

Summary
Single-cell technologies are revolutionizing the ability of researchers to infer the causes and results of biological processes. Although several studies of pluripotent cell differentiation have recently utilized single-cell sequencing data, other aspects related to the optimization of differentiation protocols, their validation, robustness, and usage are still not taking full advantage of single-cell technologies. In this review, we focus on computational approaches for the analysis of single-cell omics and imaging data and discuss their use to address many of the major challenges involved in the development, validation, and use of cells obtained from pluripotent cell differentiation.
Keywords: single cell, pluripotent cell differentiation, protocol optimization, computational approaches, stem cell applications
Introduction
Induced pluripotent stem cell (iPSC) differentiation has emerged as a promising technology for generating cells for several human tissues and organs. Cells derived from iPSCs are used for many different purposes including to study development (Zhu and Huangfu, 2013) and for therapeutics (Moradi et al., 2019). A key advantage of iPSCs over stem cells is their autologous nature, which enables their therapeutic usage. In 2014, the first iPSC therapy was initiated in the RIKEN Center in Japan for patients with macular degeneration (Wang et al., 2020). The first completed human clinical trial using iPSC-derived cells was reported in 2020 and demonstrated tolerability and safety of iPSC-derived mesenchymal stem cell (MSC) in the treatment of steroid-resistant acute graft versus host disease (Bloor et al., 2020). Case studies or clinical trials have also been initiated for a number of pathologies, including CD19+ malignancies (Elstrom, 2020), heart diseases (Sawa, 2021), or brain pathologies (Takahashi, 2020). In addition to directly using differentiated cells for clinical applications, iPSCs are also useful for drug development (Farkhondeh et al., 2019) and for personalized medicine (Chun et al., 2011). The former relies on the ability of these cells to differentiate to several different human organs to study the impact of different drugs. The latter makes use of the fact that these cells can be directly derived from individuals to tailor specific treatments on the basis of how differentiated cells from these individuals respond to the infection. Several other non-clinical applications of iPSCs have been extensively discussed including their use in studying development (Hoshina et al., 2018; Snoeck, 2015), identifying regulators and pathways (Friedman et al., 2018), and for inferring underlying causes of specific diseases (Lin et al., 2017b).
Although promising, the use of differentiated iPSCs has faced several challenges since their discovery 15 years ago. A key challenge for the use of iPSCs in clinical applications is the development of protocols that can efficiently differentiate iPSCs to various cell types and organoids. Although several protocols have been developed for differentiating iPSC-derived cells to different tissue types, including protocols for heart cells (Friedman et al., 2018; Giacomelli et al., 2020; Mummery et al., 2012), various brain cells (Gunhanlar et al., 2018), lung (Hurley et al., 2020; Mucci et al., 2018), liver (Mallanna and Duncan, 2013; Zhang et al., 2018), pancreas (Jacobson and Tzanakakis, 2017), and more, their efficiency (yield), success rates, and reproducibility vary considerably with many leading to only a small fraction of the cells differentiating to the desired tissue and/or cell type (Liu et al., 2020). Other challenges include the issue of safety and their potential to cause unregulated replication and cancer (Doss and Sachinidis, 2019). Epigenetics differences between the derived and original cells is also a major concern (Liu et al., 2020). Finally, for cells being injected, their ability to replicate within the organs they are injected to and their ability to function as the primary cells is still a major open question (Morris et al., 2014; Rezvani et al., 2016). There are also challenges associated with using iPSCs for drug development and/or personalized medicine. These include questions about how similar are derived cells to cells from the same organ of the individual and are the cell types and their frequencies similar to what we would observe in the healthy organ (Handel et al., 2016). Other issues relate to determination of the pathways, regulators, and interactions that are involved in the differentiation both within the different cell types and between cell types of the same organ/tissue.
The promise of single-cell technologies in studying iPSCs
Recent advances in genomics enable the study of biological processes at the single-cell level (Cahan et al., 2021). The most widely used technology for such studies is single-cell RNA sequencing (scRNA-seq) (Tang et al., 2009), which profiles the expression of genes in single cells. Several other related single-cell sequencing technologies have been developed for studying molecular events at the single-cell level. These include assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) (Buenrostro et al., 2015), which looks for open chromatin locations in single cells, methylation sequencing (methyl-seq), which profiles methylation at single cells (Smallwood et al., 2014), and other epigenetics profiling technologies (Ramani et al., 2017; Smallwood et al., 2014). More recently spatial single-cell level technologies for both transcriptomics and proteomics have been developed. Unlike scRNA-seq in which cells are first extracted from the sample and then sequenced, in spatial transcriptomics the location of cells is recorded and expression levels for each cell are determined (Ståhl et al., 2016). These methods extend fluorescence in situ hybridization and combines it with transcription or proteomics profiling. This enables the quantification of expression levels for several genes or proteins at a single-cell resolution while still recording the location of each of the cells in the sample. Although not all spatial transcriptomics methods are profiling at the single-cell level (Rodriques et al., 2019), many newer methods do. Examples of platforms for single-cell spatial transcriptomics include MERFISH (Chen et al., 2015), seqFISH (Shah et al., 2016), seqFISH+ (Eng et al., 2019), osmFISH (Codeluppi et al., 2018), and the 3D transcriptomics record (STARMAP) (Wang et al., 2018). Similarly, advances in spatial proteomics enable the study of protein levels at the single-cell resolution. Methods, including CODEX (Goltsev et al., 2018) and digital spatial profiling (Merritt et al., 2020), can detect the location of up to 70 different proteins in single tissue sections by using antibodies for specific proteins and imaging them directly on the slides.
Single-cell technologies provide several promising directions for solving the most challenging issues researchers face when developing iPSC-based cell types and organoids. The ability to profile the expression of single cells over time enables the study of the different states and cell types that various protocols introduce. It can also be used to identify key junctions, places in the protocol where some cells commit to a specific cell type whereas others commit to another. Such junctions can often be studied further to determine the pathways and regulators involved in such commitment and to determine where specific intervention can further optimize the protocol, leading more cells to differentiate to the desired outcome. Single-cell technologies can also be used to study in much greater details the set of resulting cells from each protocol and to compare these and their distribution with the cells observed in real tissues and organs. It can also be applied to study variations and mutations in individual cells, their longevity and the factors impacting them. Spatial profiling can further provide information on the key interactions between different cells and cell types and how these impact the ability to generate the required set of cells or organoids. When using iPSCs to generate organoids, multilineage communication plays an important role in self-organization of differentiated cells. For example, He et al. (2020) have recently used lineage-coupled spatial transcriptomics to study lineage and clonal locations during brain organoid regionalization. Cell-cell interaction might also occur in other settings, such as reprogramming to iPSCs (Shakiba et al., 2019). Spatial transcriptomics and other single-cell imaging methods can thus improve our ability to better model and understand differentiation in these systems.
In this review we discuss several aspects related to the use of single-cell technologies and analysis in iPSC differentiation studies (Figure 1). We first present single-cell technologies and computational analysis methods for designing and using time series experiments to model regulatory and signaling networks involved in the differentiation process, and their time of activation. We then discuss methods that can be used to validate the resulting cells to determine consistency or similarity to primary human cells. Finally, we discuss potential applications of single-cell technologies for reducing cancer risk and immunogenicity and for improving personal-based disease treatments.
Figure 1.

An end-to-end approach for using single cell technologies in iPSC studies
Top: computational models for optimizing cell differentiation protocols. These methods fall into four different categories. (1) Time series iPSC single-cell experimental design. (2) Clustering/cell-type annotation: using marker genes and annotation datasets to determine cell types at different stages in the analysis. (3) Trajectory inferences: reconstructing cell differentiation trajectories (from iPSCs) to identify the different fates and their onset. (4) Regulatory and signaling network inference: using epigenomics and spatial single cell data to infer the underlying regulatory network within cells and the cell-cell interaction networks. Middle: protocol validation and reproducibility. Cell alignment methods can compare datasets to human reference or between repeated studies. Reproductivity metrics are used to estimate the similarity between multiple datasets (e.g., from different runs). Barcoding methods are used to determine the cellular trajectories and timing of the cell fate decisions. Bottom: applications. Generated cells can be studied to determine longevity and potential cancer risk. Cells can then be either used to replace damaged cells or for studying the impact of treatments on individuals.
Designing single-cell experiments
Several points should be considered when designing single-cell experiments for studying iPSC differentiation protocols. In sequencing-based experiments researchers need to determine how many cells would be profiled from each time point and at what depth. The number of cells, and the sequencing depth can have a large impact on the ability to identify rare cell types and to correctly characterize subtypes. In addition, although protocols for stem cell differentiation differ in their duration, they usually span multiple days and even weeks. This makes it important to determine a subset of time points to sample. Selecting the most appropriate time points is important to identify all stages of the process while minimizing costs and the need to use large quantities of cells.
To define adequate cell numbers per time point, one must consider cell heterogeneity and expected frequency for each subpopulation. Several computational tools have been developed to determine how many cells should be profiled for different expected distributions. For example, howmanycells (https://satijalab.org/howmanycells/) and sceb (Zhang et al., 2020) (Table 1) are computational tools that can estimate the total required number of cells on the basis of user inputs for the expected subpopulation structure and cell-type frequency. Sequencing depth is often determined by the platform used for the analysis. For example, 10x genomics scRNA-seq measurements often requires at least 20k reads/cell. It is important to note that, unlike the number of cells, increasing read depth usually will not increase the cost by much. To capture genes that are less abundant but might be of importance it is recommended to profile at least 50k reads/cell. A list of recommended sequencing depth for various single-cell sequencing platforms (e.g., 10x Chromium, smart-seq) is also available for reference (Haque et al., 2017).
Table 1.
Computational tools for the processing, analysis, and modeling of single-cell data
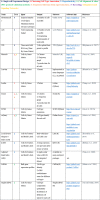 |
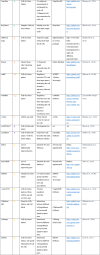 |
The tools are numbered and color coded based on the specific tasks they are intended for. Note that a number of tools can be applied to multiple tasks.
As for selecting time points, this is often a tradeoff between costs and accuracy. Obviously, the more time points the better, although exactly how many are needed and how they should be spaced (uniformly, more emphasis on the start, etc.) is often unknown. To select an optimal set of time points Kleyman et al. (2017) developed the time point selection (TPS) method (Table 1). TPS is based on oversampling (i.e., sampling at very high frequency) followed by nanostring profiling of relatively few genes in bulk samples. This procedure is often much cheaper and faster than full bulk or single-cell sequencing. Although results are best if the genes selected are relevant for the process being studied, random genes can also lead to good results. Next, computational methods are used to reconstruct the temporal profiles of the genes and to determine the optimal subset of points which are enough to correctly predict the other, unused, time points. In addition to the selected points, TPS provides a measure of accuracy that indicates the expected loss resulting from the reduction in time points sampled. TPS was recently applied to a single-cell study which aimed to optimize the protocol differentiating iPSCs to alveolar epithelial type 2 cells (AEC2s) (Hurley et al., 2020). The method selected six time points to profile and these were shown to accurately capture the different stages and transitions involved in the differentiation process.
Cell type annotation for single-cell sequencing data
The end results of specific differentiation are sets of cells that can be in one or several states (cell type). Inferring the set of cell types, their distribution, and their similarity to known human cell types is one of the most important issues when determining the success of a differentiation protocol. Accurate assignments are even more challenging for iPSC studies when compared with other scRNA-seq studies because of variability in the differentiation ability of iPSC lines that can impact consistent cell-type annotation (Cuomo et al., 2020; Guhr et al., 2018). Even canonical markers of differentiation stage can be unreliable for annotating significant portions of iPSCs that might be rapidly undergoing transcriptional changes, or are in transition between differentiation states (Cuomo et al., 2020). Further complication is due to higher variability at the mRNA level compared with expression of canonical protein markers (Carcamo-Orive et al., 2017).
Cell-type inference can be performed in an unsupervised or supervised manner. The former involves clustering of the cells (either from the last time point or using the branches obtained by the pseudo-time analysis). Next, each cluster is analyzed by looking at differentially expressed genes or known markers to determine cell types. Finally, cell types are assigned from a set of ontologies. If none of the known types matches a specific cluster it might represent a new or intermediate state. Most clustering methods used for such assignment work on a dimensionally reduced set of genes and provide visualization by using 2D projections. PCA, t-SNE (Maaten and Hinton, 2008), and UMAP (McInnes et al., 2018) are among the most widely used dimensionality reduction methods. Examples of clustering methods used for this include Monocle (Cao et al., 2019; Qiu et al., 2017), Louvain (Blondel et al., 2008), and Leiden (Traag et al., 2019) (Table 1). A variety of statistical methods, such as Student’s t test, Wilcoxon rank-sum test, and several others can be applied to the resulting clusters to identify top differential genes, which are then compared with known cell-type markers to identify the most likely cell types (or a list of probabilities for different cell types) (Zhang et al., 2019).
Another option for assigning cell types to new scRNA-seq datasets are supervised methods that either classify cell types (classification methods) or use previously annotated datasets to “transfer” labels to a new dataset (alignment methods). In classification methods, iPSCs are used as a query and a classifier trained on a labeled reference is used to predict their labels. Examples of these methods include SCNN (Lin et al., 2017a) and scQuery (Alavi et al., 2018) (Table 1). Alignment methods (Johansen and Quon, 2019) rely on various strategies and often start by identifying similar sets of anchor cells between the two datasets based on the expression of a subset or all genes. This is an important step for iPSC studies, as there are many sources of batch effects present that make comparisons of experiments difficult. As a result, adjustment for experimental batch effects is often an early step in iPSC scRNA-seq analysis (Jerber et al., 2021). Next, nearest neighbor approaches (either between or within the same dataset) are used to assign all cells in the new dataset to a cell or representation in the previously annotated dataset. Most alignment methods begin with learning a shared joint reduced dimension representation, either using various embedding neural networks or using more standard methods (e.g., PCA or SVD [Golub and Reinsch, 1971]). Next, variants of nearest neighbor algorithms are used to assign cells in the new dataset. Although some of these methods rely on linear transformations of one dataset onto the other, based on identified anchor cells (Alavi and Bar-Joseph, 2020; Haghverdi et al., 2018; Stuart et al., 2019), others utilize neural network models to embed both datasets in a shared reduced dimension (Johansen and Quon, 2019; Lopez et al., 2018). Several evaluation metrics for the accuracy of the assignment have been proposed and are used to determine if the alignment can indeed correctly match the same cell types. One such metric is local inverse Simpson’s index (LISI) (Korsunsky et al., 2019), which estimates the diversity within a small neighborhood around each cell, measuring the effective number of datasets around the cell (higher values indicate better alignments) and the effective number of cell types around the cell (lower values indicate better alignments) simultaneously. Another example is k-BET (Büttner et al., 2019), which tests the null hypothesis that the fraction of cells from a particular batch within a small neighborhood is the same as the fraction among all cells. This test is conducted for many random local neighborhoods, and a low rejection rate indicates well-mixed batches and thus a good alignment. Other metrics to assess alignments include those based on clustering agreement measures, such as the adjusted Rand index, where a clustering method is applied to the aligned cells, and this clustering is compared with cell-type labels and dataset labels (Tran et al., 2020) (Table 1).
Alignment methods are also useful for a critical question in stem cell differentiation: evaluation of the similarity of cell types generated by a differentiation protocol and cells from healthy controls for the same tissue/organ. Although individual differences will likely lead to some differences between iPSC-derived cells and control cells from healthy individuals, alignment methods can be used to overcome batch and experimental effects to determine the similarity of the differentiated cells. This is particularly important in iPSC differentiation, as batch effects, such as time point of collection, cell line of origin, and experimental batch, often dominate the variability in the data (Cuomo et al., 2020). In addition to alignment to human-derived datasets, iPSC studies can also be aligned to model species datasets. In this case the alignment method would be used to overcome both batch affects and differences in the set of genes between species (Polański et al., 2020). Such analysis might be of critical importance in cases where it is hard to obtain specific reference sets from human (e.g., specific developmental data from real embryos). As an example, we present here an alignment of iPSCs from a recent large iPSC study (Cuomo et al., 2020) that contained cells from 125 donors collected at different time points during differentiation (Figure 2).Without aligning the cells, iPSCs from the same time points are separated by donor, making it hard to compare cell types across donors (Figures 2A and 2B). However, after we integrate the cells by using Seurat (Stuart et al., 2019), we see that the batches (donors) are well mixed, while still keeping the biological signal (distinct differentiation time points) intact (Figures 2C and 2D). We also quantitatively evaluate the alignment via the LISI score and observe that the alignment indeed leads to high mixing between batches (donors), while time points remain distinct (Figure 2E).
Figure 2.

Alignment of scRNA-seq data from a large iPSC single-cell study.
Results are presented for the top three human iPSC donors in terms of cell counts (“joxm,” 1,415; “guss,” 1,093; “poih,” 1,077).
(A and B) UMAP embeddings of the unaligned cells, colored by donor ID (A) and differentiation time point (B).
(C and D) UMAP embeddings of the cells after integration with Seurat, colored by donor ID (C) and differentiation time point (D).
(E) Evaluation of the alignment, quantified using the local inverse Simpson’s index score based on PCA coordinates after integration with Seurat (the same PCA embeddings used to produce the UMAP embeddings) in (C and D). The top part shows a high mixing score for donors, indicating that the alignment successfully overcomes batch affects. The bottom score shows low mixing for time points, indicating that the alignment correctly separates cells based on their differentiation stage.
Computational tools to evaluate iPSC differentiation protocols
A number of machine learning and network analysis methods have also been developed to directly evaluate differentiation protocols. These methods, including CellNet (Morris et al., 2014), SingleCellNet (Tan and Cahan, 2019), Capybara (Kong et al., 2020), and KeyGenes (Roost et al., 2015), examine the relationship between differentiated (or reprogrammed) cells and their in vivo counterparts to quantify how closely engineered cell populations resemble target cell types (Table 1). CellNet, which was developed for bulk data, uses information on a set of cell types and associated gene regulatory networks (GRNs). For an input dataset, it predicts active GRNs by using context-likelihood and, on the basis of the set of identified GRNs, it determines probabilities for cell and tissue types. Other methods can use similar ideas for single-cell data. For example, Alavi et al. (2018) presented scQuery, a neural network-based classification method that can be used to assign cell types in new samples on the basis of previously annotated cell types. scQuery compiled a large database of annotated scRNA-seq data and combined neural networks and k nearest neighbors to predict cell types for new samples on the basis of the similarity to samples in the database. SingleCellNet uses Random Forest to achieve a similar goal of cell-type classification. Capybara (Kong et al., 2020) is another method for cell type assignment on the basis of reference data. It is based on quadratic programming and uses bulk expression signatures to deconvolve single cells as a linear combination of different cell types. Each cell is then assigned a score for each of the possible cell type. The method was further extended to incorporate single-cell annotations, which improves performance (Table 1).
Tools for trajectory inference from scRNA-seq time series data
Although analysis of the set of resulting cells is often required to determine if the protocol indeed achieves its goals, the process itself and the way cells differentiate from iPSCs to the required cell types is also of great interest. Understanding the biological events that take place as part of the differentiation process provides information on both the way specific tissues and organs develop and the way different aspects of the protocol contribute to the correct differentiation. Thus, reconstructing the set of states and transitions leading to the end result is often of great interest. When performing time series analysis, which is the major focus on many differentiation studies, scRNA-seq and scATAC-seq studies present challenges that differ from bulk experiments. Although in bulk studies it is possible to follow the (average) trajectories of genes over time by obtaining samples at specific time points, when individual cells are profiled, they are consumed and cannot be directly followed over time. In addition, single cells even at the same time point might be at different stages (some can be slightly faster or slower than others), which means that even cells sampled at the same time might not be in the same state. To overcome this issue, several methods have been developed for pseudo-time inference from time series scRNA-seq data (Trapnell et al., 2014). Most methods for pseudo-time inference rely on using dimensionality reduction followed by the construction of spanning trees or graphs to connect points in the lower dimensional space (Figure 3). A large number of variants on this basic idea have been proposed. Some of the more widely used include Monocole3 (Cao et al., 2019), Diffusion maps (Haghverdi et al., 2016), and PAGA (Wolf et al., 2019) (Table 1). Other methods, including scdiff and CSHMM, use a probabilistic approach that either assigns cells to a discrete (Weinreb et al., 2018) or infinitely many states (Lin and Bar-Joseph, 2019). Such methods can be used to infer continuous trajectories. Following cell assignments by any of the methods researchers can reconstruct the expression trajectories of individual genes for specific branches by using their levels in cells assigned along the branches.
Figure 3.

A typical dimensionality reduction-based trajectory inference pipeline
The high-dimensional cell expression profiles (blue dots, top) are first projected to a lower space (bottom, 2D in this example). Next, a trajectory is inferred by connecting anchor nodes (gray dots), which represent cell clusters. In most methods the initial (or starting) set of cells are identified by the user and serve as the starting point for the trajectory (left most gray node). Next, this point is linked to other points by edges to construct the full trajectory (edges from left to right).
Another direction for reconstructing such trajectories is RNA velocity (La Manno et al., 2018). scVelo is an example RNA velocity method that could be generalized to systems with transient cell states (Bergen et al., 2020). Unlike pseudo-time ordering methods that only look at the expression levels of genes in different cells, RNA velocity methods analyze both the levels of gene expression and the levels of unspliced introns to obtain a time derivative estimate of the direction the cell is moving. This estimate can assign a trajectory to each individual cell by comparing the unspliced RNAs in one cell to the spliced (presumably a later time point) RNA in another. By obtaining a trajectory for each cell the method can infer an overall trajectory for all cells in a dataset that is not directly based on similarity of the expression levels.
Trajectory inference by using genetic barcoding
Trajectory inference methods based on scRNA-seq (discussed above) assume that cell states change continuously over time and so can be traced by using changes in the expression of specific genes. Although this assumption seems to be correct in many cases, the sampling rates and other experimental and biological artifacts (e.g., threshold expression switches [Torres et al., 2018]) might lead to inaccuracies in the reconstructed trajectories and might prevent the identification of cell paths and branching. An alternative, which can also provide information on the specific cell type commitment time and the potential of specific cells to differentiate to multiple types at a specific time, is to use genetic barcoding to label a set of cells at a certain point in the experiment. In these experiments, cells are tagged with a specific genetic barcode at one or multiple stages of the experiments. These barcodes can be read as part of the RNA-seq of each of the cells and used to reconstruct relationships between cells profiled at different time points. Two main variants of this approach have been used in developmental and differentiation studies. The first introduces a set of barcodes from a specific library at one or more stages and uses them to identify the set of all progenitors of a cell at later time points (Hurley et al., 2020; Weinreb et al., 2020). By profiling the cells at different time points such an approach can be used to both reconstruct trajectories by combining the scRNA-seq profiles with the genetic barcodes and to determine if multiple cell types can arise from the same cell. Inserting such barcodes at multiple time points leads to a finer resolution of the reconstructed branching tree given that cells can be traced to both immediate and previous parents. Genetic barcoding has been used in several recent iPSC studies, including for hematopoietic stem cells and progenitor cell differentiation (da Rocha and Malleshaiah, 2019; Hollmann et al., 2020).
Genetic barcoding is often limited in the number of different barcodes that can be used along a specific trajectory. Thus, not all cell divisions can be recorded by using this approach and a large number of cells might end up with the exact same barcodes making it hard to fully reconstruct specific trajectories. Thus, a second, alternative approach was developed to trace a much larger number of divisions by using CRISPR mutations (Kalhor et al., 2018; Raj et al., 2018). In this method, a CRISPR-Cas9 construct is inserted into cells and random mutations accumulate with each division. When cells are sequenced, a phylogeny tree for cells can be reconstructed by using scGESTALT (Raj et al., 2018) based on the mutations accumulated for each cell, leading to a much better resolution of the branching history. However, unlike genetic barcoding, the CRISPR approach is much noisier (mutations can be reversed and/or missing leading to errors in the reconstruction) and mutations can saturate, leading to cells with the same mutations arising from different trajectories. Recent methods have integrated single-cell expression data with the CRISPR mutations to improve the tree reconstruction. These methods, including LinTMat (Zafar et al., 2020), result in improved accuracy of tree reconstruction and overcome some of the noise associated with the CRISPR mutations. Still, even when using expression to improve accuracy, the resulting tree might still contain errors and is only the maximum likelihood estimation of the process.
Inferring regulators and pathways controlling cell differentiation
The above analysis can be used to characterize the activity and resulting outcomes of iPSC differentiation protocols. Such analysis is critical for both understanding the set of states that cells take while differentiating and determining if the resulting set of cells, or at least a specific cell type, match what we are interested in. However, these analyses do not provide explicit information on how to improve protocols and how to obtain cells and differentiated organoids that match more closely the target. In several cases, even for successful differentiation protocols, the yield might be very low (sometimes less than 5% [Chun et al., 2011]). The vast majority of resulting cells often do not express the markers that are targeted by the protocol. This means that only a small subset of the cells activate the pathways that are needed for correct differentiation. Determining how and when to activate these pathways for the other cells might lead to much better results and higher yields.
A number of computational methods have been developed to model the set of regulatory networks that are activated within cells and that govern the differentiation processes. This can be largely divided into two main categories. The first relies on trajectory inference methods (discussed above) to obtain an initial temporal model. This model is then analyzed to identify genes, regulators, and pathways that correlate with the inferred trajectories for the cells (e.g., a set of genes that go up in correlation with the order assigned to cells along a specific branch). Once such genes are identified, various set enrichment methods are used to infer the pathways and functional relevance of these genes and to identify potential regulators of such genes. Example of such methods include GSEA (Subramanian et al., 2005) and PANTHER (Mi et al., 2019) (Table 1). This set can then be used to determine potential interventions that might lead to the desired outcome; for example, overexpression of a pathway that is upregulated in the set of cells that are the target of the protocol.
The second set of methods perform joint trajectory inference and pathway analysis. Unlike the standard trajectory inference methods, which mainly use the expression of genes, such joint analysis methods take into account both the set of expressed genes and the set of regulators/pathways they belong to. This allows these methods to focus on a subset of the genes rather than on all genes and to construct the resulting trajectories on the basis of these regulatory relationships. An advantage of these methods is the fact that they can pinpoint not just the regulators and pathways but also the exact time in the process in which they are activating their targets. For example, scdiff (Ding et al., 2018a) and its extension CSHMM (Lin et al., 2020) (Table 1), were shown to correctly predict specific regulators and pathways whose removal or overexpression can lead to changes in cell fate during iPSC differentiation. Such information can be used to better tailor perturbations by either repressing or overexpressing specific transcription factors or treatment with relevant small molecules at the exact time point leading to much higher yields and improved iPSC differentiation protocols. To elucidate this application, we present CSHMM performed on an iPSC scRNA-seq of differentiating alveolar epithelial type 1 (AT1) cells (GEO accession series: GSE145539 [Kanagaki et al., 2021]) (Figure 4). Although the method is unsupervised, in the CSHMM reconstructed trajectory the expression of the AT1 marker gene, HOPX, is increasing along with the differentiation stage. The method also identifies a number of transcription factors, including EGR1, as key regulators of the differentiation process and provides information on the specific stages and time points that are critical for regulating AT1 cell differentiation (Figure 4). Many of these findings agree with previous studies (Martinez et al., 2004).
Figure 4.

CSHMM results for an iPSC data on lung alveolar epithelial cell differentiation, in GEO: GSE145539
(A) UMAP plot of all clusters.
(B and C) HOPX expression (alveolar type 1 marker) in different clusters. HOPX expression is higher in clusters 2, 6, and 8 CSHMM trajectories.
(D) CSHMM reconstructed trajectories. Each dot represents a cell, while colors for cells correspond to the clusters in (A) HOPX expression is highest for later branches. TFs are assigned by the method to some edges of the differentiation tree. For example, EGR1 regulate the cellular state transitions out of the cluster 0.
Although the above discussion is focused on scRNA-seq, for modeling regulation scATAC-seq is a valuable and sometimes critical addition. A number of methods (mainly for bulk), such as iDREM (Ding et al., 2018b) and coupleNMF (Duren et al., 2018) (Table 1), have been developed to integrate RNA-seq and ATAC-seq data for regulatory network inference and, when possible, sequencing methods that can simultaneously profile both types of data in the same cells would lead to more accurate results and better models. Other methods, such as sci-CAR and snare-seq, enable the joint profiling of scRNA-seq and scATAC-seq from the same cells (Cao et al., 2018; Liu et al., 2019; Ma et al., 2020; Xing et al., 2020). These are beneficial because they allow for a joint analysis of both the levels of genes in cells and their regulation. Recent studies have begun to integrate single-cell chromatin accessibility data (scATAC-seq) with scRNA-seq for inferring fate-specific regulators. For example, Ranzoni et al. (2021) profiled hematopoietic stem and progenitor cells and mature blood cells from fetal bone marrow and liver tissues. For some of the samples they performed both scRNA-seq and scATAC-seq profiling. They initially clustered the scRNA-seq data and inferred trajectories by using PAGA (Wolf et al., 2019). Based on the scRNA-seq analysis they selected 36 markers genes. Next, they clustered the scATAC-seq results and analyzed the accessibility of the identified markers in each of the clusters. They have also performed label transfer between scRNA-seq and scATAC-seq results. Using single-cell motif discovery methods (Schep et al., 2017), they were able to identify lineage-specific transcription factors (TFs) and match these to the clusters and trajectories they regulate as part of the process. The main advantage of using scATAC-seq was in identifying TFs that are expressed at low levels and so cannot be detected by using expression data alone. Motifs for such TFs can still show significant enrichment at key target genes enabling their identification when performing joint analysis.
Inferring signaling pathways by using scRNA-seq and spatial single-cell data
The above discussion focused on internal cell controls that are often achieved by gene regulation. However, a key aspect of development and differentiation is cell-cell signaling networks. To better model the iPSC differentiation process, especially when the goal is to obtain an organoid rather than a specific cell type requires the analysis and modeling of these networks as well. scRNA-seq and other sequencing methods do not directly provide information on such interactions given that when cells are extracted for profiling their location with respect to other cells is lost and so it is not clear which cells are neighbors in the sample. Although the spatial profiling methods discussed in the introduction above provide clear advantages over the sequencing-based methods by providing a direct measure of the spatial organization of cells, they are currently limited in several ways including the number of genes they profile, the types of samples they can analyze, and in the way samples are prepared and processed.
The computational methods attempt to determine interactions between cell types by analyzing interactions between clusters obtained from the scRNA-seq data. Generally, these methods rely on the assumption that, if cells interact, one would express a set of ligands that will interact with a set of receptors in the other (Armingol et al., 2020). To identify such interactions, researchers often start with a general or tissue-specific set of such ligand-receptor pairs and attempt to determine if any of them are activated in neighboring cells. Some methods extend the analysis to look not just at neighboring receptors but also at their downstream targets (i.e., signaling pathways activated by these receptors). This is achieved by studying the set of differentially expressed genes in each cell type and intersecting them with sets of known pathways. Such pathways can then be linked to receptors that are activating them to link cells. CellphoneDB (Efremova et al., 2020), CSOmap (Ren et al., 2020), and MESSI (Li et al., 2021) (Table 1) are three such methods that can perform cell interaction analysis. These methods rely on clustering of scRNA-seq data. For each pair of clusters, they evaluate the expression pairs of ligands and their known receptor targets and obtain a summary score for all potential ligand-receptor pairs. This score is then used to predict which clusters (cell types) might be interacting.
A number of computational methods have also been developed to infer cell-cell interactions from spatial transcriptomics and proteomics data. These methods range from graph-based approaches that attempt to model cells as nodes in a graph and their interactions as edges (Yuan and Bar-Joseph, 2020) to more global inference approaches that look at pre-defined neighborhoods on the basis of distance for performing such analysis. In addition, spatial methods can also be used to compare the organization of cells between organoids and real tissues (Fleck et al., 2020). A key advantage of spatial transcriptomics methods is their ability to determine cell types and locations of cells. This can be used to study if cells in the organoid are organized in a similar way to those in real healthy tissues; for example, by computing the neighborhood distributions for specific cell types in both types of samples. Changes observed can be then traced back to the signaling networks inferred and interventions can be determined to further improve the similarity of the two samples.
Discussion and future prospects
Single-cell technologies can revolutionize the use of iPSCs and their effectiveness. All stages of iPSC-based therapeutics, including those used for directly treating individuals and those used for personalized treatment determination, can be greatly improved by utilizing such data and the associated analysis discussed above. Similarly, developmental studies that use iPSCs will gain a much better understanding of the process when using these datasets. Starting with the differentiation protocol, single-cell technologies can provide information on the set of genes and regulators that are activated at various stages of the differentiation process. Both intracellular and intercellular pathways and regulators can be identified and modeled. By analyzing these pathways and their impact, researchers would be able to identify the most likely interventions, and their timing, required to obtain the target cells or organoids. Single-cell methods can then be used to evaluate the success of the protocol at a much more detailed level than currently done. Rather than relying on a small set of markers, by profiling all genes at the single-cell level researchers can determine the exact distribution of the resulting cells and see if they match target cells both in terms of expression and in terms of cell-type frequencies. In addition, the use of such technologies and computational methods are expected to lead to important advances in a number of other key areas relevant for the use of iPSCs in treating individuals.
Use of single-cell technologies in judging cell state longevity and cancer risk of iPSC-derived cells
In this review we mainly focus on using single-cell technologies to better understand and model the iPSC differentiation process. However, even when such methods work and lead to an effective protocol that is capable of generating tissue-specific cells, the resulting cells might still not lead to an effective treatment or clinical use. An important question for iPSC-derived cells is their ability to maintain their states for long durations without becoming cancerous (Lee et al., 2013). Similarly, the ability of these cells to maintain their states for very long durations after injections into a host, and to further proliferate in the host, is a major open question. Single-cell technologies can be used to address these questions both in vitro and in vivo. For example, recent work has shown that differentiated lung cells remain very similar to their states at the completion of the differentiation protocol several months after the differentiation (Hurley et al., 2020). Similar analysis, several weeks or months after differentiation, can be used to study the in vitro potential for cancer and oncogene expression. The advantage of single-cell technologies over bulk or marker-based methods for this task is that they can identify even a very small subset of cancerous cells, which will likely go undetected when profiling groups of cells. In addition, given that the analysis profiles all genes within a cell, there is no need to preselect a set of oncogenes or other cancer markers making it easier to identify all potential cancerous cells.
Beyond the analysis of expression data, the use of epigenomics single-cell technologies, including scATAC-seq described above and methyl-seq (Barros-Silva et al., 2018), might provide further information about the potential of cells to become cancerous. Methylation of specific DNA regions and genes has been tightly linked to various forms of cancer (Baylin, 2005). Single-cell methylation analysis can be performed to identify if even a small number of cells display similar methylation patterns. Such analysis can identify not just cells that actually became cancerous but also those with the potential to become cancerous leading to much safer resulting sets of cells.
Potential use of single-cell technologies to study immunogenicity and for personalizing iPSC-based treatments
As discussed in the Introduction, several therapeutic uses of iPSC are now being studied in clinical trials. A major promise of iPSCs is their injection to treat various diseases. One of the most well-studied treatments along these lines is the injection of MSCs to treat osteoarthritis (Jevotovsky et al., 2018). Other ongoing studies include the use of human autologous iPSC-derived dopaminergic progenitors to restore motor function in Parkinson’s disease (Song et al., 2020) and transplanting retinal pigment epithelial cells differentiated from iPSCs in patients with age-related macular degeneration (Mandai et al., 2017). However, recent studies indicate that, although some iPSC-differentiated cells are usually well tolerated (De Almeida et al., 2014), others might still trigger immune response in patients (Deuse et al., 2019; Liu et al., 2017) because of the expression of immunogenic antigens that are not expressed in parental somatic cells. For instance, mutations during reprogramming or cell expansion can generate neoantigens that stimulate immune response after transplantations (Deuse et al., 2019). Single-cell technologies are very suitable for studying these issues. Comparing derived cells that do and do not trigger immune response (Zhao et al., 2015) at the single-cell level can allow the identification of both genomic and epigenomic differences between such cells and differences in the presence (or absence) of specific cell types that might trigger the response. Combined with the ability to study the tumor-induction risks discussed above, such analysis can lead to much safer treatments.
Autologous iPSCs are also a great way to study in vivo the effect of potential treatments on an individual. There has been wide interest in using cell lines and bulk data to study drug effects (Pabon et al., 2018), and work by several groups indicated that drugs can be tailored on the basis of the predicted impact of the set of genes expressed in a certain disease. Methods such as the connectivity map (Lamb, 2007) and related approaches attempted to link the expression induced by specific drugs to the expression observed in a patient to tailor a personal treatment for individuals. The methods work by looking for drugs that lead to expression patterns that “reverse” differences observed between healthy and diseased tissue for an individual (Lamb, 2007). Although the connectivity methods successfully identified drugs for several diseases, these studies have so far focused on cell lines or bulk expression data from individuals and so could not take into account the impact of drugs on individual cell types or on the distribution of cell types within a tissue/organ. These are crucial issues for treatment. Using single-cell technologies, we can extend the use of such methods to improve the ability to identify the correct drug for patients. This can be done by combining some of the computational methods described above (including those that attempt to assign cell types in samples before and after treatment, those that look for specific genes, and those that study cell-cell interactions). The ability to predict and then test drugs on autologous iPSCs can have a large impact on the response of individuals to treatments.
To conclude, here, we present a comprehensive approach for the use of new single-cell technologies in iPSC differentiation studies. Still, several challenges remain to effectively use them for improving these protocols. Many of these challenges are computational and relate to the ability to efficiently and accurately analyze the very large data that is generated by single-cell technologies (Cahan et al., 2021). As we discussed, methods are emerging to address many of these challenges and, while more work remains, we believe that the existing computational solutions are already mature enough to provide useful information that will lead to much better iPSC-based methods.
Acknowledgments
Work was also partially funded by NIH grants 1R01GM122096 and OT2OD026682 to Z.B.-J. M.R.E. is supported by an R01 from the National Institute of Biomedical Imaging and Bioengineering (EB028532), an R01 from the National Heart, Lung, and Blood Institute (HL141805), and support from the Pittsburgh Liver Research Center (NIH- NIDDK P30DK120531). J.D. is partially supported by the FRQS Junior 1 salary award (295298,295299).
Declaration of interests
The authors declare no competing interests.
References
- Alavi A., Bar-Joseph Z. Iterative point set registration for aligning scRNA-seq data. PLoS Comput. Biol. 2020;16:e1007939. doi: 10.1371/journal.pcbi.1007939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavi A., Ruffalo M., Parvangada A., Huang Z., Bar-Joseph Z. A web server for comparative analysis of single-cell RNA-seq data. Nat. Commun. 2018;9:1–11. doi: 10.1038/s41467-018-07165-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Almeida P.E., Meyer E.H., Kooreman N.G., Diecke S., Dey D., Sanchez-Freire V., Hu S., Ebert A., Odegaard J., Mordwinkin N.M. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 2014;5:1–12. doi: 10.1038/ncomms4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armingol E., Officer A., Harismendy O., Lewis N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2020;22:1–18. doi: 10.1038/s41576-020-00292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros-Silva D., Marques C.J., Henrique R., Jerónimo C. Profiling DNA methylation based on next-generation sequencing approaches: new insights and clinical applications. Genes. 2018;9:429. doi: 10.3390/genes9090429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005;2:S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. Theor. Exp. 2008;2008:P10008. [Google Scholar]
- Bloor A.J., Patel A., Griffin J.E., Gilleece M.H., Radia R., Yeung D.T., Drier D., Larson L.S., Uenishi G.I., Hei D. Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: a phase I, multicenter, open-label, dose-escalation study. Nat. Med. 2020;26:1720–1725. doi: 10.1038/s41591-020-1050-x. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P., Chang H.Y., Greenleaf W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner M., Miao Z., Wolf F.A., Teichmann S.A., Theis F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods. 2019;16:43–49. doi: 10.1038/s41592-018-0254-1. [DOI] [PubMed] [Google Scholar]
- Cahan P., Cacchiarelli D., Dunn S.-J., Hemberg M., de Sousa Lopes S.M.C., Morris S.A., Rackham O.J., Del Sol A., Wells C.A. Computational stem cell biology: open questions and guiding principles. Cell Stem Cell. 2021;28:20–32. doi: 10.1016/j.stem.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Cusanovich D.A., Ramani V., Aghamirzaie D., Pliner H.A., Hill A.J., Daza R.M., McFaline-Figueroa J.L., Packer J.S., Christiansen L. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:1380–1385. doi: 10.1126/science.aau0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcamo-Orive I., Hoffman G.E., Cundiff P., Beckmann N.D., D’Souza S.L., Knowles J.W., Patel A., Papatsenko D., Abbasi F., Reaven G.M. Analysis of transcriptional variability in a large human iPSC library reveals genetic and non-genetic determinants of heterogeneity. Cell Stem Cell. 2017;20:518–532. e519. doi: 10.1016/j.stem.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.H., Boettiger A.N., Moffitt J.R., Wang S., Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090. doi: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun Y.S., Byun K., Lee B. Induced pluripotent stem cells and personalized medicine: current progress and future perspectives. Anat. Cell Biol. 2011;44:245–255. doi: 10.5115/acb.2011.44.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codeluppi S., Borm L.E., Zeisel A., La Manno G., van Lunteren J.A., Svensson C.I., Linnarsson S. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods. 2018;15:932–935. doi: 10.1038/s41592-018-0175-z. [DOI] [PubMed] [Google Scholar]
- Cuomo A.S., Seaton D.D., McCarthy D.J., Martinez I., Bonder M.J., Garcia-Bernardo J., Amatya S., Madrigal P., Isaacson A., Buettner F. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 2020;11:1–14. doi: 10.1038/s41467-020-14457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuse T., Hu X., Agbor-Enoh S., Koch M., Spitzer M.H., Gravina A., Alawi M., Marishta A., Peters B., Kosaloglu-Yalcin Z. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat. Biotechnol. 2019;37:1137–1144. doi: 10.1038/s41587-019-0227-7. [DOI] [PubMed] [Google Scholar]
- Ding J., Aronow B.J., Kaminski N., Kitzmiller J., Whitsett J.A., Bar-Joseph Z. Reconstructing differentiation networks and their regulation from time series single-cell expression data. Genome Res. 2018;28:383–395. doi: 10.1101/gr.225979.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Hagood J.S., Ambalavanan N., Kaminski N., Bar-Joseph Z. iDREM: interactive visualization of dynamic regulatory networks. PLoS Comput. Biol. 2018;14:e1006019. doi: 10.1371/journal.pcbi.1006019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss M.X., Sachinidis A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells. 2019;8:403. doi: 10.3390/cells8050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duren Z., Chen X., Zamanighomi M., Zeng W., Satpathy A.T., Chang H.Y., Wang Y., Wong W.H. Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc. Natl. Acad. Sci. U S A. 2018;115:7723–7728. doi: 10.1073/pnas.1805681115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- Elstrom R. FT819 is in subject with B-cell malignancies. 2020. https://clinicaltrials.gov/show/NCT04629729
- Eng C.-H.L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y., Yun J., Cronin C., Karp C., Yuan G.-C. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+ Nature. 2019;568:235–239. doi: 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkhondeh A., Li R., Gorshkov K., Chen K.G., Might M., Rodems S., Lo D.C., Zheng W. Induced pluripotent stem cells for neural drug discovery. Drug Discov. Today. 2019;24:992–999. doi: 10.1016/j.drudis.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck J.S., He Z., Boyle M.J., Camp J.G., Treutlein B. Resolving brain organoid heterogeneity by mapping single cell genomic data to a spatial reference. Cell stem cell. 2020;28:1148–1159. doi: 10.1101/2020.01.06.896282. [DOI] [PubMed] [Google Scholar]
- Friedman C.E., Nguyen Q., Lukowski S.W., Helfer A., Chiu H.S., Miklas J., Levy S., Suo S., Han J.-D.J., Osteil P. Single-cell transcriptomic analysis of cardiac differentiation from human PSCs reveals HOPX-dependent cardiomyocyte maturation. Cell Stem Cell. 2018;23:586–598. e588. doi: 10.1016/j.stem.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomelli E., Meraviglia V., Campostrini G., Cochrane A., Cao X., van Helden R.W., Garcia A.K., Mircea M., Kostidis S., Davis R.P. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell. 2020;26:862–879.e11. doi: 10.1016/j.stem.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsev Y., Samusik N., Kennedy-Darling J., Bhate S., Hale M., Vazquez G., Black S., Nolan G.P. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 2018;174:968–981. e915. doi: 10.1016/j.cell.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub G.H., Reinsch C. Linear Algebra. Springer; 1971. Singular value decomposition and least squares solutions; pp. 134–151. [Google Scholar]
- Guhr A., Kobold S., Seltmann S., Wulczyn A.E.S., Kurtz A., Löser P. Recent trends in research with human pluripotent stem cells: impact of research and use of cell lines in experimental research and clinical trials. Stem Cell Reports. 2018;11:485–496. doi: 10.1016/j.stemcr.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunhanlar N., Shpak G., van der Kroeg M., Gouty-Colomer L., Munshi S., Lendemeijer B., Ghazvini M., Dupont C., Hoogendijk W., Gribnau J. A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol. Psychiatry. 2018;23:1336–1344. doi: 10.1038/mp.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Büttner M., Wolf F.A., Buettner F., Theis F.J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods. 2016;13:845. doi: 10.1038/nmeth.3971. [DOI] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel A.E., Chintawar S., Lalic T., Whiteley E., Vowles J., Giustacchini A., Argoud K., Sopp P., Nakanishi M., Bowden R. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum. Mol. Genet. 2016;25:989–1000. doi: 10.1093/hmg/ddv637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A., Engel J., Teichmann S.A., Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9:1–12. doi: 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., Gerber T., Maynard A., Jain A., Petri R., Santel M., Ly K., Sidow L., Calleja F.S., Riesenberg S. Lineage recording reveals dynamics of cerebral organoid regionalization. bioRxiv. 2020 doi: 10.1101/2020.06.19.162032. [DOI] [Google Scholar]
- Hollmann J., Brecht J., Goetzke R., Franzen J., Selich A., Schmidt M., Eipel M., Ostrowska A., Hapala J., Fernandez-Rebollo E. Genetic barcoding reveals clonal dominance in iPSC-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2020;11:1–13. doi: 10.1186/s13287-020-01619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshina A., Kawamoto T., Sueta S.-I., Mae S.-I., Araoka T., Tanaka H., Sato Y., Yamagishi Y., Osafune K. Development of new method to enrich human iPSC-derived renal progenitors using cell surface markers. Sci. Rep. 2018;8:1–11. doi: 10.1038/s41598-018-24714-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K., Ding J., Villacorta-Martin C., Herriges M.J., Jacob A., Vedaie M., Alysandratos K.D., Sun Y.L., Lin C., Werder R.B. Reconstructed single-cell fate trajectories define lineage plasticity windows during differentiation of human PSC-derived distal lung progenitors. Cell Stem Cell. 2020;26:593–608.e8. doi: 10.1016/j.stem.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson E.F., Tzanakakis E.S. Human pluripotent stem cell differentiation to functional pancreatic cells for diabetes therapies: innovations, challenges and future directions. J. Biol. Eng. 2017;11:21. doi: 10.1186/s13036-017-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerber J., Seaton D.D., Cuomo A.S., Kumasaka N., Haldane J., Steer J., Patel M., Pearce D., Andersson M., Bonder M.J. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet. 2021;53:304–312. doi: 10.1038/s41588-021-00801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevotovsky D., Alfonso A., Einhorn T., Chiu E. Osteoarthritis and stem cell therapy in humans: a systematic review. Osteoarthr. Cartil. 2018;26:711–729. doi: 10.1016/j.joca.2018.02.906. [DOI] [PubMed] [Google Scholar]
- Johansen N., Quon G. scAlign: a tool for alignment, integration, and rare cell identification from scRNA-seq data. Genome Biol. 2019;20:1–21. doi: 10.1186/s13059-019-1766-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor R., Kalhor K., Mejia L., Leeper K., Graveline A., Mali P., Church G.M. Developmental barcoding of whole mouse via homing CRISPR. Science. 2018;361:eaat9804. doi: 10.1126/science.aat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagaki S., Ikeo S., Suezawa T., Yamamoto Y., Seki M., Hirai T., Hagiwara M., Suzuki Y., Gotoh S. Directed induction of alveolar type I cells derived from pluripotent stem cells via Wnt signaling inhibition. Stem Cells. 2021;39:156–169. doi: 10.1002/stem.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyman M., Sefer E., Nicola T., Espinoza C., Chhabra D., Hagood J.S., Kaminski N., Ambalavanan N., Bar-Joseph Z. Selecting the most appropriate time points to profile in high-throughput studies. eLife. 2017;6 doi: 10.7554/eLife.18541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W., Fu Y.C., Morris S.A. Capybara: a computational tool to measure cell identity and fate transitions. bioRxiv. 2020 doi: 10.1101/2020.02.17.947390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-r., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J. The Connectivity Map: a new tool for biomedical research. Nat. Rev. Cancer. 2007;7:54–60. doi: 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- Lee A.S., Tang C., Rao M.S., Weissman I.L., Wu J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013;19:998–1004. doi: 10.1038/nm.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Ding J., Bar-Joseph Z. Identifying signaling genes in spatial single-cell expression data. Bioinformatics. 2021;37:968–975. doi: 10.1093/bioinformatics/btaa769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Bar-Joseph Z. Continuous-state HMMs for modeling time-series single-cell RNA-Seq data. Bioinformatics. 2019;35:4707–4715. doi: 10.1093/bioinformatics/btz296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Jain S., Kim H., Bar-Joseph Z. Using neural networks for reducing the dimensions of single-cell RNA-seq data. Nucleic Acids Res. 2017;45:e156. doi: 10.1093/nar/gkx681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-H., Jewell B.E., Gingold J., Lu L., Zhao R., Wang L.L., Lee D.-F. Osteosarcoma: molecular pathogenesis and iPSC modeling. Trends Mol. Med. 2017;23:737–755. doi: 10.1016/j.molmed.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Ding J., Bar-Joseph Z. Inferring TF activation order in time series scRNA-seq studies. PLoS Comput. Biol. 2020;16:e1007644. doi: 10.1371/journal.pcbi.1007644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Li W., Fu X., Xu Y. The immunogenicity and immune tolerance of pluripotent stem cell derivatives. Front. Immunol. 2017;8:645. doi: 10.3389/fimmu.2017.00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Liu C., Quintero A., Wu L., Yuan Y., Wang M., Cheng M., Leng L., Xu L., Dong G. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 2019;10:1–10. doi: 10.1038/s41467-018-08205-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., David B.T., Trawczynski M., Fessler R.G. Advances in pluripotent stem cells: history, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 2020;16:3–32. doi: 10.1007/s12015-019-09935-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Zhang B., LaFave L.M., Earl A.S., Chiang Z., Hu Y., Ding J., Brack A., Kartha V.K., Tay T. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020;183:1103–1116. e1120. doi: 10.1016/j.cell.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maaten L.v.d., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- Mallanna S.K., Duncan S.A. Differentiation of hepatocytes from pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2013;26:1G. 4.1–1G. 4.13. doi: 10.1002/9780470151808.sc01g04s26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandai M., Watanabe A., Kurimoto Y., Hirami Y., Morinaga C., Daimon T., Fujihara M., Akimaru H., Sakai N., Shibata Y. Autologous induced stem-cell-derived retinal cells for macular degeneration. New Engl. J. Med. 2017;376:1038–1046. doi: 10.1056/NEJMoa1608368. [DOI] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J.M., Baek S.J., Mays D.M., Tithof P.K., Eling T.E., Walker N.J. EGR1 is a novel target for AhR agonists in human lung epithelial cells. Toxicol. Sci. 2004;82:429–435. doi: 10.1093/toxsci/kfh272. [DOI] [PubMed] [Google Scholar]
- McInnes L., Healy J., Melville J. Umap: uniform manifold approximation and projection for dimension reduction. arXiv. 2018 arXiv:1802.03426 [Google Scholar]
- Merritt C.R., Ong G.T., Church S.E., Barker K., Danaher P., Geiss G., Hoang M., Jung J., Liang Y., McKay-Fleisch J. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020;38:586–599. doi: 10.1038/s41587-020-0472-9. [DOI] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Huang X., Ebert D., Mills C., Guo X., Thomas P.D. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v. 14.0) Nat. Protoc. 2019;14:703–721. doi: 10.1038/s41596-019-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi S., Mahdizadeh H., Šarić T., Kim J., Harati J., Shahsavarani H., Greber B., Moore J.B. Research and therapy with induced pluripotent stem cells (iPSCs): social, legal, and ethical considerations. Stem Cell Res. Ther. 2019;10:1–13. doi: 10.1186/s13287-019-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.A., Cahan P., Li H., Zhao A.M., San Roman A.K., Shivdasani R.A., Collins J.J., Daley G.Q. Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell. 2014;158:889–902. doi: 10.1016/j.cell.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucci A., Lopez-Rodriguez E., Hetzel M., Liu S., Suzuki T., Happle C., Ackermann M., Kempf H., Hillje R., Kunkiel J. iPSC-derived macrophages effectively treat pulmonary alveolar proteinosis in Csf2rb-deficient mice. Stem Cell Reports. 2018;11:696–710. doi: 10.1016/j.stemcr.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C.L., Zhang J., Ng E.S., Elliott D.A., Elefanty A.G., Kamp T.J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ. Res. 2012;111:344–358. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabon N.A., Xia Y., Estabrooks S.K., Ye Z., Herbrand A.K., Süß E., Biondi R.M., Assimon V.A., Gestwicki J.E., Brodsky J.L. Predicting protein targets for drug-like compounds using transcriptomics. PLoS Comput. Biol. 2018;14:e1006651. doi: 10.1371/journal.pcbi.1006651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polański K., Young M.D., Miao Z., Meyer K.B., Teichmann S.A., Park J.-E. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics. 2020;36:964–965. doi: 10.1093/bioinformatics/btz625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017;14:979. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B., Wagner D.E., McKenna A., Pandey S., Klein A.M., Shendure J., Gagnon J.A., Schier A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018;36:442–450. doi: 10.1038/nbt.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani V., Deng X., Qiu R., Gunderson K.L., Steemers F.J., Disteche C.M., Noble W.S., Duan Z., Shendure J. Massively multiplex single-cell Hi-C. Nat. Methods. 2017;14:263–266. doi: 10.1038/nmeth.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzoni A.M., Tangherloni A., Berest I., Riva S.G., Myers B., Strzelecka P.M., Xu J., Panada E., Mohorianu I., Zaugg J.B. Integrative single-cell RNA-seq and ATAC-seq analysis of human developmental hematopoiesis. Cell Stem Cell. 2021;28:472–487. e477. doi: 10.1016/j.stem.2020.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Zhong G., Zhang Q., Zhang L., Sun Y., Zhang Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020;30:763–778. doi: 10.1038/s41422-020-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani M., Grimm A.A., Willenbring H. Assessing the therapeutic potential of lab-made hepatocytes. Hepatology. 2016;64:287–294. doi: 10.1002/hep.28569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha E.L., Malleshaiah M. Trajectory algorithms to infer stem cell fate decisions. Comput. Stem Cell Biol. 2019;1975:193–209. doi: 10.1007/978-1-4939-9224-9_9. [DOI] [PubMed] [Google Scholar]
- Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., Macosko E.Z. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–1467. doi: 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roost M.S., Van Iperen L., Ariyurek Y., Buermans H.P., Arindrarto W., Devalla H.D., Passier R., Mummery C.L., Carlotti F., De Koning E.J. KeyGenes, a tool to probe tissue differentiation using a human fetal transcriptional atlas. Stem Cell Reports. 2015;4:1112–1124. doi: 10.1016/j.stemcr.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa Y. (2021). Clinical Trial of Human (Allogeneic) iPS Cell-derived Cardiomyocytes Sheet for Ischemic Cardiomyopathy, https://clinicaltrials.gov/show/NCT04696328.
- Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S., Lubeck E., Zhou W., Cai L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. 2016;92:342–357. doi: 10.1016/j.neuron.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiba N., Fahmy A., Jayakumaran G., McGibbon S., David L., Trcka D., Elbaz J., Puri M.C., Nagy A., van der Kooy D. Cell competition during reprogramming gives rise to dominant clones. Science. 2019;364:eaan0925. doi: 10.1126/science.aan0925. [DOI] [PubMed] [Google Scholar]
- Smallwood S.A., Lee H.J., Angermueller C., Krueger F., Saadeh H., Peat J., Andrews S.R., Stegle O., Reik W., Kelsey G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods. 2014;11:817–820. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeck H.-W. Modeling human lung development and disease using pluripotent stem cells. Development. 2015;142:13–16. doi: 10.1242/dev.115469. [DOI] [PubMed] [Google Scholar]
- Song B., Cha Y., Ko S., Jeon J., Lee N., Seo H., Park K.-J., Lee I.-H., Lopes C., Feitosa M. Human autologous iPSC-derived dopaminergic progenitors restore motor function in Parkinson’s disease models. J. Clin. Invest. 2020;130:904–920. doi: 10.1172/JCI130767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl P.L., Salmén F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902. e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J. iPS cell-based therapy for Parkinson's disease: a Kyoto trial. Regen. Ther. 2020;13:18–22. doi: 10.1016/j.reth.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y., Cahan P. SingleCellNet: a computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst. 2019;9:207–213. e202. doi: 10.1016/j.cels.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Torres J., Monti R., Moore A.L., Seimiya M., Jiang Y., Beerenwinkel N., Beisel C., Beira J.V., Paro R. A switch in transcription and cell fate governs the onset of an epigenetically-deregulated tumor in Drosophila. eLife. 2018;7:e32697. doi: 10.7554/eLife.32697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:1–12. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H.T.N., Ang K.S., Chevrier M., Zhang X., Lee N.Y.S., Goh M., Chen J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020;21:1–32. doi: 10.1186/s13059-019-1850-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Allen W.E., Wright M.A., Sylwestrak E.L., Samusik N., Vesuna S., Evans K., Liu C., Ramakrishnan C., Liu J. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361:eaat5691. doi: 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Tang Z., Gu P. Stem/progenitor cell-based transplantation for retinal degeneration: a review of clinical trials. Cell Death Dis. 2020;11:1–14. doi: 10.1038/s41419-020-02955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C., Wolock S., Tusi B.K., Socolovsky M., Klein A.M. Fundamental limits on dynamic inference from single-cell snapshots. Proc. Natl. Acad. Sci. U S A. 2018;115:E2467–E2476. doi: 10.1073/pnas.1714723115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C., Rodriguez-Fraticelli A., Camargo F.D., Klein A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. 2020;367:eaaw3381. doi: 10.1126/science.aaw3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Hamey F.K., Plass M., Solana J., Dahlin J.S., Göttgens B., Rajewsky N., Simon L., Theis F.J. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:1–9. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Q.R., El Farran C.A., Zeng Y.Y., Yi Y., Warrier T., Gautam P., Collins J.J., Xu J., Dröge P., Koh C.-G. Parallel bimodal single-cell sequencing of transcriptome and chromatin accessibility. Genome Res. 2020;30:1027–1039. doi: 10.1101/gr.257840.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Bar-Joseph Z. GCNG: graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biology. 2020;21:1–16. doi: 10.1186/s13059-020-02214-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafar H., Lin C., Bar-Joseph Z. Single-cell lineage tracing by integrating CRISPR-Cas9 mutations with transcriptomic data. Nat. Commun. 2020;11:1–14. doi: 10.1038/s41467-020-16821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.-R., Koido M., Tadokoro T., Ouchi R., Matsuno T., Ueno Y., Sekine K., Takebe T., Taniguchi H. Human iPSC-derived posterior gut progenitors are expandable and capable of forming gut and liver organoids. Stem Cell Reports. 2018;10:780–793. doi: 10.1016/j.stemcr.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.J., Ntranos V., Tse D. Determining sequencing depth in a single-cell RNA-seq experiment. Nat. Commun. 2020;11:1–11. doi: 10.1038/s41467-020-14482-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T., Zhang Z.-n., Westenskow P.D., Todorova D., Hu Z., Lin T., Rong Z., Kim J., He J., Wang M. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell. 2015;17:353–359. doi: 10.1016/j.stem.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Huangfu D. Human pluripotent stem cells: an emerging model in developmental biology. Development. 2013;140:705–717. doi: 10.1242/dev.086165. [DOI] [PMC free article] [PubMed] [Google Scholar]