell leukemia. Front Oncol 2021; 11: 2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jain P, Kanagal-Shamanna R, Konoplev S, et al. Biclonal IGHV-4-34 hairy cell leukemia variant and CLL – successful treatment with ibrutinib and venetoclax. Am J Hematol 2018; 93: 1568–1569. [DOI] [PubMed] [Google Scholar]