

Abstract
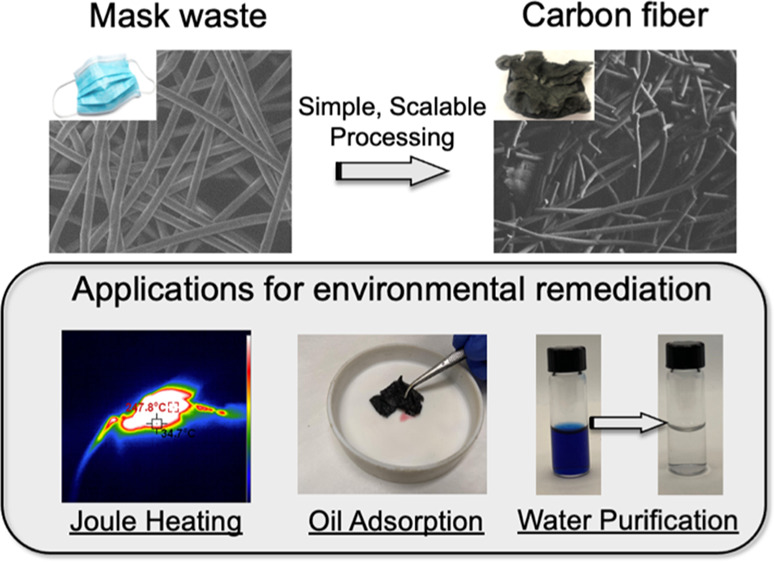
Over the past years, disposable masks have been produced in unprecedented amounts due to the COVID-19 pandemic. Their increased use imposes significant strain on current waste management practices including landfilling and incineration. This results in large volumes of discarded masks entering the environment as pollutants, and alternative methods of waste management are required to mitigate the negative effects of mask pollution. While current recycling methods can supplement conventional waste management, the necessary processes result in a product with downgraded material properties and a loss of value. This work introduces a simple method to upcycle mask waste into multifunctional carbon fibers through simple steps of thermal stabilization and pyrolysis. The pre-existed fibrous structure of polypropylene masks can be directly converted into carbonaceous structures with high degrees of carbon yield, that are inherently sulfur-doped, and porous in nature. The mask-derived carbon product demonstrates potential use in multiple applications such as for Joule heating, oil adsorption, and the removal of organic pollutants from aqueous environments. We believe that this process can provide a useful alternative to conventional waste management by converting mask waste generated during the COVID-19 pandemic into a product with enhanced value.
1. Introduction
Plastic waste is an increasing global concern, which has the generation rate that is projected to triple by the year 2060.1 Current plastic waste management practices involve multiple steps of collecting, sorting, washing, and recycling. Strained by the lack of efficient solutions and current mechanical recycling volumes, it becomes more likely that these wastes can escape into the environment where they slowly erode into microplastics over time. As a type of severe environmental threat, microplastics are known to concentrate pollutants, and the consumption of microplastics by animals can result in their entry, along with the concentrated pollutants, into the ecosystems, directly harming the human population.1 Particularly, responses to the COVID-19 pandemic further intensified this issue, leading to massive increases in the production and use of personal protective equipment.2−5 As these materials are typically designed for single use, their extensive consumption has largely exacerbated the global plastic waste issue, posing a significant strain on conventional waste management practices (i.e., landfilling and incineration).6 According to National Geographic magazine, greater than 3 billion masks have been discarded daily during 2020, and this elevated rate will likely remain throughout the following years in response to various respiratory diseases.7 In general, these disposable facial masks are produced from polypropylene (PP), and a significant amount of them is now simply leaked into the environment where they can directly threaten the wildlife and ecosystems that are present. Undoubtedly, effective solutions are required to address the global challenge of mask recycling and/or upcycling toward the development of a sustainable environment and society.
Over the past years, many types of technologies have been demonstrated for recycling the raw materials of disposable masks (i.e., plastics) to extend their associated lifetime. For example, post-cleaning and/or post-functionalization of used masks can be performed to improve their performance, allowing their efficient reuse for at least several days.8−10 Specifically, many methods of decontamination or sterilization have been previously reported,10,11 including the use of γ and electron beam irradiation.12,13 Additionally, Menon et al. demonstrated that post-functionalization of PP masks can result in effective sterilization through the use of zinc porphyrin photosensitizers for enhancing the ability of the masks to protect against influenza pathogens.14 Alternatively, masks can be collected and recycled through several processing steps, such as granulation and pelletization. While such mechanical recycling methods have been established in industry, they often result in downgraded material properties and a less valuable product due to increased yellowness and reduced melt viscosity from possible chain scission under high shear.15,16 As an example, Frache et al. reported the mechanical recycling of PP-derived surgical masks using twin-screw extrusion to compound comingled mask waste and form pellets.17 These pellets can then be used to manufacture new products through subsequent processing steps. While parts could be processed from the recycled materials, they exhibited diminished mechanical properties in comparison to virgin PP. Specifically, recycled parts exhibited similar moduli but decreased elongation at break in tensile experiments. Although it is challenging, establishing a robust and scalable technology to upcycle PP masks, converting them to higher-value products, can not only complement existing recycling methods for addressing a massive amount of plastic waste but may also stimulate interests of broader communities to improve the corresponding infrastructures for waste management.
In the early 1970s, Horikiri et al. and Postema et al. pioneered the concept of using polyolefins [e.g., polyethylene (PE)] for carbon fiber fabrication.18,19 However, compared to polyacrylonitrile (PAN),20−22 pitch-based chemicals,23−25 and lignin,26,27 the efficacy of polyolefins as carbon precursors is still underexplored, evidenced by a relatively limited number of reports regarding PE-derived carbons.28−30 It is important to note that efficient transformation of polyolefins into carbonaceous products requires the development of a process for thermally stabilizing the polyolefin chains through crosslinking prior to carbonization. Currently, the most successful stabilization/crosslinking of PE at large scales is achieved through directly soaking them in sulfuric acid at elevated temperatures to aromatize the polymer backbones. Younker et al. elucidated pyrolysis pathways of sulfonated PE through the combined use of experimental techniques [e.g., thermogravimetric analysis (TGA)] and computational approaches (e.g., density functional theory and transition state theory).31 They also found that two synergistic mechanisms, including five-centered internal elimination and radical chain reactions, are involved for detaching SO3H groups to provide reaction sites for crosslinking. While, to date, PAN is still the most common carbon precursor in carbon fiber production, it is significantly more expensive in comparison to polyolefins which are the most widely used commodity plastic, representing over 55% of total plastic production.32 Therefore, manufacturing carbon fibers from polyolefin precursors could potentially reduce production and material costs, encouraging their broad use in more diverse applications, especially when cost-effective manufacturing is strongly preferred. This approach has been investigated in recent years for applications such as the continuous production of PE-derived carbon fibers. For example, Barton et al. performed a comprehensive study using aliphatic hydrocarbons and multiple commercially available polyolefin resins as model precursors with both concentrated and fuming sulfuric acid to elucidate the chemical transformation during the sulfonation and carbonization processes.33 This work informed several subsequent studies associated with producing PE-derived carbon fibers from continuous processes and comparing their performance to conventional PAN- and pitch-derived carbon fibers.28,34 It was found that PAN-based fibers have higher moduli than those produced from the sulfonation of PE due to their improved degree of crosslinking. However, one report also suggested that PE-derived carbon fibers may be able to achieve higher moduli than PAN-derived counterparts if the precursor chains can be aligned prior to the crosslinking step.28
Capitalizing on these efforts, several research groups also extended this sulfuric acid-crosslinking strategy to upcycle PE and PP waste to carbon-based materials.35 In 2018, Pol et al. demonstrated the successful use of waste PE plastic bags for fabricating carbon products through a thermal stabilization process.36 The resulting carbons were inherently porous with a relatively high surface area of 752.3 m2/g, which can be used as anodes in lithium-ion batteries. This study also shows that both PE and PP waste can yield amorphous carbons with approximately 50% conversion rate. More recently, researchers have studied the use of the sulfonation crosslinking process to convert PP fibers from facial masks into carbonaceous materials. Hu and Lin demonstrated the synthesis of hollow carbon fibers for solid-state capacitors, which was combined with a KOH activation step to enable microporous structures.37 Lee et al. characterized the sulfonation process and elucidated the chemical transformation mechanisms for converting linear PP chains to their crosslinked analogues.35 While these inspiring works show the great potential of chemical upcycling of plastic waste to functional carbons for practical applications, their scope was primarily focused on energy storage applications. To fully unlock the potential of mask waste, more diverse applications should be developed, especially leveraging the already existing, well-defined fiber structures of disposable masks for manufacturing carbon products with improved functionality.
Herein, we report the use of sulfonation chemistry to thermally stabilize PP-based surgical masks and their conversion into carbon nanofibers without deconstruction of their defined structures. The resulting products exhibit a greater than 50% carbon yield, while retaining both the microscopic fibrous structures and macroscopic shape and size. A suite of characterization techniques have been employed to understand the nanostructures and properties of these mask-derived carbon fibers. Furthermore, we demonstrate their potential use in several practical applications, including oil sorbents, nanofillers for imparting electrical conductivity, and Joule heating behaviors of composites, as well as materials for the removal of organic contaminants from water. We believe that this work provides important and timely insights into an efficient method for upcycling mask waste, demonstrating their versatile performance and providing a potential solution to the global challenge of addressing massive facial mask waste generated in response to the pandemic.
2. Experimental Section
2.1. Materials
98 wt % sulfuric acid was purchased from Sigma-Aldrich. PP-based surgical masks were purchased from Asia Masks Inc. and used throughout this study. Deionized (DI) water was obtained by passing tap water through a Milli-Q IQ 7003 ultrapure lab water purification system from Millipore Sigma. Potassium hydroxide (KOH) was purchased from Fisher Scientific. Powder-activated carbons (PACs) used in this study were purchased from American Water Solutions with a surface area of 712 m2/g.
2.2. Methods for Converting Masks to Carbon Fibers
Surgical masks were cut to remove the elastic bands and metal nosepiece. The resulting fabric was separated into three constituent layers, including nonwoven fabrics (two outer layers) and a melt-spun mat (inner layer). In this study, only outer layers were employed for producing carbon fibers, which were cut into sections with a typical size of 8 cm by 5 cm. These mask parts (∼1 g in total) were then transferred into glass containers containing 25 mL of concentrated sulfuric acid (98 wt %). In this step, a glass slide was placed on top of the masks to keep the mask completely submerged in sulfuric acid throughout the reactions. The containers were then placed in a Thermo Scientific muffle furnace and heated to 155 °C (temperature ramp: 1 °C/min) for various amounts of time. Upon sulfonation, samples were removed from the muffle furnace and cooled down to room temperature. To wash these samples, sulfuric acid is first removed from the glass containers. Subsequently, the mask pieces were carefully placed in a quartz funnel, where it is washed at least three times with DI water in order to completely remove the residue acid. Neutralization was confirmed by pH papers. Samples were then dried by placing on a glass Petri dish in a vacuum oven for overnight. Carbonization of the sulfonated masks was performed using an MTI Corporation OTF-1200× tube furnace under a N2 atmosphere at a rate of 1 °C/min to 600 °C and thereafter 5 °C/min to 800 °C or higher. The holding time for carbonization temperatures is 3 h. The activation process was performed by physically grinding the previously produced carbon fiber product with potassium hydroxide (KOH) at a 1:2 mass ratio. After activation at 700 °C with a ramp rate of 1 °C/min for 1 h, the product was washed with DI water, centrifuged, and then dried. This process was repeated six times.
2.3. Characterization Methods
A PerkinElmer frontier attenuated total reflection Fourier transform infrared (FTIR) spectrometer was used to understand the changes in chemical compositions of sulfonated mask samples as a function of time. The scan range was 4000–600 cm–1 with 32 scans and a resolution of 4 cm–1. TGA was conducted using a Discovery Series TGA 550 (TA Instruments) to understand the mass loss of polymer precursors as a function of pyrolysis temperature. Sulfonated samples, approximately 10–20 mg in mass, were pyrolyzed under a N2 environment, replicating the carbonization procedure used in the tube furnace. Differential scanning calorimetry (DSC) was performed using a Discovery 250 (TA Instruments). An initial heating cycle to 200 °C with a ramp rate of 10 °C/min was used to erase thermal history. The sample was cooled to −90 °C at a rate of 5 °C/min and then heated to 200 °C at 10 °C/min. Data analysis was performed using Trios software. A Zeiss Ultra 60 field-emission scanning electron microscope was used to understand the morphological changes of the fiber structure both after sulfonation at different periods of time and after the carbonization process with an accelerating voltage of 10 kV. During these measurements, energy-dispersive X-ray spectroscopy (EDS) was coupled for determining the content of different elements within the materials after sulfonation and carbonization, respectively. Additionally, fiber diameters were determined and recorded using ImageJ image analysis software. X-ray photoelectron spectroscopy (XPS) experiments were performed using a Thermo Fisher ESCALAB Xi+ spectrometer equipped with a monochromatic Al X-ray source (1486.6 eV) and a MAGCIS Ar+/Arn+ gas cluster ion sputter gun. Measurements were performed using the standard magnetic lens mode and charge compensation. The base pressure in the analysis chamber during spectral acquisition was at 3 × 10–7 mbar. Spectra were collected at a takeoff angle of 90° from the plane of the surface. The pass energy of the analyzer was set at 150 eV for survey scans with an energy resolution of 1.0 eV; the total acquisition time was 220 s. Binding energies were calibrated with respect to C 1s at 284.8 eV. All spectra were recorded using the Thermo Scientific Avantage software; data files were translated to VGD format and processed using the Thermo Avantage package v5.9904. In order to determine the pore textures within the mask-derived carbon fibers, N2 adsorption and desorption isotherms were determined using a Micromeritics Tristar II 3020. Specifically, the pore size distribution of samples was estimated from the adsorption isotherms using the Barrett–Joyner–Halenda (BJH) model, whereas the surface area was determined from the typical Brunauer–Emmett–Teller analysis. Raman spectrum of carbon was obtained using a Raman spectrometer with a 532 nm laser as the excitation source (from HORIBA XploRA Plus, Japan). The Joule heating capabilities of the carbonized mask fibers were determined by connecting the fibers to a DC power supply (from Dr. Meter) using a glass slide as a support. The voltage was increased in increments of 1 V, and the temperature was measured using a thermal camera (from HTI) until the equilibrium state was reached. Water contact angle measurements were recorded and analyzed using a goniometer and Contact Angle software from Ossila. Oil adsorption studies were performed by submerging carbonized mask fibers into 20 mL of various organic solvents for at least 5 min and recording the mass adsorbed immediately after removing from the solvent. To investigate the dye adsorption performance of the carbonized mask fibers, they were first activated with KOH at a 1:2 carbon to KOH mass ratio. The adsorption performance of the activated carbon fibers and commercially available PAC was determined for a dye, basic blue 17, at concentrations of 0.07, 0.15, and 0.3 mg/mL. 20 mg of the respective carbons was added to each solution and shaken for 48 h. Aliquots were taken at multiple time points, and the concentration of the dye was measured using a Genesys 30 visible spectrometer (Thermo Scientific). The adsorption capacity, qe, of the carbons was calculated using the following equation
where C0 is the initial concentration of the dye, Ce is the final concentration of the dye, V is the volume of the solution, and M is the mass of the carbon sorbent.
3. Results and Discussion
Scheme 1A depicts our simple process to convert PP from surgical masks to carbon fibers through steps of sulfonation and carbonization. Through heating in a concentrated sulfuric acid, thermal stabilization of PP occurs, enabling such commodity plastics to become efficient carbon precursors. Subsequently, the stabilized PP is exposed to high temperatures (800–1400 °C) under a N2 atmosphere for its pyrolysis to the final carbon product. Key advantages of this method include its cost-effectiveness and high scalability. Particularly, both PP and sulfuric acid are inexpensive raw materials, and a recent review article already suggested the potential cost benefits of polyolefin-derived carbon fibers compared to those of their PAN-derived counterparts.38 Throughout this process, the initial fibril structures of the masks can be completely retained, resulting in a carbon fiber mat with mechanical flexibility. It is important to note that several studies suggested that a broad temperature range (at least above 120 °C) can be employed for crosslinking polyolefins, including both PP and PE. A very recent study from Lee et al. successfully characterized the sulfonation reaction of PP using combined techniques of FTIR and solid-state 13C nuclear magnetic resonance spectroscopy.39 The simplified stabilization mechanism of this PP crosslinking reaction is shown in Scheme 1B.
Scheme 1. (A) Schematic Illustration of Thermal Stabilization and Carbonization Processes of PP Fibers from Surgical Masks; (B) Crosslinking Mechanism of PP Is Initiated through a Sulfonation Step Which Is Followed by Olefination and Subsequent Addition/Rearrangement; PP Chains Can Then Crosslink, Followed by Ring Closure and Degradation of Functional Groups at Elevated Temperatures.
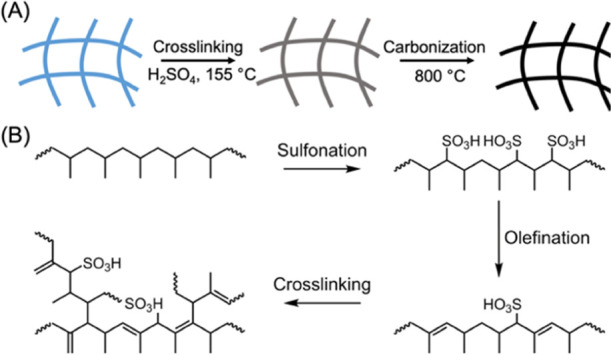
To briefly describe the thermal stabilization mechanism, the initial sulfonation reaction of PP proceeds by reacting with the secondary/tertiary carbons along the polymer backbone, followed by the homolytic dissociations of sulfonyl groups, which results in unsaturated bonds within the polymer chain. These double bonds from sulfonation can continue to react through a secondary addition, rearrangement, and dissociation, leading to the formation of radical species that can directly couple with other reactive groups from surrounding polymer chains, effectively producing crosslinked network structures.
These crosslinked polymers can then be converted to carbons upon pyrolysis, potentially stripping away functional groups upon exposure to elevated temperatures in inert atmospheres. In our process, the progress of the sulfonation reaction was monitored by two different approaches, including tracking mass gain as a function of sulfonation time, as well as through FTIR spectroscopy, which are both demonstrated in Figure 1. At short time scales, the PP mass gain as a function of time increases rapidly as the sulfonation reaction progresses (Figure 1A). After 4 h, the mass gain reaches a plateau value of 51%, which remains nearly constant even after extending the reaction time to 12 h (∼52%). FTIR spectra in Figure 1B also confirm that the sulfonation reaction results in the formation of double bonds and sulfonic acid groups in PP. Specifically, pristine PP fibers from masks exhibit peaks indicative of C–H stretching at 2920 cm–1, which diminishes as the sulfonation/crosslinking reaction progresses and completely disappears after 4 h of reaction time. Additionally, the appearance of three separate peaks can be attributed to the progress of the reaction. The broad −OH stretching peak at 3300 cm–1 emerges after 30 min, and its peak intensity increases with the increasing reaction time. Peaks from 1250 to 1000 cm–1 are attributed to the presence of sulfonic acid groups. The addition of alkenes into the PP backbone is demonstrated by the emerging peaks at 1600 cm–1. Although the masks do not gain further mass after 4 h of the reaction time, the FTIR traces suggest that the reaction continues to progress until 12 h. Compared to previous reports, the kinetics of the reaction is slightly slower, where Lee et al. demonstrated that the peak at 2920 cm–1 represents that the crosslinkable polyolefin units diminish after 2 h of the reaction time.39 Their work utilized a sulfonation process at lower temperatures (120 °C) but separated the waste masks into very small pieces. The smaller mask pieces likely enhanced reaction rates compared to the system presented here due to the increased surface area exposed to the sulfuric acid-crosslinking agent.35
Figure 1.
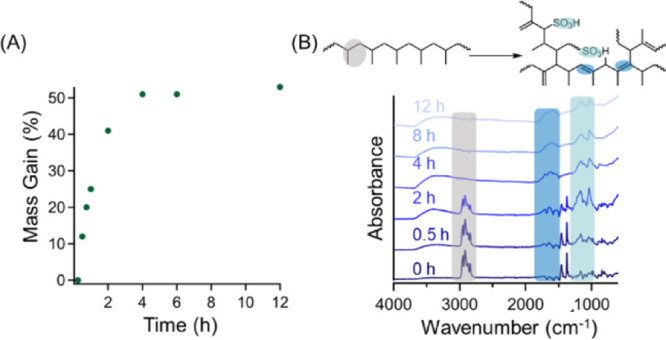
(A) Mass gain of PP masks as a function of sulfonation reaction time (at 155 °C). (B) FTIR spectra of sulfonated mask pieces at various reaction times where peaks at 2920, 1600, and 1250–1000 cm–1 are highlighted to monitor reaction progression.
The fiber structures of pristine and sulfonated masks (including after 2 and 12 h) were investigated using scanning electron microscopy (SEM). As shown in Figure 2a, the outer layers of masks were composed of PP fibers with a relative uniform diameter of 25.7 ± 0.7 μm. After our sulfonation reactions, these fibral structures were completely retained. This result is consistent with previous reports of crosslinking PE fibers using acids. Notably, even a slightly higher crosslinking temperature was employed in our systems, which approaches the onset of melting in the PP fibers (156 °C, Figure S1) but does not lead to structural collapse. After the reaction for 2 h, the fiber diameter slightly changes to 21.6 μm and remains relatively constant after 12 h of sulfonation. Interestingly, our result varies from a recent work studying a variety of PE-based fibers under similar chemical treatments, which observed that all low-density PE fibers exhibited increases in the fiber diameter at increased reaction times up to 25% of the original diameter.40 Other works also demonstrate a similar phenomenon in PE fibers under proper reaction conditions.41,42 We attributed this difference to the use of sulfuric acids with different concentrations, where the diluted ones are known to be significantly more reactive than their concentrated counterparts. Nevertheless, throughout the sulfonation process, the pre-existed fibrous structures are completely maintained. It is also found that extending the reaction time to 12 h does not alter the fiber diameters and yet can result in slight distortion and curving of the fibers, as shown in Figure 2c. Furthermore, as shown in the insets of Figure 2, the macroscopic structures are retained after each processing step. Figure 2a demonstrates the neat PP mask and its initial macroscopic structure, while the inset in Figure 2c shows that the shape is maintained throughout the sulfonation process. We then employed TGA to determine the char yield of sulfonated masks after two distinct crosslinking time, 2 and 12 h. For our control sample (unsulfonated PP), all organic components are completely degraded with 0% mass retention after exposure to 800 °C under N2. For sulfonated PP, it is found that the lower reaction time leads to a higher mass loss upon carbonization (Figure 3a), which is probably due to the incomplete crosslinking of PP throughout the entire fiber structure. Specifically, 2 h of sulfonation results in a carbon yield of 51%, while 12 h of sulfonation increases the yield to 58%, both derived from the sulfonate state. As our sulfonation reaction is a diffusion-limited process, the center part of the fibers could remain unreacted after a relatively short time, which is susceptible to decomposition or degradation.
Figure 2.

SEM images of fibers within the mask (A) prior to sulfonation and (B) after 2 and (C) 12 h of sulfonation. The average fiber diameter within the image is included in the bottom left-hand corner of the corresponding SEM image. The inset images show the macroscopic structures of these masks.
Figure 3.

(A) TGA thermograms of pristine PP and sulfonated PP (from masks) after different crosslinking times where longer reaction times increase the carbon yield. (B) SEM image of carbonized fibers after 2 h of sulfonation, leading to the decomposition of the unreacted center portions of the fiber. The inset image depicts a hollow fiber which results from insufficient crosslinking. (C) SEM image of carbonized fibers after 12 h of sulfonation which results in complete crosslinking and continuous fibers. EDX analysis highlighting (D) carbon and (E) sulfur contents in carbonized mask fibers. (F) XPS spectrum of carbonized fibers after 12 h of sulfonation.
Additionally, for 12 h samples, the TGA thermogram exhibits no secondary thermal decomposition after 100 °C, which is present in the 2 h sulfonated sample that has been attributed to the decomposition of unreacted polymer chains within the fiber. As shown in Figure 3B, carbon fibers with hollow structures were observed from samples with only 2 h of sulfonation. For PP fibers after 12 h of sulfonation, an identical carbonization condition results in carbon fibers with completely solid cores (Figure 3C). The sulfonation-crosslinking step also imparts additional functionality into the carbon fibers, such as inherent incorporation of sulfur heteroatoms into the carbon framework. Sulfur doping can enhance the functionality of associated carbon-based materials in many applications, including energy storage,43 catalysis,44 and CO2 adsorption.45 EDS was performed to determine the elemental composition of the final carbon fiber product. Figure 3D,E depicts the elemental maps that correspond to both carbon and sulfur produced through EDS. The sulfur-doping content was found to be 5.6 wt % for carbonized PP masks with 12 h, and the overlayed elemental map demonstrates that the heteroatoms are uniformly distributed within the carbon fibers. The presence of heteroatoms in the carbon framework of our mask waste-derived carbon fibers was further investigated using XPS. Figure 3F depicts the survey scan of carbonized fibers after 12 h of sulfonation, indicating the presence of carbon (284.09 eV), oxygen (532.20 eV), and sulfur (163.79 eV) atoms within the carbon framework at 96.7, 2.9, and 0.4 at. %, respectively. This lower doping content from XPS measurements compared to EDS results suggest that the fiber surface might have much lower sulfur content than the inner parts. Additionally, high-resolution XPS scans for each element can be found in Figure S2, which describe their chemical environments. Interestingly, the results of the sulfur high-resolution scans differ from the recent work from Lee et al. where most of the sulfur content was bonded to oxygen.39 In this work, the peaks at 163.5 and 164.7 eV suggest that the sulfur atoms are directly bonded to carbon as part of the framework rather than being bonded to oxygen which would be illustrated by the presence of peaks at slightly higher binding energies. Furthermore, Raman spectroscopy was employed to characterize the degree of graphitization of these carbon products. In general, carbon materials with higher degrees of graphitization can exhibit better electrical and thermal conductivity through facilitating the electron transport along the in-plane direction as opposed to the amorphous carbon counterparts. The ratio of the intensities of the disordered (at 1370 cm–1) and graphitic bands (at 1597 cm–1) is 1.21 (Figure S3). The N2 adsorption–desorption behavior of the mask-derived carbon fiber was characterized using gas physisorption measurements, which can determine pore volume, pore size distribution, and surface area of the carbon samples. As shown in Figure 4A, our carbon fibers exhibit a typical type-II isotherm, suggesting the presence of both macropores and mesopores, with a surface area of 295.46 m2/g. Furthermore, the pore size distribution is relatively uniform and centered around 12 nm (Figure 4B). We want to note that the generation of pores occurred during the carbonization process when portions of the polymer chains are thermally degraded and gases (CO, CO2, H2O, and SO2) are evolved, which can potentially react with the carbon framework. From our control experiments, we found that sulfonated fibers prior to carbonization possess no micropores (Figure S4).
Figure 4.
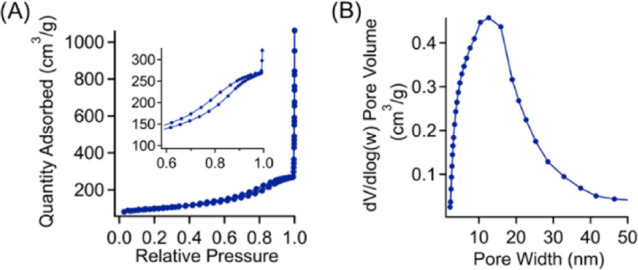
(A) Nitrogen adsorption–desorption isotherm with the inset highlighting the hysteresis that occurs at the partial pressure range from 0.6 to 0.98 and (B) the associated pore size distribution determined using the BJH method.
To further demonstrate the use of these carbon fibers in practical applications, we investigate their performance for Joule heating, oil absorption, and water remediation. The ability of a material to reach elevated temperatures upon the application of low voltages through Joule heating provides great potential in several applications, including thermotherapy,46,47 crude oil recovery,48,49 and thermochromics.50,51 Joule heating is a result of electrons colliding with atoms within a conductor and which generates heat in regions where current transmits. Equation 1 simplistically depicts the Joule heating of a current density j in an electrical field E in a material of electrical conductivity σ.52
| 1 |
This relationship demonstrates that the thermal energy produced from Joule heating is directly dictated by the conductivity of the material where enhanced conductivity results in increased output of energy in to the form of Joule heating. Figure 5A shows a simple demonstration confirming the electrically conductive nature of our carbon fibers, which can illuminate the light-emitting diode (LED) bulb intensely in the circuit. In Joule heating experiments, the carbonized mask fibers were subjected to different voltages and then allowed to be equilibrated. As shown in Figure 5B, with the application of increased voltage from 1 to 10 V, the mask-derived carbon fibers can reach a broad temperature range from 29 °C to greater than 300 °C with the application of 10 V. The thermal image in Figure 5C further clearly demonstrates that the temperature of the mask fibers was at ∼248 °C (at 9 V). The Joule heating capability of these mask-derived carbon fibers is much more robust than that of other carbon products from waste resources, specifically coal tar which required 60 V to achieve the maximum temperature demonstrated here.53 Furthermore, the heating happens rapidly due to the high conductivity of the carbon fibers, equilibrating in a matter of seconds. After the voltage is removed, heat dissipates quickly, and the fibers return to room temperature in less than 10 s. While it may be challenging to directly use these carbonized masks for making products due to their shape and brittleness, these results suggest that carbon fibers derived from mask waste could still be employed as fillers in preparing Joule heating composites.
Figure 5.

(A) Portion of a mask-derived carbon fiber mat completing a circuit and illuminating an LED light as a demonstration of the conductivity of the carbon. (B) Temperature of the carbon fiber increases with increasing voltage, achieving greater than 300 °C after the application of 10 V. (C) Image from the thermal camera demonstrating the excellent Joule heating performance of the carbon fiber mat at 9 V.
The hydrophobicity of carbon materials enables their use for oil adsorption. The favorable interactions between organic solvents and hydrophobic carbon drive the adsorption of oils to the carbon surface. The carbonized mask fibers exhibit high water contact angles (Figure S5A) but are easily wet by organic solvents, such as chloroform (Figure S5B). Figure 6A–C depicts a demonstration of the carbonized mask fibers being used to absorb organic solvents which act as surrogates for oil-based pollutants. Acetone (red) and chloroform (blue) are easily absorbed by simply placing the carbonized fibers into the solvent droplets. This behavior is consistent for many organic solvents, as demonstrated in Figure 6D. The carbon mask fibers exhibit varied adsorption capacities for different organic solvents, with a maximum amount of up to 14 g of mineral oil per g of carbon fiber. The difference in the uptake capacity against different solvents is primarily associated with the surface energy of carbon surfaces and the interactions between the surface functional groups and solvent molecules, which has been commonly observed in various oil sorbent systems.54,55 Additionally, this performance is highly cyclable, where the sorbate can be efficiently removed, and the mask can be reused in further adsorption. This advantageous property is confirmed in Figure 6E where chloroform has been repeatedly adsorbed by a carbon fiber mat, recovered, and adsorbed again. We wanted to note that while the oil adsorption capacities are much lower than other sorbent technologies such as porous foams due to their limited porosity,56 conventional methods for preparing porous sorbents require multiple steps for functionalization. Alternatively, carbon foam can be derived from direct pyrolysis of melamine foams; however, this approaches only produce 2–5% carbon yield,57,58 restricting their scalability. Our materials exhibit carbon yields which are an order of magnitude higher, through very simple processing, making them competitive from a scalability-focused perspective. These performances demonstrate the potential applications of the hydrophobic carbon fibers to act as an efficient mechanism in oil sorption and oil/water separation applications.
Figure 6.
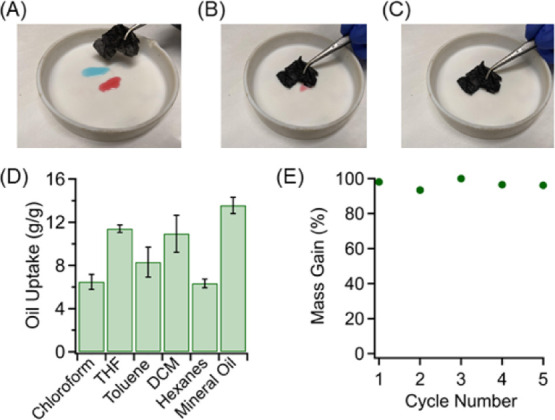
(A–C) Demonstration of carbon fibers being used to adsorb oil represented by acetone (red) and chloroform (blue). (D) Oil uptake capacity of the carbonized mask fibers given as gram of sorbate per gram of sorbent. (E) Cycling performance of oil adsorption performed by adsorbing chloroform, heating to remove the sorbate, and repeating this process for five cycles.
Porous carbon sorbents are established technologies for the removal of pollutants such as small-molecule dyes from water. Commercially, PAC is broadly employed in the treatment of wastewater because it is inexpensive to manufacture and widely available. PAC is typically produced through carbonization of biomass, containing micropores with a surface area ranging from 500 to 1500 m2/g. The presence of micropores provides sorption sites for facilitating the adsorption of pollutants from water. Herein, we used a KOH-based activation technique (Figure 7A). Specifically, the carbonized masks were activated through reacting with KOH to enhance the porosity of the carbon fibers and increase surface area. From the N2 isotherm in Figure 7B, it is evident by the large increase in the quantity of N2 adsorbed at low relative pressures (p/p0: 0–0.1) that micropores have been generated in the fibers. The activation process significantly improves the surface area of these carbon fibers from 295 to 600 m2/g. Commercially available PAC has a very similar isotherm (Figure S6), with a slightly higher surface area of 712 m2/g. As shown in Figure S7, the grinding process during blending of the activating agent disrupts the macroscopic structure of the mask, but the microscopic, fibrous shape is maintained, albeit at a shorter scale. It was also found that the oxygen content of carbon fibers increases from imperceptible amounts to 25.6 wt %, determined by the EDX measurements. To gauge the performance of the activated mask in water remediation applications, dye adsorption studies were performed with a water-soluble dye, basic blue 17, and compared to the performance of a commercially available PAC. The adsorption capacities as a function of time in three different dye concentrations were investigated, which were 0.07 mg/mL (Figure S8A), 0.15 mg/mL (Figure 7C), and 0.30 mg/mL (Figure S8B). Across all of the dye concentrations, the two sorbents exhibit very similar adsorption capacities after 48 h of exposure to the dye solution. Both PAC and the activated mask fibers had adsorption capacities of roughly 0.033, 0.09, and 0.19 mg/mg for the 0.07, 0.15, and 0.30 mg/mL solutions, respectively. At higher concentrations (0.15 and 0.30 mg/mL), our activated carbon fibers exhibit much faster adsorption kinetics than the PAC, as demonstrated in Figure S8. The dye adsorption kinetics was fit to a pseudo first-order model using the following equation
where qe is the amount of dye adsorbed at equilibrium, qt is the amount of dye adsorbed at time t, and k1 is the first-order equilibrium rate constant. At 0.15 and 0.30 mg/mL, the rate constant of the dye adsorption by the activated fibers (0.649 and 0.213 h–1, respectively) is significantly higher than the adsorption by the PAC (0.076 and 0.075 h–1, respectively), which can be attributed to the combined effects of the fibrous structures of our surgical mask-derived carbons (Figure S9) and the enhanced surface functionality through KOH activation. Previous studies suggested that these fiber structures can improve the contact between sorbent and sorbates, facilitating the sorption process.59,60 Our activated carbon fibers, with a diameter of approximately of 21 μm, can provide enhanced contact areas and improved transport and diffusion of dye molecules as opposed to powder samples with an average particle size over hundreds of micrometers. Additionally, the functional groups on material (e.g., polymers, carbons, and metal oxides) surfaces can also alter their performance in different applications.61,62 In this study, the oxygen groups introduced through KOH activation can facilitate the interactions between carbon fibers and basic blue 17 for improving the sorption kinetics. These results demonstrate that the activated carbon fibers fabricated from waste products and scalable production methods are competitive with current commercially viable materials for water remediation.
Figure 7.
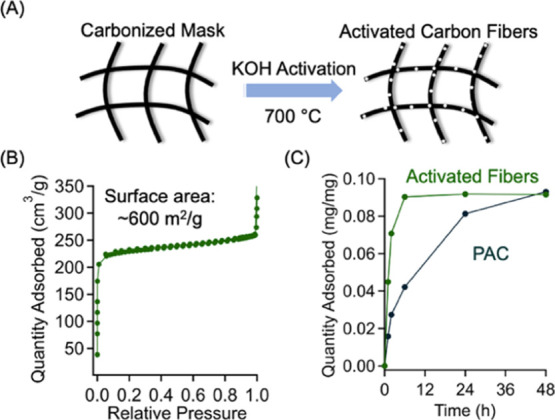
(A) Schematic illustration of KOH activation of already carbonized mask fibers to induce microporosity and increase the surface area. (B) N2 adsorption isotherm of carbonized mask fibers after the activation process. The isotherm indicates the increased surface area compared to the neat, carbonized fibers. (C) Dye adsorption study at a concentration of 0.15 mg/mL investigating the adsorption capacities as a function of time of activated carbon fibers compared to PAC.
4. Conclusions
As the Covid-19 pandemic continues to spur the production and consumption of single-use plastics, including PP masks, efficient methods are required to repurpose materials to mitigate their negative effects. This work depicts a facile and scalable approach to thermally stabilize PP masks and convert them into multifunctional carbon fibers. The thermal stabilization process imparts additional functionality into the fibers through sulfur doping the carbon framework and generating pores upon carbonization. The performance of the fibers for Joule heating materials, oil adsorption, and dye removal from aqueous solutions is demonstrated, suggesting the practical applications of these carbonaceous products derived from waste materials.
Acknowledgments
The authors would like to acknowledge Dr. Derek Patton and Surabhi Jha for assistance with XPS experiments and access to this instrumentation. Additionally, the authors would like to thank Michael Blanton for training and necessary access to perform SEM experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00711.
DSC curves, nitrogen adsorption isotherms, Raman spectrum, water contact angle measurements, and dye adsorption kinetics (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
This work was financially supported by the start-up from the University of Southern Mississippi. The purchase of the XPS instrumentation used in this work was supported by the NSF Major Research Instrumentation program (DMR-1726901).
The authors declare no competing financial interest.
Supplementary Material
References
- Lebreton L.; Andrady A. Future Scenarios of Global Plastic Waste Generation and Disposal. Palgrave Commun. 2019, 5, 6. 10.1057/s41599-018-0212-7. [DOI] [Google Scholar]
- Adyel T. M. Accumulation of Plastic Waste during COVID-19. Science 2020, 369, 1314–1315. 10.1126/science.abd9925. [DOI] [PubMed] [Google Scholar]
- Patrício Silva A. L.; Prata J. C.; Walker T. R.; Duarte A. C.; Ouyang W.; Barcelò D.; Rocha-Santos T. Increased Plastic Pollution Due to COVID-19 Pandemic: Challenges and Recommendations. Chem. Eng. J. 2021, 405, 126683. 10.1016/j.cej.2020.126683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson N. U.; Bassey D. E.; Palanisami T. COVID Pollution: Impact of COVID-19 Pandemic on Global Plastic Waste Footprint. Heliyon 2021, 7, e06343 10.1016/j.heliyon.2021.e06343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Wu P.; Schartup A. T.; Zhang Y. Plastic Waste Release Caused by COVID-19 and Its Fate in the Global Ocean. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2111530118 10.1073/pnas.2111530118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantoko D.; Li X.; Pariatamby A.; Yoshikawa K.; Horttanainen M.; Yan M. Challenges and Practices on Waste Management and Disposal during COVID-19 Pandemic. J. Environ. Manage. 2021, 286, 112140. 10.1016/j.jenvman.2021.112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- How to Stop Discarded Face Masks from Polluting the Planet. https://www.nationalgeographic.com/environment/article/how-to-stop-discarded-face-masks-from-polluting-the-planet (accessed on 1/12/2022).
- Rubio-Romero J. C.; Pardo-Ferreira M. d. C.; Torrecilla-García J. A.; Calero-Castro S. Disposable Masks: Disinfection and Sterilization for Reuse, and Non-Certified Manufacturing, in the Face of Shortages during the COVID-19 Pandemic. Saf. Sci. 2020, 129, 104830. 10.1016/j.ssci.2020.104830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. X.; Shan H.; Zhang C. M.; Zhang H. L.; Li G. M.; Yang R. M.; Chen J. M. Decontamination of Face Masks with Steam for Mask Reuse in Fighting the Pandemic COVID-19: Experimental Supports. J. Med. Virol. 2020, 92, 1971–1974. 10.1002/jmv.25921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hearn K.; Gertsman S.; Webster R.; Tsampalieros A.; Ng R.; Gibson J.; Sampson M.; Sikora L.; McNally J. D. Efficacy and Safety of Disinfectants for Decontamination of N95 and SN95 Filtering Facepiece Respirators: A Systematic Review. J. Hosp. Infect. 2020, 106, 504–521. 10.1016/j.jhin.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscusi D. J.; Bergman M. S.; Eimer B. C.; Shaffer R. E. Evaluation of Five Decontamination Methods for Filtering Facepiece Respirators. Ann. Occup. Hyg. 2009, 53, 815–827. 10.1093/annhyg/mep070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker L.; Krajnc A. P.; Malec J.; Radulović V.; Gradišek A.; Jelen A.; Remškar M.; Mekjavić I. B.; Kovač J.; Mozetič M.; Snoj L. Sterilization of Polypropylene Membranes of Facepiece Respirators by Ionizing Radiation. J. Membr. Sci. 2021, 619, 118756. 10.1016/j.memsci.2020.118756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hadyan K.; Alsbeih G.; Al-Harbi N.; Judia S. B.; Al-Ghamdi M.; Almousa A.; Alsharif I.; Bakheet R.; Al-Romaih K.; Al-Mozaini M.; Al-Ghamdi S.; Moftah B.; Alhmaid R. Effect of Gamma Irradiation on Filtering Facepiece Respirators and SARS-CoV-2 Detection. Sci. Rep. 2021, 11, 19888. 10.1038/s41598-021-99414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert T. J.; Ennis S.; Musolino S. F.; Buckley H. L.; Niikura M.; Wulff J. E.; Menon C. Covalent Functionalization of Polypropylene Filters with Diazirine–Photosensitizer Conjugates Producing Visible Light Driven Virus Inactivating Materials. Sci. Rep. 2021, 11, 19029. 10.1038/s41598-021-98280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schyns Z. O. G.; Shaver M. P.; G Schyns Z. O.; Shaver M. P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2021, 42, 2000415. 10.1002/marc.202000415. [DOI] [PubMed] [Google Scholar]
- Yin S.; Tuladhar R.; Shi F.; Shanks R. A.; Combe M.; Collister T. Mechanical Reprocessing of Polyolefin Waste: A Review. Polym. Sci. Eng. 2015, 55, 2899–2909. 10.1002/pen.24182. [DOI] [Google Scholar]
- Battegazzore D.; Cravero F.; Frache A. Is It Possible to Mechanical Recycle the Materials of the Disposable Filtering Masks?. Polymers 2020, 12, 2726. 10.3390/polym12112726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikiri S.; Iseki J.; Minobe M.. Process for production of carbon fiber. U.S. Patent 4,070,446 A, 1978.
- Postema A. R.; De Groot H.; Pennings A. J. Amorphous Carbon Fibres from Linear Low Density Polyethylene. J. Mater. Sci. 1990, 25, 4216–4222. 10.1007/bf00581075. [DOI] [Google Scholar]
- Gupta A.; Harrison I. R. New Aspects in the Oxidative Stabilization of PAN-Based Carbon Fibers. Carbon 1996, 34, 1427–1445. 10.1016/s0008-6223(96)00094-2. [DOI] [Google Scholar]
- Wangxi Z.; Jie L.; Gang W. Evolution of Structure and Properties of PAN Precursors during Their Conversion to Carbon Fibers. Carbon 2003, 41, 2805–2812. 10.1016/s0008-6223(03)00391-9. [DOI] [Google Scholar]
- Jang D.; Lee M. E.; Choi J.; Cho S. Y.; Lee S. Strategies for the Production of PAN-Based Carbon Fibers with High Tensile Strength. Carbon 2022, 186, 644–677. 10.1016/j.carbon.2021.10.061. [DOI] [Google Scholar]
- Mochida I.; Yoon S.-H.; Takano N.; Fortin F.; Korai Y.; Yokogawa K. Microstructure of Mesophase Pitch-Based Carbon Fiber and Its Control. Carbon 1996, 34, 941–956. 10.1016/0008-6223(95)00172-7. [DOI] [Google Scholar]
- Xi X.; Chung D. D. L. Colossal Electric Permittivity Discovered in Polyacrylonitrile (PAN) Based Carbon Fiber, with Comparison of PAN-Based and Pitch-Based Carbon Fibers. Carbon 2019, 145, 734–739. 10.1016/j.carbon.2019.01.069. [DOI] [Google Scholar]
- Wazir A. H.; Kakakhel L. Preparation and Characterization of Pitch-Based Carbon Fibers. New Carbon Mater 2009, 24, 83–88. 10.1016/s1872-5805(08)60039-6. [DOI] [Google Scholar]
- Li Q.; Serem W. K.; Dai W.; Yue Y.; Naik M. T.; Xie S.; Karki P.; Liu L.; Sue H.-J.; Liang H.; Zhou F.; Yuan J. S. Molecular Weight and Uniformity Define the Mechanical Performance of Lignin-Based Carbon Fiber. J. Mater. Chem. A 2017, 5, 12740–12746. 10.1039/c7ta01187c. [DOI] [Google Scholar]
- Kadla J. F.; Kubo S.; Venditti R. A.; Gilbert R. D.; Compere A. L.; Griffith W. Lignin-Based Carbon Fibers for Composite Fiber Applications. Carbon 2002, 40, 2913–2920. 10.1016/s0008-6223(02)00248-8. [DOI] [Google Scholar]
- Behr M. J.; Landes B. G.; Barton B. E.; Bernius M. T.; Billovits G. F.; Hukkanen E. J.; Patton J. T.; Wang W.; Wood C.; Keane D. T.; Rix J. E.; Weigand S. J. Structure-Property Model for Polyethylene-Derived Carbon Fiber. Carbon 2016, 107, 525–535. 10.1016/j.carbon.2016.06.032. [DOI] [Google Scholar]
- Choi D.; Yoo S. H.; Lee S. Safer and More Effective Route for Polyethylene-Derived Carbon Fiber Fabrication Using Electron Beam Irradiation. Carbon 2019, 146, 9–16. 10.1016/j.carbon.2019.01.061. [DOI] [Google Scholar]
- Frank E.; Muks E.; Ota A.; Herrmann T.; Hunger M.; Buchmeiser M. R. Structure Evolution in Polyethylene-Derived Carbon Fiber Using a Combined Electron Beam-Stabilization-Sulphurization Approach. Macromol. Mater. Eng. 2021, 306, 2100280. 10.1002/mame.202100280. [DOI] [Google Scholar]
- Younker J. M.; Saito T.; Hunt M. A.; Naskar A. K.; Beste A. Pyrolysis Pathways of Sulfonated Polyethylene, an Alternative Carbon Fiber Precursor. J. Am. Chem. Soc. 2013, 135, 6130–6141. 10.1021/ja3121845. [DOI] [PubMed] [Google Scholar]
- World Plastics Production 1950–2015; Plastics Europe, 2018.
- Barton B. E.; Patton J.; Hukkanen E.; Behr M.; Lin J.-C.; Beyer S.; Zhang Y.; Brehm L.; Haskins B.; Bell B.; Gerhart B.; Leugers A.; Bernius M. The Chemical Transformation of Hydrocarbons to Carbon Using SO3 Sources. Carbon 2015, 94, 465–471. 10.1016/j.carbon.2015.07.029. [DOI] [Google Scholar]
- Hukkanen E. J.; Barton B. E.; Patton J. T.; Schlader D. R.; Zhang Y.; Qiu X.; Brehm L.; Haskins B.; Wang W.; Horstman N.; Spalding M. A.; Hickman D. A.; Derstine C. W. A Novel Continuous Multiphase Reactor for Chemically Processing Polymer Fibers. Ind. Eng. Chem. Res. 2018, 57, 6123–6130. 10.1021/acs.iecr.8b00482. [DOI] [Google Scholar]
- Lee G.; Eui Lee M.; Kim S.-S.; Joh H.-I.; Lee S. Efficient Upcycling of Polypropylene-Based Waste Disposable Masks into Hard Carbons for Anodes in Sodium Ion Batteries. J. Ind. Eng. Chem. 2022, 105, 268–277. 10.1016/j.jiec.2021.09.026. [DOI] [Google Scholar]
- Villagómezvillagómez-Salas S.; Manikandan P.; Acuñ S. F.; Guzmán G.; Pol V. G. Amorphous Carbon Chips Li-Ion Battery Anodes Produced through Polyethylene Waste Upcycling. ACS Omega 2018, 3, 17520–17527. 10.1021/acsomega.8b02290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Lin Z. Transforming Waste Polypropylene Face Masks into S-Doped Porous Carbon as the Cathode Electrode for Supercapacitors. Ionics 2021, 27, 2169–2179. 10.1007/s11581-021-03949-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D.; Kil H.-S.; Lee S. Fabrication of Low-Cost Carbon Fibers Using Economical Precursors and Advanced Processing Technologies. Carbon 2019, 142, 610–649. 10.1016/j.carbon.2018.10.028. [DOI] [Google Scholar]
- Lee G.; Eui Lee M.; Kim S.-S.; Joh H.-I.; Lee S. Efficient Upcycling of Polypropylene-Based Waste Disposable Masks into Hard Carbons for Anodes in Sodium Ion Batteries. J. Ind. Eng. Chem. 2022, 105, 268–277. 10.1016/j.jiec.2021.09.026. [DOI] [Google Scholar]
- Eun J. H.; Lee J. S. Study on Polyethylene-Based Carbon Fibers Obtained by Sulfonation under Hydrostatic Pressure. Sci. Rep. 2021, 11, 18028. 10.1038/s41598-021-97529-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton B. E.; Behr M. J.; Patton J. T.; Hukkanen E. J.; Landes B. G.; Wang W.; Horstman N.; Rix J. E.; Keane D.; Weigand S.; Spalding M.; Derstine C. High-Modulus Low-Cost Carbon Fibers from Polyethylene Enabled by Boron Catalyzed Graphitization. Small 2017, 13, 1701926. 10.1002/smll.201701926. [DOI] [PubMed] [Google Scholar]
- Hunt M. A.; Saito T.; Brown R. H.; Kumbhar A. S.; Naskar A. K. Patterned Functional Carbon Fibers from Polyethylene. Adv. Mater. 2012, 24, 2386–2389. 10.1002/adma.201104551. [DOI] [PubMed] [Google Scholar]
- Qiang Z.; Chen Y.-M.; Xia Y.; Liang W.; Zhu Y.; Vogt B. D. Ultra-Long Cycle Life, Low-Cost Room Temperature Sodium-Sulfur Batteries Enabled by Highly Doped (N,S) Nanoporous Carbons. Nano Energy 2017, 32, 59–66. 10.1016/j.nanoen.2016.12.018. [DOI] [Google Scholar]
- Patel A.; Gaharwar A. K.; Iviglia G.; Zhang H.; Mukundan S.; Mihaila S. M.; Demarchi D.; Khademhosseini A. Highly Elastomeric Poly(Glycerol Sebacate)-Co-Poly(Ethylene Glycol) Amphiphilic Block Copolymers. Biomaterials 2013, 34, 3970–3983. 10.1016/j.biomaterials.2013.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiciński W.; Dziura A. Heteroatom-Doped Carbon Gels from Phenols and Heterocyclic Aldehydes: Sulfur-Doped Carbon Xerogels. Carbon 2014, 75, 56–67. 10.1016/j.carbon.2014.02.085. [DOI] [Google Scholar]
- Choi S.; Park J.; Hyun W.; Kim J.; Kim J.; Lee Y. B.; Song C.; Hwang H. J.; Kim J. H.; Hyeon T.; Kim D.-H. Stretchable Heater Using Ligand-Exchanged Silver Nanowire Nanocomposite for Wearable Articular Thermotherapy. ACS Nano 2015, 9, 6626–6633. 10.1021/acsnano.5b02790. [DOI] [PubMed] [Google Scholar]
- Lan W.; Chen Y.; Yang Z.; Han W.; Zhou J.; Zhang Y.; Wang J.; Tang G.; Wei Y.; Dou W.; Su Q.; Xie E. Ultraflexible Transparent Film Heater Made of Ag Nanowire/PVA Composite for Rapid-Response Thermotherapy Pads. ACS Appl. Mater. Interfaces 2017, 9, 6644–6651. 10.1021/acsami.6b16853. [DOI] [PubMed] [Google Scholar]
- Niu H.; Li J.; Wang X.; Luo F.; Qiang Z.; Ren J. Solar-Assisted, Fast, and in Situ Recovery of Crude Oil Spill by a Superhydrophobic and Photothermal Sponge. ACS Appl. Mater. Interfaces 2021, 13, 21175–21185. 10.1021/acsami.1c00452. [DOI] [PubMed] [Google Scholar]
- Niu H.; Li J.; Qiang Z.; Ren J. Versatile and Cost-Efficient Cleanup of Viscous Crude Oil by an Elastic Carbon Sorbent from Direct Pyrolysis of a Melamine Foam. J. Mater. Chem. A 2021, 9, 11268–11277. 10.1039/d1ta01133b. [DOI] [Google Scholar]
- Zhou Q.; Lyu J.; Wang G.; Robertson M.; Qiang Z.; Sun B.; Ye C.; Zhu M. Mechanically Strong and Multifunctional Hybrid Hydrogels with Ultrahigh Electrical Conductivity. Adv. Funct. Mater. 2021, 31, 2104536. 10.1002/adfm.202104536. [DOI] [Google Scholar]
- Jin Y.; Lin Y.; Kiani A.; Joshipura I. D.; Ge M.; Dickey M. D. Materials Tactile Logic via Innervated Soft Thermochromic Elastomers. Nat. Commun. 2019, 10, 4187. 10.1038/s41467-019-12161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fangohr H.; Chernyshenko D. S.; Franchin M.; Fischbacher T.; Meier G. Joule Heating in Nanowires. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 054437. 10.1103/physrevb.84.054437. [DOI] [Google Scholar]
- Lei Z.; Du Z.; Yu W.; Yan J.; Li Z.; Shui H.; Ren S.; Wang Z.; Ying K.; Kang S. Facile Synthesis of Carbon Film with High Carrier Concentration Using Coal Tar and the Application in Joule Heating. ACS Appl. Electron. Mater. 2021, 3, 3271–3277. 10.1021/acsaelm.1c00434. [DOI] [Google Scholar]
- Saleem J.; Adil Riaz M.; Gordon M. Oil Sorbents from Plastic Wastes and Polymers: A Review. J. Hazard. Mater. 2018, 341, 424–437. 10.1016/j.jhazmat.2017.07.072. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Tai N.-H. Carbon Materials as Oil Sorbents: A Review on the Synthesis and Performance. J. Mater. Chem. A 2016, 4, 1550–1565. 10.1039/c5ta08321d. [DOI] [Google Scholar]
- Niu H.; Qiang Z.; Ren J. Durable, Magnetic-Responsive Melamine Sponge Composite for High Efficiency, in-situ Oil-Water Separation. Nanotechnology 2021, 32, 275705. 10.1088/1361-6528/abef2e. [DOI] [PubMed] [Google Scholar]
- Inagaki M.; Qiu J.; Guo Q. Carbon Foam: Preparation and Application. Carbon 2015, 87, 128–152. 10.1016/j.carbon.2015.02.021. [DOI] [Google Scholar]
- Chen S.; He G.; Hu H.; Jin S.; Zhou Y.; He Y.; He S.; Zhao F.; Hou H. Elastic Carbon Foam via Direct Carbonization of Polymer Foam for Flexible Electrodes and Organic Chemical Absorption. Energy Environ. Sci. 2013, 6, 2435–2439. 10.1039/c3ee41436a. [DOI] [Google Scholar]
- Ko Y. G.; Choi U. S.; Kim J. S.; Park Y. S. Novel Synthesis and Characterization of Activated Carbon Fiber and Dye Adsorption Modeling. Carbon 2002, 40, 2661–2672. 10.1016/s0008-6223(02)00168-9. [DOI] [Google Scholar]
- Tamai H.; Yoshida T.; Sasaki M.; Yasuda H. Dye Adsorption on Mesoporous Activated Carbon Fiber Obtained from Pitch Containing Yttrium Complex. Carbon 1999, 37, 983–989. 10.1016/s0008-6223(98)00294-2. [DOI] [Google Scholar]
- Yang P.; Yang W. Surface Chemoselective Phototransformation of C–H Bonds on Organic Polymeric Materials and Related High-Tech Applications. Chem. Rev. 2013, 113, 5547–5594. 10.1021/cr300246p. [DOI] [PubMed] [Google Scholar]
- Wang D.; Ha Y.; Gu J.; Li Q.; Zhang L.; Yang P. 2D Protein Supramolecular Nanofilm with Exceptionally Large Area and Emergent Functions. Adv. Mater. 2016, 28, 7414–7423. 10.1002/adma.201506476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.