

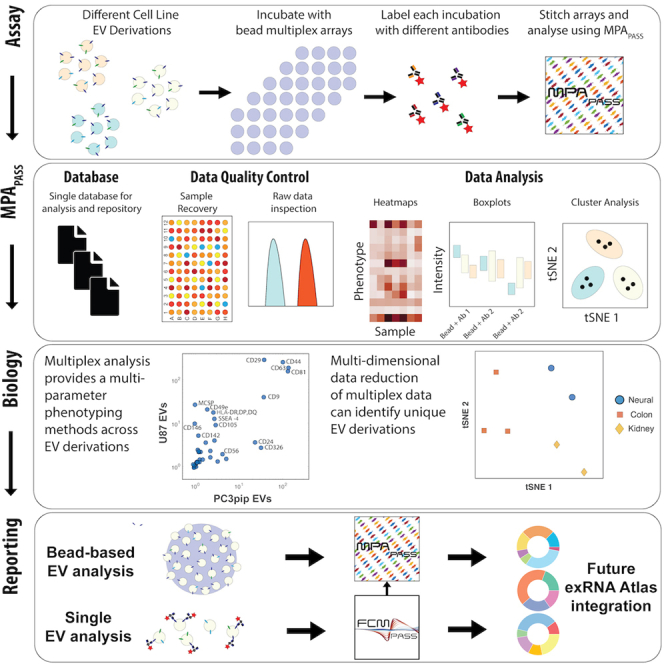
Summary
Extracellular vesicles (EVs) of various types are released or shed from all cells. EVs carry proteins and contain additional protein and nucleic acid cargo that relates to their biogenesis and cell of origin. EV cargo in liquid biopsies is of widespread interest owing to its ability to provide a retrospective snapshot of cell state at the time of EV release. For the purposes of EV cargo analysis and repertoire profiling, multiplex assays are an essential tool in multiparametric analyte studies but are still being developed for high-parameter EV protein detection. Although bead-based EV multiplex analyses offer EV profiling capabilities with conventional flow cytometers, the utilization of EV multiplex assays has been limited by the lack of software analysis tools for such assays. To facilitate robust EV repertoire studies, we developed multiplex analysis post-acquisition analysis (MPAPASS) open-source software for stitched multiplex analysis, EV database-compatible reporting, and visualization of EV repertoires.
Keywords: EV flow cytometry, multiplex, atlas, standardization, liquid biopsy methods
Graphical abstract
Highlights
-
•
High-parameter EV multiplex analysis is introduced
-
•
MPAPASS enables multiplex data stitching and multidimensional data visualization
-
•
EV multiplex experiment design, performance, analysis, and reporting are presented
Motivation
Assessment of EV repertoire profiles is a priority for many research groups, but methods for performing and analyzing EV markers with multimarker multiplex bead sets are not well established. MPAPASS enables stitched multiplex EV analyses, and our results illustrate key considerations for designing, performing, and analyzing EV multiplex studies.
Welsh et al. demonstrate the utility of EV multiplex analysis with MPAPASS software for EV repertoire studies. MPAPASS enables the standard reporting and ergonomic stitching of multiplex datasets for extracellular vesicle repertoire identification.
Introduction
Extracellular vesicles (EVs) are submicron phospholipid bilayer-enclosed spheres secreted from cells. EVs carry proteins on their surface as well as intracellular cargo in the form of proteins, nucleic acids, metabolites, lipids, and others. EV research is a rapidly developing area due to its prospective use as translational biomarkers and therapeutics (Matsuzaki and Ochiya, 2017; Reiner et al., 2017). However, the utilization of EVs is hampered by currently available analysis methods.
The majority of EVs have been demonstrated to be ≤100 nm in diameter, with a power-law distribution ranging from ∼25 to >1,000 nm (van der Pol et al., 2014; Tian et al., 2020; Lennon et al., 2019). A wide variety of detection methods have been utilized for characterizing single EVs (Issadore et al., 2011; Shao et al., 2010, 2012; Lennon et al., 2019; Tian et al., 2018; van der Pol et al., 2010, 2013, 2018). A current limitation of EV characterization in the field is the sensitivity of single-EV phenotyping assays. The low numbers of expressed proteins on the surface of EVs make the use of common high-throughput, single-particle phenotyping techniques such as flow cytometry and confocal microscopy particularly difficult due to having sensitivities ranging from >10 to 1,000 molecules of a given fluorophore (Gasecka et al., 2020; Tertel et al., 2020; Welsh et al., 2020b). More recently, a commercially available, dedicated, small-particle flow cytometer has shown the ability to enumerate the majority of EVs and demonstrate fluorescence sensitivity for <10 molecules of phycoerythrin (Zhu et al., 2014; Tian et al., 2020). This has important implications for accurately enumerating EV phenotypes and counts within clinical samples. It is only with single-molecule detection that we can confidently state that EVs may be negative for a given marker at a single particle level (Tian et al., 2018). However, this technique is limited to one color in this single-molecule sensitivity range and therefore requires prior knowledge of subset markers to identify within a sample.
A method that has been used by the EV field to semi-quantitatively enable EV phenotyping of dim markers is to use a multiplex array of fluorescently bar-coded antibody capture beads in combination with a detection antibody (Koliha et al., 2016; Wiklander et al., 2018). This allows potentially hundreds to thousands of EVs with a commonly expressed protein to attach to the surface of a single 5–8 μm bead conjugated with an antibody for that protein. A fluorescently conjugated detection antibody is then used to confirm EV-binding to any specific bead. While useful, single bead-based assays offer limited utility to understand the heterogeneous phenotypes of EVs compared with multiplex arrays. A particular advantage of bead-based assays is their ability to be run on conventional cytometers. While newer-generation cytometers with more sensitivity and a larger dynamic range are beneficial, in many cases legacy instruments can be sufficient for phenotyping EVs.
More recently, EV multiplex kits have become available, enabling the simultaneous use of up to 39 distinct antibody-capture beads (Koliha et al., 2016; Wiklander et al., 2018). While this commercially available kit is sold with three detection antibodies for tetraspanins (CD9, CD63, and CD81), the kit is compatible with any detection antibody provided it fluoresces in a region of the spectrum not occupied by the fluorophore-coated beads. However, the utilization of this method of analysis quickly generates large quantities of data. With each tetraspanin detection antibody being used separately, 111 protein combinations are generated with six bead controls per sample. Analysis of this scale of data is currently limited by a lack of software analysis tools.
Here, the use of stitched multiplex analysis, which provides a method to survey the expression of hundreds of protein combinations on EVs, is evaluated. Considerations for undertaking multiplex analysis for surveying EV repertoires are also outlined. The ability to perform stitched multiplex analysis to analyze data on this scale is demonstrated using a lab-built, open-source software package (multiplex analysis post-acquisition analysis software [MPAPASS]) that allowed for high-throughput, multiparametric quality control and data analysis of EV multiplex data. In this work, we use a commercially available EV multiplex kit, and this pipeline is compatible with any combination of bead-based assays.
Results
Interpreting EV multiplex outputs
Unlike a typical multiplex assay or an ELISA in which there is a single analyte measured per capture antibody, EV multiplex assays capture a heterogeneous set of particles with respect to size and composition. These captured particles then have their other surface analytes probed with detection antibodies (Figures 1A–1D). This, therefore, leads to differences in interpretation and analysis compared with traditional multiplex analysis. Because EVs express multiple different proteins on their surface, the signal obtained from each bead is dependent both on the relative expression and heterogeneity of EVs and on the other capture beads that are present in the multiplex set (Figure 1A). The signal intensity as well as the variation can therefore encode useful information about the epitope abundance and population heterogeneity (Figures 1E and 1F). Optimal data scaling will also differ from typical multiplex and ELISA assays, as the binding capacity of the analyte to the capture antibody is fixed, allowing calibration into standard units, such as μg. Unlike these assays, the binding capacity of the detection analyte depends on the number of captured EVs on a bead, their diameter, and their expression. These could, therefore, differ drastically between each capture bead. How signals are normalized and interpreted will greatly affect reported results, as shown in Figures 1G and 1H. It is clear from these data that a multiplex analysis using EVs and not soluble proteins, for which many are generally intended, is limited when the data are interpreted in linear units. Using a logarithmic scale regardless of the normalization algorithm allows for the differentiation of high, medium, and dim signals from a control population. While there is no clear solution to differentiate high from low coefficients of variation (CVs) with any of the algorithms when using linear scaling, using a form of separation index is more capable of differentiating dim signals from a control population compared with fold-change or background subtraction. While background subtraction may be commonly used with different assays, due to the reliance on logarithmic scaling, it poses challenges resulting from the creation of negative numbers that cannot be scaled logarithmically. While SI1 and SI2 are commonly used in the flow cytometry field, their use in interpreting multiplex data may lead to ambiguous data given the influence of CVs in differentiating the positivity of a signal. For our work, we chose to use log-scaled metrics in the form of fold-change and background subtraction that were unaffected by CVs to differentiate signals.
Figure 1.

EV multiplex array overview and signal interpretation
(A) Illustration of different scenarios leading to obtained signal from each capture bead. Capture beads are: TP, true positive; FP, false positive; FN, false negative; and TN, true negative.
(B) Gating of singlet bead populations on forward (FSC-A) and side (SSC-A) signals.
(C) Capture bead population fluorescent “bar coding” in the multiplex array using 488 nm illuminations and collection at 525/40 and 690/50.
(D) Specificity of each capture bead population shown in (C).
(E and F) Data of (E) CD63 and (F) CD81 capture beads with detection of 1 × 109 EVs from different cell derivations using CD9, CD63, and CD81 detection antibody mixture.
(G) Artificial flow cytometry signals of a control bead, along with a high-, medium-, and low-intensity bead with high and low variance.
(H) Interpretation of data from (C) using different normalization methods with linear and logarithmic scaling. Normalization formulas (separation indexes 1 and 2 [SI1,2], fold change [FC], and background subtraction [BS]) are outlined in the methods.
Signal interpretation of EV multiplex assays is not dissimilar from traditional multiplex assays and ELISAs with true positives, true negatives, false positives, and false negatives possible (Figure 1A). Like single-EV flow cytometry, false negatives may be prevalent either because not enough EVs bind to bead populations to provide a sufficient signal or the number of expressed proteins is too low to provide a sufficient signal to be detected.
EV multiplex controls: Sample titration
As with all assays, controls are important to ensure the reliability of data. Typically, assays quantifying proteins based on fluorescence intensity will use standards to create a titration curve to convert fluorescence intensity to a concentration measurement. Due to EVs differing in surface area and epitope abundance and expressing multiple proteins that will affect binding affinity to multiplex capture beads, the use of titration to quantitate the EV number is not a robust measure using EV multiplex assays. Whereas using titration for a quantitative EV measurement output is unreliable, the inclusion of titration controls is critical given the nonspecificity that can arise with detection antibodies binding to beads. A titration of PC3pip and U87 EV input material from 106 to 1010 with ∼20,000 total beads (Figures 2A and 2B; Figure S2) demonstrates the effect of titration on the signal intensity of a CD9, CD63, and CD81 detection antibody cocktail. Capture beads CD81, CD63, CD44, and CD29 show an expected increase in signal intensity with increasing EV input material across both PC3pip and U87 EVs. Most markers show little nonspecific binding with the CD9, CD63, and CD81 detection antibody cocktail; however, there does appear to be a consistently increased background signal compared with blank beads from markers SSEA-4, CD105, CD56, and CD25 irrespective of the input material (Figure S2). The appearance of staining distinguishable from the background begins at 108 and 109 total EVs for U87 and PC3pip, respectively. However, this amount of input material is relative to the ∼20,000 total beads incubated. The increase in signal intensity with input EV counts from 109 to 1010 differs depending on the capture bead and EV source, highlighting the multifactorial binding kinetics and semiquantitative nature of this assay. This is further highlighted by looking at the relationships among markers across EV cell-line derivation (Figures 2A and 2B). While the relationship of epitope abundance from 109 to 1010 is preserved, the change in intensity across markers would not necessarily be predictable without first having done a titration of input material. Given a high enough amount of input material, such as 1010 EVs, the intensity of the CD9, CD63, and CD81 detection antibody cocktail across numerous beads provides a high enough signal intensity to be able to make distinctions among expressed marker intensity EV cell lines (Figures 2A and 2B). CD81, CD63, CD44, and CD29 are highly expressed across both U87 and PC3pip EVs, whereas CD24 and CD326 show a higher abundance in PC3pip EVs and MCSP (CSPG4, NG2), CD49e, and human leukocyte antigen (HLA)-DR, -DP, and -DQ, showing a higher abundance in U87 EVs. We recommend the use of a sample titration for multiplex sample input either in a concentration- or volume-dependent manner, depending on the assay, as a method to show the antibody detection signal is titrating with the sample input.
Figure 2.

Effect of input material on signal and identifying unique marker combinations between EV derivations
(A and B) Scatterplot of bead intensities incubated with 1 × 1010 and 1 × 109 EVs from U87 and PC3pip cell lines.
(C) Heatmap of detection intensities of EVs from kidney (VOK111 and VOK151), colon (HCT116wt and mt, −/−), and neural (U87 and U251) cell derivations.
(D and E) Beads were incubated with 1 × 109 EVs and detected using an EV mix (CD9, CD63, and CD81). tSNE was performed on samples, stratifying them by tissue derivation (D) and cell line (E).
EV multiplex controls: Identifying nonspecific signals
Identifying the nonspecific binding of detection antibodies to capture beads is critical for the reproducibility and interpretation of data. A capture bead with a detection antibody alone as a control is critical in all assays. One test investigated the efficacy of 17 alternative detection antibodies (Table 1) to CD9, CD63, and CD81 using two different blocking buffers across the 39-capture bead array. U87 and U251 cell-line-derived EVs were also included as positive controls. A selection of the capture bead markers is shown in Figure 3 (full array in Figure S3). This screening of custom antibodies with the commercial bead-based assay demonstrates that nonspecific binding of the antibody can be capture-bead-dependent. For example, CD147 detection of CD44 capture beads demonstrates a high signal on U87 EVs and, to a lesser extent, U251 EVs. The bead + antibody control in this example shows minimal binding. Capture beads such as CD1c show an undetectable signal for CD147, whereas the CD31, CD133/1, and CD326 capture beads show a dim positive signal for the CD147 detection antibody and have a lower signal for the bead + sample + antibody. This indicates a small amount of nonspecific binding. This may be reduced by EVs due to the EVs blocking antibody binding to the beads before antibody incubation. In this comparison, we also included 2% EV-depleted FBS as a control. The EV-depletion method using ultracentrifugation shows that a number of positive signals can have a better result compared with Miltenyi Biotec buffer alone. It is notable that the etraspanin signals CD9, CD63, and CD81 are all lower when samples are incubated in EV-depleted FBS, and this indicates that small EVs remain after the depletion method and may be able to compete with cell-culture-derived EVs to varying degrees with cross-reactive antibodies for both capture and detection. It is notable that one antibody, CD47, bound to every capture bead regardless of buffer. These data demonstrate the need to screen detection antibodies with capture beads before use. This is also plotted by ordering the average intensity across groups with the top 75 summarized in Figure S4. For this reason, while including bead + antibody controls is essential, subtracting the bead + antibody intensity for bead + EVs +antibody is not recommended, asit may result in negative intensity data scaling.
Table 1.
Detection antibodies used
| Target | Labeling | Fluorescent | Isotype | Clone | Manufacturer | Catalog |
|---|---|---|---|---|---|---|
| CD9 | 5 μL | APC | mouse IgG1 | proprietary | Miltenyi Biotec | 130-108-813 |
| CD63 | 5 μL | APC | mouse IgG1, κ | proprietary | Miltenyi Biotec | 130-108-813 |
| CD81 | 5 μL | APC | REA | proprietary | Miltenyi Biotec | 130-108-813 |
| CTLA-4 | 0.5 μg | APC | mouse IgG2a, κ | BNI3 | BioLegend | 369612 |
| CD166 | 0.5 μg | APC | recombinant human IgG1 | REA442 | Miltenyi Biotec | 130-106-576 |
| CD147 | 0.5 μg | APC | mouse IgG1, κ | HIM6 | BioLegend | 306214 |
| CD87 | 0.5 μg | APC | recombinant human IgG1 | REA892 | Miltenyi Biotec | 130-114-851 |
| CD90 | 0.5 μg | APC | mouse IgG1, κ | 5E10 | BioLegend | 328114 |
| CD47 | 0.5 μg | APC | mouse IgG1, κ | CC2C6 | BioLegend | 323124 |
| CD38 | 0.5 μg | APC | mouse IgG1, κ | HIT2 | BioLegend | 303510 |
| CD15 | 0.5 μg | APC | mouse IgM, κ | HI98 | BioLegend | 301908 |
| CD171 | 0.5 μg | APC | recombinant human IgG1 | REA163 | Miltenyi Biotec | 130-100-684 |
| CD49F | 0.5 μg | APC | rat IgG2a, κ | GoH3 | BioLegend | 313616 |
| VEGFR | 0.5 μg | APC | mouse IgG1κ | ES8-20E6 | Miltenyi Biotec | 130-093-601 |
| EGFR | 0.5 μg | APC | mouse IgG1, κ | AY13 | BioLegend | 352906 |
| EGFRvIII | 0.5 μg | AF647 | mouse IgG1, κ | DH8.3 | Novus Biologicals | NBP2-50599AF647 |
| CD140 | 0.5 μg | APC | mouse IgG1, κ | 18A2 | BioLegend | 323608 |
| CD10 | 0.5 μg | APC | mouse IgG1κ | 97C5 | Miltenyi Biotec | 130-093-450 |
| CA9 | 0.5 μg | APC | recombinant human IgG1 | REA658 | Miltenyi Biotec | 130-110-058 |
| CD314 | 0.5 μg | APC | mouse IgG1κ | 1D11 | BioLegend | 320808 |
IgG, immunoglobulin G; VEGFR, vascular endothelial growth factor receptor; EGFRvIII, epidermal growth factor receptor variant III.
Figure 3.

Screening antibodies for nonspecific binding
A selection of capture beads when incubated with 0.5 μg of detection antibody with buffer (asterisks) was incubated as negative controls. EV samples derived from U87 (circles) and U251 (squares) cell lines were used as positive controls. The included Miltenyi buffer (shown in red) and 2% EV-depleted FBS (shown in black) were also compared.
EV multiplex analysis identifies cell-line tissue derivation
Multiplex analysis of EVs derived from neural, kidney, and colon cell lines using just a cocktail of CD9, CD63, and CD81 tetraspanins as detection antibodies was performed to identify whether EVs from different tissues had unique expression profiles (Figures 2C–2E). HCT116 cell lines show a higher intensity compared with kidney and neural cell lines with markers including CD326, CD133/1, CD81, and ROR1 being exclusively expressed and undetectable on neural and kidney EVs (Figure 2C). The variations in expression can be visualized among cell lines from the same tissue from heat map visualizations (Figure 2C).
Although all other capture beads, such as CD9, stain positively for the tetraspanin detection antibody mix, the expression levels vary considerably among EV tissue derivations. For example, EVs from the colon cell line expressing high levels captured on the CD9 bead stain brightly for the tetraspanin mix, whereas the kidney EVs stain very dimly, and the neural EVs stain at an intermediate level (Figure 2C). These differences further highlight the heterogeneity of EVs and demonstrate that CD9, CD63, and CD81 may not be present on all EVs.
Use of clustering to identify difference between biofluid and purification
The use of multiplex analysis in sample cohorts can result in large arrays of data. Our proposed method of stitched multiplex analysis further increases that size. Methods of data reduction to identify populations in large datasets are beginning to become commonplace (e.g., t-distributed stochastic neighbor embedding [tSNE]). With the accumulation of data across samples, equipment, assays, laboratories, and data reduction methods will play a role in understanding differences and similarities. For this reason, principal-component analysis (PCA) and tSNE have been built into MPAPASS. To understand the utility data reduction methods bring to stitched multiplex analysis plasma, serum, and CSF samples were analyzed using tetraspanins CD9, CD63, and CD81 individually at different input volumes and with different sample purification methods (Figure 4). Figure S5 shows the resulting heatmap of this large dataset when hierarchically clustered. At a high level,it can be seen that there appear to be areas of capture-detection marker enrichment between the plasma and CSF sample sources.
Figure 4.

Clustering of samples from differing biological fluids
(A–D) tSNE was performed on data from samples of plasma, serum, and CSF and stained with CD9, CD63, and CD81 across 39 unique capture beads and stratified by biological fluid (A), isolation method (B), and volume (C). (D) Boxplot of the top 25 capture bead and detection antibody combinations across samples showing the most significance as determined by Kruskal-Wallis.
An alternative method of visualizing similarities and differences across this dataset with a means to stratify by metadata, such as by purification or input material volume, is tSNE analysis. Using tSNE analysis, it is possible to separate CSF from plasma and serum (Figure 4A). This separation was independent of the sample purification method or input volume (Figures 4B and 4C). Serum and plasma samples prepared with qEV columns, however, cluster separately from neat and EV-clean samples. By looking at the top 25 most significant group differences (Figure 4D) it can be seen that the predominant differences are increased intensities of classical EV tetraspanin detection: CD9, CD63, and CD81, but also immune and stemness marker detection: CD8, CD14, CD24, CD40, and CD44. Serum and plasma groups both showed increased detection for platelet markers CD41b, CD42a, and CD62P. Preliminary data suggest that multiplex arrays can differentiate between samples from platelet-rich plasma (PRP) and platelet-poor plasma (PPP) and in different isolation tubes (Figure S6). These data demonstrate the utility of data reduction methods potentially for sample identification as well as batch effects such as different isolation methods among sample cohorts that may occur in aggregation methods such as the creation of an atlas.
Many plasma samples in clinical protocols are frozen while still containing platelets, which is not recommended within the EV field. The phenotypic difference of platelet depletion was investigated before samples were frozen and a comparison of fixed versus non-fixed blood isolation tubes was shown using the ratio of PPP to PRP for Streck and EDTA isolation tubes from a matched sample time point (Figure S6). It was seen in this isolated case that PPP contained a higher detection intensity for tetraspanins CD63 and CD81, platelet markers CD41b, CD42a, and CD62P, and immune markers CD8 and CD69 than did PRP. These results indicate that the thawing process and the subsequent platelet depletion protocol may remove more platelet EVs than when the platelet depletion protocol is conducted prior to freezing.
Fewer differences can be seen between the ratio of PRP to PPP plasma in fixed versus non-fixed isolations tubes. CD1c, CD14, and CD20 intensities are slightly higher in EDTA versus Streck tubes, whereas CD69 and CD4 intensities appear to be slightly higher in Streck versus EDTA tubes. Whether these changes are due to the number of particles present or the effect of the fixative on the epitope itself requires further investigation.
MPAPASS facilitates data sharing and atlas curation
MPAPASS was developed in response to the lack of commercially available or free software packages for EV multiplex analysis. As demonstrated, MPAPASS provides researchers with normalization methods, multiplex data stitching capabilities, data reduction methods, and data visualization methods in open-source software.
Along with a lack of software interfaces for EV multiplex analysis, there is also a current a lack of reporting standards for bead-based assays within the EV field. MPAPASS has attempted to facilitate this by being built on a standard database framework in the form of a spreadsheet containing key metadata criteria related to the assay (Table 2). This spreadsheet database framework (see key resources table) provides set sample, capture, and labeling metadata criteria for users to complete and allows users to further define their own metadata fields for stratification of their data analyses. Databases can also be merged to create aggregate datasets, making it possible to create personal atlases. While all metadata are found within a spreadsheet, once imported into the software, all data and metadata are then stored in a single file in order to simplify data sharing. Metadata related to cytometer acquisition are already developed in the form of MIFlowCyt. Software for the calibration of data and the automated reporting of acquisition metadata has already been established using previously developed FCMPASS software.
Table 2.
Default MPAPASS reporting framework criteria
| Criteria | Sheet | Description |
|---|---|---|
| Sample_Filename_Prefix | Sample | name of raw data file |
| Sample_Set_ID | Sample | numeric set ID of sample if stitched analyses are being used |
| Sample_ID | Sample | descriptive ID of sample |
| Sample_Grouping_ID | sample | primary sample testing group (e.g., treated/untreated) |
| Sample_Control_ID | sample | control set ID being used for normalization |
| Sample_Control_Filename | sample | name of raw data file for the control |
| Sample_Label_Mix_No | sample | detection antibody cocktail mix number |
| Incubated_Sample_Volume | sample | volume of sample incubated |
| Incubated_Sample_Concentration_per_mL | sample | concentration of sample incubated |
| Sample_Source | sample | source of sample (e.g., cell culture, plasma) |
| Sample_Isolation_Tube | sample | tube used to isolate EVs (e.g., ethylenediaminetetraacetate, heparin) |
| Sample_Purification_Method | sample | method used to purify EVs |
| Sample_Incubation_Time_With_CaptureBead | sample | sample incubation time with capture bead |
| Sample_Incubation_Time_With_Antibody | sample | sample incubation time with detection antibody |
| Antibody_Wash_Method | sample | method to wash excess antibody |
| Flow_Cytometer | sample | cytometer on which control data were acquired |
| Bead_Identifier | beads | unique identify for multiplex array |
| Bead_CaptureAntibody_Target | beads | bead capture antibody target |
| Bead_CaptureAntibody_Isotype | beads | bead capture antibody isotype |
| Bead_Capture_Antibody_Clone | beads | bead capture antibody clone |
| Bead_Wash_Buffer | beads | bead wash buffer |
| Bead_Capture_Antibody_Manufacturer | beads | bead capture antibody manufacturer |
| Bead_Capture_Antibody_CatNo | beads | bead capture antibody catalog number |
| Bead_Capture_Antibody_LotNo | beads | bead capture antibody lot number |
| Bead_Diameter | beads | bead diameter |
| Bead_Manufacturer | beads | bead manufacturer |
| Bead_Conjugation_Molecule | beads | bead conjugation method |
| Bead_Volume_Incubated | beads | bead volume incubated |
| Bead_Count_Incubated | beads | bead count incubated |
| Mix_Number | labeling | detection antibody cocktail mix number |
| Import_Column_Number | labeling | column relating to the specific detection antibody data within raw data file |
| Label_Target | labeling | target of the detection antibody |
| Label_Fluorophore | labeling | fluorophore conjugated to the detection antibody |
| Label_Isotype | labeling | isotype of the detection antibody |
| Label_Manufacturer | labeling | manufacturer of the detection antibody |
| Label_Catalogue_Number | labeling | catalog number of the detection antibody |
| Control_Filename_Prefix | controls | name of the raw data file |
| Control_Set_ID | controls | set to which control data relate |
| Control_Name | controls | name of control (e.g., blank bead) |
| Sample_Label_Mix_No | controls | mix number to which control relates |
| Control_Incubation_Time_With_Antibody | controls | detection antibody incubation time with control (if applicable) |
| Antibody_Wash_Method | controls | method used to wash excess antibody (e.g., filter plate) |
| Flow_Cytometer | controls | cytometer on which control data were acquired |
Outlined are reporting metadata database fields generated upon MPAPASS dataset creation. Each criteria field is created within the spreadsheet and is organized onto sheets within the spreadsheet relating to sample, beads, labeling, or controls.
Discussion
Here, we have demonstrated a software pipeline and controls that enable the ergonomic analysis of hundreds to thousands of protein combinations on EVs using stitched multiplex analysis. This tool is a powerful method of identifying potential markers for downstream high-sensitivity, single-EV assays, such as high-sensitivity flow cytometry or super-resolution microscopy (Lennon et al., 2019; Tian et al., 2018). Alternatively, this method could be used to identify markers for targeted subset isolation methods that could be used for subsequent downstream bulk assays such as RNA sequencing. Stratifying RNA sequencing data by protein subsets could provide novel insights into EV derivation in complex fluids building on previously developed deconvolution algorithms (Murillo et al., 2019). The MPAPASS software has been developed for the future curation of datasets into the existing ERCC exRNA Atlas in mind (Figure 5) (Murillo et al., 2019). The software is part of a package of tools developed for standardized reporting and analysis. While MPAPASS provides these tools for bead-based EV analyses, its counterpart, FCMPASS, provides standardized reporting and analysis tools for single-EV flow cytometry experiments, utilizing the MIFlowCyt-EV reporting framework (Welsh et al., 2020a, 2020d, 2020e; Welsh and Jones, 2020). FCMPASS may also be used for '.fcs' file calibration prior to MPAPASS data import for fluorescence calibration (Welsh and Jones, 2020). The use of these standardized reporting and analysis methods will aid in the transparency, reproducibility, and integration of EV repositories that will allow new approaches to EV characterization and subset identification.
Figure 5.

Pipeline for single-EV and bead-based assay EV Atlas curation
The processing of single-EV data (1) using FCMPASS and bead-based assays and (2) using MPAPASS software produces standard reporting methods and data. MPAPASS data can be calibrated with FCMPASS (3) software or other means. The product of these software packages is the integration of single-EV data (4) and bead-based EV data (5) that will enable standard comparisons and reporting for integration into online repositories such as the exRNA atlas.
One key consideration for the future of EV repertoire analysis with multiplex assays is that reproducibility and standardization across studies with these assays is an area for further development. A number of steps can be taken to improve assay quality and standardization: (1) quantification of the binding capacity of each bead in order to help standardize lots and bead types; (2) development of custom multiplex arrays that contain pathology-specific monoclonal capture antibodies, with matched individual monoclonal capture beads for validation assays and pull down; (3) potential use of Molecules of Equivalent Soluble Fluorophore (MESF) standards for calibration and comparison of data among instruments; and (4) development of a minimum set of reporting criteria based on established single-EV flow cytometry reporting frameworks (Welsh et al., 2020f). One of the largest hurdles of multiplex analysis optimization is resolving the nonspecific binding of detection antibodies to beads. Ideally, a positive control method allowing identification of EVs, rather than just a nonspecifically bound antibody, to the beads could be identified, such as using a membrane dye. Such positive control (marker-positive) EVs are not generally available, and, to date, the authors have been unable to identify a suitable membrane dye that is compatible with the multiplex beads used in this study.
In summary, we have demonstrated that the use of stitched multiplex analysis in combination with MPAPASS software is a powerful tool for creating, exploring, and analyzing data from hundreds of combinations of markers that can have great utility in guiding downstream quantitative single-EV methods or bulk EV subset analysis methods. This methodology holds utility for screening a large variety of markers simultaneously on EV-containing samples, with the main limitations being sample quantity and the semiquantitative nature of the assay itself. While super-resolution analysis (single-molecule detection) of single EVs is a highly quantitative assay with a demonstrated ability to identify clinical differences in samples, the clinical utility of such super-resolution assays is limited (Lennon et al., 2019; Tian et al., 2018) without robust EV repertoire assays. Stitched multiplex analyses, as performed with MPAPASS, provide a means for EV repertoire analysis so that particular pathology-specific subsets can then be further quantified at a single-EV level as part of a systematic multiplex-to-single EV (MtSEA) pipeline. As such, MPAPASS will streamline novel EV biomarker identification and the study of EV subset functions in health and disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD9 | Miltenyi Biotec | Cat# 130-108-813 |
| CD63 | Miltenyi Biotec | Cat# 130-108-813 |
| CD81 | Miltenyi Biotec | Cat# 130-108-813 |
| CTLA-4 | BioLegend | Cat# 369612; RRID: AB_2632873 |
| CD166 | Miltenyi Biotec | Cat# 130-106-576; RRID: AB_2655529 |
| CD147 | BioLegend | Cat# 306214; RRID: AB_2750170 |
| CD87 | Miltenyi Biotec | Cat# 130-114-851; RRID: AB_2726804 |
| CD90 | BioLegend | Cat# 328114; RRID: AB_893431 |
| CD47 | BioLegend | Cat# 323124; RRID: AB_2716203 |
| CD38 | BioLegend | Cat# 303510; RRID: AB_314362 |
| CD15 | BioLegend | Cat# 301908; RRID: AB_314200 |
| CD171 | Miltenyi Biotec | Cat# 130-100-684; RRID: AB_2655584 |
| CD49F | BioLegend | Cat# 313616; RRID: AB_1575047 |
| VEGFR | Miltenyi Biotec | Cat# 130-093-601; AB_10828920 |
| EGFR | BioLegend | Cat# 352906; AB_11150410 |
| EGFRvIII | Novus Biologicals | Cat# NBP2-50599AF647; RRID: AB_2904021 |
| CD140 | BioLegend | Cat# 323608; RRID: AB_2162787 |
| CD10 | Miltenyi Biotec | Cat# 130-093-450; RRID: AB_10828545 |
| CA9 | Miltenyi Biotec | Cat# 130-110-058; RRID: AB_2651327 |
| CD314 | BioLegend | Cat# 320808; RRID: AB_492962 |
| Biological samples | ||
| Human cerebral spinal fluid | National Institutes of Health (Dr. Steven Jacobson, Dr. Jennifer Jones) | N/A |
| Human plasma and serum | National Institutes of Health (Dr. Steven Jacobson, Dr. Jennifer Jones) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 |
| RPMI 1640 Medium | ThermoFisher Scientific | 11875093 |
| RPMI 1640 Medium (no phenol red) | ThermoFisher Scientific | 11835030 |
| McCoy's 5A (Modified) Medium | ThermoFisher Scientific | 16600082 |
| Critical commercial assays | ||
| MACSPlex Exosome Kit, human | Miltenyi Biotec | 130-108-813 |
| Deposited data | ||
| Example database | This publication | https://doi.org/10.5281/zenodo.5725951 |
| Experimental models: Cell lines | ||
| PC3 | Laboratory of Hisataka Kobayashi, NCI, NIH | N/A |
| PC3pip | Laboratory of Hisataka Kobayashi, NCI, NIH | N/A |
| U87 | Laboratory of Kevin Camphausen, NCI, NIH | N/A |
| U251 | Laboratory of Kevin Camphausen, NCI, NIH | N/A |
| HCT116 wt | Laboratory of Curt Harris, NCI, NIH | N/A |
| HCT116 mt | Laboratory of Curt Harris, NCI, NIH | N/A |
| HCT116 -/- | Laboratory of Curt Harris, NCI, NIH | N/A |
| UOK111 | Laboratory of W. Marston Linehan, NCI, NIH | N/A |
| UOK151 | Laboratory of W. Marston Linehan, NCI, NIH | N/A |
| Software and algorithms | ||
| MATLAB | Mathworks, Inc | https://www.mathworks.com/ |
| MPAPASS Software | NIH | Original Code https://zenodo.org/record/5725534#.YZ7r-b3MIUE Standalone Software https://nano.ccr.cancer.gov/mpapass/ |
| Other | ||
| qEVOriginal 70 nm | Izon Biosciences | N/A |
| qEV-10 70 nm | Izon Biosciences | N/A |
| JumboSep | PALL Corporation | FD100K65 |
| Nanosight LM10 | Malvern | N/A |
| CytoFLEX S | Beckman Coulter | N/A |
| Aurora | Cytek Biosciences | N/A |
| Optima XE ultracentrifuge | Beckman Coulter | B10049 |
| 45 titanium rotor | Beckman Coulter | 339160 |
Resource availability
Lead contact
Requests for further information and requests for resources or reagents in this manuscript should be directed to the lead contact author, Jennifer Jones (jennifer.jones2@nih.gov).
Materials availability
No unique reagents were generated in this study.
Experimental model and subject details
Ethics statement
Serum, plasma, and CSF samples used in this study were collected from the subject followed at the National Institute of Neurologic Disorders and Stroke under protocols # 98-N-0047, 89-N-0045, 13-N-0017, 13-N-0149. Prior to study inclusion, written informed consent was obtained from all subjects in accordance with the Declaration of Helsinki. Sample metadata for donors can be found in data database (see key resources table). Further statistical analysis of biofluid data was not performed, as the samples were deidentified, and age and sex information was not available for this study. Cell line models (PC3, PC3pip, U87, U251, UOK111, UOK151, HCT116 wt, HCT116 mt, HCT116 −/−) were used to produce EVs. Further details on culture conditions and source can be found in ‘STAR Methods’ and the ‘key resources table’, respectively.
Method details
Human sample isolation and storage
CSF samples were obtained by lumbar puncture. After centrifugation at 1,300×g for 10 minutes, the supernatants were collected into cryotubes and immediately frozen at −80°C until use. Whole blood was collected using serum separation tubes and centrifuged at 2000×g for 10 minutes. After centrifugation, the supernatants were collected into cryotubes and immediately frozen at −80°C until use. Plasma samples were collected in EDTA vacutainers. Samples were centrifuged at 900×g for 7 minutes. After centrifugation, the supernatants were collected into cryotubes and immediately frozen at −80°C until use. All samples were thawed at 37°C for 10 minutes. Plasma samples were centrifuged at 2500×g for 10 minutes twice to remove residual platelets.
EV isolation from human samples
Platelet-poor plasma (PPP) samples were created by centrifuging 2 mL of plasma twice at 2000×g for 10 minutes twice with the supernatant isolated. Multiplex samples referred to as ‘neat’ were aliquoted from PPP into multiplex bead mixture in the volumes outlined in the database (see key resources table) and results sections. EVs isolated from PPP using size exclusion chromatography (qEV-5, Izon Bioscience) were performed by adding 500 μL to the top of a qEV with fractions collected in 500 μL volumes. Fraction 7–9 were pooled, and the volumes incubated with multiplex beads are outlined in the database (see key resources table) and results sections. EV-Clean, a multimodal resin composition, in this case CaptoCore 700 (Cytiva Life Sciences, Cat. 17548101), prepared EVs were created by incubating 50 μL of PPP with 100 μL of resin for 30 minutes at room temperature, as previously described (Welsh et al., 2020c). The supernatant was removed from the resin and incubated with multiplex beads in the volumes indicated within the database (see key resources table) and results sections. 2 mL of CSF was concentrated to ∼50 μL using 100 kDa filters (Nanosep, PALL, Cat. OD100C34).
FBS depletion of EVs
To deplete FBS of EVs, 13 mL of fetal bovine serum (FBS) was added to 52 mL cell line media and added to a 65 mL ultracentrifugation tube. Tubes were placed in a pre-cooled 45 titanium (Ti) rotor and centrifuged at 100,000×g (k-factor 312.6) for 18 hours at 4°C using an Optima XE centrifuge (Beckman Coulter). The top 50 mL of supernatant from each tube was immediately removed and added to an equal volume of media to create a 10% EV-depleted FBS media stock.
Cell culture
PC3 and PC3pip cell lines were cultured in ∼35 mL 0.22 μm-filtered, phenol red, RPMI-1640, 10% EV-depleted FBS, 1% Pen/Strep, and 1% L-glutamine. HCT116 wild-type (wt), HCT116 p53 knock-out (−/−), and HCT116 mutant (mt) lines were cultured in ∼35 mL 0.22 μm-filtered, phenol red, McCoy's modified medium, 10% EV-depleted FBS, 1% Pen/Strep, and 1% L-glutamine. Cell lines were cultured in Falcon T175 tissue culture flasks with ambient (21%) oxygen and 5% CO2. U87 and U251 cell lines were cultured in Dulbecco-modified Eagle medium, with 4.5 mg mL−1 glucose, L-glutamine, 110 mg L−1 sodium pyruvate, 10% FBS. U87 and U251 cell lines were cultured in hypoxic conditions with 5% oxygen. Prior to EV collection cell lines were transferred to phenol red free media with 10% EV-depleted FBS for 24–48 hours.
EV isolation from cell culture
EV containing cell culture supernatants were aspirated from tissue culture flasks and transferred to 50 mL tubes. The tubes were centrifuged in 50 mL aliquots at 2000×g twice for 10 minutes. Cell-free supernatant was aliquoted into 60 mL JumboSep canisters (PALL Corporation) and concentrated using 100 kDa filters (JumboSep, PALL Corporation) until ∼5 mL was left. The 5 mL of cell-free concentrate was then run on a 10 mL 70 nm size exclusion column (qEV, Izon Science) with 5 mL fractions collected. The first 15 fractions were collected starting immediately after the sample was pipetted onto the column. EV concentration and protein content of fractions were approximated using nanoparticle tracking analysis (LM10, Malvern) and NanoDrop (Thermo Fisher Scientific), respectively, Figure S1. Fractions 7–9 of each preparation were mixed and used for multiplex experiments.
Multiplex assay
EVs were incubated with 15 μL of MACSPlex Exosome Kit (Miltenyi Biotec, Cat No. 130-108-813) in 500 μL low protein binding tubes overnight at 21°C and protected from light. EV inputs were based on total particle numbers obtained using NTA. The total counts of particles incubated are outlined for each figure. A 1.2 μm filter plate was washed using 150 μL of MACSPlex buffer (Miltenyi Biotec) and cleared using a vacuum manifold. Experiments comparing blocking buffers replaced MACSPlex buffer at all steps with 2% EV-depleted FBS. FBS depletion of EVs was described previously, with the 10% resulting stock diluted to 2% with Dulbecco phosphate buffered saline (Gibco, Cat. No. 14190144). 50 μL of MACSPlex (Miltenyi Biotec) buffer was added to each well before incubated EV-capture bead mixture was aliquoted into each of the wells. The EV-capture bead mixture was washed using vacuum manifold and immediately resuspended in 150 μL buffer to each well. Wells were reverse pipetted to resuspend beads. Antibody (information including volume, concentration, manufacturers, catalogue numbers can be found in Table 1 and respective MPAPASS .xls database files for each assay) was then added to each well and incubated, protected from light and shaking, for two hours at room temperature. Samples were reverse pipetted before washing using the vacuum manifold and immediately resuspending in 150 μL MACSPlex buffer, this was repeated. Each well was resuspended with 75 μL MACSPlex buffer, reverse pipetted and transferred to 96-well V-bottom polypropylene plates. This was repeated to ensure maximal bead recovery from the 1.2 μm filter plates, giving a final volume of 150 μL. Samples were then analyzed using flow cytometry. A detailed protocol and template for applying the multiplex samples to cell culture EVs can be found in Methods S1) and at: https://doi.org/10.17504/protocols.io.be7yjhpw;
Flow cytometry
Beads were triggered using a forward light scatter threshold and optimal gains for each detector (CytoFLEX S, Beckman Coulter, USA; Aurora, Cytek Bioscience, USA) were found by performing voltration on 8-peak beads (Cat. 422903, BioLegend, USA). A detailed protocol for bead gating strategies using FlowJo Software can be found in Methods S2 and at: https://doi.org/10.17504/protocols.io.bm3gk8jw. Briefly, Figure 1 outlines the types of signal that can originate from the multiplex assays (Figure 1A), shows how singlets are gated (Figure 1B), and the fluorescent ‘bar-coding’ of the multiplex array (Figure 1C) and the specificity of each population (Figure 1D).
Nanoparticle tracking analysis
Detectable sample concentration was approximated using a NanoSight LM10 instrument (Malvern, UK), equipped with a 405 nm LM12 module and EMCCD camera (DL-658-OEM-630, Andor). Video acquisition was performed with NTA software v3.4, using a camera level of 14. Three 30 second videos were captured per sample. Post-acquisition video analysis used the following settings: minimum track length = 5, detection threshold = 4, automatic blur size = 2-pass, maximum jump size = 12.0. Exported datasets were compiled and plotted using scripts written in MATLAB (v9.7.0.1261785 (R2019b) Update 3, The Mathworks Inc, Natick, MA).
MPAPASS software
A standalone software package for multiplex analysis was developed using MATLAB (v9.7.0.1261785 (R2019b) Update 3, The Mathworks Inc, Natick, MA). Compiled standalone software and GitHub repository links are available from nanopass.ccr.cancer.gov/mpapass. Maintained protocols for the use of the software can be found at: https://doi.org/10.17504/protocols.io.bm3gk8jw. An example import database for the software can be found at the "example database" link provided in the key resources table.
Normalization methods
Normalization methods shown in Figure 1C are as shown below and named separation index 1 and 2 (SI1-2), fold change (FC), and background subtraction (BS). Each method uses the median of positive bead, , and negative bead, . All methods are available for use within the MPAPASS package. Results in the manuscript used fold-change to avoid the creation of negative numbers thereby allowing logarithmic transformation of data for conducting downstream analyses without the need to alter or exclude datapoints.
Quantification and statistical analysis
Multiplex data was analyzed using MPAPASS software, normalization methods are noted in figure legends and formulas used can be found in ‘normalization methods’ section of STAR Methods.
Limitations of the study
Despite being a powerful method to identify markers useful for immune-affinity isolation methods or downstream single particle analysis, multiplex analysis itself is semi-quantitative and the changes in signal intensity can occur for a variety of reasons. For this reason, statistical analysis methods using post-hoc tests have been excluded from the MPAPASS software, which has been designed to primarily inspect data quality; stitch, normalize, and explore large multiplex dataset; perform data-reduction methods; and semi-qualitatively identify markers and associations that appear between samples.
Acknowledgments
We thank the translational clinical programs and staff at the United States National Institutes of Health (NIH) National Cancer Institute (NCI) and the National Institute of Neurological Disorders and Stroke (NINDS) Clinics. J.W., T.T., B.K., J.K., J.S., J.L.G., J.B.T., M.M., T.G, T.C., C.H., A.A., H.M., M.L, K.C., J.G, A.L., J.B., E.R., K.A., and J.J. were supported by the Intramural Research Program of the NIH, NCI, Center for Cancer Research. S.J., S.M., and M.P. were supported by the intramural NIH, NINDS program. I.G., A.M., A.A-M., and J.J. were supported by the NIH Common Fund. J.J. specifically acknowledges NIH ZIA BC011502, ZIA BC011503, U01 HL126497, and UG3 TR002881, the Prostate Cancer Foundation, and the NCI-Curie Partnership Program (J.J. and C.T.). A.L. acknowledges NIH Z01-BC006161. F.R. was funded by the Deutsche Forschungsgemeinschaft (RI2616/3-1). A.I. acknowledges support by NIH 1R01GM120272 (A.I.) and R01CA218500 (A.I., I.G., and J.J.). J.W. is an International Society for Advancement of Cytometry (ISAC) Marylou Ingram Scholar (2019-2023).
Author contributions
J.A.W. and J.C.J. conceived this research and designed the experiments. All authors participated in sample and data acquisition, data analysis, and editing of the manuscript.
Declaration of interests
A.G. has an affiliation with Evox Therapeutics, Ltd. All other authors declare no competing interests.
Published: January 24, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100136.
Supplemental information
Data and code availability
-
•
Multiplex data has been shared in Supplementary Data 1. Any further data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. https://doi.org/10.5281/zenodo.5725533
-
•
Any additional information for reanalysis of the data reported in this paper may be requested from the lead contact.
References
- Gasecka A., Nieuwland R., Budnik M., Dignat-George F., Eyileten C., Harrison P., Lacroix R., Leroyer A., Opolski G., Pluta K., et al. Ticagrelor attenuates the increase of extracellular vesicle concentrations in plasma after acute myocardial infarction compared to clopidogrel. J. Thromb. Haemost. 2020;18:609–623. doi: 10.1111/jth.14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issadore D., Min C., Liong M., Chung J., Weissleder R., Lee H. Miniature magnetic resonance system for point-of-care diagnostics. Lab Chip. 2011;11:2282–2287. doi: 10.1039/c1lc20177h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliha N., Wiencek Y., Heider U., Jungst C., Kladt N., Krauthauser S., Johnston I.C., Bosio A., Schauss A., Wild S. A novel multiplex bead-based platform highlights the diversity of extracellular vesicles. J. Extracell Vesicles. 2016;5:29975. doi: 10.3402/jev.v5.29975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon K.M., Wakefield D.L., Maddox A.L., Brehove M.S., Willner A.N., Garcia-Mansfield K., Meechoovet B., Reiman R., Hutchins E., Miller M.M., et al. Single molecule characterization of individual extracellular vesicles from pancreatic cancer. J. Extracell Vesicles. 2019;8:1685634. doi: 10.1080/20013078.2019.1685634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki J., Ochiya T. Circulating microRNAs and extracellular vesicles as potential cancer biomarkers: a systematic review. Int. J. Clin. Oncol. 2017;22:413–420. doi: 10.1007/s10147-017-1104-3. [DOI] [PubMed] [Google Scholar]
- Murillo O.D., Thistlethwaite W., Rozowsky J., Subramanian S.L., Lucero R., Shah N., Jackson A.R., Srinivasan S., Chung A., Laurent C.D., et al. exRNA atlas analysis reveals distinct extracellular RNA cargo types and their carriers present across human biofluids. Cell. 2019;177:463–477.e15. doi: 10.1016/j.cell.2019.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A.T., Witwer K.W., van Balkom B.W.M., de Beer J., Brodie C., Corteling R.L., Gabrielsson S., Gimona M., Ibrahim A.G., de Kleijn D., et al. Concise review: developing best-practice models for the therapeutic use of extracellular vesicles. Stem Cells Transl. Med. 2017;6:1730–1739. doi: 10.1002/sctm.17-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H., Chung J., Balaj L., Charest A., Bigner D.D., Carter B.S., Hochberg F.H., Breakefield X.O., Weissleder R., Lee H. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med. 2012;18:1835–1840. doi: 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H., Yoon T.J., Liong M., Weissleder R., Lee H. Magnetic nanoparticles for biomedical NMR-based diagnostics. Beilstein J. Nanotechnol. 2010;1:142–154. doi: 10.3762/bjnano.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tertel T., Bremer M., Maire C., Lamszus K., Peine S., Jawad R., Andaloussi S.E.L., Giebel B., Ricklefs F.L., Gorgens A. High-resolution imaging flow cytometry reveals impact of incubation temperature on labeling of extracellular vesicles with antibodies. Cytometry A. 2020;97:602–609. doi: 10.1002/cyto.a.24034. [DOI] [PubMed] [Google Scholar]
- Tian Y., Gong M., Hu Y., Liu H., Zhang W., Zhang M., Hu X., Aubert D., Zhu S., Wu L., Yan X. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. J. Extracell Vesicles. 2020;9:1697028. doi: 10.1080/20013078.2019.1697028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y., Ma L., Gong M., Su G., Zhu S., Zhang W., Wang S., Li Z., Chen C., Li L., et al. Protein profiling and sizing of extracellular vesicles from colorectal cancer patients via flow cytometry. ACS Nano. 2018;12:671–680. doi: 10.1021/acsnano.7b07782. [DOI] [PubMed] [Google Scholar]
- van der Pol E., Coumans F., Varga Z., Krumrey M., Nieuwland R. Innovation in detection of microparticles and exosomes. J. Thromb. Haemost. 2013;11:36–45. doi: 10.1111/jth.12254. [DOI] [PubMed] [Google Scholar]
- van der Pol E., Coumans F.A., Grootemaat A.E., Gardiner C., Sargent I.L., Harrison P., Sturk A., van Leeuwen T.G., Nieuwland R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014;12:1182–1192. doi: 10.1111/jth.12602. [DOI] [PubMed] [Google Scholar]
- van der Pol E., de Rond L., Coumans F.A.W., Gool E.L., Boing A.N., Sturk A., Nieuwland R., van Leeuwen T.G. Absolute sizing and label-free identification of extracellular vesicles by flow cytometry. Nanomedicine. 2018;14:801–810. doi: 10.1016/j.nano.2017.12.012. [DOI] [PubMed] [Google Scholar]
- van der Pol E., Hoekstra A.G., Sturk A., Otto C., van Leeuwen T.G., Nieuwland R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J. Thromb. Haemost. 2010;8:2596–2607. doi: 10.1111/j.1538-7836.2010.04074.x. [DOI] [PubMed] [Google Scholar]
- Welsh J.A., Horak P., Wilkinson J.S., Ford V.J., Jones J.C., Smith D., Holloway J.A., Englyst N.A. FCMPASS software aids extracellular vesicle light scatter standardization. Cytometry A. 2020;97:569–581. doi: 10.1002/cyto.a.23782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J.A., Jones J.C. Small particle fluorescence and light scatter calibration using FCMPASS software. Curr. Protoc. Cytometry. 2020;94:e79. doi: 10.1002/cpcy.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J.A., Jones J.C., Tang V.A. Fluorescence and light scatter calibration allow comparisons of small particle data in standard units across different flow cytometry platforms and detector settings. Cytometry A. 2020;97:592–601. doi: 10.1002/cyto.a.24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J.A., Killingsworth B., Kepley J., Traynor T., Mckinnon K., Savage J., Appel D., Aldape K., Camphausen K., Berzofsky J.A., et al. A simple, high-throughput method of protein and label removal from extracellular vesicle samples. bioRxiv. 2020 doi: 10.1039/D0NR07830A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J.A., Tang V.A., van der Pol E., Gorgens A. MIFlowCyt-EV: the next chapter in the reporting and reliability of single extracellular vesicle flow cytometry experiments. Cytometry A. 2020;99A:365–368. doi: 10.1002/cyto.a.24268. [DOI] [PubMed] [Google Scholar]
- Welsh J.A., van der Pol E., Arkesteijn G.J.A., Bremer M., Brisson A., Coumans F., Dignat-George F., Duggan E., Ghiran I., Giebel B., Gorgens A. MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles. 2020;9:1713526. doi: 10.1080/20013078.2020.1713526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J.A., van der Pol E., Arkesteijn G.J.A., Bremer M., Brisson A., Coumans F., Dignat-George F., Duggan E., Ghiran I., Giebel B., et al. MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles. 2020;9:1713526. doi: 10.1080/20013078.2020.1713526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklander O.P.B., Bostancioglu R.B., Welsh J.A., Zickler A.M., Murke F., Corso G., Felldin U., Hagey D.W., Evertsson B., Liang X.M., et al. Systematic methodological evaluation of a multiplex bead-based flow cytometry assay for detection of extracellular vesicle surface signatures. Front. Immunol. 2018;9:1326. doi: 10.3389/fimmu.2018.01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Ma L., Wang S., Chen C., Zhang W., Yang L., Hang W., Nolan J.P., Wu L., Yan X. Light-scattering detection below the level of single fluorescent molecules for high-resolution characterization of functional nanoparticles. ACS Nano. 2014;8:10998–11006. doi: 10.1021/nn505162u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Multiplex data has been shared in Supplementary Data 1. Any further data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. https://doi.org/10.5281/zenodo.5725533
-
•
Any additional information for reanalysis of the data reported in this paper may be requested from the lead contact.