

Abstract

MASP-1 and MASP-2 are key activator proteases of the complement lectin pathway. The first specific mannose-binding lectin-associated serine protease (MASP) inhibitors had been developed from the 14-amino-acid sunflower trypsin inhibitor (SFTI) peptide by phage display, yielding SFTI-based MASP inhibitors, SFMIs. Here, we present the crystal structure of the MASP-1/SFMI1 complex that we analyzed in comparison to other existing MASP-1/2 structures. Rigidified backbone structure has long been accepted as a structural prerequisite for peptide inhibitors of proteases. We found that a hydrophobic cluster organized around the P2 Thr residue is essential for the structural stability of wild-type SFTI. We also found that the same P2 Thr prevents binding of the rigid SFTI-like peptides to the substrate-binding cleft of both MASPs as the cleft is partially blocked by large gatekeeper enzyme loops. Directed evolution removed this obstacle by replacing the P2 Thr with a Ser, providing the SFMIs with high-degree structural plasticity, which proved to be essential for MASP inhibition. To gain more insight into the structural criteria for SFMI-based MASP-2 inhibition, we systematically modified MASP-2-specific SFMI2 by capping its two termini and by replacing its disulfide bridge with varying length thioether linkers. By doing so, we also aimed to generate a versatile scaffold that is resistant to reducing environment and has increased stability in exopeptidase-containing biological environments. We found that the reduction-resistant disulfide-substituted l-2,3-diaminopropionic acid (Dap) variant possessed near-native potency. As MASP-2 is involved in the life-threatening thrombosis in COVID-19 patients, our synthetic, selective MASP-2 inhibitors could be relevant coronavirus drug candidates.
Introduction
Practically, all physiological and pathological processes manifest through transient, specific protein–protein interactions (PPIs). Because of their utmost importance, revealing the fundamental rules that govern the strength, persistence time, and specificity of PPIs has been a central topic of molecular biology. The better we understand these interactions, the more opportunity we have to control them, allowing the design of highly selective, mechanism-based therapeutics.
One of the classic PPI model systems is the interaction between relatively simple serine proteases of the digestive system, for example, trypsin and chymotrypsin and their substrate-like, reversible, canonical inhibitors. A general view emerged that upon complex formation, both the enzyme and the inhibitor retain their essentially rigid structure.1−3 Such a binding mechanism is associated with a minimal conformational entropy decrease-related energetic cost, which then maximizes the binding affinity. Moreover, a rigid inhibitor can better avoid or withstand proteolytic cleavage without losing its native structure.
Canonical serine protease inhibitors emerged at least 18 times through convergent evolution. Each independent family has a distinct, unrelated overall fold, but all carry a loop occupying the substrate-binding cleft of the target protease in invariant, that is, canonical conformation.2,3 A general model for their action was introduced by Michael Laskowski, referred to as the standard mechanism.1 According to this model, the canonical inhibitory loop occupies the substrate-binding cleft of the target protease that catalyzes the slow hydrolysis of the scissile bond between the P1–P1′ residues (Schechter–Berger nomenclature, see Figure 1).4 The cleavage itself does not lead to complex dissociation; the two new termini created on the canonical loop are held in close proximity by loop-stabilizing noncovalent inter- and intramolecular interactions and usually also by intramolecular disulfide bridge(s). Since the enzyme catalyzes peptide bond religation as well, this leads to a thermodynamic equilibrium of cleaved and intact forms, both securely blocking the substrate-binding site.5,6
Figure 1.

Applying the traditional Schechter–Berger nomenclature for identifying the 14 residue positions on the SFTI-family substrate-like canonical protease inhibitors, SFTI, SFMI1, and SFMI2. The structure of acyclic SFTI (PDB-code: 1JBN(27)) is used as an example. P3 and P6′ carry disulfide-forming cysteines, while P3′ is a conserved cisPro, which is essential for the formation of the β-hairpin turn. The scissile bond is located between the P1 and P1′ residues. The substrate/inhibitor-recognition sites are numbered accordingly. For example, the main specificity-determinant P1 residue becomes buried in the S1 pocket of the protease.
As mentioned earlier, the canonical loop is supported by the rest of the inhibitor, that is, the “scaffold”, through various intramolecular interactions. The interscaffolding additivity model originally proposed that while the canonical loop dictates the affinity and specificity of the inhibitor, the scaffold has a mere nonspecific structure-stabilizing contribution.7,8 We have recently demonstrated—by swapping loops between unrelated scaffolds—that the loop and the scaffold form a single functional unit brought about by co-evolution,9 also implying that when developing a novel inhibitor against a protease, the choice of the scaffold can strongly influence the potency and selectivity of the resulting compound.
Most trypsin-like proteases are not simple broad specificity digestive enzymes but are highly selective regulators playing vital roles in numerous life processes including blood coagulation, fibrinolysis, or the complement system, which is a powerful effector arm of the innate immunity.10−12 It can recognize, label, and eliminate invading pathogenic microorganisms and dangerously altered self-structures (e.g., apoptotic and necrotic cells, cancer cells). There are three proteolytic complement activating pathways: the classical pathway, the lectin pathway, and the alternative pathway. The lectin pathway provides first-line defense against infections. It relies on pattern recognition molecules (PRMs) such as mannose-binding lectin, ficolins, and other collectins that circulate in complex with associated trypsin-like serine proteases called mannose-binding lectin-associated serine proteases, MASPs.
MASP-1 and MASP-2 are responsible for lectin pathway activation, while MASP-3 has a central role in alternative pathway activation.13 The complement system is indispensable for maintaining immune homeostasis, but its uncontrolled regulation can cause serious self-tissue damage.14−18 Very recently, it was also shown that the lectin pathway, specifically MASP-2, plays a central pathologic role in life-threatening widespread thrombotic microangiopathy developing in COVID-19 patients.19−24
Unlike trypsin, highly specific MASP-1 and MASP-2 carry large gatekeeper loops that partially cover their substrate-binding cleft. MASP-specific inhibitors had been developed through directed evolution starting from the smallest natural canonical trypsin inhibitor, the 14-amino acid sunflower trypsin inhibitor (SFTI), which does not inhibit the MASPs.25−29 Seven positions, 2, 4–7, 10, and 12, corresponding to P4, P2–P2′, P5′, and P7′ of phage-displayed SFTI, were evolved yielding SFMI1 and SFMI2 (Figure 1). SFMI1 turned out to be a potent 65 nM affinity MASP-1 (KI) inhibitor, which also inhibited MASP-2 with micromolar affinity, while SFMI2 proved to be the very first monospecific MASP-2 inhibitor, inhibiting the enzyme with a 180 nM affinity.25 SFMI1 and SFMI2 differ from SFTI at 5–6 out of the 14 positions, while they differ from each other at only 3 positions, which, importantly, do not include the energetically most important P1, P2, and P1′ residues or the structure-stabilizing disulfide bridge. In the case of the ultrasmall SFTI and SFMI peptides, the canonical loop and the inhibitory scaffold have almost the same number of residues, and inhibitory and structure-stabilizing functions of these regions cannot be clearly separated. In line with this, designing potent and selective SFTI-based protease inhibitors had been usually carried out by simultaneously optimizing direct enzyme contacts and maintaining structural integrity of the inhibitor.30
In this work, we aimed to map general principles underlying small-peptide-based inhibition of serine proteases having structurally restricted substrate-binding clefts. The surprising finding of our study was that directed evolution managed to meet the strict selection requirement for binding to the partially blocked substrate-binding cleft of MASP-1 or MASP-2 by liberating the rigid SFTI-like inhibitor conformation, which enabled an induced-fit binding mechanism.
While we successfully solved the crystal structure of the MASP-1/SFMI1 complex, we were unable to crystallize the MASP-2/SFMI2 complex. To promote crystallization by slightly altering the complex, we introduced moderate modifications to SFMI2 by capping its N- and C-termini and by systematically replacing the disulfide bridge with increasing length non-natural linkers. In the latter case, we also aimed to test whether upon disulfide substitution, the SFMI2 architecture could be preserved enough to maintain MASP-2 inhibiting potency. If so, then this method might be suitable for developing binders against intracellular or industrial targets existing in a reductive environment. Although this approach did not yield crystallizable complexes with MASP-2, these studies provided important new insights into the nature of interactions between MASP enzymes and their peptide ligands. Most importantly, we found that only those inhibitor variants remained functional that mimicked the evolved structural adaptability of SFMI2.
Results and Discussion
Crystal Structure of the MASP-1/SFMI1 Complex
To analyze the interactions formed between MASPs and their respective SFMI inhibitors, we set out to crystallize MASP-1/SFMI1 and MASP-2/SFMI2 complexes. While the MASP-2/SFMI2 complex could not be crystallized, we were able to solve the structure of the MASP-1/SFMI1 complex and refined it to 2.4 Å resolution.
The structure shows the inhibitor bound according to the canonical binding mode, with its P1 residue (Arg5) immersed in the S1 pocket of the enzyme (Figure 2). The conformation and binding topology of the inhibitor are very similar to both the trypsin-SFTI inhibitor complex (PDB ID: 1SFI(31)), with a 0.89 Å inhibitor-backbone root-mean-square-deviation (rmsd) (after superimposing the enzyme structures), and the solution structure of both the natural cyclic SFTI inhibitor and its acyclic variant (PDB IDs: 1JBL, 1JBN(32)), with backbone rmsd values of 1.25 and 1.31 Å, respectively (Figure 2a). The electron density for the 2 C-terminal inhibitor residues could not be observed, indicating their high flexibility (Figure 2d).
Figure 2.

Crystal structure of the MASP-1/SFMI1 complex shows that the inhibitor binds via canonical interactions. (a) Comparison of the conformation of the inhibitors in the trypsin (yellow)/SFTI (blue) and MASP-1 (green)/SFMI1 (orange) complexes after superimposing the enzyme structures reveals similar positioning of the core segment of the inhibitors. Active site enzyme residues and the P1 residue of the inhibitors are shown as sticks. (b) Comparison of loop conformations in uncomplexed MASP-1 (purple) in the MASP-1/SGMI-1 complex (yellow and dark blue, respectively) and in the MASP-1/SFMI1 complex (green and orange). (c) Intra- and intermolecular hydrogen bonds stabilizing the inhibitor in the crystal structure of the MASP-1/SFMI1 complex (listed in Supporting Information Table 2). (d) 2Fo – Fc type electron density map contoured at a 1.0σ electron density level in the SFMI1-binding region.
The canonical binding mode of the inhibitor33 is stabilized by numerous intra- and intermolecular hydrogen bonds (Figure 2c and Supporting Information Table 2). The side chain of the P1 arginine of SFMI1 forms a salt bridge with the side chain of Asp640 (Asp189 by chymotrypsinogen numbering) positioned at the bottom of the S1 pocket and forms two other hydrogen bonds with main chain oxygens. The carbonyl oxygen of P1 is bound to the oxyanion hole by Gly644 (Gly193), Asp645 (Asp194), and Ser646 (Ser195), while the main chain amide forms a hydrogen bond with the carbonyl oxygen of Ser667. The canonical loop of the inhibitor forms a short antiparallel β-sheet with residues 668–671 of MASP-1. The N-terminus of the inhibitor forms salt bridges with Asp670 and Asp671 of MASP-1.
The conformation of the enzyme remains practically unchanged by inhibitor binding (the backbone rmsd of the free and inhibitor-bound form is 0.55 Å). The only significant difference between the complexed and the uncomplexed (PDB ID: 3GOV) forms is in the conformation of the flexible and partially disordered loop B (489–515; enzyme loops are labeled as defined in Perona and Craik34). Even this appears to arise from different crystal contacts.
The structure of MASP-1 in complex with the 14-amino-acid SFMI1 peptide is also similar to that in complex with our larger, 35-amino-acid, second-generation protein inhibitor, SGMI1 (PDB ID: 4DJZ(26)). The only notable difference is that while in the MASP-1/SFMI1 complex the position of the gatekeeper loop 3 (610–624) is nearly identical to that of the free enzyme (PDB ID: 3GOV(35)), in the MASP-1/SGMI1 complex, the position of this segment is shifted (Figure 2b).
As the MASP-2/SFMI2 complex could not be crystallized, we aimed to introduce moderate modifications to SFMI2 that might promote crystallization through slightly altered binding to the enzyme. We chose alterations that, at the same time, could potentially increase the metabolic stability of the inhibitors: capping the termini could make them exopeptidase-resistant, while replacing the disulfide bridge with non-natural linkers provides resistance against reduction. Although we could not crystallize MASP-2 in complex with any of these variants, their analysis provided important insights into the nature of the MASP enzyme–peptide ligand interactions.
Synthesis of Modified Variants of SFMI2 and Their Inhibitory Efficiency
The N- and/or C-terminal functional groups of SFMI2 were protected by acetylation, amidation, or both (Ac-SFMI2, SFMI2-NH2, or SFMI2cap, respectively). The highest affinity toward the MASP-2 enzyme was observed in the case of SFMI2cap (Table 1), where both termini of the peptide were blocked.
Table 1. Inhibition of MASP-2 by SFMI2 and Its Terminally Capped and Thioether-Linked Derivatives.
| variant | KI* (μM) | KI (μM) |
|---|---|---|
| SFMI2 | 0.30 ± 0.01 | 0.21 |
| Ac-SFMI2 | 0.20 ± 0.01 | 0.14 |
| SFMI2-NH2 | 0.17 ± 0.00 | 0.12 |
| SFMI2cap | 0.10 ± 0.01 | 0.07 |
| SFMI2cap-Dap fraction 1a | 0.69 ± 0.05 | 0.49 |
| SFMI2cap-Dap fraction 2a | 6.35 ± 0.44 | 4.52 |
| SFMI2cap-Dab | 57 ± 2 | 41 |
| SFMI2cap-Orn | 84 ± 19 | 60 |
| SFMI2cap-Lys | 58 ± 1 | 41 |
| SFMI2cap-Agl | 827 ± 129 | 589 |
For SFMI2cap-Dap, two isomers could be isolated (see Supporting Information Results, Characterization of SFMI2 and derived peptides by HPLC-MS). KI* stands for apparent inhibitory constant values with standard error of measurement (SEM) values indicated. The KI values are called apparent because they are biased by the substrate competing with the inhibitor for enzyme binding. The genuine KI is calculated as follows: KI = KI*/(1 + 1/KM), where KM is the Michaelis constant. Details of experimental procedures are provided in the Supporting Information.
The structure of SFTI and SFMIs is stabilized with a conserved disulfide bridge between Cys3 and Cys11 (P3 and P6′). It was shown that the removal of the disulfide bridge from SFTI and its acyclic variant greatly reduces its inhibitory activity and significantly increases its rate of proteolytic degradation.5,36,37 Disulfide bonds are sensitive to reducing agents; therefore, their replacement by mimetics could increase the pharmaceutical applicability of these bioactive peptides. Several disulfide bond mimetics were developed and tested in various model systems, some of which retained biological activity of the parent compound.38−41 Here, we replaced the disulfide bridge of SFMI2 with thioether linkers of various lengths (as listed in Figure 3). All thioether-linked inhibitors were prepared with both termini protected.
Figure 3.

Sequences of the inhibitors appearing in this study (with the linkers explicitly formulated in italics) and one example showing an insertion of five extra atoms (as compared to SFMI2) when introducing a thioether linker between Lys3 and Cys11.
To create the thioether linker, Cys3 was replaced with Lys and its side chain amino group was chloroacetylated. Chemoselective thioether ligation was carried out between the chloroacetylated Lys and the thiol group of Cys11. Because this long thioether bridge significantly lowered the binding affinity, we gradually shortened the bridge by incorporating diamino acids with incrementally shorter side chains. The linker contained 5, 4, 3, 2, or only 1 extra atom in the case of Lys-, Orn-, Dab-, Dap-, and Agl-containing peptides, respectively, with the last one creating a (−NH–CO–CH2–S–CH2−) bridge.
As listed in Table 1, both N-terminal and C-terminal blocking of SFMI2 increased MASP-2 inhibitory potency, and their effects were additive in the SFMI2cap variant, which is threefold more potent than SFMI2. While four out of the five thioether linkers were detrimental, the Dap-derived thioether linker provided a near-original inhibitor potency.
Serum Stability of SFMI2 Variants Assessed Through Their Lectin Pathway Inhibiting Potency
In order to test whether capping the termini and replacing the disulfide affect serum stability of the inhibitors, we tested six SFMI2 variants in a serum assay. The four SFMI2 variants differing only in the presence or absence of caps at the two termini as well as SFMI2cap-Dap (fraction 1) and SFMI2cap-Lys were analyzed. Serial dilutions of these inhibitors were incubated in 100-fold diluted human serum for 30 min and then loaded on ELISA plates containing immobilized mannan, which triggers the lectin complement pathway. The extent of lectin pathway activation was monitored through the deposition of C3 fragments (Figure 4). The IC50 values for SFMI2, Ac-SFMI2, SFMI2-NH2, and SFMI2cap were 819, 624, 473, and 303 nM, respectively, while SFMI2cap-Dap had an IC50 value of 3.4 μM and SFMI2cap-Lys had an IC50 value of 18.5 μM. The KI and the IC50 values reveal the same ranks in the in vitro and the serum test, suggesting that at least at this low serum concentration and half an hour incubation, the chemical modifications did not affect serum stabilities of the compounds.
Figure 4.

MASP-2 inhibiting potencies of six SFMI2 variants assessed in human serum. Serum stability of the indicated SFMI2 variants was tested in lectin-pathway activation inhibition ELISA. The IC50 rank of the variants is the same as their KI value rank in Table 1, suggesting that neither capping of the termini nor replacing the disulfide with the thioether linker affected serum stability of the peptides.
Solution Structures of SFMI2 and Its Variants Assessed by Far-UV ECD
Far-UV electronic circular dichroism (ECD) spectra of acyclic SFTI21 (measured as a reference) closely resembled that of the wild-type, cyclic SFTI,42 which has a stable β-hairpin structure32 (Figure 5). This is consistent with the observation that acyclic and cyclic SFTI have highly similar stable β-hairpin structures.32
Figure 5.

ECD spectra of MASP-2 inhibitors and an acyclic variant of SFTI measured at 25 °C.
ECD spectra of SFMI2 and its variants contain a negative minimum at around 200 nm, indicating high overall flexibility and a lack of a stable β-hairpin solution structure. A small shoulder at 220 nm and the positive ellipticity at wavelengths shorter than 190 nm indicate the presence of some kind of an ordered structure, presumably due to the disulfide/thioether linker, which prevents complete unfolding of the peptides. Indeed, upon incubating SFMI2cap with 0.5 mM (tris(2-carboxyethyl)phosphine) (TCEP) at 37 °C, these spectral features disappeared, verifying the crucial structural role of the disulfide. In contrast, as the thioether-containing linker in SFMI2cap-Dap cannot be reduced, TCEP treatment did not alter the corresponding spectrum (Supporting Information Figure 8).
MD Simulations of the Uncomplexed Inhibitors
Our modeling methodology was tested using the NMR structure of uncomplexed acyclic SFTI having a stable β-hairpin solution structure (PDB ID:1JBN;32). When starting from its NMR-derived conformation, free SFTI retained its folded, β-pleated structure for the entirety of the 4000 ns simulation time and regained it when we started from the common starting structure we used for all inhibitors studied here (Figure 6, Supporting Information Figure 4). In sharp contrast, SFMI1, SFMI2, and the capped and thioether-bridged SFMI2 variants behaved entirely differently: instead of retaining their modeled β-hairpin starting conformation during molecular dynamics (MD) simulations, these peptides lost this structure within a few hundred nanoseconds. The hairpin did not reappear during the rest of the simulations, and the structures relaxed to an ensemble of conformers. The only exceptions were the two isomers of SFMI2cap-Dap, which also had relaxed conformations—but mostly sampled the β-hairpin-like structure (Figure 6).
Figure 6.

(a) Conformers of the last 600 ns of the equilibrium trajectories of the studied inhibitors [mid-structures of the backbone clusters (using a 1.5 Å cutoff) representing 80% of all snapshots]. P4–P4′ (residues 2–9) were superimposed, and linker residues are shown explicitly (without nonpolar hydrogens). (b,c) Secondary structure timelines of the entire trajectories of SFMI2cap (b) and SFMI2cap-transDap (c), showing a slow fluctuation between unstructured conformations and the infrequently appearing β-stranded conformers in the case of SFMI2cap (b) and the predominantly β-stranded conformation of SFMI2cap-transDap (c). Secondary structure elements are represented by the following color codes: turn—green, extended strand—yellow, isolated β-bridge—dark brown, α-helix—magenta, 310 helix—blue, π helix—red, coil—white.
We identified a particular structural element as a likely source of the observed difference in conformational dynamics. Wild-type and acyclic SFTI contains a hydrophobic, tripartite, β-hairpin-stabilizing Thr4-Phe12-Ile10 interaction cluster, which is missing from all SFMIs. In both SFMI1 and SFMI2, the bulky Phe12 (P7′ according to Schechter–Berger nomenclature) is replaced with an Ile, and the methyl group containing Thr4 (the P2 residue) is replaced with a Ser. In addition, instead of Ile10, SFMI2 contains a shorter Val. For acyclic SFTI, it had been shown that the interaction of the Thr4 methyl group with the hydrophobic residue at position 10 contributes to the overall rigidity of the peptide.43 Moreover, it was found that even in the case of the well-fixed backbone of cyclic SFTI, certain position 10 residues introduce conformational heterogeneity.44,45 Based on these findings, we argued that the alterations present in the SFMIs could entirely eliminate the stabilizing effect of the hydrophobic cluster.
To test this notion, we carried out simulations with the Ser4Thr and Ile12Phe single mutants and the Ser4Thr/Ile12Phe double mutant of SFMI1 and SFMI2. While the double mutants retained the β-hairpin structure during the simulations, the Ile12Phe single mutants relaxed to an ensemble of structures similar to the original SFMI inhibitors (Supporting Information Figure 5). In the case of the Ser4Thr single mutants, the β-hairpin structure reappeared but only for a few hundred nanoseconds, after which the peptides lost their ordered structure again.
These results strongly suggest that directed evolution-driven loss of the hydrophobic cluster eliminated the ordered solution structure of the phage-evolved SFMIs. It also shows that while Thr4 and Phe12 alone provide only small or marginal stabilization, together they establish a stable β-hairpin structure.
MD Simulations of Various MASP/SFMI Complexes
We investigated the selection criteria of the MASP enzymes by carrying out MD simulations of the following systems: MASP-1, MASP-1/SFMI1, MASP-1/SFMI2, MASP-2, MASP-2/SFMI1, MASP-2/SFMI2, and MASP-2/SFMI2cap. Based on the measured inhibitory constants (Table 1), the affinity is the highest for the MASP-1/SFMI1 complex and the lowest for the MASP-1/SFMI2 complex. Using the calculated equilibrium ensembles of the complexes and the corresponding free enzymes and free ligands, binding enthalpies were calculated for each complex. These were in agreement with the experimentally established ranking of “binding free energies” expressed as standard Gibbs energy change (Figure 7). Moreover, the MD-derived structural ensemble of the MASP-1/SFMI1 complex could be fitted to the crystal structure with an overall 1.30 Å rmsd for the determined Gly1–Cys11 segment of SFMI1 and for MASP-1 residues 449–696 omitting loop B (Figure 7).
Figure 7.

MD simulation-based calculated binding energies of modeled MASP/SFMI complexes as a function of experimentally determined inhibitory constant (KI)-based binding free-energy values calculated as RT ln(KI) for the enzyme–inhibitor interactions.
Structural Model of the Highest-Affinity MASP-1/SFMI1 Complex
Although we experimentally determined the crystal structure of the MASP-1/SFMI1 complex, we also calculated its structure partly to verify that the modeling methodology we apply for all other complexes is able to reproduce the experimental results, and also because some segments of the crystal structure remained unresolved (Figure 8a). To be able to compare H-bonding patterns and binding conformations among all complexes, we had to augment the experimentally determined structure with the missing residues and atom groups and allow the solvent-phase relaxation of the overall structure to proceed similarly to all other models we calculated and aimed to compare it to.
Figure 8.

Comparison of the crystal structure and the mid-structure of the most populated cluster from the MD simulation of the MASP-1/SFMI1 complex (a). MD derived structures of the MASP-1/SFMI (b) and MASP-2/SFMI (c) complexes. The mid-structure of the most populated cluster of each simulation is shown, with the N- and C-termini, the disulfide bridge, the P1 and P4 residues of the inhibitor, and Leu621 of MASP-1 and Arg609of MASP-2 shown in sticks.
In the highest-affinity MASP-1/SFMI1complex (Figure 8b), an average of 14.1 H-bonds are formed between the enzyme and the inhibitor during the simulation, in good agreement with the 14 intermolecular H-bonds found in the crystal structure. Binding of the inhibitor does not perturb the enzyme structure: the uncomplexed and complexed forms (derived by MD) can be fitted for the core structure with an rmsd of 0.82 Å and along the full backbone with an rmsd of 1.41 Å (see the Supporting Information). The inhibitor adopts a hairpin conformation, stabilized by an average of five intramolecular H-bonds, while the P4 residue, Ile2, is anchored in the hydrophobic pocket created between the gatekeeper loop 3 (618–628) of the enzyme and the outer wall of the S1 pocket. The P1 residue is anchored by an average of 6.8 H-bonds within the S1 pocket. Most of the fingerprint H-bond motif of SFTI-type inhibitors is present in the majority of the structures of the equilibrium trajectory (Supporting Information Table 4).
Structural Model of the Lowest-Affinity MASP-1/SFMI2 Complex
In the case of the computed model of the unmeasurably weak MASP-1/SFMI2 complex (Figure 8b), an average of 11.8 H-bonds is formed between the inhibitor and the enzyme, with 6.3 of these between the P1 residue and the enzyme. Both values are the lowest ones within the five studied complexes. Tyr2 (P4) of the inhibitor docks into a hydrophobic pocket created by Trp668, Leu621, Tyr618, Phe549, and the aliphatic chain of Lys623 (the S4 subsite). Steric clash with Leu621 is avoided by restructuring of the entire loop 3 (618–628) of the gatekeeper region of MASP-1. The structure of the complex can be fitted to that of the uncomplexed form with a core structure rmsd of 0.92 Å and a full backbone rmsd of 1.76 Å. As explained below, we found that potent inhibitors do not perturb the structure of the MASP enzymes upon complexation. Therefore, the observed steric incompatibility appears to be the major reason why SFMI2 does not inhibit MASP-1.
Structural Model of the Micromolar Affinity MASP-2/SFMI1 Complex
In the MASP-2/SFMI1 complex (Figure 8c), an average of 12.3 H-bonds is formed between the enzyme and the inhibitor, and the internal H-bond system of the inhibitor is also extensive (having an average of 8.8 H-bonds, see Supporting Information Table 4). In 67% of the snapshots, the N- and C-termini of the inhibitor form a H-bond, mimicking the circular backbone of SFTI-type inhibitors. The guanidine moiety of the P1 Arg residue (Arg5) is bound through an average of 3.4 H-bonds by the enzyme, while its main-chain amino and carbonyl groups are also bound by 3.4 H-bonds. Ile2 is immersed into the hydrophobic S4 pocket lined by Ile12 of the inhibitor and by Phe529, Tyr602, Tyr607, Pro608, and Trp655 of the enzyme; the N-terminal segment of the inhibitor lines up close to the enzyme surface. This closeness initiates interactions by pulling a loop (656–666) (loop 2) toward the inhibitor, resulting in the formation of two main-chain H-bonds between Gly656 of the enzyme and the P3 Cys of the inhibitor. However, the very same loop forms one of the walls of the S1 cavity, which is widened by this subtle rearrangement, resulting in the loss of some contacts within the cavity. Apparently, these adjustments make the MASP-2/SFMI1 interaction about sixfold weaker than the phage-evolved 180 nM MASP-2/SFMI2 interaction.
Structural Model of the High-Nanomolar Affinity MASP-2/SFMI2 Complex
SFMI2 binds to MASP-2 in a similar mode as SFMI1 (Figure 8c) (13.3 H-bonds form between the enzyme and inhibitor, 6.3 within the inhibitor, distributed in the same fashion as in the case of SFMI1), but it does not line up quite as close to the primary contact surface of the enzyme as SFMI1. This is due to the P4 residue Tyr2, which—instead of docking into the hydrophobic S4 pocket—pushes the N-terminal segment of the inhibitor a bit farther than Ile2 of SFMI1, while forming a H-bond with the carbonyl oxygen of Gly656 in nearly all (99.9%) of the snapshots. The guanidine moiety of the P1 Arg of SFMI2 forms an average of 4.2 H-bonds with the enzyme. In nearly 40% of the snapshots, there is an extra H-bond between the guanidine moiety of Arg5 and Ser657 of loop 2 (656–666)—an interaction rarely present in the MASP-2/SFMI1 complex. This appears to be a trade-off: instead of forming a H-bond at the water-accessible, open binding surface of the cleft, a H-bond is formed within the S1 binding pocket, resulting in a higher-affinity complex in the case of SFMI2.
Another significant difference in the binding mode of the two inhibitors is that in the MASP-2/SFMI2 complex, the C-terminal residue (Asp14) is in a position from where it can reach Arg609 of the enzyme forming (usually more than one) H-bonds with it (in 72% of the snapshots). This interaction is only present in less than 2% of the equilibrium population of the MASP-2/SFMI1 complex.
Structural Background of the Differing Selectivity of SFMI1 and SFMI2
When SFTI was subjected to phage-display-based directed evolution to yield MASP-1 and MASP-2 inhibitors, a subset of the MASP-2-selected inhibitor-phage clones bound only to MASP-2, that is, were MASP-2-selective, while the rest bound to MASP-1 as well, that is, were nonselective. Interestingly, all MASP-1-selected clones were nonselective, binding to both enzymes. Out of the seven randomized positions, the MASP-2-selective and nonselective (MASP-1 and MASP-2 binding) clones possessed similar sets of amino acids at the P2, P1, P1′, and P7′ positions.25 When SFMI1 and SFMI2 were designed based on the sequence logos of the above two clone sets (Figure 9), at each of these four positions, the common most preferred amino acids were introduced: P2 Ser, P1 Arg, P1′ Ser, and P7′ Ile. Therefore, differences between SFMI1 and SFMI2 are confined to the P4, P2′, and P5′ positions, where SFMI1 carries the nonselective consensus P4 Ile, P2′ Leu, and P5′ Ile, while SFMI2 carries the MASP-2-selective consensus P4 Tyr, P2′ Tyr, and P7′ Val residues.
Figure 9.

WebLogo diagram of phage-selected sequences. Framed positions were randomized. Position heights represent conservation degrees. Letter heights indicate normalized amino acid frequencies. Phage-selected clones were grouped in two subsets: MASP-2-selective and nonselective, with the latter ones binding both MASP-1 and MASP-2. Dotted areas represent amino acid distributions characteristic to MASP-2-selective clones. Checkered areas represent amino acid distributions characteristic to the nonselective subset. Striped areas highlight amino acid patterns shared by the two functionally distinct subsets. The figure was originally published in The Journal of Immunology: Kocsis, A.; Kékesi, K. A.; Szász, R.; Végh, B. M.; Balczer, J.; Dobó, J.; Závodszky, P.; Gál, P.; and Pál, G. (2010). Selective inhibition of the lectin pathway ofcomplement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation. J. Immunol.185, 4169–4178, Copyright 2010 by The American Association of Immunologists, Inc.25
In possession of the MASP-1/SFMI1 crystal structure and the calculated models of the other corresponding complexes, we can provide a consistent model on how differences at these three positions result in the relatively low selectivity of SFMI1 versus high selectivity of SFMI2.
At the P4 position, the selective MASP-2-binding clones carried bulky, hydrophobic residues (Tyr, Met, Trp) and the most preferred Tyr was introduced into SFMI2. In contrast, clones that could bind both MASP-1 and MASP-2 contained mostly small beta-branched hydrophobic residues (Ile, Val) and a P4 Ile was incorporated in SFMI1. The observed difference in P4 preference can be explained with the different length of loop 3 in MASP-1 (19 residues) and MASP-2 (23 residues) (Supporting Information Table 5 and Figure 7). Due to the 4-residue longer loop 3 of MASP-2, the P5–P3 segment of the inhibitor is kept further away from the enzyme; therefore, while Val and Ile are accepted, larger side chains can also be accommodated by the hydrophobic S4 pocket of the enzyme. On the other hand, the 4-residue shorter loop 3 of MASP-1 allows the formation of backbone hydrogen bonds between the enzyme and the P5 residue of SFMI1. In this structural context, only small hydrophobic P4 residues can be accommodated in the apolar S4 pocket.
Position P2′ of the SFTI scaffold was already demonstrated to be important for selectivity against a large variety of proteases.46 At the P2′ site of evolved SFTI, nonselective clones carried mostly the hydrophobic Leu, but the negatively charged Glu and Asp also occurred. Most of the MASP-2-selective clones, on the other hand, carried the bulkier Tyr and Phe side chains. Remarkably, the same P2′ preference was also observed when we developed MASP-1 and MASP-2 inhibitors on the unrelated SGPI-2 scaffold.26 It resulted in MASP-1-specific SGMI1 having a Leu P2′- and MASP-2-specific SGMI2 having a Trp P2′ residue.
The observed differential P2′ preference of the two enzymes is explained by their different S2′ pockets. While the apolar S2′ of MASP-2 formed by Leu575 and Leu581 can accommodate a Leu P2′, it is also spacious enough for larger hydrophobic Tyr, Phe, and Trp. In contrast, the MASP-1 S2′ pocket, lined by Lys591 and Phe597, is both shallower and has a positive charge, and therefore, it accepts only smaller residues, such as the hydrophobic Leu, and the acidic Asp and Glu.
Position P5′ was also reported to contribute to the selectivity of SFTI-based protease inhibitors.44 At this position, in contrast to a variety of other enzymes that favor longer P5′ residues,44 both MASP-1 and MASP-2 preferred the beta-branched Ile and Val, suggesting a similar S5′ binding site. While MASP-2-selective clones carried the beta-branched Val and Ile in equal proportions, in the nonselective clone set, the larger Ile dominated, suggesting that MASP-1 slightly prefers Ile over Val. Therefore, we introduced a P5′ Ile in SFMI-1 and a Val in SFMI-2, respectively. The P5′ residue interacts with loop B of the enzyme. While most serine proteases contain a 9–10-residue loop B, MASP-1 and MASP-2 have a 27- and a 15-residue loop B, respectively (Supporting Information Table 5 and Figure 7). This provides a plausible explanation for their preference toward relatively small P5′ side chains.
Structural Effects of Capping SFMI2
In agreement with the experimental results, modeling also indicated that placing capping groups on both termini of SFMI2 improves the affinity of the inhibitor (Figure 8c). In the MASP-2/SFMI2cap complex, Arg609 of the enzyme, which normally interacts with the uncapped C-terminal carboxyl of SFMI2, flips toward the solvent and by doing so opens a more spacious entrance to the hydrophobic S4 pocket, allowing for a better accommodation of the bulky Tyr2 (P4) of the inhibitor. Moreover, while Tyr2 of uncapped SFMI2 forms a H-bond with Gly656, this interaction is also lost in the capped derivative. Therefore, SFMI2cap lines up closer to the enzyme surface than uncapped SFMI2 but not as close as the inhibitor in the MASP-2/SFMI1 complex. As a cumulative result, in the SFMI2cap/MASP-2 complex, the loop 2 (656–660) segment of the S1 pocket retains its original position and the undisturbed S1 pocket forms a total of 7.6 H-bonds bonds with the P1 residue, which is the highest value among the MASP-2 complexes studied here.
MD Simulations of the Complexes of MASP-2 and Thioether-Linked SFMI2 Variants
MD simulations were also carried out for all thioether-linked SFMI2 inhibitors in complex with their target enzyme, MASP-2. These inhibitors differ only in the length of their disulfide-replacing bridge, yet their inhibition constants range over 4 orders of magnitude. Thus, together with the previously studied five variants, they provide an ideal basis for pinpointing structural features that define inhibitor potency.
The thioether-linked peptides bound to the enzyme in the expected canonical, β-hairpin-like binding mode and formed a total of 12–15 hydrogen bonds with the enzyme. The guanidinium group of the P1 Arg formed on average 3.5–4.5 hydrogen bonds within the S1 pocket in all complexes. The conformations of the peptides in the complex differ significantly from their uncomplexed conformations. Potent inhibitors form well-defined structures with the enzyme with most of the canonical H-bonds of the inhibitor remaining intact (Supporting Information Table 4). As the inhibitor potency decreases, the structural heterogeneity within the complex increases in parallel.
We found that inhibitor efficiency does not necessarily depend on how similar the solution-state free structure of the inhibitor is to the one it adopts in the complex (Figure 10a). On the other hand, inhibitor potency positively correlates with inherent capacity of the inhibitor to deform and this way adapt to the substrate-binding cleft without perturbing its structure (Figure 10b).
Figure 10.

Inhibitor activity related to structural dynamics of the inhibitors and complexes. (a) rmsd calculated for the entire backbone of the inhibitor (light gray) and for the canonical binding segment (P4′–P4) (dark gray) between the free and enzyme-bound forms of the inhibitor. The extent of the structural change of the inhibitor upon complex formation shows no correlation with inhibitor potency. (b) The number of clusters required for representing 90% of the equilibrium trajectories of various MASP-2 complexes (light gray) and rms deviation of backbone atoms of the gatekeeper loops in the complexes (dark gray) as compared to the respective crystal structures of the free enzymes (PDB IDs: 3GOV, 1Q3X)35,47 are shown. Both flexibility of the inhibitor within the binding pocket and the extent of enzyme distortion increase with decreasing inhibitor potency.
Assessing the Specificities of Modified SFMI2 Variants
As both capping and disulfide replacement affected the conformational heterogeneity of the variants, we tested whether this also affected their specificities. The inhibitory potency of the four SFMI2 variants differing only in the presence or absence of caps at the two termini as well as SFMI2cap-Dap (fraction 1) was tested on trypsin, thrombin (Table 2), and MASP-1.
Table 2. Potency of SFMI2 Variants on Trypsin and Thrombin and the Corresponding Specificity Ratios.
| KI* (μM) | KI* (μM) | specificity | KI* (μM) | specificity | |
|---|---|---|---|---|---|
| MASP-2a | trypsin | KI Tr/KI MASP-2 | thrombin | KI Thr/KI MASP-2 | |
| SFMI2 | 0.30 | 0.11 | 0.4 | 384 | 1280 |
| Ac-SFMI2 | 0.20 | 0.07 | 0.4 | 434 | 2170 |
| SFMI2-NH2 | 0.17 | 0.11 | 0.6 | 258 | 1518 |
| SFMI2cap | 0.10 | 0.09 | 0.9 | 713 | 7130 |
| SFMI2cap-Dap fraction 1 | 0.69 | 0.48 | 0.7 | ND | ND |
Data for MASP-2 are from Table 1. Note that inhibitor potency values are provided for all three enzymes as apparent inhibitory constants (KI*). Details of experimental procedures are provided in the Supporting Information. ND indicates that inhibition was undetectable.
Equilibrium inhibition constant values of five SFMI2 variants were determined on trypsin and thrombin and are listed in Table 2. Trypsin has a fully open substrate-binding cleft, and all tested SFMI2 variants are practically equally potent on trypsin and MASP-2. Note, however, that wild-type SFTI is about 1000-fold more potent on trypsin, while it does not inhibit MASP-2 at all.25 On one hand, the data show that capping both termini increased inhibitor potency significantly only on the original target enzyme of SFMI2 evolution, MASP-2. Trypsin slightly preferred N-terminally capped SFMI2, while thrombin slightly preferred the C-terminally capped version. As a result, capping of both termini provided an about twofold specificity increase of SFMI2 against trypsin and thrombin. The most potent thioether derivative, SFMI2cap-Dap, had practically the same specificity against trypsin as its parental molecule, SFMI2cap, but because its interaction with thrombin was too weak to be determined, a specificity value could not be calculated. While it was shown previously that SFMI2 is practically inactive on MASP-1,25 we tested the specificities of the 5 SFMI2 variants on MASP-1 as well, but to observe any effect, we had to apply the inhibitors in 1500-fold molar excess. In these tests, 10 nM MASP-1 was incubated with 15 μM inhibitor, and SFMI2 provided 31%, Ac-SFMI2 17%, SFMI2-NH2 27%, SFMI2cap 20%, and SFMI2cap-Dap 11% inhibition. The data indicate that these chemical modifications did not affect the specificity of SFMI2 against MASP-1.
Thioether-Linked SFMI2cap-Dap Has Increased Susceptibility for Cleavage by MASP-2
While SFMI2cap-Dap is only an about twofold weaker MASP-2 inhibitor than SFMI2, compared to its parental SFMI2cap, its affinity drop is approximately sevenfold. In the case of these two SFMI2 variants, we tested whether this difference is related to a different level of susceptibility for proteolytic cleavage. For both inhibitors, a 1:1 enzyme/inhibitor ratio was applied and the enzyme and inhibitor concentrations were set to be 10-fold of the respective KI value to drive the equilibrium toward complex formation. After 1 h incubation, the proportion of the intact and cleaved inhibitor forms was determined by HPLC-MS (Figure 11). It turned out that 6% of SFMI2cap and 93% of SFMI2cap-Dap were cleaved, and the cleavage occurred exclusively between the P1–P1′ positions. Note that SFMI2cap was optimized by directed evolution for MASP-2 inhibition in the structural context of the wild-type disulfide. Replacing this disulfide with a nonisosteric thioether might have caused structural rearrangement of the original SFMI2 residues, which promoted cleavage of the canonical loop. We studied this possibility by modeling MASP-2 complexes of the cleaved inhibitors.
Figure 11.

Proteolytic stability of SFMI2cap and SFMI2cap-Dap. (a,b) High-resolution mass spectra of intact SFMI2cap (a) and intact SFMI2cap-Dap (b). (c,d) High-resolution mass spectra of cleaved SFMI2cap (c) and cleaved SFMI2cap-Dap (d) peptides showing the doubly protonated molecules with the expected mass shifts. (e,f) LC–MS intensity of the intact and cleaved forms of SFMI2cap (e) and SFMI2cap-Dap (f) after 1 h incubation with MASP-2.
MD Simulations of the Cleaved SFMI2cap and SFMI2cap-Dap Inhibitors in Complex with MASP-2
To determine the structural reasons of the significantly lower proteolytic stability of SFMI2cap-Dap at the P1–P1′ site as compared to SFMI2cap, we carried out MD simulations starting from the mid-structure of the most populated cluster from the MD simulations of the intact peptides. Before the simulations, we introduced new N- and C-termini at the cleavage site and then equilibrated the new structures using the same protocol we employed for the simulations of the complexes with the intact peptides.
The cleaved forms of SFMI2cap and SFMI2cap-Dap adopted nearly identical binding conformations during the 1000 ns long simulations. The new N- and C-termini remained in close proximity to each other and to the catalytic Ser633 and His483 residues (Ser195 and His57 by chymotrypsinogen numbering) of the enzyme, forming new H-bonds with both, with the newly formed N-terminus inserted between them (Supporting Information Figure 9).
The fact that the cleaved forms of the two studied peptides formed structurally more similar complexes with MASP-2 than the intact peptides suggested that structural differences between the intact inhibitors could be responsible for their different proteolytic stability. Indeed, in their MASP-2 complexes, intact SFMI2cap formed slightly more intramolecular hydrogen bonds (4.7 vs 4.3 in SFMI2cap-Dap) and had lower B-factors (16.6 vs 18.7 nm2, in SFMI2cap-Dap), indicating a slightly more rigid conformation. In all, the longer linker of SFMI2cap-Dap apparently results in a less self-stabilized bound conformation, leading to lower proteolytic stability.
MASP-2 seems especially efficient in stabilizing the cleaved inhibitor (and presumably substrate) as its Thr466-Thr467 segment anchors the P2′–P3′ residues with an average of 2.1 H-bonds, providing extra stability to the C-terminal inhibitor segment even after proteolysis. This interaction is enabled by the extended first β-hairpin turn of the N-terminal beta-barrel domain of MASP-2, providing ideal proximity. The corresponding regions are similarly shaped in cathepsin G, chymase, and factor XIIa but differ in MASP-1, the KLK proteases, trypsin, and thrombin. In these latter enzymes, the hairpin is severely bent and its backbone is removed from the proximity of the P2′–P3′ residues. This could explain why compared to the MASP-2/SFMI2 complexes, in the crystal structure of the KLK4/SFTI-FCQR(Asn14)[1,14] complex, P1–P1′ peptide bond cleavage resulted in much greater destabilization of the inhibitor.48
Conclusions and Outlook
Using directed evolution, novel binders can be developed against practically any target. With a proper balance between “common knowledge”-based considerations and perfectly unbiased sequence space exploration, directed evolution can reproducibly yield functional clones and deliver unexpected solutions for selectable functional traits. In this study, we encountered such new insights.
Since its identification, cyclic SFTI has proved to be an ideal vehicle for the design of potent and specific inhibitors against various proteases by use of knowledge-based systematic mutation, grafting, incorporation of non-natural amino acids, or chemical modifications of the backbone structure, while preserving the rigid, cyclized fold of the parent molecule (for recent examples, see refs (44)–46 and (49)–52). Here, we found that when selecting binders from an SFTI-based library against the structurally restricted substrate-binding sites of MASP-1 and MASP-2,25in vitro evolution replaced only a few residues but globally rewired the structural dynamics and binding mode of the parental SFTI molecule (Figure 12).
Figure 12.

(a) Superimposed mid-structures of the clusters representing 90% of the equilibrium trajectories of various MASP-2 complexes. (b) Close-up of the inhibitor-binding cleft of MASP-2 with SFMI2 and all the capped and thioether bridged variants. Structural heterogeneity of the complexes is reflected in the number of displayed conformers. The uncomplexed MASP-2 crystal structure superimposed is shown in gray for reference. Anchoring P1 Arg and P4 Tyr residues as well as the linker of the inhibitors are shown with sticks.
Both MASP-1 and MASP-2 contain a trypsin-like serine protease domain consisting of two anti-parallel beta-barrel subdomains and a substrate-binding cleft located between them. While the substrate-binding cleft of trypsin is readily accessible, in the MASP enzymes, it has restricted accessibility due to two gatekeeper loops, loop B at the first beta-barrel and loop 3 at the second beta-barrel. The usual length of loop B in trypsin-like serine proteases is around 9–10 residues. In contrast, MASP-1 and MASP-2 contain 27- and 15-residue loop B, respectively. In the latter one, loop B contains a 10-residue helix, followed by a short loop. Loop 3 of trypsin-like enzymes usually contains 16–18 residues, while in MASP-1 and MASP-2, it has 19 and 23 residues, respectively (see Supporting Information Table 5 and Figure 7).
The notion that long gatekeeper loops of MASP enzymes limit the access to the substrate-binding cleft has been clearly verified by the crystal-structures of nanomolar-affinity MASP complexes formed with the 35-amino acid, phage-evolved second-generation inhibitors, SGMI-1 and SGMI-2. These revealed that complex formation required reshaping of these loops, extending the binding interface and liberating the partially blocked S2 pocket, requiring energy investment that could have limited the affinity of these interactions.26 Note that against several enzymes having nonrestricted substrate-binding clefts, the same parent molecule, SGPI-2 was successfully evolved to yield picomolar inhibitors.53,54
As it turns out, the 14-amino acid SFTI and its phage-evolved homologs are simply too small for inducing the same large-scale movements of the gatekeeper loops. This also means that Phe549 in MASP-1 and Phe529 in MASP-2 that block the S2 pocket of these enzymes remain in place even when SFMI inhibitors occupy the substrate-binding cleft. This obviously limits the size of the acceptable P2 residues. In such a structural boundary condition, directed evolution selected a P2 Ser instead of the Thr. This is rather surprising as natural evolution conserved a P2 Thr both in the Pacifastin family of the SGMIs and in the Bowman–Birk family of SFTI homologs (for a detailed explanation, see the Supporting Information).
While one might think that functional consequences of this subtle Thr to Ser replacement remain confined at the P2–S2 interaction, in reality, it came with unexpectedly complex and important consequences. Our modeling studies showed that this replacement disrupted the original tripartite, hydrophobic, beta-sheet-stabilizing side-chain cluster formed by Thr4-Phe12-Ile10 in parental SFTI. The result is a fuzzy ensemble of free structures and an induced fit mechanism of binding. It is overwhelmingly the inhibitor that goes through large-scale conformational adaptation optimizing enzyme–inhibitor structural complementarity in the complex, while the largely unchanged shape of the substrate-binding cleft guides the fuzzy ensemble of inhibitor conformations into a hairpin-like fold (Figure 12).
The modeling studies also identified an inhibitor-specificity-affecting interplay of the P4 and P2′ residues and the S1 pocket, mediated by loop 3 of MASP-1 and MASP-2. Both P4 and P2′ are stabilized by hydrophobic interactions in the complex. SFMI1 carries Ile and Leu at the P4 and P2′ sites, while SFMI2 has Tyr at both. The shape and size of these residues seem to play a crucial role in selectivity as these adjust the distance between the inhibitor and the substrate-binding cleft and also affect the shape of the S1 substrate specificity pocket.
Trypsin-like enzymes having typical loop B and loop 3 lengths have a readily accessible substrate-binding cleft, which contributes to a relatively relaxed, mostly P1/S1 compatibility-driven substrate and substrate-like inhibitor specificity. The most potent inhibitors of these enzymes will be those that have a stable solution structure which does not change upon complex formation, and are also capable of forming a great number of stabilizing interactions with the enzyme. (Note that refined models of this lock-and-key-type binding mechanism are compatible with small amplitude fluctuation of the canonical loop conformation as long as these fluctuations cover the conformation adopted by the bound form, and the related conformational rearrangements are quicker than the association rate with the protease.55)
Based on our results, we conclude that potent SFTI-based MASP inhibitors face different requirements. They need to have a highly flexible solution structure that can accommodate the substrate-binding cleft without significantly perturbing the gatekeeper loops of the enzyme, and they need to maximize new H-bond formation—both inter- and intramolecular—during the transition from the solution state to the enzyme-bound state.
We also found that introducing capping groups to both termini of SFMI2 further increased the affinity of the in vitro evolved peptide, but only against MASP-2, their original target. This reflects how SFMIs were evolved as an internal segment of a fusion protein displayed on the phage, that is, being already “capped” at both ends. Therefore, in terms of structural properties, our capped SFMI2cap peptide meets the original selection criteria better than SFMI-2 having free termini.
We developed a version of capped SFMI2 having the original disulfide replaced with an artificial, thioether bond containing a stabilizing bridge, which only slightly decreased the MASP-2 inhibitory efficiency. We suggest that replacing the disulfides with l-2,3-diaminopropionic acid (Dap) bridges can be a general approach to transform disulfide-containing peptides to reduction-resistant variants that preserve their original functionality. This might open new possibilities in all applications that engage peptides from drug development against intracellular targets to any other industrial applications that require reducing conditions.
Materials and Methods
Preparation of the MASP Catalytic Fragments
The recombinant catalytic fragments of human MASP-1 and MASP-2, consisting of the CCP1–CCP2-SP domains, were produced and purified as described earlier,56,57 except that benzamidine was omitted from the last purification step of the MASP-1 catalytic fragment.
Synthesis of MASP-2-Inhibiting SFMI2 Peptide Derivatives
All amino acid derivatives, Fmoc-Asp(OtBu)-2-ClTrt resin, and Rink-Amide MBHA resin were purchased from Iris Biotech GmBH (Marktredwitz, Germany), except Fmoc-d-Agl(Boc)-OH (also called Boc-L-Alg(Fmoc)-OH), which was purchased from Bachem (Bubendorf, Switzerland). Chemicals for the syntheses [N,N′-diisopropylcarbodiimide (DIC), 1-hydroxybenzotriazole (HOBt), piperidine, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), N-diisopropylethylamine (DIEA), trifluoroacetic acid (TFA), triisopropylsilane (TIS), and acetic anhydride (Ac2O)] were obtained from Sigma-Aldrich Kft. (Budapest, Hungary), while the solvents [dichloromethane (DCM), N,N-dimethylformamide (DMF), acetonitrile (MeCN), and diethyl ether] were purchased from Molar Chemicals (Budapest, Hungary). All reagents and solvents were of analytical grade or the highest available purity.
The MASP-2 inhibitor peptide (SFMI2: NH2-GY[CSRSYPPVC]IPD-COOH) and its terminally blocked derivatives (either N-terminal acetylated or C-terminal amidated or both) were prepared by standard solid-phase peptide synthesis using the Fmoc/tBu strategy. For the synthesis of peptide amides, Rink-Amide MBHA resin (0.54 mmol/g capacity) was used as a solid support, while peptides with a free carboxyl group at the C-terminus were built up on Fmoc-Asp(OtBu)-loaded 2-ClTrt resin (0.40 mmol/g capacity) to avoid any diketopiperazine formation and racemization. The following Fmoc-protected amino acid derivatives were applied Fmoc-Asp(OtBu)-OH, Fmoc Arg(Pbf)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Ser(tBu)-OH, and Fmoc-Tyr(tBu)-OH. The protocol of the synthesis was as follows: (i) DMF washing (3 × 0.5 min), (ii) Fmoc deprotection with 2% DBU, 2% piperidine in DMF (4 times; 2 + 2 + 5 + 10 min), (iii) DMF washing (8 × 0.5 min), (iv) coupling of Fmoc-protected amino acid derivative: DIC: HOBt (3 equiv each for the resin capacity) in DMF (1 × 60 min), (v) DMF washing (2 × 0.5 min), (vi) DCM washing (2 × 0.5 min), and (vii) ninhydrin test. After coupling of the last amino acid derivative, the N-terminal Fmoc group was removed, and in cases of preparation of acetylated peptides, the amino group was acetylated with the Ac2O/DIEA/DMF (1:1:3, v/v/v) mixture at room temperature (RT) for 30 min. The prepared peptides were cleaved from the resin in a parallel manner with the removal of protecting groups using a mixture of 95% TFA, 2.5% TIS, and 2.5% water (v/v/v) for 2.5 h at RT and then precipitated with ice-cold diethyl ether, washed three times with diethyl ether, and dissolved in 10% acetic acid prior to freeze drying. The crude products were purified by preparative reverse-phase high-performance liquid chromatography (RP-HPLC) and analyzed by mass spectrometry prior to disulfide bond formation.
The intramolecular disulfide bond was formed in 0.1 M TRIS pH 8.1 buffer at RT for 48 h using a 0.2 mg/mL peptide concentration. The reaction mixture was acidified to pH 2 afterward, followed by RP-HPLC purification. The purity of the peptides was checked by analytical RP-HPLC, and the compounds were characterized by HPLC-MS.
Synthesis of Thioether Bond-Containing SFMI2 Peptides
The linear precursors of thioether bond-containing peptides were synthesized on Rink-Amide MBHA resin with the same protocol described above. However, in position 3, the Cys derivative was replaced with Nα,Nω-Fmoc-diamino acid (l-lysine, l-ornithine (Orn), l-2,4-diaminobutyric acid (Dab), Dap) derivatives with Dde protection on the side chain. d-α-amino-glycine (Agl) was incorporated as Fmoc-d-Agl(Boc)-OH. After the coupling of the last amino acid, the Fmoc group was removed and the N-terminus was acetylated, followed by deprotection with 2% hydrazine/DMF solution six times for 5 min. The free amino group was functionalized with the chloroacetyl (ClAc) group by using chloroacetic acid pentachlorophenyl ester (ClAc-OPcp) prepared in our laboratory. (The chloroacetylation was done in solution after removal of the peptide from the resin in the case of the Agl-containing peptide). The chloroacetylated peptides were cleaved from the resin with the same cleavage mixture mentioned above, and after purification of the crude compounds, the thioether bond formation was carried out in 0.1 M TRIS buffer at pH 8.1. The peptides were added to the buffer solution in portions within an hour, and the final peptide concentration was 10 mg/mL. The reaction was continued for an additional hour; then, the pH of the solution was adjusted to pH 2 with TFA, and the reaction mixture was injected directly to the RP-HPLC system.
Analysis and Purification by RP-HPLC of SFMI2 Derivatives
Analytical RP-HPLC was performed on a KNAUER (H. Knauer, Bad Homburg, Germany) system using a Phenomenex Luna C18 column (250 mm × 4.6 mm) with 5 μm silica (100 Å pore size) (Torrance, CA) as a stationary phase. A linear gradient elution (0 min 0% B; 5 min 0% B; 50 min 90% B) with eluent A (0.1% TFA in water) and eluent B [0.1% TFA in acetonitrile–water (80:20, v/v)] was used at a flow rate of 1 mL/min. Peaks were detected at λ = 220 nm. The crude products were purified on a preparative Phenomenex Luna C18 column (250 mm × 21.2 mm) with 10 μm silica (100 Å pore size). An isocratic elution with 5% of eluent B (using the same eluents) was applied from 0 to 5 min; then, from 5 to 50 min, a gradient elution of 5–50% of eluent B was used with a 9 mL/min flow rate. Peaks were detected at λ = 220 nm.
Electrospray Ionization Mass Spectrometry Analysis
The identification of the products was achieved by mass spectrometry. Electrospray ionization mass spectrometry was performed with a Bruker Daltonics Esquire 3000 Plus (Bremen, Germany) ion trap mass spectrometer, operating in continuous sample injection at a 10 μL/min flow rate. The peptides were dissolved in a 50% acetonitrile—50% water mixture containing 0.1% formic acid (v/v). Mass spectra were recorded in the positive ion mode in the m/z 200–1500 range.
HPLC-MS analysis was performed on a Thermo Scientific Q Exactive Focus, high-resolution and high-mass accuracy, hybrid quadrupole-orbitrap mass spectrometer (Bremen, Germany) using online ultra-high-pressure liquid chromatography (UHPLC) coupling. UHPLC separation was performed on a Dionex 3000 UHPLC system using a Waters Acquity C18 column (2.1 × 150 mm, 1.7 μm). A linear gradient elution (0 min 2% B, 2 min 2% B, 6 min 100% B) with eluent A (0.1% formic acid in water, v/v) and eluent B (0.1% formic acid in acetonitrile/water, 80:20, v/v) was used at a flow rate of 0.4 mL/min at 55 °C. High-resolution mass spectra were acquired in the 200–2000 m/z range. (High-resolution HPLC-MS chromatograms of the peptides are shown in Supporting Information Figure 2).
Synthesis and Analysis of the Acyclic SFTI Variant
The acyclic variant of the wild-type SFTI (H-GRCTKSIPPICFPD-OH) was synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry on TG-R Wang resin. The first amino acid (Fmoc-Asp(OtBu)-OH) was coupled manually; then, the rest of the peptide was synthesized using an automatic continuous flow peptide synthesizer. The raw product was solubilized in 0.1 M Tris buffer (pH 8.1); then, the intramolecular disulfide bridge was formed via air oxidation at RT at a peptide concentration of 0.2 mg/mL by stirring the solution for 48 h. After lyophilization and purification using preparative RP-HPLC, the purity of the peptide was verified using analytical HPLC, and the pure peptide was characterized by HPLC-MS measurements (Supporting Information Figure 3).
Electronic Circular Dichroism Spectroscopy
Far-UV ECD measurements were carried out on a Jasco J1500 spectrophotometer in 1.0 mm quartz cuvettes. We used a spectral scanning speed of 50 nm/min with a 1 nm bandwidth and a 0.2 nm step resolution over wavelength ranges of 185–250 nm and four scans averaged for each spectrum. The temperature of the cuvette was controlled using a Peltier-type heating system. The raw ellipticity data were converted into mean residue molar ellipticity units ([θ]MR/deg*cm2*dmol–1). ECD spectra were measured in water at pH 7 at a peptide concentration of 60–80 μM. For ECD measurements under reducing conditions, TCEP was added to the samples in a 0.5 mM concentration; then, the samples were incubated at 37 °C for 2 h.
NMR Study of SFMI2
Homonuclear TOCSY and NOESY spectra of SFMI2 were acquired at pH = 3.04 and T = 300 K on a Bruker AVANCE 500 spectrometer. Resonance assignment could be achieved for all residues except the N-terminal glycine (G1). With the exception of the guanidino group of Arg5 and the HD1 methyl group of Ile12, chemical shifts of all nonexchanging hydrogen-containing side chain groups could be determined. NMR chemical shifts of SFMI2 have been deposited in the BMRB under accession number 50529.
Modeling and MD Simulations
The serine protease domain
of the crystal structures of the uncomplexed form of both MASP-1 and
MASP-2 (PDB ID: 3GOV,35 and 1Q3X,47 respectively),
their complexes with SGPI-derived 35 amino acid small protein inhibitors
(PDB IDs: 1DJZ and 3TVJ,26 respectively), and the crystal structure described
herein were used for model building. In all cases, we modeled only
the SP domain of the MASP enzymes. Initial geometry optimization was
carried out using the Schrödinger software suite (MacroModel,
Schrödinger, LLC, New York, NY, 2019). Simulations of complexes
were started from identical conformers by simply mutating the appropriate
residues of SFMI1 and SFMI2 and keeping the backbone fixed. MD simulations
were carried out as implemented in GROMACS58 using the Amber99sb-ildnp* force field.59 Systems were solvated in dodecahedral boxes with an either 8 Å
or 10 Å buffer distance using TIP3P water molecules, the total
charge of the system was neutralized, and the physiological salt concentration
(0.15 M) was set using Na+ and Cl– ions.
Energy minimization of starting structures was followed by sequential
relaxation of constraints on protein atoms in three steps and an additional NVT step (all of 100 ps) to stabilize the pressure. When
the protein–inhibitor snapshots were collected along the last
300 ns of the 600 ns NPT simulations for further
analysis (where heavy atom–hydrogen bonds were constrained
using the LINCS algorithm58), the temperature
was kept at 310 K (applying the velocity rescale algorithm60), and the pressure was
kept at 1 bar (using a Berendsen barostat). Simulations concerning
the free peptides were of 1500–4000 ns long. Clustering of
conformations61 was carried out using a
cutoff of 1.5 Å for 300 ns of the equilibrated trajectories.
To estimate the binding energy of the complexes, the mid-structures
of the most populated clusters of the trajectories of the free enzyme,
free inhibitor, and complex were energy-minimized using MacroModel
(MacroModel, Schrödinger, LLC, New York, NY, 2019) and the
OPLS3 force field62 and weighted sums were
calculated for each state 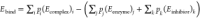 , where i, j, and k are the respective indices
of the cluster
mid-structures and p is their probability weight.
Similarly, when comparing MD-derived structures, rmsd was calculated
as the cluster weighted average of all deviations between the mid-structures
of the most populated clusters.
, where i, j, and k are the respective indices
of the cluster
mid-structures and p is their probability weight.
Similarly, when comparing MD-derived structures, rmsd was calculated
as the cluster weighted average of all deviations between the mid-structures
of the most populated clusters.
Simulations of thioether-linked SFMI2 variants were started from the same backbone conformation as that of the disulfide-bridge containing variant. Two non-natural residues were incorporated into each thioether-linked inhibitor: a cysteine modified in its side chain with a CH2–CHO group and a Lys/Orn/Dap/Dab/Agl residue modified in its side chain with an amino group. The two residues were linked with a standard amide bond.
For parametrization, two conformers were used for each residue, one in an α-helical conformation (φ = −65.1°, ψ = −30.5°) and one in a β-sheet conformation (φ = −179.7°, ψ = −179.7°). The N- and C-terminal ends and the side chains were capped with acetyl- and N-methyl amide groups. The R.E.D. server63 was used to obtain partial charges (RESP) using the HF/6-31G(d) level of theory. Other force field parameters were generated by the R.E.D. server using the parameters present in the Amber ff99SB and the generalized Amber force fields (GAFF).64
pKa estimation was carried out as implemented in Maestro (Schrödinger, LLC, New York, NY, 2019).
For comparison of structural ensembles, cluster-weighted averages were calculated using the cluster mid-structures and their probability weight (see above). rmsd was calculated for either all the backbone atoms or those of the so-called core of the structures, referring to those segments that are part of a secondary structural element. Thus, in the case of MASP-1, “core” stands for residues 462–469, 474–481, 483–487, 513–518, 530–540, 553–559, 584–589, 600–607, 610–619, 628–633, 650–653, 659–669, and 678–696, while in the case of MASP-2, the “core” residues are 448–451, 458–462, 467–473, 476–490, 496–499, 510–519, 534–539, 567–573, 583–591, 594–602, 615–619, 633–642, 646–672, and 675–685.
Crystallographic Study
The recombinant MASP-1 catalytic fragment (containing the CCP1–CCP2-SP domains) at a 9.6 mg/mL concentration in 50 mM NaCl, 5 mM Tris, 0.5 mM ethylenediaminetetraacetic acid (EDTA), pH 8.8 buffer, and SFMI1 (2.96 mg/mL, in water) were premixed to give final concentrations of 8.5 mg/mL (187 μM) and 0.35 mg/mL (240 μM), respectively, equivalent to an enzyme to inhibitor ratio of approximately 1:1.3. The crystallization was carried out using the hanging drop vapor diffusion method at RT: 1 μL of the MASP-1—SFMI1 solution was mixed with 1 μL of the reservoir solution. Initial crystals were obtained with the PEGRx 1 screen (Hampton Research) with 0.1 M N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), pH 7.5, 42% PEG-200 as the reservoir solution. RP-HPLC analysis confirmed that the crystals contained the inhibitory peptide (data not shown). After optimization, the largest crystals were obtained by mixing 1 μL of the MASP-1 catalytic fragment (9.6 mg/mL, 211 μM), 1 μL of SFMI1 (2.96 mg/mL, 2 mM), and 1 μL of the optimized reservoir solution (0.1 M HEPES, pH7.5, 38% PEG-200). The final conditions corresponded to an enzyme to inhibitor ratio of approximately 1:9.5. The sizes of the largest single crystals were up to 300 μm × 300 μm × 300 μm (see Supporting Information Figure 1). Crystals were flash-cooled in liquid nitrogen without requiring any further treatment for cryoprotection.
Structure of the MASP-2/SFMI2 complex could not be determined due to unsuccessful crystallization. Diffraction data were collected at beamline X12 at EMBL-Hamburg at 100 K (wavelength 0.9769 Å). The data set was processed using the XDS package.65 The phase problem was solved by molecular replacement with Phaser66 using SP and CCP1-CCP2 fragments of the structure of the uncomplexed activated form of MASP-1 (PDB ID: 3GOV(35)) as search models. Manual model building was carried out using Coot.67 The structure was refined using the Phenix68 and Buster69 refinement packages. Refinement included TLS refinement (TLS groups were generated automatically by phenix.refine) and refinement of coordinates and isotropic B-factors. The model was validated using the Phenix package and MolProbity.70 Water molecules were added to the model manually. Protease–inhibitor interactions were analyzed using the PISA server.71
The resolution limit used for data processing was initially determined based on the CC1/2 values as 2.4 Å.72 The use of weak data up to a 2.4 Å resolution and radiation damage at the end of data collection resulted in unconventionally high Rmeas values in the higher resolution shells. CC1/2 values and improvements of the electron density maps (tested during the initial step of the refinement) indicate however that these resolution shells contain useful data.
The final structure contained 1 MASP-1/SFMI1 complex, 1 diethylene glycol molecule, and 26 water molecules. The C-terminal residues of the inhibitor as well as two loops in MASP-1 were disordered; therefore, these residues were not included in the final model.
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (http://wwpdb.org/) with accession code 7ARX. Data collection and refinement statistics are shown in Supporting Information Table 1.
Determination of Equilibrium Inhibitory Constants (KI)
The binding affinity of the SFMI2 variants for MASP-2 (produced as described above in Preparation of the MASP Catalytic Fragments), bovine cationic trypsin (Worthington Biochemical, LS003740), and human thrombin (Merck, 605190-M) was determined by measuring the equilibrium inhibitory constant values (KI) according to Empie and Laskowski.73 The lyophilized inhibitors were dissolved in water, and the concentration of each variant was determined based on their absorbance at 280 nm. Increasing amounts of the inhibitor were preincubated with fixed concentrations of the enzyme for 1 h to reach equilibrium; then, the appropriate substrate [250 μM Z-L-Lys-SBzl (Sigma-Aldrich, C3647) for MASP-2, 500 μM Z-Gly-Pro-Arg-pNA (Bachem, 4000768) for thrombin, and 5 μM Z-Gly-Pro-Arg-AMC (PeptaNova, 3208-v)] was added to the mixtures.
The experiments were carried out in 20 mM HEPES pH 7.6, 145 mM NaCl, 5 mM CaCl2, 0.05% Triton-X 100 for thrombin, and the same buffer supplemented with 500 μM DTNB (5,5-dithio-bis-(2-nitrobenzoic acid), Sigma-Aldrich, D8130) (as a co-substrate) for MASP-2. The affinity of the peptides for trypsin was determined in 50 mM Tris-HCl pH 8.0, 10 mM CaCl2, 0.005% Triton X-100.
The initial velocity of the reactions was measured
using a BioTek
Synergy H4 hybrid microplate reader. The measurements were performed
in triplicates (MASP-2) or duplicates (trypsin and thrombin). The
following equation was fitted to the data: 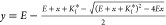 , where x designates
the
total inhibitor concentration, y represents the free
protease concentration in equilibrium, KI* stands for the
apparent equilibrium inhibitory constant, and E is
the total protease concentration. In the case of MASP-2, the measured KI values were corrected for competition with the substrate according
to the following equation:
, where x designates
the
total inhibitor concentration, y represents the free
protease concentration in equilibrium, KI* stands for the
apparent equilibrium inhibitory constant, and E is
the total protease concentration. In the case of MASP-2, the measured KI values were corrected for competition with the substrate according
to the following equation:  , where KI represents
the equilibrium inhibitory constant, [S] stands for
the total substrate concentration, and KM is the Michaelis–Menten constant of the substrate. The 618
μM value of KM used for the corrections
was determined earlier.74
, where KI represents
the equilibrium inhibitory constant, [S] stands for
the total substrate concentration, and KM is the Michaelis–Menten constant of the substrate. The 618
μM value of KM used for the corrections
was determined earlier.74
The ability of selected SFMI2 variants to inhibit MASP-1 was tested in a similar fashion as described for MASP-2 but only at a single 15 μM inhibitor concentration.
Assessing Serum Stability of SFMI2 Variants through Lectin Pathway Inhibition Potency
Serum stability of the inhibitor variants was assessed via lectin pathway-specific ELISA as described previously,25 with modifications. ELISA plates (Greiner Bio-One, #655061) were coated with 10 μg/mL mannan in 50 mM sodium carbonate, pH 9.6, for 12 h at 4 °C. Wells were blocked with 1% BSA in TBS, pH 7.4, for 1.5 h at 37 °C and then washed with TBS, 5 mM CaCl2, 0.1% Tween 20. Pooled normal human serum (NHS) was diluted 100-fold in 20 mM HEPES, 145 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 0.1% Tween 20, pH 7.4 containing serial dilutions of inhibitors. The mixtures were preincubated in single loose tubes (National Scientific Supply Company) for 30 min at RT and then were transferred onto the mannan-coated and BSA-blocked ELISA plates. After incubating for 30 min at 37 °C, the polyclonal rabbit anti-human C3c antibody (DakoCytomation, A0062) was used as the primary antibody in 2000-fold dilution and the horseradish peroxidase-conjugated anti-rabbit antibody (Sigma, A1949) as the secondary antibody in 40000-fold dilution. 1 mg/mL o-phenylenediamine dihydrochloride in 50 mM K-citrate, 0.1% H2O2, pH 5.0, was used as the chromogenic substrate, and the signal intensity was read at 490 nm. The signal produced by the noninhibited NHS was considered to be 100% activity, while that produced by NHS treated with 20 mM EDTA was considered to be 0%. The results were obtained from two parallel measurements. IC50 values were obtained via the OriginPro 8 software, fitting the DoseResp equation (“Pharmacology” built-in equation set) onto the data set.
Acknowledgments
These studies were supported by grants (VEKOP-2.3.3-15-2017-00020, VEKOP-2.3.2-16-2017-00014, and VEKOP-2.3.3-15-2017-00018) from the European Union and the State of Hungary, co-financed by the European Regional Development Fund from the Ministry of Human Capacities in Hungary in the frame of Institutional Excellence Program for Higher Education. Project no. 2018-1.2.1-NKP-2018-00005 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the 2018-1.2.1-NKP funding scheme and by the National Research, Development and Innovation Office (Hungarian Scientific Research Fund) grants NK100769, NK100834, K116305, K119374, K119386, K135289, and KH130376. Simulations were carried out using the infrastructure provided by the Governmental Agency for IT Development (KIFÜ-NIIF). We thank EMBL in Hamburg for providing facilities on beamline X12. We thank V. Farkas for his assistance in the ECD measurements. We also thank V. Farkas and K. Ferentzi for their assistance in the synthesis of the acyclic variant of SFTI and L. E. Dókus for his assistance in the synthesis of SFMI2cap and SFMI2cap-Lys. We are grateful to M. Sahin-Tóth for critical reading of the manuscript and providing insightful advices.
Glossary
Abbreviations
- MBL
mannan-binding lectin,
- MASP
MBL-associated serine protease
- COVID-19
coronavirus disease 19
- SFTI
sunflower trypsin inhibitor
- SFMI
SFTI-based MASP inhibitor
- SGPI-2
Schistocerca gregaria protease inhibitor 2
- SGMI
SGPI-based MASP inhibitor
- Orn
l-ornithine
- Dab
l-2,4-diaminobutyric acid
- Dap
l-2,3-diaminopropionic acid
- Agl
d-α-aminoglycine
- PRM
pattern recognition molecule
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00114.
Crystallographic data collection and refinement statistics, HPLC–MS data of all synthesized peptides, NMR study of SFMI2, ECD data of peptides under reducing conditions, and additional MD simulations and structure analysis (PDF)
Accession Codes
The atomic coordinates and structure factors of the MASP-1/SFMI1 complex have been deposited in the Protein Data Bank (http://wwpdb.org/) with accession code 7ARX. NMR chemical shifts of SFMI2 have been deposited in the BMRB under accession number 50529 (NMR results are described in the Supporting Information file).
The authors declare no competing financial interest.
Supplementary Material
References
- Laskowski M.; Kato I. Protein Inhibitors of Proteinases. Annu. Rev. Biochem. 1980, 49, 593–626. 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- Laskowski M.; Qasim M. A. What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes?. Biochim. Biophys. Acta 2000, 1477, 324–337. 10.1016/s0167-4838(99)00284-8. [DOI] [PubMed] [Google Scholar]
- de Veer S. J.; Swedberg J. E.; Akcan M.; Rosengren K. J.; Brattsand M.; Craik D. J.; Harris J. M. Engineered protease inhibitors based on sunflower trypsin inhibitor-1 (SFTI-1) provide insights into the role of sequence and conformation in Laskowski mechanism inhibition. Biochem. J. 2015, 469, 243–253. 10.1042/bj20150412. [DOI] [PubMed] [Google Scholar]
- Schechter I.; Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Colgrave M. L.; Korsinczky M. J. L.; Clark R. J.; Foley F.; Craik D. J. Sunflower trypsin inhibitor-1, proteolytic studies on a trypsin inhibitor peptide and its analogs. Biopolymers 2010, 94, 665–672. 10.1002/bip.21415. [DOI] [PubMed] [Google Scholar]
- Marx U. C.; Korsinczky M. L. J.; Schirra H. J.; Jones A.; Condie B.; Otvos L.; Craik D. J. Enzymatic cyclization of a potent bowman-birk protease inhibitor, sunflower trypsin inhibitor-1, and solution structure of an acyclic precursor peptide. J. Biol. Chem. 2003, 278, 21782–21789. 10.1074/jbc.m212996200. [DOI] [PubMed] [Google Scholar]
- Qasim M. A.; Ganz P. J.; Saunders C. W.; Bateman K. S.; James M. N. G.; Laskowski M. Interscaffolding additivity. Association of P1 variants of eglin c and of turkey ovomucoid third domain with serine proteinases. Biochemistry 1997, 36, 1598–1607. 10.1021/bi9620870. [DOI] [PubMed] [Google Scholar]
- Kelly C.; Laskowski M.; Qasim M. The Role of Scaffolding in Standard Mechanism Serine Proteinase Inhibitors. Protein Pept. Lett. 2005, 12, 465–471. 10.2174/0929866054395383. [DOI] [PubMed] [Google Scholar]
- Boros E.; Sebák F.; Héja D.; Szakács D.; Zboray K.; Schlosser G.; Micsonai A.; Kardos J.; Bodor A.; Pál G. Directed Evolution of Canonical Loops and Their Swapping between Unrelated Serine Proteinase Inhibitors Disprove the Interscaffolding Additivity Model. J. Mol. Biol. 2019, 431, 557. 10.1016/j.jmb.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Ricklin D.; Hajishengallis G.; Yang K.; Lambris J. D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle N. S.; Church S. E.; Fremeaux-Bacchi V.; Roumenina L. T. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle N. S.; Noe R.; Halbwachs-Mecarelli L.; Fremeaux-Bacchi V.; Roumenina L. T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobó J.; Pál G.; Cervenak L.; Gál P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol. Rev. 2016, 274, 98–111. 10.1111/imr.12460. [DOI] [PubMed] [Google Scholar]
- Walsh M. C.; Bourcier T.; Takahashi K.; Shi L.; Busche M. N.; Rother R. P.; Solomon S. D.; Ezekowitz R. A. B.; Stahl G. L. Mannose-Binding Lectin Is a Regulator of Inflammation That Accompanies Myocardial Ischemia and Reperfusion Injury. J. Immunol. 2005, 175, 541–546. 10.4049/jimmunol.175.1.541. [DOI] [PubMed] [Google Scholar]
- Schwaeble W. J.; Lynch N. J.; Clark J. E.; Marber M.; Samani N. J.; Ali Y. M.; Dudler T.; Parent B.; Lhotta K.; Wallis R.; et al. Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 7523–7528. 10.1073/pnas.1101748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osthoff M.; Katan M.; Fluri F.; Schuetz P.; Bingisser R.; Kappos L.; Steck A. J.; Engelter S. T.; Mueller B.; Christ-Crain M.; et al. Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLoS One 2011, 6, e21338 10.1371/journal.pone.0021338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari E.; Farrar C. A.; Lynch N.; Ali Y. M.; Roscher S.; Stover C.; Zhou W.; Schwaeble W. J.; Sacks S. H. Mannan-binding lectin-associated serine protease 2 is critical for the development of renal ischemia reperfusion injury and mediates tissue injury in the absence of complement C4. FASEB J. 2014, 28, 3996–4003. 10.1096/fj.13-246306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini F.; Chrysanthou E.; Dudler T.; Cummings W. J.; Takahashi M.; Fujita T.; Demopulos G.; De Simoni M.-G.; Schwaeble W. Mannan binding lectin-associated serine protease-2 (MASP-2) critically contributes to post-ischemic brain injury independent of MASP-1. J. Neuroinflammation 2016, 13, 213. 10.1186/s12974-016-0684-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magro C.; Mulvey J. J.; Berlin D.; Nuovo G.; Salvatore S.; Harp J.; Baxter-Stoltzfus A.; Laurence J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klok F. A.; Kruip M. J. H. A.; van der Meer N. J. M.; Arbous M. S.; Gommers D. a. M. P. J.; Kant K. M.; Kaptein F. H. J.; van Paassen J.; Stals M. a. M.; Huisman M. V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z.; Flammer A. J.; Steiger P.; Haberecker M.; Andermatt R.; Zinkernagel A. S.; Mehra M. R.; Schuepbach R. A.; Ruschitzka F.; Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. 10.1016/s0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T.; Hu M.; Zhang X.; Li H.; Zhu L.; Liu H.; Dong Q.; Zhang Z.; Wang Z.; Hu Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. 2020, medRxiv. 10.1101/2020.03.29.20041962. [Google Scholar]
- Ramlall V.; Thangaraj P. M.; Meydan C.; Foox J.; Butler D.; Kim J.; May B.; De Freitas J. K.; Glicksberg B. S.; Mason C. E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609. 10.1038/s41591-020-1021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Java A.; Apicelli A. J.; Liszewski M. K.; Coler-Reilly A.; Atkinson J. .; Kim A. H.; Kulkarni H. S. The complement system in COVID-19: friend and foe?. JCI Insight 2020, 5, e140711 10.1172/jci.insight.140711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis A.; Kékesi K. A.; Szász R.; Végh B. M.; Balczer J.; Dobó J.; Závodszky P.; Gál P.; Pál G. Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation. J. Immunol. 2010, 185, 4169–4178. 10.4049/jimmunol.1001819. [DOI] [PubMed] [Google Scholar]
- Héja D.; Harmat V.; Fodor K.; Wilmanns M.; Dobó J.; Kékesi K. A.; Závodszky P.; Gál P.; Pál G. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and-2 Are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J. Biol. Chem. 2012, 287, 20290. 10.1074/jbc.M112.354332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Héja D.; Kocsis A.; Dobó J.; Szilágyi K.; Szász R.; Závodszky P.; Pál G.; Gál P. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 10498–10503. 10.1073/pnas.1202588109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács D.; Kocsis A.; Szász R.; Gál P.; Pál G. Novel MASP-2 inhibitors developed via directed evolution of human TFPI1 are potent lectin pathway inhibitors. J. Biol. Chem. 2019, 294, 8227–8237. 10.1074/jbc.ra119.008315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobó J.; Szakács D.; Oroszlán G.; Kortvely E.; Kiss B.; Boros E.; Szász R.; Závodszky P.; Gál P.; Pál G. MASP-3 is the exclusive pro-factor D activator in resting blood: the lectin and the alternative complement pathways are fundamentally linked. Sci. Rep. 2016, 6, 31877. 10.1038/srep31877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg J. E.; de Veer S. J.; Sit K. C.; Reboul C. F.; Buckle A. M.; Harris J. M. Mastering the Canonical Loop of Serine Protease Inhibitors: Enhancing Potency by Optimising the Internal Hydrogen Bond Network. PLoS One 2011, 6, e19302 10.1371/journal.pone.0019302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckett S.; Garcia R. S.; Barker J. J.; Konarev A. V.; Shewry P. R.; Clarke A. R.; Brady R. L. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 1999, 290, 525–533. 10.1006/jmbi.1999.2891. [DOI] [PubMed] [Google Scholar]
- Korsinczky M. L. J.; Schirra H. J.; Rosengren K. J.; West J.; Condie B. A.; Otvos L.; Anderson M. A.; Craik D. J. Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J. Mol. Biol. 2001, 311, 579–591. 10.1006/jmbi.2001.4887. [DOI] [PubMed] [Google Scholar]
- Tong L.; Qian C.; Massariol M.-J.; Déziel R.; Yoakim C.; Lagacé L. Conserved mode of peptidomimetic inhibition and substrate recognition of human cytomegalovirus protease. Nat. Struct. Biol. 1998, 5, 819–826. 10.1038/1860. [DOI] [PubMed] [Google Scholar]
- Perona J. J.; Craik C. S. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J. Biol. Chem. 1997, 272, 29987–29990. 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- Dobó J.; Harmat V.; Beinrohr L.; Sebestyén E.; Závodszky P.; Gál P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J. Immunol. 2009, 183, 1207–1214. 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
- Guo X.; Shi J.; Tang Z.; Cui D.; Zhang Y. Synthesis and Biological Activity of Seleno Sunflower Trypsin Inhibitor Analog. Chem. Biol. Drug Des. 2006, 68, 341–344. 10.1111/j.1747-0285.2006.00457.x. [DOI] [PubMed] [Google Scholar]
- Zabłotna E.; Kaźmierczak K.; Jaśkiewicz A.; Stawikowski M.; Kupryszewski G.; Rolka K. Chemical synthesis and kinetic study of the smallest naturally occurring trypsin inhibitor SFTI-1 isolated from sunflower seeds and its analogues. Biochem. Biophys. Res. Commun. 2002, 292, 855. 10.1006/bbrc.2002.6746. [DOI] [PubMed] [Google Scholar]
- Veer S. J.; White A. M.; Craik D. J. Sunflower Trypsin Inhibitor-1 (SFTI-1): Sowing Seeds in the Fields of Chemistry and Biology. Angew. Chem., Int. Ed. 2021, 60, 8050–8071. 10.1002/anie.202006919. [DOI] [PubMed] [Google Scholar]
- White A. M.; Veer S. J.; Wu G.; Harvey P. J.; Yap K.; King G. J.; Swedberg J. E.; Wang C. K.; Law R. H. P.; et al. Application and Structural Analysis of Triazole-Bridged Disulfide Mimetics in Cyclic Peptides. Angew. Chem., Int. Ed. 2020, 59, 11273–11277. 10.1002/anie.202003435. [DOI] [PubMed] [Google Scholar]
- Gori A.; Gagni P.; Rinaldi S. Disulfide Bond Mimetics: Strategies and Challenges. Chem.—Eur. J. 2017, 23, 14987. 10.1002/chem.201703199. [DOI] [PubMed] [Google Scholar]
- Tugyi R.; Mezö G.; Fellinger E.; Andreu D.; Hudecz F. The effect of cyclization on the enzymatic degradation of herpes simplex virus glycoprotein D derived epitope peptide. J. Pept. Sci. 2005, 11, 642–649. 10.1002/psc.669. [DOI] [PubMed] [Google Scholar]
- Chen Y.-Q.; Chen C.-C.; He Y.; Yu M.; Xu L.; Tian C.-l.; Guo Q.-X.; Shi J.; Zhang M.; Li Y.-M. Efficient synthesis of trypsin inhibitor SFTI-1 via intramolecular ligation of peptide hydrazide. Tetrahedron Lett. 2014, 55, 2883–2886. 10.1016/j.tetlet.2014.03.093. [DOI] [Google Scholar]
- Costa J. R.; Yaliraki S. N. Role of rigidity on the activity of proteinase inhibitors and their peptide mimics. J. Phys. Chem. B 2006, 110, 18981–18988. 10.1021/jp0575299. [DOI] [PubMed] [Google Scholar]
- Li C. Y.; de Veer S. J.; White A. M.; Chen X.; Harris J. M.; Swedberg J. E.; Craik D. J. Amino Acid Scanning at P5′ within the Bowman–Birk Inhibitory Loop Reveals Specificity Trends for Diverse Serine Proteases. J. Med. Chem. 2019, 62, 3696–3706. 10.1021/acs.jmedchem.9b00211. [DOI] [PubMed] [Google Scholar]
- Shariff L.; Zhu Y.; Cowper B.; Di W.-L.; Macmillan D. Sunflower trypsin inhibitor (SFTI-1) analogues of synthetic and biological origin via N→S acyl transfer: potential inhibitors of human Kallikrein-5 (KLK5). Tetrahedron 2014, 70, 7675–7680. 10.1016/j.tet.2014.06.059. [DOI] [Google Scholar]
- de Veer S. J.; Wang C. K.; Harris J. M.; Craik D. J.; Swedberg J. E. Improving the Selectivity of Engineered Protease Inhibitors: Optimizing the P2 Prime Residue Using a Versatile Cyclic Peptide Library. J. Med. Chem. 2015, 58, 8257–8268. 10.1021/acs.jmedchem.5b01148. [DOI] [PubMed] [Google Scholar]
- Harmat V.; Gál P.; Kardos J.; Szilágyi K.; Ambrus G.; Végh B.; Náray-Szabó G.; Závodszky P. The Structure of MBL-associated Serine Protease-2 Reveals that Identical Substrate Specificities of C1s and MASP-2 are Realized Through Different Sets of Enzyme–Substrate Interactions. J. Mol. Biol. 2004, 342, 1533–1546. 10.1016/j.jmb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Riley B. T.; Ilyichova O.; de Veer S. J.; Swedberg J. E.; Wilson E.; Hoke D. E.; Harris J. M.; Buckle A. M. KLK4 Inhibition by Cyclic and Acyclic Peptides: Structural and Dynamical Insights into Standard-Mechanism Protease Inhibitors. Biochemistry 2019, 58, 2524–2533. 10.1021/acs.biochem.9b00191. [DOI] [PubMed] [Google Scholar]
- Li C. Y.; Yap K.; Swedberg J. E.; Craik D. J.; de Veer S. J. Binding Loop Substitutions in the Cyclic Peptide SFTI-1 Generate Potent and Selective Chymase Inhibitors. J. Med. Chem. 2020, 63, 816–826. 10.1021/acs.jmedchem.9b01811. [DOI] [PubMed] [Google Scholar]
- de Veer S. J.; Li C. Y.; Swedberg J. E.; Schroeder C. I.; Craik D. J. Engineering potent mesotrypsin inhibitors based on the plant-derived cyclic peptide, sunflower trypsin inhibitor-1. Eur. J. Med. Chem. 2018, 155, 695–704. 10.1016/j.ejmech.2018.06.029. [DOI] [PubMed] [Google Scholar]
- Swedberg J. E.; Li C. Y.; de Veer S. J.; Wang C. K.; Craik D. J. Design of Potent and Selective Cathepsin G Inhibitors Based on the Sunflower Trypsin Inhibitor-1 Scaffold. J. Med. Chem. 2017, 60, 658–667. 10.1021/acs.jmedchem.6b01509. [DOI] [PubMed] [Google Scholar]
- Tian S.; Swedberg J. E.; Li C. Y.; Craik D. J.; de Veer S. J. Iterative Optimization of the Cyclic Peptide SFTI-1 Yields Potent Inhibitors of Neutrophil Proteinase 3. ACS Med. Chem. Lett. 2019, 10, 1234–1239. 10.1021/acsmedchemlett.9b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó A.; Héja D.; Szakács D.; Zboray K.; Kékesi K. A.; Radisky E. S.; Sahin-Tóth M.; Pál G. High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4′ acidic residues. J. Biol. Chem. 2011, 286, 22535–22545. 10.1074/jbc.m111.235754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros E.; Szabó A.; Zboray K.; Héja D.; Pál G.; Sahin-Tóth M. Overlapping Specificity of Duplicated Human Pancreatic Elastase 3 Isoforms and Archetypal Porcine Elastase 1 Provides Clues to Evolution of Digestive Enzymes. J. Biol. Chem. 2017, 292, 2690–2702. 10.1074/jbc.m116.770560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gáspári Z.; Várnai P.; Szappanos B.; Perczel A. Reconciling the lock-and-key and dynamic views of canonical serine protease inhibitor action. FEBS Lett. 2010, 584, 203–206. 10.1016/j.febslet.2009.11.058. [DOI] [PubMed] [Google Scholar]
- Ambrus G.; Gál P.; Kojima M.; Szilágyi K.; Balczer J.; Antal J.; Gráf L.; Laich A.; Moffatt B. E.; Schwaeble W.; et al. Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: a study on recombinant catalytic fragments. J. Immunol. 2003, 170, 1374–1382. 10.4049/jimmunol.170.3.1374. [DOI] [PubMed] [Google Scholar]
- Dobó J.; Harmat V.; Sebestyén E.; Beinrohr L.; Závodszky P.; Gál P. Purification, crystallization and preliminary X-ray analysis of human mannose-binding lectin-associated serine protease-1 (MASP-1) catalytic region. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2008, 64, 781–784. 10.1107/s174430910802294x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Aliev A. E.; Kulke M.; Khaneja H. S.; Chudasama V.; Sheppard T. D.; Lanigan R. M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins: Struct., Funct., Bioinf. 2014, 82, 195–215. 10.1002/prot.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Daura X.; van Gunsteren W. F.; Mark A. E. Folding–unfolding thermodynamics of a β-heptapeptide from equilibrium simulations. Proteins: Struct., Funct., Bioinf. 1999, 34, 269–280. . [DOI] [PubMed] [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Vanquelef E.; Simon S.; Marquant G.; Garcia E.; Klimerak G.; Delepine J. C.; Cieplak P.; Dupradeau F.-Y. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511. 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 352–367. 10.1107/s0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc E.; Roversi P.; Vonrhein C.; Flensburg C.; Lea S. M.; Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER–TNT. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2210–2221. 10.1107/s0907444904016427. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/s0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Karplus P. A.; Diederichs K. Linking Crystallographic Model and Data Quality. Science 2012, 336, 1030. 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empie M. W.; Laskowski M. Thermodynamics and kinetics of single residue replacements in avian ovomucoid third domains: effect on inhibitor interactions with serine proteinases. Biochemistry 1982, 21, 2274–2284. 10.1021/bi00539a002. [DOI] [PubMed] [Google Scholar]
- Nagy Z. A.; Szakács D.; Boros E.; Héja D.; Vígh E.; Sándor N.; Józsi M.; Oroszlán G.; Dobó J.; Gál P.; et al. Ecotin, a microbial inhibitor of serine proteases, blocks multiple complement dependent and independent microbicidal activities of human serum. PLoS Pathog. 2019, 15, e1008232 10.1371/journal.ppat.1008232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.