

Summary
Oncogenic mutations in KRAS can be recognized by T cells on specific class I human leukocyte antigen (HLA-I) molecules, leading to tumor control. To date, the discovery of T cell targets from KRAS mutations has relied on occasional T cell responses in patient samples or the use of transgenic mice. To overcome these limitations, we have developed a systematic target discovery and validation pipeline. We evaluate the presentation of mutant KRAS peptides on individual HLA-I molecules using targeted mass spectrometry and identify 13 unpublished KRASG12C/D/R/V mutation/HLA-I pairs and nine previously described pairs. We assess immunogenicity, generating T cell responses to nearly all targets. Using cytotoxicity assays, we demonstrate that KRAS-specific T cells and T cell receptors specifically recognize endogenous KRAS mutations. The discovery and validation of T cell targets from KRAS mutations demonstrate the potential for this pipeline to aid the development of immunotherapies for important cancer targets.
Keywords: cancer, HLA, immunotherapy, mass spectrometry, neoantigen, PRM, T cell, TCR, RAS
Graphical abstract
Highlights
-
•
A pipeline is developed to evaluate whether T cells can recognize cancer targets
-
•
Mass spectrometry is used to identify distinct KRAS mutation/HLA-I pairs
-
•
Nearly all KRAS mutation/HLA-I pairs tested can lead to a T cell response
-
•
TCRs lead to cytotoxicity of tumor cells with the cognate KRAS mutation/HLA-I pair
Motivation
We created a pipeline for the systematic discovery and validation of T cell targets for the development of targeted cancer immunotherapies. This pipeline addresses the two critical aspects of a T cell target: presentation on an HLA molecule and immunogenicity. Applying our methodologies to the comprehensive analysis of common KRAS mutations, we demonstrated the therapeutic potential of our pipeline to increase patient populations that can be reached with T cell-based therapies.
T cell-based therapies for cancer rely on targets that are both presented on specific HLA molecules and are immunogenic. Choi et al. establish a systematic pipeline to evaluate both aspects sensitively and apply it to common KRAS mutations to broadly explore T cell targets from this oncogene.
Introduction
Globally, mutations in Ras genes account for around 3.4 million cancer diagnoses each year (Prior et al., 2020). A recently published meta-analysis of the four preeminent cancer genetics databases (i.e., COSMIC, cBioPortal, ICGC, and TCGA) revealed that RAS mutations are associated with approximately 19% of yearly cancer diagnoses in the United States, with KRAS mutations comprising roughly 75% of that total (Prior et al., 2020). KRAS mutations frequently occur in several common cancers, including non-small cell lung cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. Missense mutations at codon 12 are most common, promoting carcinogenesis through a gain-of-function action that leads to an increase in cell growth and survival (Simanshu et al., 2017).
KRAS mutations are detectable in a sizable portion of human cancers, are tumor specific, and critical to tumor cell biology, making them attractive therapeutic targets. There have been several attempts to drug mutant RAS proteins since their discovery in the 1980s, with substantial progress being made in recent years (Bar-Sagi et al., 2020). Small-molecule drugs that target the cysteine amino acid from the KRAS codon 12 mutation (KRASG12C) are the most advanced (Hong et al., 2020; Mullard, 2019). While KRASG12C is the most common KRAS mutation in non-small-cell lung cancer, this mutation is less prominent in other cancers. Accordingly, there is a significant need to develop therapies that target KRAS mutations without the limitations inherent to some small-molecule drug and amino acid combinations.
Within the past decade, immunotherapies have become an increasingly effective way to treat cancer, and this therapeutic modality has been shown to control tumor cells with KRAS mutations. In particular, seminal work from the Rosenberg and Yang laboratories at the National Cancer Institute established the potential of T cell immunotherapy targeting KRASG12D/V mutations in the context of specific human leukocyte antigen (HLA) molecules (Tran et al., 2016; Wang et al., 2016; Yossef et al., 2018). Their data demonstrate that HLA molecules can present KRAS mutant peptides (also known as “neoantigens”) at levels recognizable by T cells and establish a paradigm for the therapeutic utility of T cells targeting KRAS mutations.
While important, these discoveries relied on isolating T cells reactive to KRAS mutations from patient samples, such as peripheral blood mononuclear cells (PBMCs) or tumors, or by using transgenic mouse models, which are only available for a limited number of alleles. A recent report from Bear et al. (2021) overcame some of these constraints by using engineered cell lines and T cell induction assays from healthy donor cells, but only profiled four HLA-I alleles. Here, we established a systematic cell-based pipeline to discover and validate therapeutically relevant KRAS T cell targets across any HLA-I allele and applied this pipeline to 10 HLA-I alleles. We evaluated the HLA-I presentation of mutant KRAS peptides using targeted mass spectrometry (MS) to detect peptides isolated from cells engineered to simultaneously express four KRAS codon 12 mutations and a specific class I HLA (HLA-I) molecule. Using a T cell induction protocol, we generated naive T cell responses against nearly all mutant KRAS peptides that were detected by MS, confirming their immunogenicity.
Through this antigen discovery pipeline, we identified 13 unpublished KRASG12C/D/R/V mutation/HLA-I molecule pairs that have not been reported in the literature, thereby more than doubling the number of T cell targets from KRAS mutations that can potentially be targeted therapeutically. Moreover, cytotoxicity assays demonstrated that the induced T cells or T cell receptor (TCR)-transduced T cells could recognize the KRASG12V neoantigens on target tumor cell lines that naturally express the KRASG12V mutation. These T cells do not cross-react with the wild-type KRAS peptide presented on the same HLA-I molecule. We believe that this scalable discovery and validation pipeline will help drive the development of focused immunotherapies for KRAS and other important cancer targets.
Results
Establishment of a pipeline to evaluate KRAS neoantigens
There have been multiple efforts to discover KRAS neoantigens through epitope prediction or structural modeling of peptide-HLA interactions (Bai et al., 2021; Castle et al., 2019; Wu et al., 2018). However, these primarily leverage affinity-based predictors, which rely on traditional binding assays and a priori knowledge of the peptides to be assayed. As an alternative approach, we used HLAthena (Sarkizova et al., 2020), an MS-based predictor trained using endogenous ligands processed and presented by cells. All 8-11mers from KRASG12C/D/R/V with the mutated amino acid were predicted for the 12 most common HLA-A, B, and C alleles in the United States population (Data S1). As an initial evaluation, HLA-I alleles for which there was at least one KRAS-mutated 9mer or 10mer peptide predicted to bind with a 2% rank or lower were selected for further validation (Table S1). These included all four HLA-I alleles previously shown to present at least one KRAS neoantigen (Bear et al., 2021; Douglass et al., 2021; Tran et al., 2016; Wang et al, 2016, 2019).
To evaluate the predicted HLA-I neoantigens, we established a systematic pipeline to assess the fundamental criteria of an antigen target for a T cell, including whether the neoantigen is presented on cells with the mutation and if the neoantigen can elicit a T cell response (Figure 1). First, we evaluated the presentation of the reported neoantigens using targeted MS. Next, we used a T cell induction protocol to elicit naive T cell responses. To expedite the evaluation of these criteria, we engineered the malignant melanoma-derived A375 cell line to stably express each of the four most common KRAS codon 12 mutations simultaneously and an HLA-I molecule of interest. Together, we generated multiple double-transduced A375 cell lines containing KRASG12C/D/R/V mutations and a specific HLA-I allele. For a subset of the neoantigens, we assessed both induced T cell and cloned TCR functionality using cytotoxicity assays with target cancer cells that naturally harbor the KRASG12V mutation.
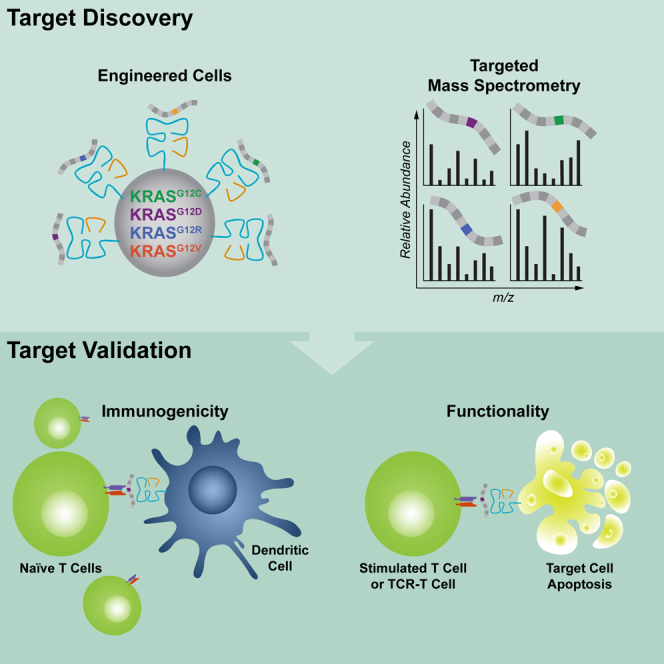
Figure 1.

Establishment of a pipeline to discover and validate KRAS neoantigens
Overview of the cell-based systematic pipeline established to discover and validate KRAS neoantigens using targeted MS (top) and an in vitro T cell stimulation protocol (middle). A375 cells were engineered to simultaneously express four KRASG12C/D/R/V mutations and an HLA-I molecule of interest. After purifying HLA-I peptides, targeted MS was used to detect individual KRAS neoantigens (lines with a colored box) in the presence of endogenous peptides that are also presented by the transduced HLA-I molecules (gray lines). An in vitro stimulation protocol was used to elicit naive T cell responses against the detected KRAS neoantigens. For a subset of the detected neoantigens (dashed arrows represent expanded pipeline), a cytotoxicity assay was performed to show that induced T cells can kill tumor cell lines that naturally express the KRASG12V mutation. Cytotoxicity assays were also used to demonstrate that cloned TCRs can kill tumor cells that naturally present the KRASG12V neoantigen.
Targeted MS of HLA-I peptides
Parallel reaction monitoring (PRM) is a targeted MS-based data acquisition approach that utilizes high-resolution mass spectrometers to selectively detect predefined peptides in complex samples (Peterson et al., 2012). We used PRM to detect both previously reported KRAS neoantigens (Bear et al., 2021; Douglass et al., 2021; Tran et al., 2016; Wang et al, 2016, 2019) and unpublished KRAS neoantigens since profiling-based MS data acquisition approaches, such as data-dependent acquisition can miss low abundance peptides (Peterson et al., 2012). For this, we engineered A375 cells to concurrently and stably express mutant KRAS gene segments (i.e., KRASG12C/D/R/V) with the first 35 amino acids of each variant, then further engineered the cells to express individual HLA-I molecules stably. In the absence of specific antibodies for each HLA-I allele, we inferred robust expression either from the expression of HLA-C or from decreased expression of HLA-A∗02:01, which implies out competition for β2 microglobulin by the transduced allele over the endogenous (Figure S1A). For most HLA-I alleles studied, the HLA-I molecules contained a biotin acceptor peptide (BAP) affinity tag located on the heavy chain that facilitated the isolation of the tagged HLA-I molecule using an approach termed mono-allelic purification with tagged allele constructs (MAPTAC) (Abelin et al., 2019). Immunoprecipitation was used to isolate an HLA-I molecule that did not have a BAP tag and endogenously expressed HLA-I molecules of interest when present. We also used synthetic peptide standards to confirm each detection by PRM.
Overall, we detected 30 individual KRASG12C/D/R/V neoantigens across HLA-A, HLA-B, and HLA-C alleles by PRM (Table 1). A list of neoantigens that we did not detect by PRM is also included in Table S1. These results are notable for several reasons. First, 20 of these discoveries are KRAS neoantigens presented by common HLA-I alleles that have not been previously reported. For example, the unpublished KRAS neoantigens include KRASG12V on HLA-A∗30:01, HLA-A∗68:01, HLA-C∗01:02, HLA-C∗03:03, and HLA-C∗03:04, at least one of which is present in roughly one-third of the United States population. Second, most HLA-I alleles present neoantigens from only one or two of the four evaluated KRAS mutations, even in cases where the mutated amino acid resides at a position that is not expected to strongly affect binding to the HLA-I molecule (i.e., anchor residues) (Figure S2). Third, we did not detect KRAS neoantigens presented on HLA-A∗02:01 from any of the evaluated mutations despite a previous publication suggesting possible presentation of a mutant KRAS epitope on this common allele (Kubuschok et al., 2006). Our evaluation included looking for HLA-A∗02:01 neoantigens in engineered cell lines and tumor cell lines and is concordant with recent publications (Bear et al., 2021; Douglass et al., 2021).
Table 1.
Summary of KRAS neoantigens detected by targeted MS
| HLA-I | KRAS mutation | Peptide | Peptide length | Reference |
|---|---|---|---|---|
| A∗03:01 | wild-type | ∗VVVGAGGVGK | 10 | this article |
| G12C | VVVGACGVGK | 10 | this article | |
| G12D | VVVGADGVGK | 10 | Wang et al. (2019) and this article | |
| G12R | VVVGARGVGK | 10 | Bear et al. (2021) (9mer) and this article (10mer) | |
| G12V | ∗VVVGAVGVGK | 10 | Douglass et al. (2021) and this article | |
| G12V | ∗VVGAVGVGK | 9 | Douglass et al. (2021) and this article | |
| A∗11:01 | G12C | VVVGACGVGK | 10 | this article |
| G12D | VVVGADGVGK | 10 | Wang et al. (2016) and this article | |
| G12R | VVVGARGVGK | 10 | Bear et al. (2021) (9mer) and this article (10mer) | |
| G12V | ˆVVVGAVGVGK | 10 | Wang et al. (2016) and this article | |
| G12V | VVGAVGVGK | 9 | Wang et al. (2016) and this article | |
| A∗30:01 | G12R | VVVGARGVGK | 10 | this article |
| G12R | VVGARGVGK | 9 | this article | |
| G12V | VVVGAVGVGK | 10 | this article | |
| G12V | VVGAVGVGK | 9 | this article | |
| A∗68:01 | G12C | VVVGACGVGK | 10 | this article |
| G12D | VVVGADGVGK | 10 | this article | |
| G12R | VVVGARGVGK | 10 | this article | |
| G12V | VVVGAVGVGK | 10 | this article | |
| G12V | VVGAVGVGK | 9 | this article | |
| B∗07:02 | G12D | GADGVGKSAL | 10 | Bear et al. (2021) and this article |
| G12R | GARGVGKSAL | 10 | Bear et al. (2021) and this article | |
| C∗01:02 | G12V | AVGVGKSAL | 9 | this article |
| C∗03:03 | G12V | GAVGVGKSAL | 10 | this article |
| G12V | GAVGVGKSA | 9 | this article | |
| C∗03:04 | G12C | GACGVGKSA | 9 | this article |
| G12D | GADGVGKSAL | 10 | this article | |
| G12V | GAVGVGKSAL | 10 | this article | |
| G12V | GAVGVGKSA | 9 | this article | |
| C∗08:02 | G12D | GADGVGKSAL | 10 | Tran et al. (2016) and this article |
| G12D | GADGVGKSA | 9 | Tran et al. (2016) and this article |
KRAS G12C/D/R/V neoantigens detected by PRM in this article as well as those reported in other publications are indicated (first report referenced). Peptides detected in the NCH-H44I cell line are marked with (∗), and the peptide detected in the SW620 cell line is marked with (ˆ).
To demonstrate that we could detect KRAS neoantigens at physiological levels by MS, we targeted KRAS neoantigens in two different cell lines that naturally express KRAS mutations. Both the SW620 colon cancer cell line and the NCI-H441 lung cancer cell line naturally harbor the KRASG12V mutation. However, we engineered the SW620 cells to express the BAP-tagged HLA-A∗11:01 allele, whereas the NCI-H441 cells endogenously express HLA-A∗03:01. Following the isolation of HLA-I molecules, we targeted KRASG12V neoantigens in the SW620A∗11:01 and non-engineered NCI-H441 cells. We detected the KRASG12V neoantigens in both cell lines and a wild-type peptide in NCI-H441 cells (Table 1). Since heavy isotope-labeled internal standards were spiked into the SW620 and NCI-H441 samples, it enabled us to calculate the amount of each neoantigen that was recovered from sample processing and detected by MS. We estimate that we detected approximately 124 amol of the 10mer KRASG12V neoantigen in the SW620 sample and around 894 and 109 amol of the 10mer and 9mer KRASG12V neoantigens from the NCI-H441 sample, respectively (Data S1). Extrapolating these recovered peptide values, we estimate that we detected <10 peptides per cell of both the 10mer and 9mer KRASG12V neoantigens (Data S1), which is consistent with a recent study for the 10mer KRASG12V peptide in the same cells (Douglass et al., 2021). Figure 2 shows the spectra for the 10mer KRASG12V neoantigen in the engineered A375 cells and the two cancer cell lines. The mass spectra for all the KRAS neoantigen peptides detected by MS and the corresponding synthetic peptides used to confirm the detections are in Data S2 and the extracted ion chromatogram values used to create the spectral visualizations are included in Data S1. Together, these data highlight the sensitivity of targeted MS assays that facilitated the discovery of KRAS neoantigens predicted to be presented by specific HLA-I molecules.
Figure 2.

Representative targeted MS data
Targeted MS was used to detect the 10mer KRASG12V HLA-A3 superfamily neoantigen VVVGAVGVGK in (A) A375 cells engineered to express both the KRASG12V mutation and HLA-A∗11:01, (B) SW620 cells that naturally express the KRASG12V mutation but were engineered to express HLA-A∗11:01, and (C) NCI-H441 cells that naturally express both the KRASG12V mutation and HLA-A∗03:01. The top panels specify the KRAS mutation and HLA-I molecule present in each sample. The middle panels contain the mass spectra for the peptide detected in each sample (top), and the mass spectra for the heavy isotope-labeled synthetic peptide (bottom) used to confirm the detections. The bottom panels show mass error in parts per million (ppm) of the a-ions (purple), b-ions (blue), and y-ions (red) for the endogenous (top) and heavy isotope-labeled synthetic (bottom) mass spectra. Data from the same heavy isotope-labeled synthetic peptide was used in this figure. See also Data S2.
KRAS mutant-specific T cells can be induced to epitopes detected via MS
To evaluate whether the epitopes observed via targeted MS can also elicit naive T cell responses, we used a T cell induction protocol using healthy human donors with at least one HLA-I allele of interest for a subset of MS-observed epitopes (i.e., HLA-A∗03:01, HLA-A∗11:01, HLA-A∗68:01, HLA-B∗07:02, HLA-C∗01:02, HLA-C∗03:04, or HLA-C∗08:02). T cell responses were evaluated after one to two rounds of stimulation with minimal epitope peptides and assessed via peptide-HLA-I multimers (Hadrup et al., 2009). For nearly all KRAS neoantigens tested, we induced T cell responses in at least one healthy donor (Figures 3A and S3; Table S2). For KRASG12C on HLA-A∗68:01, we did not detect a T cell response in the one healthy donor tested. Combined with the MS analyses, these data confirm that there are bona fide immunogenic neoantigen targets from all four KRAS mutations studied on the common HLA-I alleles we tested.
Figure 3.

Evaluation of KRAS neoantigen immunogenicity in healthy donors
(A) Flow cytometry plots of pHLA staining of KRAS neoantigens after naive T cell inductions in healthy donors with the appropriate HLA-I molecules. Multimer-positive populations are circled in red. Naive T cell inductions and multimer analysis were performed for all neoantigens detected by MS, except for KRASG12C on HLA-A∗68:01. See also Figure S3.
(B) Cytotoxicity assay evaluating the specificity of multiple independent T cell cultures stimulated against KRASG12V on HLA-A∗11:01. T cell cultures with identifiable multimer-positive populations (left) were co-cultured with A375 cells transduced to express HLA-A∗11:01 and loaded with increasing amounts of the KRASG12V/HLA-A∗11:01 neoantigens (red dots) or a single concentration of the matching wild-type peptides (blue dots). Data are presented as mean ± SD.
(C–E) Cytotoxicity assay evaluating the ability of a representative T cell culture to recognize endogenously processed and presented KRASG12V neoantigens.
(C) Total HLA-I expression was evaluated on SW620 cells transduced to express HLA-A∗11:01 (red) compared with non-transduced SW620 cells (blue) or isotype control-stained cells (orange). See also Figure S1.
(D and E) Cytotoxicity assays used to evaluate the ability of representative T cell cultures to recognize endogenously processed and presented KRASG12V neoantigens where (D) T cells were co-cultured with SW620A∗11:01 cells and annexin V was measured over time (red) compared with SW620A∗11:01 cells alone (blue) and (E) T cells were co-cultured with NCI-H441 cells, and annexin V was measured over time (red) compared with NCI-H441 cells alone (blue).
Data are presented as mean ± SD and are representative of two independent experiments. See also Figure S4.
We induced T cell responses in >50% of healthy donors for most epitopes, except for HLA-A∗03:01 (Table S2). For this HLA-I allele, T cell responses were induced in 20%–55% of healthy donors. By contrast, we can induce T cell responses in >75% of HLA-A∗11:01+ donors, another commonly expressed member of the HLA-A3 superfamily. Although the same epitopes bind to these HLA-I molecules with a strong affinity (most at <500 nM), the epitopes are more stable on HLA-A∗11:01 (Table S3). This increased stability for the same epitopes on HLA-A∗11:01 versus HLA-A∗03:01 may explain the increased response to the former, as the stability of epitopes on HLA-I molecules has previously been correlated with immunogenicity (Harndahl et al., 2012).
To evaluate the ability of T cells to distinguish the mutant epitope from the wild-type version, we performed a kinetic annexin V-based cytotoxicity assay with T cell populations from independent cell cultures recognizing a KRASG12V neoantigen on HLA-A∗11:01. Using target cells without peptide loading as a baseline, all the T cell responses tested killed target cells loaded with KRASG12V mutant peptide, but not with KRAS wild-type peptide, even at relatively high concentrations (Figure 3B). By titrating the KRASG12V mutant peptide, we found the half-maximal effective concentration of all tested responses to be less than 5 nM (Figure 3B).
To determine if the functional avidities of the TCRs from the induced T cells are sufficient for recognition of KRAS neoantigens at physiological levels, we performed cytotoxicity assays with the same two human cell lines that naturally contain KRASG12V mutations that were used for MS analysis, SW620, and NCI-H441. Although SW620 cells were transduced to express HLA-A∗11:01, the overall surface expression of HLA-I molecules is unchanged relative to the parental cell line with minimal downregulation of the endogenously expressed HLA-A∗02, demonstrating that the transgenic HLA-A∗11:01 is expressed at comparable levels with endogenous HLA-I molecules (Figures 3C and S1B). Significant cytotoxicity of the HLA-A∗11:01-expressing SW620 cells was observed only upon co-culture with a representative T cell induction against KRASG12V on HLA-A∗11:01 (Figure 3D). Importantly, this same T cell population induced significant cytotoxicity in an A375 cell line engineered to express HLA-A∗11:01 and KRASG12V, but not HLA-A∗11:01 and KRASG12D, confirming the specificity for the KRAS mutation/HLA-I molecule pair (Figure S4A). Furthermore, T cells induced against KRASG12V on HLA-A∗11:01 only led to the cytotoxicity of SW620A∗11:01 cells, but not control SW620 cells without HLA-A∗11:01 (Figure S4B). This confirms the tumor specificity of this cytotoxicity. An analogous experiment with the non-engineered NCI-H441 cell line led to target cell cytotoxicity only upon co-culture with a population of T cells stimulated to recognize KRASG12V on HLA-A∗03:01 (Figure 3E). Together, these data demonstrate that naive T cells can be induced that recognize MS-detected KRAS neoantigens. Importantly, these T cells can distinguish mutant from wild-type epitopes, and the avidity of their TCRs is potent enough to recognize the natural level of the neoantigen on tumor cells. This supports the concept of T cell-based therapies to target a range of KRAS neoantigens.
Characterization of TCRs specific to common KRAS neoantigens
The 30 peptides detected by targeted MS (Table 1) cover 22 distinct KRAS mutation/HLA-I molecule pairs (i.e., four KRAS mutations on HLA-A∗03:01, HLA-A∗11:01, and HLA-A∗68:01, three KRAS mutations on HLA-C∗03:04, two KRAS mutations on HLA-A∗30:01 and HLA-B∗07:02, and one KRAS mutation on HLA-C∗01:02, HLA-C∗03:03, and HLA-C∗08:02). Of these 22 KRAS mutation/HLA-I molecule pairs, patient T cell responses have previously been published for only two (KRASG12V on HLA-A∗11:01 and KRASG12D on HLA-C∗08:02) (Tran et al., 2015, 2016; Cafri et al., 2019). Although these epitopes are immunogenic in an ex vivo naive T cell induction assay, the paucity of patient T cell responses reported in the literature may indicate that responses to KRAS neoantigens are unlikely to occur without intervention. This supports an approach where KRAS-specific T cell responses are generated systematically, such as with vaccination or TCR-T-engineered T cells.
An understanding of physiologically relevant KRAS neoantigens allows for the focused discovery of physiologically relevant TCRs. As proof of principle for the discovery of TCRs using our pipeline, we leveraged the naive T cell responses to KRAS neoantigens induced from healthy donors. We sorted antigen-specific T cells and sequenced the TCRs (Figure S5A). We stably transduced all TCRs into Jurkat cells expressing the CD8 alpha chain and characterized the avidity of the TCRs using peptide titration and co-culture with target cells expressing the relevant HLA-I alleles. For example, we were able to isolate TCRs specific for KRASG12V and KRASG12D on HLA-A∗11:01 (Figures 4A and 4C). TCRs specific for these two neoantigens are currently being tested in clinical trials (Chatani and Yang, 2020). In addition, we isolated TCRs specific for these same mutations on HLA-A∗03:01 (Figures 4B and 4D), the more prevalent member of the HLA-A3 superfamily in the United States. All four TCRs displayed avidities ranging from 10 to 140 nM (Figures 4A–4D). These TCRs demonstrate no reactivity to wild-type KRAS epitopes even at high peptide concentrations (Figure S5B). Some of these TCRs’ avidities are in the same range as those targeting KRAS neoantigens isolated from cancer patients, including ones that elicited an anti-tumor response upon expansion and reinfusion (Sim et al., 2020; Tran et al., 2016). Screening additional TCRs will be necessary to identify those with therapeutic avidities for the remaining KRAS neoantigens validated in our pipeline.
Figure 4.

Characterization of KRAS-specific TCRs on common alleles
(A–D) TCRs isolated from healthy donor T cells stimulated against different KRAS neoantigens were transduced into JurkatCD8 cells. TCR avidity was evaluated by titration of each cognate peptide presented on A375 cells transduced to express the cognate HLA-I molecule. TCR stimulation was assessed by IL-2 secretion or CD69 expression, and half-maximal effective concentration was calculated. (A) TCR specific for KRASG12V on HLA-A∗11:01. (B) TCR specific for KRASG12V on HLA-A∗03:01. (C) TCR specific for KRASG12D on HLA-A∗11:01. (D) TCR specific for KRASG12D on HLA-A∗03:01. Data are presented as mean ± SD and are representative of at least two independent experiments. See also Figure S5.
(E) The TCR from (A) was transduced into PBMCs and used in a cytotoxicity assay against SW620A∗11:01 across a range of specific effector to target (E:T) ratios (red). The ratio was calculated using the number of TCR-transduced CD8+ T cells in the well. As a control, an equivalent number of PBMCs transduced with an irrelevant TCR was co-cultured with SW620A11:01 cells (blue). Both target cell growth (top) and target cell death (bottom) were measured. Data are presented as mean ± SD and are representative of three independent experiments.
(F) The TCR from (B) was transduced into PBMCs and used in a cytotoxicity assay against SW620A∗03:01 (red), and cell death of the target cell was measured. As a negative control, PBMCs transduced with the TCR from (A) were co-cultured with SW620A∗03:01 (blue). Data are presented as mean ± SD and are representative of two independent experiments.
(G) The TCR from (B) was transduced into PBMCs and used in a cytotoxicity assay against NCI-H441 (red), and cell death of the target cell was measured. As a negative control, PBMCs transduced with the TCR from (A) were co-cultured with NCI-H441 (blue). Data are presented as mean ± SD and are representative of two independent experiments.
To confirm the potency of the KRASG12V-specific TCRs, we performed analogous cytotoxicity assays to those performed with the induced T cell populations with TCR-transduced Jurkat cells (Figures 4E–4G). We titrated the number of specific effector cells (T cells expressing the transgenic TCR) to evaluate a range of effector to target (E:T) ratios. For the TCR specific to KRASG12V on HLA-A∗11:01, we used HLA-A∗11:01-transduced SW620 target cells with endogenous expression of the KRASG12V mutation as target cells in the cytotoxicity assays. We observed a decrease in target cell growth based on target cell GFP expression and an increase in expression of the apoptotic marker annexin V on target cells starting at a ratio of 1.3:1. Complete target cell killing was observed at a ratio of 2.6:1 (Figure 4E). Furthermore, for the TCR specific to KRASG12V on HLA-A∗03:01, we established that effector cells expressing this TCR could also kill SW620 cells transduced with HLA-A∗03:01 at a similar E:T ratio of 2.5:1 (Figure 4F). Finally, we assessed whether T cells expressing this TCR could kill the NCI-H441 target cell line with endogenous expression of a KRASG12V mutation and HLA-A∗03:01. We observed an increase in expression of annexin V on the target cells at an E:T ratio of 0.8:1, demonstrating recognition and cytotoxic potential by this TCR of a natural KRASG12V neoantigen (Figure 4G). Although significant, the level of cytotoxicity with this TCR is less substantial than with the TCR recognizing KRASG12V on HLA-A∗11:01, which is likely due to the weaker functional avidity of the former (Figure 4A). Characterization of additional TCRs may yield others with a stronger functional avidity. Together, these data indicate that TCRs identified through this pipeline can recognize KRAS neoantigens at physiological levels.
Discussion
Adoptive T cell therapy targeting KRAS mutations can have a profound therapeutic effect in the clinic (Tran et al., 2016). However, only nine validated KRAS T cell targets (KRAS mutation/HLA-I molecule pairs) have been published (Bear et al., 2021; Douglass et al., 2021; Tran et al., 2016; Wang et al, 2016, 2019). The systematic pipeline that we established allowed us to rapidly evaluate many KRAS mutation/HLA-I molecule pairs and identify additional KRAS T cell targets. Our work substantially increases the number of patients with KRASG12C/D/R/V mutation-derived cancers that can be targeted with a T cell-based therapy. Based on HLA-I frequencies in the United States, approximately 46% of cancer patients with the KRASG12C mutation, 60% of patients with the KRASG12D mutation, 52% of patients with the KRASG12R mutation, and 53% of patients with the KRASG12V mutation present at least one KRAS neoantigen. When considering previously reported KRAS neoantigens, our work contributes to a 10%–20% increase in patients harboring a neoantigen among those with KRASG12D, KRASG12R, and KRASG12V mutations. Since this is the first report of HLA-I molecules presenting KRASG12C neoantigens, our work reveals a sizable portion of cancer patients with these KRAS mutations who may benefit from targeted immunotherapies.
The increase in the fraction of patients with a validated KRAS neoantigen emphasizes the importance of a comprehensive analysis of potential T cell targets (i.e., across more HLA-I molecules) that does not rely on patient samples. Although there is value in observations of natural KRAS-specific T cell responses in patient samples because they alone validate the target neoantigen, such responses seem infrequent. For example, in two recent publications evaluating the T cell reactivity to KRAS mutations from tumors or peripheral blood, 14 patients had at least one of the KRAS mutation/HLA-I molecule pairs that we validated with our pipeline (Cafri et al., 2019; Parkhurst et al., 2019). Only 3 of 14 patients had T cells that were reactive to KRAS mutations, as demonstrated by tumor-infiltrating lymphocyte analysis and study of the memory T cell compartment from peripheral blood. These results convincingly confirm these specific mutation/HLA-I molecule pairs, and they also demonstrate the inefficiency of this route of target validation. By using a pipeline that does not rely on cancer patient samples, we can evaluate T cell targets from KRAS mutations systematically, more rapidly, and with high sensitivity.
The relative infrequency of spontaneous responses to targetable KRAS mutation/HLA-I molecule pairs in patients likely limits the effect of immune editing during the development of these tumors that has been proposed across multiple oncogenes (Marty et al., 2017). This also supports a therapeutic approach that systematically induces T cells that recognize these neoantigens. Neoantigen vaccines are potent at eliciting T cell responses in patients (Ott et al., 2017; Sahin et al., 2017). In patients with a large tumor burden, a cell-based approach, including the use of KRAS-specific TCRs, may be more effective. In our pipeline, targeted MS increases the efficiency of T cell target discovery by providing direct evidence that a predicted KRAS neoantigen is processed within a cell then presented by an HLA-I molecule. This removes false positives due to the immunogenicity of peptides that are not processed endogenously (Strønen et al., 2016). Other groups have overcome this issue through the use of immunogens that have to be processed endogenously (Ali et al., 2019). However, targeted MS reduces false negatives inherent in all immunogenicity-based approaches due to inefficient T cell inductions to specific peptides. For example, T cell responses to KRAS mutations on HLA-A∗03:01 were generally less frequent in our study than on other molecules. By leveraging highly sensitive targeted MS as the first arm of our pipeline, we prioritized and evaluated MS-detected targets in T cell inductions across many healthy donors. Weakly immunogenic targets may be ideal for TCR-T therapies, whereby many healthy donors can be screened to discover a TCR with preferred clinical characteristics.
During the preparation of this article, a similar workflow was published that also focused on discovery and validation of common KRAS neoantigens (Bear et al., 2021). Applying analogous technologies, such as epitope prediction, targeted MS, and immunogenicity assays, the authors reported positive T cell responses for five KRAS mutation/HLA-I pairs across three common alleles. Although both studies yielded a few overlapping findings, the differences in our pipeline enabled the detection and validation of significantly more KRAS mutation/HLA-I pairs across seven common alleles. First, our use of affinity pull-down and MAPTAC technology (Abelin et al., 2019) enabled the rapid profiling of HLA-I alleles, increasing the breadth of KRAS mutation/HLA-I pairs that can be analyzed by targeted MS. The number of alleles profiled in our study is more than twice what has been reported in the literature and results in the discovery of 13 unpublished KRAS mutation/HLA-I pairs and nine pairs already reported in the literature. Second, our targeted MS workflow incorporated a cysteine modification step that enabled the discovery of four KRASG12C epitopes across four unique alleles. We believe our study is the first time that direct evidence of processing and presentation of cysteine-containing epitopes have been reported. Finally, we leveraged a library of peptide-HLA-I multimers (pHLA) to sensitively detect T cell responses after our in vitro immunogenicity assay. This allowed us to characterize the immunogenicity of these neoantigens more broadly and deeply, and we were able to evaluate and detect T cell responses to 16 total KRAS mutation/HLA-I pairs across seven alleles. By contrast, Bear et al. used IFN-γ ELISPOT assays to detect T cell responses after their stimulation and confirmatory pHLA multimers only in cases where positive responses were detected in the initial screen. For these reasons, we detected responses to 11 more KRAS neoantigens than Bear et al., seven that they did not screen and four that they screened but to which they did not detect responses.
We applied this pipeline to focus on KRAS mutations, but it can be applied broadly to study other classes of antigens. For example, other common mutations or even patient-specific mutations can be rapidly evaluated because this pipeline does not rely on limited patient samples or available animal models. Common mutations, tumor-associated antigens, or viral antigens can be assessed across various common HLA-I alleles to identify therapeutically relevant and practical targets. Furthermore, this systematic pipeline provides an efficient manner to evaluate less common HLA-I alleles, where the infrequency of the HLA-I allele compounds the relative infrequency of spontaneous T cell responses to any given antigen. Overall, this pipeline supports the ability to discover and validate peptides presented by HLA-I molecules for any HLA-I allele.
Limitations of the study
The presented pipeline has some limitations. It was primarily constructed to address whether any given epitope can be presented on a given HLA-I allele and subsequently immunogenic. By leveraging engineered cell lines with overexpressed KRAS mutations, HLA-I alleles, or both, we were able to confirm that all listed epitopes can be processed and presented, but we only confirmed that a subset is naturally presented without overexpression.
The use of targeted MS for validation of processing and presentation relies on the integrity and accuracy of the initial epitope prediction. This approach biases the analysis toward targeting the canonical HLA-I peptide repertoire and potentially misses neoantigens with atypical binding motifs. In addition, epitope prediction has the potential to create large numbers of candidate peptides for targeted MS. Although MS acquisition techniques have advanced the number of peptides that can be assayed in a single analysis (hundreds), PRM methods are limited in scope and rely on upfront assay development with heavy isotopically labeled peptides.
The immunogenicity assay was used to evaluate whether T cell responses could be reproducibly generated to these neoantigens. Although the T cell responses are specific, they may not always be sensitive enough to detect the natural levels of the target neoantigens. Dedicated screens would be required to generate multiple TCRs per target to identify ones that can recognize the natural level of the neoantigen. In addition, the induced T cells or TCR-T cells were not screened in mouse tumor models to evaluate whether these T cells can recognize the natural level of the neoantigens during in vivo growth.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HLA-A2 | Abcam | Clone: BB7.2; Cat# ab74674; RRID:AB_1280940 |
| HLA-I | Abcam | Clone: W6/32; Cat# ab22432 |
| HLA-I (in-house) | ATCC | HB-95 hybridoma |
| HLA-ABC-APC | BioLegend | Clone: W6/32; Cat# 311410; RRID:AB_314879 |
| HLA-A02-PE | Invitrogen | Clone: BB7.2; Cat# 12-9876-42; RRID:AB_2784636 |
| HLA-C-BV711 | BD Biosciences | Clone: DT-9; Cat# 747594; RRID:AB_2744163 |
| mTCRβ-PE | BioLegend | Clone: H57-597; Cat# 109208; RRID:AB_313431 |
| hCD4-AF700 | BD Biosciences | Clone: RPA-T4; Cat# 557922; RRID:AB_396943 |
| hCD8-FITC | BioLegend | Clone: SK1; Cat# 344704; RRID:AB_1877178 |
| hCD14-AF700 | BD Biosciences | Clone: M5E2; Cat# 557923; RRID:AB_396944 |
| hCD16-AF700 | BD Biosciences | Clone: 3G8; Cat# 557920; RRID:AB_396941 |
| hCD19-AF700 | BD Biosciences | Clone: HIB19; Cat# 557921; RRID:AB_396942 |
| hCD69-BV786 | BD Biosciences | Clone: FN50; Cat# 563834; RRID:AB_2738441 |
| CD14 MicroBeads, human | Miltenyi Biotec | Cat# 130-050-201; RRID:AB_2665482 |
| CD25 MicroBeads, human | Miltenyi Biotec | Cat# 130-092-983 |
| Biological samples | ||
| Human PBMCs from healthy donors | AllCells | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Heavy isotope-labeled synthetic peptides | Vivitide and BioNTech US Inc. (in-house) | N/A |
| FLT3L | Cellgenix | 1015-050 |
| TNF-α | Cellgenix | 1006-050 |
| IL-1β | Cellgenix | 1011-050 |
| PGE1 | Cayman Pharma | CAS: 745-65-3 |
| IL-7 | Cellgenix | 1010-050 |
| IL-15 | Cellgenix | 1013-050 |
| IncuCyte Annexin V red reagent | Sartorius | 4641 |
| Critical commercial assays | ||
| Chromium Single Cell 5’ Library & Gel Bead Kit | 10x Genomics | PN-1000006 |
| Chromium Single Cell V(D)J Enrichment Kit, Human T Cell | 10x Genomics | PN-1000005 |
| Chromium Single Cell A Chip Kit | 10x Genomics | PN-120236 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| PhiX Control | Illumina | FC-110-3001 |
| MiSeq Reagent Kit v2 (300-cycles) | Illumina | MS-102-2002 |
| U-PLEX Custom Biomarker (hu) Assays (IL-2, IFN-g, TNFa) | Meso Scale Diagnostics | K15067M-2 |
| Deposited data | ||
| Mass spectrometry RAW files | https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp | MSV000087987 |
| TCR sequences | https://www.ncbi.nlm.nih.gov/nucleotide/ | GenBank: MZ819175, MZ819176, MZ819177, MZ819178, MZ819179, MZ819180, MZ819181, MZ819182 |
| R code for HLA-I sequence logo generation | Zenodo | https://doi.org/10.5281/zenodo.5222292 |
| Experimental models: Cell lines | ||
| SW620 | ATCC | Cat# CCL-227; RRID:CVCL_0547 |
| A375 | ATCC | Cat# CRL-1619; RRID:CVCL_0132 |
| NCI-H441 | ATCC | Cat# CRM-HTB-174; RRID:CVCL_1561 |
| J.RT3-T3.5 | ATCC | Cat# TIB-153; RRID:CVCL_1316 |
| Lenti-X 293T | Takara Bio | Cat# 632180; RRID:CVCL_4401 |
| Recombinant DNA | ||
| pCDH-CMV-MCS Lenti vector | System Biosciences | CD500B-1 |
| pCDH-SFFV-Puro Lenti vector | In-house | N/A |
| pCDH-CMV-BSD Lenti vector | In-house | N/A |
| pCDH-CMV-Puro Lenti vector | In-house | N/A |
| MISSIOM lentiviral packaging plasmid mix | Sigma-Aldrich | SHP001 |
| Software and algorithms | ||
| R | The R Foundation | Version 4.0.4 |
| RStudio | RStudio | Version 1.4.1106 |
| GraphPad Prism | GraphPad Software | Version 9.1.0; RRID:SCR_002798 |
| Excel | Microsoft | Version 21.03; RRID:SCR_016137 |
| HLAthena | Sarkizova et al. (2020) | http://hlathena.tools/ |
| Interactive Peptide Spectral Annotator tool | Brademan et al. (2019) | http://www.interactivepeptidespectralannotator.com/PeptideAnnotator.html |
| Xcalibur | Thermo Scientific | Version 2.1.1565.23, 3.1.2412.25, or 3.3.2782.34; RRID:SCR_014593 |
| Skyline-daily | University of Washington | Version 19.1.1.283 or newer; RRID:SCR_014080 |
| Qual Browser Thermo Xcalibur | Thermo Scientific | Version 4.0.27.42 |
| FreeStyle | Thermo Scientific | Version 1.6.75.20 |
| FlowJo | BD Biosciences | Version 10.6.1; RRID:SCR_008520 |
| Cell Ranger | 10x Genomics | Version 2.0.2; RRID:SCR_017344 |
| Loupe V(D)J Browser | 10x Genomics | Version 2.0; RRID:SCR_018555 |
| FACSDIVA | BD Biosciences | Version 8.0.1; RRID:SCR_018555 |
| IncuCyte S3 | Sartorius | Version 2018C; RRID:SCR_019874 |
| MiSeq Control Software | Illumina | Version 2.6.2.1; RRID:SCR_016379 |
| Meso Scale Discovery | Meso Scale Diagnostics | Version 4.0.12.1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Vikram Juneja (vikram.juneja@biontech.us).
Materials availability
All newly generated materials associated with this paper are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell culture
SW620 (ATCC), Lenti-X 293T (Takara Bio), and A375 (ATCC) cell lines were cultured in DMEM media containing 10% FBS, penicillin, and streptomycin. NCI-H441 (ATCC) and Jurkat that has the TCR beta-chain-deficient mutant (J.RT3-T3.5, ATCC) were cultured in RPMI-1640 containing 10% FBS, penicillin, and streptomycin.
Generation of double-transduced A375 cells or single-transduced 293T cells
The A375 cell line (ATCC) was used to generate cells that contained KRASG12C/D/R/V mutations and individual HLA-I molecules (i.e., HLA-A∗03:01, HLA-A∗11:01, HLA-A∗30:01, HLA-A∗68:01, HLA-B∗07:02, HLA-C∗01:02, HLA-C∗03:03, HLA-C∗03:04, or HLA-C∗08:02). One pCDH-CMV-BSD lentivirus vector encoded KRASG12C/D/R/V segments and a second pCDH-CMV-Puro lentivirus vector encoded each BAP tagged HLA-I molecule (HLA-A∗68:01 was not BAP tagged). A375 cells were co-transduced with the KRASG12C/D/R/V lentivirus followed by each HLA-I lentivirus. In one instance, 293T cells were transduced with a KRASG12R lentivirus, and the endogenous alleles including HLA-A∗02:01 and HLA-B∗07:02, were evaluated. The double-transduced A375 cells were maintained in complete media and selected under 5 g/L blasticidin (to select for KRASG12C/D/R/V expressing cells) and 1 g/L puromycin (to select for HLA-I expressing cells) in complete media for more than seven days. The single-transduced 293T cells were maintained in complete media and selected under 5 g/L blasticidin (to select for KRASG12R expressing cells). Cells were harvested with 0.5% trypsin then washed twice with cold PBS. 5 x 105 cells were used for HLA-I expression analysis while the rest of the cells were pelleted, frozen in a dry ice and 70% ethanol slurry, then stored in -80°C prior to HLA-I pull-down.
T cell inductions against KRAS neoantigens in healthy donors
Human PBMCs from healthy donors (HLA-A∗03:01, HLA-A∗11:01, HLA-A∗68:01, HLA-B∗07:02, HLA-C∗01:02, HLA-C∗03:04, and/or HLA-C∗08:02) were isolated using Ficoll separation from apheresis material (AllCells). A total of 28 healthy donors were used (22 males ranging in age from 22-56 and 6 females ranging in age from 21-55). Naive T cell inductions were performed with some modifications as previously described (Lissina et al., 2016). In brief, CD14+ and CD25+ cells were depleted and the negative fraction was resuspended in AIM V media with serum (FBS or Human Serum) and cultured in the presence of FLT3L (Cellgenix), then loaded with 9-10mer KRAS neoantigen peptides (2 mM) matched to the healthy donor HLA-I alleles. APCs in the culture were matured with TNF-α (Cellgenix), IL-1β (Cellgenix), PGE1 (Cayman Pharma), and IL-7 (Cellgenix) and further T cell growth was promoted through the addition of IL-7 and IL-15 (Cellgenix). Cells were restimulated after two weeks with an identical cell culture at a 1:1 mix and continued to be supplemented with IL-7 and IL-15. KRAS neoantigen peptides were synthesized using SPPS on a computer-controlled high-throughput peptide synthesizer. The peptides were assembled on resins by employing Fmoc chemistry. The chemical cleavages of peptides from the resins were performed by using TFA solution containing scavengers followed by peptide precipitation and washes with diethyl ether. Crude pellets were resuspended in acetonitrile and water and dried on a parallel centrifugal evaporator. When necessary peptides were resuspended in DMSO and purified via automated prep HPLC systems equipped with UV and MS detectors. The HPLC fractions that met the molecular weight (MW) and purity criteria based on UPLC-UV/MS analysis were combined and dried using either parallel centrifugal evaporators or lyophilized. Crude or purified peptides were reconstituted in DMSO at a final concentration of 20 mM.
Transfection and lentivirus production
Lenti-X 293T cells (Takara Bio) were cultured in DMEM culture media (DMEM containing 10% FBS) to produce lentivirus. The day before transfection, 2.3x105 293T cells were plated per well of six-well plate. The culture media was replaced with DMEM media without FBS at the day of transfection. 0.5 mg of lentiviral construct plasmid and 4.6 mL of the MISSION lentiviral packaging plasmid mix (Sigma-Aldrich) were mixed in Opti-MEM (Thermo Fisher Scientific). The mixture was combined with 10 mL of FuGENE HD (Promega) and added directly to the cells. 24 hours later, the media was replaced with fresh RPMI-1640 contained 10% FBS. The supernatant contained lentivirus was harvested at 72 hours after transfection and was concentrated 10x by the Lenti-X concentrator (Takara Bio). The concentrated lentivirus supernatants were stored in -80°C.
CD8 transduced Jurkat cells
The TCR beta-chain-deficient mutant Jurkat cell line (J.RT3-T3.5, ATCC) was cultured in complete RPMI-1640 media contained 10% FBS, 10 unit/mL penicillin and 10 μg/ml of streptomycin. 5x105 of the cells were plated per well in a six-well plate in 1.5 mL of RPMI-1640 containing 6 μg/mL polybrene before 0.5 mL of CD8 lentiviral supernatant was added. The CD8 transduced Jurkat (CD8-Jurkat) cells were sorted one week after transduction and maintained in complete RPMI-1640 media.
Recombinant TCR transduced CD8-Jurkat cells or PBMCs
5x105 of CD8-Jurkat cells or PBMCs (activated by Dynabeads Human CD3/CD28 (Gibco) in 100 μL of RPMI-1640 contained 6 μg/mL polybrene and 10% FBS) were plated per well in 96-well plate, and 25 μL of concentrated lentivirus was added. The cells were centrifuged at 2,400 rpm for 1 hour and incubated in a CO2 incubator. The cells were transduced again in fresh media with polybrene and FBS and 25 μL of concentrated lentivirus. The media was replaced with RPMI-1640 containing 10% FBS and penicillin and streptomycin at 24 hr after second transduction. Puromycin treatment started at day four after transduction as 1 μg/mL concentration.
Method details
HLA-I epitope prediction
Mutant KRASG12C/D/R/V peptides were predicted using the HLAthena tool (Sarkizova et al., 2020) (http://hlathena.tools/). An input file containing the first 25 amino acids of each mutant KRAS protein was uploaded to HLAthena, then HLA-I epitope predictions were made for the top-12 most abundant HLA-A, HLA-B, and HLA-C alleles in the United States population. Default query parameters were used for the predictions: peptides were assigned to alleles by ranks, the threshold was set to 0.1, and the aggregate by peptide setting was set to yes. Both 9mer and 10mer neoantigens with ≤2% rank scores were targeted by MS in engineered or natural cell lines with relevant HLA-I alleles. Three predicted neoantigens that did not meet our criteria were targeted in cells engineered to express all four neoantigens. Those peptides are marked as “extrapolated” in Table S1.
Validation of HLA-I transduction
The HLA-I transduced A375 cells were stained with an HLA-ABC-APC antibody (W6/32; BioLegend), an HLA-A02-PE (BB7.2; Invitrogen), and an HLA-C BV711 antibody (DT-9; BD Biosciences). The stained cells were analyzed using a LSRFortessa Flow Cytometer (BD Biosciences) and analyzed with FlowJo software (BD Biosciences).
HLA-I sequence logo generation
Sequence logos were created using R software (version 4.0.4) with filtered peptide lists that were detected from monoallelic cell lines reported by Sarkizova et al. (2020) in Table S1E. The sequence logos were generated using unique 9mer peptide sequences. Letter heights are proportional to the frequency of the amino acid at that position, and positions with the lowest variation (lowest entropy) are colored. The different colors loosely correspond to amino acid properties.
Isolation of HLA-I peptide complexes
HLA-I-bound peptides were isolated as described in Abelin et al. (2019). Frozen cell pellets expressing either untagged or biotin acceptor peptide (BAP)-tagged HLA molecules were thawed on ice for 20 min. Cells were gently lysed by hand pipetting in cold lysis buffer [20 mM Tris-Cl pH 8, 100 mM NaCl, 6 mM MgCl2, 1.5% (v/v) Triton X-100, 60 mM octyl B-D-glucopyranoside, 0.2 mM of 2-Iodoacetamide, 1 mM EDTA pH 8, 1 mM PMSF, 1X complete EDTA-free protease inhibitor cocktail (Roche)] at a ratio of 1.2 mL lysis buffer per 50x106 cells. Lysates were incubated end-over-end at 4°C for 15 min with Benzonase nuclease (Millipore-Sigma) at a ratio of ≥250 units Benzonase per 50x106 cells to degrade DNA and RNA. Lysates were then cleared of insoluble cellular debris by centrifugation at 15,000 x g at 4°C for 20 min. For samples containing a BAP tagged HLA-I molecule (i.e., HLA-A∗03:01, HLA-A∗11:01, HLA-A∗30:01, HLA-B∗07:02, HLA-C∗01:02, HLA-C∗03:03, HLA-C∗03:04, and HLA-C∗08:02), cleared supernatants were transferred to new tubes, and the BAP tag was biotinylated by incubating the samples with 0.56 μM biotin, 1 mM ATP, and 1 or 3 μM BirA biotin ligase end-over-end at room temperature for 10 min. Biotinylated HLA complexes were enriched by incubation with Pierce High-Capacity NeutrAvidin Agarose (Thermo Fisher Scientific) at a ratio of 200 μL resin slurry per 50x106 cells at 4°C for 30 min.
Antibody immunoprecipitation was used to isolate HLA-A∗02:01, HLA-A∗68:01, and HLA-B∗07:02 molecules. For this, GammaBind Plus Sepharose Beads (GE Healthcare) were pre-charged with the BB7.2 anti-HLA-A2 antibody (Abcam, ab74674) or W6/32 anti-HLA-I antibody [produced in-house (ATCC HB-95 hybridoma) or purchased from Abcam (ab22432)] at a ratio of 75 μL of resin and 10 or 15 μg of antibody per 50x106 cells. Following the cell lysis and centrifugation described above, cleared supernatants were added to the antibody-loaded beads and incubated end-over-end at 4°C for 3 hr.
HLA-I-peptide filtration and desalting
HLA bound peptides were purified and desalted as described in Abelin et al. (2019) or by using a high-throughput 96-well plate purification approach adapted from Chong et al. (2018). When using the plate workflow, individual wells in a customized 96-well filter plate (Agilent) were rinsed and conditioned with 2 x 1 mL of methanol, 1 mL of acetonitrile, and 1 mL of 1% (v/v) formic acid. HLA-bound resin was transferred from tubes to the plate, washed four times with 1 mL of cold wash buffer [20 mM Tris-Cl pH 8, 100 mM NaCl, 60 mM octyl B-D-glucopyranoside, 0.2 mM of 2-Iodoacetamide, 1 mM EDTA pH 8], then rinsed four times with 1 mL of cold 10 mM Tris-Cl pH 8. All washes and rinses were performed on a customized Positive Pressure-96 Processor (Waters) manifold with nitrogen gas applied at 3-5 PSI.
Peptides were eluted directly into a pre-conditioned 96-well AcroPrep Advanced 10K molecular weight cut-off (MWCO) filter plate (Pall). Prior to use, the MWCO filter plate was passivated with 1% (v/v) of BSA in PBS for at least 1 hr, rinsed with 2 x 400 μL of water, then acidified with 400 μL of 10% (v/v) acetic acid. Peptides were eluted from HLA-bound beads by incubating with 2 x 200 μL of 10% (v/v) acetic acid for 5 min per round. During the first round of acetic acid elution, 100 fmol Pierce Retention Time Calibration Mixture was added to each sample as a loading control. The beads were then rinsed with 200 μL of 1.5M Trizma base solution (Sigma), and the rinse was pooled with the eluate in the MWCO filter plate. Elutions were pushed into the MWCO plate by applying nitrogen gas at 1-3 PSI. The MWCO filter plate was stacked on top of a 2 mL Protein LoBind 96-well collection plate (Eppendorf) and peptides were filtered by centrifuging at 2,100 x g for 30 min. An additional 400 μL of 1.5M Trizma base solution was added to the retentate in the MWCO plate and filtered into the same collection plate by centrifuging at 2,100 x g for 30-60 min.
Filtered peptides were reduced with 5 mM of Bond-Breaker TCEP Solution (Thermo Fisher Scientific) at 60°C for 30 min while shaking at 1,000 rpm in a ThermoMixer. Reduced thiols were alkylated with 15 mM 2-Iodoacetamide at room temperature for 30 min in the dark min while shaking at 300 rpm. The alkylation reaction was quenched with an additional 5 mM of TCEP for 15 min at room temperature in the dark. Samples were acidified by adding 100 μL of 100% formic acid. For samples that were not reduced and alkylated, 1% (v/v) formic acid replaced the 1.5M Trizma base used in the bead rinsing and MWCO filtration steps above.
Filtered peptides were desalted on a 10 mg Sep-Pak 37-55 mm particle size tC18 96-well μElution plate (Waters). Unless otherwise noted, all desalting steps were performed on a positive pressure manifold at 6 PSI. The C18 plate was activated with 2 x 500 μL of methanol followed by 250 μL of 99.9% (v/v) acetonitrile/0.1% (v/v) formic acid. The C18 plate was then washed 4 x 500 μL of 1% (v/v) formic acid, and acidified peptide samples were loaded at ∼3 PSI. After sample loading, the collection plate was rinsed with 2 x 500 μL of 1% (v/v) formic acid and loaded onto the C18 plate. The C18 plate was then washed with 4 x 500 μL of 1% (v/v) formic acid. Peptides were eluted into hard-shell low-profile thin-wall 96-well skirted PCR plate (Bio-Rad Laboratories) using a step gradient of 20 μL 15% (v/v) acetonitrile/1% (v/v) formic acid followed by 2 x 20 μL cuts of 50% (v/v) acetonitrile/1% (v/v) formic acid. The elution steps were performed by centrifugation at 300 x g at room temperature for 1 min. Eluted peptides were then frozen and dried in a vacuum centrifuge.
Tandem Mass Tag zero (TMTzero) reagent (Thermo Fisher Scientific) was used to facilitate the detection of specific peptides that were not retained on C18 columns in their native unmodified form. When TMTzero labeling was performed, the above protocol was modified as follows: Prior to peptide elution, the HLA-bound resin was rinsed three times with 1 mL of cold water to remove residual Tris buffer that could interfere with TMTzero labeling. Furthermore, 1% (v/v) formic acid replaced the 1.5 M Trizma base used in the bead rinsing and MWCO filtration steps. Following MWCO filtration, samples were transferred to a 1.5mL tube and vacuum centrifuged to dryness. Dried peptides were solubilized in 15 μL of 50 mM HEPES buffer pH 8.5, combined with 5 μL of 6.67 μg/μL TMTzero diluted in 100% anhydrous acetonitrile, and incubated at 25°C for 1 hr while shaking at 400 rpm. The TMTzero labeling reaction was quenched by adding 1 mL of 100 mM Tris-Cl pH 8. After labeling with TMTzero, reduction, alkylation, and desalting were performed as described above.
HLA-I-peptide sequencing by MS
PRM analyses on the RAS peptides employed the liquid chromatography separation conditions described below. Peptides were separated using an EASY-nLC 1200 System (Thermo Fisher Scientific) fitted with a PicoFrit 75 μm inner diameter and 10 μm emitter nanospray column (New Objective) packed at ∼1,000 PSI to ∼30 cm with ReproSil-Pur 120Ǻ C18-AQ 1.9 μm packing material (Dr. Maisch GmbH) and heated at 60°C during separation. The column was equilibrated with 10X bed volume of solvent A [3% (v/v) acetonitrile/0.1% (v/v) formic acid], samples were loaded in 3% (v/v) acetonitrile/5% (v/v) formic acid, and peptides were eluted with linear gradients of 0-30% or 0-40% or 6-40% Solvent B [80% (v/v) acetonitrile/0.1% (v/v) formic acid] over 84 min or 95 min. Linear gradients were run at a flow rate of 200 nL/min or 250 nL/min. Peptides were eluted into an Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific) equipped with a Nanospray Flex Ion source (Thermo Fisher Scientific) with a positive spray voltage set at 2.4 or 2.5 kV. A full-scan MS was acquired at a resolution of 50k or 60k with an automatic gain control (AGC) target of 4e5 and either 50 millisecond or 86 millisecond maximum injection times. Each MS scan was followed by targeted MS/MS scans according to mass lists comprising the calculated monoisotopic ion masses of the targeted peptide epitopes and their predicted charge states. We included additional m/z values for peptides if they met the following criteria: i) multiple charge states were expected due to presence of one or more basic residues in the sequence and/or ii) cysteine and methionine residues were expected to be modified during sample processing. Maximum injection times varied to maintain cycle times of approximately 3 seconds. MS/MS scans were acquired at a resolution of 15k or 30k from using an isolation width of 0.4 m/z, 0.7 m/z, or 1 m/z, normalized HCD collision energy of 34 or 38, and an AGC target of 1e5. In some instances, heavy synthetic peptides were spiked into samples to simplify data analysis and facilitate the relative quantification of endogenous peptides.
MS data analysis
Data analysis was performed using Skyline-daily software (version 19.1.1.283 or newer), and spectral visualizations were generated using the Interactive Peptide Spectral Annotator tool (Brademan et al., 2019) with extracted ion chromatogram (XIC) data exported using Qual Browser Thermo Xcalibur software (version 4.0.27.42) or Thermo FreeStyle software (version 1.6.75.20). Both Skyline and the Peptide Spectral Annotator tool were used to confirm endogenous peptide detections by comparing endogenous and synthetic peptide spectra. In some instances, the same synthetic peptide spectra were used to confirm endogenous peptide detections.
Combicoding multimer analysis and sorting
pHLA multimers were used to measure peptide-specific T cell expansion in the immunogenicity assays. Mulitmers were prepared as previously described, with the exception that the HLA monomers were purified by anion exchange chromatography instead of gel filtration chromatography (Hadrup et al., 2009). For the assessment, multimers are added to 1x106 cells in PBS. Cells were incubated at 37°C in the CO2 incubator for 15 min. hCD4-AF700, hCD8-FITC, hCD14-AF700, hCD16-AF700, and hCD19-AF700 antibodies (BD Biosciences) were added to the cells then incubated at 4°C for 30 min. Cells were washed and resuspended with cold FACS buffer. Cells were acquired on a LSRFortessa Flow Cytometer (BD Biosciences) and analyzed with FlowJo software (BD Biosciences). For isolation of TCR sequences, the tetramer positive and CD8+ cells were sorted by use of FACSAria Fusion (BD Biosciences) to analyze single cell TCR sequences.
Barcoding singles by 10X genomics and sequencing by MiSeq
The sorted T cells were immediately washed with PBS and 0.04% FBS. Single cell barcoding was performed on a Chromium Controller (10X Genomics) according to the manufacturer's protocol. The generating library for paired TCR alpha and beta sequence analysis was processed according to Chromium Single Cell V(D)J reagent kit (10X Genomics). 4 nM of the library was denatured with 0.2 N NaOH and mixed with 1% PhiX control (Illumina). The library sequencing was conducted using a MiSeq system according to the manual (Illumina). Single cell TCR V(D)J sequences were analyzed using the Cell Ranger (v2.0.2) and Loupe V(D)J Browser v2.0 (10X Genomics) software to process multiplexing, barcoding, and gene counting. The dominant TCR paired alpha and beta sequences were codon optimized and designed for the lentiviral construct.
Recombinant TCR lentiviral vector construction
The lentiviral construct, pCDH-SFFV-Puro vector, was utilized to insert TCR alpha and beta variable region sequences linked with mouse TCR alpha and beta constant region sequences after codon optimization by gene-synthesis service (Genscript). The TCR Lentiviral vector contained a furin-cleave site (RIKR), a spacer (SGSG), and a F2A sequence in between the TCR beta and TCR alpha sequences followed by T2A and a Puromycin sequence. The CD8a (NCBI reference sequence NM_001768.5) gene was cloned into the pCDH-CMV-MCS vector (System Biosciences) for lentivirus production.
Cytotoxicity assay
5x104 of target cells per well were seeded in 100 μL of culture media on 96-well flat bottom plate and cultured for overnight in an incubator set at in 37°C with 5% CO2. 50 μL of fresh media containing IncuCyte Annexin V red reagent (Sartorius) with or without antigen peptides were added on the target cells. 1-2x105 of effector cells, TCR transduced PBMCs, or peptide induced PBMCs, were co-cultured with target cells. The co-cultured cells were incubated and monitored using an IncuCyte system (Sartorius).
IL-2 release assay and CD69 assay
5x104 of target cells and 2.5x105 of effector cells per well were seeded and cultured with or without antigen peptides on a 96-well plate and cultured for 48 hr in an incubator set a 37°C with 5% CO2 incubator. The 50 μL supernatants of co-culture were harvested and assessed IL-2 concentration with V-PLEX Human IL-2 assays according to the manufacturer's protocol (Meso Scale Discovery). The cells of co-culture were harvested and stained with mTCRb-PE (H57-597; BD Biosciences), hCD8-FITC (SK1; BioLegend) and hCD69-BV786 (FN50; BD Biosciences) antibodies. Cells were acquired on a LSRFortessa Flow Cytometer (BD Biosciences) and analyzed with FlowJo software (BD Biosciences). CD69 positivity was assessed on hCD8 positive and mTCR positive cells.
Peptide affinity and stability measurements
Peptide affinity for specific HLA-I molecules was measured as previously described (Sidney et al., 2013). Peptide stability on specific HLA-I molecules was measured as previously described (Harndahl et al, 2011).
Quantification and statistical analysis
Statistical analysis
All statistical analyses, including EC50 calculations, were performed with GraphPad Prism (Version 9.1.0). Data in Figures 3B, 3D, 3E, 4, S4, and S5B are presented as mean ± SD as denoted in the figure legends. All data presented are representative of two or more independent experiments.
Peptide abundance calculations
The abundance of KRAS peptides detected in the HCI-H441 and SW620 samples was calculated using fragment ion abundances exported from Skyline. For this, the fragment ion signal for each peptide was summed, and the ratio of endogenous peptide to synthetic peptide was multiplied by the amount of synthetic peptide added to the sample (Data S1). Peptide per cell calculations were performed by multiplying the amount of detected endogenous peptide by Avogadro's number, followed by dividing the number of calculated peptide molecules by the number of cells used in the IP (Data S1).
Acknowledgments
We dedicate this paper to our friend and colleague Ying Sonia Ting (1984–2019), whose memory inspires us to think critically and innovate; we miss her immensely. We are grateful to all the members of BioNTech US Inc. for their support and assistance with this research. We would like to specifically thank Yvonne Ware, Jonathan McGee, Daniel Kallin, Yuting Huang, and Jesse Dong for supplying some synthetic peptides used in these studies; Janani Sridar for BirA reagent generation; and Kerry Chios and Amanda Zarzycki for lab operation support and biobank management. We would also like to thank John Welle from Acumen Medical Communications for assistance with figure generation.
Author contributions
Conceptualization, J.C., S.P.G., M.S.R., R.B.G., J.G.A., T.A.A., and V.R.J.; methodology, J.C., S.P.G., B.P.C., J.L.D., J.K., D.L., A.B., D.A.R., K.C.F., M.M.v.B., J.G.A., T.A.A., and V.R.J.; validation, A.B. and K.C.F.; formal analysis, J.C., S.P.G., and B.P.C.; investigation, J.C., S.P.G., B.P.C., C.D.McG., J.L.D., A.B., and K.C.F.; resources, P.J.T. and J.R.S.; writing – original draft, J.C., S.P.G., T.A.A., and V.R.J.; writing – review & editing, J.C., S.P.G., B.P.C., C.D.McG., J.L.D., J.K., D.L., A.B., D.A.R., P.J.C., J.R.S., K.C.F., M.S.R., M.M.v.B., R.G.B., J.G.A., T.A.A., and V.R.J.; visualization, J.C., S.P.G., T.A.A., and V.R.J.; supervision and project administration, J.C., S.P.G., T.A.A., and V.R.J.
Declaration of interests
J.C., S.P.G., B.P.C., J.L.D., J.K., D.L., D.A.R., P.J.T., J.R.S., K.C.F., M.S.R., M.M.v.B., R.B.G., T.A.A., and V.R.J. are employees and shareholders of BioNTech SE. C.D.McG., A.B., and J.G.A. are former employees of Neon Therapeutics (acquired by BioNTech SE). C.D.McG. is a graduate student at the University of Washington. A.B. is an employee and shareholder of TScan Therapeutics. J.G.A. is an employee of the Broad Institute of MIT and Harvard. R.B.G. is a member of the board of directors at Alkermes plc and Infinity Pharmaceuticals, and a member of the scientific advisory board at Leap Therapeutics.
Published: September 16, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100084.
Supplemental information
HLAthena predictions of KRAS neoantigens, KRASG12V neoantigen abundance calculations and fragment ion m/z values from Skyline for NCI-H441 and SW620 cells, and synthetic and endogenous peptide extracted ion chromatogram values for peptides.
Data and code availability
-
•
The RAW MS data was uploaded to the MassIVE repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and has the following identifier: MSV000087987. TCR sequences were deposited in the NCBI GenBank nucleotide database (GenBank: MZ819175, MZ819176, MZ819177, MZ819178, MZ819179, MZ819180, MZ819181, MZ819182). Remaining data is available in the supplemental information.
-
•
This paper does not report original code; however, we deposited the R code used to generate HLA-I sequence logos in Zenodo. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abelin J.G., Harjanto D., Malloy M., Suri P., Colson T., Goulding S.P., Creech A.L., Serrano L.R., Nasir G., Nasrullah Y., et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51:766–779.e17. doi: 10.1016/j.immuni.2019.08.012. [DOI] [PubMed] [Google Scholar]
- Ali M., Foldvari Z., Giannakopoulou E., Böschen M.-L., Strønen E., Yang W., Toebes M., Schubert B., Kohlbacher O., Schumacher T.N., et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019;14:1926–1943. doi: 10.1038/s41596-019-0170-6. [DOI] [PubMed] [Google Scholar]
- Bai P., Zhou Q., Wei P., Bai H., Chan S.K., Kappler J.W., Marrack P., Yin L. Rational discovery of a cancer neoepitope harboring the KRAS G12D driver mutation. Sci. China Life Sci. 2021 doi: 10.1007/s11427-020-1888-1. Published online March 16, 2021. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D., Knelson E.H., Sequist L.V. A bright future for KRAS inhibitors. Nat. Cancer. 2020;1:25–27. doi: 10.1038/s43018-019-0016-8. [DOI] [PubMed] [Google Scholar]
- Bear A.S., Blanchard T., Cesare J., Ford M.J., Richman L.P., Xu C., Baroja M.L., McCuaig S., Costeas C., Gabunia K., et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat. Commun. 2021;12:4365. doi: 10.1038/s41467-021-24562-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brademan D.R., Riley N.M., Kwiecien N.W., Coon J.J. Interactive peptide spectral annotator: a versatile web-based tool for proteomic applications. Mol. Cell Proteomics. 2019;18:S193–S201. doi: 10.1074/mcp.TIR118.001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafri G., Yossef R., Pasetto A., Deniger D.C., Lu Y.-C., Parkhurst M., Gartner J.J., Jia L., Ray S., Ngo L.T., et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat. Commun. 2019;10:449. doi: 10.1038/s41467-019-08304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle J.C., Uduman M., Pabla S., Stein R.B., Buell J.S. Mutation-derived neoantigens for cancer immunotherapy. Front. Immunol. 2019;10:1856. doi: 10.3389/fimmu.2019.01856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatani P.D., Yang J.C. Mutated RAS: targeting the “Untargetable” with T cells. Clin. Cancer Res. 2020;26:537–544. doi: 10.1158/1078-0432.CCR-19-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C., Marino F., Pak H., Racle J., Daniel R.T., Müller M., Gfeller D., Coukos G., Bassani-Sternberg M. High-throughput and sensitive immunopeptidomics platform reveals profound interferonγ-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol. Cell Proteomics. 2018;17:533–548. doi: 10.1074/mcp.TIR117.000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass J., Hsiue E.H.-C., Mog B.J., Hwang M.S., DiNapoli S.R., Pearlman A.H., Miller M.S., Wright K.M., Azurmendi P.A., Wang Q., et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021;6:eabd5515. doi: 10.1126/sciimmunol.abd5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadrup S.R., Bakker A.H., Shu C.J., Andersen R.S., van Veluw J., Hombrink P., Castermans E., Thor Straten P., Blank C., Haanen J.B., et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods. 2009;6:520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- Harndahl M., Rasmussen M., Roder G., Buus S. Real-time, high-throughput measurements of peptide-MHC-I dissociation using a scintillation proximity assay. J. Immunol. Methods. 2011;374:5–12. doi: 10.1016/j.jim.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harndahl M., Rasmussen M., Roder G., Dalgaard Pedersen I., Sørensen M., Nielsen M., Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity: antigen processing. Eur. J. Immunol. 2012;42:1405–1416. doi: 10.1002/eji.201141774. [DOI] [PubMed] [Google Scholar]
- Hong D.S., Fakih M.G., Strickler J.H., Desai J., Durm G.A., Shapiro G.I., Falchook G.S., Price T.J., Sacher A., Denlinger C.S., et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020;383:1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubuschok B., Neumann F., Breit R., Sester M., Schormann C., Wagner C., Sester U., Hartmann F., Wagner M., Remberger K., et al. Naturally occurring T-cell response against mutated p21 ras oncoprotein in pancreatic cancer. Clin. Cancer Res. 2006;12:1365–1372. doi: 10.1158/1078-0432.CCR-05-1672. [DOI] [PubMed] [Google Scholar]
- Lissina A., Briceño O., Afonso G., Larsen M., Gostick E., Price D.A., Mallone R., Appay V. Priming of qualitatively superior human effector CD8+ T cells using TLR8 ligand combined with FLT3 ligand. J. Immunol. 2016;196:256–263. doi: 10.4049/jimmunol.1501140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty R., Kaabinejadian S., Rossell D., Slifker M.J., van de Haar J., Engin H.B., de Prisco N., Ideker T., Hildebrand W.H., Font-Burgada J., et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171:1272–1283.e15. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. Cracking KRAS. Nat. Rev. Drug Discov. 2019;18:887–891. doi: 10.1038/d41573-019-00195-5. [DOI] [PubMed] [Google Scholar]
- Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst M.R., Robbins P.F., Tran E., Prickett T.D., Gartner J.J., Jia L., Ivey G., Li Y.F., El-Gamil M., Lalani A., et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 2019;9:1022–1035. doi: 10.1158/2159-8290.CD-18-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A.C., Russell J.D., Bailey D.J., Westphall M.S., Coon J.J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell Proteomics. 2012;11:1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior I.A., Hood F.E., Hartley J.L. The frequency of ras mutations in cancer. Cancer Res. 2020;80:2969–2974. doi: 10.1158/0008-5472.CAN-19-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B., et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L., et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J., Southwood S., Moore C., Oseroff C., Pinilla C., Grey H.M., Sette A. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr. Protoc. Immunol. 2013;Chapter 18:18.3. doi: 10.1002/0471142735.im1803s100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim M.J.W., Lu J., Spencer M., Hopkins F., Tran E., Rosenberg S.A., Long E.O., Sun P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. U S A. 2020;117:12826–12835. doi: 10.1073/pnas.1921964117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu D.K., Nissley D.V., McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strønen E., Toebes M., Kelderman S., van Buuren M.M., Yang W., van Rooij N., Donia M., Böschen M.-L., Lund-Johansen F., Olweus J., et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- Tran E., Ahmadzadeh M., Lu Y.-C., Gros A., Turcotte S., Robbins P.F., Gartner J.J., Zheng Z., Li Y.F., Ray S., et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E., Robbins P.F., Lu Y.-C., Prickett T.D., Gartner J.J., Jia L., Pasetto A., Zheng Z., Ray S., Groh E.M., et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.J., Yu Z., Griffith K., Hanada K.-i., Restifo N.P., Yang J.C. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 2016;4:204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Douglass J., Hwang M.S., Hsiue E.H.-C., Mog B.J., Zhang M., Papadopoulos N., Kinzler K.W., Zhou S., Vogelstein B. Direct detection and quantification of neoantigens. Cancer Immunol. Res. 2019;7:1748–1754. doi: 10.1158/2326-6066.CIR-19-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Zhao W., Zhou B., Su Z., Gu X., Zhou Z., Chen S. TSNAdb: a database for tumor-specific neoantigens from immunogenomics data analysis. Genomics Proteomics Bioinformatics. 2018;16:276–282. doi: 10.1016/j.gpb.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yossef R., Tran E., Deniger D.C., Gros A., Pasetto A., Parkhurst M.R., Gartner J.J., Prickett T.D., Cafri G., Robbins P.F., et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. 2018;3:e122467. doi: 10.1172/jci.insight.122467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HLAthena predictions of KRAS neoantigens, KRASG12V neoantigen abundance calculations and fragment ion m/z values from Skyline for NCI-H441 and SW620 cells, and synthetic and endogenous peptide extracted ion chromatogram values for peptides.
Data Availability Statement
-
•
The RAW MS data was uploaded to the MassIVE repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and has the following identifier: MSV000087987. TCR sequences were deposited in the NCBI GenBank nucleotide database (GenBank: MZ819175, MZ819176, MZ819177, MZ819178, MZ819179, MZ819180, MZ819181, MZ819182). Remaining data is available in the supplemental information.
-
•
This paper does not report original code; however, we deposited the R code used to generate HLA-I sequence logos in Zenodo. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.