

Abstract
DNA repair vulnerabilities are present in a significant proportion of cancers. Specifically, germline alterations in DNA repair not only increase cancer risk but are associated with treatment response and clinical outcomes. The therapeutic landscape of cancer has rapidly evolved with the FDA approval of therapies that specifically target DNA repair vulnerabilities. The clinical success of synthetic lethality between BRCA deficiency and poly ADP-ribose polymerase or PARP inhibition has been truly revolutionary. Defective mismatch repair has been validated as a predictor of response to immune checkpoint blockade associated with durable responses and long-term benefit in many cancer patients. Advances in next generation sequencing technologies and their decreasing cost have supported increased genetic profiling of tumors coupled with germline testing of cancer risk genes in patients. The clinical adoption of panel testing for germline assessment in high-risk individuals has generated a plethora of genetic data, particularly on DNA repair genes. Here, we highlight the therapeutic relevance of germline aberrations in DNA repair to identify patients eligible for precision treatments such as PARP inhibitors (PARPis), immune checkpoint blockade, chemotherapy, radiation therapy and combined treatment. We also discuss emerging mechanisms that regulate DNA repair.
Keywords: germline, cancer, precision oncology, therapeutic response, DNA repair, DNA damage response
Introduction.
It is estimated that ~5–10% of all cancers are due to pathogenic variants (PV) inherited in the germline.[1] For high-risk families, early identification of a cancer-predisposing germline PV is critical, as subsequent genetic counseling can encourage and motivate patients to adhere to risk-reducing interventions.[1] Germline PVs in DNA repair genes are known to not only increase cancer risk but are also relevant for guiding cancer treatment. DNA repair is crucial for genome stability, with multiple specialized pathways existing in the cell to repair different types of DNA lesions (Figure 1). These pathways include homologous recombination repair (HRR), non-homologous end-joining (NHEJ), Fanconi anemia (FA), microhomology-mediated end joining (MMEJ), nucleotide excision repair (NER), base excision repair (BER), mismatch repair (MMR), and replication repair.[2]
Figure 1. A simplified view of DNA damage response and repair: cancer risk and approved therapeutic biomarkers or therapies.
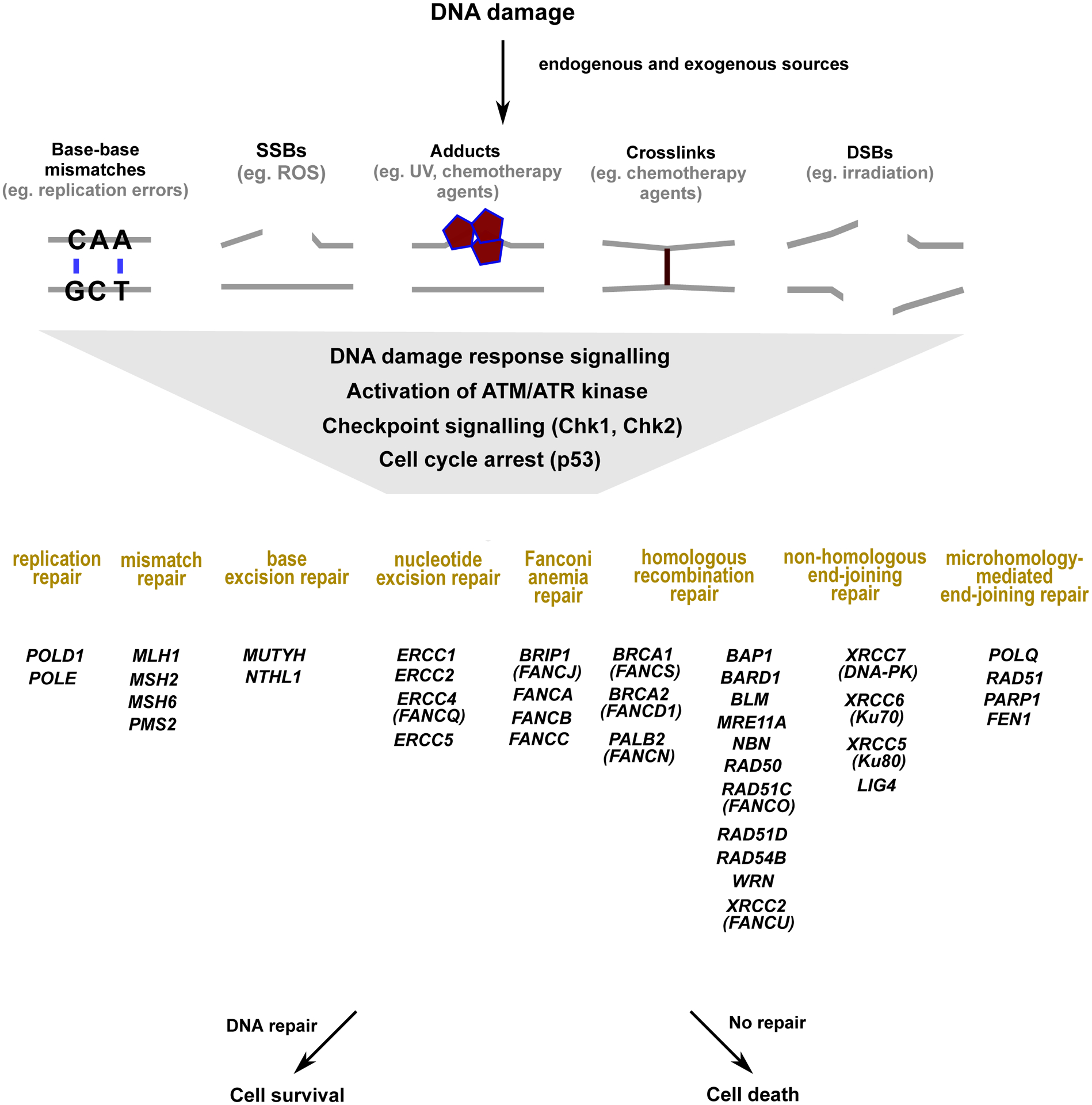
DNA damage may be caused by multiple endogenous (metabolites, replication errors) or exogenous (irradiation, UV light, chemotherapy agents) sources.[195, 196] Multiple forms of DNA damage, such as replication errors, single stranded and double stranded breaks can activate the DNA damage response signaling and the checkpoint response. The DNA damage response signaling involves the activation of the sensory kinases, ATM and ATR. The signals from these sensory kinases are amplified by the checkpoint kinases, Chk1 and Chk2, with cells arresting cell cycle in a p53 dependent manner to either repair the damage or to proceed to cell death via apoptosis.[196–198] Different types of DNA damage are repaired by specialized DNA repair pathways, with pathway members listed below that are associated with increased cancer risk and/or currently tested on germline cancer gene panels. These specialized repair pathways are HRR, FA, MMEJ, and NHEJ for DSBs, BER for SSBs, NER for bulky DNA adducts, MMR and replication-repair for base-base mismatches.[197–204]
Tumors with DNA repair defects are particularly sensitive to DNA damaging agents, such as platinum-based chemotherapeutic drugs and radiation therapy. Table 1 shows DNA repair genes that are associated with therapeutic benefit and cancer risk in preclinical and/or clinical studies. In the early 2000s, it was observed that inhibition of poly ADP-ribose polymerase (PARP) in tumor cells that carry genetic changes damaging BRCA (BRCA1 or BRCA2) function led to synthetic lethality.[3] In 2014, olaparib (Lynparza, AstraZeneca) was the first PARPi that was approved by the Federal Drug Administration (FDA). Olaparib monotherapy was first indicated for advanced ovarian cancer (OC) patients with germline PVs in BRCA who had been treated with three or more prior lines of chemotherapy.[4] Since the first approval, the next generation of PARPis (veliparib, niraparib, rucaparib, and talazoparib) have not only demonstrated increase potency but also expanded to the treatment of tumor types that include PVs in other DNA repair genes.[5] Talazoparib (Talzenna, Pfizer Inc.), which is significantly more potent than olaparib, was approved by the FDA on October 16th 2018 for HER-2 negative locally advanced or metastatic breast cancer (BC) patients with germline PVs in BRCA.[6–8] The POLO trial in patients with metastatic pancreatic cancer and the PROfound trial in patients with metastatic castration-resistant prostate cancer led to the approval of olaparib for patients with germline PVs in BRCA genes and/or other specific HRR genes respectively.[9–12]
Table 1.
DNA repair genes that are associated with therapeutic benefit and cancer risk in preclinical and/or clinical studies.
| Pathway | Gene | Function |
|---|---|---|
| BER | MUTYH | Excises inappropriately paired adenine bases from DNA backbone to initiate repair[210] |
| NTHL1 | Releases damaged DNA base from DNA through cleavage of N-glycosidic bond, leaves an AP site[211] | |
| DDR | ARID1A | Interacts with ATR and recruited to DSBs[179] |
| ATM | Associated with sensitivity to DNA damaging agents, ataxia telangiectasia mutated[212] | |
| BLM | Associated with sensitivity to DNA damaging agents, Bloom syndrome helicase[212] | |
| CHEK1 | Serine/threonine-protein kinase required for checkpoint-mediated cell cycle arrest and activation of DNA repair in response to DNA damage/unreplicated DNA[213] | |
| CHEK2 | Serine/threonine-protein kinase required for checkpoint-mediated cell cycle arrest, activation of DNA repair and apoptosis in response to DNA DSBs[214] | |
| RECQL4 | Associated with sensitivity to DNA damaging agents, Rothmund-Thompson syndrome[212] | |
| WRN | Werner syndrome helicase / 3’ - exonuclease[212] | |
| DNA Replication-Repair | POLE | Component of DNA polymerase epsilon complex for leading DNA strand synthesis[215] |
| POLD1 | Component of DNA polymerase delta complexes for lagging DNA strand synthesis and DNA repair[216] | |
| FA | BRIP1 (or FANCJ) | DNA helicase & BRCA1-interacting[212] |
| FANCA | Tolerance & repair of DNA crosslinks & other DNA adducts[212] | |
| FANCB | Tolerance & repair of DNA crosslinks & other DNA adducts[212] | |
| PALB2 (or FANCN) | Recruit BRCA2 and RAD51 to DNA breaks[217] | |
| HRR | BAP1 | Promotes DSB repair by interacting with BARD1[181, 182] |
| BARD1 | E3 ubiquitin-protein ligase[218] | |
| BRCA1 (or FANCS) | E3 ubiquitin-protein ligase, mediates formation of ‘Lys-6’-linked polyubiquitin chains and plays role in repair by facilitating cellular responses to DNA damage[219] | |
| BRCA2 (or FANCD1) | Binds RAD51, promotes assembly of RAD51 onto single-stranded DNA to potentiate HRR[220] | |
| MRE11A | 3’ exonuclease, defective in ATLD (ataxia-telangiectasia-like disorder)[212] | |
| NBN | Component of MRN complex which plays critical role in cellular response to DNA damage and maintenance of chromosome integrity[221] | |
| RAD50 | ATPase in complex with MRE11A, NBS1[212] | |
| RAD51C (FANCO) | Binds single-stranded DNA and double-stranded DNA, DNA-dependent ATPase activity[222] | |
| RAD51D | Rad51 homolog[212] | |
| RAD54B | Involved in DNA repair and mitotic recombination[223] | |
| XRCC2 (or FANCU) | DNA break and crosslink repair[212] | |
| MMR | MLH1 | Heterodimerizes with PMS2 to form MutL alpha[224] |
| MSH2 | Forms two different heterodimers: MutS alpha and MutS beta[224] | |
| MSH6 | Heterodimerizes with MSH2 to form MutS alpha[224] | |
| PMS2 | Heterodimerizes with MLH1 to form MutL alpha[224] | |
| NER | ERCC1 | Structure-specific endonuclease component[225] |
| ERCC2 | 5’ to 3’ DNA helicase[212] | |
| ERCC4 (or FANCQ) | Structure-specific endonuclease component, removes interstrand cross-link[226] | |
| ERCC5 | Structure-specific DNA endonuclease involved in DNA excision repair, 3’ excision[227] | |
| MMEJ | POLQ | DNA polymerase that promotes MMEJ and limits RAD51 accumulation at resected ends[155] |
| FEN1 | 5’ nuclease[212] |
AP, apurinic/apyrimidinic site; BER, base excision repair; DDR, DNA damage response; DSB, double-strand breaks; FA, Fanconi anemia; HRR, homologous recombination repair; MMEJ, microhomology-mediated end joining; MMR, mismatch repair; NER, nucleotide excision repair; NHEJ, non-homologous end-joining; pol, polymerase.
In the last few years, immune checkpoint inhibitors (ICI) have transformed the treatment of solid tumors such as melanoma, lung, head neck, breast, renal, and bladder cancers.[13] Currently, the approved ICIs are monoclonal antibodies targeting programmed cell death protein (PD-1), programmed cell death protein ligand 1 (PD-L1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) which increase T-cell activation, proliferation and mount an anti-tumor response.[13] It is well-appreciated that a key biomarker of response to PD-L1 inhibitors is MMR deficiency.[14, 15] Notably, tumors with defects in MMR exhibit a high tumor mutation burden (TMB), high neoantigen load, and T-cell infiltration. The cancer agnostic FDA approval of MMR deficiency as a biomarker of PD-L1 inhibitors on May 23rd, 2017 was a landmark decision for treating tumors based solely on the genetic profile regardless of tumor type.[13] Recent studies are finding that other DNA repair defects are also associated with an increased TMB and tumor neoantigen load.[16, 17] These findings are paving the way for identifying DNA repair defects that may serve as biomarkers for ICI response.[18] This review will summarize the current literature on DNA repair defects in the germline and their relevance to personalized cancer treatment. In this review we also discuss emerging paradigms of germline DNA repair vulnerabilities that could be potentially exploited as therapeutic targets. This review does not discuss the biology of the loss of the normal allele in cancer or the nature of the PV (e.g., hyper or hypomorphic alleles), rather focuses on the clinical significance of germline aberrations in specific DNA repair genes as a biomarker for treatment strategies as well as clinical benefit of germline genetic testing for treatment decision.
Germline testing for cancer risk.
In 1895, a family was reported with a spectrum of abdominal cancers in different generations, demonstrating a familial predisposition to cancer.[19] In 1985, Dr. Lynch characterized the first of multiple cancer syndromes as Hereditary Non-Polyposis Colorectal Cancer, subsequently designated as Lynch Syndrome.[19, 20] Our current understanding of the genetic basis of Lynch Syndrome is due to the groundbreaking research in the 1990s by Bert Vogelstein, Albert de la Chapelle, Manuel Perucho, Fishel Kolodner, and others (extensively reviewed in[20]). It is now well-appreciated that Lynch Syndrome is defined by germline PVs in MMR genes.[21]
In the 1940s, Sir David Smithers noted that BC can cluster in families.[22] In 1990, a large case-control study with 4,730 BC cases and 4,688 matched controls concluded that age of onset was a strong indicator of genetic predisposition to BC.[23] In the early 1990s, Dr. Marie-Clair King’s studies linked a region on chromosome 17q21 to increased risk of early-onset BC, identifying BRCA1.[24–26] Subsequently, Dr. Michael Stratton linked BRCA2, localized to chromosome 13q12-q13, with increased BC risk.[26] Further groundbreaking work, led by multiple groups over several years, confirmed and elucidated that the BRCA genes are indeed associated with increased risk of BC and OC.[25–34]
In the early 1990s, BRCA gene sequence was commercially patented by Myriad Genetic Laboratories, which then developed and marketed the first widely available commercial DNA sequencing test for hereditary BC and OC risk based on sequencing of BRCA1, followed shortly thereafter by the addition of BRCA2 (the BRCAnalysis test). In 2013, a landmark ruling by the Supreme Court that, ‘isolated human genes cannot be patented’, rescinded the patents on BRCA and this changed the landscape of genetic testing for cancer diagnostics. A period of rapid innovation through next-generation sequencing (NGS), the introduction of multiple new commercial entities into the genetic testing marketplace, and expansion of genetic testing criteria followed.[35] It must also be appreciated that the cloning and widescale marketing of BRCA testing in the 2000s to evaluate cancer risks in women laid the foundation for enormous growth in the field of clinical cancer genetics. Clinical testing has been proven in this time to not only help guide cancer risk assessment for the carrier and their family members but also in its potential to guide patient management and treatment.[36–38]
Like the BRCA genes, multiple other cancer susceptibility genes function in DNA repair pathways (genes in Figure 1 and Table 1). Today, multigene hereditary cancer panels that include a number of the DNA repair genes listed in Table 1 are used in the clinic to test for germline PVs in cancer susceptibility genes.[36, 39–52] An individual with a family history of cancers may undergo germline testing to quantify their cancer risk. If a PV is found in an individual, their physician(s) can perform cancer screening for early detection, develop preventive management plans, or prevention procedures; or patients may personally maintain their health through behavioral modification and risk mitigation. The results of germline testing may also guide patient treatment.[53] Perhaps equally important, the identification of a PV can motivate family members to undergo testing for cancer risk assessment and risk stratification.[8, 53, 54]
Patients are selected for genetic testing based on personal and family cancer history, the assessment of which has been codified in numerous practice guidelines. However, there is increasing evidence to suggest that germline sequencing may also be beneficial for patients who do not meet guidelines for genetic testing. Advanced stage (stage III-IV) cancer patients (n=1040) were sequenced for 76 cancer predisposition genes and ~17.5% of these patients (182/1040) had clinically actionable germline PVs in cancer risk genes.[55] Among the 17.5% patients with germline PVs, over half (~9.7%, 101/1040) did not meet clinical criteria for testing, and these PVs would not have been detected based on the current guidelines. In pediatric cancer patients (n=1120), it was found that ~8.5% had a PV or a likely PV.[56] Here, only 40% of the pediatric patients with cancer-predisposing PVs had a family history of cancer. Analysis of whole-exome data from 49,738 participants in the UK Biobank found that ~1% (441/49738) of the cohort harbored PVs in genes associated with familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and Lynch syndrome. These individuals in the cohort with the PVs (or likely PVs) were at increased risk for the associated diseases and were not reliably detected by their family history alone.[57] Germline testing in a prospective cohort of patients with solid tumors found that multigene panel testing identified PVs in 13.3% (397/2,984) of the patients. Here, treatment strategies were modified for 28.2% (42/397) of the patients based on the germline findings.[58] Manickam et. al. found that exome sequencing based screening strategies identify 5 times as many individuals with BRCA GPVs than screening based on family history.[59] Beitsch et. al. found that testing based on current guidelines misses nearly half of the BC patients with a GPV.[60]
Although the current literature suggests benefit of population-based screening, there are several barriers to genetic testing such as lack of initial referral from the oncologist, poor knowledge of genetics, access to care, health insurance, medical costs, and confidentiality concerns. [61] [62] [63] Recent studies are focusing on understanding and addressing these barriers so that more patients can benefit from genetic testing.[62, 63] In conclusion, the increased utilization of NGS technology in the clinic, significant reduction in the cost, and broader community acceptance have made it possible to identify clinically actionable germline PVs.[57, 58, 64, 65]
Interpretation of germline variants.
The American College of Medical Genetics and Genomics and the Association for Molecular Pathology have established guidelines for the classification of variants identified through clinical genetic testing further grounding the wide applicability of cancer risk assessment. Variants are classified under five categories of increasing disease severity: benign, likely benign, variants of uncertain significance (VUS), likely pathogenic, and pathogenic.[66] Clinical testing has identified a huge number of rare variants that are currently classified as VUS, and it is not known whether these impact cancer risk.[66] For example, while the clinical significance of a germline PV in an MMR gene is straightforward, the interpretation of clinical significance is not straightforward when a patient harbors a germline VUS in an MMR gene. These VUS account for ~20–30% of variants in MMR genes.[67] Thus, a critical aspect in the field of cancer risk and treatment decision is to evaluate the clinical significance of VUS in DNA repair genes.
The interpretation of VUS is challenging and inclusion of multiple lines of evidence has been suggested, for example, the inclusion of data such as presence in the general population (rare variants with minor allele frequency <1%), segregation of the variant with the disease, in silico prediction of variant effect as predicted-pathogenic or predicted likely-pathogenic, results from structural modeling, mutation rates, and signatures in the tumor, biochemical assays of the variant.[67] As PVs in MMR and DNA polymerase genes are known to be associated with increased mutation rates, the analysis of tumor sequence data has become increasingly important when a VUS is found in these genes.[68] Intriguingly, primary lymphocytes from familial early-onset cancer patients, that carried predicted-pathogenic germline variants in DNA repair genes, exhibited a constitutional defect in the suppression of double strand breaks (DSBs).[69] [70] These results suggest functional assays in primary lymphocytes may assist in providing the various lines of evidence required to clinically interpret the relevance of specific germline VUS in DNA repair genes.
Overall, knowing that a variant is indeed a PV or is benign may aid clinicians in interpreting results from germline testing and guiding patients for their cancer risk as well as personalized treatment.[67] Recent studies suggest implementation of multidisciplinary molecular tumor board teams to recommend customized therapeutic solution(s) for cancer patients that have failed existing treatment.[71, 72] The multidisciplinary team recommends therapeutic strategies based on a comprehensive integrated review of results from genetic testing (germline and/or somatic, TMB etc.), other laboratory results (imaging, pathology, biomarker etc.), patient’s clinical and family history along with potentially available clinical trials. [71, 72] Kato and colleagues showed that patients who received molecular tumor board recommended treatment had better clinical outcomes. [73] Currently, there is a need for global standardization of molecular tumor boards to deliver therapeutic recommendations that can facilitate the goals of precision oncology.[71, 74]
Therapeutic implications for ICIs.
Conventionally, the knowledge of germline PVs in DNA repair genes has informed our understanding of cancer risk. More recently, DNA repair pathways have emerged as playing a major role in the selection of patients for ICI therapy. In 2017, the PD-1 inhibitor pembrolizumab received FDA approval for adult and pediatric patients with metastatic or unresectable tumors with microsatellite instability-high (MSI-H) or MMR deficiency (dMMR) phenotype that has progressed following previous treatments or for whom no other treatment options are available. Five studies contributed data for the FDA application, with a total of 149 patients that had MSI-H or dMMR tumors.[75–77] Of the 149 patients, 90 patients had metastatic CRC, and 59 patients had other cancers. Further, out of the 149 patients, 59 patients responded to treatment (objective response rate (ORR)=39.6%, 95% CI, 31.7–47.9), with a 7% complete response rate.[75–77]
The landmark tumor agnostic FDA approval of pembrolizumab was based on the understanding of the biology of dMMR/MSI-H which occurs due to germline or somatic PVs in MMR genes (MLH1, MSH2, MSH6, PMS2). The MMR proteins maintain genome stability by correcting single base nucleotide insertions or deletions.[78] Germline PVs impairing MMR are associated with the autosomal dominant condition, Lynch Syndrome, which increases the risk of CRC, endometrial cancer (EC), and several other cancers, such as OC, brain, skin, pancreatic, and other gastrointestinal cancers.[79, 80] Currently, ~1–7% of CRCs, and ~13–25% of ECs are associated with germline PVs in MMR genes.[81–83] Somatic PVs in MMR are also common in tumors; for example, in ~12–15% of localized CRC, ~3–4% of metastatic CRC, and up to 25% of ECs.[80, 84, 85] Epigenetic silencing of the MLH1 promoter due to hypermethylation can also contribute to dMMR tumors.[86, 87] Other sources of dMMR include deletions at the 3’ end of EPCAM which causes the inactivation of the nearby MSH2 gene.[86] Although rare, it is also possible to inherit two MMR variants (one from each parent), which can result in either a compound heterozygote or, if the variants are in the same MMR gene, a rare recessive inherited syndrome known as Constitutional MMR Deficiency (cMMRD) leading to a wide spectrum of childhood malignancies.[78] In a case study, two cMMRD siblings with recurrent glioblastoma were treated with PD-1 inhibitor nivolumab (Opdivo, Bristol-Myers Squibb) which led to a significant clinical benefit.[88] Recently, anti-PD-1 monoclonal antibody, dostarlimab (Jemperli, GlaxoSmithKline) showed clinical benefit in patients with dMMR EC[89, 90]. Based on the results of the GARNET trial, dostarlimab was approved by the FDA for patients with recurrent or advanced dMMR EC[89, 90].
It is well-appreciated that dMMR leads to increased TMB and tumor neoantigen production, potentially contributing to the better response to ICIs.[91–93] Infact, testing for dMMR or MSI-H also can identify patients with TMB-H. In June 2020, accelerated FDA approval for pembrolizumab was indicated for the treatment of TMB-high (or TMB-H, ≥10 mutations/megabase) adult and pediatric unresectable or metastatic tumors that have either progressed on prior therapy or for which no other treatment options are available.[94] Germline PVs in DNA polymerase genes, POLE (typically in exonuclease domain, ExoD) and POLD1, increase the risk of CRCs, ECs, OC, other malignancies, and have been associated with ultra-hypermutation in the tumor (i.e. tumors with >100 mutations/megabase).[68, 95–97] Latest data suggest that germline PVs in POLE and POLD1 lead to ~0.1–0.4% of familial cancers.[68, 98] While several studies have focused on variants in the ExoD, increased TMBs have also been noted with variants outside the ExoD.[99, 100] Patients with POLE/POLD1 variants had significantly better clinical outcomes following treatment with ICIs when compared to patients without them.[101, 102] No difference in clinical outcomes was observed in patients with POLE/POLD1 variants in the ExoD or outside the ExoD.[102]
Several trials are underway to determine the efficacy of ICIs in combination with PARPis (Supplemental Table 1).[103, 104] The rationale for this combination arises from the observations that PARP inhibition increases DNA damage, which is particularly pronounced in cells that carry DNA repair defects such as BRCA deficiency. This damage not only leads to increased priming of the immune system in the tumor microenvironment but also increases the tumor intrinsic immunogenicity by modulating the expression of surface markers such as PD-L1 (reviewed in[105]). In models of small cell lung cancer, it was shown that PARP (and Chk1) inhibition leads to a strong anti-tumor immune response by recruitment of cytotoxic T-lymphocytes via the activation of the STING pathway.[106] Based on these observations, an ongoing non-interventional, prospective clinical study (NCT03495544, Supplemental Table 1) is evaluating the association between germline PVs in hereditary BC genes (such as BRCA1, BRCA2, CHEK2) and PD-L1 expression in breast tumor cells and immune cells. This study will evaluate the relationship between germline PVs, and PD-L1 expression to allow precision selection of ICIs in these patients. Interestingly, a recent preclinical study showed that BRCA deficiency differentially modulates the tumor microenvironment.[107] While BRCA1 mutations may drive immune responses that limit benefit from ICI, BRCA2 mutations increased tumor immunogenicity and showed benefit from ICI therapy.[107] These observation suggest a mechanistic difference between the BRCA genes and warrant further investigation.[107]
Therapeutic implications for PARPi therapy.
PARPis prevent the repair of single-strand breaks (SSBs), and in HRR-deficient cells these SSBs are converted to DSBs which ultimately leads to unrepaired damage and consequent cell death.[108–110] Proof-of-concept studies in 2005 and 2007 provided the rationale for the use of PARPis in cancer therapy.[4, 111] In 2009, a phase I trial evaluated the antitumor activity of the PARPi, olaparib, for cancer patients with either wild-type or BRCA (BRCA1 or BRCA2) PVs.[112] In the expansion phase of the trial, only BRCA (BRCA1 or BRCA2) PV carriers with OC, BC, and prostate cancer were enrolled and these patients showed clinical benefit from olaparib.[112] In 2014, olaparib monotherapy received FDA approval for advanced OC patients with a germline PV in BRCA who have had three or more lines of prior chemotherapy.[4]
The efficacy and tolerability of olaparib in advanced OC and BC patients with germline BRCA PVs were assessed in two proofs-of-concept multicenter phase II studies.[113–116] In 2018, the FDA approved olaparib therapy for patients with germline BRCA PVs in HER2-negative metastatic BC patients based on the results from the OlympiAD trial (Supplemental Table 2).[54, 116, 117] While no statistically significant change or improvement in median overall survival (OS) was observed, an OS benefit was observed in patients who had not previously been treated for metastatic BC (HR 0.51, 95%CI: 0.29–0.90).[118] Overall, treatment with olaparib significantly increased the ORR and OS of the metastatic BC patients. A pooled analysis was performed to explore the benefit of olaparib in OC patients carrying germline PVs in BRCA who had received multiple lines of prior chemotherapy. Here, all patients had received olaparib monotherapy at relapse, and 91% (273/300) patients were evaluated for response.[119] In the pooled analysis, the ORR was 36% (95% CI, 30–42) and the median duration of response (DoR) was 7.4 months (95% CI 5.7–9.1).[112] In the subset of patients with measurable disease at baseline (205/300) who had received ≥3 lines of prior chemotherapy, the ORR was 31% (95% CI 25–38) and median DoR was 7.8 months (95% CI 5.6–9.5). This analysis demonstrated that olaparib monotherapy was safe and effective not only for patients at baseline but also for the patients who underwent multiple prior chemotherapies (both platinum-sensitive and resistant).[119] In Study 19, a phase II trial of olaparib maintenance monotherapy in relapsed OC patients (n=265) with BRCA PVs (germline or somatic), olaparib-treated patients had a higher PFS when compared to the patients in the placebo arm (median, 8.4 months vs. 4.8 months; P < 0.00001).[120] A retrospective analysis of these patients found that the benefit of olaparib versus placebo was greater in patients with BRCA PV when compared to patients without BRCA PVs.[120]
The PROfound trial in metastatic castration-resistant prostate cancer patients with specific HRR gene PVs evaluated their response to olaparib.[11, 12, 121] ORR was 33% (n=16/49; 95% Cl, 20% to 48%), and homozygous and/or deleterious somatic or germline aberrations in BRCA, ATM, FANCA, and CHEK2 were identified in 16 out of 49 (33%) patients.[121] The POLO trial assessed the efficacy of olaparib maintenance therapy in metastatic pancreatic cancer patients with germline BRCA PVs.[9] This trial showed that the olaparib maintenance therapy can extend PFS in metastatic pancreatic cancer patients.[9] Recently, two parallel phase II studies concluded that olaparib may be therapeutically efficacious for pretreated advanced pancreatic cancer patients with PVs in other DNA repair genes (Supplemental Table 2).[122]
Following the success of olaparib, several next generation PARPis have also received FDA approval including veliparib, niraparib, rucaparib, and talazoparib. Mechanistic studies have increased our understanding of how PARPis exert their anti-cancer activities (Figure 2), however, this is still not completely understood. Initially, PARPis were thought to exert anti-tumor activity by inhibiting the catalytic activity of PARP1/2.[5, 123] Subsequently, it was shown that PARPis effectively ‘trap’ the PARP1- and PARP2- DNA complexes in pre-clinical testing (Figure 2).[5] Olaparib, veliparib, niraparib, rucaparib, and talazoparib all have been shown to have PARP-trapping activity.[5] Mechanistically, these inhibitors function similarly, however it has been shown in biochemical assays that veliparib functions as a catalytic inhibitor with slight trapping activity, while olaparib, niraparib and rucaparib function as PARP trappers, ~100-fold more efficiently in comparison to veliparib (Figure 2).[5, 123] Talazoparib is by the far the most potent PARP trapper with ~100-fold more PARP-trapping efficiency than olaparib, rucaparib, and niraparib (Figure 2).[5, 123] Both olaparib and rucaparib (Rubraca, Clovis Oncology) have been approved for treatment-refractory OCs with germline BRCA PVs. Niraparib (Zejula, GlaxoSmithKline) and olaparib have both received FDA-approval for maintenance therapy in patients with recurrent OC.[124, 125] Niraparib was approved for women with recurrent OC in March 2017, for late-line treatment of women with recurrent OC in October 2019, and as the first-line maintenance monotherapy in April 2020 for women with advanced OC regardless of biomarker status (PRIMA trial).[126] In the PRIMA trial, newly diagnosed advanced OC patients were matched to either niraparib or the placebo. A statistically significant PFS improvement and higher OS in the niraparib cohort were observed compared to the placebo, regardless of HRR deficiency status.[126, 127]
Figure 2. A simplified view of PARP1 function in DDR and PARP1 inhibition mechanisms.

When a single-strand break occurs, PARP1 binds to the damaged site and uses NAD+ as a substrate for PARylation and auto-PARylation. PARylation leads to the recruitment of DNA repair proteins, and auto-PARylation decreases the affinity of PARP1 to DNA. PARP1 dissociates from DNA and BER proteins repair DNA. PARPis differ significantly in their ability to trap PARP1 and/or inhibition of PARP1 catalytic activity.[205, 206] There are two known mechanisms of PARP1-inhibition, PARP1-trapping (left) and inhibition of PARP1 catalytic activity (right). During PARP1 trapping, the PARPi binds directly to the PARP1 active site, and prevents dissociation of PARP1 from DNA, leading to replication fork stalling and DSBs.[205] Here, the DNA damage might be repaired via HRR, FA, template switching, and other repair proteins.[207, 208] In the second mechanism, the binding of NAD+ to PARP1 is prevented and thereby PARP1 loses the ability to use its substrate and perform catalysis with downstream replication fork staling and DSBs.[207, 209] Here, the DNA damage might be repaired via HRR.[207, 208]
The BrighTNess trial assessed the addition of PARPi, veliparib, plus carboplatin to standard neoadjuvant paclitaxel chemotherapy or carboplatin alone to standard neoadjuvant chemotherapy in stage II-III triple-negative BC patients (n=634) (Supplemental Table 2).[38] The pathological complete response (pCR) was higher in patients that received the combination of veliparib, carboplatin, and paclitaxel (53%, n=68/316) versus the patients that received paclitaxel alone (31%, n=49/158), but not higher when compared to the group that received carboplatin alone with paclitaxel (58%, n=92/160).[38] Here, 51% of the patients (47/92) with a germline BRCA PV achieved a pCR versus 48% of the patients (262/542) without a germline BRCA PV that achieved a pCR.[38] In the GeparOla trial, early HER-2 negative breast cancer patients with HRR deficiency (germline or somatic BRCA1 or BRCA2 PV and/or HRR deficiency) received either paclitaxel plus olaparib with standard chemotherapy or paclitaxel alone with standard chemotherapy. The addition of olaparib with paclitaxel showed a significantly higher rate of pCR in hormone receptor-positive patients that were less than 40 years of age, warranting further investigation in primary BCs.[128] Ongoing trials (NCT02227082, NCT01562210, NCT02229656, Supplemental Table 2) are assessing PARPis as radiosensitizers and assessing the safety and tolerability of the combination treatment.
Several trials are assessing the activity of PARPis in tumors with mutations in other DNA repair genes.[129–132] Yurgelun et. al. showed that pancreatic ductal adenocarcinoma patients with germline PVs in other HRR genes had a better OS when compared to patients without them.[133] A phase II trial (NCT04508803, Supplemental Table 2) is assessing the combination of an anti-PD1 monoclonal antibody, HX008, in combination with the PARPi, niraparib in metastatic BC patients with germline PVs in multiple DNA repair genes apart from BRCA. Another phase II, interventional trial (NCT03967938, Supplemental Table 2) is assessing the efficacy of olaparib in advanced cancer patients with germline or somatic PVs in HRR genes. With the availability of germline multigene panel data from cancer patients, several studies are finding that germline PVs in DNA repair genes are overrepresented in cancers where the use of PARPis has not been established.[134–136]
While PARPi therapy has been revolutionary, de novo or acquired resistance to PARPi has been observed in patients. Several mechanisms have been proposed such as alterations in PARP1 leading to increase in PARP1 catalytic activity or removal of PARP trapping, restoration of HRR via BRCA reversion mutations or demethylation of BRCA promoter as reviewed by Lee and Matulonis.[137] It is anticipated that the number of patients that are resistant to PARPis will continue to rise and thus it is critical to understand the mechanisms of PARPi resistance for optimizing treatment strategies such as targeted combination therapy.[137] In summary, several PARPis have been approved as therapeutic and maintenance therapy for multiple cancer types, typically based on the presence of germline or somatic BRCA PVs. Furthermore, PARPi therapy is currently being investigated in patients with alterations in other DNA repair genes and combination with other treatments so that a broader range of patients can achieve clinical benefit.
Implications for chemotherapy and radiation therapy.
It is now well-appreciated that BRCA deficiency can increase sensitivity to DNA crosslinking agents (such as platinum-based agents) and ionizing radiation.[138] In 2010, a pilot study assessed a small group of women (n=12) with BC and a germline BRCA1 PV.[139] Here, 10 of the 12 (~83%) women had a pCR to neoadjuvant cisplatin. These findings were validated in a larger study in 2014 in women with early-stage BC and a BRCA1 germline PV. Here, 61% of the women experienced a pCR to the neoadjuvant cisplatin.[139] In a phase II trial, patients (n=28) with stage II and stage III triple-negative BC (TNBC) were treated with neoadjuvant cisplatin. Then, after undergoing definitive surgery, these patients received standard adjuvant chemotherapy and radiation therapy. Here, 22% (6/28) of the patients experienced a pCR. Interestingly, of the 6 patients with pCR, only 2 patients had a germline BRCA1 PV. Additionally, 64% of patients had a complete or partial response. These studies showed that while patients with a germline BRCA1 PV have a good response to cisplatin, there is a subset of patients with wild-type BRCA1 and TNBC that also respond to cisplatin.[140] In a phase II non-randomized trial, women with metastatic BC and a germline BRCA1 PV were treated with cisplatin chemotherapy. An ORR of 80% (16/20) was observed, with 45% (9/20) of the patients exhibiting complete clinical response, and 7/20 (35%) patients exhibited a partial response.[141] This study demonstrated that cisplatin was highly effective in women with metastatic BC and BRCA1 PVs.
The INFORM trial prospectively assessed pCR in newly diagnosed HER2-negative BC patients with germline BRCA PVs with neoadjuvant cisplatin versus anthracycline-based therapy (Supplemental Table 3). The study patients were randomly assigned to neoadjuvant single-agent cisplatin or to doxorubicin-cyclophosphamide.[142] The pCR rates for neoadjuvant cisplatin and doxorubicin-cyclophosphamide were 18% and 26% respectively. This study did not find any differences in pCR and residual cancer burden by either agent in patients with TNBC, consistent with those reported by the BrighTNess and Geparsixto trials.[38, 142, 143] The results from the INFORM trial suggest that BRCA PV status might predict sensitivity to any DNA damaging agents and not exclusively to platinum-based agents. Recently, a small trial of neoadjuvant talazoparib without chemotherapy reported a pCR of 53% in 20 patients with operable BC and a germline PV in BRCA, warranting larger investigation including comparison with neoadjuvant chemotherapy.[144]
A 2006 retrospective study compared the outcomes in BC patients with germline BRCA PVs to patients with sporadic BC. All patients in the study had breast conservation surgery and received radiation therapy for their stage I or II BC with no significant differences observed in in-breast tumor recurrence in the carriers versus the non-carriers. This study also did not find any significant difference in recurrence in the carriers versus the non-carriers who underwent bilateral oophorectomy. However, the recurrence rates were twice as high in the carriers who did not undergo oophorectomy. Also, as expected, the study found an increased incidence of contralateral BCs in the carriers versus the non-carriers. While tamoxifen significantly reduced the incidence of contralateral BCs in carriers, the incidence remained higher than sporadic non-carriers.[145] Overall, this study suggests that hormonal intervention and prophylactic bilateral oophorectomy are associated with reduced recurrence or reduced risk of new primary BCs and fewer contralateral BCs in carriers. Despite this, carriers still have a high risk of contralateral BCs, warranting the need for additional strategies in carriers undergoing breast conservation surgery to reduce their risk. A prospective study found that an equal proportion of carriers chose breast conservation surgery and bilateral prophylactic mastectomy for risk reduction, thus warranting assessment of additional risk-reduction strategies for breast conservation surgery.[144]
Beyond BC, women with OC who are carriers of a germline BRCA1 or BRCA2 PV have also shown high response rates to platinum-based therapy. In a study by Gorodnova et al., 34 BRCA1 and 1 BRCA2 germline PV carriers were identified. The total clinical response in the patients with germline PVs in BRCA to platinum-based therapy was determined to be 34% (n=12/35). In contrast, the clinical response rate of the patients with somatic PVs in BRCA was determined to be 4% (8/190).[146] In a retrospective study, men with castration-resistant prostate cancer, with or without germline BRCA2 PVs, were assessed for response to platinum-based therapy. These patients were treated with carboplatin and docetaxel and were categorized based on the germline BRCA2 PV status. This study found a significant association between the germline BRCA2 PV status and the response to platinum-based therapy.[147] Overall, both chemotherapy and radiation therapy act by inducing DNA damage, and vulnerabilities in DNA repair, either germline or somatic, impacts response to these therapies.
Therapeutic implications of germline variants in other DNA repair genes.
Growing evidence suggests that germline variants in other DNA repair genes may also impact response to cancer therapeutics. Defects in the Fanconi Anemia (FA) pathway lead to impaired ability to perform homology-directed repair and thereby increased sensitivity to DNA cross-linking agents.[148] Tumors carrying germline defects in the FA are also sensitive to PARPis.[148] There are 22 FA or FA-like genes including FANCA, B, C, D1 (or BRCA2), D2, E, F, G, I, J (or BRIP1), L, M, N (or PALB2), O (or RAD51C), P (or SLX4), Q (or ERCC4), R, S (or BRCA1), T (or UBE2T), U (or XRCC2), V (or REV7) and W (or RFWD3). [149–151] The BRCA genes and several associated HRR genes are important members of the FA pathway. FANCD2-FANCI monoubiquitinylation is a critical step in the activation of the FA pathway and can be assessed by nuclear staining of monoubiquitylated FANCD2.[152] In NSCLC, it was observed that a subset of tumors was negative for FANCD2 foci staining (22%, n=23/104) and thus was considered deficient in FA pathway.[148] These results suggest that at least a subset of lung cancers may be sensitive to DNA damaging therapy and/or PARPi therapy due to their limited ability to perform FA repair.[148] In a clinical study, 643 tumors (including BC, OC, CRC, EC, NSCLC, and several others) were evaluated for FANCD2 nuclear foci and 28.7% (185/643) were found to be negative.[153] 61 patients with FANCD2-foci negative tumors received either veliparib alone or in combination with mitomycin C, and 6 patients in the combination arm showed clinical benefit. Subsequent tumor sequencing found germline alterations in the FA pathway. These data suggest that PARPi alone or in combination with a DNA damaging therapy is safe and can lead to clinical benefit in some patients with FA-deficient tumors.[153]
The MMEJ pathway is emerging as a promising therapeutic target; it begins after exposure of microhomology (i.e., short complementary sequence) regions for alignment of DNA ends.[154] Following the alignment, the resulting 5’ flaps are processed, the gaps are filled by a polymerase and then ligated. PARP1 is an essential component of the MMEJ pathway and PARPi can also inhibit the MMEJ pathway.[154] Furthermore, loss of expression of MMEJ proteins such as POLQ and FEN1 in HRR-deficient cells was found to cause synthetic lethality.[154, 155] While MMEJ pathway is emerging as a promising biomarker to guide cancer treatment, only the therapeutic potential of somatic tumor alterations has been assessed so far. Brandalize et. al. identified a germline polymorphism in the promoter region of POLQ that was associated with increased risk of inherited BC, warranting further studies on cancer risk and therapy outcomes.[156]
The core NER genes include ERCC1, 2 (XPD), 3 (XPB), 4 (XPF), 5 (XPG), 6 (CSB), 8 (CSA), XPA, XPC, TFIIH, LIG1, RPA, and DDB1.[157] The NER pathway excises bulky DNA lesions such as those generated by platinum-based agents.[158] Targeting the core NER protein complex, the ERCC1-ERCC4 heterodimer has been shown to improve cisplatin cytotoxicity in several cancer cell line models.[159] Additionally, it has also been suggested that low nuclear ERCC1 expression may correlate with improved treatment response and confer sensitivity to radiation therapy in HNSCCs.[160] A meta-analysis of 836 NSCLC patients found that ERCC1 protein levels could predict better PFS and OS to platinum-based therapy.[158, 161, 162] Three regulatory SNPs in NER genes were determined to predict better PFS and OS following treatment with cisplatin.[158] NER genes were also shown to be differentially expressed in CRC and normal colon tissues, with the expression of ERCC2, ERCC4, and XPC associated with CRC prognosis.[163] Aiello et al. evaluated ERCC1 T19007C and C8092A SNPs in tumor specimens from two independent cohorts of NSCLC patients who had received nivolumab. Here, a better response to nivolumab was observed in patients with ERCC1 C8092A SNP.[164] Overall, preclinical studies suggest that defects in several DNA repair genes could potentially predict response to therapy, however it remains to be determined how effective they are for patient stratification.
Therapeutic implications of germline variants in genes that regulate DNA repair.
Several genes that do not directly mediate DNA repair are emerging as key regulators of DNA repair pathways. Recent work is defining the molecular circuits that govern the relationship between androgen receptor (AR) signaling and DNA repair.[165, 166] Combined therapy of androgen deprivation and radiation has been the standard treatment for prostate cancers showing improved outcomes for high-risk prostate cancer patients.[167–169] Previously, the mechanisms that led to this outcome benefit were not known. It was recently shown that AR activity is induced by DNA damage, and in turn, promotes transcription of DNA repair genes.[165, 166] Intriguingly, patients at a high risk for familial prostate cancer frequently carry germline variants in DNA repair and AR signaling genes and function-testing of their primary lymphocytes showed increased sensitivity to DNA damaging agents.[70]
VHL germline PVs cause Von Hippel-Lindau syndrome and strongly predispose individuals to RC.[170–172] It is appreciated that VHL is important for the ubiquitination and proteasomal degradation of hypoxia-inducible factor-alpha (HIF1-alpha and HIF-2alpha). Inactivation or loss of VHL leads to the constitutive expression of HIFalpha, which freely translocates to the nucleus and activates the expression of multiple genes that are relevant for tumorigenesis. Interestingly, several DNA repair genes, including HRR and MMR, are down-regulated during hypoxic stress.[173–175] In fact, hypoxia has been specifically shown to downregulate the expression of BRCA1 and RAD50, which are critical for HRR.[176, 177] Investigation of VHL-deficient RC cell lines (786-O, RCC4) found reduced expression of HRR and MMR genes, the persistence of radiation-induced DSBs, and increased sensitivity to PARPis.[178]
ARID1A and BAP1 are tumor suppressor genes, and their protein products play important roles in DNA repair.[179, 180] ARID1A is recruited to DSB sites via an interaction with DNA damage response signaling kinase ATR. Studies in both in vitro and in vivo models of cancer have shown that ARID1A deficiency sensitizes cells to PARPis.[179] BAP1 interacts with BARD1 and promotes HRR to repair DSBs.[181, 182] No known therapeutics that exploit ARID1A deficiency have been discovered. However, phase II trials focusing on the effect of PARPis on patients with BAP1 PVs are underway (NCT03207347) alongside mechanistic studies that focus on the specific nuclear processes that involve BAP1.[181, 183] Several other clinical trials (NCT03425201, NCT03207347, NCT03786796, NCT03741426, NCT03682289, NCT04068831, NCT03875313, NCT03729245, NCT04195750) are also currently underway to test a combination of PARPis with other treatment strategies in RC.[180]
Germline PVs in the citric acid cycle genes FH and SDHx are associated with cancer predisposition syndromes.[184–187] Heterozygous loss-of-function PVs in FH are associated with hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome and these patients are predisposed to aggressive papillary RCC which are refractory to standard therapies.[186] Similarly, heterozygous loss-of-function PVs in SDHx (SDHA, SDHB, SDHC, SDHD) are associated with SDH-related hereditary paraganglioma and pheochromocytoma (SDH PGL/PCC). Patients with certain familial RCs, and familial gastrointestinal stromal tumors also harbor germline PVs in FH and SDHx.[134, 188, 189] Loss of FH and SDH function leads to the cellular accumulation of metabolites, with fumarate observed in patients with HLRCC syndrome and succinate in patients with SDH-PGL/PCC.[187] Sulkowski et. al. found that primary tumors from HLRCC and SDH PGL/PCC patients showed elevated DSBs. They also showed that fumarate or succinate can competitively inhibit lysine demethylases KDM4A and KDM4B due to structural similarities. KDM4A and B are important regulators of DNA repair, specifically the repair of DSBs by HRR.[187] In vitro assessment in HEK293FT renal cells showed that deregulation of SDHB and FH led to the accumulation of metabolites and DSBs. Downregulation of SDHB or FH conferred increased sensitivity to DNA damaging therapies and PARPis in preclinical studies. Overall, this is an exciting new area of crosstalk between metabolism and DNA repair, and germline defects in these genes could be relevant for response to therapies that target DNA repair.
Finally, regulation of histone modifications is essential for genomic stability and this process is often aberrant in multiple cancers.[190] Several lysine demethylase inhibitors are being developed as potential agents to enhance response to radiotherapy or genotoxic chemotherapy.[191] The lysine methylation modifications regulate gene expression based on the position of methylation and methylation state.[192] For example, methylations in H3K4, H3K36, and H3K79 are generally associated with active transcription, while methylations in H3K9, H3K27, and K4K20 are associated with repressed transcription. These lysine methylations are regulated by histone methyltransferases and demethylases; defects in these have been associated with several diseases. [192] The pattern of histone methylations not only serves as a focal point for DSB repair proteins (such as HRR and NHEJ proteins) to assemble but also selects the pathway to repair the DSBs.[193] For example, H3K9 demethylases remove repressive H3K9 methylations and have been shown to play a role in DSB repair.[191] Some H3K9 demethylases (such as JMJD1A, PHF2) upregulate the expression of DNA repair factors involved in HRR and/or NHEJ. Other H3K9 demethylases (such as JMJD1C, KDM4D, PHF8) directly regulate DSB repair by migrating to the lesion and regulating the recruitment or function of DSB repair factors.[191] While some H3K9 demethylases (KDM4D, PHF8) are required for DSB repair, their precise mechanism with which they alter DNA repair has not been clearly defined.[191] Currently, it is not known how germline variants in core histone proteins may impact DNA repair and response to DNA damaging therapies.[194]
Conclusions
The massive use of NGS technology in the clinic has revealed a rich landscape of germline defects in DNA repair genes. The availability of FDA-approved therapies that specifically target DNA repair defects such as PARPis or ICIs that show benefit in patients with DNA repair defects have expanded the clinical options for cancer patients with durable responses and long-term benefits. Given the significance of DNA repair defects for cancer risk and treatment response, it is important in the future to determine if germline vulnerabilities in mediators of DNA repair pathways can be exploited therapeutically. Overall, it is the right time to fully understand the therapeutic extent of germline vulnerabilities in DNA repair and assess their clinical benefit in cancer patients.
Supplementary Material
Acknowledgments.
We express gratitude to Dr. Erica Golemis (Fox Chase Cancer Center) for valuable input on the article.
Funding.
All Fox Chase Cancer Center affiliated authors are in part supported by the NCI Core Grant, P30 CA006927, to the Fox Chase Cancer Center. M.J.E. was supported by the DOD W81XWH-18-1-0196. S.A. was supported by the DOD W81XWH-18-1-0148, and a CEP grant from the Yale Head and Neck Cancer SPORE. R.W.L. was supported by an Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship. M.J.H. was supported by funding from the American Cancer Society. M.B.D was supported by the NIH U01 CA164920, R01 CA207365 grants. J.E.M. was supported by the Colorectal Cancer Alliance and Varian Medical Systems.
Declaration of Interests.
M.J.E. has served on scientific advisory board for Biomarker strategies, as an advisor for Windmil Therapeutics, Sanofi/Regeneron, and Syndax; data safety monitoring boards for Astra Zeneca, Seattle Genetics, Takeda, GSK and has received clinical research funding (to the institution) from Apexigen, BMS, Nektar, Precision Oncology, Windmil and Merck. M.J.H. performs collaborative research (with no funding) with the following: Myriad Genetics, Invitae Corporation, Ambry Genetics, Foundation Medicine, Inc. He also performs collaborative research (with no funding) and is part of a Precision Oncology Alliance funded by Caris Life Sciences (cover travel and meals at meetings). S.A. performs collaborative research (with no funding) with Caris Life Sciences, Foundation Medicine, Inc., Ambry Genetics and Invitae Corporation. J.E.M. performs collaborative research (with no funding) with Foundation Medicine, Inc. and Caris Life Sciences. S.A.’s spouse is employed by Akoya Biosciences and has stocks in Akoya Biosciences, HTG Molecular Diagnostics, Abcam Plc., and Senzo Health. S.A., M.J.H., R.W.L., J.E.M. have patents and/or pending patents related to cancer diagnostics/treatment. M.J.E. has pending patent for radiopharmaceuticals to treat small-cell lung cancer. All other authors declare no competing interests.
Abbreviations.
- AR
androgen receptor
- BC
breast cancer
- BER
base excision repair
- CI
confidence interval
- CR
complete response
- CRC
colorectal cancer
- DDR
DNA damage response
- dMMR
mismatch repair deficiency
- DoR
duration of response
- DSB
double-strand breaks
- EC
endometrial cancer
- ExoD
exonuclease domain
- FDA
federal drug administration
- FA
Fanconi Anemia
- HLRCC
hereditary leiomyomatosis and renal cell cancer
- HNSCC
head and neck squamous cell carcinoma
- HR
hazard ratio
- HRR
homologous recombination repair
- ICI
immune checkpoint inhibitor
- MMEJ
microhomology-mediated end-joining
- MMR
mismatch repair
- MSI
microsatellite instability
- MSS
microsatellite stable
- NER
nucleotide excision repair
- NGS
next generation sequencing
- NHEJ
non-homologous end joining
- OC
ovarian cancer
- ORR
objective response rate
- OS
overall survival
- PA
patients analyzed
- PAR
poly ADP ribose
- PARP
poly-ADP ribose polymerase
- PARPi
PARP inhibitor
- PC
pancreatic cancer
- pCR
pathological complete response
- PFS
progression free survival
- PV
pathogenic variant
- RC
renal cancer
- SCLC
small cell lung cancer
- SD
stable disease
- SSB
single strand breaks
- TMB
tumor mutation burden
- TNBC
triple negative breast cancer
- UC
urothelial carcinoma
- VUS
variants of uncertain significance
Main Text References
- [1].Hall ET, Parikh D, Caswell-Jin JL, Gupta T, Mills MA, Kingham KE, et al. Pathogenic Variants in Less Familiar Cancer Susceptibility Genes: What Happens After Genetic Testing? JCO Precision Oncology. 2018:1–10. [DOI] [PubMed] [Google Scholar]
- [2].O’Connor J, Mark. Targeting the DNA Damage Response in Cancer. Molecular Cell. 2015;60:547–60. [DOI] [PubMed] [Google Scholar]
- [3].Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Molecular Oncology. 2011;5:387–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim G, Ison G, Mckee AE, Zhang H, Tang S, Gwise T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clinical Cancer Research. 2015;21:4257–61. [DOI] [PubMed] [Google Scholar]
- [5].Shen Y, Aoyagi-Scharber M, Wang B. Trapping Poly(ADP-Ribose) Polymerase. Journal of Pharmacology and Experimental Therapeutics. 2015;353:446–57. [DOI] [PubMed] [Google Scholar]
- [6].FDA. FDA approves talazoparib for gBRCAm HER2-negative locally advanced or metastatic breast cancer. 2018.
- [7].Ettl J, Quek RGW, Lee KH, Rugo HS, Hurvitz S, Gonçalves A, et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Annals of oncology. 2018;29:1939–47. [DOI] [PubMed] [Google Scholar]
- [8].Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee K-H, et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. New England Journal of Medicine. 2018;379:753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019;381:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. New England Journal of Medicine. 2020;383:2345–57. [DOI] [PubMed] [Google Scholar]
- [12].De Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. New England Journal of Medicine. 2020;382:2091–102. [DOI] [PubMed] [Google Scholar]
- [13].Arora S, Velichinskii R, Lesh RW, Ali U, Kubiak M, Bansal P, et al. Existing and Emerging Biomarkers for Immune Checkpoint Immunotherapy in Solid Tumors. Adv Ther. 2019;36:2638–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Duffy MJCJ. Biomarkers for Predicting Response to Immunotherapy with Immune Checkpoint Inhibitors in Cancer Patients. 2019. [DOI] [PubMed]
- [15].FDA. FDA Approves First-Line Immunotherapy for Patients with MSI-H/dMMR Metastatic Colorectal Cancer. 2020.
- [16].Sillo TOBA, Morton DG, Middleton G. Mechanisms of immunogenicity in colorectal cancer. 2019. [DOI] [PMC free article] [PubMed]
- [17].Chae YK, Anker JF, Oh MS, Bais P, Namburi S, Agte S, et al. Mutations in DNA repair genes are associated with increased neoantigen burden and a distinct immunophenotype in lung squamous cell carcinoma. Scientific Reports. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bever KM, Le DT. DNA repair defects and implications for immunotherapy. Journal of Clinical Investigation. 2018;128:4236–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Boland CR, Lynch HT. The History of Lynch Syndrome. Fam Cancer. 2013;12:145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boland CR. Evolution of the Nomenclature for the Hereditary Colorectal Cancer Syndromes. Familial Cancer. 2005;4:211–8. [DOI] [PubMed] [Google Scholar]
- [21].Douglas JA, Gruber SB, Meister KA, Bonner J, Watson P, Krush AJ, et al. History and molecular genetics of Lynch syndrome in family G: a century later. Jama. 2005;294:2195–202. [DOI] [PubMed] [Google Scholar]
- [22].Smithers DW. Family Histories of 459 Patients with Cancer of the Breast. British Journal of Cancer. 1948;2:163–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Claus EB, Risch NJ, Thompson WD. AGE AT ONSET AS AN INDICATOR OF FAMILIAL RISK OF BREAST CANCER. American Journal of Epidemiology. 1990;131:961–72. [DOI] [PubMed] [Google Scholar]
- [24].Murthy P, Muggia F. Women’s cancers: how the discovery of BRCA genes is driving current concepts of cancer biology and therapeutics. Ecancermedicalscience 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. [DOI] [PubMed] [Google Scholar]
- [26].Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–92. [DOI] [PubMed] [Google Scholar]
- [27].Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998. p. 676–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stratton MR, Ford D, Neuhasen S, Seal S, Wooster R, Friedman LS, et al. Familial male breast cancer is not linked to the BRCA1 locus on chromosome 17q. Nature Genetics. 1994;7:103–7. [DOI] [PubMed] [Google Scholar]
- [29].FitzGerald MG MD, Krainer M, et al. Germ-line BRCA1 mutations in Jewish and non-Jewish women with early-onset breast cancer.: N Engl J Med; 1996. [DOI] [PubMed] [Google Scholar]
- [30].Lenoir G. Familial breast-ovarian cancer locus on chromosome 17q12-q23. The Lancet. 1991;338:82–3. [DOI] [PubMed] [Google Scholar]
- [31].Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. American journal of human genetics. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- [32].Albertsen HM, Smith SA, Mazoyer S, Fujimoto E, Stevens J, Williams B, et al. A physical map and candidate genes in the BRCA1 region on chromosome 17q12–21. Nature Genetics. 1994;7:472–9. [DOI] [PubMed] [Google Scholar]
- [33].Futreal P, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–2. [DOI] [PubMed] [Google Scholar]
- [34].Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317:2402. [DOI] [PubMed] [Google Scholar]
- [35].Klusty T, Weinmeyer R. Supreme Court to Myriad Genetics: Synthetic DNA is Patentable but Isolated Genes Are Not. AMA Journal of Ethics. 2020;17:849–53. [DOI] [PubMed] [Google Scholar]
- [36].Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genetics in Medicine. 2015;17:630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].NCI. Discovery – BRCA Connection to Breast and Ovarian Cancer - National Cancer Institute. 2014.
- [38].Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, Mckee MD, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. The Lancet Oncology. 2018;19:497–509. [DOI] [PubMed] [Google Scholar]
- [39].Gracia-Aznarez FJ, Fernandez V, Pita G, Peterlongo P, Dominguez O, De La Hoya M, et al. Whole Exome Sequencing Suggests Much of Non-BRCA1/BRCA2 Familial Breast Cancer Is Due to Moderate and Low Penetrance Susceptibility Alleles. PLoS ONE. 2013;8:e55681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Park DJ, Tao K, Le Calvez-Kelm F, Nguyen-Dumont T, Robinot N, Hammet F, et al. Rare Mutations in RINT1 Predispose Carriers to Breast and Lynch Syndrome-Spectrum Cancers. Cancer Discovery. 2014;4:804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Park DJ, Lesueur F, Nguyen-Dumont T, Pertesi M, Odefrey F, Hammet F, et al. Rare Mutations in XRCC2 Increase the Risk of Breast Cancer. The American Journal of Human Genetics. 2012;90:734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Basourakos SP, Li L, Aparicio AM, Corn PG, Kim J, Thompson TC. Combination Platinum-based and DNA Damage Response-targeting Cancer Therapy: Evolution and Future Directions. Curr Med Chem. 2017;24:1586–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kiiski JI, Pelttari LM, Khan S, Freysteinsdottir ES, Reynisdottir I, Hart SN, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proceedings of the National Academy of Sciences. 2014;111:15172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hilbers FS, Meijers CM, Laros JFJ, Van Galen M, Hoogerbrugge N, Vasen HFA, et al. Exome Sequencing of Germline DNA from Non-BRCA1/2 Familial Breast Cancer Cases Selected on the Basis of aCGH Tumor Profiling. PLoS ONE. 2013;8:e55734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stadler ZK, Schrader KA, Vijai J, Robson ME, Offit K. Cancer Genomics and Inherited Risk. Journal of Clinical Oncology. 2014;32:687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Castéra L, Krieger S, Rousselin A, Legros A, Baumann J-J, Bruet O, et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. European Journal of Human Genetics. 2014;22:1305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited Mutations in 17 Breast Cancer Susceptibility Genes Among a Large Triple-Negative Breast Cancer Cohort Unselected for Family History of Breast Cancer. Journal of Clinical Oncology. 2015;33:304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kurian AW, Hare EE, Mills MA, Kingham KE, Mcpherson L, Whittemore AS, et al. Clinical Evaluation of a Multiple-Gene Sequencing Panel for Hereditary Cancer Risk Assessment. Journal of Clinical Oncology. 2014;32:2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Laduca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genetics in Medicine. 2014;16:830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of mutations in individuals with breast cancer referred forBRCA1andBRCA2testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25–33. [DOI] [PubMed] [Google Scholar]
- [52].Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DYH, Tothill RW, et al. Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLoS Genetics. 2012;8:e1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pal T, Agnese D, Daly M, Spada AL, Litton J, Wick M, et al. Points to consider: is there evidence to support BRCA1/2 and other inherited breast cancer genetic testingfor all breast cancer patients? A statement of the American College of Medical Geneticsand Genomics (ACMG). Genetics in Medicine. 2019;22:681–5. [DOI] [PubMed] [Google Scholar]
- [54].Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. http://dxdoiorg/101056/NEJMoa1706450. 2017. [DOI] [PubMed]
- [55].Mandelker DZL, Kemel Y, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. 2018. [DOI] [PMC free article] [PubMed]
- [56].Zhang JWM, Wu G, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. 2015. [DOI] [PMC free article] [PubMed]
- [57].Patel AP, Wang M, Fahed AC, Mason-Suares H, Brockman D, Pelletier R, et al. Association of Rare Pathogenic DNA Variants for Familial Hypercholesterolemia, Hereditary Breast and Ovarian Cancer Syndrome, and Lynch Syndrome With Disease Risk in Adults According to Family History. JAMA Network Open. 2020;3:e203959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Samadder NJ, Riegert-Johnson D, Boardman L, Rhodes D, Wick M, Okuno S, et al. Comparison of Universal Genetic Testing vs Guideline-Directed Targeted Testing for Patients With Hereditary Cancer Syndrome. JAMA Oncology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Manickam K, Molecular and Human Genetics Department NCsH, Columbus, Ohio, Genomic Medicine Institute G, Danville, Pennsylvania, Buchanan AH, Genomic Medicine Institute G, Danville, Pennsylvania, Schwartz MLB, et al. Exome Sequencing–Based Screening for BRCA1/2 Expected Pathogenic Variants Among Adult Biobank Participants. JAMA Network Open. 2020;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Beitsch PD, Whitworth PW, Hughes K, Patel R, Rosen B, Compagnoni G, et al. Underdiagnosis of Hereditary Breast Cancer: Are Genetic Testing Guidelines a Tool or an Obstacle? Journal of Clinical Oncology. 2019;37:453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Manchanda R, Gaba F. Population Based Testing for Primary Prevention: A Systematic Review. Cancers. 2018;10:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].National Academies of Sciences E, and Medicine, Division HaM, Policy BoHS, Health RoGaP. Understanding Disparities in Access to Genomic Medicine: Proceedings of a Workshop. Washington (DC): National Academies Press (US); 2018. [PubMed] [Google Scholar]
- [63].Swink A, Nair A, Hoof P, Matthews A, Burden C, Johnson K, et al. Barriers to the utilization of genetic testing and genetic counseling in patients with suspected hereditary breast and ovarian cancers. Baylor University Medical Center Proceedings. 2019;32:340–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tan O, Shrestha R, Cunich M, Schofield DJ. Application of next-generation sequencing to improve cancer management: A review of the clinical effectiveness and cost-effectiveness. Clinical Genetics. 2018;93:533–44. [DOI] [PubMed] [Google Scholar]
- [65].Grzymski JJ, Elhanan G, Morales Rosado JA, Smith E, Schlauch KA, Read R, et al. Population genetic screening efficiently identifies carriers of autosomal dominant diseases. Nature Medicine. 2020;26:1235–9. [DOI] [PubMed] [Google Scholar]
- [66].Esterling L, Wijayatunge R, Brown K, Morris B, Hughes E, Pruss D, et al. Impact of a Cancer Gene Variant Reclassification Program Over a 20-Year Period. JCO Precision Oncology. 2020:944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Arora S, Huwe P, Sikder R, Shah M, Browne A, Lesh R, Nicolas E, Deshpande S,, Hall M, Dunbrack R, & Golemis E Functional analysis of rare variants in mismatch repair proteins augments results from computation-based predictive methods. Cancer Biology & Therapy; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mur P, García-Mulero S, Del Valle J, Magraner-Pardo L, Vidal A, Pineda M, et al. Role of POLE and POLD1 in familial cancer. Genetics in Medicine. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Arora S, Yan H, Cho I, Fan H-Y, Luo B, Gai X, et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology. 2015;149:1872–83.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nicolas E, Arora S, Zhou Y, Serebriiskii IG, Andrake MD, Handorf ED, et al. Systematic evaluation of underlying defects in DNA repair as an approach to case-only assessment of familial prostate cancer. Oncotarget. 2015;6:39614–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Luchini C, Lawlor RT, Milella M, Scarpa A. Molecular Tumor Boards in Clinical Practice. Trends Cancer. 2020;6:738–44. [DOI] [PubMed] [Google Scholar]
- [72].Patel M, Kato SM, Kurzrock R. Molecular Tumor Boards: Realizing Precision Oncology Therapy. Clin Pharmacol Ther. 2018;103:206–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kato S, Kim KH, Lim HJ, Boichard A, Nikanjam M, Weihe E, et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun. 2020;11:4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Křížová U, Petruželka L. Next generation sequencing and the molecular tumor board from the point of view of oncologists. Cesk Patol. 2021;57:144–6. [PubMed] [Google Scholar]
- [75].Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clinical Cancer Research. 2019;25:3753–8. [DOI] [PubMed] [Google Scholar]
- [76].Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine. 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Westdorp H, Kolders S, Hoogerbrugge N, De Vries IJM, Jongmans MCJ, Schreibelt G. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Letters. 2017;403:159–64. [DOI] [PubMed] [Google Scholar]
- [79].Pande M, Wei C, Chen J, Amos CI, Lynch PM, Lu KH, et al. Cancer spectrum in DNA mismatch repair gene mutation carriers: results from a hospital based Lynch syndrome registry. Familial Cancer. 2012;11:441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sinicrope FA. DNA mismatch repair and adjuvant chemotherapy in sporadic colon cancer. Nature Reviews Clinical Oncology. 2010;7:174–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nagle CM, O’Mara TA, Tan Y, Buchanan DD, Obermair A, Blomfield P, et al. Endometrial cancer risk and survival by tumor MMR status. Journal of Gynecologic Oncology. 2018;29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Abedalthagafi M. Constitutional mismatch repair-deficiency: current problems and emerging therapeutic strategies. Oncotarget. 2018;9:35458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shirts BH, Konnick EQ, Upham S, Walsh, Ranola JMO, Jacobson AL, et al. Using Somatic Mutations from Tumors to Classify Variants in Mismatch Repair Genes. The American Journal of Human Genetics. 2018;103:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pearlman R, Haraldsdottir S, De La Chapelle A, Jonasson JG, Liyanarachchi S, Frankel WL, et al. Clinical characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. Journal of Medical Genetics. 2019;56:462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncology. 2017;3:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lee V, Murphy A, Le DT, Diaz LA. Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. The Oncologist. 2016;21:1200–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Viale G, Trapani D, Curigliano G. Mismatch Repair Deficiency as a Predictive Biomarker for Immunotherapy Efficacy. BioMed Research International. 2017;2017:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bouffet E, Larouche V, Campbell BB, Merico D, De Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Journal of Clinical Oncology. 2016;34:2206–11. [DOI] [PubMed] [Google Scholar]
- [89].Oaknin A, Tinker AV, Gilbert L, Samouëlian V, Mathews C, Brown J, et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020;6:1766–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Oaknin A, Tinker AV, Gilbert L, Samouëlian V, Mathews C, Brown J, et al. Clinical activity and safety of the anti-PD-1 monoclonal antibody dostarlimab for patients with recurrent or advanced dMMR endometrial cancer. Future Oncol. 2021;17:3781–5. [DOI] [PubMed] [Google Scholar]
- [91].Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. New England Journal of Medicine. 2017;377:2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].FDA approves pembrolizumab for adults and children with TMB-H solid tumors. 2020.
- [95].Rayner E, Van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nature Reviews Cancer. 2016;16:71–81. [DOI] [PubMed] [Google Scholar]
- [96].Palles C, Cazier J-B, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genetics. 2013;45:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, Ter Haar N, et al. Prognostic Significance of POLE Proofreading Mutations in Endometrial Cancer. JNCI: Journal of the National Cancer Institute. 2015;107:dju402-dju. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Siraj AK, Bu R, Iqbal K, Parvathareddy SK, Masoodi T, Siraj N, et al. POLE and POLD1 germline exonuclease domain pathogenic variants, a rare event in colorectal cancer from the Middle East. Molecular Genetics & Genomic Medicine. 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Arora S, Xiu J, Sohal D, Lou E, Goldberg RM, Weinberg BA, et al. The landscape of POLE variants in colorectal and endometrial tumors: Correlation with microsatellite instability (MSI) and tumor mutation burden (TMB). Journal of clinical oncology :. 2020;38:e13538–e. [Google Scholar]
- [100].Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, De Borja R, et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell. 2017;171:1042–56.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Silberman R, Steiner DF, Lo AA, Gomez A, Zehnder JL, Chu G, et al. Complete and Prolonged Response to Immune Checkpoint Blockade in POLE-Mutated Colorectal Cancer. JCO Precision Oncology. 2019:1–5. [DOI] [PubMed] [Google Scholar]
- [102].Wang F, State Key Laboratory of Oncology in South China CICfCM, Sun Yat-sen University Cancer Center, Guangzhou, China, Zhao Q, State Key Laboratory of Oncology in South China CICfCM, Sun Yat-sen University Cancer Center, Guangzhou, China, Wang Y-N, State Key Laboratory of Oncology in South China CICfCM, Sun Yat-sen University Cancer Center, Guangzhou, China, et al. Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncology. 2020;5:1504–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wang Z, Zhao J, Wang G, Zhang F, Zhang Z, Zhang F, et al. Comutations in DNA Damage Response Pathways Serve as Potential Biomarkers for Immune Checkpoint Blockade. Cancer Res. 2018;78:6486–96. [DOI] [PubMed] [Google Scholar]
- [104].Li A, Yi M, Qin S, Chu Q, Luo S, Wu K. Prospects for combining immune checkpoint blockade with PARP inhibition. Journal of Hematology & Oncology. 2019;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Stewart RA, Pilie PG, Yap TA. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018;78:6717–25. [DOI] [PubMed] [Google Scholar]
- [106].Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019;9:646–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Samstein RM, Krishna C, Ma X, Pei X, Lee K-W, Makarov V, et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nature Cancer. 2020;1:1188–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Morales JC, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit Rev Eukaryot Gene Expr. 2014;24:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kyle S, Thomas HD, Mitchell J, Curtin NJ. Exploiting the Achilles Heel of Cancer: The Therapeutic Potential of poly(ADP-ribose) Polymerase Inhibitors in BRCA2-defective Cancer. The British journal of radiology. 2008;81 Spec No 1. [DOI] [PubMed] [Google Scholar]
- [110].Cannan WJ, Pederson DS. Mechanisms and Consequences of Double-strand DNA Break Formation in Chromatin. J Cell Physiol. 2016;231:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature. 2005;434. [DOI] [PubMed] [Google Scholar]
- [112].Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) Polymerase in Tumors From BRCA Mutation Carriers. The New England journal of medicine. 2009;361. [DOI] [PubMed] [Google Scholar]
- [113].Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. The Lancet. 2010;376:245–51. [DOI] [PubMed] [Google Scholar]
- [114].Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The Lancet Oncology. 2011;12:852–61. [DOI] [PubMed] [Google Scholar]
- [115].Wiggans AJ, Cass GK, Bryant A, Lawrie TA, Morrison J. Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer. Cochrane Database of Systematic Reviews. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. The Lancet. 2010;376:235–44. [DOI] [PubMed] [Google Scholar]
- [117].FDA. FDA approves olaparib for germline BRCA-mutated metastatic breast cancer 2018.
- [118].Robson ME, Tung N, Conte P, Im S-A, Senkus E, Xu B, et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Annals of Oncology. 2019;30:558–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Annals of Oncology. 2016;27:1013–9. [DOI] [PubMed] [Google Scholar]
- [120].Ledermann JA, Pujade-Lauraine E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther Adv Med Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. The New England journal of medicine. 2015;373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Javle M, Shacham-Shmueli E, Xiao L, Varadhachary G, Halpern N, Fogelman D, et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer With DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants. JAMA Oncology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Murai J, Huang S-yN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. The New England journal of medicine. 2016;375. [DOI] [PubMed] [Google Scholar]
- [125].FDA approves olaparib tablets for maintenance treatment in ovarian cancer. 2017.
- [126].FDA. FDA approves niraparib for first-line maintenance of advanced ovarian cancer. 2020.
- [127].González-Martín A, Pothuri B, Vergote I, Depont Christensen R, Graybill W, Mirza MR, et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. New England Journal of Medicine. 2019;381:2391–402. [DOI] [PubMed] [Google Scholar]
- [128].Fasching PA, Jackisch C, Rhiem K, Schneeweiss A, Klare P, Hanusch C, et al. GeparOLA: A randomized phase II trial to assess the efficacy of paclitaxel and olaparib in comparison to paclitaxel/carboplatin followed by epirubicin/cyclophosphamide as neoadjuvant chemotherapy in patients (pts) with HER2-negative early breast cancer (BC) and homologous recombination deficiency (HRD). Journal of clinical oncology :. 2019;37:506–. [Google Scholar]
- [129].Staff N. PARP Inhibitors Show Promise as Initial Treatment for Ovarian Cancer. NCI; 2019. [Google Scholar]
- [130].Morgan RD, Clamp AR, Evans DGR, Edmondson RJ, Jayson GC. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemotherapy and Pharmacology. 2018;81:647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Nakamura K, Aimono E, Tanishima S, Imai M, Nagatsuma AK, Hayashi H, et al. Olaparib Monotherapy for BRIP1-Mutated High-Grade Serous Endometrial Cancer. JCO Precision Oncology. 2020:283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Moore KM, Markham JM. Management of ovarian cancer associated with BRCA and other genetic mutations. UpToDate; 2021. [Google Scholar]
- [133].Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med. 2019;21:213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Hartman TR, Demidova EV, Lesh RW, Hoang L, Richardson M, Forman A, et al. Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer. Sci Rep. 2020;10:13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Carlo MI, Mukherjee S, Mandelker D, Vijai J, Kemel Y, Zhang L, et al. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients With Advanced Renal Cell Carcinoma. JAMA Oncol. 2018;4:1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Nassar AH, Abou Alaiwi S, Aldubayan SH, Moore N, Mouw KW, Kwiatkowski DJ, et al. Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma. Genetics in Medicine. 2020;22:709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lee EK, Matulonis UA. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers. 2020;12:2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Mylavarapu S, Das A, Roy M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Frontiers in Oncology. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Research and Treatment. 2014;147:401–5. [DOI] [PubMed] [Google Scholar]
- [140].Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. 2010. [DOI] [PMC free article] [PubMed]
- [141].Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. 2012. [DOI] [PMC free article] [PubMed]
- [142].Tung N, Arun B, Hacker MR, Hofstatter E, Toppmeyer DL, Isakoff SJ, et al. TBCRC 031: Randomized Phase II Study of Neoadjuvant Cisplatin Versus Doxorubicin-Cyclophosphamide in Germline BRCA Carriers With HER2-Negative Breast Cancer (the INFORM trial). Journal of Clinical Oncology. 2020;38:1539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Hahnen E, Lederer B, Hauke J, Loibl S, Kröber S, Schneeweiss A, et al. Germline Mutation Status, Pathological Complete Response, and Disease-Free Survival in Triple-Negative Breast Cancer. JAMA Oncology. 2017;3:1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Schwartz MD, Lerman C, Brogan B, Peshkin BN, Hughes Halbert C, Demarco T, et al. Impact ofBRCA1/BRCA2Counseling and Testing on Newly Diagnosed Breast Cancer Patients. Journal of Clinical Oncology. 2004;22:1823–9. [DOI] [PubMed] [Google Scholar]
- [145].Pierce LJ, Levin AM, Rebbeck TR, Ben-David MA, Friedman E, Solin LJ, et al. Ten-Year Multi-Institutional Results of Breast-Conserving Surgery and Radiotherapy in BRCA1/2-Associated Stage I/II Breast Cancer. Journal of Clinical Oncology. 2006;24:2437–43. [DOI] [PubMed] [Google Scholar]
- [146].Gorodnova TV, Sokolenko AP, Ivantsov AO, Iyevleva AG, Suspitsin EN, Aleksakhina SN, et al. High response rates to neoadjuvant platinum-based therapy in ovarian cancer patients carrying germ-line BRCA mutation. Cancer Letters. 2015;369:363–7. [DOI] [PubMed] [Google Scholar]
- [147].Pomerantz MM, Spisák S, Jia L, Cronin AM, Csabai I, Ledet E, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer. 2017;123:3532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Duan W, Gao L, Aguila B, Kalvala A, Otterson GA, Villalona-Calero MA. Fanconi Anemia Repair Pathway Dysfunction, a Potential Therapeutic Target in Lung Cancer. Frontiers in Oncology. 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362:1909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Popp I, Punekar M, Telford N, Stivaros S, Chandler K, Minnis M, et al. Fanconi anemia with sun-sensitivity caused by a Xeroderma pigmentosum-associated missense mutation in XPF. BMC Med Genet. 2018;19:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Liang CC, Li Z, Lopez-Martinez D, Nicholson WV, Venien-Bryan C, Cohn MA. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat Commun. 2016;7:12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Villalona-Calero MA, Duan W, Zhao W, Shilo K, Schaaf LJ, Thurmond J, et al. Veliparib Alone or in Combination with Mitomycin C in Patients with Solid Tumors With Functional Deficiency in Homologous Recombination Repair. J Natl Cancer Inst. 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Patterson-Fortin J, D’Andrea AD. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Research. 2020;80:4593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Brandalize APC, Schüler-Faccini L, Hoffmann J-S, Caleffi M, Cazaux C, Ashton-Prolla P. A DNA repair variant in POLQ (c.-1060A > G) is associated to hereditary breast cancer patients: a case–control study. BMC Cancer. 2014;14:850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–81. [DOI] [PubMed] [Google Scholar]
- [158].Bowden NA. Nucleotide excision repair: Why is it not used to predict response to platinum-based chemotherapy? Cancer Letters. 2014;346:163–71. [DOI] [PubMed] [Google Scholar]
- [159].Arora S, Kothandapani A, Tillison K, Kalman-Maltese V, Patrick SM. Downregulation of XPF–ERCC1 enhances cisplatin efficacy in cancer cells☆. DNA Repair. 2010;9:745–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Mehra R, Zhu F, Yang D-H, Cai KQ, Weaver J, Singh MK, et al. Quantification of Excision Repair Cross-Complementing Group 1 and Survival in p16-Negative Squamous Cell Head and Neck Cancers. Clinical Cancer Research. 2013;19:6633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Bepler G, Williams C, Schell MJ, Chen W, Zheng Z, Simon G, et al. Randomized International Phase III Trial of ERCC1 and RRM1 Expression–Based Chemotherapy Versus Gemcitabine/Carboplatin in Advanced Non–Small-Cell Lung Cancer. Journal of Clinical Oncology. 2013;31:2404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Chen S, Zhang J, Wang R, Luo X, Chen H. The platinum-based treatments for advanced non-small cell lung cancer, is low/negative ERCC1 expression better than high/positive ERCC1 expression? A meta-analysis. Lung Cancer. 2010;70:63–70. [DOI] [PubMed] [Google Scholar]
- [163].Liu J, Li H, Sun L, Feng X, Wang Z, Yuan Y, et al. The Differential Expression of Core Genes in Nucleotide Excision Repair Pathway Indicates Colorectal Carcinogenesis and Prognosis. BioMed Research International. 2018;2018:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Aiello MM, Solinas C, Santoni M, Battelli N, Restuccia N, Latteri F, et al. Excision Repair Cross Complementation Group 1 Single Nucleotide Polymorphisms and Nivolumab in Advanced Non-Small Cell Lung Cancer. Frontiers in Oncology. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Bolla M, Gonzalez D, Warde P, Dubois JB, Mirimanoff RO, Storme G, et al. Improved survival in patients with locally advanced prostate cancer treated with radiotherapy and goserelin. N Engl J Med. 1997;337:295–300. [DOI] [PubMed] [Google Scholar]
- [168].Zelefsky MJ, Reuter VE, Fuks Z, Scardino P, Shippy A. Influence of local tumor control on distant metastases and cancer related mortality after external beam radiotherapy for prostate cancer. J Urol. 2008;179:1368–73; discussion 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Jones CU, Hunt D, McGowan DG, Amin MB, Chetner MP, Bruner DW, et al. Radiotherapy and short-term androgen deprivation for localized prostate cancer. N Engl J Med. 2011;365:107–18. [DOI] [PubMed] [Google Scholar]
- [170].Nordstrom-O’Brien M, Van Der Luijt RB, Van Rooijen E, Van Den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, et al. Genetic analysis of von Hippel-Lindau disease. Human Mutation. 2010:n/a-n/a. [DOI] [PubMed] [Google Scholar]
- [171].Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A Randomized Trial of Bevacizumab, an Anti–Vascular Endothelial Growth Factor Antibody, for Metastatic Renal Cancer. New England Journal of Medicine. 2003;349:427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Maher ER, Neumann HP, Richard S. von Hippel–Lindau disease: A clinical and scientific review. European Journal of Human Genetics. 2011;19:617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Bindra RS, Glazer PM. Genetic instability and the tumor microenvironment: towards the concept of microenvironment-induced mutagenesis. Mutat Res. 2005;569:75–85. [DOI] [PubMed] [Google Scholar]
- [174].Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Scanlon SE, Glazer PM. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst). 2015;32:180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005;65:11597–604. [DOI] [PubMed] [Google Scholar]
- [177].Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, Roth ME, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24:8504–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Scanlon SE, Hegan DC, Sulkowski PL, Glazer PM. Suppression of homology-dependent DNA double-strand break repair induces PARP inhibitor sensitivity in VHL-deficient human renal cell carcinoma. Oncotarget. 2018;9:4647–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discovery. 2015;5:752–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Klaassen Z. ASCO GU 2020: Emerging Therapies in RCC: From PARP Inhibitors to Novel Immunotherapeutics. UroToday. 2020. [Google Scholar]
- [181].Carbone M, Harbour JW, Brugarolas J, Bononi A, Pagano I, Dey A, et al. Biological Mechanisms and Clinical Significance of BAP1 Mutations in Human Cancer. Cancer Discovery. 2020;10:1103–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Kobrinski DA, Yang H, Kittaneh M. BAP1: role in carcinogenesis and clinical implications. Translational Lung Cancer Research. 2020;9:S60–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].George TJ, DeRemer DL, Parekh HD, Lee J-H, Markham MJ, Daily KC, et al. Phase II trial of the PARP inhibitor, niraparib, in BAP1 and other DNA damage response (DDR) pathway deficient neoplasms including cholangiocarcinoma. Journal of clinical oncology :. 2020;38:TPS591–TPS. [Google Scholar]
- [184].Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72:106–16. [DOI] [PubMed] [Google Scholar]
- [185].Linehan WM, Rouault TA. Molecular Pathways: Fumarate Hydratase-Deficient Kidney Cancer--Targeting the Warburg Effect in Cancer. Clinical Cancer Research. 2013;19:3345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kerins MJ, Milligan J, Wohlschlegel JA, Ooi A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Science. 2018;109:2757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Sulkowski PL, Sundaram RK, Oeck S, Corso CD, Liu Y, Noorbakhsh S, et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nature Genetics. 2018;50:1086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Postow MA, Robson ME. Inherited gastrointestinal stromal tumor syndromes: mutations, clinical features, and therapeutic implications. Clinical Sarcoma Research. 2012;2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Nicolas E, Demidova EV, Iqbal W, Serebriiskii IG, Vlasenkova R, Ghatalia P, et al. Interaction of germline variants in a family with a history of early-onset clear cell renal cell carcinoma. Mol Genet Genomic Med. 2019;7:e556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Jambhekar A, Anastas JN, Shi Y. Histone Lysine Demethylase Inhibitors. Cold Spring Harbor Perspectives in Medicine. 2017;7:a026484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Role of H3K9 demethylases in DNA doublestrand break repair. Journal of Cancer Biology. 2020;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Wei S, Li C, Yin Z, Wen J, Meng H, Xue L, et al. Histone methylation in DNA repair and clinical practice: new findings during the past 5-years. J Cancer. 2018;9:2072–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Chen D, Jin C. Histone variants in environmental-stress-induced DNA damage repair. Mutation Research/Reviews in Mutation Research. 2019;780:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nature Reviews Cancer. 2016;16:20–33. [DOI] [PubMed] [Google Scholar]
- [196].Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environmental and Molecular Mutagenesis. 2017;58:235–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Ciccia A, Elledge SJ. The DNA Damage Response: Making It Safe to Play with Knives. Molecular Cell. 2010;40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews Molecular Cell Biology. 2017;18:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Santivasi WL, Xia F. Ionizing Radiation-Induced DNA Damage, Response, and Repair. Antioxidants & Redox Signaling. 2014;21:251–9. [DOI] [PubMed] [Google Scholar]
- [201].Wallace SS. Base excision repair: A critical player in many games. DNA Repair. 2014;19:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Zhu J, Su F, Mukherjee S, Mori E, Hu B, Asaithamby A. FANCD2 influences replication fork processes and genome stability in response to clustered DSBs. Cell Cycle. 2015;14:1809–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Freudenthal BD, Beard WA, Perera L, Shock DD, Kim T, Schlick T, et al. Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature. 2015;517:635–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Li Z, Pearlman AH, Hsieh P. DNA mismatch repair and the DNA damage response. DNA Repair. 2016;38:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Min A, Im S-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers. 2020;12:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Murai J, Huang S-YN, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Molecular Cancer Therapeutics. 2014;13:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Murai J, Huang S-YN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Research. 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Ottaviani D, Lecain M, Sheer D. The role of microhomology in genomic structural variation. Trends in Genetics. 2014;30:85–94. [DOI] [PubMed] [Google Scholar]
- [209].Dziadkowiec KN, Gąsiorowska E, Nowak-Markwitz E, Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Menopausal Review. 2016;4:215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].D’Agostino VG, Minoprio A, Torreri P, Marinoni I, Bossa C, Petrucci TC, et al. Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA repair. 2010;9:700–7. [DOI] [PubMed] [Google Scholar]
- [211].Eide L, Luna L, Gustad EC, Henderson PT, Essigmann JM, Demple B, et al. Human Endonuclease III Acts Preferentially on DNA Damage Opposite Guanine Residues in DNA†. Biochemistry. 2001;40:6653–9. [DOI] [PubMed] [Google Scholar]
- [212].Wood R, Lowery M. Human DNA Repair Genes.
- [213].Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, et al. Activation of mammalian Chk1 during DNA replication arrest. Journal of Cell Biology. 2001;154:913–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27:3977–85. [DOI] [PubMed] [Google Scholar]
- [215].Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, et al. Three DNA Polymerases, Recruited by Different Mechanisms, Carry Out NER Repair Synthesis in Human Cells. Molecular Cell. 2010;37:714–27. [DOI] [PubMed] [Google Scholar]
- [216].Li H, Xie B, Zhou Y, Rahmeh A, Trusa S, Zhang S, et al. Functional Roles of p12, the Fourth Subunit of Human DNA Polymerase δ. Journal of Biological Chemistry. 2006;281:14748–55. [DOI] [PubMed] [Google Scholar]
- [217].Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Molecular Cell. 2006;22:719–29. [DOI] [PubMed] [Google Scholar]
- [218].Morris JR, Solomon E. BRCA1 : BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Human Molecular Genetics. 2004;13:807–17. [DOI] [PubMed] [Google Scholar]
- [219].Hiraike H, Wada-Hiraike O, Nakagawa S, Koyama S, Miyamoto Y, Sone K, et al. Identification of DBC1 as a transcriptional repressor for BRCA1. British Journal of Cancer. 2010;102:1061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–5. [DOI] [PubMed] [Google Scholar]
- [221].Trujillo KM, Yuan S-SF, Lee EY-HP, Sung P. Nuclease Activities in a Complex of Human Recombination and DNA Repair Factors Rad50, Mre11, and p95. Journal of Biological Chemistry. 1998;273:21447–50. [DOI] [PubMed] [Google Scholar]
- [222].Rodrigue A, Coulombe Y, Jacquet K, Gagné J-P, Roques C, Gobeil S, et al. The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. Journal of Cell Science. 2013;126:348–59. [DOI] [PubMed] [Google Scholar]
- [223].Miyagawa K. A role for RAD54B in homologous recombination in human cells. The EMBO Journal. 2002;21:175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Kansikas M, Kariola R, Nyström M. Verification of the three-step model in assessing the pathogenicity of mismatch repair gene variants. Human Mutation. 2011;32:107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Friboulet L, Postel-Vinay S, Sourisseau T, Adam J, Stoclin A, Ponsonnailles F, et al. ERCC1 function in nuclear excision and interstrand crosslink repair pathways is mediated exclusively by the ERCC1–202 isoform. Cell Cycle. 2013;12:3298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, Elledge SJ, et al. Mammalian BTBD12/SLX4 Assembles A Holliday Junction Resolvase and Is Required for DNA Repair. Cell. 2009;138:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Walsh CS, Ogawa S, Karahashi H, Scoles DR, Pavelka JC, Tran H, et al. ERCC5 Is a Novel Biomarker of Ovarian Cancer Prognosis. Journal of Clinical Oncology. 2008;26:2952–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.