

Summary
The recent development of single-cell multiomics analysis has enabled simultaneous detection of multiple traits at the single-cell level, providing deeper insights into cellular phenotypes and functions in diverse tissues. However, currently, it is challenging to infer the joint representations and learn relationships among multiple modalities from complex multimodal single-cell data. Here, we present scMM, a novel deep generative model-based framework for the extraction of interpretable joint representations and crossmodal generation. scMM addresses the complexity of data by leveraging a mixture-of-experts multimodal variational autoencoder. The pseudocell generation strategy of scMM compensates for the limited interpretability of deep learning models, and the proposed approach experimentally discovered multimodal regulatory programs associated with latent dimensions. Analysis of recently produced datasets validated that scMM facilitates high-resolution clustering with rich interpretability. Furthermore, we show that crossmodal generation by scMM leads to more precise prediction and data integration compared with the state-of-the-art and conventional approaches.
Keywords: single-cell multiomics, deep generative model, dimensionality reduction, multimodal data integration, multimodal prediction
Graphical abstract
Highlights
-
•
scMM learns low-dimensional joint representations from single-cell multiomics data
-
•
scMM detects previously overlooked cell populations in single-cell multimodal data
-
•
Pseudocell generation enables scMM to learn interpretable latent dimensions
-
•
scMM accurately predicts missing modalities by crossmodal generation
Motivation
Revolutionary single-cell multiomics technologies have enabled acquiring characteristics of individual cells across multiple modalities, such as transcriptome, epigenome, and surface proteins. However, computational methods for integrated analysis of complex and high-dimensional multimodal single-cell data are currently limited. Here, we present scMM, a mixture-of-experts deep generative model for integrated analysis of single-cell multiomics data. scMM effectively infers interpretable joint representations from multimodal single-cell data. In addition, scMM learns underlying relationships across modalities, enabling crossmodal generation of single-cell data.
Minoura et al. report the development of scMM, a multimodal deep generative model-based framework for analyzing single-cell multiomics data. scMM extracts biologically interpretable joint representations from high-dimensional multimodal data that can be used for downstream analyses. In addition, it learns relationships among single-cell modalities, enabling many-to-many prediction of missing modalities.
Introduction
Recent technological advances have enabled simultaneous acquisitions of multiple omics data at the resolution of a single cell, thus producing “multimodal” single-cell data (Zhu et al., 2019, 2020; Cao et al., 2018; Chen et al., 2019; Ma et al., 2020). These technologies offer additional measurements, such as immunophenotypes or chromatin accessibility in conjunction with transcriptome information. Research studies using emerging multimodal single-cell technologies have contributed to exciting, biologically important discoveries in various fields, including immune cell profile and cell fate decision, which could not have been elucidated with the use of only one modality (Hao et al., 2021; Ma et al., 2020).
Conversely, some obstacles need to be overcome to computationally extract useful knowledge from highly complex, single-cell multimodal data. First, it is challenging to infer the low-dimensional joint representations from multiple modalities that can be used for downstream analyses such as clustering. Second, although multimodal single-cell data allow the learning of relationships among modalities that could be used to train prediction models, many-to-many predictions of single-cell data (e.g., from single-cell transcriptome to chromatin accessibility) with high accuracy remains an unsolved problem. These problems are mainly attributed to the difficulties associated with capturing latent common factors and relationships across modalities, which differ significantly in characteristics, including data distribution, dimensionality, and sparsity.
Several existing methods have been recently developed for the analysis of single-cell multimodal data. Although they aim to address tasks such as latent feature extraction, their performance is currently limited in different aspects. Methods based on generalized linear models, such as Seurat and scAI, often fail to capture complex structures in single-cell data (Hao et al., 2021; Jin et al., 2020). One powerful approach to capture nonlinear latent structures is the use of expressive variational autoencoders (VAEs), which consist of a pair of neural networks wherein one encodes data into the latent space, and the other decodes them to reconstruct the data distribution (Kingma and Welling., 2013; Lopez et al., 2018). scMVAE and totalVI are the currently available VAE-based methods for single-cell multimodal data analysis (Zuo and Chen, 2020; Gayoso et al., 2021). Nevertheless, scMVAE requires a simplified conversion of chromatin accessibility to the transcriptome before training, which is known to lead to the non-negligible loss of epigenetic information (Jin et al., 2020). In addition, these models suffer from the “black-box” nature of deep learning models, making the interpretations of latent variables difficult. Finally, none of these VAE-based methods were designed for predictions across modalities.
To address these limitations, we have developed scMM, a novel statistical framework for single-cell multiomics analysis specialized for interpretable joint representation inference and predictions across modalities. scMM is based on a mixture-of-experts (MoE) multimodal deep generative model and achieves end-to-end learning by modeling raw count data in each modality based on different probability distributions (Shi et al., 2019). Using recently published datasets produced by cellular indexing of transcriptomes and epitopes with sequencing (CITE-seq) and a simultaneous high-throughput assay for transposase-accessible chromatin and RNA expression with sequencing (SHARE-seq), we demonstrate that scMM effectively extracts biologically meaningful latent variables encoding multimodal information. We show that these latent variables enable high-resolution clustering to reveal cellular heterogeneity that was not discovered in the original report (Hao et al., 2021; Ma et al., 2020). By leveraging the generative nature of the model, scMM provides users with multimodal “regulatory programs” that are associated with latent dimensions, thus aiding the interpretation of the results. Finally, exploration of the crossmodal generation of single-cell data by scMM demonstrated that it outperformed the state-of-the-art prediction tool and contributes to more accurate integration of single-cell data from different modalities.
Results
The scMM model
scMM takes multimodal single-cell data as input, which contains measurements for multiple modalities across each cell. Let xn,m be the feature vector for the mth modality in cell n. Theoretically, m can be any arbitrary number, although this study primarily focuses on the dual-omics analysis because most recently developed multiomics methods deal with information of two modalities. We modeled xn,m with probability distributions capturing the characteristics of data distributions for each modality. For transcriptome and surface protein data, a negative binomial (NB) distribution was selected to explain non-negative counts with overdispersion (Gayoso et al., 2021). In addition, chromatin accessibility data are non-negative count data; however, this exhibits extreme sparsity due to poor signal (only two loci exist for each diploid cell), limited coverage, and closed chromatin. Therefore, we chose the zero-inflated negative binomial (ZINB) distribution for chromatin accessibility data. Although transcriptome data also show high sparsity, recent reports showed that NB distribution is sufficient to explain the abundance of zeros in the transcriptome data (Svensson, 2020; Grønbech et al., 2020). In contrast to some recently developed probabilistic models for chromatin accessibility involving binarization, scMM models raw peak counts, allowing the natural increase of peak counts with sequencing depth (Xiong et al., 2019; González-Blas et al., 2019).
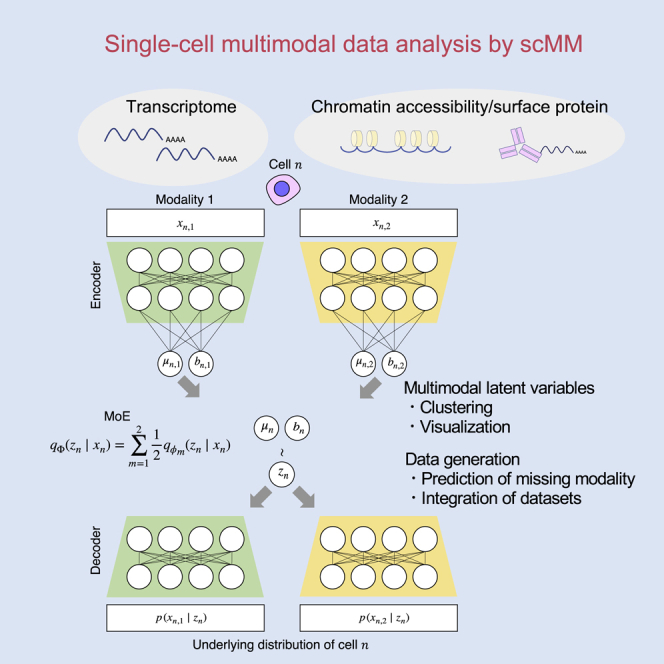
A conceptual view of scMM is shown in Figure 1. The scMM model for dual-omics analysis consists of four neural networks in which an encoder-decoder pair is present in each modality. Let z be the set of low-dimensional vectors of latent variables (here, set to ten dimensions). Encoders are used to infer the variational posterior , from which zm is sampled. Conversely, decoders calculate the parameters of NB or ZINB distributions, which can be written as . Herein, and denote the parameters for the encoder and decoder for the mth modality, respectively. The scMM uses an MoE to factorize the joint variational posterior (see the STAR Methods). Accordingly, multimodal latent variables encoding information on two modalities can be obtained from MoE: .
Figure 1.

Conceptual view of single-cell multimodal data analysis by scMM
(A) The underlying model of scMM. scMM takes feature vectors of each modality as input to separate encoders. The model is then trained to learn low-dimensional joint variational posterior factorized by an MoE. Decoders reconstruct the underlying probability distributions for data in each modality from latent variables. During the training processes, latent variables from one modality are used to reconstruct data in both modalities.
(B) Schematic view of procedure for finding latent dimension-associated multimodal features by independently traversing each dimension.
(C) Schematic view of crossmodal generation by trained scMM model when one modality is missing. For further details, see the STAR Methods section.
The ability to determine which features in each modality are associated with each latent dimension is beneficial in terms of the interpretability of the output of the model. One of the downsides of deep generative models is the difficulty of interpreting latent variables compared with linear models, such as principal-component analysis (Svensson et al., 2020). We overcame this limitation by using the generative nature of VAE (Figure 1B) (see the STAR Methods). By sequentially generating pseudocells from different latent values in one dimension with remaining fixed values, we calculated the Spearman correlation for each latent dimension and set of features in each modality. This enabled the visualization of strongly associated features with each latent dimensions, which can be interpreted as multimodal regulatory programs governing them.
A unique learning procedure of scMM involves the training of encoders to infer latent variables that can reconstruct the probability distributions not only for their own modalities but also for others, as it learns to maximize the expectation (see STAR Methods). Therefore, the trained scMM model can generate data associated with the missing modality from unimodal single-cell data in both directions, thus achieving crossmodal generation (Figure 1C). Notably, unlike conventional prediction methods, crossmodal generation by scMM can be performed in both directions across modalities.
scMM extracts biologically meaningful latent variables from multimodal data on single-cell transcriptomes and surface proteins
To validate the performance of scMM in the joint analysis of multimodal single-cell data, we applied our proposed method to a recently published CITE-seq dataset of peripheral blood mononuclear cells (PBMCs) from vaccinated patients, which consisted of the transcriptome and 224 surface protein measurements for over 160,000 cells (Hao et al., 2021). In total, 80% of the cells were randomly selected as training data, and the remaining 20% were used as testing data. After training the model, all cells were mapped in the latent space and clustering on latent variables was performed with PhenoGraph (Levine et al., 2015). Latent variables for each modality and multimodal latent variables were visualized with UMAP (McInnes et al., 2018) (Figures 2A–2C). To eliminate the possibility of overfitting, we confirmed that the training and testing datasets were embedded in shared latent space (Figure S1). Clustering by PhenoGraph discovered 54 cell populations that can be matched with known cell populations (Figure 2C). Abbreviations for cell types were assigned as shown parenthetically in the following list of types considered: CD4-positive T cell (CD4 T), CD8-positive T cells (CD8 T), gamma-delta T cells, double-negative T cells, mucosal-associated invariant T cells, B cells, natural killer (NK) cells, CD14-positive monocytes (CD14 Mono), CD16 Mono, classical dendritic cell 1 (cDC1), cDC2, plasmacytoid dendritic cells, hematopoietic stem and progenitor cells (HSPCs), and erythrocytes. Interestingly, compared with the weighted nearest neighbor (WNN) analysis by Seurat, latent variables inferred by scMM separated CD4 and CD8 T into two distinct subgroups (Figure 1D) (Hao et al., 2021). We found that these subgroups have differential expressions of surface proteins that are known to be associated with T cell activation, such as CD30, CD275, and Podoplanin (Figures 1D and S2A). Furthermore, scMM discovered a clear heterogeneity in CD14 Mono populations that was not revealed by Seurat (Figure S2B). The superior performance of the proposed model might be attributed to the rich expressive power of the neural networks used in scMM, whereas the WNN analysis was based on a linear model with limited expressive power. It was thus unable to capture complex structures in single-cell multimodal data.
Figure 2.

scMM analysis on the PBMC CITE-seq dataset
(A–C) UMAP visualization of unimodal latent variables for transcriptome, surface protein, and multimodal latent variables, respectively. Each dot represents a single cell and is color coded according to the clustering performed on multimodal latent variables.
(D) CD8 and CD4 T cells annotated in scMM and Seurat analysis are color coded in the UMAP visualization.
(E) Top: multimodal UMAP visualization colored according to the latent dimension values. Shown in the middle and on the bottom is a UMAP visualization colored according to the transcriptome and protein counts for cell type markers, respectively.
(F) Genes (left) and surface proteins (right) associated with latent dimension 1. Each feature was aligned on the basis of the Spearman correlation coefficient. The y axis represents the absolute correlation coefficient, and red and blue represent positive and negative correlations, respectively.
(G) NB mean parameters reconstructed from surface protein or transcriptome counts were plotted against original surface protein counts for each cell. The mean parameters were plotted for all available proteins. Pearson correlation coefficients are shown in the plots.
(H) Heatmap constructed from the original (left), unimodal generation (middle), and crossmodal generations. Rows and columns represent the measured 224 surface proteins and 54 clusters discovered by PhenoGraph, respectively.
Next, we compared the performance of our model on dimensionality reduction against totalVI, which is also a VAE-based method that can directly analyze multimodal data comprising transcriptome and surface protein data. scMM and totalVI showed similar evidence lower bound values (Figure S3A). In addition, modularity scores were calculated by PhenoGraph, which is indicative of how well cells were grouped in the latent space, and the number of clusters detected was also similar in both models (Figure S3A). However, a population of NK cells was merged to CD8 T population in the UMAP projection of latent variables inferred by totalVI, whereas they were clearly separated in the scMM result (Figure S3B). To compare how well latent variables inferred by scMM and totalVI preserved structures in the original transcriptome and surface protein space, we calculated the Jaccard index (JI) (Sun et al., 2019). A higher JI indicates that neighborhoods in the original spaces were better preserved in the latent space. The results showed that, although the performances were similar for transcriptome modality, the neighborhood structures were better preserved in scMM compared with totalVI for surface protein modality (Figure S3C). Together, our results suggest that dimensionality reduction performance of scMM is comparable with that of totalVI. It is worth noting that, as discussed below, scMM is implemented with a method to support latent dimension interpretation and also capable of crossmodal generation of missing modalities, both of which are unavailable in totalVI.
scMM supports result interpretation by providing multimodal features associated with latent dimensions
scMM uses a Laplace prior with different scale values in each dimension, which encourages disentanglement of information by learning axis-aligned representations (Shi et al., 2019) (see STAR Methods). Visualizing values of latent variables revealed similar patterns with canonical gene and surface protein markers (Figure 2E). This might indicate axis-aligned encoding of information related to certain cell types. For example, low values in latent dimension 1 were concentrated to B cell and plasmablast clusters (7, 17, 31, and 44), as indicated by the expression of the CD19 gene and protein. This might suggest that latent dimension 1 encodes information on cellular characteristics of B cells and plasmablasts. Figure 2E shows multimodal features strongly associated with latent dimension 1. Genes showing negative correlations include those related to immunoglobulin (Iglv1-44), chemotaxis (Cxcr5), and membrane trafficking (Rab30), and their expressions in B cell and plasmablast clusters were confirmed (Figures 2E and S4A). Surface proteins are well known to be associated with the B cell and plasmablast phenotypes, such as CD19, CD20, and CD22, which were also detected in negatively correlated features (Figures 2E and S4B). Collectively, these results validate the utility of interpretable latent representations learned by scMM.
Crossmodal generation by scMM accurately predicts surface protein measurements from transcriptome data
A trained scMM model can generate surface protein measurements conditioned on transcriptome observations (and vice versa, although transcriptome counts are generally acquired simultaneously when measuring surface proteins with sequencing technologies) through crossmodal generation. Using held-out test data, estimates of mean parameters for NB distributions were plotted against original surface protein counts, which exhibited a high correlation not only in transcriptome-to-transcriptome but also in transcriptome-to-protein crossmodal estimation. Through these NB distributions, surface protein measurements were sampled for each cell, and heatmaps were generated for 54 clusters (Figure 2H). Thus, the heatmap of transcriptome data showed a high resemblance to that of the original, confirming the performance of crossmodal generative data in scMM.
This feature of scMM can be used to predict surface protein measurements from unimodal single-cell datasets, which comprise transcriptome information only. We validated the performance of scMM by predicting surface protein abundance by using data from different experimental batches. To compare predicted versus ground-truth data, we chose bone marrow mononuclear (BMNC) CITE-seq data, containing approximately 30,000 cells with transcriptome and 25 with surface protein information (Stuart et al., 2019). With scMM trained with the PBMC training data, latent variables were obtained from transcriptome measurements of BMNC data and visualized by using UMAP (Figure 3A). Therefore, BMNC data were successfully embedded in the latent space learned from the PBMC training data. Notably, scMM correctly illustrated the enrichment of CD34-positive HSPCs in the bone marrow, where this population is scarce in peripheral blood (Figures 3B and S5A). In addition, it is noteworthy that scMM embedded CD8 and CD4 T cells in BMNC datasets with activated, CD30-positive T cell subsets found in the PBMC dataset (Figures S5B–S5E). This finding is reasonable given that CD30 marks memory T cells, and they reside mainly in the bone marrow (Rosa and Pabst, 2005; Kennedy et al., 2006).
Figure 3.

Prediction of surface protein measurements for the BMNC dataset
(A) Joint UMAP embedding of transcriptome latent variables inferred from PBMC training data and the BMNC dataset. The black arrowhead indicates the HSPC population.
(B) UMAP embedding of the BMNC dataset was colored according to the protein expression levels of CD34.
(C) Heatmaps constructed from original (left) and predicted (right) surface protein counts. Rows and columns represent 24 shared surface protein markers and clusters discovered by PhenoGraph, respectively.
(D) Benchmarking on surface protein prediction performance of scMM against Seurat. Centered log-transformed data were used to calculate the sum of squared error (SSE) per cell and plotted for each prediction result. Statistical analysis was performed with the two-sided Wilcoxon signed-rank test.
(E) Heatmap constructed with the predicted 224 surface protein markers. Rows and columns represent surface protein markers and clusters, respectively. Black arrowheads denote B cell clusters, and their markers are indicated.
Subsequently, crossmodal data generation was performed by sampling from NB distributions for surface proteins. Out of 25 surface proteins analyzed in the BMNC dataset, 24 were shared with the PBMC dataset. For 19 clusters discovered by PhenoGraph clustering, expression levels of the shared surface proteins were visualized by using a heatmap. The result shows that surface protein data generated by scMM captured the characteristics of the original data well (Figure 3C). We benchmarked the prediction accuracy of scMM against Seurat, which is currently the state-of-the-art method to predict surface proteins from single-cell transcriptomes (Stuart et al., 2019). For comparison, we trained scMM and Seuratby using the PBMC training data and used them to predict surface proteins of the BMNC dataset. The sum of squared error per cell indicated that the proposed method was more accurate than Seurat in predicting surface proteins (Figure 3D). The higher variation in scMM might be attributed to stochastic sampling processes. Figure 3E illustrates all 224 surface proteins predicted by scMM. Notably, prediction by scMM recovered crucial features of cell populations. For instance, B cell clusters (cluster 2 and 14) characterized by CD19 expression in the original data were predicted to have high expression levels of known B cell markers that were missing from the original data (CD72, CD73, CD22, CD20, CD21, CD24, IgD, and IgM) (Figure 3E).
scMM analysis of single-cell transcriptome and chromatin accessibility multimodal data
Next, we applied scMM to recently reported mouse skin single-cell transcriptome and chromatin accessibility multimodal data obtained by SHARE-seq (Ma et al., 2020). As described above, training and testing data were obtained by an 80%/20% random split. Latent variables for transcriptome and chromatin accessibility, and multimodal latent variables were visualized by UMAP (Figures 4A–4C). In addition, embeddings of training and testing datasets into a shared latent space were confirmed (Figure S1B). PhenoGraph clustering on multimodal latent variables showed clusters corresponding to known cell types present in epidermis and hair follicles (Joost et al., 2020). As above, abbreviations for cell types are given parenthetically as follows: cycling interfollicular epidermis (IFE C), basal IFE, suprabasal IFE, upper hair follicle, sebaceous gland, outer bulge, outer root sheath, companion layer, germinative layer (GL), inner root sheath (IRS), cortex/cuticle, medulla (MED), fibroblast, dermal sheath, dermal papilla, macrophage, endothelial cell, vascular smooth muscle, melanocyte. Visualization of latent variables per dimension revealed similar patterns with certain gene expression levels, thus indicating axis-aligned encoding of information associated with cell types (Figure 4D). For example, latent dimension 9 seemed to correlate positively with the DNA topoisomerase gene Top2a expression levels. Top2a is a marker for cells entering mitosis and is upregulated in proliferative keratinocyte subsets including IFE C, GL, MED, and IRS. By sequentially generating pseudocells while independently traversing latent dimensions, we found genes and peaks strongly associated with the latent dimension 9 (Figure 4E). Consistent with the cell annotations, genes closely related to the cell cycle, such as Stil, Brca1, and Cdca2, were found in positively associated features. We then sought motif enrichment in detected peaks to reveal latent dimension-associated motifs. Motif enrichment analysis discovered significantly enriched motifs, including FOS:JUNB (p = 6.08 × 10−21), TP63 (p = 2.86 × 10−13), POUF3F3 (p = 1.16 × 10−11), and MEOX2 (p = 1.39 × 10−4) (Figure 4F). By visualizing expression levels and motif scores, we confirmed enrichment of associated genes and motifs in proliferative keratinocyte subsets (Figures S6A and S6B).
Figure 4.

scMM analysis on mouse skin SHARE-seq dataset
(A–C) UMAP visualization of unimodal latent variables for transcriptome, chromatin accessibility, and multimodal latent variables. Each dot represents a single cell and is color coded according to the clustering performed on multimodal latent variables.
(D) Top: multimodal UMAP visualization color coded on the basis of latent dimension values. Shown in the middle and on the bottom is a UMAP visualization colored according to transcriptome counts for cell type markers.
(E) Genes (left) and peaks (right) associated with latent dimension 9. Each feature was aligned on the basis of Spearman correlation coefficient. The y axis represents the absolute correlation coefficient, where red and blue colors represent positive and negative correlations, respectively.
(F) Motif plot for representative motifs significantly enriched in peaks positively associated with latent dimension 9.
Crossmodal generation of transcriptome measurements contribute to accurate data integration
The prediction of chromatin accessibility from the transcriptome and vice versa is a more difficult task than the prediction of the surface proteins, because of their high dimensionality and sparsity. Specifically, there are only limited methods available for the prediction of chromatin accessibility. A conventional method for predicting the transcriptome from chromatin accessibility is performed by merely summing peak counts within the +2 kb upstream of the gene transcription start site (TSS), which returns the “gene activity matrix (GAM)” (Stuart et al., 2019). Although GAM corresponds with the transcriptome status of cells to some extent, it is associated with an inevitable loss of information because it ignores the distant interaction of enhancers and TSSs (Jin et al., 2020).
Regarding the current limitations associated with the prediction of transcriptome and chromatin accessibility from one information set to another, we sought to achieve crossmodal generation by scMM in these modalities. The plot of the estimated means of the NB parameters against the original transcriptome counts showed high correlations in both transcriptome-to-transcriptome and accessibility-to-transcriptome reconstruction (Figure 5A). Figure 5B shows heatmaps for 25 clusters on 1,126 statistically significant deferentially expressed genes. The heatmap for crossmodal generation showed similar patterns to those of the original transcriptome data, suggesting that generated data captured the characteristics of the original clusters well.
Figure 5.

Crossmodal generation from chromatin accessibility to transcriptome leads to better data integration
(A) NB mean parameters reconstructed from transcriptome or chromatin accessibility counts are plotted against original transcriptome counts for each cell. Pearson correlation coefficients are shown in the plots.
(B) Heatmaps constructed from original, unimodal generation, and crossmodal generation transcriptome data. Rows and columns represent deferentially expressed (DE) genes and clusters discovered by PhenoGraph, respectively.
(C) Joint visualization of original and predicted single-cell transcriptome data integrated by LIGER and Seurat. For the prediction, either scMM or GAM was used.
(D) Boxplot showing entropy of batch mixing for each integration. Statistical test performed with two-sided Wilcoxon rank-sum test. ∗p < 2.2 × 10−22.
Integration of single-cell data from different modalities is among the most important goals of modern computational biology. Recently developed single-cell integration tools, including LIGER and Seurat, require the conversion of chromatin accessibility to transcriptome by creating GAM to perform integration (Stuart et al., 2019; Welch et al., 2019). Recent research using single-cell multimodal data as ground-truth has reported that this approach often fails to identify corresponding cells correctly (Jin et al., 2020). We reasoned that the use of crossmodal generated transcriptome data by scMM might lead to more accurate integration, as it considers all chromatin sites upon prediction. First, we obtained predicted transcriptome measurements for each cell in test data by either crossmodal generation of scMM or by the construction of GAM. Then, we integrated predicted and original single-cell transcriptome data into space shared by LIGER and Seurat. Quantitative evaluation of the integration was performed by calculating the entropy of batch mixing, which is a measurement of how well samples from two batches are integrated, where a higher entropy indicates better integration (Haghverdi et al., 2018). With both LIGER and Seurat, integration of cells generated by scMM resulted in better embedding with original cells compared with those performed with GAM (Figures 5C and 5D). Collectively, these results suggest that scMM has a promising potential in generating transcriptome data that precisely reflect chromatin accessibility, and significantly contributes to single-cell integration analysis when used in combination with existing methods.
scMM achieves chromatin accessibility prediction
Next, we investigated the prediction of chromatin accessibility from the transcriptome. In contrast to the transcriptome measurements, where counts continue to increase with the abundance of mRNA in each cell, there are theoretically only two states of chromatin accessibility: open or closed. Therefore, larger peak counts only reflect sequences with favorable Tn5 binding, or they are just random events. Thus, the prediction model is required to discriminate zero and nonzero values, rather than predict the absolute counts. Figure 6A shows the estimated mean parameters of the ZINB distribution for held-out test datasets against the original peak counts. Generally, estimated mean parameters were lower than the original peak counts, which might reflect the low detection rate of open chromatin. Of note, for the peaks with zeros of the original count, the mean ZINB parameter concentrated at zero, thus indicating that scMM accurately captured the closed chromatin (Figure 6A). Unimodal and crossmodal generation of chromatin accessibility measurements for test datasets was performed by sampling the estimated ZINB distributions. Interestingly, 4,018 statistically significant deferentially accessible heatmap peaks showed high similarities for crossmodal generation and original data (Figure 6B). In addition, Motif scores in original clusters were accurately recovered by crossmodal generation (Figure 6C). To investigate the crossmodal generated chromatin accessibility data, we analyzed coverage peaks in the Lef1 and Krt1 gene regions, which are essential markers for anagen hair follicle keratinocytes and permanent epidermis keratinocytes, respectively. Coverage plots showed chromatin accessibility data generated by scMM reconstructed peaks specifically detected in the keratinocyte subsets, further confirming the crossmodal data generation performance of scMM (Figure 6D). Notably, crossmodal generation by sampling from predicted ZINB distributions allows the formulation of sparse representations of high-dimensional chromatin accessibility data that are memory efficient compared with dense representations. For 6,955 cells in the test dataset, the memory sizes of the two representations were 504 MB for sparse representations (sampled peak counts) versus 10.6 GB for dense representations (ZINB mean parameters).
Figure 6.

scMM accurately predicts chromatin accessibility from transcriptome data
(A) ZINB mean parameters reconstructed from transcriptome or chromatin accessibility counts are plotted against original chromatin accessibility counts for each cell. Twenty-five million data points were randomly selected for plotting.
(B) Heatmaps constructed from original, unimodal generation, and crossmodal generation chromatin accessibility data. Rows and columns represent deferentially accessible (DA) peaks and clusters discovered by PhenoGraph, respectively.
(C) Heatmaps of motif scores for original and crossmodal generation data. Rows and columns represent motif scores and clusters, respectively.
(D) Coverage plots for representative clusters within regions of Lef1 and Krt1.
Discussion
The rapidly evolving field of single-cell multimodal analysis requires the development of methods for the joint analysis of the obtained data. scMM was designed to meet this demand. In this study, we have shown that scMM extracts low-dimensional latent variables from multimodal single-cell data that are useful for downstream analysis, such as clustering.
We have also shown that scMM was able to identify cell populations that were difficult to detect by existing methods. The expressive deep generative model captured complex, nonlinear structures that could not be captured by the linear model used in Seurat. The improved performance of scMM compared with totalVI might be attributed to the incorporation of the MoE posterior. scMM estimates posterior distributions independently for each modality and then mixes them equally by MoE. In contrast, totalVI estimates the single posterior by taking both modalities as input. This might lead to the undesirable dominance of a certain modality that can mask information in the other modality.
In addition, we leveraged the data generative nature of scMM to compensate for the difficulties in interpreting deep generative models. Exploring the multimodal regulatory programs associated with each latent dimension is expected to provide deeper insights into the clusters discovered in scMM analyses.
Furthermore, the experimental results show that crossmodal generation with scMM achieved accurate prediction of measurements in different modalities. In addition, these predictions can be used for integrating multiple unimodal datasets. Benchmarking of scMM against the state-of-the-art prediction tool and conventional integration approaches demonstrated the superiority of scMM on these tasks. These features of scMM will lead to the effective utilization of accumulating unimodal single-cell databases that are annotated and well characterized.
One of the strengths of scMM is its extensibility, as it can be applied to any modality by constructing the model with different distributions. In addition to the modalities considered in this study, applications to other multimodal data, such as the single-cell transcriptome and DNA methylome, are promising directions for future research (Hu et al., 2016). Extending scMM to several multimodal single-cell data might decipher novel cellular states or functions regarding transcriptomes, epigenomes, and proteomes. Application to single-cell data with spatial information would be an exciting research question because, in contrast to other modalities, coordinates in spatial data are meaningful only in the context of positional relationships with other cells (Rodriques et al., 2019). In essence, we expect that the proposed model will establish a foundation for deep generative models for multimodal single-cell data from the scope of interpretable latent feature extraction and crossmodal generation.
Limitations of study
A limitation of scMM is that it might be challenging to generate cell populations that were not present in data used to train the model. It is expected that, as the construction of large-scale multimodal single-cell atlas progresses, more training data will become available, which would mitigate the problem (Bakken et al., 2020).
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Teppei Shimamura (shimamura@med.nagoya-u.ac.jp)
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data used in this study is publicly available. PBMC and BMNC CITE-seq data are available at the official website of Seurat https://satijalab.org/seurat/. Mouse skin SHARE-seq data is available under the NCBI GEO accession number GSE140203.
The scMM model was implemented with Python using PyTorch deep learning library, and code is available at https://github.com/kodaim1115/scMM. All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Variational autoencoder model
Variational autoencoder (VAE) models are deep generative neural networks widely used to perform dimensionality reduction and data generation. Let x be data and z be the set of low-dimensional latent variables. VAE consists of a pair of encoder and decoder neural networks, which parametrize and , respectively. Here, is a variational posterior that approximates true posterior , which is intractable. Additionally, is a likelihood of the data given a sample from the variational posterior. In the VAE objective function, maximization of the marginal likelihood p(x) is approximated by maximizing the ELBO, which can be written with a reconstruction term and Kullback-Leibeler (KL) divergence regularization term:
| (Equation 1) |
| (Equation 2) |
where p(z) is a prior. After optimization, an encoder learns a non-linear embedding of data into a low-dimensional latent space and a decoder learns generation of data from a given low-dimensional representations.
Mixture-of-experts multimodal VAE model for single-cell multiomics data
This section describes MoE multimodal VAE (MMVAE), which scMM is based on (Shi et al., 2019). Suppose there is single-cell multimodal dataset x1:M. For current multimodal single-cell data, M = 2 in general. MMVAE aims to learn a multimodal generative model (m = 1, …,M), where represents the likelihood for each of mth modality parameterized by the decoder's deep neural network. The training objective is to maximize the marginal likelihood , which is approximated by optimizing the ELBO by stochastic gradient descent (SGD). As shown in Equation 1, formulation of the ELBO requires approximation of the true posterior by joint variational posterior .
| (Equation 3) |
The key idea of MMVAE is to factorize the joint variational posterior with a MoE; , αm = 1/M, where is the variational posterior for the m-th modality parameterized by the encoder deep neural network. Using stratified sampling (Robert and Casella, 2004), the ELBO can be formulated as:
| (Equation 4) |
| (Equation 5) |
Intuitively, the first term could be interpreted as the goodness of reconstruction for all M modalities from latent variables for the mth modality. Note that data for all modalities are reconstructed from latent variables for each modality, which enables cross-modal generation of data. The second term regularizes the model so that variational posterior follows the prior distribution.
For the prior p(z), we used the Laplacian distribution with a zero mean and a scaling constraint (, wherein bd is a scale parameter for the dth dimension, and D is the number of latent dimensions) (Shi et al., 2019). Scale parameters for prior were learned from data through SGD.
Likelihoods were selected according to the data distribution characteristics of each single-cell modality. An NB distribution was used for transcriptome and surface protein counts, and ZINB distribution was used for chromatin accessibility peak counts.
For all modalities, row counts were normalized by dividing with the sequencing depth of each cell, multiplying with the scale factor (10,000) and used as input to encoders. Mean parameters estimated by decoders were processed with reverse processes, and log likelihoods were calculated with raw count data.
Model architecture and optimization
Optimization was performed with an Adam optimizer with AMSGrad (Reddi et al., 2019). Hyperparameter optimization was performed by Optuna (Akiba et al., 2019). For CITE-seq data, three hidden layers with 200 hidden units were used for both modalities. For SHARE-seq data, three hidden layers with 500 units for transcriptome and 100 hidden units for chromatin accessibility were used. Learning rates were set to 2 × 10−3 and 1 × 10−4 for CITE-seq and SHARE-seq data, respectively. Minibatch sizes of 128 and 64 were used for CITE-seq and SHARE-seq data, respectively. We used a deterministic warm-up learning scheme for 25 and 50 epochs, with maximum of 50 and 100 epochs for CITE-seq and SHARE-seq data, respectively (Sønderby et al., 2016). After deterministic warm-up, early stopping with a tolerance of 10 epochs was applied. We observed that minor changes in hyperparameters did not significantly affect the analyzed results.
Data preproccessing
For transcriptome count data, 5000 most variable genes were first selected by applying the Seurat FindMostVariable function to log-normalized counts. Raw counts were used for model input. For chromatin accessibility data, the top 25% peaks were selected for input using Seurat's FindTopFeatures function. No preprocessing and feature selection were performed on surface protein count data.
Cluster analysis
Clustering was performed with the R package Rphenograph with nearest neighbor numbers k = 20 and k = 15 for human PBMC/BMNC CITE-seq data and mouse skin SHARE-seq data, respectively.
For CITE-seq data, cell types were manually annotated with known surface protein markers and by referring to the original report (Hao et al., 2021). For SHARE-seq data, manual annotations were performed with the mouse skin single-cell data portal http://kasperlab.org/mouseskin.
Heatmaps were generated with the R package pheatmap. For gene, surface protein, and chromatin accessibility, z-scores for total feature counts normalized by the total sequencing depth per cluster were used to generate heatmaps. For motif scores, z-scores for median values were used.
Visualization of latent representations
Mean parameters for variational posteriors in each modality and MoE were used as latent variables. Latent variables obtained from trained models were visualized on the two-dimensional space using the “umap” package in R.
Detection of latent dimension-associated features
We generated series of pseudocells by independently traversing latent dimensions. This approach was inspired by the study on the original MMVAE paper (Shi et al., 2019). Using the learned standard deviation for the dth dimension σd, with other dimensions fixed to zero, we linearly changed the dth dimension from −5σd to 5σd at a rate of 0.5σd. The obtained latent vectors were then decoded for M modalities and resulted in 20 pseudocells. Spearman's correlation coefficients were calculated for traversed latent dimensions and features in each modality. Latent dimension-associated features were selected using p value thresholds that produced a reasonable number of associated features, namely, p < 1 × 10−12, p < 1 × 10−3, and p < 1 × 10−21 for genes, proteins, and peaks, respectively.
Motif analysis
Motifs enriched in latent dimension-associated peaks were obtained by the FindMotifs function in Signac (Stuart et al., 2020). Motif scores were calculated using the chromVAR wrapper function RunChromVAR in Signac (Schep et al., 2017).
Benchmarking of scMM
Surface protein prediction by Seurat was performed following the official tutorial. Specifically, anchors between PBMC training data and BMNC data were calculated, and prediction was performed with the MapQuery function. Given that Seurat returns the centered log-transformed measurements, the predicted results of scMM were also transformed to compare SSEs per cell.
For JI calculation, 10,000 cells were randomly chosen and k nearest neighbors in the original feature space (set A) and the latent space (set B) were identified. We tested k = 10, 20, and 30. JI was calculated as the cardinality of neighbor sets: . JI for sampled cells were averaged to obtain mean JI. This process was repeated 20 times with different randomly chosen cells.
GAM was generated by the GeneActivity function of Signac. Cross-modal transcriptome reconstruction by scMM and GAM were integrated with original data by LIGER and Seurat following official tutorials with default parameters. The entropy of batch mixing was calculated as described in a previous study (Haghverdi et al., 2018). Briefly, for 100 randomly chosen cells, their 100 nearest neighbors were used to calculate the batch proportion xi, where x1+x2 = 1. Regional entropy was estimated according to E = x1logx1+x2logx2, and entropy of batch mixing was calculated as their sum. For boxplot, this process was repeated 100 times with different randomly chosen cells.
Quantification and statistical analysis
The function findMarkers of the R package scran was applied on log-normalized gene counts to detect DE genes (p value ¡ 0.05, FDR ¡ 0.1). For detection of DA peaks, the function FindMarkers in the R package Signac was used with a logistic regression mode (p value ¡ 0.05, log2-fold change ¿ 0.5). Wilcoxon signed-rank test and sum rank test were performed with the function wilcox.exact in the R package exactRankTests.
Acknowledgments
This research was supported by JSPS Grant-in-Aid for Scientific Research (grant nos. 20H04841, 20H04281, and 20K21832). It was also supported by the Japan Agency for Medical Research and Development (AMED) (grant no. JP20ek0109488 and JP21wm0425007), and the Japan Science and Technology Agency (JST) (Moonshot R&D, grant no. JPMJMS2025). The computational resource of AI Bridging Cloud Infrastructure (ABCI) was provided by the National Institute of Advanced Industrial Science and Technology (AIST), and SHIROKANE by the Human Genome Center, the University of Tokyo.
Author contributions
K.M. designed and performed the experiments under supervision of H.N. and T.S. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 15, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100071.
Supplemental information
References
- Akiba T., Sano S., Yanase T., Ohta T., Koyama M. Optuna: a next-generation hyperparameter optimization framework. arXiv. 2019:1907.10902. [Google Scholar]
- Bakken T.E., Jorstad N.L., Hu Q., Lake B.B., Tian W., Kalmbach B.E., Crow M., Hodge R.D., Krienen F.M., Sorensen S.A., et al. Evolution of cellular diversity in primary motor cortex of human, marmoset monkey, and mouse. bioRxiv. 2020 doi: 10.1101/2020.03.31.016972. [DOI] [Google Scholar]
- Cao J., Cusanovich D.A., Ramani V., Aghamirzaie D., Pliner H.A., Hill A.J., Daza R.M., McFaline-Figueroa J.L., Packer J.S., Christiansen L., et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:eaau0730. doi: 10.1126/science.aau0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Lake B.B., Zhang K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019;37:1452–1457. doi: 10.1038/s41587-019-0290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso A., Steier Z., Lopez R., Regier J., Nazor K.L., Streets A., Yosef N. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods. 2021:1–11. doi: 10.1038/s41592-020-01050-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Blas C.B., Minnoye L., Papasokrati D., Aibar S., Hulselmans G., Christiaens V., Davie K., Wouters J., Aerts S. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods. 2019;16:1–4. doi: 10.1038/s41592-019-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grønbech C.H., Vording M.F., Timshel P.N., Sønderby C.K., Pers T.H., Winther O. scVAE: variational auto-encoders for single-cell gene expression data. Bioinformatics. 2020;36:4415–4422. doi: 10.1093/bioinformatics/btaa293. [DOI] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;84:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Huang K., An Q., Du G., Hu G., Xue J., Zhu X., Wang C.-Y., Xue Z., Fan G. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016;17:88. doi: 10.1186/s13059-016-0950-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S., Zhang L., Nie Q. scAI: an unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles. Genome Biol. 2020;21:25. doi: 10.1186/s13059-020-1932-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S., Annusver K., Jacob T., Sun X., Dalessandri T., Sivan U., Sequeira I., Sandberg R., Kasper M. The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell. 2020;26:441–457.e7. doi: 10.1016/j.stem.2020.01.012. [DOI] [PubMed] [Google Scholar]
- Kennedy M.K., Willis C.R., Armitage R.J. Deciphering CD30 ligand biology and its role in humoral immunity. Immunology. 2006;118:143–152. doi: 10.1111/j.1365-2567.2006.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Welling M. Auto-encoding variational Bayes. arXiv. 2013:1312.6114. [Google Scholar]
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Zhang B., LaFave L.M., Earl A.S., Chiang Z., Hu Y., Ding J., Brack A., Kartha V.K., Tay T., et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020;183:1103–1116.e20. doi: 10.1016/j.cell.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L., Healy J., Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv. 2018:1802.03426. [Google Scholar]
- Reddi S.J., Kale S., Kumar S. On the convergence of Adam and beyond. arXiv. 2019:1904.09237. [Google Scholar]
- Robert C.P., Casella G. Springer Texts Statistics; 2004. Monte Carlo Statistical Methods; pp. 511–543. [DOI] [Google Scholar]
- Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., Macosko E.Z. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–1467. doi: 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa F.D., Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. 2005;26:360–366. doi: 10.1016/j.it.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Siddharth N., Paige B., Torr P.H.S. Variational mixture-of-experts autoencoders for multi-modal deep generative models. arXiv. 2019:1911.03393. [Google Scholar]
- Sønderby C.K., Raiko T., Maaløe L., Sønderby S.K., Winther O. Ladder variational autoencoders. arXiv. 2016:1602.02282. [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Srivastava A., Lareau C., Satija R. Multimodal single-cell chromatin analysis with Signac. bioRxiv. 2020 doi: 10.1101/2020.11.09.373613. [DOI] [Google Scholar]
- Sun S., Zhu J., Ma Y., Zhou X., Sun S., Zhu J., Ma Y., Zhou X. Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis. Genome Biol. 2019;20:269. doi: 10.1186/s13059-019-1898-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 2020;38:147–150. doi: 10.1038/s41587-019-0379-5. [DOI] [PubMed] [Google Scholar]
- Svensson V., Gayoso A., Yosef N., Pachter L. Interpretable factor models of single-cell RNA-seq via variational autoencoders. Bioinformatics. 2020;36:3418–3421. doi: 10.1093/bioinformatics/btaa169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J.D., Kozareva V., Ferreira A., Vanderburg C., Martin C., Macosko E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177:1873–1887.e17. doi: 10.1016/j.cell.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L., Xu K., Tian K., Shao Y., Tang L., Gao G., Zhang M., Jiang T., Zhang Q.C. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat. Commun. 2019;10:4576. doi: 10.1038/s41467-019-12630-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C., Yu M., Huang H., Juric I., Abnousi A., Hu R., Lucero J., Behrens M.M., Hu M., Ren B. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol. 2019;26:1063–1070. doi: 10.1038/s41594-019-0323-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C., Preissl S., Ren B. Single-cell multimodal omics: the power of many. Nat. Methods. 2020;17:11–14. doi: 10.1038/s41592-019-0691-5. [DOI] [PubMed] [Google Scholar]
- Zuo C., Chen L. Deep-joint-learning analysis model of single cell transcriptome and open chromatin accessibility data. Brief. Bioinform. 2020;22:bbaa287. doi: 10.1093/bib/bbaa287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study is publicly available. PBMC and BMNC CITE-seq data are available at the official website of Seurat https://satijalab.org/seurat/. Mouse skin SHARE-seq data is available under the NCBI GEO accession number GSE140203.
The scMM model was implemented with Python using PyTorch deep learning library, and code is available at https://github.com/kodaim1115/scMM. All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.