

Abstract

Two mononuclear oxidovanadium(V) complexes type of [VVO(L1)(OMe)(MeOH)] (1), [VVO(L2)(OMe)(MeOH)] (2) and two [V2O3]4+ core of μ-oxidodioxidodivanadium(V) complexes (L1)(O)VV–O–VV(O)(L1) (3) and (L2)(O)VV–O–VV(O)(L2) (4) and two complexes [VVO(L1)(8-Hq)] (5) and [VVO(L2)(8-Hq)] (6) incorporating 8-hydroxyquinoline (8-hq) as co-ligand have been reported where L1 [(E)-N′-(2-hydroxybenzylidene)cinnamohydrazide] and L2 [(2E,N′E)-N′-(2-hydroxybenzylidene)-3-(naphthalen-1-yl)acrylohydrazide] are the dianionic forms of the conjugated keto-imine functionalized substituted hydrazone ligands. The μ-oxidodioxidodivanadium complexes are generated upon switching the solvent from methanol to acetonitrile. The X-ray analysis showed octahedral geometry for the mononuclear complexes 1, 2 and 5 but oxido-bridged dinuclear complexes 3 and 4 formed penta-coordinated square-pyramidal geometry about metal atoms. Two mixed-valence species of type II, 3a and 4a, of general formulae (L)(O)VIV–O–VV(O)(L), are being generated upon constant potential electrolysis (CPE) of 3 and 4 respectively. Frozen solution EPR spectra have 13 hyperfine lines, revealing the unpaired electron is majorly localized on one of the two vanadium centres. All these complexes have been well characterized by physio-chemical techniques and the density functional theory (DFT) calculations were applied to obtain further insight into the electronic structure of this type of molecule. The oxidomethoxido complexes 1 and 2 were taken to investigate the catechol oxidase mimicking activity following the oxidation of 3,5-di-tert-butyl catechol (3,5-DTBC) to 3,5-di-tert-butyl benzoquinone (3,5-DTBQ).
Introduction
The coordination chemistry of vanadium has provided an impetus in a variety of catalytic processes,1−4 biochemical processes such as peroxidase mimicking activity,5 insulin mimicking activities,6 cytotoxic activities,7 nitrogen fixation,8 haloperoxidation,9 epoxidation,10 inhibition of phosphate-metabolizing enzymes,11 alleviation of diabetes mellitus symptoms12 and so on. Besides this, the literature reveals oxidovanadium complexes of salicylaldimine and salicylaldehyde-hydrazones as potential anti-cancer drugs13 for brain cancer treatment, 8-hydroxyquinoline vanadium complexes as anti-tuberculosis and anti-proliferative metallodrug.14a So, vanadium chemistry has certainly attracted medicinal attention.14b
On the other hand, nitrogen-oxygen donor ligands containing hydrazone moieties are a very popular choice among the researcher for its facile synthesis, easily tunable electronic properties, conformational diversity,15 and widespread biological applicability.16 These types of ligands play an important role in determining the overall charge of the metal complexes and therefore their stability depending on factors like oxidation state of the metal ions, reaction conditions, nature of the substituents on hydrazone skeleton and tautomerisation.17 Again, recent research on multiple oxidation states of vanadium in solution and its interconversion is expected to generate immense potential usage in making vanadium flow battery (VFB).18
Moreover, the importance of monooxido-bridged binuclear vanadium(V/V) species of the type (VVOL)2O, likelihood as precursors of mixed-valence V(IV, V) species is worthwhile.19 The role of the electronic environment of ligand in the electron localisation or delocalisation across the metal centre is an important aspect of the applicability of mixed-valence complex in molecular electronics and molecular computing.20 The rich coordination chemistry of mixed-valence vanadium21 incorporating acyl hydrazone21c ligands is evident in the context of odd electron behaviour in solid and solution in different biological processes. In comparison to the well-known oxidovanadium complexes containing VIVO2+ or VVO3+ moieties,22 relatively few mixed-valence divanadium (IV/V) complexes have been chemically synthesised and characterised, for for example chemically synthesised mixed-valence oxovanadium (IV/V) citrates and homocitrate with co-ligand 1,10-phenanthroline.23 In the present context, we want to mention that the chemistry of chemically-synthesised mixed-valence divanadium (IV/V) complexes is reasonably developed compare to a few reported examples of isolated electro-synthesised compounds. Herein, we are trying to explore the underlying chemistry of type II mixed-valence oxidovanadium (IV/V) species and their precursors.
We are going to present six vanadium complexes, [VO(L1)(OMe)(MeOH)] (1, 2), [VV2O3(L1)2] (3, 4), [VVO(L1)(8-Hq)] (5, 6), depicted in Chart 1 where L1 is the dianionic form of (E)-N′-(2-hydroxybenzylidene)cinnamohydrazide and L2 is that of (2E,N′E)-N′-(2-hydroxybenzylidene)-3-(naphthalen-1-yl)acrylohydrazide.
Chart 1. Schematic representation of the synthesis of the complexes 1, 2, 3, 4, 5, and 6.

All compounds were characterized with the help of different physio-chemical techniques. Solid-state structures of the complexes 1–5 were confirmed using single crystal X-ray diffraction technique. Redox sites of the complexes were investigated by cyclic voltammetry. Electro-generation of the reduced species was monitored by spectro-electrochemical measurements and theoretically supported by density functional theory (DFT) calculations. Dinuclear complexes were found to be EPR silent both at solid and in CH2Cl2 solution (298, 77 K), indicating binuclear (VVVV) moiety. In contrast, the electro-generated complexes (3a and 4a) showed 13 hyperfine lines spectra at 77 K in the frozen solution, consistent with the formation of dinuclear type II mixed-valence oxidovanadium (IV/V) compounds, where the unpaired electron largely localised on one of the vanadium centres. Extensive studies on catecholase activity for many model first transition metal complexes including vanadium are well documented.24 It is noteworthy that the synthesised small molecules mimic enzyme like cores and their mode of binding with the substrate is extremely significant. In this context, to identify these complexes as functional templates for catechol oxidase we investigated their efficacy to catalyse the oxidation of catechol to quinone by utilizing a well-known model substrate 3,5-di-tert-butylcatechol. As expected, the complexes 5 and 6 were inactive, whilst complexes 1 and 2 were active oxidant and their reactivities are similar to their corresponding dimer 3 and 4.
Results and Discussion
Synthesis
Ligand H2L1 was prepared by following the literature method.25 H2L2 was prepared following the synthesis route of H2L1. Choice of solvent is very important for the coordination environment of the vanadium centre. We have already experienced that polar protic solvent like methanol or ethanol always gives the solvent coordinated product. Stoichiometric reactions of methanolic solution of H2L1 with VO(acac)2 at room temperature afforded complexes 1 and 2 in good yields. We got dinuclear species 3 and 4 in acetonitrile medium. Noteworthy observation is that single crystals of same dinuclear complexes were also formed by slow evaporation of complex 1 and 2 in acetonitrile solvent. Complexes 5 and 6 were afforded in presence of 8-hydroxyquinoline as co-ligand in methanol. The details of the synthetic procedures are illustrated in Scheme 1. Dinuclear type II mixed-valence (VIVVV) species, that is complexes 3a and 4a were synthesised through the constant potential electrolysis (CPE) of complex 3 & 4 respectively at 298 K. The electro-generated complexes showed characteristic EPR spectra.
Scheme 1. Schematic Representation for the Synthesis of the Complexes 1, 2, 3, 4, 5, and 6.

IR Spectroscopy
The IR stretching peak in the region 1000–850 cm–1 indicate the presence of V=O moiety. The IR spectra of complexes 1–2 in solid state depict distinct V=O stretching peaks at ∼956 and ∼951 cm–1 respectively. The complex 3 and 4 exhibit distinct peaks at ∼752 and 763 cm–1 respectively, which might be authenticate the ν (V–O–V) mode.19 Replacement of coordinated methanol molecule and methoxide group by an aromatic co-ligand further strengthen the system in case of 5 and 6 as evident from occurrence of slightly higher V=O stretching at 966 and 968 cm–1 compare to 1 and 2. Details of the procedures and relevant spectral data are outlined in the Experimental Section.
Crystal Structure Description
Single crystal X-ray diffractometric data indicated that both the complexes 1 and 2 crystallized in the triclinic crystal system with P-1 space group adopting a distorted octahedral geometry. In these complexes, charge balance requires the coordination of the solvent (methanol) molecule and binding of the metal centre through the iminolate anion by losing the N–H proton (ONO2–). This is evident from the bond lengths N2–C4 (1.313 (2) Å) and C4–O2 (1.299 (2) Å). The equatorial coordination plane of the octahedron is defined by the methoxy group and three donors from the ligand. The oxo group and methanol molecule reside in the axial positions with O3–V1–O4 bond angle of 174.5 (7) for complex 1 and 175.37 (6) for complex 2, forming distorted octahedron. The coordinated methanol and methoxy groups are cis to each other and they exhibit typical short V1–O5 (V1-methoxy) bond distances of 1.771 (15) Å and elongated V1–O4 (V1-methanol) bond of 2.364 (16) Å. The elongation may also occur due to axial placement of methanol with respect to V=O moiety forming the shortest V1–O3 bond 1.583 (17) Å. The other bonds are within their normal ranges.19,25 The solid state structure of 1 is naturally stabilized through several hydrogen bonding (HB) interactions (V=O/imine H, phenolic O/Ar–H, amine N/OHMe). For complex 2, in addition to HB interaction (amine N/OHMe), two CH···π interactions are also present forming a 2D network (Figure S1). For most of the cases a five membered chelate ring can be observed for mononuclear oxo vanadium complexes incorporating hydrazone moiety.14b,19b But in our case, the distorted octahedral coordination arrangements around V(V) for each of the complexes are observed which are quite similar with the other reported complexes of naphthohydrazide.26
Complex 3 was crystallized in the monoclinic crystal system with the C2/c space group. The asymmetric unit consists of one vanadium atom, V1, one oxo group O3 and one tridentate (ONO2–) Schiff base ligand binding to the vanadium (V) centre. The numbering scheme of the symmetry related atoms are n and n_a in complex 3, for example V1–O1 and V1_a–O1_a. The basal plane around each vanadium centre is defined with three donors from the ligand, that is phenolic oxygen atom, O1, imine nitrogen atom, N1, and an iminolate oxygen atom, O2 along with the bridging oxygen atom, O4 for both the complexes 3 and 4. The remaining apical position of the square pyramid is occupied by the oxo oxygen O4. The short V1=O3 bond distance in the range ∼1.6 Å is comparable with other structurally characterized oxidovanadium complexes.19 The relative disposition of the two V=O groups in 3 is almost trans with the O3=V1···V1_a=O3_a torsion angle of 107.733° and V1···V1_a separation of 2.9728 Å. However, such a type of symmetry is not observed in the case of 4. Complex 4 crystallized in the orthorhombic crystal system with the Pbcn space group. The asymmetric unit of 4 consists of one vanadium atom, V2, one oxo group O3 along with tridentate Schiff base ligand (L2). Other molecular arrangements around the vanadium centres and the numbering scheme are same as that of complex 3. The relative disposition of the two V=O groups in 4 is almost trans with the O3=V2···V2_a=O3_a torsion angle of 97.21° and V2···V2_a separation 3.077 Å. The τ value is found to be ∼0.01 for complex 3 and ∼0.07 for 4, indicating almost ideal square pyramidal geometry around the vanadium(V) centres. In 3, no such significant supramolecular interactions are present except weak CH···π interaction. In contrast, a supramolecular 2D chain is formed in 4 due to the participation of the two V=O groups in HB and CH···π interactions (Figure S1).
Complex 5 also crystallized in the triclinic crystal system with the P-1 space group. The four coordination sites of the metal, three from the ligand and one from the oxo group, are similar to complex 1 (ONO2–). The remaining two sites are occupied by the oxygen and nitrogen atom from co-ligand (NO–) 8-hq moiety forming an overall (ONO2––NO–) arrangement. The equatorial coordination plane of the octahedron is defined by three donors from the ligand (ONO), and oxygen atom from 8-hq. The oxo group and nitrogen atom of 8-hq lie in the axial positions with O3–V1–N3 bond angle of 175.14 (1)° and V1–N3 bond length (2.3521 (2) Å) being the largest. The other bonds are within normal ranges. Self-assembly by three types of HB interactions (V=O/Ar–H, V=O/imine H, phenolic O/Ar–H) links the molecule into a two-dimensional network (Figure S1). Considering all the compounds 1, 2, 3, 4, and 5 the overall V–O bond lengths follow the order V–O(oxido) < V–O(alkoxido) < V–O(quinolato) < V–O(enolato). These data indicate stronger binding of the alkoxido moiety compared to those of quinolato and enolato O atoms. The ortep diagram of the complexes 1–5 are shown in Figure 1 and crystallographic parameters are given in Table S1.
Figure 1.

ORTEP diagram (50% probability) ellipsoid and atom-numbering scheme for complexes 1, 2, 3, 4 and 5.
Electronic Spectroscopy and Redox Properties
The UV–vis absorption spectra of complexes (1–6) were recorded in CH2Cl2 at 298 K by employing concentrations in the range ∼20 μM and they are being illustrated in Figure S2. In the UV–vis spectra, the complexes followed a particular pattern with respect to their structural motif. Here observed three distinct sets of absorption spectra in general. The monoxido mononuclear vanadium(V) complexes 1, 2 and monoxido bridged-di-vanadium (V,V) complexes 3, 4 were nearly identical in appearance. The first set, complexes 1 and 3, showed closely spaced twin humps ∼305 and 335 nm. For the oxidovanadium(V) complexes no d–d bands are expected. These low energy twin humps can be attributed to ligand-centred charge transfer (LCT) bands. With the second set of complexes 2 and 4, the presence of naphthylic conjugation induces a significant red shift of twin humps (LCT band) to single bands at around 344 and 351 nm respectively. Moreover, for all the complexes 1–4, a broad shoulder in the range 405–416 nm was observed in the spectra. This region can be assigned to set of common LMCT bands. The third set of spectra, in the case of intense violet complexes 5 and 6, is distinctly different in peak pattern and position. The additional π-conjugated NO– donor unit of 8-hq (8-hq as co-ligand) induces a significant bathochromic shift of the LMCT absorption band at ∼445 and 546 nm respectively along with a relatively intense peak at 316 and 359 nm. A careful look into the spectra of 5 and 6 reveals the presence of three distinct absorption channels in the molecules which is aptly supported by its co-ordination environment. For Complexes, 3a and 4a a characteristic feature of the [V2O3]3+ is also observed in the near-IR region between 1200 and 1500 nm. It is indicative of intra-valence charge transition (IVCT) between mixed valence vanadium nuclei.19,27 (Figure 2).
Figure 2.

Absorption spectra of 3a and 4a in CH2Cl2.
The redox activities of 1–6 were investigated by cyclic voltammetry in CH2Cl2 using [N(n-Bu)4]PF6 as supporting electrolyte at 298 K. The cyclic voltammograms with the redox potential data referenced to ferrocinium/ferrocine (Fc+/Fc) redox couple is illustrated in Figure 3. All the experiments were performed in the nitrogen atmosphere. The cyclic voltammograms of all the mononuclear vanadium complexes (1, 2, 5 and 6) displayed reversible cathodic waves in the range −0.08 to −0.56 V assigned to VV/VIV reduction couple. The nature of the graph authenticated the redox non-innocent role of the ligand framework towards the coordination of the metal centre. Complex 1 and 2 exhibits reversible cathodic waves at −0.082 and −0.15 V respectively due to the VVO/VIVO redox couple as shown in Figure 3a,b. The redox activity of 5 and 6 are quite similar to those of 1 and 2 as depicted in Figure 3e,f respectively. The cyclic voltammograms of 5 and 6 displayed the cathodic waves at −0.55 and −0.56 V respectively due to VVO/VIVO redox couple. The introduction of NO– donor aromatic co-ligand, for complexes 5 and 6, causes vanadium(V) centre less susceptible to reduction in comparison to complexes 1 and 2. Hence, a higher potential is utilized to reduce the metal centre. The redox activities of 3 and 4 are notably different from those of 1, 2, 5 and 6. Complex 3 exhibits two reversible cathodic waves [Figure 3c] due to the presence of VV^ÔVV/VIV^ÔVV and VIV^ÔVV/VIV^ÔVIV redox couples at −0.20 and −1.25 V respectively. Similarly, 4 also shows the similar redox properties as 3 depicted in Figure 3d, two reversible cathodic waves arises at −0.25 and −1.30 V. The very slight increment of the reduction potential values in complex 4 over complex 3 can be rationalised by the presence of extra π conjugation of naphthalene ring in otherwise similar structural motifs.
Figure 3.

Cyclic voltammograms of (a) 1, (b) 2, (c) 3, (d) 4, (e) 5 and (f) 6 in CH2Cl2 at 298 K. Conditions: scan rate, 100 mV s–1; 0.20 M [N(n-Bu)4]PF6 supporting electrolyte; platinum working electrode.
Spectroelectrochemistry
To evaluate spectroscopic signatures of the reduced species in monomeric complexes 1, 2, 5, 6 we have conducted spectro-electrochemical experiments in the CH2Cl2/0.3 M [N(n-Bu)4]PF6 system. In the case of the monomeric monooxido (ONO2–) complex 1, the reduction under constant potential conditions resulted in an intensity decrease of the high-energy twin humps at 305 and 336 nm and the formation of a distinct peak with increased intensity at 406 nm. Isobestic crossover points at 386 and 450 nm further proved the transformation of reduced species through a common intermediate [Figure 4a]. For complex 2, the UV–vis spectral transformations during reduction look similar to those observed in complex 1. The intensity of the higher energy band at 344 nm decreased gradually and that at 410 nm increased gradually to form a distinct peak [Figure 4b]. Moreover, two almost similar isobestic points at 390 and 450 nm confirmed the structural and electrochemical uniformity of complexes 1 and 2.
Figure 4.

Change of absorption spectra during (a) 1 → 1– and (b) 2 → 2– (c) 5 → 5– (d) 6 → 6– conversions in CH2Cl2 achieved by constant potential spectroelectrochemical measurements at 298 K.
The single-electron reduction for complex 5 resulted in a decrease in the intensities of 316 and 546 nm bands and the formation of a new band at 414 nm [Figure 4c] with the appearance of two isobestic points at 384 nm and 446 nm respectively. As expectedly, the electrochemical reduction of complex 6 resulted in a decrease in the intensity of two bands at 342 and 550 nm [Figure 4d]. A closer look at the spectral features of all the reduced species unveils that all the complexes undergo a reduction process through common pathways (isobestic points ∼385 and ∼450 nm). Thus, spectroscopic signatures of all reduced species are characterized by spectroelectrochemical experiments.
EPR Study
All the six complexes were found to be EPR silent hence diamagnetic in nature. The electro-reduced species were found to be paramagnetic. The EPR spectra of all the species were recorded in CH2Cl2 solution at ambient temperature. The EPR spectrum of all the electro-generated mononuclear anionic complexes exhibited eight line spectrum due to S = 1/2 and hyperfine coupling of 51V (I = 7/2). Spectra were compatible with octahedral coordination sphere of vanadium (IV) complexes. This change of diamagnetic [VVO]2+ to paramagnetic [VIVO]2+ is also supported by spin density data from DFT calculations.
Electrochemically generated mixed-valence [{VVVIVO2(L)2}-μ-O] species (3a and 4a) were paramagnetic. For divanadium(IV,V) complexes, 15-hyperfine profile on the EPR time scale suggest a type II or III character according to Robin and Day classification. Type III, when the unpaired electron is uniformly delocalized over both of the vanadium centers and type II when the electron is partially delocalized, that is weakly interacting.19,21 An 8-line pattern, suggests a valence-trapped situation for the odd electron (type I). The EPR spectra of solid solution of 3a and 4a consist of eight lines pattern at room temperature (Figure S14, Table S8). In contrast, the frozen glass solution of the same showed 13 lines at 77 K (Figure 5, Table 1). The absence of 15 line patterns in case of 3a and 4a indicate that the complexes are not exactly mixed valance of type III as it requires complete delocalization of electronic spin between the two vanadium nuclei. In this case, from the EPR pattern it can be concluded that the unpaired electron spin is mostly localized above one of the vanadium(IV) center and partially mixed with adjacent vanadium nuclei, thus defined by the [{VVO(L)}{VIVO(L)}-μ-O] ↔ [{VIVO(L)}{VVO(L)}-μ-O]. This type of anisotropic distribution of unpaired electron spin in the EPR spectra reveals that 3a and 4a are type II mixed valance complexes.28
Figure 5.

X-band EPR spectra of [V2O3L1–22]3+, (a) 3a, (b) 4a in frozen solution at 77 K (black-experimental; red-simulated).
Table 1. X-Band EPR Spectral Parameter of Complexes 3a and 4a.
| complex | matrix | g∥ | g⊥ | gav | A∥ (G) | A⊥(G) | Aav (G) | lw (mT) |
|---|---|---|---|---|---|---|---|---|
| 3a | CH2Cl2, 77 K | 1.978 | 1.947 | 1.957 | 141.78 | 75.87 | 97.84 | 1.35 |
| 4a | CH2Cl2, 77 K | 1.965 | 1.925 | 1.938 | 153.85 | 64.23 | 94.10 | 1.8 |
DFT Calculation
To elucidate spectroscopic and redox properties of the monomeric complexes 1, 2, 5, and 6 as well as dimers 3 and 4, we have conducted DFT calculations of all the target complexes.
First, occupied frontier orbitals are indicative of the spin free d0 configuration of the vanadium centres. In particular, in the case of monomeric complexes 1, 2, 5, and 6, the highest occupied molecular orbital (HOMO) is largely ligand centric. The metal orbitals are almost pure dx2–y2 or dxy type with some p contribution from ligand moiety whereas the LUMO is admixture of metal and ligand contribution (Figure S15). Again, for these monomeric complexes, on reduction spin polarization was observed from mainly oxido ligand to vanadium centre (Figure 6). It confirms that the first reduction is metal-centered with no significant participation of ligand moiety supported by EPR spectra of electro-reduced mononuclear complexes.
Figure 6.

Spin density plot of (a) 1– (b) 2–, (c) 3a, (d) 4a, (e) 5– and (f) 6–
Second, the lowest unoccupied molecular orbital (LUMO) of the dinuclear compounds [{VVO(L1)}2-μ-O] (3) and [{VVO(L2)}2-μ-O] (4) is centred at the V atoms, oxido ligands, and bridging O atom whereas the HOMO mainly ligand centric (Figure 7). In the singly reduced complex [VVO(L1)–O–VIVO(L1)]−3a, with the ground state optimized geometries, the spin density is mostly localized at one of the V centre [1.09 e; Figure 6c]. The negative spin density values at the bridging O atom, carbonyl O atom, hydrazine N atom and one of the dioxo ligand (−0.01 to −0.14 e) indicate some spin polarization and delocalization between the metal centre with O and N atoms.
Figure 7.

Isodensity plot of selected frontier orbitals of (a) 3 and (b) 4.
All of these data clearly indicate that reduction affect largely one metal centre which weakly interacts with another vanadium centre and thus lead to the formation of a mixed-valence complex of type II. Overall the spin density data indicate that the electronic spin is anisotropically distributed over two vanadium centre which is well supported by EPR data showing 8 line in solid solution at room temperature (Figure S15, Table S8) and 13 line (Figure 5, Table 1) in frozen glass solution at 77 K. (It would be perfect 15 line spectra in case of complete delocalization.) Similar phenomena regarding electron spin density is being observed for complex 4a too.
Catechol Oxidation
Employing 3,5-di-tert-butyl catechol as the substrate, catecholase activity of all the six complexes has been checked. Complexes 1, 2 found to be catalytically active with respect to catechol oxidation reaction. Complexes 3 and 4 behaved similarly to their respective monomers that is 3 with 1 and 2 with 4. As expectedly, the complexes 5 and 6 turned out to be inactive towards the above reaction due to strong ligating environment. The rate of catechol oxidation reaction for 1 and 2 was investigated using time dependent UV–vis absorption spectra in 1:1 CH3OH and CH2Cl2 under atmospheric air. To check the ability of the complexes to oxidize 3,5-DTBC, the reaction was initiated by adding 2 × 10–4 M solutions of 1 and 2 with 0.01 M of 3,5-DTBC at 25 °C. The immediate spectral run exhibited two peaks around ∼596 and ∼415 nm. The low energy broad band appearing at 596 nm (for 1) and 598 nm (for 2) may be assigned to charge transfer (CT) bands from phenolate to vanadium(dπ), diminished gradually with the advancement of reaction time (5 min interval) [Figure 8]. Meanwhile, the growing quinone band resulted blue shifting of the spectra at around 414 nm (for 1) and 417 nm (for 2). It finally saturated to ∼400 nm indicating the formation of 3,5-di-tert-butyl quinone (3,5-DTBQ). These changes were accompanied by the formation of isosbestic point at 440 nm (for 1) and 428 nm (for 2) with a colorimetric transformation of the reaction mixture from bluish green to deep brown. The brown solution so obtained after completion of the reaction was purified by column chromatography to get the yield of 3,5-DTBQ. The yield for complex 2 was higher (∼62%) than that of the complex 1 (∼54%). To obtain the kinetic parameters, 2 × 10–4 M solution of complexes were treated with 0.001–0.01 M solution of the substrate and considering the absorption at quinone band ∼400 nm, the rates of the reactions were determined. The rate of the reaction was found to depend on substrate—catalyst ratio as depicted in Figure 9. The Michaelis–Menten and Lineweaver–Burk equations were applied to determine the kinetic parameters for complexes as follows. [Complex 2: K2 (Turnover no) = 9.8 min–1, KM = 2.25 × 10–3 and Vmax = 2.0 × 10–3 M min–1 and complex 1: K2 (Turnover no) = 11.3 min–1, KM = 2.57 × 10–3 and Vmax = 2.22 × 10–3 M min–1] [Figures S3 and S4].
Figure 8.
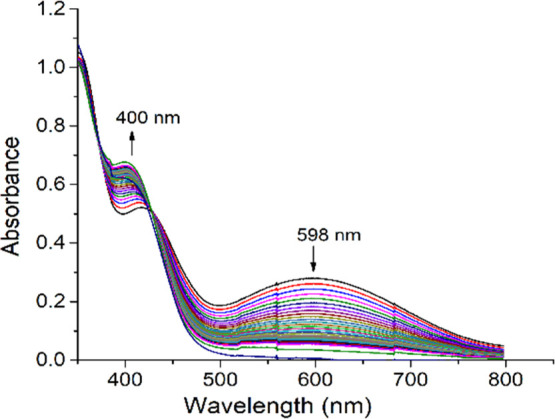
Absorption spectra of a solution containing 2 × 10–4 M of complex 2 and 0.01 M of 3,5-DTBC, recorded at the interval of 5 min.
Figure 9.
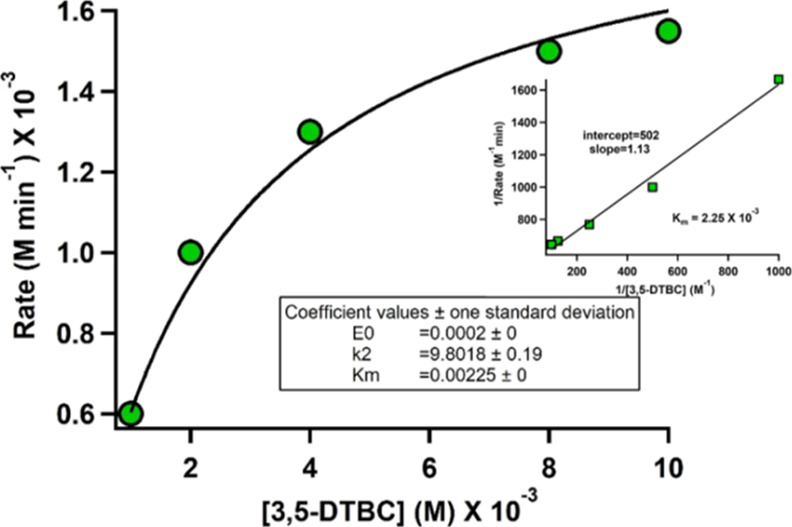
Plot of rate vs concentration of 3,5-DTBC for the oxidation reaction catalysed by complex 2 (Inset: Lineweaver–Burk plot).
Reaction Pathway Monitoring through Mass Spectrometry
To elucidate the reaction pathway, we have chosen complex [VVO(L2)(OMe)(MeOH)] (2) as a model (Scheme 2). The presence of two weak V–O bonds, one from the attached methoxy group and the other from the solvent moiety, makes it labile and susceptible to attack by catechol derivatives. The progress of the reaction was monitored by ESI-Mass spectrometry. Free complex 2 has a peak at m/z = 443. After initial reaction the base peaks was detected at m/z = 601, corresponding to intermediate II. After completion of reaction, the reaction mixture exhibited two characteristics peaks at m/z 221 and 443 corresponding to 3,5-DTBQ and complex 2 (Figures S11–S13). This means after completion of the reaction compound 2 is regenerated.
Scheme 2. Proposed Reaction Pathway for Catecholase Oxidation for Complex 2.

Experimental Section
Materials and Methods
All reagents or analytical grade materials were purchased from commercial sources and used without further purification. The synthetic precursor VO(acac)2 was prepared according to the literature.22a Spectroscopic and electrochemical measurements were carried out using spectroscopic grade solvents. The C, H, and N contents of the compounds were performed on Perkin–Elmer 2400 Series II analyzer. IR spectra (4000–400 cm–1) were recorded with Perkin–Elmer L-0100 spectrophotometer. Electronic absorption spectra were measured on a PerkinElmer Lambda 750 spectrophotometer. 1H NMR was conducted using Bruker FT 300/400 MHz spectrometer with TMS as an internal reference. Electrospray ionization mass spectrometry (ESI-MS positive) spectra were obtained on a MicromassQ-Tof YA 263 mass spectrometer or Shimadzu LCMS 2020 mass spectrometer equipped with electrospray ionization (ESI) ion source. Cyclic voltammetry experiments were performed on electroanalytical instrument BASi Epsilon-EC in CH2Cl2 solutions containing 0.2 M tetrabutylammoniumhexafluorophosphate as supporting electrolyte. Experiments were performed using a BASi platinum working electrode, platinum auxiliary electrode, and Ag/AgCl reference electrode. The redox potential data are referenced to ferrocenium/ferrocene (Fc+/Fc) couple. A BASi SEC-C thin-layer quartz glass spectroelectrochemical cell kit (light path length of 1 mm) with a platinum gauze working electrode and an SEC-C platinum counter electrode was used for the spectroelectrochemistry measurements. The X-band EPR spectra were recorded with a Magnettech GmbH MiniScope MS400 spectrometer (equipped with a TC H03 temperature controller), and the microwave frequency was measured with an FC400 frequency counter.
Computational Details
The geometrical structure were optimized without any symmetry constraints by the DFT29 method with non-local correlation functional of Lee–Yang–Parr (B3LYP).30 Calculation approach31 associated with the effective core potential (ECP) approximation of Hay and Wadt was used for describing the (1s22s22p6) core electron for vanadium whereas the associated “double-ξ”quality basis set LANL2DZ was used for the valence shell.32 For H atoms, we used 6-31+G basis set, for non-hydrogen atoms C, N and O, we employed 6-311+G basis set. All the calculations were performed with the Gaussian 09W software package33 supported by Gauss View 5.1 software.
Crystallographic Studies
The single crystals suitable for X-ray crystallographic analysis of the complexes were obtained by slow evaporation of the respective solution system. The X-ray intensity data were collected on Bruker AXS SMART APEX CCD diffractometer equipped with a Mo target rotating-anode X-ray source and a graphite monochromator (Mo Kα, λ = 0.71073 Å) at 293 K. The data were reduced in SAINTPLUS and empirical absorption correction was applied using the SADABS package.34 Full matrix least-square procedure on F2 was used for structure refinement. All non-hydrogen atoms were refined anisotropically. All calculations reported in this article were performed using the SHELXTL V 6.14 program package.35 Molecular structure plots were drawn using the ORTEP and Mercury softwares.36
Constant Potential Electrolysis
In order to utilise the monooxido bridged divanadium (VV/VV) complexes as precursor to mixed valence species [{VVVIVO2(L)2}-μ-O], CPE was performed at potential selected with respect to the experimental cyclic voltammograms value for the reduction of complex 3 and 4. The coulometric process was undertaken in accordance with the redox potential range of the complexes. Then it was interrupted, and a sample of the electrolyzed solution was collected and frozen for further studies. Another sample of the same solution was evaporated quickly to get a solid residue. Then, EPR experiment was performed with both the frozen solution and solid residue.
Synthesis of Ligands
Ligand H2L1 was prepared by following the literature method.24 The ligand H2L2 was synthesized using the same procedure as for H2L1, except that 3-(1-napthyl)acrylic acid hydrazide was used instead of cinnamic acid hydrazide. Yield: 22 g (72%). Anal. Calcd for C20H16N2O2: C, 75.93%; H, 5.10%; N, 8.86%. Found: C, 75.82%; H, 5.08%; N, 8.68%. 1H NMR {300 MHz, DMSO-d6, δ (ppm)}: 12.032 (NH, s), 11.222 (OH, s), 8.430 (1H, s), 8.277–8.260 (1H, d), 8.041–7.997 (2H, m), 7.894–7.879 (1H, d),7.681–7.7.574 (6H, m), 7.337–7.306 (1H, t), 6.971–6.929 (1H, m).6.917–6.890 (1H, d). IR (cm–1): ν(O–H) 3188; ν(N–H) 3008; ν(C=O) 1655, ν(C=N) 1604.
Synthesis of Complexes
Synthesis of Oxidomethoxidovanadium(V) Complexes [VVO(L1–2)(OMe)(MeOH)], (1 and 2)
A 2 mL methanolic solution of [VO(acac)2] (5 mmol) was added to a 20 mL methanolic solution of ligand (H2L1 or H2L2) (5 mmol) and stirred for 15 min. The colour immediately changed to deep brown. After few days brown coloured crystals suitable for X-ray diffraction analysis were obtained after slow evaporation of the solvent.
[VVO(L1)(OMe)(MeOH)] (1)
Yield: 81%. Anal. Calcd for C18H19N2O5V: C, 54.83%; H, 4.86%; N 7.10%. Found: C, 55.05%; H, 4.94%; N, 7.18%. 1H NMR {300 MHz, CDCl3, δ (ppm)}: 8.551 (1H, s, −N=CH), 7.843–7.763 (2H, m, Ar–H), 7.740–7.642 (1H, m, Ar–H), 7.620–7.122 (5H, m), 6.899–6.876 (1H, d, Ph–CH=CH), 6.770–6.718 (1H, d, Ph–CH=CH), 6.634–6.585 (1H, t, Ar–H). IR (KBr, νmax/cm–1): 1638 (imine C=N), 1211 (C–O)enolic, 2323 (broad from bound MeOH moiety), 956 (V=O).
[VVO(L2)(OMe)(MeOH)] (2)
Yield: 75%. Anal. Calcd for C22H21N2O5V: C, 59.47%; H, 4.76%; N, 6.30%; Found: C, 59.62%; H, 4.88%; N, 6.42%. 1H NMR {300 MHz, DMSO-d6, δ (ppm)}: 8.447 (1H, s, −N=CH), 7.835–7.736 (3H, m, Ar–H), 7.637–7.614 (1H, d, Ar–H), 7.546–7.518 (1H, d, Ar–H), 7.463–7.348 (3H, m, Ar–H), 7.169–7.119 (1H, t, Ar–H), 6.898–6.872 (1H, d, Ph–CH=CH), 6.763–6.713 (1H, d, Ph–CH=CH), 6.627–6.579 (1H, t, Ar–H). IR (KBr, νmax/cm–1): 1634 (imine C=N), 1227 (C–O)enolic, 2284 (broad from bound MeOH moiety), 951 (V=O), 760 (V–O), 559 (V–N).
Synthesis of μ-Oxidodioxidodivanadium(V) Complexes [VV2O3(L1–2)2] (3 and 4)
When [VO(acac)2] was allowed to react with CH3CN solution of ligand (H2L1 or H2L2) and other reaction conditions were kept the same as that of methanol solution, we have got brown coloured crystals of binuclear μ-oxidovanadium(V) complexes 3 and 4 from H2L1 and H2L2 respectively. Moreover, the same binuclear complexes were formed when the corresponding mononuclear complexes were dissolved in acetonitrile and kept for slow evaporation.
[VV2O3(L1)2] (3)
Yield: 64%. Anal. Calcd for C32H24N4O7V2: C, 56.65%; H, 3.57%; N, 8.26%. Found: C, 56.82%; H, 3.66%; N, 8.34%. 1H NMR {300 MHz, CDCl3, δ (ppm)}: 8.451 (1H, s, −N=CH), 7.842–7.740 (2H, m, Ar–H), 7.642–7.620 (1H, d, Ar–H), 7.554–7.524 (1H, d, Ar–H), 7.469–7.387 (3H, m, Ar–H), 7.174–7.121 (1H, t, Ar–H), 6.899–6.875 (1H, d, Ph–CH=CH), 6.770–6.617 (2H, d, Ph–CH=CH), 6.633–6.584 (1H, t, Ar–H), IR (KBr, νmax/cm–1): 1596 (imine C=N), 1256 (C–O)enolic, 984 (V=O), 752 (V–O–V), 664 (V–O), 464 (V–N).
[VV2O3(L2)2] (4)
Yield: 64%. Anal. Calcd for C40H28N4O7V2: C, 61.71%; H, 3.62%; N, 7.30%; Found: C, 61.82%; H, 3.72%; N, 7.41%. 1H NMR {300 MHz, CDCl3, δ (ppm), 8.451 (1H, s, −N=CH), 7.954–7.442 (10H, m, Ar–H), 7.204–7.149 (1H, t, Ar–H), 6.770–6.717 (1H, d, Ph–CH=CH), 6.585–6.545 (1H, d, Ph–CH=CH), IR (KBr, νmax/cm–1): 1619 (imine C=N), 1265 (C–O)enolic, 995 (V=O), 762 (V–O), 695 (V–O–V), 483 (V–N).
Synthesis of 8-Hydroxyquinolato vanadium(V) Complexes [VVO(L1–2)(8-hq)], (5 and 6)
[VO(acac)2] (5 mmol) was added to a 20 mL methanolic solution of ligand (H2L1 or H2L1) (5 mmol) and stirred for 15 min. Then 8-hydroxyquinoline was added in situ and the colour changed immediately from brown to blackish. The solution was filtered off and kept for slow evaporation. After few days black crystals of complex 5 were obtained used directly for SCXRD structure determination. Moreover, one observation was that same complexes 5 and 6 were also obtained from complex 1 and 2 respectively on addition of 8-hydroxyquinoline in methanol solvent.
[VVO(L1)(8-hq)] (5)
Yield: 85%. Anal. Calcd for C25H18N3O4V: C, 63.17%; H, 3.82%; N, 8.84%. Found: C, 63.22%; H, 3.94%; N, 8.96%. 1H NMR {300 MHz, CDCl3, δ (ppm)}: 8.870 (1H, s, −N=CH), 8.212–8.209 (1H, d, Ar–H), 8.185–8.181 (1H, d, Ar–H), 8.075–7.013 (12H, m, Ar–H), 6.989–6.963 (1H, t, Ar–H), 6.835–6.807 (1H, d, Ph–CH=CH), 6.628–6.577 (1H, d, Ph–CH=CH). IR (KBr, νmax/cm–1): 1606 (imine C=N), 1276 (C–O)enolic, 966 (V=O), 756 (V–O), 498 (V–N).
[VVO(L2)(8-hq)] (6)
Yield: 76%. Anal. Calcd for C29H20N3O4V: C, 66.29%; H, 3.84%; N, 8.00%. Found: C, 66.35%; H, 3.95%; N, 8.14%. 1H NMR {300 MHz, CDCl3, δ (ppm)}: 8.907 (1H, s, −N=CH), 8.219–8.192 (1H, t, Ar–H), 8.115–8.105 (1H, d, Ar–H), 8.070–7.304 (14H, m, Ar–H), 7.283–7.000 (1H, t, Ar–H), 6.974–6.855 (1H, d, Ph–CH=CH), 6.827–6.721 (1H, d, Ph–CH=CH). IR (KBr, νmax/cm–1): 1629 (imine C=N), 1257 (C–O)enolic, 968 (V=O), 772 (V–O), 497 (V–N).
Conclusions
This article deals with the syntheses and characterisation of four mononuclear and two binuclear vanadium complexes with the ligands H2L1 [(E)-N′-(2-hydroxybenzylidene)cinnamohydrazide] and H2L2 [(2E,N′E)-N′-(2-hydroxybenzylidene)-3-(naphthalen-1-yl)acrylohydrazide]. Both monomeric and dimeric complexes can be synthesised by suitable tuning of solvents. Furthermore, mononuclear (ONO2–) complexes 1 and 2 directly combined with NO– donor (as 8hq) to form a mixed ligand (ONO2––NO–) complexes 5 and 6. Dimeric complexes (V/V) 3 and 4 have been treated as precursor to mixed-valence vanadium (V/IV) complexes 3a and 4a, generated through coulometric reduction of 3 and 4. The compounds are characterised in solution state through UV–vis and EPR spectroscopy. Frozen solution EPR results demonstrate the electron is partly localized on one of the two vanadium centre in the electro reduced complexes. Theoretically, electronic spin density calculation of these complexes also supports this fact. Thus they can be best described as mixed valance of type II on the EPR time scale. These types of mixed-valence complexes have high degree of importance in exploring the redox and electronic behaviour of vanadium compounds. Complex 1 and 2 appear to be an active catalyst for catechol oxidation.
Acknowledgments
R.P. acknowledges the Council of Scientific and Industrial Research for fellowship. S.M. acknowledges U.G.C. for D.S. Kothari Fellowship Grant (F.4-2/2006 (BSR)/CH/18-19/0305, Dated October 16, 2019). K.K.R. thanks the CSIR, New Delhi vide project No: 01(3066)/21/EMR-II and Department of Chemistry, Jadavpur University for infrastructure and support. We gratefully acknowledge Dr. Prasanta Ghosh, Department of Chemistry, R. K. Mission Residential College, Narendrapur, Kolkata 103, West Bengal, India for instrumental support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c06713.
Electronic Supporting Information (ESI) available: X-ray crystallographic file in CIF format for 1, 2, 3, 4, 5; CCDC reference number: 1979232, 1998586, 2074860, 2074861 & 2078613 (PDF)
Author Contributions
R.P. and S.M. contributed equally. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Schober L.; Sako M.; Takizawa S.; Gröger H.; Sasai H. Catalytic and Enantioselective oxa-Piancatelli Reaction Using Chiral Vanadium Complex. Chem. Commun. 2020, 56, 10151–10154. 10.1039/d0cc02621b. [DOI] [PubMed] [Google Scholar]
- Sako M.; Takizawa S.; Sasai H. Chiral vanadium complex-catalyzed oxidative coupling of arenols. Tetrahedron 2020, 76, 131645. 10.1016/j.tet.2020.131645. [DOI] [Google Scholar]
- Griffin S. E.; Schafer L. L. Vanadium Pyridonate Catalysts: Isolation of Intermediates in the Reductive Coupling of Alcohols. Inorg. Chem. 2020, 59, 5256–5260. 10.1021/acs.inorgchem.0c00071. [DOI] [PubMed] [Google Scholar]
- a Sutradhar M.; Martins L. M. D. R. S.; Guedes da Silva M. F. C.; Pombeiro A. J. L. Vanadium complexes: Recent progress in oxidation catalysis. Coord. Chem. Rev. 2015, 301–302, 200–239. 10.1016/j.ccr.2015.01.020. [DOI] [Google Scholar]; b Ta S.; Ghosh M.; Ghosh K.; Brandão P.; Félix V.; Hira S. K.; Manna P. P.; Das D. Exploring Anticancer and (Bio)catalytic Activities of New Oxovanadium(V), Dioxomolybdenum(VI), and Copper(II) Complexes of Amide–Imine Conjugates. ACS Appl. Bio Mater. 2019, 2, 2802–2811. 10.1021/acsabm.9b00226. [DOI] [PubMed] [Google Scholar]
- Maurya M. R.; Tomar R.; Avecilla F.; Ribeiro N.; Carvalho M. F. N. N.; Kuznetsov M. L.; Correia I.; Pessoa J. C. Trinuclear vanadium(iv) and vanadium(v) complexes derived from 2,4,6-triacetylphloroglucinol and study of their peroxidase mimicking activity. Dalton Trans. 2020, 49, 2589–2609. 10.1039/c9dt04415a. [DOI] [PubMed] [Google Scholar]
- Pessoa J. C.; Etcheverry S.; Gambino D. Vanadium compounds in medicine. Coord. Chem. Rev. 2015, 301–302, 24–48. 10.1016/j.ccr.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reytman L.; Braitbard O.; Hochman J.; Tshuva E. Y. Highly Effective and Hydrolytically Stable Vanadium(V) Amino Phenolato Antitumor Agents. Inorg. Chem. 2016, 55, 610–618. 10.1021/acs.inorgchem.5b02519. [DOI] [PubMed] [Google Scholar]; b Pessoa J. C.; Correia I. Misinterpretations in Evaluating Interactions of Vanadium Complexes with Proteins and Other Biological Targets. Inorganics 2021, 9, 17. 10.3390/inorganics9020017. [DOI] [Google Scholar]; c Ribeiro N.; Bulut I.; Cevatemre B.; Teixeira C.; Yildizhan Y.; André V.; Adão P.; Pessoa J. C.; Acilan C.; Correia I. Cu(II) and V(IV)O complexes with tri- or tetradentate ligands based on (2-hydroxybenzyl)- L-alanines reveal promising anticancer therapeutic potential. Dalton Trans. 2021, 50, 157. 10.1039/d0dt03331f. [DOI] [PubMed] [Google Scholar]
- Eady R. Current status of structure function relationships of vanadium nitrogenase. Coord. Chem. Rev. 2003, 237, 23–30. 10.1016/s0010-8545(02)00248-5. [DOI] [Google Scholar]
- Butler A.; Walker J. V. Marine Haloperoxidases. Chem. Rev. 1993, 93, 1937–1944. 10.1021/cr00021a014. [DOI] [Google Scholar]
- a Hoshino Y.; Yamamoto H. Novel α-Amino Acid-Based Hydroxamic Acid Ligands for Vanadium-Catalyzed Asymmetric Epoxidation of Allylic Alcohols. J. Am. Chem. Soc. 2000, 122, 10452–10453. 10.1021/ja002602o. [DOI] [Google Scholar]; b Pesiri D. R.; Morita D. K.; Walker T.; Tumas W. Vanadium-Catalyzed Epoxidations of Olefinic Alcohols in Liquid Carbon Dioxide. Organometallics 1999, 18, 4916–4924. 10.1021/om990467m. [DOI] [Google Scholar]
- Dennis Chasteen N.The biochemistry of vanadium. Copper, Molybdenum, and Vanadium in Biological Systems; Structure and Bonding; Springer, 1983; Vol. 53, pp 105–138. [Google Scholar]
- Thompson K. H.; Orvig C. Vanadium in diabetes: 100 years from Phase 0 to Phase I. J. Inorg. Biochem. 2006, 100, 1925–1935. 10.1016/j.jinorgbio.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Levina A.; Pires Vieira A.; Wijetunga A.; Kaur R.; Koehn J. T.; Crans D. C.; Lay P. A. A Short-Lived but Highly Cytotoxic Vanadium(V) Complex as a Potential Drug Lead for Brain Cancer Treatment by Intratumoral Injections. Angew. Chem. 2020, 59, 15834–15838. 10.1002/anie.202005458. [DOI] [PubMed] [Google Scholar]
- a Correia I.; Adão P.; Roy S.; Wahba M.; Matos C.; Maurya M. R.; Marques F.; Pavan F. R.; Leite C. Q. F.; Avecilla F.; Costa Pessoa J. Hydroxyquinoline derived vanadium(IV and V) and copper(II) complexes as potential anti-tuberculosis and anti-tumor agents. J. Inorg. Biochem. 2014, 141, 83–93. 10.1016/j.jinorgbio.2014.07.019. [DOI] [PubMed] [Google Scholar]; b Banerjee A.; Dash S. P.; Mohanty M.; Sahu G.; Sciortino G.; Garribba E.; Carvalho M. F. N. N.; Marques F.; Costa Pessoa J.; Kaminsky W.; Brzezinski K.; Dinda R.; Dinda R. New VIV, VIVO, VVO, and VVO2 Systems: Exploring their Interconversion in Solution, Protein Interactions, and Cytotoxicity. Inorg. Chem. 2020, 59, 14042–14057. 10.1021/acs.inorgchem.0c01837. [DOI] [PubMed] [Google Scholar]
- Su X.; Aprahamian I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963–1981. 10.1039/c3cs60385g. [DOI] [PubMed] [Google Scholar]
- a Kalinowski D. S.; Sharpe P. C.; Bernhardt P. V.; Richardson D. R. Structure–Activity Relationships of Novel Iron Chelators for the Treatment of Iron Overload Disease: The Methyl Pyrazinylketone Isonicotinoyl Hydrazone Series. J. Med. Chem. 2008, 51, 331–344. 10.1021/jm7012562. [DOI] [PubMed] [Google Scholar]; b Bouzayani N.; Marque S.; Djelassi B.; Kacem Y.; Marrot J.; Ben Hassine B. Enantiopure Schiff bases of amino acid phenylhydrazides: impact of the hydrazide function on their structures and properties. New J. Chem. 2018, 42, 6389–6398. 10.1039/c7nj04597b. [DOI] [Google Scholar]; c De P.; Baltas M.; Bedos-Belval F. Cinnamic Acid Derivatives as Anticancer Agents-A Review. Curr. Med. Chem. 2011, 18, 1672–1703. 10.2174/092986711795471347. [DOI] [PubMed] [Google Scholar]
- a Mukiza J.; Habarurema G.; Gerber T. I. A.; Hosten E.; Betz R.; Umumararungu T. Rhenium(I) and (V) complexes of aroylhydrazone derivatives: Synthesis, spectroscopic and crystallographic studies. Polyhedron 2020, 175, 114192. 10.1016/j.poly.2019.114192. [DOI] [Google Scholar]; b Joseph B.; Kurup M. R. P.. Studies on Transition Metal Chelates of some Tridentate Aroylhydrazones. Ph.D. thesis, 2014. [Google Scholar]
- Ding C.; Zhang H.; Li X.; Liu T.; Xing F. Vanadium Flow Battery for Energy Storage: Prospects and Challenges. J. Phys. Chem. Lett. 2013, 4, 1281–1294. 10.1021/jz4001032. [DOI] [PubMed] [Google Scholar]
- a Mondal A.; Sarkar S.; Chopra D.; Guru Row T. N.; Pramanik K.; Rajak K. K. Family of Mixed-Valence Oxovanadium(IV/V) Dinuclear Entities Incorporating N4O3-Coordinating Heptadentate Ligands: Synthesis, Structure, and EPR Spectra. Inorg. Chem. 2005, 44, 703–708. 10.1021/ic048919x. [DOI] [PubMed] [Google Scholar]; b Dinda R.; Sengupta P.; Sutradhar M.; Mak T. C. W.; Ghosh S. Solution Study of a Structurally Characterized Monoalkoxo-Bound Monooxo-Vanadium(V) Complex: Spontaneous Generation of the Corresponding Oxobridged Divanadium(V,V) Complex and its Electroreduction to a Mixed-Valence Species in Solution. Inorg. Chem. 2008, 47, 5634–5640. 10.1021/ic702172p. [DOI] [PubMed] [Google Scholar]; c Mondal S.; Ghosh P.; Chakravorty A. A Family of α-Amino Acid Salicylaldiminates Incorporating the Binuclear V2O33+ Core: Electrosynthesis, Structure, and Metal Valence. Inorg. Chem. 1997, 36, 59–63. 10.1021/ic960671r. [DOI] [Google Scholar]
- Albino A.; Benci S.; Tesi L.; Atzori M.; Torre R.; Sanvito S.; Sessoli R.; Lunghi A. First-Principles Investigation of Spin–Phonon Coupling in Vanadium-Based Molecular Spin Quantum Bits. Inorg. Chem. 2019, 58, 10260–10268. 10.1021/acs.inorgchem.9b01407. [DOI] [PubMed] [Google Scholar]
- a Maurya M. R. Probing the synthetic protocols and coordination chemistry of oxido-, dioxido-, oxidoperoxido-vanadium and related complexes of higher nuclearity. Coord. Chem. Rev. 2019, 383, 43–81. 10.1016/j.ccr.2018.12.010. [DOI] [Google Scholar]; b Dutta S. K.; Samanta S.; Kumar S. B.; Han O. H.; Burckel P.; Pinkerton A. A.; Chaudhury M. Mixed-Oxidation Divanadium(IV,V) Compound with Ligand Asymmetry: Electronic and Molecular Structure in Solution and in the Solid State. Inorg. Chem. 1999, 38, 1982–1988. 10.1021/ic980743+. [DOI] [PubMed] [Google Scholar]; c Mondal B.; Ghosh T.; Sutradhar M.; Mukherjee G.; Drew M. G. B.; Ghosh T. Synthesis, structure and solution chemistry of a family of dinuclear hydrazonato-vanadium(V) complexes with [OV(μ-O)VO]4+ core. Polyhedron 2008, 27, 2193–2201. 10.1016/j.poly.2008.03.018. [DOI] [Google Scholar]
- a Majumder M.; Rajak K. K. Oxidovanadium (V and IV) complexes incorporating coumarin based ÔN̂O ligand: synthesis, structure and catalytic activities. Polyhedron 2019, 176, 114241. 10.1016/j.poly.2019.114241. [DOI] [Google Scholar]; b Chakravarty J.; Dutta S.; Chandra S. K.; Basu P.; Chakravorty A. Chemistry of variable-valence oxovanadium VOz+ (z = 2, 3) complexes: synthesis structure, and metal redox of new VVO(ONO)/(ON) and VIVO(ONO)/(NN) families. Inorg. Chem. 1993, 32, 4249–4255. 10.1021/ic00072a017. [DOI] [Google Scholar]
- Chen C.-Y.; Zhou Z.-H.; Chen H.-B.; Huang P.-Q.; Tsai K.-R.; Chow Y. L. Formations of Mixed-Valence OxovanadiumV,IV Citrates and Homocitrate with N-Heterocycle Chelated Ligand. Inorg. Chem. 2008, 47, 8714–8720. 10.1021/ic800553p. [DOI] [PubMed] [Google Scholar]
- a Salonen P.; Peuronen A.; Lehtonen A. Oxidovanadium(V) amine bisphenolates as epoxidation, sulfoxidation and catechol oxidation catalysts. Inorg. Chem. Commun. 2017, 86, 165–167. 10.1016/j.inoche.2017.10.017. [DOI] [Google Scholar]; b Banu K. S.; Chattopadhyay T.; Banerjee A.; Bhattacharya S.; Suresh E.; Nethaji M.; Zangrando E.; Das D. Catechol Oxidase Activity of a Series of New Dinuclear Copper(II) Complexes with 3,5-DTBC and TCC as Substrates: Syntheses, X-ray Crystal Structures, Spectroscopic Characterization of the Adducts and Kinetic Studies. Inorg. Chem. 2008, 47, 7083–7093. 10.1021/ic701332w. [DOI] [PubMed] [Google Scholar]; c Dey S. K.; Mukherjee A. Catechol oxidase and phenoxazinone synthase: Biomimetic functional models and mechanistic studies. Coord. Chem. Rev. 2016, 310, 80–115. 10.1016/j.ccr.2015.11.002. [DOI] [Google Scholar]; d Rath S. P.; Rajak K. K.; Chakravorty A. Synthesis, Structure, and Catecholase Reaction of a Vanadate Ester System Incorporating Monoionized Catechol Chelation. Inorg. Chem. 1999, 38, 4376–4377. 10.1021/ic990551u. [DOI] [PubMed] [Google Scholar]; e Salonen P.; Savela R.; Peuronen A.; Lehtonen A. Vanadium aminophenolates in catechol oxidation: conformity with Finke’s common catalyst hypothesis. Dalton Trans. 2021, 50, 6088. 10.1039/d1dt00419k. [DOI] [PubMed] [Google Scholar]
- Patra R.; Rajak K. K. Modulation of an Alpha-Beta Unsaturated N-Acylhydrazone as Phase Selective Dual Efficient Chemosensor for Zn(II) and Al(III). ChemistrySelect 2020, 5, 9477–9485. 10.1002/slct.202002097. [DOI] [Google Scholar]
- You Z.-L.; Shi D.-H.; Zhang J.-C.; Ma Y.-P.; Wang C.; Li K. Synthesis, structures, and urease inhibitory activities of oxovanadium(V) complexes with Schiff bases. Inorg. Chim. Acta 2012, 384, 54–61. 10.1016/j.ica.2011.11.039. [DOI] [Google Scholar]
- a Dutta S.; Basu P.; Chakravorty A. Chemistry of Mononuclear and Binuclear Oxidic VV, VVVV, and VVVIV Azophenolates. Inorg. Chem. 1993, 32, 5343–5348. 10.1021/ic00075a063. [DOI] [Google Scholar]; b Zabierowski P.; Radoń M.; Szklarzewicz J.; Nitek W. Mixed-valence VIV/VV tetrametallate core {V4N2O14} cluster containing tris(hydroxymethyl)aminomethane and acetylacetone. Inorg. Chem. Commun. 2014, 41, 72–75. 10.1016/j.inoche.2013.12.034. [DOI] [Google Scholar]
- Mahroof-Tahir M.; Keramidas A. D.; Goldfarb R. B.; Anderson O. P.; Miller M. M.; Crans D. C. Solution and Solid State Properties of [N-(2-Hydroxyethyl)iminodiacetato]vanadium(IV), -(V), and -(IV/V) Complexes1. Inorg. Chem. 1997, 36, 1657–1668. 10.1021/ic9610243. [DOI] [PubMed] [Google Scholar]
- Runge E.; Gross E. K. U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. 10.1103/physrevlett.52.997. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- a Casida M. E.; Jamorski C.; Casida K. C.; Salahub D. R. Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold. J. Chem. Phys. 1998, 108, 4439–4449. 10.1063/1.475855. [DOI] [Google Scholar]; b Stratmann R. E.; Scuseria G. E.; Frisch M. J. An Efficient Implementation of Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules. J. Chem. Phys. 1998, 109, 8218–8224. 10.1063/1.477483. [DOI] [Google Scholar]; c Bauernschmitt R.; Ahlrichs R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. 10.1016/0009-2614(96)00440-x. [DOI] [Google Scholar]
- Hay P. J.; Wadt W. R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]
- Frisch M. J.; Trucks W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.. Gaussian 09, Revision A. 1; Gaussian; 2009.
- SMART; SAINT; SADABS; XPREP; SHELXTL; Bruker AXS Inc.: Madison WI, 1998.
- a Sheldrick G. M.The SHELX-97 Manual. SHELXL-97, Program for Crystal Structure Refinement; University of Göttingen: Germany. 1997. [Google Scholar]; b Sheldrick G. M. Crystal Structure Refinement with {\\it SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. K.ORTEP Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge TN,1976.; b Macrae C. F.; Edgington P. R.; McCabe P.; Pidcock E.; Shields G. P.; Taylor R.; Towler M.; van de Streek J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. 10.1107/s002188980600731x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.