

Summary
Patient-derived cell lines are often used in pre-clinical cancer research, but some cell lines are too different from tumors to be good models. Comparison of genomic and expression profiles can guide the choice of pre-clinical models, but typically not all features are equally relevant. We present TumorComparer, a computational method for comparing cellular profiles with higher weights on functional features of interest. In this pan-cancer application, we compare ∼600 cell lines and ∼8,000 tumor samples of 24 cancer types, using weights to emphasize known oncogenic alterations. We characterize the similarity of cell lines and tumors within and across cancers by using multiple datum types and rank cell lines by their inferred quality as representative models. Beyond the assessment of cell lines, the weighted similarity approach is adaptable to patient stratification in clinical trials and personalized medicine.
Keywords: cancer genomics, cell lines, weighted similarity, oncogenic alterations, TCGA, CCLP, cancer therapy, decision support, patient stratification, web application
Graphical abstract
Highlights
-
•
TumorComparer uses tumor and cell line profiles to guide experimental model choice
-
•
User-selected feature weights allow customization to specific research questions
-
•
The suitability of cell lines for modeling 24 cancer types is assessed
-
•
A user-friendly web application and R package are available
Motivation
Cancer is a genetic disease, typically marked by widespread somatic alterations (e.g., mutations, copy-number alterations, and gene expression changes). However, not all changes are functionally important—few genes can promote oncogenesis (also termed “cancer drivers”), whereas other altered genes have little effect on the phenotype (termed “passengers”). Furthermore, many research questions focus on particular genes and their activity (e.g., specific signaling pathways, drug targets, etc.). This motivates the need for a flexible method of comparing tumors with potential cell line models by using researcher-selected properties. We present TumorComparer, a computational comparison method based on weighted features to allow expert- and knowledge-driven comparison of tumors and experimental models, such as cell lines or organoids. We apply TumorComparer to the comparison of ∼8,000 tumors and ∼600 cell lines across 24 cancer types as an initial application to provide a general, pan-cancer resource based on knowledge of oncogenic alterations gained from The Cancer Genome Atlas program (TCGA). TumorComparer is a generally applicable method suitable for pre-clinical cancer research and personalized medicine applications where sets of samples need to be assessed for similarity.
Sinha et al. present TumorComparer, a flexible method to assess the similarity of cancer models and clinical samples based on multiple datum types. TumorComparer uses a weighted similarity approach to incorporate prior knowledge and investigator interest and can also be used for patient stratification and guiding treatment decisions.
Introduction
Immortalized cancer cell lines, derived from patient tumors and grown and maintained in vitro, are the most commonly used experimental model in cancer research. Cell lines preserve many properties of tumors and have been of immense value in advancing our understanding of cancer biology and developing novel therapies over the past decades (Masters, 2000; Wistuba et al., 1998, 1999). However, there are important differences, both in general and in particular tumor types, between molecular and genetic profiles of cell lines and tumors, which are the subject of this study.
Although cell lines retain many features of tumors, they also typically acquire additional alterations during the process of immortalization, and during growth and maintenance in culture. Several studies have reported differences between cell lines and tumors with respect to gene expression (Ross et al., 2000; Sandberg and Ernberg, 2005), DNA methylation (Hennessey et al., 2011; Houshdaran et al., 2010; Smiraglia, 2001), and copy-number alterations (CNAs) (Greshock et al., 2007; Tsuji et al., 2010). Such differences are important for the cancer research community to understand so we can assess the translatability of findings from cell lines to patients.
Cell lines generally have more genomic alterations than primary tumors. There are several possible explanations for this. First, there is a bias toward using cell lines derived from metastatic tumors, which tend to have more genomic alterations than primary and especially early-stage tumors (Masters, 2000). Also, cell lines typically do not represent all subtypes of cancers nor do they reflect tumor heterogeneity. In particular, tumor subtypes with the fewest genetic alterations tend to be under-represented (Kao et al., 2009; Klijn et al., 2015; van Staveren et al., 2009). Finally, mutations and CNAs might be acquired during the immortalization process, and subpopulations of cell lines with more genomic alterations might be selected for during long periods of growth and maintenance in vitro (Masters, 2000). Germline mutations are typically filtered out of primary tumor data by using matched normal samples. Matched normal samples are not available for most commercially available cell lines, so it is only possible to filter out common germline variants.
Given these differences, selecting the most suitable cell line(s) for a laboratory study becomes a technical challenge of practical interest. In general, cell lines with molecular profiles similar to tumor samples are more suitable for use than outlier cell lines that differ significantly from the corresponding tissue of origin by observations in one or more molecular or genetic profiles. However, in some cases, it might be important to consider particular features that are required for cell lines to “phenocopy” aspects of tumors, such as oncogenic mutations or alterations in signaling pathways, and focusing on these features might provide a more useful assessment of similarity (Elias et al., 2015). Thus, beyond overall genetic similarity, the choice of an appropriate cell line for a specific scientific project crucially depends on the goal and context of the study and comparison algorithms should take the investigator’s interest into account. For example, one might want to choose a cell line that is most similar to a set of tumors in terms of alterations in signaling pathways, such as protein phosphorylation cascades; or, in terms of mutations in particular pathways; or, in terms of the overall level of alterations in known oncogenic pathways.
We, therefore, developed a general method for identifying appropriate cell lines that allows investigators to adapt the selection criteria to the specific biological question at hand. A simple yet powerful approach is to incorporate feature weights into the measure of similarity of molecular profiles. For example, alterations in genes involved in a signaling process of particular relevance to drug action might get a higher weight in relation to other genes.
A very simple choice of weights on molecular and genetic features is 1.0 (chosen) and 0.0 (ignored), but a more refined choice of weights is real numbers 0.0 ≤ w ≤ 1.0. Here, as a baseline method for the selection of cell lines for experiments focused on oncogenic processes, we use sets of real number weights that emphasize potentially oncogenic genomic alterations (“driver” mutations), while de-emphasizing alterations that are likely to be “passengers” in tumors. We fine-tuned such weights by using The Cancer Genome Atlas (TCGA) tumor profiles, and then applied them to compute the weighted similarity between tumors and cell lines. We compared tumors from 24 different cancer types from TCGA (Collins and Barker, 2007) to cell lines from the Cancer COSMIC Cell Line Project (CCLP) (Iorio et al., 2016), and identified good, moderate, and poor matches between cell lines and tumors, as well as outlier cell lines, to guide cell line selection for laboratory experiments focused on oncogenic processes.
Results
TumorComparer: A weighted similarity framework for comparing cancer samples by comparison of molecular profiles
Given that the similarity of cell lines and tumors (and, in general, any tumor-derived material) can vary by gene sets, data type, and number of tumors compared, we developed TumorComparer, a tool and framework for flexible comparison of tumor-derived genomic profiles. The method uses a weighted similarity to flexibly incorporate a variable emphasis on genomic features and gene expression, according to data and/or investigator knowledge and interest.
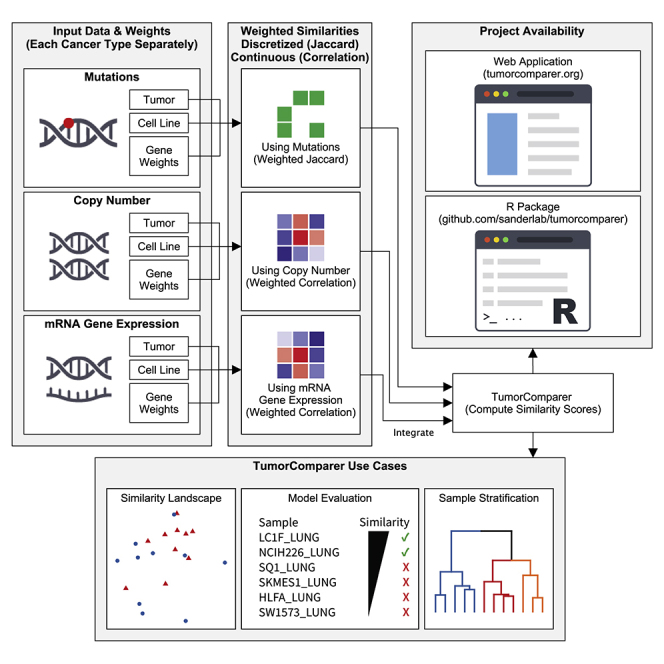
TumorComparer compares tumors and cell lines on the basis of multiple datum types, such as mutations, DNA CNAs, and mRNA gene expression, by using weights to emphasize events relevant to the specific biological question at hand, e.g., the more frequent and/or known oncogenic events (Figure 1A). The method is publicly available as an R package (https://github.com/sanderlab/tumorcomparer), and as an interactive web application (http://projects.sanderlab.org/tumorcomparer). We applied our method to compare cell lines and tumors for 24 different cancer types by using genomic data from 594 CCLP cell lines and 7,975 TCGA tumor samples (Figure 1B) and by using weights emphasizing recurrent alterations or differential expression in tumors. Weights are real numbers between 0 and 1, where a value closer to 1 indicates greater importance. Here, we use a weighting scheme that emphasizes key alterations and gene expression changes in each particular cancer with a secondary emphasis on pan-cancer alterations (see the STAR Methods). We inspected the resultant landscape of tumor model similarities and identified cell lines that best match tumors of various types, as well as poor matches and outlier cell lines.
Figure 1.

TumorComparer workflow and available tumor samples and cell lines
(A) Weighted similarity between pairs of cancer material is computed using data type-specific datum matrices and weights for each molecular data point (e.g., a mutation in a specific gene). Weights are either derived from data or provided by the user, reflecting an emphasis on particular genomic alterations. The weighted similarities for each data type are then normalized and combined into a final weighted similarity score. To compare cell lines and tumors, we used mutations, CNAs, and gene expression (mRNA) values, and chose weights for these features based on the recurrence of cancer type-specific (or pan-cancer) events in sets of tumors samples, as a proxy for the likelihood of the feature to be functional (e.g., to be “drivers”); for expression, we chose the log-fold change in expression in relation to pooled normals. Top: mutated, green; wild type, white. Middle: gains, light and dark red; losses, light and dark blue; diploid, white. Bottom: overexpressed, red; underexpressed, blue.
(B) The number of TCGA tumors and CCLP cell lines for each cancer type included in this study. CCLP, Cancer Cell Line Project; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B cell lymphoma; ESCA, esophageal adenocarcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TCGA, The Cancer Genome Atlas; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma.
To gain an overall perspective on tumor cell line similarity across cancer types, we first compared the average profiles of cell lines and tumors across cancer types (Figure 2), by using the 5,000 most variable genes for gene expression (Figure 2A) and CNAs (Figure 2B), 299 significantly mutated genes across the TCGA pan-cancer dataset for mutations (Figure 2C), and the normalized rank, which was derived by converting the similarity scores of each cancer type into ranks, and dividing by the number of cancer types to obtain a score in the [0,1] range. Although the majority of cancer types showed the highest similarity of average cell line profiles to average tumor profiles matching cancer types by using gene expression (Figure 2A), this was often not the case for CNAs and mutations (Figures 2B and 2C). However, conversion of similarity scores into ranks revealed that, even when the similarity between average profiles of the matching cancer types was not the highest, it was often the second or third highest (Figure 2D). This makes sense, especially when there are related cancer types present, e.g., lung squamous cell carcinoma (LUSC) and cervical squamous cell carcinoma and endocervical adenocarcinoma, LUSC and lung adenocarcinoma (LUAD), high-grade serous ovarian cancer (HGSOC), and a subset of uterine corpus endometrial carcinoma, and so on. Furthermore, although several cancer types have high similarity between cell lines and tumors when using all three datum types, some only have high similarity when using one or two of the three datum types. In particular, the average mutational profiles of acute myeloid leukemia (LAML) cell lines and the average CNA profiles of thyroid carcinoma (THCA), brain lower grade glioma (LGG), and mesothelioma (MESO) are not more similar to the average profiles of their respective cancer subtypes than to other cancer (sub)types.
Figure 2.

Average similarities between cell lines and tumor samples by cancer subtype and data type
(A) Top left: similarity of CCLP cell lines (rows) and TCGA tumors (columns) using the mean gene expression levels of the 5,000 most variable genes across the tumors (correlation coefficient). The similarity is highest for matching cancer (sub)type in the vast majority (18 out of 24 = 75%; dark red or orange on the diagonal) of cases.
(B) Top right: similarity of CCLP cell lines (rows) and TCGA tumors (columns) using the mean copy-number changes of the 5,000 most variable genes across the tumors (correlation coefficient). Unlike for expression, only a minority (7 out of 24 = 29%) of the closest matches are for the same cancer (sub)type.
(C) Bottom left: similarity of CCLP cell lines (rows) and TCGA tumors (columns) using the mutation frequencies of the 299 most frequently mutated genes across the tumors (correlation coefficient of average mutation frequencies). Similar to CNA, only a minority of closest matches are for the same cancer type (9 out of 24 = 37.5%). The low percentage in (B and C) indicate that CNAs and somatic mutations contain less tissue-specific information than gene expression.
(D) Bottom right: ranked similarity between CCLP cell lines (rows) and TCGA tumors of the same cancer type, based on the similarities in the rows of (A–C) above. A top rank indicates the cell lines of a certain cancer (sub)type are on average well matched to the tumors of the same (sub)type. In several cases, when the same tissue type does not provide the closest match, it might be among the top few matches, as might be expected in cases where other related tissues are present. Although several cancer types have high similarity between cell lines and tumors when using various datum types, some only have high similarity when using one or two of the three datum types.
The landscape of cell line-tumor similarity across 24 cancer types
TCGA studied 33 different cancer types and, using detailed annotation available from the Cell Model Passports resource (van der Meer et al., 2019), we report results on 594 cell lines corresponding to 24 cancer types studied in TCGA. As a first application, we surveyed cell line-tumor similarity by using weights emphasizing recurrent genomic alterations in tumors. For mutations and CNAs, we gave the highest weight to genes that are recurrently altered in the specific cancer type (MutSig SMGs, i.e., significantly [by recurrence] mutated genes [Lawrence et al., 2013]; and genes found in GISTIC peaks, i.e., recurrent CNAs [Mermel et al., 2011]); an intermediate weight to genes that are recurrently altered across many cancer types (“pan-cancer”), but not in the specific cancer type; and a lower “background” weight all other alterations. For gene expression, a gene was weighted by the log of the ratio of the mean expression level of a gene over samples of the specific cancer type in relation to the mean expression level over a pool of normal samples (“log-fold-change,” scaled to the interval 0.0–1.0; see the STAR Methods).
Weights were chosen to emphasize features of importance in tumors only (rather than tumors and cell lines), to avoid giving high weights to features that are only recurrent in cell lines or giving low weights to features that are recurrent in tumors, but not in a combined set of tumors and cell lines. This is similar in spirit to (Iorio et al., 2016), who published a comprehensive comparison of cell lines and primary tumors at the sample population level and looked at cancer cell lines under the lens of genomic features derived from primary tumors only, thus discarding features unlikely to be clinically relevant.
Exploratory analysis using all three datum types as well as overall average weighted similarity allows us to identify cell lines that have high or low similarity to tumors across datum types, as well as those that have high similarity when using only one datum type. For instance, ovarian cancer cell lines, such as OAW-28 and Kuramochi, have a high ranking across all three datum types, whereas cell lines, such as PA-1 and TYK-nu, are poor matches across multiple datum types, as assessed in terms of similarity over the most varying or recurrently altered genes; in contrast, Caov-4 has high similarity to tumors by gene expression, but not by mutations and CNAs (Figure 3). This demonstrates the usefulness of using multiple datum types for a broader perspective.
Figure 3.

The distribution of feature-weighted similarities between ovarian cancer cell lines and ovarian (HGSOC) tumors
(A–D) (A) The overall similarity represents the average of the feature-weighted similarity over the three datum types: (B) mRNA expression (with higher weights on genes with the largest expression changes); (C) mutations; and (D) copy-number alterations (with higher weights on the most recurrently altered genes in (C and D)). The distributions of feature-weighted similarities using mutations reveal striking differences between cell lines, with low, high, and intermediate similarities to tumors for three groups of cell lines. Cell lines, such as OAW-28 and Kuramochi have a high overall similarity to tumors ranking as they rank highly across all three datum types, whereas cell lines, such as PA-1 and TYK-nu, are poor matches to tumors across multiple datum types in terms of genomic similarity over the genes used in the feature-weighted similarity measure.
Similarly, the representativeness of a tumor model can vary by gene set, i.e., the specific genes or genomic alterations that form the basis of comparison with tumors. To illustrate this, we compared melanoma and liver cancer cell lines from CCLP to skin cutaneous melanoma (SKCM) and liver hepatocellular carcinoma (LIHC) tumors from TCGA, using two different gene sets—the RTK-RAS pathway and the WNT pathway. We chose these cancer types and pathways because SKCM tumors show frequent alterations in the RTK-RAS pathway but not the WNT pathway; whereas LIHC tumors show more frequent alterations in the WNT pathway than the RTK-RAS pathway. Consistent with that, we see that SKCM cell lines show similar or better similarity scores when using the RTK-RAS pathway than with the WNT pathway, whereas LIHC cell lines show lower scores with the RTK-RAS pathway than with the WNT pathway (Figure 4).
Figure 4.

The similarity of cell lines and tumors varies by gene set—the best matches might be quite different for different gene sets/pathways
The top two panels show the similarity scores of SKCM tumors and melanoma cell lines when using uniform weights on all features, and genes from (A) RTK-RAS pathway and (B) WNT pathway. Similarly, the bottom two panels (C and D) show corresponding scores for liver cancer cell lines, compared with TCGA LIHC tumors. SKCM cell lines show similar/better similarity scores when using the RTK-RAS pathway than the WNT pathway, whereas LIHC cell lines show lower scores with the RTK-RAS pathway than with the WNT pathway—this is consistent with the frequency of alterations in the member genes of the RTK-RAS and WNT pathways in these cancer types.
SKCM, COAD, READ, GBM, and KIRC have the highest proportion of good cell lines
Applying TumorComparer across 24 cancer types from TCGA and CCLP, we compared tumors from each cancer type with all cell lines and used ranks based on the similarities to compare across cancer types (Figure 5). Most cancer types have a mix of cell lines with low, intermediate, and high similarity, also called a match, to tumors. A normalized rank of 0.9 means that a cell line is more similar to the tumors of its parental type than 90% of CCLP cell lines. Ideally, cell lines of the matching cancer type should have ranks close to 1.0. For example, given 50 cell lines of the matching type out of a total of 1,000 cell lines, one would expect most of the matching cell lines to have a normalized percentile rank larger than 0.95 = (1,000–50)/1,000.
Figure 5.

Mean weighted similarity of CCLP cell lines to parental tumor types across 25 TCGA cancer (sub)types
Each dot is a cell line (depicting its mean similarity to matching tumors), each boxplot summarizes the mean similarity ranks of cell lines and tumors of a given cancer type, and cancer types are ordered by increasing median average weighted similarity ranks. The overall similarity (A) is the average of the weighted similarity by each data type (B–D) mRNA: expression, copy-number alterations, mutations. Feature weights were chosen to emphasize the most significant recurrent mutations, copy-number alterations, and overexpression in relation to normal samples. Most tumor (sub)types have a mix of good, moderate, and poor matches to tumors among cell lines, as reflected by high, moderate, and low similarity scores, except for DLBC, THCA, PRAD, and UCEC, which have a high proportion of poor matches (with PRAD, UCEC, and THCA also having relatively few cancer cell lines in CCLP).
In particular, given that none of the cancer types included in this study had >56 matching cell lines, we can reasonably expect cell lines to have normalized ranks >0.9 when compared with their parental tumor type. In reality, cell lines of most cancer types have varying degrees of similarity to tumors, with good and moderate matches as well as some poor matches or even candidate outliers (Figure 5). Melanoma has the highest proportion of cell lines with a normalized rank >0.9, at least partly driven by the recurrent BRAF V600E mutation occurring in most melanoma cell lines. Other cancer types with a high proportion of well-matching cell lines with high normalized ranks are colon adenocarcinoma (COAD), esophageal adenocarcinoma (ESCA), kidney renal clear cell carcinoma (KIRC), and glioblastoma multiforme. On the other hand, cancer types, such as ovarian serous cystadenocarcinoma (OV), stomach adenocarcinoma (STAD), LUAD, and THCA have a relatively low proportion of highly ranked cell lines. We found 18 very well-matched cell lines (out of 594 cell lines), belonging to 9 different cancer types (out of 24 cancer types), with normalized ranks >0.9 for all three datum types (7 cell lines SKCM, 3 rectum adenocarcinoma [READ], 2 KIRC, and 1 each from breast invasive carcinoma [BRCA], COAD, ESCA, LAML, LGG, and lymphoid neoplasm diffuse large B cell lymphoma). On the other hand, we find 14 extreme outlier cell lines with normalized ranks <0.5 for all three datum types (Figure 6; 3 each from BRCA, OV, and LUAD, 2 LIHC, and 1 each from prostate adenocarcinoma [PRAD], STAD, and head and neck squamous cell carcinoma [HNSC]).
Figure 6.

Cell lines that consistently score high/low across all datum types
(A) Cell lines that rank in the top 10% for mutations, CNAs and gene expression. These might be considered good representatives of their respective tumor types.
(B) Cell lines that rank in the bottom 50% for mutations, CNAs, and gene expression. These are poor representatives of their respective tumor types. Ranks were based on weighted similarity computations when using the most variable (in tumors) genes for each data type, and feature weights emphasizing recurrent alterations (mutations, CNAs) or expression change in relation to a pool of normal samples. Circle sizes reflect the rank values.
A cell line can be a good match when using one data type, but a poor match when using others
To assess the usefulness of using multiple datum types, we compared the normalized ranks of cell lines across datum types. Several cell lines had a rank of >0.9 for one data type only—this includes, for example, cell lines that have a high gene expression-based rank (indicating retention of tissue-specific expression), but lack characteristic mutations and/or CNAs. In particular, of cell lines that had a normalized rank >0.99 when using expression data, 30 cell lines had a normalized rank <0.5 when using mutation data, and 23 had a rank <0.5 when using CNA data. This underscores the importance of considering multiple datum types when assessing cell line-tumor similarity.
Most cancer types have only a few outlier cell lines
We assessed outlier status in two different ways by comparing the similarity values of a query cell line and tumors of the same cancer type to (1) the similarity values of all cell lines and tumors of the query cell line’s cancer type (“within cancer type”), and (2) the similarity of all cell lines, including those of other cancer types, to tumors of the query cell lines cancer type (“across cancer types”). In other words (for example) an OV cell line will be evaluated by comparing its similarity with all OV tumors to (1) the similarity of all OV cell lines to all OV tumors and (2) all cell lines (OV as well as non-OV) to OV tumors.
We defined the threshold for being an outlier as a combined score rank <0.5, i.e., an outlier is a cell line with a lower overall similarity score than half of all cell lines when compared with its parental tumor type. Overall, there are 69 outlier cell lines (out of 594, ∼12%). The number of outlier cell lines varies by cancer type; most cancer types have 1–5 outlier cell lines. OV (11), LUAD (11), and STAD (7) are the only cancer types with more than 5 outlier cell lines. The high number of outlier cell lines for OV is at least partially explained by the fact that the TCGA OV tumors (Cancer Genome Atlas Research Network, 2011) are all of one particular subtype of ovarian cancer (i.e., HGSOC), whereas the CCLP OV cell lines are a mix of subtypes.
Several highly cited cell lines are genomic outliers
Even though most cancer types have only a few outlier cell lines, it is important to be aware of outliers, especially if they are widely used and might thus seriously impact scientific research. To address this, we estimated citation counts for the cell lines (see STAR Methods). The exact number of times a given cell line has been cited can be challenging to determine because of various factors, including the usage of several alternative names for a given cell line (e.g., OVCAR5, OVCAR-5, OVCAR.5, OV-CAR-5, OVCA5, or NIHOVCAR5), some cell lines being named after a person (Kuramochi, Ishikawa, Becker, or Kelly), some cell lines having names that match names or terms commonly used otherwise (TEN, K2, or NY), and so on. Nevertheless, we can determine which cell lines are the most highly cited ones. Using a threshold of ≥1,000 citations, we identified 31 highly cited cell lines that were outliers based on a low normalized rank and a variety of atypical characteristics, including lack of mutations and/ or CNAs typical of their cancer type, presence of mutations/CNAs atypical of their cancer type (and more common in other cancer types), unusually high or low mutational or copy-number burden, low gene expression-based similarity to the parental tumor type, and so on (Table 1 details such atypical characteristics for each highly cited outlier). Some of these cell lines have also previously been identified as outliers (Domcke et al., 2013).
Table 1.
Thirty-one highly cited outlier cell lines from 11 cancer types
| Cancer type or subtype | Cell line | Citation count | Alterations in the cell line atypical of the parent cancer type | TumorComparer score notes |
|---|---|---|---|---|
| LAML | HL-60 | ∼109,000 | lacks cancer type-specific mutations and CNAs, has mutations in CDKN2A and NRAS | poor scores for mutations and CNAs |
| BRCA | MDA-MB-231 | ∼102,000 | mutations in BRAF and NF2; deep deletions in CDKN2B, CDKN2B-AS1, PTPRD | bottom 50% overall, as well as for all 3 datum types |
| CESC | HELA | ∼86,000 | lacks cancer type-specific mutations and amplifications | bottom 10% by mutations |
| PRAD | DU145 | ∼42,000 | many mutations in pan-cancer genes, lacks cancer-specific amplifications, has a KRAS deep deletion | bottom 10% overall, <0.5 for all 3 daum types |
| LAML | KG1 | ∼16,000 | lacks cancer type-specific mutations and CNAs | bottom 10% by mutations |
| LUAD | NCI-H23 | ∼11,000 | high number of mutations in cancer-specific genes, pan-cancer mutations in DNMT3A and EEF1A1 | bottom 50% overall, as well as for all 3 datum types |
| PRAD | 22Rv1 | ∼7,700 | many mutations in pan-cancer genes, lacks cancer-specific CNAs | bottom 50% overall, as well as for mutations and CNAs |
| OV | SK-OV-3 | ∼7,600 | many mutations in pan-cancer genes | bottom 10% by CNAs, <0.5 for all 3 datum types |
| LUAD | NCI-H522 | ∼7,000 | lacks cancer-specific CNAs | bottom 50% overall as well as for expression and CNAs |
| STAD | MKN28 | ∼6,600 | lacks cancer-specific amplifications | poor scores for mutations and CNAs |
| KIRC | CAKI-1 | ∼5,800 | lacks cancer-specific mutations and amplifications, has a CDKN2A deep deletion | outlier by mutation |
| LIHC | SK-HEP-1 | ∼5,600 | lack of cancer-specific amplifications, has many pan-cancer deep deletions | bottom 50% overall, <0.5 for all 3 daum types |
| OV | IGROV-1 | ∼5,000 | excessive mutation count, many mutations in pan-cancer genes | poor scores for mutations and CNAs |
| OV | ES-2 | ∼4,100 | mutations in BRAF, KMT2D, and MAP2K1 | poor score overall, and for all datum types |
| CESC | C-33A | ∼4,100 | excessive mutation count, many mutations in pan-cancer genes | poor score overall, and for all datum types |
| DLBC | CRO-AP2 | ∼3,500 | lacks cancer-specific mutations and amplifications | poor scores overall, as well as for mutations and CNAs |
| LUAD | NCI-H82 | ∼3,500 | poor score overall, and for all datum types | |
| OV | PA-1 | ∼3,200 | lacks a TP53 mutation and cancer-specific amplifications | poor score overall, as well as for mutations and CNAs |
| OV | OVCAR-8 | ∼3,000 | lacks a TP53 mutation, has several pan-cancer mutations and amplifications | poor score by mutation |
| OV | OVCAR-5 | ∼2,900 | lacks a TP53 mutation, has a KRAS mutation as well as deep deletion | poor score overall, as well as for expression and CNAs |
| LAML | CESS | ∼2,900 | lacks cancer-specific mutations and amplifications | poor score overall, as well as for mutations |
| BLCA | UM-UC-3 | ∼2,800 | has several pan-cancer deep deletions | poor score overall, as well as for expression and mutations |
| KIRC | TK-10 | ∼2,000 | lacks cancer-specific mutations and CNAs | poor score overall, as well as for mutations and CNAs |
| KIRC | U-031 | ∼1,900 | lacks cancer-specific mutations | poor score for mutations |
| GBM | LN-229 | ∼1,900 | lacks cancer-specific mutations | poor score for mutations |
| STAD | HGC-27 | ∼1,720 | mutations in CDK12 and SMARCA4 | poor score overall, as well as for expression and CNAs |
| LUAD | SW1573 | ∼1,500 | poor score overall, as well as for expression and mutations | |
| LUAD | NCI-H1793 | ∼1,400 | poor score overall, as well as for mutations and CNAs | |
| MESO | NCI-H28 | ∼1,400 | lacks cancer-specific mutations and amplifications | poor score overall, as well as for expression and mutations |
| LUAD | A-427 | ∼1,200 | poor score for each data type | |
| LUAD | DU4475 | ∼1,100 | lacks cancer-specific mutations and amplifications | poor score overall, as well as for expression and mutations |
The genomic profiles of these cell lines are not well matched to tumors from the annotated parent cancer type. These cell lines are probably not good models for tumors. Details of alterations for each cell line are in Table S1. Poor score overall refers to all three data types. Citation numbers (>1,000 required) were estimated using Google Scholar as of June 2020, and have been binned next to the nearest multiple of 100 for those less than 1,000 and to the nearest multiple of 1,000 for those greater than 10,000.
Abbreviations are as follows: BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B cell lymphoma; GBM, glioblastoma multiforme; KIRC, kidney renal clear cell carcinoma; LAML, acute myeloid leukemia; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; OV, ovarian serous cystadenocarcinoma; PRAD, prostate adenocarcinoma; STAD, stomach adenocarcinoma.
At first glance, it might seem surprising that widely used cell lines could be poor representatives of their tumor type. However, it stands to reason that convenience is a factor in deciding which cell lines are most widely used—and cell lines that are the most convenient to work with tend to be those which are commercially or locally available, easy to manage, grow quickly, and so on. Furthermore, there might be a “founder effect” of sorts, wherein the cell lines that have already been used in many studies will tend to be used by more and more researchers, to build upon earlier findings. These will also tend to be more easily available, and reinforcing effects will combine to create a relatively small set of widely used and highly cited cell lines.
Another explanation for some outliers is that they are mislabeled, and are actually of a different tissue type than what was initially assumed (or is widely believed)—the issue of misidentification of cell lines is an important and well-known challenge in the field (Lorenzi et al., 2009). The widespread use of cell lines with a low genomic resemblance to tumors is concerning, although they could potentially still be good representatives for certain purposes.
Comparison with related studies
We compared our findings with those reported by related studies that evaluate cell lines as tumor models. Melanoma is the cancer type with the highest proportion of cell lines with high genomic similarity to tumors in our analysis. Vincent and Postovit (2017) compared the transcriptomes of 42 melanoma cell lines with TCGA tumors and single melanoma cell lines. Of the top well-matching 5 cell lines reported by Vincent and Postovit, 2 (COLO-849 and 537MEL) are missing from our dataset—the remaining 3 have high normalized ranks by expression in our analysis (SKMEL30, 0.996; UACC257, 1; A375, 0.96), but A375, in particular, has a very low normalized rank by CNAs, which are not used by Vincent and Postovit. Jiang et al. (2016) evaluated 68 breast cancer cell lines by using mutations, CNAs, gene expression, and protein expression, and nominated BT-483, T47D, and MDA-MB-453 as the cell lines with the highest similarity to tumors. In our analysis, all three cell lines indeed have expression-based ranks of ≥ 0.99, and MDA-MB-453 and BT-483 also have overall ranks ≥ 0.98; however, T47D has a slightly lower overall rank of 0.84 because of lower mutation-based and CNA-based ranks. Ronen et al. (2019) used deep learning on CNAs, gene expression, and point mutations to evaluate colorectal cancer subtypes and cell lines, and nominated CL-40, SW1417, and CW-2 as the top matches between tumors and cell lines. We also find all three to have high expression-based ranks but, although SW1417 and CL-40 have high overall ranks, CW-2 is an outlier in the CNA-based comparison, with a normalized rank of 0.01.
Yu et al. (2019) did a transcriptomic analysis of cell lines from the Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019) across 22 tumor types. They proposed a new cancer cell line panel, the “TCGA-110,” consisting of the top 5 cell lines from each of the 22 cancer types, as a new panel for pan-cancer studies. Our results on the subset of TCGA-110 present in CCLP show that, although most have a high expression-based normalized rank in our analysis, some are not highly ranked on the basis of mutation, CNA, or both. In particular, of the 110 CCLE cell lines in TCGA-110, 71 are included in CCLP, and 62 have mutation, gene expression, and CNA data available. The distribution of these 62 cell lines across the 22 cancer types is uneven: BRCA and LAML are the only cancer types with all 5 cell lines from TCGA-110 present in our analysis, whereas none of the 5 cell lines from STAD and HNSC are included—all other cancer types have 1–4 cell lines in our dataset. Although most of the TCGA-110 cell lines had a high expression-based normalized rank in our analysis, surprisingly, two—ML-1 (LAML, expression rank 0.02) and DU-145 (PRAD, expression rank 0.16)—were in fact outliers based on expression data. This could be due to differences in the genes used for the comparison or the platform (TCGA-110 is based on CCLE RNA sequencing data, we used microarray data from CCLP), or possibly a difference in the actual cell lines used by CCLE and CCLP. In our analysis, ML-1 does have a high rank by mutation and CNA data. Overall, 25 of the TCGA-110 cell lines have a mutation-based ranking of <0.5, 14 have a CNA rank <0.5, and 4 have a rank of <0.5 for both datum types. Although we find overall agreement with the assessments reflected in TCGA-110, our analysis provides a more refined ranking, as we not only use transcript-based expression values, but also CNAs and mutations. In particular, cell lines that are good matches to tumors in terms of gene expression profiles might not be good matches in terms of oncogenic mutations and/or copy-number changes.
Recently, while this manuscript was in preparation, Najgebauer et al. (2020) published a complementary study using CELLector, an R package and R Shiny application that allows cell line selection on the basis of genomic subtypes. CELLector identifies recurrent alterations across patient samples in mutation, CNA, and methylation data, and divides patient samples into subtypes on the basis of shared recurrent alterations. It then selects representative cell lines for each subtype on the basis of shared genomic alterations and ranks cell lines on the basis of two factors—the length of a subtype-associated genomic signature present in the cell line, and the proportion of patient samples represented by that cell line. Although this focus on recurrent alterations is a very reasonable choice, TumorComparer has a flexible, more general mechanism for weighting different features either in a data-driven or investigator-defined fashion (for example, using pathway-oriented weights as in Figure 4).
As for coverage of tumor types and cell lines, Najgebauer et al. (2020) applied CELLector to 16 tissue types, encompassing 4,550 tumors and 499 cell lines. TumorComparer includes all of these cancer types, and an additional 7 cancer types (we count COAD and READ as distinct tumor types, whereas Najgebauer et al. count them as one type, “COREAD”). Furthermore, although both CELLector and TumorComparer used mutations and CNAs, only CELLector used methylation data, whereas only TumorComparer used gene expression data. CELLector and TumorComparer represent two complementary and independently useful methods of genomics-guided cell line selection. The more general data-driven or investigator-defined weighted similarity approach of TumorComparer has many potential applications, with cell line evaluation being one use case, as highlighted in this study.
Discussion
By applying TumorComparer to pan-cancer data from TCGA and CCLP, we have identified both good and poor genomic matches between cell lines and tumors, as well as outliers among the cell lines of the 24 cancer types. Several of the outliers and poor matches were cell lines that lacked cancer-specific recurrent alterations as reported by TCGA. Notably, although we found 11 outlier cell lines for OV (5 widely used) and LUAD (7 widely used), we found only 1–7 outliers in the other cancer types. Overall, ∼12% of all cell lines were found to be poor genomic matches to their tumor type. Thus the vast majority of cell lines, including most of the widely used ones, bear at least a moderate resemblance to tumors, in terms of sharing cancer type-specific recurrent alterations, not having an unusually high or low number of alterations, and matching the gene expression patterns characteristic of the parental tumor type.
This study has generalized our previous work on evaluating cell lines via comparison of genomic profiles in ovarian cancer (Domcke et al., 2013), using weighted similarity with a set of weights chosen to emphasize important genomic alterations when computing pairwise similarity/distance. Although the main conclusions of our previous study were reproduced by this more general approach, the ranking of individual cell lines (barring a few outliers) might be different in this work, depending on the choice of similarity/distance measure, and the features emphasized. In particular, the study on HGSOC tumors and ovarian cell lines in the previous study used TP53 mutation status, hyper-mutant status, and mutation status in seven “non-HGSOC” genes, along with correlation with the mean CNA profile of tumors. The weighted similarity approach introduced here is more general and systematic in that all 24 cancer types and/or subtypes were studied by using a consistent approach. We also compared our results with other recent studies and, although our results generally agreed with those reported by others, we found cell lines, which matched tumors closely based on just a single data type and were not a good match using other or several datum types.
Our method is widely applicable to comparisons of genomic profiles, including, but not limited to tumor-tumor, tumor cell line, and cell line-cell line comparisons. A particularly promising application is assessing patient-patient similarity, which is a critical component in personalized cancer therapy. As we acquire more molecular and clinical patient data along with treatment outcomes, meaningful measures of similarity to previously treated patients will be an invaluable guide for treatment strategies. By emphasizing, via a choice of weights, determinants of response, and resistance to anti-cancer drugs, our approach can be adapted to patient-patient comparisons for use in prognosis, assignment to clinical trials, and choice of therapy. If such patient data are not available, one can exploit large drug sensitivity screens conducted in cell line panels (Barretina et al., 2012; Basu et al., 2013; Garnett et al., 2012; Iorio et al., 2016; Klijn et al., 2015; Shoemaker, 2006) to draw inferences regarding potential response to particular drug therapy based on cell lines most similar to a particular patient’s tumor.
Limitations of the study
Although the results presented here provide a generally useful resource for evaluation of a large number of widely available cell lines, there are a few caveats. The TCGA project focused on the genomics of primary surgical samples. Non-primary tumors—including metastases and recurrences—are known to have distinct characteristics not necessarily represented in this study. Also, the CCLP collection does not include all commercially available cell lines—e.g., some cancer (sub)types, such as endometrial cancer (10 cell lines), are under-represented. Therefore, summary conclusions based on a comparison of TCGA tumor and CCLP cell line data might not be directly applicable outside the datasets examined here. Furthermore, feature weights based on the frequency of occurrence of somatic alterations in tumor samples tend to emphasize major cancer subtypes and might not be equally meaningful for subtypes with only a few TCGA samples (e.g., for triple-negative breast cancer tumors that comprise only ∼10% of TCGA breast cancer tumors). In addition, tumors with substantial intra-tumor heterogeneity cannot be well represented by a single cell line, unless one subpopulation of cells dominates. In cases where single-cell alteration and expression information about a tumor is available, we recommend that each tumor cell subpopulation be separately compared with cell lines. Notwithstanding these caveats, the weighted similarity approach can be applied to genomic and molecular profiles to assess sample similarity beyond the TCGA and CCLP datasets analyzed here, including non-primary tumors, other collections of cell lines, and other types of tumor models (e.g, xenografts, organoids, and tumor spheroids).
Future extensions of the method and tool might include machine learning optimization of the distribution of weights over the feature set; addition of other molecular datum types, such as DNA methylation, protein expression, and modifications, in particular histone modifications and protein phosphorylation, as well as metabolites. For patient-patient comparisons, clinical parameters from electronic health records can be incorporated. Future updates of the TumorComparer database of tumor and cell line profiles are possible in crowdsourcing mode, engaging the cancer genomics researcher community, similar to the CIViC, WikiPathways or BioFactoid/Pathway Commons projects (Griffith et al., 2017; Kelder et al., 2012; Rodchenkov et al., 2020; Wong et al., 2020).
In summary, our approach informs the comparison via the incorporation of features and weights reflecting the features of interest to a researcher or clinician. Such features might include the expression of specific biomarkers, alterations in the members of a particular pathway, or features indicative of proliferation or response to particular therapies (Elias et al., 2015). The main thrust of the weighted similarity approach is to improve the comparison of cancer samples, be they in vivo or in vitro, in a flexible and data-driven manner.
The TumorComparer tool is publicly available as an open-source R package at https://github.com/sanderlab/tumorcomparer and as an interactive web application at http://projects.sanderlab.org/tumorcomparer. Researchers with new cell lines of interest with genomic and mRNA expression profiles should contact the authors for normalization and format requirements for input data.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| The Cancer Genome Atlas (TCGA) | Collins and Barker, 2007 | (synapse.org/#!Synapse:syn3241074, Data Freeze 1.3.1) |
| COSMIC Cell Line Project (CCLP) | Hoadley et al., 2018; Tate et al., 2019 | cancer.sanger.ac.uk/cell_lines |
| Cell Model Passports | van der Meer et al., 2019 | https://cellmodelpassports.sanger.ac.uk |
| Cellosaurus | Bairoch, 2018 | https://web.expasy.org/cellosaurus/ |
| Data Matrices used in this paper | This Paper | Zenodo (https://doi.org/10.5281/zenodo.4627644). |
| Software and algorithms | ||
| ANNOVAR | Wang et al., 2010 | https://annovar.openbioinformatics.org/en/latest/ |
| GISTIC2 | Mermel et al., 2011 | https://www.genepattern.org/modules/docs/GISTIC_2.0 |
| TumorComparer | This Paper | http://projects.sanderlab.org/tumorcomparer; https://github.com/sanderlab/tumorcomparer |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Chris Sander (tumorcells@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and software availability
The TumorComparer tool and data are publicly available as the TumorComparer R package at https://github.com/sanderlab/tumorcomparer with package dependencies described in the DESCRIPTION file of the package. Additionally, TumorComparer is available as an R Shiny web application at http://projects.sanderlab.org/tumorcomparer, and the datum matrices used in this study are available via Zenodo (https://doi.org/10.5281/zenodo.4627644).
Method details
Data acquisition and pre-processing
Data were pre-processed using the R programming language (R Core Team, 2015). Tumor data were obtained from the TCGA (The Cancer Genome Atlas) via the TCGA pan-cancer data resource (synapse.org/#!Synapse:syn3241074, Data Freeze 1.3.1) and cell line data werw obtained from the CCLP (COSMIC Cell Line Project) website (cancer.sanger.ac.uk/cell_lines) (Hoadley et al., 2018; Tate et al., 2019). Detailed annotation of cell lines was obtained from the Cell Model Passports website (https://cellmodelpassports.sanger.ac.uk). To focus on the mutations most likely to be functional, we excluded various categories of mutations. For TCGA, in a first step mutations were removed in these categories: 3′ Flank, 3′ UTR, 5′ Flank, 5′ UTR, Intron, RNA, Silent, and for CCLP: ‘Complex - compound substitution’, ‘Substitution - coding silent’, Unknown. Mutations were further flagged to enrich for putative functional mutations, by the following criteria. A mutation was flagged as functional if one of three criteria was true: (1) The mutation was considered deleterious; deleterious categories: TCGA: "Frame_Shift_Del", "Frame_Shift_Ins", "In_Frame_Del", "In_Frame_Ins", "Nonsense_Mutation", "Nonstop_Mutation", "Splice_Site", and "Translation_Start_Site", CCLP: "Complex - deletion inframe", "Complex - frameshift", "Complex - insertion inframe", "Deletion - Frameshift", "Deletion - In frame", "Insertion - Frameshift", "Insertion - In frame", and "Nonstop extension", "Substitution - Nonsense"; (2) the mutation had at least 10 studies describing its impact in cancer in the COSMIC database (v81); (3) the mutation received a high score for functional mutation impact from the MutationAssessor algorithm (Reva et al., 2011) used via ANNOVAR (Wang et al., 2010); otherwise a mutation was excluded.
Comparison of average genomic profiles across 24 cancer types
Gene expression data of tumors and cell lines was compared using the 2000 most variable genes across TCGA tumor samples. Similarly, for CNA data, we used the 2000 genes with the most variable copy number data across TCGA tumor samples. The variance was computed over all TCGA samples across all tumor types. For mutations, we used the frequency of mutations of the 299 genes, which are reported as significantly recurrently mutated across the TCGA pan-cancer cohort (Bailey et al., 2018).
GISTIC2 on CCLP copy number alteration (CNA) data
Then GISTIC2 (Mermel et al., 2011) method was run using the GenePattern (Reich et al., 2006) website, using the CCLP segmented copy number data, with default parameters for filtering, except “confidence,” which was increased from 0.75 to 0.99. We used both the continuous as well as the discretized 5-valued (-2,-1,0,1,2) gene-wise data produced by the GISTIC2 algorithm for copy-number analysis.
Gene expression data
RNASeq data for TCGA (Hoadley et al., 2018; Sanchez-Vega et al., 2018) and microarray data for CCLP (Tate et al., 2019) were obtained from the respective websites (see above). Quantile normalization (Bolstad et al., 2003) was applied to bring the expression values of TCGA and CCLP samples on the same scale. We chose not to apply batch correction methods since batch correction would also potentially remove real biological differences (Leek et al., 2010).
Quantification and statistical analysis
Assignment of weights to features
In general, feature weights are to be determined depending on the particular interest of the investigator and the question(s) being asked. Here, we chose a particular set of weights focused on recurrent genomic alterations across many cancer samples. We assigned each of the features a weight between 0.0 and 1.0. Genes in the results from the recurrence analysis programs MUTSIG or GISTIC, as given in the pan-cancer resource (synapse.org/#!Synapse:syn3241074, Data Freeze 1.3.1), for specific tumor types were assigned weights as follows: (i) weight of 1.0 for each gene with a cancer-specific MUTSIG q-value ≤ 0.1 (Lawrence et al., 2013), and (ii) weight of 1/N for each gene in a GISTIC peak that spans N genes (Mermel et al., 2011). In addition, since alterations in cancer genes that have no statistically significant recurrence in a particular cancer type may still be of biological interest, we gave all known cancer genes (which were not recurrent in the given cancer type) from the TCGA pan-cancer studies a weight of 0.1.
Thus, all genes with significant recurrence of mutation events according to the MUTSIG method, and all genes in singleton peaks according to the GISTIC method, have the maximum possible weight of 1. Other GISTIC peak genes have a weight inversely proportional to the size of the peak. All remaining known cancer genes have a weight of 0.1, and the remaining alterations, assumed to be passengers, have a weight of 0.01.
For gene expression, we used log2(expression ratio) relative to pooled normal samples from TCGA (since most TCGA cancer types have gene expression data for only a small number of normal samples) - the expression ratios (‘fold changes’) were converted to a range of 0-1 using a min-max transformation. This particular set of weights is meant to focus primarily on genes known to be likely functional contributors. Users can use different sets of weights depending on their particular question of interest.
Weighted matching for discrete data
Most genes do not have functional alterations in most samples, since only a small fraction of genes are mutated (or have high-level CNAs) in a typical tumor; therefore, 0-0 feature matches between two samples are the “default” or expected case, and not very informative. We, therefore, computed the weighted similarity of two samples after discarding the 0-0 matches.
Samples are represented by feature vectors
and and a weight vector for feature weights . Their weighted similarity is calculated as
that is, 0-0 matches are discarded, and the similarity is calculated as the ratio of the sum of weights of features for which the two samples have the same value, to the sum of weights of all features for which at least one of the samples has a non-zero value. This is similar to the widely used Jaccard Index for binary data, in which 0-0 matches are discarded, and the similarity is calculated as the ratio of the intersection to the union of the subsets of features for which at least one of the samples has non-zero values.
Weighted correlation for continuous data
For weighted similarity using continuous data, we used a weighted correlation.
Weighted correlation incorporates weights into the computation of correlation coefficients by using weighted versions of the mean and covariance. If we represent samples by feature vectors as above,
The weighted means are computed as
The weighted variances are given by
the weighted covariance is
and finally, the weighted correlation is computed as
Computation of overall weighted similarity scores
Since the different datum types (“-omics”) produce similarity scores with different numerical ranges and distributions, they should be normalized in some manner before being combined. One approach is to convert them all to [0,1] using a min-max conversion. Another is to use percentile ranks, where we rank the values, and then divide by the number of samples, N, to get scores in the [1/N,1] range (or subtract 1 and divide by N-1 to get values in [0,1]); however, this assigns the same fixed values to any N similarity values, regardless of their actual distribution. We preferred the min-max conversion for the analyses presented here. Lastly, the TumorComparer tool allows different weighting for the different datum types; in this report, we have used identical weighting for mutations, CNAs and mRNA expression.
Ranking of cell lines using mean similarity to tumors
In order to compare cell line - tumor similarities across multiple cancer types, we compared all cell lines to each tumor type in turn. For each tumor type, we compared all cell lines to the tumors, which produced a baseline of cell line-tumor similarity scores for the given tumor type. We then computed the mean of the pairwise similarity scores between the tumors and each cell line and used it to rank the cell lines. By dividing this rank by the number of cell lines, we obtained a “normalized percentile rank” between 0 and 1 for each cell line - this was done separately for each data type, and then the overall scores were computed as a weighted average of the data type-specific ranks/scores, followed by a conversion to normalized ranks. Intuitively, cell lines of the matching cancer type should have ranks close to 1 - given, say, 50 cell lines of the matching type out of a total of 1000 cell lines, wed expect most of them to have a normalized percentile rank > (1000-50)/1000 = 0.95.
Generating citation estimates
Estimates of literature citation counts were produced using R code available at: https://github.com/cannin/cellline_citations. To find literature about cell lines, one has to address the potential ambiguity in cell line names. CCLP cell line names were first mapped to Cellosaurus synonyms (Bairoch, 2018) by examining cell line information for the 'Cosmic-CLP' collection extracted from Cellosaurus. CCLP cell line names not found programmatically in Cellosaurus were manually added to a list of cell line search terms that included at least 1 entry for all cell lines used in the TumorComparer analysis.
Next, Google Scholar was searched iteratively for all cell line search terms. The Google Scholar resource for scientific articles was chosen for its ability to identify search terms in the full-text of articles (when available). Each search contained flags to remove search results related to dissertations, theses, and patents. The search pattern used was: "cancer cell line CELL_LINE_SEARCH_TERM CANCER_TYPE" where CELL_LINE_SEARCH_TERM are terms from the list generated using Cellosaurus (along with manually added terms) and CANCER_TYPE is derived from the cancer type information also provided from Cellosaurus. Additionally, terms with fewer than 4 characters or where all the characters of the term were numbers were excluded. During the search, terms that were identified as part of the author name string on Google Scholar were flagged for later filtering.
Next, Google Scholar search results were filtered for 1) synonyms to common names (e.g., "scott") and acronyms (e.g., "lcms") and 2) short synonyms composed of only numbers. Lastly, citation counts of synonyms were merged to produce a final estimate that was binned into groups.
Acknowledgments
We thank JianJiong Gao, Giovanni Ciriello, Yasin Senbabaoglou, Debora Marks, and members of the Sander lab at MSKCC, DFCI, and HMS for discussions, Debra Bemis at MSKCC and Laura Kleiman at DFCI for manuscript editing and valuable feedback. Funding for R.S., C.S., and N.S. was provided by the US National Cancer Institute for the TCGA Genome Data Analysis Center (NCI-U24CA143840 and NCI-R21CA135870) and the National Resource for Network Biology (NIH-P41 GM103504). Funding for A.L. was provided by a Ruth L. Kirschstein National Research Service Award (F32 CA192901).
Author contributions
C.S. initiated and managed the project. R.S., C.S., and N.S. conceived the approach. R.S. developed and implemented the method, analyzed the data, and drafted the manuscript. A.L. led the development of the R package and online tool based on the method and developed and implemented the method to count citations per cell line. R.S., C.S., N.S., and A.L. interpreted the results and completed the manuscript.
Declaration of interests
The authors declare no competing interest.
Published: June 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100039.
Supplemental information
References
- Bailey M.H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M.C., Kim J., Reardon B., et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385.e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A. The Cellosaurus, a cell-line knowledge resource. J. Biomol. Tech. 2018;29:25–38. doi: 10.7171/jbt.18-2902-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A., Bodycombe N.E., Cheah J.H., Price E.V., Liu K., Schaefer G.I., Ebright R.Y., Stewart M.L., Ito D., Wang S., et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–1161. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad B.M., Irizarry R.A., Astrand M., Speed T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F.S., Barker A.D. Mapping the cancer genome. Sci. Am. 2007;296:50–57. [PubMed] [Google Scholar]
- Domcke S., Sinha R., Levine D.A., Sander C., Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias K.M., Emori M.M., Papp E., MacDuffie E., Konecny G.E., Velculescu V.E., Drapkin R. Beyond genomics: critical evaluation of cell line utility for ovarian cancer research. Gynecol. Oncol. 2015;139:97–103. doi: 10.1016/j.ygyno.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J., et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greshock J., Nathanson K., Martin A.-M., Zhang L., Coukos G., Weber B.L., Zaks T.Z. Cancer cell lines as genetic models of their parent histology: analyses based on array comparative genomic hybridization. Cancer Res. 2007;67:3594–3600. doi: 10.1158/0008-5472.CAN-06-3674. [DOI] [PubMed] [Google Scholar]
- Griffith M., Spies N.C., Krysiak K., McMichael J.F., Coffman A.C., Danos A.M., Ainscough B.J., Ramirez C.A., Rieke D.T., Kujan L., et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017;49:170–174. doi: 10.1038/ng.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessey P.T., Ochs M.F., Mydlarz W.W., Hsueh W., Cope L., Yu W., Califano J.A. Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PLoS One. 2011;6:e20584. doi: 10.1371/journal.pone.0020584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V., et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304.e6. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houshdaran S., Hawley S., Palmer C., Campan M., Olsen M.N., Ventura A.P., Knudsen B.S., Drescher C.W., Urban N.D., Brown P.O., et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One. 2010;5:e9359. doi: 10.1371/journal.pone.0009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H., et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G., Zhang S., Yazdanparast A., Li M., Pawar A.V., Liu Y., Inavolu S.M., Cheng L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics. 2016;17(Suppl 7):525. doi: 10.1186/s12864-016-2911-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao J., Salari K., Bocanegra M., Choi Y.-L., Girard L., Gandhi J., Kwei K.A., Hernandez-Boussard T., Wang P., Gazdar A.F., et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4:e6146. doi: 10.1371/journal.pone.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelder T., van Iersel M.P., Hanspers K., Kutmon M., Conklin B.R., Evelo C.T., Pico A.R. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2012;40:D1301–D1307. doi: 10.1093/nar/gkr1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klijn C., Durinck S., Stawiski E.W., Haverty P.M., Jiang Z., Liu H., Degenhardt J., Mayba O., Gnad F., Liu J., et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015;33:306–312. doi: 10.1038/nbt.3080. [DOI] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A., et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T., Scharpf R.B., Bravo H.C., Simcha D., Langmead B., Johnson W.E., Geman D., Baggerly K., Irizarry R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi P.L., Reinhold W.C., Varma S., Hutchinson A.A., Pommier Y., Chanock S.J., Weinstein J.N. DNA fingerprinting of the NCI-60 cell line panel. Mol. Cancer Ther. 2009;8:713–724. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters J.R.W. Human cancer cell lines: fact and fantasy. Nat. Rev. Mol. Cell Biol. 2000;1:233–236. doi: 10.1038/35043102. [DOI] [PubMed] [Google Scholar]
- van der Meer D., Barthorpe S., Yang W., Lightfoot H., Hall C., Gilbert J., Francies H.E., Garnett M.J. Cell Model Passports—a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res. 2019;47:D923–D929. doi: 10.1093/nar/gky872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najgebauer H., Yang M., Francies H.E., Pacini C., Stronach E.A., Garnett M.J., Saez-Rodriguez J., Iorio F. CELLector: genomics-guided selection of cancer in vitro models. Cell Syst. 2020;10:424–432.e6. doi: 10.1016/j.cels.2020.04.007. [DOI] [PubMed] [Google Scholar]
- R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. https://www.R-project.org/ [Google Scholar]
- Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J.P. GenePattern 2.0. Nat. Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Reva B., Antipin Y., Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodchenkov I., Babur O., Luna A., Aksoy B.A., Wong J.V., Fong D., Franz M., Siper M.C., Cheung M., Wrana M., et al. Pathway Commons 2019 Update: integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020;48:D489–D497. doi: 10.1093/nar/gkz946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronen J., Hayat S., Akalin A. Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci. Alliance. 2019;2:e201900517. doi: 10.26508/lsa.201900517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross D.T., Scherf U., Eisen M.B., Perou C.M., Rees C., Spellman P., Iyer V., Jeffrey S.S., Van de Rijn M., Waltham M., et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat. Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., Dimitriadoy S., Liu D.L., Kantheti H.S., Saghafinia S., et al. Oncogenic signaling pathways in the Cancer Genome Atlas. Cell. 2018;173:321–337.e10. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R., Ernberg I. The molecular portrait of in vitro growth by meta-analysis of gene-expression profiles. Genome Biol. 2005;6:R65. doi: 10.1186/gb-2005-6-8-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Smiraglia D.J. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum. Mol. Genet. 2001;10:1413–1419. doi: 10.1093/hmg/10.13.1413. [DOI] [PubMed] [Google Scholar]
- van Staveren W.C.G., Weiss Solís D.Y., Hébrant A., Detours V., Dumont J.E., Maenhaut C. Human cancer cell lines: experimental models for cancer cells in situ? For cancer stem cells? Biochim. Biophys. Acta. 2009;1795:92–103. doi: 10.1016/j.bbcan.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji K., Kawauchi S., Saito S., Furuya T., Ikemoto K., Nakao M., Yamamoto S., Oka M., Hirano T., Sasaki K. Breast cancer cell lines carry cell line-specific genomic alterations that are distinct from aberrations in breast cancer tissues: comparison of the CGH profiles between cancer cell lines and primary cancer tissues. BMC Cancer. 2010;10:15. doi: 10.1186/1471-2407-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent K.M., Postovit L.-M. Investigating the utility of human melanoma cell lines as tumour models. Oncotarget. 2017;8:10498–10509. doi: 10.18632/oncotarget.14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistuba I.I., Behrens C., Milchgrub S., Syed S., Ahmadian M., Virmani A.K., Kurvari V., Cunningham T.H., Ashfaq R., Minna J.D., et al. Comparison of features of human breast cancer cell lines and their corresponding tumors. Clin. Cancer Res. 1998;4:2931–2938. [PubMed] [Google Scholar]
- Wistuba I.I., Bryant D., Behrens C., Milchgrub S., Virmani A.K., Ashfaq R., Minna J.D., Gazdar A.F. Comparison of features of human lung cancer cell lines and their corresponding tumors. Clin. Cancer Res. 1999;5:991–1000. [PubMed] [Google Scholar]
- Wong J., Franz M., Siper M.C., Fong D., Durupinar F., Dallago C., Luna A., Giorgi J.M., Rodchenkov I., Babur Ö. et al. 2020 doi: 10.7554/eLife.68292. https://www.biorxiv.org/content/10.1101/2021.03.10.382333v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K., Chen B., Aran D., Charalel J., Yau C., Wolf D.M., van ’t Veer L.J., Butte A.J., Goldstein T., Sirota M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019;10:3574. doi: 10.1038/s41467-019-11415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TumorComparer tool and data are publicly available as the TumorComparer R package at https://github.com/sanderlab/tumorcomparer with package dependencies described in the DESCRIPTION file of the package. Additionally, TumorComparer is available as an R Shiny web application at http://projects.sanderlab.org/tumorcomparer, and the datum matrices used in this study are available via Zenodo (https://doi.org/10.5281/zenodo.4627644).