

Summary
Single-cell Strand-seq generates directional genomic information to study DNA repair, assemble genomes, and map structural variation onto chromosome-length haplotypes. We report a nanoliter-volume, one-pot (OP) Strand-seq library preparation protocol in which reagents are added cumulatively, DNA purification steps are avoided, and enzymes are inactivated with a thermolabile protease. OP-Strand-seq libraries capture 10%–25% of the genome from a single-cell with reduced costs and increased throughput.
keywords: Strand-seq, structural genomic variation, inversions, haplotyping, one-pot library construction, thermolabile protease, single-cell sequencing, nanoliter dispensing
Graphical abstract
Highlights
-
•
We describe a one-pot (OP) protocol to construct single-cell Strand-seq libraries
-
•
The OP protocol uses nanoliter volumes of reagents in open nanoliter arrays
-
•
OP-Strand-seq libraries can be made in large numbers and at low cost
-
•
OP-Strand-seq libraries capture 10%–25% of the DNA template strands in a cell
Motivation
Strand-seq's directional genomic information is invaluable, but the library preparation protocol is difficult, costly, and slow. Moreover, the libraries typically have low complexity; that is, they capture at most 5% of the genome in a single cell. We developed a scalable, high-throughput protocol that is suitable for the centralized production of large numbers of Strand-seq libraries with improved complexity.
Single-cell Strand-seq libraries are used for phasing, structural variant discovery, and genome assembly. Hanlon et al. describe an updated protocol in nanoliter volumes that improves the throughput and cost of Strand-seq libraries, as well as the fraction of the genome captured.
Introduction
Recent analytic advances in haplotype-resolved genome assembly and comprehensive calls of structural variants (SVs) rely on the chromosome-length phase information in single-cell DNA template strand sequencing (Strand-seq) data (Ebert et al., 2021; Porubsky et al., 2020a). Strand-seq exploits the semi-conservative nature of DNA replication to incorporate the thymidine analog bromodeoxyuridine (BrdU) into the newly synthesized strand, which is nicked at those sites upon exposure to UV light (Falconer et al., 2012). Thus, all reads from a particular chromosome in a Strand-seq library originate from the DNA template strand used for DNA replication in the parental cell, which determines their orientation when mapped to the reference genome. By processing single human nuclei containing just 7.2 pg of DNA without prior amplification, Strand-seq avoids the uneven coverage and unreliable base calls associated with whole-genome amplification (reviewed in Ellis et al., 2021). However, the conventional low-throughput Strand-seq library construction protocol for microliter volumes (Sanders et al., 2017) is costly and yields sparse, low-complexity libraries, such that >40 libraries/sample are required for most analyses (Porubsky et al., 2017).
Results and discussion
We developed one-pot (OP)-Strand-seq by pursuing three improvements to the conventional protocol. First, we reasoned that 500- to 1,000-fold smaller reagent volumes would improve the efficiency of enzymatic reactions by increasing the relative concentration of DNA fragments. During the ligation of DNA fragments to Illumina adapters, for example, this allowed us to add less adapter and thereby reduce the formation of adapter dimers, which are preferentially amplified during PCR and displace informative library fragments during sequencing (Gao et al., 2020; Sanders et al., 2017). Second, we reduced the variability in the fragmentation of genomic DNA by treating intact nuclei with micrococcal nuclease (MNase) in bulk (Gao et al., 2020), rather than individually, followed by fixation with formaldehyde (Figures 1 and 2A). Over-digestion with MNase results in GC-biased libraries, which correlates with uneven coverage and a higher proportion of non-directional reads (background, Figures 2C and S1A). Finally, as DNA is inevitably lost upon purification, we replaced bead clean-ups in the conventional protocol with thermolabile protease treatments in a base buffer system that is compatible with all enzymes used during the procedure.
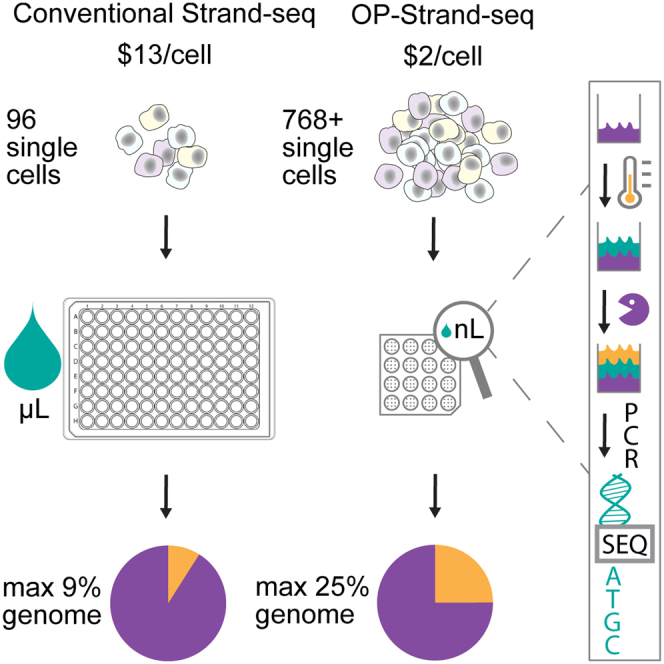
Figure 1.

Comparison of the conventional Strand-seq protocol with the OP-Strand-seq method
The labels A–I correspond to subheadings in the STAR Methods.
Figure 2.

Characteristics of OP-Strand-seq libraries
(A) DNA fragment size of nuclei digested with MNase (0–5,000 gel units/mL). The sizes of ladder bands (L) are given in base pairs. The lanes marked (∗) are correctly digested.
(B) A mixture of nuclei from human cells cultured with or without BrdU and stained with propidium iodide (PI) and Hoechst dye. Nuclei with BrdU show quenched Hoechst fluorescence.
(C) Properties of 78 OP-Strand-seq libraries and 78 conventional Strand-seq libraries. We used the outer coordinates of fragments from unique, properly paired reads to calculate the GC content, fragment length, and coverage. We used breakpointR (Porubsky et al., 2020b) to calculate the background, and we calculated spikiness (see results and discussion) as described in Bakker et al. (2016). The two clusters of conventional Strand-seq libraries in the fragment size plots are from different batches.
(D) Complexity curves for individual libraries (gray lines) produced with OP-Strand-seq or conventional Strand-seq, and their mean and SD (colored lines and ribbons). The dashed vertical lines and bands represent the means and SDs of the sequencing effort used (OP-Strand-seq on right, conventional on left). Breadth of coverage is the fraction of the haploid reference genome covered by at least one read fragment. Complexity estimates were made using Preseq (Daley and Smith, 2014).
In brief, cells are cultured in the presence of BrdU and 5-fluorodeoxyuridine (5-FdU), lysed, digested with MNase, and fixed with formaldehyde before BrdU-positive nuclei with 2N DNA are sorted by fluorescence-activated cell sorting (FACS), as shown in Figure 2B. Following storage at 4°C, individual nuclei are dispensed in 0.3 nL droplets using the cellenONE (Cellenion), a commercially available robotic liquid handler, into open 115 nL wells of a bespoke aluminum nanoarray (16 clusters of 49 libraries, 784 total; Figure S1B; Data S1). Alternatively, a commercially available ICELL8 nanoarray can be used (up to 5,184 libraries; Takara Bio; Figure S2A). In all subsequent steps, the cellenONE is used to dispense buffer-enzyme master mixes into wells, which are reversibly sealed with a non-adhesive film before incubations on a thermocycler, as described previously (Laks et al., 2019). First, MNase-digested DNA fragments of ∼150 bp are liberated from nuclei using heat treatment to reverse formaldehyde cross-links, followed by protease digestion of proteins. The fragments are then end repaired and A-tailed (polished) in a single step using a variant of a protocol by Neiman et al. (2012), followed by the ligation of forked Illumina adapters. Next, Hoechst dye is added during a protease cleanup, enabling the ablation of the BrdU-substituted strand during a 150 second exposure to UV light (365 nm). Unique pairs of dried indexed PCR primers, dispensed previously into a second nanoarray, are rehydrated, frozen, and transferred to the library nanoarray by clamping aligned nanoarrays face-to-face before centrifugation. Finally, after 13 PCR cycles, each cluster of 49 libraries is collected by centrifugation in a PCR tube (recovery volume 2.4 μL). OP-Strand-seq, like conventional Strand-seq, takes ∼2 days for MNase, cell sorting, and library construction. However, OP-Strand-seq yields 6× to 16× more libraries at ∼15% of the cost per library, excluding sequencing (OP-Strand-seq costs US$1–US$2 per library with throughput of 768 cells; conventional Strand-seq costs ∼US$13 per library with throughput of 96 cells; Sanders et al., 2017). Most of these savings come from smaller reagent volumes and the elimination of plastic consumables, such as pipette tips (Table S1).
We sequenced 96 OP-Strand-seq libraries for HapMap sample NA12878 to compare with 288 conventional Strand-seq libraries previously reported for the same individual (Porubsky et al., 2016). First, we excluded libraries with poor Strand-seq characteristics: those with more than 5% non-directional reads (background >0.05, Figure S1A), fewer than 50 reads per Mb, or with chromosome-arm deletions (Porubsky et al., 2016), which can result from too many cycles of BrdU incorporation. We retained 78 (81.3%) OP-Strand-seq libraries. We also retained 78 conventional Strand-seq libraries, randomly selected from the 148 libraries (51.4%) with good Strand-seq characteristics. The libraries were sequenced deeply, that is, to high duplication rates, for both conventional (mean 39.2%, SD 17.1%) and OP-Strand-seq libraries (mean 34.1%, SD 12.7%).
The OP-Strand-seq libraries have ∼4-fold greater complexity on average than those produced with conventional Strand-seq (Figure 2D), capturing up to 25% of the haploid genome per cell. OP-Strand-seq libraries also have reproducible fragment sizes and low background. Moreover, the GC content of OP-Strand-seq reads is nearer the 40.9% reported for the human genome (Piovesan et al., 2019). Libraries with higher GC content have more uneven coverage, that is, increased bin-to-bin variation in read depth (spikiness, Figure 2C), which can confound copy number variant (CNV) analysis. This is likely due to the uniformity of bulk MNase digestion, the choice of KAPA HiFi HotStart DNA polymerase rather than Phusion polymerase (Quail et al., 2012), and the reduced number of PCR cycles (Aird et al., 2011). We also compared OP-Strand-seq libraries with those produced with DLP+, a tagmentation-based library preparation method that uses similar open nanoarrays (Laks et al., 2019). OP-Strand-seq libraries had comparable or greater complexity than DLP+ libraries produced for EBV-transformed B cells (Figure S2B).
We found that adding 5-FdU to cell cultures improved flow cytometry resolution for nuclei stained with Hoechst 33258 (Figure 3). This likely explains why libraries made with 5-FdU had higher ASHLEYS quality scores than those without (Gros et al., 2021), as it should help exclude cells with either slightly less or slightly more than one cycle of BrdU incorporation. In the former case, some genomic regions in a cell incorporate BrdU into neither strand and yield non-Strand-seq scWGS reads, whereas in the latter case, some regions incorporate BrdU into both strands and yield no reads at all—or very few—creating apparent deletions. Overall, 49% more libraries with 5-FdU had at a quality score of at least 0.5, which is the threshold we use to distinguish good from poor Strand-seq, effectively increasing the number of informative libraries in datasets created with 5-FdU (Table S1). Libraries made with 5-FdU also had lower background (Figure S1A; mean 0.007, SD 0.004 versus mean 0.011, SD 0.006). This is consistent with the prediction that by blocking thymidine kinase, 5-FdU should reduce the availability of dTTP in cells. The slightly higher complexity observed for libraries without 5-FdU may reflect the inclusion of more cells with less than one cycle of BrdU incorporation, which increases the number of unique DNA fragments available for sequencing but results in poorer-quality libraries.
Figure 3.

Effects of 5-FdU on OP-Strand-seq libraries
(A) Bivariate flow cytometry plots of nuclei from human peripheral blood mononuclear cells treated with BrdU and 0, 0.3, or 1 uM of 5-FdU. Nuclei were stained with Hoechst 33258 (excited by a 405 nm laser and detected using a 450/50 BP filter) and propidium iodide (excited by a 561 nm laser and detected using a 610/20 BP filter on a Fortessa flow cytometer). Greater separation between the +BrdU (left) and −BrdU (right) populations is observed when 5-FdU is used.
(B) Properties of OP-Strand-seq libraries after shallow sequencing (as in Table S1) with and without 5-FdU. Only libraries with at least 5,000 mapped reads are included; 108 of 144 libraries with 5-FdU met this criterion, as did 98 of 144 libraries without 5-FdU. For the background plot, we also excluded libraries with an ASHLEYS quality score below 0.5 (Gros et al., 2021). “Complexity”, here, means predicted breadth of coverage at 1 Gb sequencing effort. Significance: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. Boxplots indicate the median, interquartile range, minimum and maximum (whiskers), and outliers.
Deeply sequenced OP-Strand-seq libraries contain more information than conventional Strand-seq libraries, which can improve analyses such as single-nucleotide variant (SNV) phasing with StrandPhaseR, an R package that exploits the fact that forward and reverse reads originate from different homologs for roughly half the chromosomes in each Strand-seq library (Porubsky et al., 2017). Using the 78 conventional Strand-seq libraries, above, we could phase 1,124,384 (54.1%) of 2,078,238 high-confidence heterozygous SNVs provided by the Genome in a Bottle Consortium, v3.2 (Zook et al., 2014). By contrast, using the 78 OP-Strand-seq libraries, we could phase 2,007,500 of the SNVs (96.6%). Similarly, OP-Strand-seq libraries with high complexity should improve the detection of small, low-frequency SVs, increase the resolution of SV breakpoint calls, and facilitate studies of DNA repair. For the analysis of somatic SVs, such as low-frequency CNVs, OP-Strand-seq provides high-resolution information that cannot be obtained with conventional Strand-seq. As a demonstration, we used AneuFinder (Bakker et al., 2016) to identify complex somatic CNVs on 16q in one OP-Strand-seq library with 20.5% depth of coverage (Figure S1C), and we repeated the analysis after down-sampling the library to the mean coverage depth of the 78 conventional Strand-seq libraries (2.7%). For four consecutive CNV breakpoint locations, the 99% confidence intervals were 4- to 6-fold larger in the down-sampled library (average size 14.1 versus 70.6 kb). The smaller confidence intervals allowed better characterization of the gene content of CNV breakpoints as well as microhomology between pairs of breakpoints, which is implicated in several mechanisms of DNA repair and the formation of most SVs (Carvalho and Lupski, 2016; Table S1).
The OP library construction approach described here greatly increases the fraction of the single-cell genome that can be sequenced. The principles of the method can be applied to other investigations in molecular biology where the number of cells or the amount of available DNA is limiting. With reduced costs and increased throughput, we expect that OP library construction will make data from complex, picogram-input library construction protocols such as Strand-seq more accessible, enabling cutting-edge genomic research into SV discovery, haplotyping, and genome assembly.
Limitations of the study
Future improvements to library construction in nanoliter volumes should address sources of variability in our method (e.g., variable coverage, Figure 2C), such as evaporation. Open library wells are sensitive to ambient temperature and humidity, diluting or concentrating reagents with condensation or evaporation. The reduced number and duration of steps in the protocol, careful sealing, and temperature control are used to mitigate these problems. Nevertheless, dispensing primers during library preparation takes too long (4 min/primer, ∼4.5 h total) to consistently prevent evaporation and maintain reagent volumes. This motivated us to pre-dispense primers and transfer them into library wells by centrifugation: the transfer, however, risks leaking primers into non-target wells unless both nanoarrays are positioned and clamped correctly. Mechanisms such as these may contribute to the observed variability in library quality and complexity between clusters and wells in the nanoliter arrays (Table S1).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Bromodeoxyuridine (BrdU, 5-bromo-2′-deoxyuridine) | Sigma-Aldrich | Cat. #: B5002 |
| 5-fluorodeoxyuridine (5-FdU, floxuridine, 5-fluoro-2′-deoxyuridine) | Sigma-Aldrich | Cat. #: F0503 |
| Micrococcal nuclease (MNase) | New England Biolabs | Cat. #: M0247 |
| Qiagen protease | Qiagen | Cat. #: 19155 |
| Hoechst 33258 (Hoechst) | ThermoFisher Scientific | Cat. #: H3569 |
| Propidium iodide (PI) | Sigma-Aldrich | Cat. #: P4864 |
| Deposited data | ||
| FASTQ files | This paper | SRA: SRP326369; BioProject: PRJNA742746 |
| Experimental models: Cell lines | ||
| Human: EBV-transformed B cells for NA12878 | Coriell | RRID: CVCL_7526 |
| Oligonucleotides | ||
| Sequencing adapters and index primers; see Table S1 | This paper | N/A |
| Software and algorithms | ||
| Preseq gc_extrap and bam2mr | (Daley and Smith, 2014) | RRID: SCR_018664 |
| AneuFinder | (Bakker et al., 2016) | N/A |
| BreakpointR | (Porubsky et al., 2020b) | N/A |
| ASHLEYS quality control | (Gros et al., 2021) | N/A |
| Bowtie2 | (Langmead and Salzberg, 2012) | RRID:SCR_016368 |
| Picard Tools | Broad Institute | RRID: SCR_006525 |
| Other | ||
| cellenONE X1i | SCIENION, Cellenion | N/A |
| Bespoke nanoarrays; see Figure S1B and Data S1 (or use ICELL8 chips, below) | This paper | N/A |
| Parylene-C coating services | Plasma Ruggedized Solutions | N/A |
| ICELL8 150V chips | Takara Bio | Cat. #: 640013 |
| Bespoke primer transfer device | This paper | N/A |
| Microseal ‘A’ Sealing Film | Bio-Rad | Cat. #: MSA5001 |
| BlueWave MX-250 Emitter, RediCure (365 nm) | Dymax | Cat. #: C-42806, FR0045 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter Lansdorp (plansdor@bccrc.ca).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Reference EBV-transformed female B cells for NA12878 were obtained from Coriell (Camden, NY; RRID CVCL_7526). They were cultured in RPMI 1640 with 10% FBS, 50 U/ml of Penicillin-Streptomycin, and 2 mM L-Glutamine. The cells were grown at 37°C with 5% CO2 at a density of 0.25×106/mL. The cell line was not authenticated.
Method details
A. Preparing nuclei for OP-Strand-seq libraries
Semi-confluent cultures of NA12878 cells in logarithmic phase were pulsed for 20-24 hrs either with 40 μM BrdU ± 1 μM 5-FdU or with no further additions and then spun down at 258 x g for 5 min before the cell pellet was resuspended in phosphate buffered saline (PBS). Viable cells were counted with a hemocytometer, then cells were spun down again, PBS was removed, and they were resuspended at 107cells/mL in cold lysis buffer (LB; Brind’Amour and Lansdorp, 2011) consisting of 15 mM Tris-HCl (pH 7.5), 10 mM NaCl, 80 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.1% (v/v) 2-mercaptoethanol, 0.1% Triton X-100, 0.2 mM spermine, and 0.5 mM spermidine (Falconer et al., 2012; Sanders et al., 2017). After 15 minutes on ice, nuclei were spun at 250 x g for 4 min at 4°C, 90% of the supernatant was aspirated, and pelleted nuclei were gently resuspended at 107/mL in cold H4-10 buffer (4 mM HEPES, pH 7.5, 10 mM CaCl2, 0.05% Tween-20), which prevents DNA leaking out of nuclei after MNase digestion (Meers et al., 2019). These wash steps in H4-10 were performed three times in total prior to an incubation at 37°C for 15 min with RNase A in H4-10 (Sigma, R6513, 0.1 mg/mL final). Aliquots of 106 nuclei in 100 μL were then incubated with 5 μL of selected dilutions of MNase (NEB, M0247, 2 x 106 gel units/mL) in H4-10 buffer at 37°C for 15 min. After digestion, nuclei were fixed with 3% formaldehyde (final concentration) in H4-10 buffer at room temperature for 15 min. Throughout MNase treatment and fixation, nuclei were kept in suspension by flicking tubes every 5 minutes. Following fixation, nuclei were spun at 250 x g for 4 min at 4°C followed by aspiration of 90% of the supernatant and resuspension of the pelleted nuclei in cold LB at 107/mL. The wash steps with LB were then repeated twice more. To monitor MNase digestion, DNA from aliquots of MNase-digested, fixed nuclei was purified using 1.8X Ampure XP beads and analyzed on an Agilent 2100 Bioanalyzer (e.g., Figure 2A; Sanders et al., 2017). For this purpose, formaldehyde crosslinks in the fixed nuclei were reversed at 72°C for 10 min followed by digestion of proteins with 2 mg/mL Qiagen protease (QP) at 50°C for 15 min and 80°C for 15 min. Based on the results for the NA12878 cells, we chose nuclei digested with 500 gel units/mL MNase for sorting. Next, the selected sample of MNase-predigested, fixed nuclei was filtered through a 35 μm cell strainer and incubated on ice for at least 15 minutes with Hoechst 33258 (3 μg/mL final) and propidium iodide (3 μg/mL final; Sanders et al., 2017). 30,000 nuclei with BrdU in the G1 phase of the cell cycle were sorted using a mixture of nuclei with and without BrdU (Sanders et al., 2017) as shown in Figure 2B. Nuclei were collected in a tube with 500 μL LB and kept at 4°C. Sorted nuclei that were predigested with MNase and fixed could be kept at 4°C for several weeks in LB before dispensing with the CellenONE (SCIENION US Inc; Phoenix, AZ 85042). After centrifugation at 100 x g for 10 min at 4°C or overnight sedimentation, we removed the supernatant such that nuclei were resuspended at a concentration of 1-1.5 x 105/mL for efficient dispensing of single nuclei.
B. Dispensing nuclei with the CellenONE
Prior to dispensing single nuclei, the CellenONE instrument was turned on and its piezo dispense capillary (PDC) was primed with filtered and degassed water following the manufacturer’s instructions. To prevent evaporation of reagents dispensed in nanoliter volumes during library construction, the dispense platform of the CellenONE instrument was set at 3°C below the ambient dewpoint. An unused 5,184-well nanoarray (ICELL8 150v Chip; 640013, Takarabio.com) or a bespoke nanoarray chip (see below; Figure S1B; Data S1) was placed on the CellenONE dispense deck for 1 minute to allow for temperature equilibration. The precise positions of wells were determined by the fiducial markings on the nanoarray chip with the built-in CellenONE head camera. Next, release buffer (20 mM Tris-acetate, pH 7.9, 50 mM potassium acetate, 0.1% bovine serum albumin, 0.1% Triton X-100, 0.05% Tween-20, 1 mM EGTA, 50 mM NaCl) was aspirated from a 384-well plate with the PDC and 7.4 nL of this buffer was dispensed into each well. For ICELL8 nanoarrays, a subset of the 5,184 wells was filled according to the number of required libraries. Next, a suspension of the nuclei was aspirated and single nuclei (in 0.3 nL) were dispensed into wells prefilled with release buffer (Data S2 and additional information at Cellenion.com). We omitted dispensing of nuclei in some of the wells as negative controls. Nuclei dispensing can be repeated with different nuclei suspensions and control solutions for each cluster in a nanoarray (e.g., different cell types or donors). Alternatively, up to 5,184 nuclei can be dispensed in a single ICELL8 150v nanoarray. Following dispensing, the nanoarray was sealed with non-adhesive film (Microseal ‘A’ Sealing Film, MSA5001, Bio-Rad, with the clear layer replaced with Adhesive PCR Plate Foil, AB0626, ThermoFisher; Data S2) pressed twice into the nanoarray chip using a bespoke pneumatic press (Laks et al., 2019), which at 32 psi generates 265 lbs (1,180 N) on the nanoarray (Data S2). The sealed nanoarray was next spun at 1,700 x g for 1 min at 4°C and incubated at 72°C for 10 min on a thermal cycler to reverse the formaldehyde crosslinks (Data S2).
The bespoke nanoarrays used in library construction, above, were made from fine-finishing 2011 aluminum alloy (40 x 40 x 2.3 mm; Figure S1B; Data S1). 2011 is an exceptionally free machining alloy and so minimizes the growth of burrs when drilling small holes. The use of aluminum allows for good thermal conductivity and reduces the build-up of static electricity. The nanoarrays were made successfully on two different CNC milling machines (an in-house Haas VM-3 and a Matsurra 5 axis machine at a local job shop). We used uncoated miniature drills (Harvey Tool; Rowley, MA 01969; part numbers 20138 for 350 μm holes and 20098 for 250 μm holes). Both library and primer nanoarrays have 16 clusters (4 x 4) containing 49 wells (7 x 7), for a total of 784 wells. The library nanoarrays have 115 nL wells (350 μm diameter, 1,200 μm depth) in clusters surrounded by a trench, which fit 200 μL PCR tubes for collecting libraries. The primer nanoarrays have 31 nL wells (250 μm diameter, 1,000 μm depth), without a surrounding trench, designed to transfer pre-spotted index primers into the library nanoarray. After CNC machining, nanoarrays were cleaned with 70% isopropanol to remove residual oil and debris. Once dry, nanoarrays underwent a wet bath silane adhesion promotion and plasma treatment and were then coated with 3-4 μm of Parylene-C to ensure biocompatibility (Plasma Ruggedized Solutions; San Jose, California 95131).
C. Removal of proteins from DNA fragments
After cooling to 4°C, the nanoarray was returned to the CellenONE deck, the seal was removed, and 0.3 nL of 5 mg/mL QP in release buffer was dispensed into each well. The final concentration of the protease was 0.2 mg/mL and the total reaction volume was 8 nL. Next, the nanoarray was sealed and centrifuged as before (Data S2) and incubated at 50°C for 15 min (protease active) and 80°C for 15 min (to inactivate the protease) before cooling to 4°C on the thermal cycler. At this point, the nanoarray can be stored at 4°C.
D. End repair and A-tailing of DNA fragments
A fresh 3X polishing mastermix was prepared by mixing 10.4 μL polishing buffer (23.1 mM Tris-acetate, pH 7.9, 57.7 mM potassium acetate, 34.6 mM magnesium acetate, 0.058% Tween-20, 34.6 mM DTT, 0.346 mM dNTP mix, 3.12 mM dATP, 4.62 mM ATP) and 1.6 μL polishing enzyme mix (0.6 U/μL T4 DNA Polymerase, 6.75 U/μL T4 Polynucleotide Kinase, 0.625 U/μL Taq DNA Polymerase). The total volumes of all mastermixes should be adjusted based on the number of wells used in the nanoliter array. 4 nL of this solution was dispensed into each well for a reaction volume of 12 nL. Following sealing and centrifugation of the nanoarray as before, it was incubated on a thermocycler at 12°C for 15 min, 37°C for 15 min (end repair) and 72°C for 20 min (A-tailing by Taq polymerase), followed by cooling to 4°C.
E. Ligation to forked adapters
A fresh 4X adapter ligation mastermix was prepared by mixing 9.6 μL ligation buffer (25 mM Tris-acetate, pH 7.9, 62.5 mM potassium acetate, 12.5 mM magnesium acetate, 0.0625% Tween-20, 12.5 mM DTT, 1.66 mM ATP), 0.8 μL pre-annealed iTru Adapters (24 μM; Table S1; Glenn et al., 2019), and 1.6 μL ligase. T4-type ligases were from Codexis (Redwood City, CA 94063) or NEB (Quick ligase). For ligation with Quick ligase (NEB), an adapter ligation mastermix was prepared by mixing 4.8 μL 2X ligation buffer (50 mM Tris-acetate, pH 7.9, 125 mM potassium acetate, 25 mM magnesium acetate, 0.125% Tween-20, 25 mM DTT, 3.33 mM ATP), 0.8 μL pre-annealed iTru Adapters (24 μM; Table S1 μL Ultrapure distilled water, and 3.2 μL Quick ligase (Figure S2C). The sealed nanoarray was returned to the cooled dispense platform of the CellenONE, the seal was removed and 4 nL of the mastermix was dispensed into each well of the nanoarray resulting in a reaction volume for ligation of 16 nL per well. Following sealing and centrifugation as before, the nanoarray was incubated at 20°C for 30 min for ligation (alternatively: overnight at 16°C) and cooled to 4°C on a thermal cycler.
F. Enzyme inactivation by protease
A fresh mastermix of Hoechst 33258 and QP was prepared by mixing 0.56 μL of 100 μg/mL Hoechst 33258, 2.78 μL of 5 mg/mL QP, and 4.16 μL of Ultrapure distilled water. Similar to step C above, the nanoarray was placed on the cooled dispense platform, the seal was removed, and 2.5 nL of the thermolabile protease and Hoechst 33258 was dispensed into each well of the nanoarray. The concentration of protease was 0.25 mg/mL and the concentration Hoechst dye was 1 μg/mL, with a total reaction volume of 18.5 nL per well. Following this dispense step, the nanoarray, sealed and centrifuged as before, was incubated on the thermocycler at 50°C for 15 min and 80°C for 15 min prior to cooling to 4°C.
G. UV treatment to nick DNA with BrdU
Following return to the cooled CellenONE deck, the seal was removed, a 365 nm wavelength UV lamp (Dymax BlueWave MX-250 LED UV) was placed 2.5 cm above the nanoarray in a dedicated lamp holder, and the nanoarray was irradiated at 10% intensity for 150 seconds (Data S2).
H. Transfer of index primers for PCR
Each library well requires a unique pair of Tru5 and Tru7 index primers (Glenn et al., 2019; Table S1). Lyophilized index primers in master plates were reconstituted in buffer EB (10 mM Tris-HCl, pH 8.5) at a concentration of 100 μM. Primers were diluted in working plates to 10 μM in 10 mM Tris-HCl, pH 8.5 and 0.1 mM EDTA. Prior to library construction, 4.8 nL of an Tru5 index primer and 4.8 nL of an Tru7 index primer were pre-spotted in each well of a bespoke primer-specific nanoarray, at positions that were the mirror image of the desired positions in the library nanoarray (i.e., rows and columns exchanged; Table S1). Alternatively, if an ICELL8 library nanoarray was used, a second ICELL8 nanoarray was used for index primer transfer. After each primer was dispensed, the PDC was rinsed with 0.1% Tween-20. The CellenONE temperature was set to 2°C above the ambient dewpoint to prevent condensation from forming on primer nanoarrays while still maintaining minimal evaporation of working stock primers in the 384-well plate. The dispense platform accommodates up to six primer nanoarrays (Data S2). After spotting all index primers (16 x Tru5 and 49 x Tru7), the wells were allowed to dry out in a laminar flow hood and then overlayed with non-adhesive film, wrapped with Parafilm, and stored at -20°C.
On the day of library construction, a primer nanoarray was thawed and primer mixtures were reconstituted by dispensing 7.5 nL water into each well. The primer nanoarray was sealed and centrifuged as before (Data S2), then incubated at 37°C for 30 min to reconstitute the primers. Details of the transfer of primers from the primer nanoarray to the library nanoarray are shown in a video contained in Data S2. Briefly, after cooling both nanoarrays on the CellenONE deck and removal sealing films, the nanoarrays were placed face-to-face in a bespoke primer transfer device. They were carefully aligned and immobilized using position pins, clamping, and a lid secured by screws, then frozen rapidly on dry ice. The transfer device was placed in a centrifuge adapter and centrifuged at 4,000 rpm for 10 min at 0°C. Upon reaching a centrifuge speed of 4,000 rpm, centrifugation was continued at 12°C, resulting in transfer of 7.5 nL of Tru5 and Tru7 index primers into the corresponding mirrored well in the library nanoarray for a volume of 26 nL per well. The primer transfer device was returned to the CellenONE dispense platform for temperature equilibration before the library nanoarray was removed.
A fresh PCR mastermix was prepared by mixing 42 μL PCR buffer (2.29X KAPA HiFi Fidelity Buffer, 0.51 mM dCTP, 0.51 mM dGTP, 0.51 mM dTTP, 7.62 mM EDTA) with 2 μL of KAPA HiFi HotStart DNA polymerase. dATP was not added because enough persists after its addition in the polishing step until PCR. 22 nL of PCR mastermix was dispensed into each well for a final reaction volume of 48 nL per well. Following sealing and centrifuging as before, the library nanoarray was incubated at 95°C for 3 min, 13 cycles of (98°C for 20 s, 57°C for 30 s, 72°C for 30 s), 72°C for 5 min, and 4°C hold on a thermal cycler. Lids were removed from PCR tubes, which were placed in a collection device (Figure S1B). The seal from the nanoarray was removed and the nanoarray was placed face down on top of the PCR tubes, then centrifuged at 4,000 rpm for 5 min at 4°C. Pools of up to 49 libraries from each cluster were collected in a volume of 2.4 μL, which was topped to 5 μL with Ultrapure distilled water prior to analysis and purification (final concentrations of reagents shown in Table S1).
I. Size selection, QC, and sequencing
To examine library quality, 1 μL of each library pool sample was analyzed on a Bioanalyzer using the High Sensitivity DNA Assay. DNA fragments with a size of >200 bp were selected for sequencing after running for 15 min on a 2% E-Gel EX Agarose Gel and purification with the Zymoclean Gel DNA Recovery Kit to remove primer and adapter dimer contamination as previously described (Sanders et al., 2017). We then determined the average library DNA size and confirmed primer and adapter dimer removal with a High Sensitivity DNA Assay on the Bioanalyzer. The DNA concentration in the purified sample was measured using a Qubit fluorometer prior to sequencing. Libraries were prepared for loading onto a NextSeq 550 system following Illumina guidelines for denaturation and dilution (76 bp PE reads, dual 8 bp indexes; Table S1). For the OP-Strand-seq NA12878 libraries, a mid-output NextSeq kit was used for initial shallow sequencing, and two clusters were then selected for a high-output run.
J. Bioinformatic analysis
For the NA12878 libraries, we removed adapters from demultiplexed FASTQ files using Cutadapt (Martin, 2011), discarded reads shorter than 30 bp, and trimmed bases with quality less than 15 from the ends of reads. We then aligned reads to the GRCh38 human reference with Bowtie2 (Langmead and Salzberg, 2012), kept only those reads with MAPQ of at least 10 mapping to chromosomes 1-22, X, and Y, and marked duplicate reads with Picard (https://github.com/broadinstitute/picard/). Nearly the same procedure was used for the comparisons with data from Laks et al. (2019) and Porubsky et al. (2016), although for the Laks data we did not restrict the analysis to chromosomes 1-22, X, and Y (this has a negligible effect on the estimated complexity). For the complexity analysis, we used default settings with bam2mr and gc_extrap from the Preseq suite (Daley and Smith, 2014; Figure S3). We identified libraries with good Strand-seq characteristics manually or with ASHLEYS (Gros et al., 2021). For the SNV phasing analysis, we chose Watson-Crick (WC; i.e., with reads in both orientations) regions using a bin size of 1 Mb, and we ran StrandPhaseR on chromosomes 1-22 and X with num.iterations set to 4 (Porubsky et al., 2017). For the CNV analysis, AneuFinder (Bakker et al., 2016) was run with binsize 200 kb, use.bamsignals set to FALSE, method set to ‘edivisive’, strandseq set to TRUE, confint set to 0.99, and refine.breakpoints set to TRUE. The variable-width reference was composed of good-quality OP-Strand-seq NA12878 libraries with WC strand state for chr16 and no large CNVs (confirmed with previous AneuFinder runs). Regions of the genome with anomalously high or low coverage in a previous experiment (data not shown) were blacklisted.
Quantification and statistical analysis
Statistics for the comparisons of OP-Strand-seq libraries with and without 5-FdU can be found in the results and discussion and Figure 3. P-values are from two-sided, unpaired t-tests with unequal variance, and p-values below 0.05 are considered significant.
Acknowledgments
We thank Stephen Pleasance, Hans Zahn, Steven Henikoff, Sheila Teves, and Geraldine Aubert for discussions. Yas Oloumi, Hans Thole, Collin Mazeppa, and Scott Young are thanked for making custom parts. Christian Steidl and Liz Chavez are thanked for discussions and use of Illumina sequencers. This work was funded by a program project grant (#1074) from the Terry Fox Research Foundation and a research grant (#159787) from the Canadian Institutes of Health Research.
Author contributions
P.M.L. conceived the approach. V.C.T.H., D.D.C., Z.H., Y.W., D.C.J.S., and P.M.L. contributed to protocol development and troubleshooting. R.J.N.C. made the bespoke nanoliter arrays and other bespoke equipment. V.C.T.H. and C.-A.M. analyzed the sequence data. V.C.T.H., D.D.C., D.C.J.S., and P.M.L. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Published: January 24, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100150.
Contributor Information
Vincent C.T. Hanlon, Email: vhanlon@bccrc.ca.
Peter M. Lansdorp, Email: plansdor@bccrc.ca.
Supporting citations
The following references appear in the supplemental information: Chiaromonte et al. (2002).
Supplemental information
A description of each item is contained in the Data S1 file.
A detailed description of each video is contained in the Data S2 file.
Data and code availability
-
•
The single-cell Strand-seq data have been deposited in the NCBI Sequence Read Archive and are publicly available. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.
References
- Aird D., Ross M.G., Chen W.S., Danielsson M., Fennell T., Russ C., Jaffe D.B., Nusbaum C., Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12 doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker B., Taudt A., Belderbos M.E., Porubsky D., Spierings D.C.J., de Jong T.V., Halsema N., Kazemier H.G., Hoekstra-Wakker K., Bradley A., et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 2016;17 doi: 10.1186/s13059-016-0971-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brind’Amour J., Lansdorp P.M. Analysis of repetitive DNA in chromosomes by flow cytometry. Nat. Methods. 2011;8:484–486. doi: 10.1038/nmeth.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho C.M.B., Lupski J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaromonte F., Yap V.B., Miller W. Scoring pairwise genomic sequence alignments. Pac. Symp. Biocomput. 2002:115–126. doi: 10.1142/9789812799623_0012. [DOI] [PubMed] [Google Scholar]
- Daley T., Smith A.D. Modeling genome coverage in single-cell sequencing. Bioinformatics. 2014;30:3159–3165. doi: 10.1093/bioinformatics/btu540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert P., Audano P.A., Zhu Q., Rodriguez-Martin B., Porubsky D., Bonder M.J., Sulovari A., Ebler J., Zhou W., Mari R.S., et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372 doi: 10.1126/science.abf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis P., Moore L., Sanders M.A., Butler T.M., Brunner S.F., Lee-Six H., Osborne R., Farr B., Coorens T.H.H., Lawson A.R.J., et al. Reliable detection of somatic mutations in solid tissues by laser-capture microdissection and low-input DNA sequencing. Nat. Protoc. 2021;16:841–871. doi: 10.1038/s41596-020-00437-6. [DOI] [PubMed] [Google Scholar]
- Falconer E., Hills M., Naumann U., Poon S.S.S., Chavez E.A., Sanders A.D., Zhao Y., Hirst M., Lansdorp P.M. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods. 2012;9:1107–1112. doi: 10.1038/nmeth.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W., Lai B., Ni B., Zhao K. Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq. Nat. Protoc. 2020;15:68–85. doi: 10.1038/s41596-019-0243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn T.C., Nilsen R.A., Kieran T.J., Sanders J.G., Bayona-Vásquez N.J., Finger J.W., Pierson T.W., Bentley K.E., Hoffberg S.L., Louha S., et al. Adapterama I: Universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext) PeerJ. 2019;7 doi: 10.7717/peerj.7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros C., Sanders A.D., Korbel J.O., Marschall T., Ebert P. ASHLEYS: automated quality control for single-cell Strand-seq data. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laks E., McPherson A., Zahn H., Lai D., Steif A., Brimhall J., Biele J., Wang B., Masud T., Ting J., et al. Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell. 2019;179:1207–1221. doi: 10.1016/j.cell.2019.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Meers M.P., Bryson T.D., Henikoff J.G., Henikoff S. Improved cut&run chromatin profiling tools. Elife. 2019;8 doi: 10.7554/eLife.46314.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman M., Sundling S., Grönberg H., Hall P., Czene K., Lindberg J., Klevebring D. Library preparation and multiplex capture for massive parallel sequencing applications made efficient and easy. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan A., Pelleri M.C., Antonaros F., Strippoli P., Caracausi M., Vitale L. On the length, weight and GC content of the human genome. BMC Res. Notes. 2019;12 doi: 10.1186/s13104-019-4137-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D., Sanders A.D., Van Wietmarschen N., Falconer E., Hills M., Spierings D.C.J., Bevova M.R., Guryev V., Lansdorp P.M. Direct chromosome-length haplotyping by single-cell sequencing. Genome Res. 2016;26:1565–1574. doi: 10.1101/gr.209841.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D., Garg S., Sanders A.D., Korbel J.O., Guryev V., Lansdorp P.M., Marschall T. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-01389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D., Ebert P., Audano P.A., Vollger M.R., Harvey W.T., Marijon P., Ebler J., Munson K.M., Sorensen M., Sulovari A., et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D., Sanders A.D., Taudt A., Colomé-Tatche M., Lansdorp P.M., Guryev V. BreakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics. 2020;36:1260–1261. doi: 10.1093/bioinformatics/btz681. [DOI] [PubMed] [Google Scholar]
- Quail M.A., Otto T.D., Gu Y., Harris S.R., Skelly T.F., McQuillan J.A., Swerdlow H.P., Oyola S.O. Optimal enzymes for amplifying sequencing libraries. Nat. Methods. 2012;9:10–11. doi: 10.1038/nmeth.1814. [DOI] [PubMed] [Google Scholar]
- Sanders A.D., Falconer E., Hills M., Spierings D.C.J., Lansdorp P.M. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc. 2017;12:1151–1176. doi: 10.1038/nprot.2017.029. [DOI] [PubMed] [Google Scholar]
- Zook J.M., Chapman B., Wang J., Mittelman D., Hofmann O., Hide W., Salit M. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 2014;32:246–251. doi: 10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A description of each item is contained in the Data S1 file.
A detailed description of each video is contained in the Data S2 file.
Data Availability Statement
-
•
The single-cell Strand-seq data have been deposited in the NCBI Sequence Read Archive and are publicly available. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.