

Abstract
Guillain-Barré syndrome (GBS) is a rapidly progressive, monophasic, and potentially devastating immune-mediated neuropathy in humans. Preceding infections trigger the production of cross-reactive antibodies against gangliosides concentrated in human peripheral nerves. GBS is elicited by at least five distinct common bacterial and viral pathogens, speaking to the notion of polymicrobial disease causation. This Opinion emphasizes that GBS is the best-supported example of true molecular mimicry at the B-cell level. Moreover, we argue that mechanistically, single and multiplexed microbial carbohydrate epitopes induce IgM, IgA, and IgG subclasses in ways that challenge the classic concept of thymus-dependent (TD) versus thymus-independent (TI) antibody responses in GBS. Finally, we discuss how GBS can be exemplary for driving innovation in diagnostics and immunotherapy for other antibody-driven neurological diseases.
GBS, true molecular mimicry at the B-cell level
In molecular mimicry, the antigenic structures of pathogens and humans are sufficiently similar to induce an autoreactive response of T or B lymphocytes after infection, contributing to disease pathogenesis [1]. A well-known example of a disease likely induced via molecular mimicry is rheumatic fever caused by Streptococcus pyogenes [2]. Although the concept of molecular mimicry is intuitive and mechanistically appealing, it is exceedingly hard to prove [3, 4]. Historically, much work on molecular mimicry focuses on T-cell cross-reactivity, and much less on B cells or antibodies.
Guillain-Barré syndrome (GBS) is an immune-mediated neuropathy causing a rapidly progressive weakness that may affect respiratory muscles for which patients need ventilation at an ICU (Box 1 Clinician’s corner, and websites for patients, neurologists and other resourcesi, ii, iii, iv). GBS can develop within 1–3 weeks after infection with several commonly found pathogens, including viruses and bacteria. These include Campylobacter jejuni, involved in approximately 30% of GBS cases and a common cause of bacterial gastroenteritis, Mycoplasma pneumoniae, hepatitis E virus (HEV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), and Zika virus (ZIKV)[5]. The associations between these pathogens and GBS have all been proven in comparative case-control studies [6, 7]. Other infections may trigger GBS as well, including influenza virus [8], but case-control studies are lacking. Recent studies have indicated that there is a small risk that SARS-CoV-2 infections may precede GBS [9, 10], which may be higher than after vaccination against SARS-CoV2 [9]. Of note, GBS is a rare disease with an incidence rate of approximately 1–2 cases per 100,000 per year, worldwide. However, pandemic or outbreaks of infections might increase the incidence of GBS temporarily or locally, as was observed in French Polynesia during the ZIKV outbreak and recently, in Peru with C. jejuni infection[6, 11]. Nevertheless, the risk for developing GBS following C. jejuni infection is estimated to be only 1 in 1,000–5,000 people, indicating that pathogen and host factors crucially determine susceptibility for developing GBS [12, 13].
Box 1. Clinician’s corner.
Guillain-Barré syndrome is a highly diverse disorder in terms of clinical presentation, course, and outcomes.
Preceding infections, cross-reactive anti-glycolipid antibodies, as well as complement activation are the three key factors in the pathogenesis and diversity of GBS.
Current treatments with immunoglobulins or plasmapheresis/exchange are insufficient for most GBS patients.
Biomarkers are required to support early GBS diagnosis, and to personalize and monitor patient treatments.
Advances in immunological and biochemical technologies are allowing the development of combinatorial antigen assays to measure antibody properties for the diagnosis and subtyping of GBS patients.
We anticipate that new therapies for GBS might include inhibitors that target neuropathogenic antibodies (cleaving enzymes, extracorporeal or in vivo-capturing of antibodies), as well as complement proteins.
There are several clinical variants of GBS that relate to the type of peripheral nerves involved. For instance, the classic sensorimotor form of GBS causes limb muscle weakness as well as sensory deficits, and is the most frequent manifestation in the Western world [14]. The pure motor form of GBS causes only muscle weakness of the limbs, whereas in the Miller Fisher syndrome (MFS), weakness is restricted to muscles involved in eye movements which cause double vision [14]. Ataxia is also a prominent feature in MFS. In approximately half of GBS patients, serum antibodies against various gangliosides and other glycolipids are found [15–17]. Gangliosides are a family of sialylated glycolipids abundantly expressed in human cell membranes. Crucial to GBS is their high concentration in pre-synaptic membranes of the neuromuscular junction and their presence in the axolemma at the nodes of Ranvier which allow saltatory nerve conduction [18]. Peripheral nerves vary in their ganglioside composition, which contributes to explain the association between a patient’s clinical variant and the specificity of the anti-ganglioside antibodies detected (Figure 2). For example the motor variant of GBS is highly associated with serum antibodies to the GM1a and GD1a gangliosides present in motor nerves [19]; by contrast, MFS is highly associated with the presence of antibodies directed against the GQ1b ganglioside present in oculomotor nerves [20]. In addition, prior C. jejuni infections are associated with the presence of antibodies to GM1, GD1a, and GQ1b [17]. The rare occurrence of GBS following a relatively common infection such as C. jejuni suggests that the B-cell response to gangliosides is strictly controlled, and that a series of factors need to coalesce to develop a pathogenic cross-reactive antibody response. GBS has a typical monophasic clinical course which manifests as a rapid disease progression (within days to weeks) followed by a slow recovery (within weeks to months) which parallels the reduction in antibody titers [21]. Although in GBS, active disease lasts less than a week in most patients, nerve recovery is frequently incomplete and often results in residual disability and complaints.
Figure 2. Distinct symptom patterns of GBS variants that have been associated with elicited antibodies.
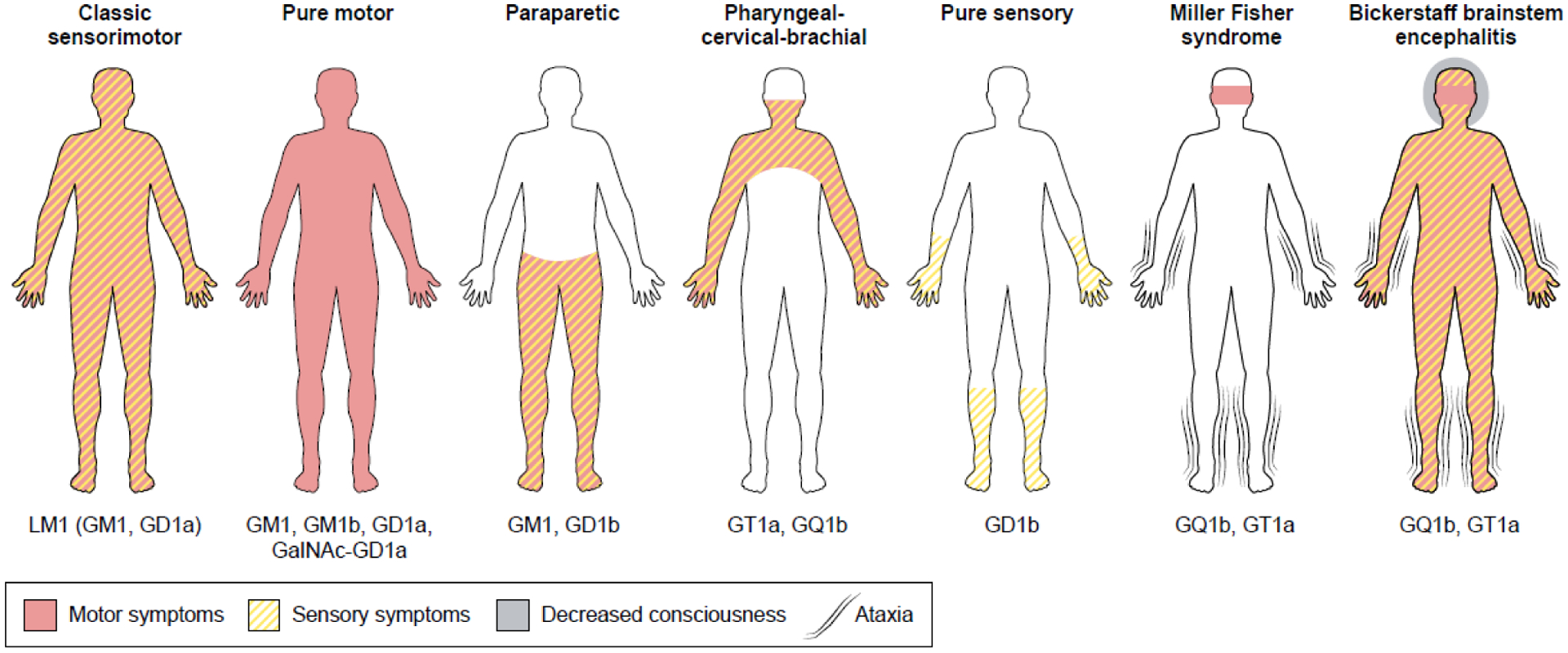
The gangliosides indicated below the clinical variants represent the predominant targets of the antibodies found in the serum of these GBS patients. Antibodies against GM1 and GD1a are associated with the pure motor form of GBS, but also occur in other clinical variants, such as the paraparetic form in which only lower limbs are affected. The pharyngeal-cervical-brachial variant (affecting upper limbs), Miller Fisher syndrome and Bickerstaff brainstem encephalitis are associated with antibodies against GQ1b and GT1a. The figure is modified from [83].
While GBS is clearly immune-mediated, it lacks some typical general features of autoimmune diseases such as chronic disease course and association with certain HLA class I or II alleles (Box 2) [22]. Instead, GBS behaves more like a post-infectious disease because of the strong relationship with a preceding infection and the monophasic disease course [5–8]. Indeed, we have previously proposed that GBS is one of the best-documented examples of a disease caused by molecular mimicry at the B-cell level because all revisited Witebsky’s postulates [23] – from epidemiological disease association to reproduction of disease in animal models – have been fulfilled [24]. Thus, in the case of GBS, similar epitopes in pathogens and human hosts have resulted in actual pathology stemming from cross-reactive antibodies; mimicry epitopes have been firmly established for C. jejuni within the lipo-oligosaccharide (LOS) inducing pathogenic antibodies to host gangliosides [25]. As shown in Figure 3, it is the carbohydrate part of LOS (and not the lipid part), that mimics the ganglioside. Of note, LOS from C. jejuni isolated from patients with pure motor GBS mimic GM1 and GD1a, while LOS from C. jejuni isolated from patients with MFS mimics GQ1b [25]. Similar cross-reactive antibodies to those found in GBS patients have been induced in mice and rabbits after immunization with LOS from C. jejuni isolates derived from GBS patients; moreover, such antibodies against several gangliosides have induced complement-mediated neural damage [25,26]. For other pathogens related to GBS, specific criteria remain to be fulfilled. However, in the case of M. pneumoniae, the epidemiological association between preceding infection and the development of GBS is firmly established and cross-reactive antibodies against galactocerebrosides that are present in human nerves have been identified [26, 27]; nevertheless, the exact microbial mimics remain to be identified.
Box 2. Distinguishing features of GBS.
GBS is not a typical autoimmune disease
GBS is a typical post-infectious disease
More than 95% of patients have a monophasic disease course [62]
Two-thirds of patients have symptoms of a recent respiratory or gastro-intestinal infection or a vaccination
Serological evidence for a recent infection is present in 50–60% of patients [5]
Characteristic clinical features are associated with an eliciting pathogen
Disease prognosis is associated with an eliciting pathogen
Figure 3. Molecular mimicry of LOS and gangliosides in GBS caused by Campylobacter jejuni.
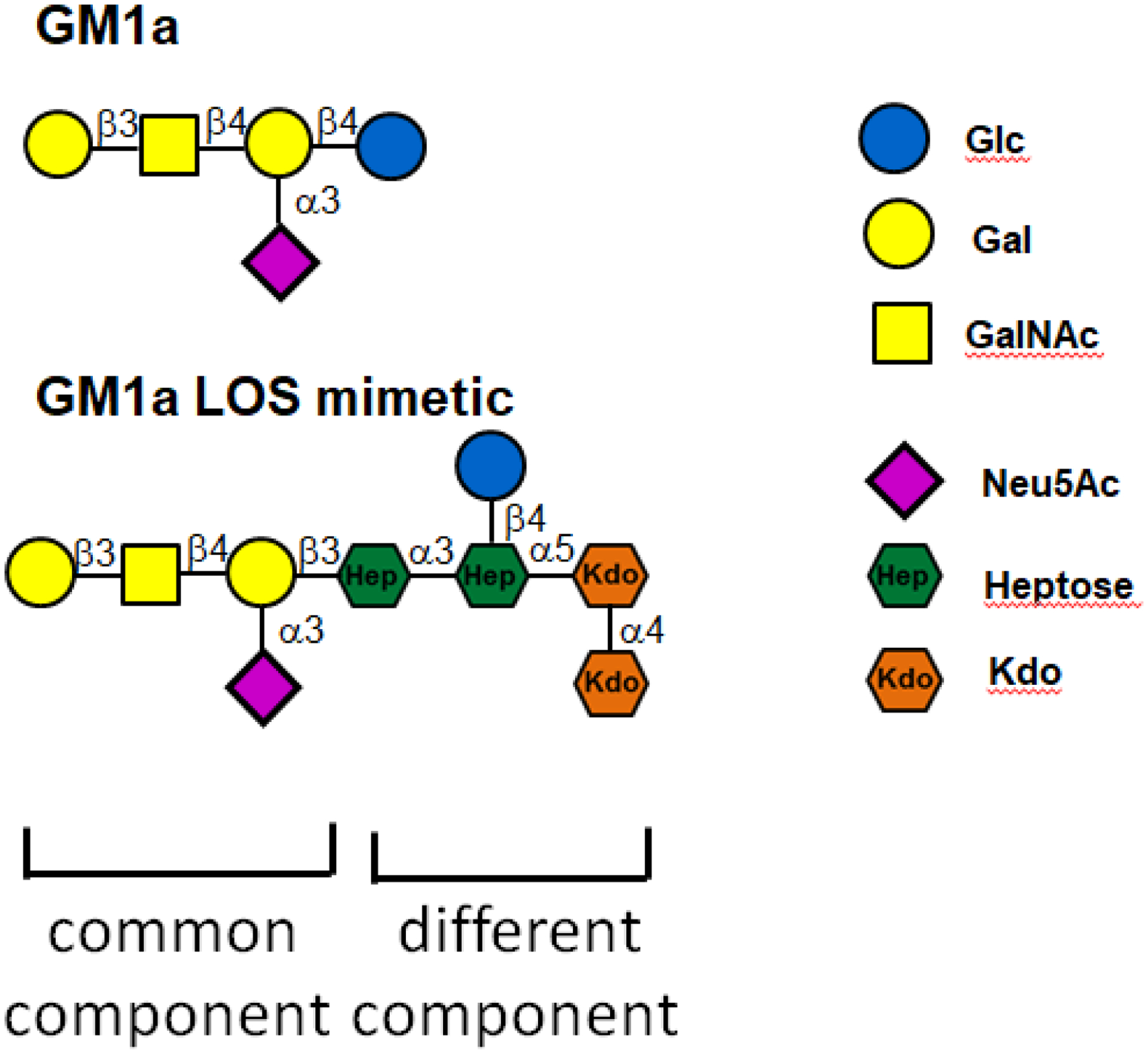
The upper part of the figure shows the oligosaccharide moiety of GM1a. The lipo-oligosaccharide (LOS) of C. jejuni mimics this structure. The terminal parts of the molecules are shared (common component) whereas LOS also contains a different component representing the inner core. The host ganglio-oligosaccharide has a glucose residue whereas the bacterial LOS has a heptose moiety at the same position. Furthermore, the heptose of LOS is linked to an inner core saccharide composed of additional heptose and ketodeoxyoctonic acid (KDO) moieties [25, 46].
Recently, several studies have deepened our understanding of the pathogenesis of GBS in terms of the role of carbohydrate mimicry, the cross-reactive antibody response, and its effect on nerves. In this Opinion, we describe how novel chemoenzymatic approaches can be used to create host epitopes and mimetics as well as multiplex assays; we argue that this approach can lead to a deeper understanding of the mechanism by which molecular mimicry can induce autoimmunity. Furthermore, we discuss how antibody responses against gangliosides shed new light on the classic TI-TD paradigm. Finally we outline recent progress in other neuroinflammatory disorders in which molecular mimicry may be involved.
Antibodies to complexes of heteromeric glycolipids
A groundbreaking discovery in 2004 documented (via ELISA) that patients with GBS could harbor serum antibodies against a heterodimer complex of GD1a and GD1b, instead of harboring antibodies against each individual ganglioside alone [28]. Furthermore, the strength of antibody binding to a particular ganglioside could either be enhanced or reduced by close proximity to other gangliosides [26]. The relevance of this finding was demonstrated in a murine ex vivo muscle-nerve preparation showing that certain antibodies targeting GM1 did not bind to live nerve terminals due to the presence of other gangliosides in close proximity to GM1 [29]. Similar modifying effects have been described for other (glyco-)lipids, including cholesterol, galactocerebroside, and sulfatide [30], suggesting that this phenomenon is not restricted to gangliosides alone. Moreover, the optimal spacing of glycolipids might also be important for the formation of IgG hexamers against gangliosides, allowing proper binding of C1q complement [31]. This is relevant as complement activation contributes to nerve damage in GBS via the formation of the membrane attack complex (MAC) [32].
Protective effect of anti-glycolipid antibody endocytosis in cells
In GBS, antibodies to gangliosides bind both the peripheral nerve terminal and the nodes of Ranvier where binding is not prevented by myelin or Schwann cells [33]. Notably, neural tissues appear to be particularly sensitive to antibodies binding targets embedded in myelin and axonal membranes (evidence summarized in Box 3). Some nerve regions however are more vulnerable than others to damage, and in a landmark study, [34] researchers built on evidence that neuronal membranes with high endocytic activity, including in nerve terminals, were less vulnerable to damage due to the rapid endocytosis of bound antibodies [35]. Specifically, anti-ganglioside antibody administered to transgenic mice expressing gangliosides exclusively in neurons (GalNAcT−/−Tg(neuronal) mice) was rapidly cleared by endocytic uptake at nerve terminals. Subsequently, the antibody was transported in retrograde manner to the cell body of the motor neuron in the spinal cord and locally secreted [20]. This might contribute to explain why patients with MFS harboring anti-GQ1b antibodies initially present with peripheral oculomotor neuropathy followed by brainstem encephalitis (‘Bickerstaff encephalitis’) [36].
Box 3. Neural tissue vulnerability to antibody attack.
Neural tissue is particularly sensitive to loss of function, has low resilience and permissiveness to changes in the microenvironment, and has limited repair capabilities.
The detailed molecular composition of local microenvironments along the soma, extended axon, and synapse of neurons, controls vulnerability to antibodies [29].
In humans and mice, gangliosides are concentrated and accessible to antibodies at the nodes of Ranvier which are key structures for normal saltatory nerve conduction [18].
Target-mediated clearance of antibodies by neuronal endocytosis can limit the damage to axon terminals. Mouse ex vivo studies indicate that the highly dynamic nature of the neurological synapse -- with constant vesicle release as well as uptake of antibodies -- is much less vulnerable than the static nature of membranes at the nodes of Ranvier [34, 35]
Antibodies can access their molecular targets despite the presence of the blood-nerve barrier in the peripheral nervous system (PNS) and blood-brain barrier in the CNS, as the barrier can be leaky at certain locations, e,g, at nerve roots, ganglia, and nerve terminals [18].
Pathogenic antibodies access the NMOSD self-antigen aquaporin-4 (AQP4) expressed on astrocyte podocytes at the blood-brain barrier. Gut microbes are thought to provide cross-reactive mimetics of AQP4 [59].
In chronic neuropathies such as autoimmune nodopathy, IgG4 binding to Schwann cells can impair the function of the latter in the absence of complement activation; passive transfer of human antibody against neurofascin-155 into mice has resulted in reduced NF155 protein content in nerve roots (via immunoblotting), suggesting depletion of target antigens and interference with the formation of paranodal loops [63].
In MS, autoantibodies do not necessarily need to access the CNS parenchyma for pathogenic action since they can promote antigen presentation in lymph nodes as demonstrated in the EAE mouse model for MS [64]. This is contrary to the common assumption that autoantibodies need to exclusively engage their target in the CNS.
Novel insight into antibody responses to glycolipids
The finding that anti-glycolipid antibodies are removed from circulation after endocytosis [34] also prompts re-evaluation of the concept of B-cell tolerance to gangliosides. For a long time, immunization of rodents with self-glycolipids to induce antibodies has proven difficult, requiring extensive immunization regimens, or the use of transgenic animals; many of these obstacles have been attributed to B-cell tolerance mechanisms [37, 38]. However, a study demonstrating anti-GD1b antibody- secreting B cells (via ELISPOT) following immunization of wildtype mice with GD1b-containing liposomes [34], indicates that B cells can respond to gangliosides, at least in mice. Also in humans, antibodies against gangliosides can be comprised in the normal immune repertoire, as evidenced by the presence of IgM antibodies against GM1 in healthy individuals [39]. Because B-cell tolerance to gangliosides may not be complete, regulatory mechanisms are likely present that limit the risk of developing autoimmunity to self-glycolipids and this may be impaired in patients with GBS. In support of this, GBS patients were recently found to exhibit defective signaling from the inhibitory receptor Siglec-10 (expressed by B cells); consequently such impaired signaling could have resulted or contributed to aberrant B-cell responses against gangliosides or sialic-acid bearing molecules such as LOS [40].
Therefore, how are B cells activated to produce anti-glycolipid antibodies in GBS? The antibody response to glycolipids in GBS does not conform to the standard TD-TI definitions [41], because these glycolipid antibodies recognize repetitive carbohydrates but are dominated by IgG1 and IgG3 subclasses whereas typical anti-carbohydrate antibodies are IgM or IgG2 (Box 3) [42]. However, somatic hypermutations reported in the anti-GM1 IgM of a GBS patient suggest that anti-glycolipid antibodies in GBS patients are generated in a TD-manner [43]. Since antibody responses to glycolipids are relatively short-lived in GBS patients (i.e. antibody titers decline within weeks to months), it is possible that class switching is induced by lymphocyte subsets other than T cells. In mice immunized with the haptenated-lipid antigen 4-Hydroxy-3-nitrophenylacetyl (NP)-α-GalCer, invariant NK T cells have been reported that promote TD-associated IgG subclasses in an IL-21 and CD1d-dependent manner; however, in this study, no memory B-cell responses were induced [44]. As memory responses to both TD and TI antigens require a germinal center reaction [45], it remains to be investigated whether a germinal center response to gangliosides is induced in mice and humans.
Recent breakthroughs in understanding molecular mimicry in GBS
C. jejuni strains associated with GBS often produce a mixture of LOS molecules mimicking the saccharide moieties of several gangliosides, such as GM1a and GD1a [25, 46]. The antibodies of GBS patients that recognize ganglioside complexes cross-react with purified LOS of these strains in vitro [47, 48], suggesting that antibodies to heteromeric complexes can also be induced via molecular mimicry. However, purified LOS contains a mixture of molecules which complicates detailed comparisons of antibody specificities and binding strengths. Moreover, the chemical structure of the saccharide component of gangliosides and the oligosaccharide of LOS of C. jejuni differ [25]. In particular, the formation of ganglio-oligosaccharides is initiated by a glucose residue followed by galactose, whereas LOS of GBS-associated pathogenic strains contains a heptose moiety attached to galactose (Figure 3) [25]. Furthermore, this LOS heptose is linked to an additional heptose and to ketodeoxyoctonic acid (KDO) moieties which together, form the inner core of LOS, further increasing their dissimilarity with gangliosides [46]. Of note, heptose is only expressed by gram-negative bacteria and its metabolite heptose-1,7-bisphosphate was recently identified as a bacterial pathogen-associated molecular pattern (PAMP) [49], demonstrating that human innate immunity has evolved to respond to, rather than tolerate, this oligosaccharide. We hypothesize that this could also be the case for adaptive immunity, in particular for B cells.
To test this hypothesis and develop a more robust platform to diagnose and monitor GBS disease, we exploited advances in chemoenzymatic synthesis to prepare a panel of well-defined and homogeneous oligosaccharides composed of the inner core of the LOS of C. jejuni extended by various ganglioside carbohydrate mimics (Figure 4a) [50]. Similar oligosaccharides derived for gangliosides were prepared. All synthetic carbohydrates were equipped with an artificial amino-containing linker which made it possible to immobilize them on glass slides bearing amino reactive succinamate esters. The resulting glycan microarray provided a convenient platform to examine binding specificities of lectins, anti-ganglioside antibodies, and serum antibodies from GBS patients. Although lectins and monoclonal anti-ganglioside antibodies did not differentiate between ganglio-oligosaccharides and corresponding LOS mimics, GBS patient serum antibodies bound more strongly to particular LOS derived structures than to ganglio-oligosaccharides [50] (Figure 4b). The data suggest that antibodies were elicited against a foreign epitope containing a heptosyl residue which is unique to bacterial LOS, and to an oligosaccharide component mimicking that of gangliosides. The antibodies could cross-react with particular gangliosides because these represent partial epitopes [50]. In summary, these findings suggest that a certain degree of structural dissimilarity is required to break immuno-tolerance and presumably, to develop autoimmunity via molecular mimicry.
Figure 4. Chemoenzymatic diagnostics for monomeric and heteromeric-multiplexed antigens.
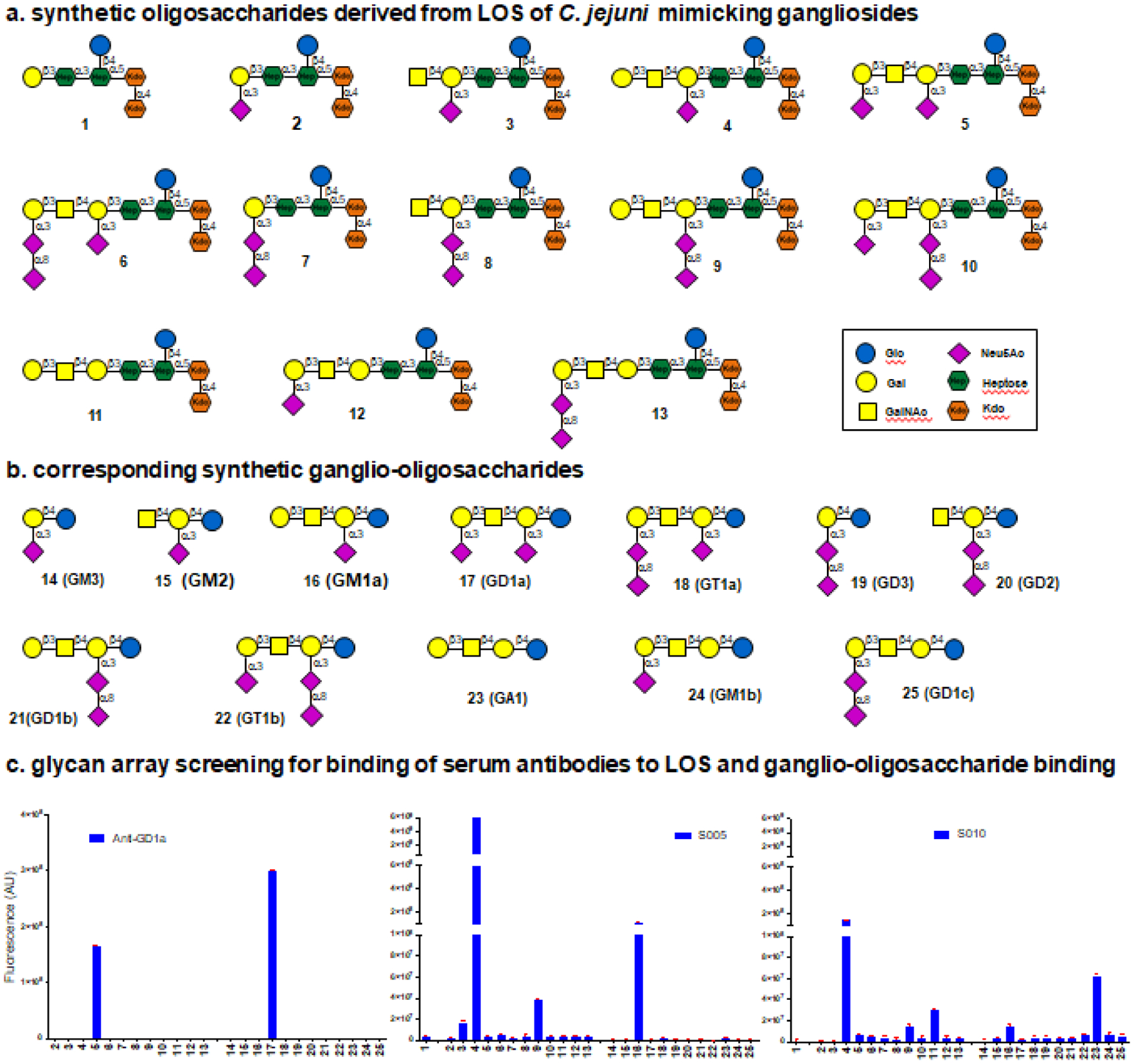
Novel combinatorial diagnostics based on immunofluorescence detection are being developed to finely discriminate antibodies cross-reacting between host structures and pathogen mimetics. Panel a and b show examples of various oligosaccharides that have been synthesized [50]. Panel c shows examples of probed oligosaccharide arrays with a monoclonal antibody against GD1a, and the sera of two GBS patients (S005 and S010). Bound antibodies were visualized using fluorescently-conjugated anti-human IgG secondary antibodies and fluorescence intensities were quantified using a microarray scanner [50]. The numbers on the x-axis correspond to the structures displayed in a and b. These diagnostic systems can also be used to identify antibodies directed against epitopes being formed by heteromeric-multiplexed gangliosides. The figure is modified from [50].
Molecular mimicry in other neuroinflammatory diseases
Classic as well as recent work has significantly deepened our understanding of molecular mimicry driving human neurological diseases other than GBS [51–55], notably multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD) and narcolepsy. Table 1 provides a comprehensive selection of putative mimicry epitopes, with variable levels of experimental evidence derived from studies in animal models and patient samples.
Table 1.
Neurological autoimmune diseases and molecular mimicry
| Disease | Pathogen | Pathogen epitope | Host epitope | Host | References |
|---|---|---|---|---|---|
| MS | Epstein-Barr virus | EBNA1 (386–405) SQSSSSGSPPRRPPPGRRPF | Glial Cellular Adhesion Molecule (Glial)CAM (370–389) ATGRTHSSPPRAPSSPGRSR |
Human | [54] |
| MS | Epstein-Barr virus | EBNA1 (431–440) PGAIEQGPAD | Anoctamin 2 (140–147) PGDIELGPLD | Human | [58] |
| MS – EAE | Epstein-Barr virus | FARQAVWLRE | RASGRP2 (78–87) | Human | [53] |
| MS | Human herpesvirus 6 | U24(1–13) MDPPRTPPPSYSE | MBP (92–104) IVTPRTPPPSQGK | Human | [75] |
| MS – EAE | Cytomegalovirus | UL86; mcp (986–993) WLRSPFSR | MOG (39–46) WYRPPFSR | Rhesus monkey | [76] |
| MS – EAE | Akkermansia muciniphila | LSVGWISGQY | RASGRP2 (78–87) | Human | [53] |
| MS – EAE | Chlamydia pneumoniae | Cpn0483 gene YGCLLPRNPRTEDQN | MBP (68–86) YGSLPQKSQRTQDEN | Lewis rat | [77] |
| MS | Chlamydia pneumoniae | Cpn0442 gene KNLFPPYEPPP | MBP (84–102) KNIVTPRTPPP | Lewis rat | [77] |
| MS | Acinetobacter calcoaceticus | 3-oxoadipate COA-transferase subunit A (83–97) DSYVFDELYRAGKIE | MOG (43–57) PFSRVVHLYRNGKDQ | Human | [78] |
| MS | Acinetobacter calcoaceticus | 4-carboxy-muconolactone decarboxylase (38–52) QNFISRFAWGEVNSR | MBP (110–124) GLSLSRFSWGAEGQR | Human | [78] |
| MS | Pneumocystis aeruginosa | γ-carboxy-muconolactone decarboxylase (38–52) QEMITRHAWGDIWTR | MBP(110–124) GLSLSRFSWGAEGQR | Human | [78] |
| MS – EAE | Lactobacillus reuteri | UvrA peptide TIKREGFVRVQVD | MOG(38–50) GWYRSPFSRVVHL | Mouse C57Bl/6 | [51] |
| NMOSD | Clostridium perfringens | ABC transporter permease(204–217) FIILPVSMVLISLV | AQP4 (63–76) EKPLPVDMVLISLC | Human | [79] |
| NMOSD | Human T-lymphotropic virus | TAX1BP1 protein (219–233) EFKKRFSDATSKAHQ | AQP4 (6–20) EFKRRFKEAFSKAAQ | Human | [80] |
| Narcolepsy | Influenza virus H1N1 | pHA273–287 AMERNAGSGIIISDT | Hypocretin/orexin 54–66 HGAGNHAAGILTL | Human | [81, 82] |
Bold letters indicate amino acids identical in both epitopes.
MS: multiple sclerosis, NMOSD: neuromyelitis optica spectrum disorder, GBS: Guillain-Barré syndrome, HCRT: hypocretin/orexin
Although MS mimicry research mostly focuses on T-cell epitopes, an exciting study from 2022 [54] provides evidence of a human antibody that is cross-reactive between the EBV nuclear antigen 1 (EBNA1) epitope 386–405 and the central nervous system (CNS) protein glial cell adhesion molecule (GlialCAM). It has been suggested that such antibodies might potentially contribute to brain tissue damage in MS; immunization with this specific EBNA1 epitope aggravated symptoms of experimental autoimmune encephalomyelitis (EAE; model for MS) in SJL/J mice, and appeared to be driven by proteolipid protein (PLP) peptide 139–151 [54].
In MS patients, recent evidence of mimicry at the T-cell level against EBV and Akkermansia muciniphila has been reported [53]; A. muciniphila is a gut commensal with features that might limit or promote MS and gut inflammation. Epitopes from A. muciniphila are presented by HLA-DR15 molecules, and autoreactive T-cell clones cross-react with HLA-DR-derived self-peptides, peptides derived from EBV and A. muciniphila, as well as autoantigens [53]. HLA-II polymorphisms are considered a major genetic risk factor for developing MS [56]. A similar study with MS patient CD4+ T cells identified a novel candidate autoantigen peptide from RAS Guanyl Releasing Protein 2 (RASGRP2) that is expressed in neurons and B cells [57]. Thus, identifying a putative microbial mimic of RASGRP2 would be most valuable. Another example of molecular mimicry is the presence, in some MS patients, of IgG antibodies binding both the EBV protein EBNA-1 and anoctamin 2, a chloride channel protein on neurons in the CNS [58].
Finally, using the EAE model in C57BL/6 mice immunized with MOG35–55 peptide, one study showed that two bacterial species in the gut microbiota jointly activated myelin oligodendrocyte glycoprotein (MOG)-specific CD4+ T cells [51]. Specifically, Lactobacillus reuteri expressed mimicry peptides in its UvrABC system protein A (UvrA) protein, while Erysipelotrichaceae bacteria elicited pro-inflammatory factors IL-23 and serum amyloid A (SAA) which enhanced the effector function of pathogenic Th17 cells, exacerbating EAE [51]. Although in need of confirmation in humans, these findings suggest that two key constituent gut commensal bacteria activate CNS-specific T cells, where one species yields mimicry epitopes (e.g. MOG), and the other elicits inflammatory responses (e.g. IL-23 and SAA).
In NMOSD, there is evidence for an antibody mimicry epitope expressed by Clostridium perfringens and a host epitope of aquaporin-4 (AQP4), expressed by astrocytes [59]. Particular Clostridia clusters (IV, XIVa and XVII) promote T-cell regulation [59]. Overabundance of C. perfringens appears to generate AQP-specific T and B cells, and plasma cells secrete pathogenic antibodies against AQP4. Moreover, the ABC-TP of C. perfringens sequence 204–217 has homology with AQP463–76 [59].
In type 1 narcolepsy, autoantibodies contributing to pathophysiology have not been consistently identified despite large efforts, and the current consensus is that a T-cell mediated response that includes mimicry epitopes is likely contributing to the pathogenic mechanism [60].
In summary, accruing evidence in certain neurological diseases other than GBS suggests that there are key contributions to neural tissue damage that may be mediated by both T- and B-cell mimicry epitopes.
Concluding remarks
By numbers, GBS is a minor disease; however, it provides new insight into basic immunology in terms of TD and TI antigens, antibody and complement functionality, and the fascinating but complex molecular mimicry concept.
The insights and technologies discussed in this Opinion article contribute to the arsenal needed to achieve rapid and updated progress in immunopathogenesis, diagnostics, and innovative treatments for patients suffering from GBS and related diseases in which molecular mimicry is implicated. Indeed, with newly developed innovative technologies such as microarrays of synthetic oligosaccharides, the community can further examine multiplexed carbohydrate antigen-forming antibody epitopes, as well as molecular neighbor and crypto effects on antibody binding. This is important for clarifying the neurotoxic potential of anti-glycolipid antibodies and define the relationship between these antibodies and disease severity and outcomes. Indeed, the international research consortia such as the International GBS Outcome Study (IGOS)iii is promoting rapid worldwide diffusion of such insights and technologies, and aims to interpret these in the context of regional variation for GBS clinical phenotypes (e.g. [14]).
Many limitations and questions remain (see outstanding questions), the answer to which can help propel the GBS field forward, and will hopefully also cross-fertilize our understanding, diagnostic approaches, and therapy endeavors for other immune-mediated neurological diseases.
Outstanding questions.
To what extent can controlled measures that can reduce the incidence of infections with Campylobacter jejuni, also reduce the incidence of GBS globally? GBS incidence appears to be less frequent in Singapore since the COVID-19 pandemic [74], perhaps due to social distancing with reduced exposure to contaminated food sources. Further identification of new pathogens causing GBS might also contribute to exposure prevention.
Can novel systems such as oligosaccharide microarrays that are used to detect at high sensitivity and specificity the full spectrum of antibodies against multiplexed carbohydrate antigens be relevant for improving GBS diagnosis, subtyping, and prognostication?
Which pathogens, other than those currently identified, are associated with GBS and which mimicry epitopes are involved?
What are the contributions of antibodies against multiplexed antigens and differential functions of IgM versus the four subclasses of IgG in GBS?
What are the targets of neuropathogenic antibodies (or T cells) in GBS patients not harboring anti-ganglioside antibodies, especially those presenting with the demyelinating form of GBS?
Which environmental conditions or host factors activate T- and B-cells via mimicry epitopes provided by the gut microbiota and/or infectious pathogens, and which contribute to the pathogenesis of certain neuroimmunological diseases? If so, which specific diseases?
Which innovative therapies such as B-cell depletion, complement inhibition, or IgG depletion by extracorporeal immune-absorption are safe and effective for GBS patients? For example, pharmacological treatment with Streptococcus enzyme (imlifidase) to reduce ganglioside IgG titers is currently being tested in an ongoing open-label, single arm, multi-center Phase II trial (clinicaltrials.gov NCT03943589)v in 30 GBS patients receiving standard of care intravenous immunoglobulin (primary endpoints, safety and efficacy).
Can GBS treatments be optimized (dosing, treatment regimens) using sophisticated antibody monitoring technologies that include sensitive antibody affinity measurements?
Key Figure, Figure 1. Overview of the pathogenesis of Guillain-Barré syndrome (GBS).
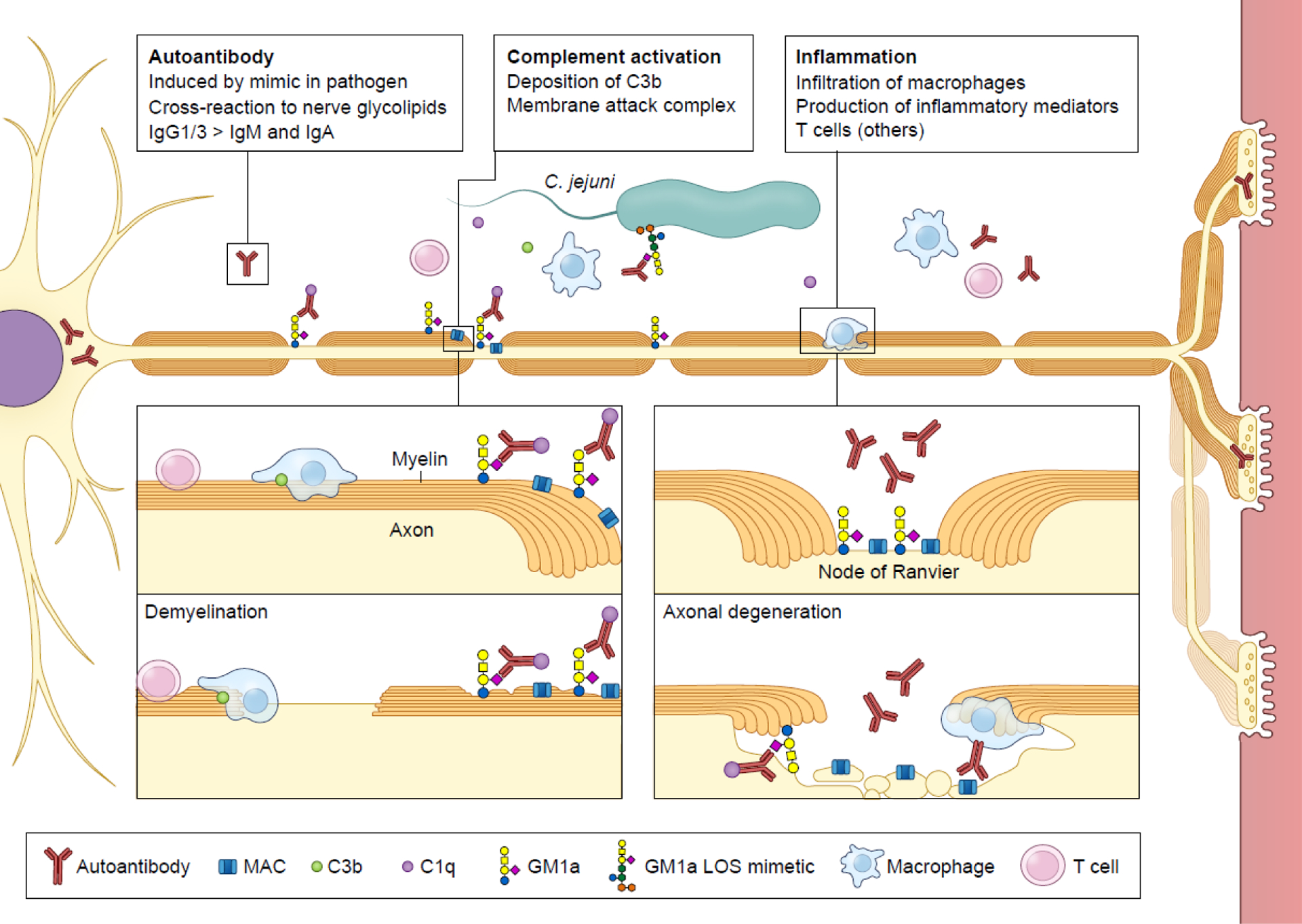
GBS is an acute immune-mediated peripheral neuropathy usually triggered by a preceding infection, the predominant type being Campylobacter jejuni (~30%). Antibodies raised against C. jejuni lipo-oligosaccharide (LOS) during infection via carbohydrate mimicry may cross-react with various human nerve gangliosides, including GM1a. The specificity of the anti-ganglioside antibodies is associated with the type of clinical GBS variant, reflecting the distribution of the targeted gangliosides in peripheral nerves. GBS is caused by axonal degeneration and/or demyelination of the nerves. Antibody depositions are found in axonal GBS at the nerve axons, especially the nodes of Ranvier, and in demyelinating GBS, at the myelin sheaths. Serum antibodies are usually IgG1 of IgG3 (less frequently IgM or IgA) and the highest titers are found in patients upon hospital admission. Binding of these antibodies results in local deposition of complement factors (including C1q, C3b) and formation of membrane attack complex (MAC). Macrophages infiltrate the nerve at the site of damage and additional T cells may be found in nerves. GBS is a monophasic disorder reflecting transient immune-mediated damage; most patients begin to clinically improve with the disappearance of anti-ganglioside antibodies. Subsequent nerve regeneration is a slow process and is often incomplete, explaining the high proportion of patients with residual disability or complaints.
Box 4. Antibody responses against glycolipids: tolerance, T-cell help, and memory.
Classic distinctions between TI (repetitive carbohydrates, inducing IgM and IgG2 in humans) and TD (protein; inducing IgG1, IgG3, and IgG4 in humans) responses do not fully apply to antibody responses against glycolipids because in GBS, anti-glycolipid antibodies are mainly IgG1 and IgG3 [42, 65].
TD and TI type 1 and type 2 responses are formally distinguished by using athymic nude mice (Foxn1 mutation), and CBA/N mice with a mutation in Bruton’s tyrosine kinase (Btk) [41]. In mice, TD responses are mediated by T cells or NKT cells; the latter give rise to class-switched but short-term antibody responses [44, 66].
Wildtype mice develop low titers of serum antibodies to self-gangliosides only [37]. However, knockout of the GalNac-transferase (GalNAcT−/−) in mice abrogates this ‘tolerance’ as these mice lack all complex gangliosides such as GM1 or GQ1b. Upon immunization with gangliosides or gangliosides mimicking LOS in mice, antibodies against GM1 or GQ1b undergo class switching to TD-dependent IgG isotypes and B cell memory [37], suggesting that a TD B-cell response to glycolipids can develop in vivo.
In humans, low titers of IgM antibodies to gangliosides develop only after birth [39], suggesting that antigenic stimulation is required for their induction.
In GBS patients, a TD response is strongly suggested by the profiles of antibody isotypes and subclasses (IgG1, IgG3, and IgA in addition to IgM). GBS-associated LOS causes strong activation of human dendritic cells, as evidenced by a higher production of cytokines and higher expression of co-stimulatory molecules in its presence; it may also polarize T cells towards a Th2 phenotype [67, 68]
An IgM-producing hybridoma reactive to GM1 from a GBS patient has provided evidence for somatic hypermutation and affinity maturation of patient antibodies, similar to what has been observed in chronic neuropathies such as multifocal motor neuropathy, which is mediated by IgM antibodies against GM1 [43, 69, 70]. However, the mutation status of IgG antibodies from GBS patients remains unknown.
The monophasic nature and rapid decline of antibody titer in GBS patients, mostly within months [21], suggests that there might probably be little to none memory B cell formation, but this remains to be proven.
Murine studies suggest that anti-glycolipid antibodies originate from an innate type of B cell, since anti-ganglioside antibodies are not mutated, and polyreactive and hybridoma cells express CD5 [71]. Whether this is similar in humans is unknown. Recent studies suggest that memory B cells and plasmablasts are expanded in the peripheral blood of GBS patients [72, 73].
Highlights.
Guillain-Barré syndrome (GBS) following Campylobacter jejuni infection is a true case of molecular mimicry-driven disease
The immunopathogenesis of GBS sheds new light on the multimolecular identities of self-antigens
The glycan-nature of mimicry epitopes that drives antibody class switching to IgG subclasses challenges rigid concepts of thymus-dependent and - independent B cell responses because anti-glycolipid antibodies are mainly IgG1 and IgG3, albeit short-lasting.
Combinatorial chemo-enzymatic technologies and arrays allow the development of novel diagnostic and research tools to identify antibodies against mono- and multimeric carbohydrate epitopes
A certain degree of dissimilarity may be required for the occurrence of molecular mimicry in Campylobacter jejuni-associated GBS.
Acknowledgments
This work was supported by the Dutch MS Research Foundation (J.D.L. and the Zabawas Foundation (J.D.L.). B.C.J was supported by Prinses Beatrix Spierfonds, GBS-CIDP Foundation International, Annexon, CSL-Behring, Grifols and Hansa Biopharma. RH was funded by GBS-CIDP Foundation International and the T2B collaboration project funded by PPP Allowance made available by Top Sector Life Sciences & Health to Samenwerkende Gezondheidsfondsen (SGF) under project number LSHM18055-SGF to stimulate public-private partnerships and co-financing by health foundations that are part of the SGF. G.J.B was funded by the National Institute of General Medical Sciences from the National Institutes of Health (Grant U01GM120408). The authors thank BSc Margot van der Maarel for a first version of Table 1 and Anna Sieben for artwork of several figures.
Glossary
- Astrocyte podocytes
end-feet of astrocyte extensions; contribute to the formation and function of the CNS blood-brain barrier.
- Axolemma
axon plasma membrane.
- Bickerstaff brainstem encephalitis (BBE)
rare variant of GBS presenting as peripheral neuropathy (usually the Miller Fisher syndrome) with weakness of muscles involved in eye movements, but progressing to limb weakness and lowered consciousness due to involvement of the brainstem as part of the CNS. BBE, like MFS, is associated with the presence of anti-ganglioside GQ1b antibodies.
- Blood-nerve barrier
cell and tissue structures controlling access of soluble molecules, including pathogenic autoantibodies to the nerve. This barrier does not prevent infiltration of leukocyte subsets since these behave distinctly from soluble molecules. The BNB is less tight at nerve roots, ganglia, and nerve terminals.
- C1q complement factor
8-polypeptide chain subcomponent of C1, the first component of the complement protein cascade, with a characteristic six-tulip like shape. The binding of the Fc-components of closely arrayed antibody molecules to C1q initiates the classic pathway of complement activation, including activation of the membrane attack complex (MAC). In humans, C1q can be bound by IgM, IgG1, IgG2 and IgG3, but not by other isotypes.
- Gangliosides
sialic acid-containing glycosphingolipids, composed of ceramide and oligosaccharide; enriched in lipid rafts and highly expressed in nervous tissue. Frequent attachment site for microbial toxins.
- Guillain-Barré syndrome (GBS)
immune-mediated peripheral neuropathy, usually triggered by a preceding infection; clinically characterized by rapidly progressive bilateral muscle weakness. GBS is clinically diverse and includes the predominant sensorimotor form, pure motor form, Miller Fisher syndrome (MFS), and other forms. The peripheral neuropathy is characterized by demyelination, axonal degeneration, or both. The specificity of the antibodies to gangliosides is associated with the clinical form and neuropathy subtype.
- Lipo-oligosaccharide (LOS)
endotoxin expressed by C. jejuni. LOS is chemically distinct from lipopolysaccharide (LPS) since it lacks the repetitive O-antigen. LOS consists of lipid A and an inner and outer core of oligosaccharides. Sialylated LOS strongly activates toll-like receptor TLR4.
- Miller Fisher syndrome
Clinical variant of GBS characterized by oculomotor neuropathy and weakness of muscles involved in eye movements causing complaints in double vision; associated with anti-ganglioside GQ1b antibodies.
- Molecular mimicry
Formal medical definition used in this Opinion is the structural similarities of host epitopes and pathogen epitopes which elicit autoreactive T and/or B cells driving pathogenesis. Postulates of Koch and Witebsky can be productively applied to validate suspected molecular mimicry.
- Neuromyelitis optica spectrum disorder
In the CNS, dominated by inflammation of the optic nerve (optic neuritis) and the spinal cord (myelitis). Previously known as Devic disease or neuromyelitis optica.
- Node of Ranvier
Incisures in the myelin sheath allowing saltatory pulse conduction. In the PNS, gangliosides are concentrated and/or more accessible to autoantibodies at the nodes of Ranvier.
- Paranodal loops or spirals
At both sides of the node of Ranvier, the myelin sheath is not compacted, and is filled with cytoplasm of the myelinating Schwann cell (PNS) or oligodendrocyte (CNS), spirally wrapped around the axon. Paranodal spirals resemble loops in cross-section.
- Pathogen-associated molecular patterns
conserved molecular motifs such as lipopolysaccharides or lipoproteins, present on pathogens and recognized by pattern recognition receptors.
- Pure motor form of GBS
Clinical GBS variant; patients have muscle weakness but no sensory deficits; associated with anti-gangliosides GM1 and GD1a antibodies.
- Saltatory conduction
occurs along myelinated axons; involves the propagation of electrical pulses from one node of Ranvier to another. Conduction velocity along myelinated fibers is much faster than along non-myelinated fibers (80–120 m/s versus 0.5 to 2.0 m/s).
- Sensorimotor form of GBS
Predominant clinical GBS form; patients have sensory deficits in combination with muscle weakness.
- Somatic hypermutation
mutations in the variable domains of immunoglobulin genes that occur during germinal center responses.
- Schwann cells
cells of the PNS that myelinate one axon, while CNS oligodendrocytes insulate and provide saltatory pulse conduction by wrapping cytoplasmic extensions around up to 70 axons. Schwann cells also surround small-diameter axons that are non-myelinated.
- Thymus-dependent (TD) versus thymus-independent (TI) antibody responses
requirements for B-cell activation and induction of antibody responses against antigens, based on classic thymectomy experiments and transgenic mice. As NK cell development also occurs in the thymus, a TD-response can also occur without T cell help.
- Witebsky’s postulates
a disease can be regarded as autoimmune based on: Direct evidence that it can be transferred by a pathogenic antibody or T cells; Indirect evidence based on the development of autoimmune disease in experimental animals; Conditional evidence from clinical signs and patient symptoms.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
European and transcontinental websites for patients: https://www.gbs-cidp.org/
International websites for clinical neurologists on diagnosis, prognosis and treatment: https://rede.tghn.org/gbs-flowchart-sample/gbs-main-sub/
International GBS Outcome Study IGOS-consortium: https://gbsstudies.erasmusmc.nl/
Explanation of GBS for the general public by Dr. Ruth Huizinga: https://www.youtube.com/watch?v=XwosOoagyeg
Clinical trial (see Outstanding questions Box)
REFERENCES
- 1.Rose NR (2017) Negative selection, epitope mimicry and autoimmunity. Curr Opin Immunol 49, 51–55 [DOI] [PubMed] [Google Scholar]
- 2.Cunningham MW (2014) Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol 33, 314–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitton JL and Fujinami RS (1999) Viruses as triggers of autoimmunity: facts and fantasies. Curr Opin Microbiol 2, 392–397 [DOI] [PubMed] [Google Scholar]
- 4.Deshpande SP, et al. (2001) Herpes simplex virus-induced keratitis: evaluation of the role of molecular mimicry in lesion pathogenesis. J Virol 75, 3077–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobs BC, et al. (1998) The spectrum of antecedent infections in Guillain-Barré syndrome: a case-control study. Neurology 51, 1110–1115 [DOI] [PubMed] [Google Scholar]
- 6.Cao-Lormeau VM, et al. (2016) Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van den Berg B, et al. (2014) Guillain-Barré syndrome associated with preceding hepatitis E virus infection. Neurology 82, 491–497 [DOI] [PubMed] [Google Scholar]
- 8.Lehmann HC, et al. (2010) Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis 10, 643–651 [DOI] [PubMed] [Google Scholar]
- 9.Patone M, et al. (2021) Neurological complications after first dose of COVID-19 vaccines and SARS-CoV-2 infection. Nat Med 27, 2144–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luijten LWG, et al. (2021) Guillain-Barré syndrome after SARS-CoV-2 infection in an international prospective cohort study. Brain 144, 3392–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos AP, et al. (2021) Guillain-Barré syndrome outbreak in Peru 2019 associated with Campylobacter jejuni infection. Neurol Neuroimmunol Neuroinflamm 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tam CC, et al. (2006) Incidence of Guillain-Barré syndrome among patients with Campylobacter infection: a general practice research database study. J Infect Dis 194, 95–97 [DOI] [PubMed] [Google Scholar]
- 13.Orlikowski D, et al. (2011) Guillain-Barré syndrome following primary cytomegalovirus infection: a prospective cohort study. Clin Infect Dis 52, 837–844 [DOI] [PubMed] [Google Scholar]
- 14.Doets AY, et al. (2018) Regional variation of Guillain-Barré syndrome. Brain 141, 2866–2877 [DOI] [PubMed] [Google Scholar]
- 15.Lleixa C, et al. (2021) Autoantibody screening in Guillain-Barré syndrome. J Neuroinflammation 18, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halstead SK, et al. (2016) Microarray screening of Guillain-Barré syndrome sera for antibodies to glycolipid complexes. Neurol Neuroimmunol Neuroinflamm 3, e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caudie C, et al. (2011) Preceding infections and anti-ganglioside antibody profiles assessed by a dot immunoassay in 306 French Guillain-Barré syndrome patients. J Neurol 258, 1958–1964 [DOI] [PubMed] [Google Scholar]
- 18.Willison HJ and Yuki N (2002) Peripheral neuropathies and anti-glycolipid antibodies. Brain 125, 2591–2625 [DOI] [PubMed] [Google Scholar]
- 19.Kim JK, et al. (2014) Prevalence of anti-ganglioside antibodies and their clinical correlates with Guillain-Barré syndrome in Korea: a nationwide multicenter study. J Clin Neurol 10, 94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiba A, et al. (1993) Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 43, 1911–1917 [DOI] [PubMed] [Google Scholar]
- 21.Odaka M, et al. (2003) Longitudinal changes of anti-ganglioside antibodies before and after Guillain-Barré syndrome onset subsequent to Campylobacter jejuni enteritis. J Neurol Sci 210, 99–103 [DOI] [PubMed] [Google Scholar]
- 22.Geleijns K, et al. (2005) HLA class II alleles are not a general susceptibility factor in Guillain-Barré syndrome. Neurology 64, 44–49 [DOI] [PubMed] [Google Scholar]
- 23.Rose NR and Bona C (1993) Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today 14, 426–430 [DOI] [PubMed] [Google Scholar]
- 24.Ang CW, et al. (2004) The Guillain-Barré syndrome: a true case of molecular mimicry. Trends in immunology 25, 61–66 [DOI] [PubMed] [Google Scholar]
- 25.Godschalk PC, et al. (2007) Structural characterization of Campylobacter jejuni lipooligosaccharide outer cores associated with Guillain-Barré and Miller Fisher syndromes. Infect Immun 75, 1245–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kusunoki S and Kaida K (2011) Antibodies against ganglioside complexes in Guillain-Barré syndrome and related disorders. J Neurochem 116, 828–832 [DOI] [PubMed] [Google Scholar]
- 27.Meyer Sauteur PM, et al. (2016) Mycoplasma pneumoniae triggering the Guillain-Barré syndrome: A case-control study. Ann Neurol 80, 566–580 [DOI] [PubMed] [Google Scholar]
- 28.Kaida K, et al. (2004) Ganglioside complexes as new target antigens in Guillain-Barré syndrome. Ann Neurol 56, 567–571 [DOI] [PubMed] [Google Scholar]
- 29.Greenshields KN, et al. (2009) The neuropathic potential of anti-GM1 autoantibodies is regulated by the local glycolipid environment in mice. J Clin Invest 119, 595–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rinaldi S, et al. (2013) Antibodies to heteromeric glycolipid complexes in Guillain-Barré syndrome. PLoS One 8, e82337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanaka S, et al. (2019) On-membrane dynamic interplay between anti-GM1 IgG antibodies and complement component C1q. Int J Mol Sci 21, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hafer-Macko CE, et al. (1996) Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol 39, 625–635 [DOI] [PubMed] [Google Scholar]
- 33.Griffin JW, et al. (1996) Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barré syndrome. J Neurocytol 25, 33–51 [DOI] [PubMed] [Google Scholar]
- 34.Cunningham ME, et al. (2016) Anti-ganglioside antibodies are removed from circulation in mice by neuronal endocytosis. Brain 139, 1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fewou SN, et al. (2012) Anti-ganglioside antibody internalization attenuates motor nerve terminal injury in a mouse model of acute motor axonal neuropathy. J Clin Invest 122, 1037–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odaka M, et al. (2001) Anti-GQ1b IgG antibody syndrome: clinical and immunological range. J.Neurol.Neurosurg.Psychiatry 70, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bowes T, et al. (2002) Tolerance to self gangliosides is the major factor restricting the antibody response to lipopolysaccharide core oligosaccharides in Campylobacter jejuni strains associated with Guillain-Barré syndrome. Infect Immun 70, 5008–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lunn MP, et al. (2000) High-affinity anti-ganglioside IgG antibodies raised in complex ganglioside knockout mice: reexamination of GD1a immunolocalization. J Neurochem 75, 404–412 [DOI] [PubMed] [Google Scholar]
- 39.Alaniz ME, et al. (2004) Normally occurring human anti-GM1 immunoglobulin M antibodies and the immune response to bacteria. Infect Immun 72, 2148–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alborzian Deh Sheikh A, et al. (2021) A Guillain-Barré syndrome-associated SIGLEC10 rare variant impairs its recognition of gangliosides. J Autoimmun 116, 102571. [DOI] [PubMed] [Google Scholar]
- 41.Swanson CL, et al. (2013) Division of labor during primary humoral immunity. Immunol Res 55, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogino M, et al. (1995) IgG anti-GM1 antibodies from patients with acute motor neuropathy are predominantly of the IgG1 and IgG3 subclasses. J Neuroimmunol 58, 77–80 [DOI] [PubMed] [Google Scholar]
- 43.Paterson G, et al. (1995) Analysis of anti-GM1 ganglioside IgM antibodies cloned from motor neuropathy patients demonstrates diverse V region gene usage with extensive somatic mutation. J Immunol 155, 3049–3059 [PubMed] [Google Scholar]
- 44.King IL, et al. (2011) Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat Immunol 13, 44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, et al. (2022) T-independent antigen induces humoral memory through germinal centers. J Exp Med 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilbert M, et al. (2002) The genetic bases for the variation in the lipo-oligosaccharide of the mucosal pathogen, Campylobacter jejuni. Biosynthesis of sialylated ganglioside mimics in the core oligosaccharide. J Biol Chem 277, 327–337 [DOI] [PubMed] [Google Scholar]
- 47.Kuijf ML, et al. (2007) Origin of ganglioside complex antibodies in Guillain-Barré syndrome. J Neuroimmunol 188, 69–73 [DOI] [PubMed] [Google Scholar]
- 48.Koga M, et al. (2015) Complex of GM1- and GD1a-like lipo-oligosaccharide mimics GM1b, inducing anti-GM1b antibodies. PLoS One 10, e0124004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaudet RG and Gray-Owen SD (2016) Heptose sounds the alarm: innate sensing of a bacterial sugar stimulates immunity. PLoS Pathog 12, e1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li T, et al. (2020) Chemoenzymatic synthesis of Campylobacter jejuni lipo-oligosaccharide core domains to examine Guillain-Barré syndrome serum antibody specificities. J Am Chem Soc 142, 19611–19621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyauchi E, et al. (2020) Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature 585, 102–106 [DOI] [PubMed] [Google Scholar]
- 52.Planas R, et al. (2018) GDP-l-fucose synthase is a CD4(+) T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci Transl Med 10, 462. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, et al. (2020) HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell 183, 1264–1281 e1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lanz TV, et al. (2022) Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature, DOI: 10.1038/s41586-41022-04432-41587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meier UC, et al. (2021) Cumulative roles for Epstein-Barr virus, human endogenous retroviruses, and human herpes virus-6 in driving an inflammatory cascade underlying MS pathogenesis. Front Immunol 12, 757302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.International Multiple Sclerosis Genetics Consortium (2019) Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jelcic I, et al. (2018) Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell 175, 85–100 e123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tengvall K, et al. (2019) Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc Natl Acad Sci USA 116, 16955–16960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zamvil SS, et al. (2018) The gut microbiome in neuromyelitis optica. Neurotherapeutics 15, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo G, et al. (2021) T cell reactivity to regulatory factor X4 in type 1 narcolepsy. Sci Rep 11, 7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Koningsveld R, et al. (2004) Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barré syndrome: randomised trial. Lancet 363, 192–196 [DOI] [PubMed] [Google Scholar]
- 62.Kuitwaard K, et al. (2009) Recurrent Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 80, 56–59 [DOI] [PubMed] [Google Scholar]
- 63.Manso C, et al. (2019) Anti-Neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J Clin Invest 129, 2222–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kinzel S, et al. (2016) Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol 132, 43–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koga M, et al. (2003) Anti-GM1 antibody IgG subclass: a clinical recovery predictor in Guillain-Barré syndrome. Neurology 60, 1514–1518 [DOI] [PubMed] [Google Scholar]
- 66.Vinuesa CG and Chang PP (2013) Innate B cell helpers reveal novel types of antibody responses. Nat Immunol 14, 119–126 [DOI] [PubMed] [Google Scholar]
- 67.Kuijf ML, et al. (2010) TLR4-mediated sensing of Campylobacter jejuni by dendritic cells is determined by sialylation. J Immunol 185, 748–755 [DOI] [PubMed] [Google Scholar]
- 68.Bax M, et al. (2011) Campylobacter jejuni lipooligosaccharides modulate dendritic cell-mediated T cell polarization in a sialic acid linkage-dependent manner. Infect Immun 79, 2681–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Hanlon GM, et al. (1996) Anti-GM1 ganglioside antibodies cloned from autoimmune neuropathy patients show diverse binding patterns in the rodent nervous system. J Neuropathol Exp Neurol 55, 184–195 [DOI] [PubMed] [Google Scholar]
- 70.Willison HJ, et al. (1996) A somatically mutated human antiganglioside IgM antibody that induces experimental neuropathy in mice is encoded by the variable region heavy chain gene, V1–18. J Clin Invest 97, 1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boffey J, et al. (2004) Innate murine B cells produce anti-disialosyl antibodies reactive with Campylobacter jejuni LPS and gangliosides that are polyreactive and encoded by a restricted set of unmutated V genes. J Neuroimmunol 152, 98–111 [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, et al. (2017) Memory B cells in Guillain-Barré syndrome. J Neuroimmunol 305, 1–4 [DOI] [PubMed] [Google Scholar]
- 73.Brem MD, et al. (2019) IVIg-induced plasmablasts in patients with Guillain-Barré syndrome. Ann Clin Transl Neurol 6, 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Umapathi T, et al. (2021) Guillain-Barré syndrome decreases in Singapore during the COVID-19 pandemic. J Peripher Nerv Syst 26, 235–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tejada-Simon MV, et al. (2003) Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol 53, 189–197 [DOI] [PubMed] [Google Scholar]
- 76.Brok HP, et al. (2007) The human CMV-UL86 peptide 981–1003 shares a crossreactive T-cell epitope with the encephalitogenic MOG peptide 34–56, but lacks the capacity to induce EAE in rhesus monkeys. J Neuroimmunol 182, 135–152 [DOI] [PubMed] [Google Scholar]
- 77.Lenz DC, et al. (2001) A Chlamydia pneumoniae-specific peptide induces experimental autoimmune encephalomyelitis in rats. J Immunol 167, 1803–1808 [DOI] [PubMed] [Google Scholar]
- 78.Hughes LE, et al. (2003) Cross-reactivity between related sequences found in Acinetobacter sp., Pseudomonas aeruginosa, myelin basic protein and myelin oligodendrocyte glycoprotein in multiple sclerosis. J Neuroimmunol 144, 105–115 [DOI] [PubMed] [Google Scholar]
- 79.Varrin-Doyer M, et al. (2012) Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 72, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kampylafka EI, et al. (2011) Fine specificity of antibodies against AQP4: epitope mapping reveals intracellular epitopes. J Autoimmun 36, 221–227 [DOI] [PubMed] [Google Scholar]
- 81.Luo G, et al. (2018) Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc Natl Acad Sci USA 115, E12323–E12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang W, et al. (2019) In vivo clonal expansion and phenotypes of hypocretin-specific CD4(+) T cells in narcolepsy patients and controls. Nat Commun 10, 5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leonhard SE, et al. (2019) Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol 15, 671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]