

Abstract

In silico methods to identify novel drug–target interactions (DTIs) have gained significant importance over conventional techniques owing to their labor-intensive and low-throughput nature. Here, we present a machine learning-based multiclass classification workflow that segregates interactions between active, inactive, and intermediate drug–target pairs. Drug molecules, protein sequences, and molecular descriptors were transformed into machine-interpretable embeddings to extract critical features from standard datasets. Tools such as CHEMBL web resource, iFeature, and an in-house developed deep neural network-assisted drug recommendation (dNNDR)-featx were employed for data retrieval and processing. The models were trained with large-scale DTI datasets, which reported an improvement in performance over baseline methods. External validation results showed that models based on att-biLSTM and gCNN could help predict novel DTIs. When tested with a completely different dataset, the proposed models significantly outperformed competing methods. The validity of novel interactions predicted by dNNDR was backed by experimental and computational evidence in the literature. The proposed methodology could elucidate critical features that govern the relationship between a drug and its target.
1. Introduction
Identifying novel drug–target interactions (DTIs) is considered a stagnant and labor-intensive process. A conventional drug discovery and development workflow can extend to about 14 years and drain almost a billion USD in capital.1,2 Lead identification, optimization, screening, and characterization are a few of the many steps in an assay-based drug discovery workflow. With the advent of big data and computational advances, in silico methods have found utility in predicting novel DTIs, ultimately aiding the process of drug discovery.3,4 While traditional workflows fare better than in silico alternatives in terms of reliability and robustness, analysis and characterization of vast volumes of data are not possible due to their inherent throughput limitations.
Computer-aided DTI estimation methods roughly fall into two classes—biophysical models and statistical methods. Biophysical methods such as molecular dynamics try to replicate a biological arrangement in silico under a set of physical constraints and infer DTIs at a molecular level.5,6 Such methods are limited by the availability of molecular structures and computational restraints.7 Alternatively, statistical approaches such as support vector machines, kernel learning, and supervised bipartite graphs extract information from interaction data and make logical inferences.8 Until recently, using these solutions, most DTI studies were limited to the datasets wherein drug–target pairs with no known affinity were included, thus neglecting any measure of binding affinity altogether.8−12
With the recent advances in high-throughput assays, chemoproteomic approaches such as thermal profiling have allowed the quantification of compound potency on a broader scale.13 DTI is generally quantified using measures such as dissociation constant (Kd), inhibition constant (Ki), or half-maximal inhibitory concentration (IC50). With the availability of large, curated DTI datasets such as DrugBank in the public domain, numerous computer-aided methods have been developed that complement the process of drug discovery.14−17
Deep learning methods find their application in almost any research field that generates one or another form of data. It has found its relevance in computer vision, natural language processing (NLP), genomics, and drug discovery.18,19 The most significant advantage of deep learning architectures is that they can model nonlinear relationships in data and generate a better representation that ultimately aids the learning process.20,21 In sequence form, drug and protein data have been used with convolutional neural networks (CNNs) to extract local residue patterns and predict binding affinities.22 However, the functioning/characteristics of a protein or drug depend on the order of its elements rather than the elements themselves.
To include this vital concept into a DTI prediction system, it was proposed to harness the potential of state-of-the-art machine learning (ML) algorithms such as bi-directional long short-term memory (biLSTM) and graph CNN (gCNN). These algorithms efficiently incorporate sequential information to make sense of its ordering and can be trained to predict the type of interaction between a given drug–target pair. In addition to the sequential information, small molecules have an inherent graphical structure, which is lost when represented and processed in one-hot encodings of text-based formats such as SMILES. Recent studies suggested that retaining the underlying graphical structure of a drug molecule could allow the model to represent them in an efficient manner.23,24 The combination of gCNN and biLSTM blocks was introduced to process the structure of chemical compounds from simplified molecular input line entry system (SMILES) and protein sequences, respectively. Moreover, molecular descriptors for proteins and drugs were also used to train the complete network. The generated representations and molecular descriptors were then fed into a fully connected feed-forward neural network to make sense of a multiclass classification problem.
2. Materials and Methods
2.1. Formulation of the Problem
For a list of probable drug–target pairs, the aim was to segregate the samples into (i) Class-I: Active, (ii) Class-II: Intermediate, or (iii) Class-III: Inactive. The proposed methodology followed a three-step process: (i) processing and labeling DTI data into predefined classes, (ii) developing and training a multiclass classification model for given drug–target pairs, and (iii) inference and validation using an external dataset to infer real-world performance.
A multiclass classification approach was chosen to efficiently understand DTIs rather than the more conventional binary classification because (i) most of the binary classification tasks tend to label nontested drug–target combinations as a negative data point and (ii) even in the case where we have the activity profile of the drug–target pair in terms of IC50, Kd, or Ki, a single activity threshold is not uniformly followed in the literature. Furthermore, the conventional binary classification task has some inherent drawbacks and inadequacies. The most evident is the need for a predefined binarization threshold, often arbitrarily decided.
To mitigate the issues mentioned earlier, binding affinities were segregated into three categories based on the magnitude of their value. The choice of activity thresholds was central to the overall objective of the proposed method. Current literature was extensively explored to formalize static thresholds that are a good indicator of the activity or inactivity of a drug–target pair. An IC50 value of <0.1 μM was a good indicator of an active DTI.25 Similarly, DTIs with an IC50 value of >30 μM can be considered inactive pairs. The remaining DTI data points were grouped under the intermediate category. We have previously explored this segregation methodology in a similar study.26 Following this criterion, a total of 7057 active (Class-I) interactions, 24,752 intermediate (Class-II) interactions, and 28,748 (Class-III) inactive interactions were fed into deep neural network-assisted drug recommendation (dNNDR) models. The overall input statistics are summarized in Figure 1B.
Figure 1.

General overview of the overall methodology. (A) Primary DTI data was collected, processed, screened, and encoded. The statistic of the screened dataset is summarized in (B). (C) An attention-biLSTM network was constructed for the protein sequences and (D) a gCNN derivative was employed for drug SMILES. (E) A fully connected feed-forward neural network taking inputs from attention-biLSTM and gCNN and (F) molecular descriptors were trained for a (G) multiclass classification problem in a fivefold cross-validated setup.
2.2. Chemical Datasets
The primary evaluation of dNNDR models was done on the drugs from the Kinase dataset Davis and KIBA dataset.27,28 These datasets have previously been used in similar DTI prediction tasks and serve as a benchmark in DTI prediction tasks.29,30
In the form of SMILE strings, drug molecules were retrieved for KIBA drugs using in-house programs in tandem with ChEMBL web services.31 The SMILE string for each drug was encoded as an undirected graph to feed into a compatible ML framework. Moreover, we calculated 111 drug descriptors for the retrieved molecules using RDKit, an open-source cheminformatics framework available at http://www.rdkit.org. Applying these operations on all the drugs provided us with two feature matrices to describe sequence information and chemical characteristics.
Similarly, a data retrieval pipeline was built to extract protein sequences for the retrieved drug–target accession identifiers using UNIPROT’s web framework and stored as FASTA files.32 Amino acid sequences for each target were encoded as a one-hot vector representing it in a machine-interpretable form. In addition to the sequence information, feature matrices for all the protein targets were also retrieved using iFeature. This Python package simplified the process of computing sequence level characteristics such as amino acid composition (AAC), composition/transition/distribution (CTD), among others, for any given protein molecule.33 Also, to further simplify the process of feature extraction, a graphical user interface was developed. We termed it dNNDR-Featx, and it can be used to extract multiple types of protein and drug descriptors without any command-line tool. A general interface and basic functionalities of dNNDR-featx are depicted in Figure S1. Also, it is an open-source utility freely made available at https://github.com/TeamSundar/dNNDR-featx.
2.3. Pharmacological Data
The KIBA dataset aggregates the bioactivity of drug–target pairs as a custom-unified metric, combining the values from IC50, Kd, and Ki measures.28 As the activity thresholds were not well defined in the KIBA dataset, IC50 values for the interacting pairs were extracted directly from ChEMBL. Although a large pool (∼0.2 M) of DTIs was retrieved from ChEMBL, a healthy chunk of it was filtered out due to nonstandard/missing activity values and incomplete information. Of 30,474 compounds, 981 targets and 61,624 interactions were finally screened after all the preprocessing steps. All the datasets used are summarized in Table 1.
Table 1. General Summary of All the Datasets Used in the Study.
| proteins | compounds | interactions | |
|---|---|---|---|
| KIBAa (IC50) | 961 | 30,474 | 61,624 |
| Davis Metzb | 237 | 18 | 4255 |
| Davis Anastasiadisb | 154 | 24 | 2575 |
Note: Proteins for drugs listed in the KIBA dataset were extracted manually from CHEMBL.
Used as an external validation dataset.
2.4. Model Architecture
All the models were executed in Python, whereas Scikit-learn, TensorFlow-Keras, and Spektral were used to implement the ML algorithms. The metrics such as accuracy, auROC, auPR, and macro-averaged F1-score was employed to quantify the performance of the proposed and baseline models.
A modular ML architecture was designed for the problem at hand. As protein sequences and drug SMILES have a fundamental difference in carrying the information forward, different ML methods were employed to handle them. The first method employed a biLSTM architecture clubbed with attention (att-biLSTM) for sequential data such as SMILES and protein sequences.34 In cases where the molecular structure of a drug was being used in the network, gCNN was employed due to its ability to represent molecules efficiently. These methods were seamlessly integrated into three different model architectures which were termed as dNNDRa (att-biLSTM), dNNDRb (att-biLSTM + descriptors), and dNNDRc (biLSTM + gCNN) thereafter. A clear description of all the models is summarized in Table 2, while a graphical representation of all the model architectures is compiled in Figures S2–S6.
Table 2. Key Characteristics of All the Models Tested in the Study.
| method | architecture highlights | Data |
|---|---|---|
| sequence-baseda | 1-D convolution | SMILES and protein sequences |
| CTD-baseda | 1-D convolution | SMILES, protein sequences, and CTD features |
| dNNDRa | Att-biLSTM | SMILES and protein sequences |
| dNNDRb | Att-biLSTM | SMILES, protein sequences, and molecular descriptors |
| dNNDRc | biLSTM and gCNN | SMILES and protein sequences |
Baseline methods.
2.5. Input Representation
To apply any mathematical operation on the sequences, they must be converted into a machine-compatible format. The sequences were represented in the form of integer encoding. Forty-four unique categories of characters from SMILES and 21 unique characters from protein sequences were encoded with a unique integer (e.g., “C”:1, “N”:2, “O”:3). For instance, “CN=C=O” was encoded as [ C N=C=O] = [1 2 35 1 35 3]. Using a similar process, protein sequences were also encoded by 21 unique characters. The sequence length limits were decided based on exploratory data analysis (EDA). Based on mean sequence lengths calculated by EDA, maximum lengths of 150 and 1000 were decided for SMILES and protein sequences, respectively. The same can be visualized from the distribution of sequence lengths for all the included datasets (Figure 2C). All the sequences were trimmed or padded to match the decision criteria. Additional features were concatenated with the existing ones for the models where descriptors were included. Details of the input dimensions are summarized in Table 3.
Figure 2.

Overall statistics of the datasets used in the study. (A,B) A general overview of multiple interactions in the datasets is depicted for drugs and targets. (C) Estimation of the sequence thresholds for drug SMILES and protein sequences. The length of SMILE strings and protein sequences does not exceed a value of about 150 and 2000 for any dataset. (D) Number of data points and an estimate of their activities for each of the three interaction groups.
Table 3. Summary of All the Models under Study and the Final Dimensions of the Data after Processing.
| method | drug feature dimension | protein feature dimension |
|---|---|---|
| sequence-based | 150 × 44 | 1000 × 21 |
| CTD-based | 150 × 44 | (1000 × 21) + 273 |
| dNNDRa | 150 × 44 | 1000 × 21 |
| dNNDRb | (150 × 44) + (150 × 111) | 1000 × 21 |
| dNNDRc | (123 × 123) + (123 × 17) + (123 × 3) | 1000 × 21 |
2.6. biLSTM with Attention and 1-D Convolution
LSTM and biLSTM are two of the best and most-used modifications of recurrent neural networks (RNNs) in the field of NLP.35−38 They try to learn long-term dependencies between sequence data such that the model can pass on critical information to the terminal layers of the network. By doing so, they have been proven to be a stable and powerful way of modeling long-term dependencies, as in the case of long amino acid sequences. On the other hand, attention helps the network focus on the given input and extract the most important information iteratively. biLSTM was preferred over vanilla LSTM because (i) it learns faster than conventional LSTM and (ii) it has a better contextual understanding.35 dNNDR models had an embedding layer to start with, followed by biLSTM and attention layers.
2.6.1. Embedding Layer
To draw out the semantic information of the amino acid/SMILE sequences, each one of them was represented as a sequence of embeddings to start with.
| 1 |
where vector e1→ denotes the vector of the ith amino acid/atom with a dimension d and s the whole sequence as a global vector.39
2.6.2. biLSTM Layer
biLSTM is a gradual extension of the traditional LSTM network used to obtain high-level features with sequential information. For the model to have a sense of the sequence order, both forward and backward LSTM outputs were employed. Therefore, the final output was calculated using the element-wise sum of both forward and backward outputs for each character. A detailed description of the working of the LSTM network is out of the scope of this study and can be found elsewhere.34
2.6.3. Attention Layer
It is widely known and accepted that functional and structural importance regions exist in any protein sequence.40 By its nature, the attention mechanism can pinpoint locations of relevance in a long protein/SMILE sequence such that the succeeding layers get the most critical information. The attention mechanism can be formally summarized as follows.
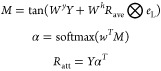 |
2 |
where Y is the output vector coming from biLSTM (refer to section 5.4.2), Rave is the output from the mean pooling layer, α is the attention vector, and Ratt is the attention-weighted sequence representation. After all these operations, Ratt is fed to the final layers of the networks for further training.
2.6.4. gCNN Branch for Drugs
gCNN is an extension of conventional CNNs that can learn a graphical representation.41 gCNN has attracted considerable attention, especially in drug discovery, due to the inherent compatibility of molecules to be represented as undirected graphs.42−44 Molecular structures can be efficiently modeled as a graph, and various studies have demonstrated the effectiveness of such representations.45 Given a molecule with its spatial coordinates, three matrices were created for its graphical representation: an edge matrix, a node matrix, and an adjacency matrix. More specifically, a subtle variation of gCNN called graph edge conditioned CNN (gECCNN) was employed for all practical purposes in this study. These three matrices served as inputs to the gECCNN network, followed by a series of edge convolutions. The complete gECCNN model was built with the help of Spektral, which is an open-source library for graph deep learning.46 The complete network architecture is available in Supporting Information File 1.
2.7. Loss and Model Optimization
Two fully connected layers of varying neuron size followed information propagating from the att-biLSTM or gECCNN branches. The neuron sizes for the dense layers were optimized for performance using a manual grid search. Training time depended on the type of model being trained but remained under a day for all the models except dNNDRc.
Being a multiclass classification problem, categorical cross-entropy was used as the loss function. It was defined as a sum of losses for each class label coming out of a SoftMax function and is mathematically given by
| 3 |
Here, N is the number of classes (three in this case), i denotes the data point, yi, cis the binary target indicator [0,1], and p is the model prediction.
2.8. Evaluation Metrics
The auROC, the auPR, accuracy, and macro-averaged F1-score were used to evaluate the performances for comparative analysis. As identifying false-negatives and false-positives are vital for a drug–target estimation system, the F1-score was included as an evaluation metric. It is mathematically computed as described in eq 4.
| 4 |
auROC and auPR are independent of imbalances and serve as a better indicator of performance than accuracy as they tend to become unreliable with class imbalances in training data. Moreover, to efficiently balance bias and variance trade-off, fivefold cross-validation was employed. It works by randomly splitting the dataset into five equal parts, training on four and testing on one, iteratively over five training rounds.
2.9. Baseline Models for Comparison
Sequence-based methods aim to generalize the relationship between drug–target pairs based on their sequence characteristics. These methods generally rely on a similarity-based learning approach wherein similarity matrices of drugs and targets are constructed to infer novel interactions. The kernel regression, bipartite local method, and pairwise kernel method are some of those where these matrices are employed.8,47 However, for heuristic approaches such as deep learning, most of the architectures of sequence-based models have a similar backbone. Similarly, molecular descriptor-based methods generally use CTD to represent a protein and its properties.48,49 On similar lines, two architectures that represented sequence and descriptor-based approaches such as FRnet-DTI, DeepConv-DTI, and DeepDTA were used.22,50,51 Of note, none of these methods were multiclass classification problems, and hence, a direct comparison was not possible. However, an indirect comparison through similar model architectures could be a valid approach for such cases.
3. Results
With comparative analysis against baseline methods, three model architectures are proposed in this study. The seed architecture over which all three models were built is described in Figure 1. The proposed models differ in the choice of data combinations and the way in which they were processed. For the first model dNNDRa, we used biLSTM with the attention layer on SMILES and protein sequences along with 1-D convolution. For the second model dNNDRb, 111 types of molecular descriptors were included along with the sequences. The architecture was similar to dNNDRa with an addition of a parallel descriptor input.
Similarly, the third model dNNDRc used gCNN to handle drug data and biLSTM layers for protein data. The shape of inputs and outputs varied accordingly for each variation and are summarized in Table 3. The results suggested that the inclusion of biLSTM, attention, and gCNN into the architecture improved the performance over models that used only 1-D convolution for sequence features and similar transformations for numerical features. Table 4 reported the average value of all the performance metrics used in all the models under consideration. Further, a benchmarking experiment was performed with the qm9 dataset wherein a set of ∼100 k small molecules were trained with LSTMs and gNNs to predict eight quantum chemical properties.52 The comparative analysis (Table S1) clearly indicated that gNNs outperformed LSTMs in terms of mean absolute error and coefficient of determination (R2). This provides significant evidence in support of utilizing the underlying graphical structure of small molecules, in addition to the sequence-based processing methods for building such predictive models. All the proposed methods outperformed baseline models with high accuracy and a greater auROC, auPR, and F1-score (Figure 3). It must be emphasized that auROC for all the dNNDR variants was better than baseline models for all three types of interactions (Figure 3A–C). Therefore, the proposed model accurately predicted the interactions and avoided giving false-negative and false-positive inferences to an extent.
Table 4. Performance of All Models under Consideration Are Summarizeda.
| auROC |
auPR | validation accuracy | |||
|---|---|---|---|---|---|
| I | II | III | |||
| seq based | 0.86 (0.003) | 0.90 (0.006) | 0.85 (0.006) | 0.83 (0.007) | 0.74 (0.008) |
| CTD | 0.87 (0.005) | 0.90 (0.004) | 0.86 (0.003) | 0.83 (0.003) | 0.75 (0.005) |
| dNNDRa | 0.87 (0.007) | 0.91 (0.005) | 0.86 (0.004) | 0.84 (0.005) | 0.76 (0.005) |
| dNNDRb | 0.88 (0.005) | 0.93 (0.005) | 0.87 (0.007) | 0.86 (0.007) | 0.77 (0.008) |
| dNNDRc | 0.90 (0.002) | 0.94 (0.001) | 0.89 (0.002) | 0.88 (0.003) | 0.79 (0.004) |
All the models were fivefold cross-validated with the standard deviation mentioned alongside every result. The best-performing models among the cross-validated ones are marked in bold. Note: dNNDRa: att-biLSTM, dNNDRb: att-biLSTM + descriptors, dNNDRc: biLSTM + gCNN.
Figure 3.

Comparison of performances among all the methods under observation. ROC curves for the three classes, namely, (A) active, (B) intermediate, and (C) inactive, indicate the effectiveness of dNNDR over baseline methods in predicting the type of interaction for a given drug–target pair. (D) Validation accuracy, (E) precision-recall curves, and (F) macro-averaged F1-score also follow a similar trend and reinforce the utility of the proposed methods.
3.1. External Validation Reinforces the Robustness of the Approach
Though the methodology demonstrated a significant improvement in performance over other methods (Figure 3), the ultimate criterion for effectiveness is generalizability for any ML solution. Therefore, two entirely different datasets were prepared for external validation of all the methods under consideration. For a fair comparison with benchmark methods, model weights were updated to be compatible with a binary classification task. It is emphasized that datapoints belonging to only the active and inactive classes of the original training data were employed for updating the model weights. This was necessary as the model architecture was optimized for differentiating active and inactive datapoints from the intermediate class and including such datapoints in either region (active/inactive) would not be ideal. Minor modifications were done to the final layer of the model architecture for achieving this moderation. Validation datasets from KIBA were processed using the same procedure as followed previously to segregate datapoints into three classes and inference was made (again using only the datapoints from active and inactive classes) for the updated binary classification models. Outputs from all the baseline and existing methods were binarized if required (Table 5).
Table 5. External Validation Results Reinforcing the Effectiveness of the Proposed Modelsa.
| external
validation set |
||||||
|---|---|---|---|---|---|---|
| Davis Anastasiadis | Davis Metz | |||||
| method | accuracy | precision | F1-score | accuracy | precision | F1-score |
| sequence-based | 0.69 | 0.60 | 0.64 | 0.66 | 0.56 | 0.60 |
| CTD | 0.19 | 0.03 | 0.06 | 0.22 | 0.05 | 0.08 |
| dNNDRa | 0.47 | 0.67 | 0.51 | 0.53 | 0.51 | 0.52 |
| dNNDRb | 0.76 | 0.58 | 0.66 | 0.70 | 0.59 | 0.58 |
| DeepDTA | 0.70 | 0.56 | 0.68 | 0.64 | 0.55 | 0.57 |
| DeepConv-DTI | 0.72 | 0.61 | 0.64 | 0.68 | 0.56 | 0.61 |
The best-performing method has been marked in bold. 2. dNNDRa: Att-biLSTM, dNNDRb: Att-biLSTM + descriptors, dNNDRc: biLSTM + gCNN.
3.2. Predicting Unknown DTIs
After the external validation of dNNDR models, prediction runs for a set of proteins and drugs from a gold-standard dataset were performed.53 The goal was to predict all potential interactions that were not present in the dataset. dNNDRb was used for making the predictions. Salicylic acid, acetaminophen, mesalamine, and sodium salicylate were among the most commonly occurring compounds in the interactions predicted by the model. The majority of compounds were anti-inflammatory, COX inhibitors, analgesics, or neuropsychiatric agents.
Similarly, proteins associated with purine metabolism, cGMP-PKG signaling pathway, steroid hormone biosynthesis, linoleic acid metabolism, and chemical carcinogenesis were abundant in the novel interactions. It must be emphasized that most of the compounds designated as an interacting partner showed the properties of a drug molecule, such as aromaticity and active centers (Table 6). A complete list of predicted interactions is available on the GitHub repository.
Table 6. Novel DTI Predicted by dNNDRba.

A subset of the novel predictions from drugs and proteins in the gold-standard dataset is reported here.
4. Discussion
In this study, a data-driven approach was attempted to overcome the demerits of framing DTI prediction as a binary classification task by introducing an intermediate region between an active and inactive interaction. This makes for a more realistic and practical solution while staying uncompromised on the performance front at the same time. As with any sequential dataset, realizing the order of individual elements in the data is as crucial as the data itself. Therefore, much emphasis was given upon the information aspect by using attention, gCNN, and biLSTM networks. As described in the earlier section, dNNDR models consistently outperformed related methods in most performance metrics. While the other methods tend to drastically deter when tested on an entirely new dataset (external validation), dNNDR models showed exceptional effectiveness levels (Table 5). Furthermore, a graphical interface was also designed to complement feature extraction for any DTI prediction task. The dNNDR-featx program accepts plain text files containing drug–target pairs and extracts the molecular descriptors selected by the user. A general overview of the interface is shown in Figure 1. Although experimental screening methods can only verify the validation of the binding energies of putative DTIs, these results indicated that the proposed models could mature into promising methods for the identification of novel DTIs.
Ultimately, the proposed integrative approach recommended a set of promising DTIs that could be experimentally validated as promising leads for novel cancer therapies. However, experimental and computational evidence in the existing literature supported dNNDR’s DTI predictions (Supporting Information file 1). Acetaminophen has been shown to bind with CYP1A1 (cytochrome P450 family 1 subfamily A member 1), which was predicted by the proposed model.54 CYP1A1 metabolizes acetaminophen to N-acetyl-p-benzoquinone imine (NAPQI), along with 3-hydroxy acetaminophen.54 Salicylic acid, predicted to bind to mammalian carbonic anhydrases, has been reported to inhibit carbonic anhydrase I.55 Aspirin (acetyl derivative of salicylic acid) is computationally predicted to interact with phosphodiesterase 7B (PDE7B) in another report.56 Evidence of salicylamide inhibiting monoamine oxidase activity in rat liver and brain fractions has also been reported.57 Such experimental studies that validate some of the predictions from dNNDR’s methodology reinforced confidence in the predictive ability of dNNDR. Although experimentation is the ultimate validation tool for binding characteristics of model recommendations and for any possible clinical applications, these results indicated that the proposed models could mature into promising methods for the identification of novel DTIs.
It was observed that although dNNDR models performed relatively well, there is much scope for improvement. In the case of dNNDRc, while it performed significantly better than most of the other methods, the size of the model exponentially increased with the inclusion of high-dimensional representations in the form of a graph matrix, a node matrix, and an adjacency matrix (Table 1 and Figure 1). This led to a massive increase in training time and limited the ability to optimize the model efficiently. Hence, it must be noted that although dNNDRc showed great promise, it was excluded from the final inference.
5. Conclusions
Using the ordered information present in SMILES and protein sequences was central to the idea of dNNDR. To solve a multiclass classification problem, att-biLSTM and gCNN were employed to learn representations from raw sequence data and molecular descriptors. These methods were compared extensively with baseline methods on various measures of performance. The results obtained in this study reinforced the idea of using representations that try to capture the underlying order in sequential data. Including att-biLSTM and gCNN served the same purpose, and as a result, it was observed that there was a significant improvement in the performance compared to the baseline methods.
Moreover, dNNDR’s effectiveness was evident in the external validation setup, where they consistently outperformed the baseline models with a healthy margin. In the future, dNNDRc can be made more efficient in terms of hardware utilization, focusing on the interpretability of the proposed models. It is also proposed to integrate the proposed ML models into the dNNDR-featx interface in the near future to make it a standalone drug recommendation program. Analyzing the details of what the model is learning can be of great utility in improving the methodology further. Furthermore, the idea of using structural information of proteins for DTI prediction remains to be of immense utility.58 Therefore, the idea is to represent the spatial information provided by the protein 3D structure to improve the proposed methodology further. Although experimental screening methods can only verify the validation of the binding energies of putative DTIs, these results indicated that the proposed models could mature into promising methods for the identification of novel DTIs.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00424.
Schematic representation of all the model architectures used in the study (PDF)
(PDF)
Author Contributions
Y.K.: conceptualization, methodology, software, data curation, writing original draft; S.Y.: conceptualization, methodology, data curation, writing-reviewing, and editing; and D.S.: conceptualization, writing-reviewing, editing, and supervision. Y.K. and S.Y. contributed equally.
The authors declare no competing financial interest.
Notes
The source codes, implementation, datasets, and all prediction results for dNNDR are available at https://github.com/TeamSundar/dNNDR. Source code for the data processing module dNNDR-featx is available at https://github.com/TeamSundar/dNNDR-featx.
Supplementary Material
References
- Moses H. 3rd; Dorsey E. R.; Matheson D. H.; Thier S. O. Financial anatomy of biomedical research. JAMA 2005, 294, 1333–1342. 10.1001/jama.294.11.1333. [DOI] [PubMed] [Google Scholar]
- Myers S.; Baker A. Drug discovery--an operating model for a new era. Nat. Biotechnol. 2001, 19, 727–730. 10.1038/90765. [DOI] [PubMed] [Google Scholar]
- Brogi S.; Ramalho T. C.; Kuca K.; Medina-Franco J. L.; Valko M. Editorial: In silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. 10.3389/fchem.2020.00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muster W.; Breidenbach A.; Fischer H.; Kirchner S.; Müller L.; Pähler A. Computational toxicology in drug development. Drug Discovery Today 2008, 13, 303–310. 10.1016/j.drudis.2007.12.007. [DOI] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- Koes D. R.; Baumgartner M. P.; Camacho C. J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rarey M.; Kramer B.; Lengauer T.; Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- Bleakley K.; Yamanishi Y. Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 2009, 25, 2397–2403. 10.1093/bioinformatics/btp433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D. S.; Liu S.; Xu Q. S.; Lu H. M.; Huang J. H.; Hu Q. N.; Liang Y. Z. Large-scale prediction of drug-target interactions using protein sequences and drug topological structures. Anal. Chim. Acta 2012, 752, 1–10. 10.1016/j.aca.2012.09.021. [DOI] [PubMed] [Google Scholar]
- Cobanoglu M. C.; Liu C.; Hu F.; Oltvai Z. N.; Bahar I. Predicting Drug–Target Interactions Using Probabilistic Matrix Factorization. J. Chem. Inf. Model. 2013, 53, 3399–3409. 10.1021/ci400219z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönen M. Predicting drug–target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310. 10.1093/bioinformatics/bts360. [DOI] [PubMed] [Google Scholar]
- Öztürk H.; Ozkirimli E.; Özgür A. A novel methodology on distributed representations of proteins using their interacting ligands. Bioinformatics 2018, 34, i295–i303. 10.1093/bioinformatics/bty287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Reinhard F. B.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Martinez Molina D.; Jafari R.; Dovega R. B.; Klaeger S.; Kuster B.; Nordlund P.; Bantscheff M.; Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Ou-Yang S. S.; Lu J. Y.; Kong X. Q.; Liang Z. J.; Luo C.; Jiang H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. 10.1038/aps.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano J. D.; Tatonetti N. P. Informatics and Computational Methods in Natural Product Drug Discovery: A Review and Perspectives. Front. Genet. 2019, 10, 368. 10.3389/fgene.2019.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsila T.; Spyroulias G. A.; Patrinos G. P.; Matsoukas M.-T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. 10.1016/j.csbj.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Cheng D.; Shrivastava S.; Tzur D.; Gautam B.; Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S.; Lee B.; Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2017, 18, 851–869. 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- Gawehn E.; Hiss J. A.; Schneider G. Deep Learning in Drug Discovery. Mol. Inform. 2016, 35, 3–14. 10.1002/minf.201501008. [DOI] [PubMed] [Google Scholar]
- LeCun Y.; Bengio Y.; Hinton G. Deep learning. Nature 2015, 521, 436–444. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Lenselink E. B.; ten Dijke N.; Bongers B.; Papadatos G.; van Vlijmen H. W. T.; Kowalczyk W.; IJzerman A. P.; van Westen G. Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. Aust. J. Chem. 2017, 9, 45. 10.1186/s13321-017-0232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öztürk H.; Özgür A.; Ozkirimli E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Wu Z.; Hsieh C.-Y.; Chen G.; Liao B.; Wang Z.; Shen C.; Cao D.; Wu J.; Hou T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform. 2021, 13, 12. 10.1186/s13321-020-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Gao K.; Nguyen D. D.; Chen X.; Jiang Y.; Wei G.-W.; Pan F. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nat. Commun. 2021, 12, 3521. 10.1038/s41467-021-23720-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvat R. S.; Parker A. S.; Choi Y.; Bailey-Kellogg C.; Griswold K. E. Mapping the Pareto Optimal Design Space for a Functionally Deimmunized Biotherapeutic Candidate. PLoS Comput. Biol. 2015, 11, e1003988 10.1371/journal.pcbi.1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalakoti Y.; Yadav S.; Sundar D. TransDTI: Transformer-Based Language Models for Estimating DTIs and Building a Drug Recommendation Workflow. ACS Omega 2022, 7, 2706. 10.1021/acsomega.1c05203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Tang J.; Szwajda A.; Shakyawar S.; Xu T.; Hintsanen P.; Wennerberg K.; Aittokallio T. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. 10.1021/ci400709d. [DOI] [PubMed] [Google Scholar]
- He T.; Heidemeyer M.; Ban F.; Cherkasov A.; Ester M. SimBoost: a read-across approach for predicting drug-target binding affinities using gradient boosting machines. Aust. J. Chem. 2017, 9, 24. 10.1186/s13321-017-0209-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahikkala T.; Airola A.; Pietila S.; Shakyawar S.; Szwajda A.; Tang J.; Aittokallio T. Toward more realistic drug-target interaction predictions. Brief Bioinform. 2015, 16, 325–337. 10.1093/bib/bbu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M.; Nowotka M.; Papadatos G.; Dedman N.; Gaulton A.; Atkinson F.; Bellis L.; Overington J. P. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. 10.1093/nar/gkv352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S. A.; Jolma A.; Campitelli L. F.; Das P. K.; Yin Y.; Albu M.; Chen X.; Taipale J.; Hughes T. R.; Weirauch M. T. The Human Transcription Factors. Cell 2018, 172, 650–665. 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Shi W.; Tian J.; Qi Z.; Li B.; Hao H.; Xu B.. Attention-Based Bidirectional Long Short-Term Memory Networks for Relation Classification. In Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers): aug 2016; Association for Computational Linguistics: Berlin, Germany, 2016; pp. 207–212. [Google Scholar]
- Schuster M.; Paliwal K. Bidirectional recurrent neural networks. IEEE Trans. Signal Process. 1997, 45, 2673–2681. 10.1109/78.650093. [DOI] [Google Scholar]
- Huang Z.; Xu W.; Yu K.. Japa: Bidirectional LSTM-CRF models for sequence tagging. arXiv:1508.01991v1, 2015.
- Melamud O; Goldberger J.; Dagan I.. context2vec: Learning generic context embedding with bidirectional lstm. In Proceedings of the 20th SIGNLL conference on computational natural language learning: 2016, 2016; pp. 51–61.
- Hochreiter S.; Schmidhuber J.. LSTM can solve hard long time lag problems. In NeurIPS Proceedings, 1996.
- Mikolov T.; Chen K.; Corrado G.; Dean J.. Efficient Estimation of Word Representations in Vector Space. In Proceedings of Workshop at ICLR 2013, 2013.
- Friedberg I.; Margalit H. Persistently conserved positions in structurally similar, sequence dissimilar proteins: roles in preserving protein fold and function. Protein Sci. 2002, 11, 350–360. 10.1110/ps.18602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli F.; Gori M.; Tsoi A. C.; Hagenbuchner M.; Monfardini G. The graph neural network model. IEEE Trans. Neur. Netw. 2009, 20, 61–80. 10.1109/TNN.2008.2005605. [DOI] [PubMed] [Google Scholar]
- Kearnes S.; McCloskey K.; Berndl M.; Pande V.; Riley P. Molecular graph convolutions: moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubaki M.; Tomii K.; Sese J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019, 35, 309–318. 10.1093/bioinformatics/bty535. [DOI] [PubMed] [Google Scholar]
- Li X.; Yan X.; Gu Q.; Zhou H.; Wu D.; Xu J. DeepChemStable: Chemical Stability Prediction with an Attention-Based Graph Convolution Network. J. Chem. Inf. Model. 2019, 59, 1044–1049. 10.1021/acs.jcim.8b00672. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Jin W.; Rogers L.; Jamison T. F.; Jaakkola T. S.; Green W. H.; Barzilay R.; Jensen K. F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. 10.1039/C8SC04228D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattarola D.; Alippi C. Graph Neural Networks in TensorFlow and Keras with Spektral. IEEE Comput. Intell. Mag. 2020, 16, 99–106. 10.1109/MCI.2020.3039072. [DOI] [Google Scholar]
- Jacob L.; Vert J. P. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics 2008, 24, 2149–2156. 10.1093/bioinformatics/btn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meher P. K.; Sahu T. K.; Mohanty J.; Gahoi S.; Purru S.; Grover M.; Rao A. R. nifPred: Proteome-Wide Identification and Categorization of Nitrogen-Fixation Proteins of Diaztrophs Based on Composition-Transition-Distribution Features Using Support Vector Machine. Front. Microbiol. 2018, 9, 1100. 10.3389/fmicb.2018.01100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubchak I.; Muchnik I.; Holbrook S. R.; Kim S. H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 8700–8704. 10.1073/pnas.92.19.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayhan F.; Ahmed S.; Mousavian Z.; Farid D. M.; Shatabda S. FRnet-DTI: Deep convolutional neural network for drug-target interaction prediction. Heliyon 2020, 6, e03444 10.1016/j.heliyon.2020.e03444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I.; Keum J.; Nam H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129 10.1371/journal.pcbi.1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum L. C.; Reymond J.-L. 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- Yamanishi Y.; Araki M.; Gutteridge A.; Honda W.; Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Deshmukh R. S.; Ericksen S. S.; Tu Y.; Szklarz G. D. Preferred binding orientations of phenacetin in CYP1A1 and CYP1A2 are associated with isoform-selective metabolism. Drug Metab. Dispos. 2012, 40, 2324–2331. 10.1124/dmd.112.047308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti A.; Vullo D.; Scozzafava A.; Supuran C. T. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I-XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg. Med. Chem. 2008, 16, 7424–7428. 10.1016/j.bmc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Balasundaram A.; David D. C. Molecular modeling and docking analysis of aspirin with pde7b in the context of neuro-inflammation. Bioinformation 2020, 16, 183–188. 10.6026/97320630016183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byczkowski J. Z.; Korolkiewicz K. Z. Inhibition of monoamine oxidase activity by phenacetin and salicylamide. Pharmacol. Res. Commun. 1976, 8, 477–483. 10.1016/0031-6989(76)90057-6. [DOI] [PubMed] [Google Scholar]
- Malhotra S.; Karanicolas J. Correction to When Does Chemical Elaboration Induce a Ligand To Change Its Binding Mode?. J. Med. Chem. 2017, 60, 5940. 10.1021/acs.jmedchem.7b00868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.