

Summary
Single-cell ATAC sequencing (scATAC-seq) is a powerful and increasingly popular technique to explore the regulatory landscape of heterogeneous cellular populations. However, the high noise levels, degree of sparsity, and scale of the generated data make its analysis challenging. Here, we present PeakVI, a probabilistic framework that leverages deep neural networks to analyze scATAC-seq data. PeakVI fits an informative latent space that preserves biological heterogeneity while correcting batch effects and accounting for technical effects, such as library size and region-specific biases. In addition, PeakVI provides a technique for identifying differential accessibility at a single-region resolution, which can be used for cell-type annotation as well as identification of key cis-regulatory elements. We use public datasets to demonstrate that PeakVI is scalable, stable, robust to low-quality data, and outperforms current analysis methods on a range of critical analysis tasks. PeakVI is publicly available and implemented in the scvi-tools framework.
Keywords: single-cell genomics, deep learning, single-cell chromatin accessibility, single-cell ATAC-seq
Graphical abstract
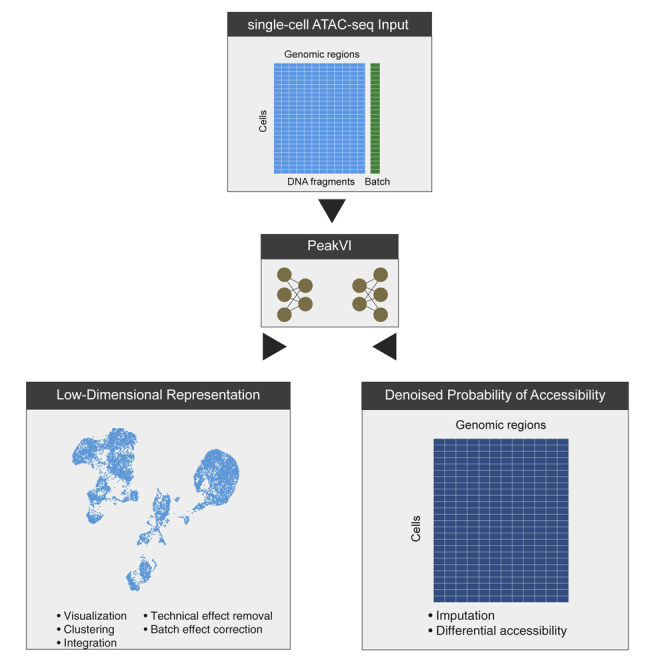
Highlights
-
•
PeakVI is a deep generative model for single-cell chromatin accessibility data
-
•
PeakVI preserves biological heterogeneity while correcting batch effects
-
•
PeakVI enables identification of differential accessibility at single-region resolution
-
•
PeakVI accurately projects query data onto a pre-analyzed reference dataset
Motivation
Single-cell chromatin accessibility assays are an increasingly popular approach for expanding the understanding of the role chromatin plays in gene expression regulation, but the combination of high sparsity and noise characteristic of single-cell assays with the binary nature of accessibility makes the data challenging to fully leverage. Here, we present PeakVI, a deep generative model that explicitly accounts for the unique properties of these data. PeakVI learns an informative low-dimensional representation of the cells, denoises the original observations, and provides functionality for differential accessibility analysis at single-region resolution. PeakVI is continuously supported and publicly available in the scvi-tools suite.
Ashuach et al. present PeakVI, a deep generative model for analyzing single-cell chromatin accessibility data. PeakVI learns informative low-dimensional representations of single cells that capture biological differences while controlling batch and technical artifacts. PeakVI provides functionality for denoising data, which can then be used to robustly identify differentially accessible loci.
Introduction
Regulatory elements in the genome tend to reside in regions of open chromatin, making the landscape of chromatin accessibility a valuable target of study. Several molecular assays have been developed to support this effort (Schones et al., 2008; Boyle et al., 2008; Crawford et al., 2006), among them ATAC sequencing (ATAC-seq) (Buenrostro et al., 2015a), in which accessible regions are fragmented, and the corresponding DNA fragments are sequenced and mapped back to the reference genome, accumulating in areas of open chromatin. Recent advances in sequencing technologies enable performing this assay in single cells (Buenrostro et al., 2015b), thereby allowing the study of chromatin variability at a single-cell resolution. Application of single-cell ATAC-seq (scATAC-seq) has led to promising results in discerning sources of variation, beyond those observed at the transcriptional level (Satpathy et al., 2019; Preissl et al., 2018), and allowed for high-resolution characterization of the regulation of in continuous processes, e.g., in immunity (Satpathy et al., 2019).
Despite the potential of scATAC-seq, analyzing the resulting data remains challenging. scATAC-seq assays have generally limited sensitivity, detecting 5%–15% of accessible regions (Preissl et al., 2018), a common issue for single-cell genomics. In addition, the coverage of these data is limited a priori since each genomic region has at most two copies in a single cell. Finally, scATAC-seq is extremely high dimensional, often consisting of hundreds of thousands of genomic regions. These challenges require specialized processing and analysis methods that are designed to account for the specific properties of scATAC-seq data.
One common task for analyzing scATAC-seq is dimensionality reduction: transforming the data to a low-dimensional space that preserves the meaningful information in the original data. This step is crucial to make some downstream analyses, such as clustering and visualization, less noisy, more stable, and computationally tractable. Existing methods use various approaches to achieve this task. Some use methods developed for natural language processing (e.g., latent Dirichlet allocation used by cisTopic [González-Blas et al., 2019] and latent semantic analysis [LSA] used by ArchR [Granja et al., 2021]) that inherently handle sparse high-dimensional data but do not inherently account for confounding factors that do not have an analog in textual language, such as batch effects. Other methods reduce dimensionality by first aggregating individual regions in the scATAC-seq data to easily interpretable features, such as binding motif scores in the case of chromVAR (Schep et al., 2017) or gene activity scores in the case of Cicero (Pliner et al., 2018), which makes the data easier to analyze but masks the fine-grain single-region resolution provided by scATAC-seq. These methods have been demonstrated to be under-powered in capturing the true heterogeneity in the original data (Chen et al., 2019). Finally, recent methods use deep generative models (e.g., SCALE [Xiong et al., 2019]) but do not account for technical factors and suffer from model over-fitting due to the dimensionality of the data in contrast with the limited number of samples.
Another common task is differential accessibility analysis. The ability to identify chromatin regions that are preferentially accessible in one population compared with another is foundational to characterizing the chromatin remodeling between cellular identities and states. However, specialized methods to perform this task in the context of scATAC-seq data have not yet been developed. Methods that rely on aggregation of individual regions, such as chromVAR and Cicero, perform differential analyses in the aggregated space, thereby losing the single-region resolution. Other methods use linear models developed for RNA-seq data (Fang et al., 2021) or standard statistical tests (Granja et al., 2021). These approaches often suffer from numerical instability due to the sparsity of the data and being statistically overpowered due to the large sample size.
Some recent processing pipelines, such as SnapATAC (Fang et al., 2021) and ArchR (Granja et al., 2021), offer comprehensive end-to-end analysis pipelines that resolve many issues with processing scATAC-seq data, such as sensitive peak calling, promoter-enhancer association, and doublet detection. However, for the fundamental tasks mentioned above, these pipelines rely on methods that were not optimized for scATAC-seq data and can therefore be improved upon.
Here, we present PeakVI, a deep generative model that learns a probabilistic low-dimensional representation of single cells from their chromatin accessibility landscape. PeakVI accounts for technical biases in the data stemming from batch effects, variation in sequence coverage, and bias due to the width of DNA regions and creates a representation of the data that minimizes these effects. The representation is provided at two levels. One part of the model infers a representation for each cell in a latent low-dimensional space. This latent representation and the space it is embedded in can be used directly for downstream analyses: integration of datasets, identification of cellular sub-populations, and visualization. A second part of the model provides a corrected, probabilistic representation of the raw data. This high-dimensional representation enables statistically robust inference of single-region-level differential accessibility and cell state annotation. We demonstrate PeakVI’s performance on published data and benchmark it against state-of-the-art published methods on a range of analysis tasks. We show that PeakVI is a powerful addition to the arsenal of scATAC-seq methods and provides capabilities that can help unlock the full potential of scATAC-seq data analysis. PeakVI is publicly available as part of the scvi-tools (Gayoso et al., 2021) suite of deep generative models for single-cell genomics.
Results
PeakVI model
PeakVI leverages variational inference with deep neural networks to model scATAC-seq data. For each cell, PeakVI estimates the probability of each chromatin region being accessible, as well as technical factors that affect the probability of an accessible region being observed. The standard output of most scATAC-seq preprocessing pipelines (including those employed here; see STAR Methods) is a table of N cells and K genomic regions. The regions typically correspond to DNA segments with enriched accessibility that are inferred through peak calling over cell aggregates (Buenrostro et al., 2015b; Fang et al., 2021; Granja et al., 2021).
The starting point of PeakVI is therefore a matrix X where is the number of reads from cell i that map to region j. While these observations are counts, the underlying biology is mostly binary (a region is either accessible or not). Therefore, PeakVI models the observations as samples from a Bernoulli distribution , where is the probability of region j being accessible in cell i, is a region-specific scaling factor, and is a cell-specific scaling factor (Figure 1A). Conceptually, these components are related to the three molecular events that are required for a region to be observed as accessible: (1) the region must be accessible in the cell, which largely depends on the cell state and identity, captured by ; (2) the accessible region must be tagmented by the transposase that underlies the ATAC-seq protocol, a process which may be skewed by region-specific factors such as width (in base pairs) and sequence biases, captured by ; and (3) finally, the corresponding fragment must be captured and sequenced, which may also depend on library-specific factors, such as sequencing depth and efficacy of the library preparation, captured by .
Figure 1.

PeakVI model overview
(A) Conceptual model illustration. The input region-by-cell count matrix (left) is estimated as the product of region-specific effects (center top), cell-specific effects (center), and accessibility probability estimates (center bottom). The observation probability matrix (right) is used to calculate the likelihood of the data for optimization.
(B) The region-specific factor is assigned higher values for wider regions, indicating a higher probability of those regions being fragmented.
(C) The cell-specific factor increases with the number of fragments up to a saturation point. Cells with sufficient fragments are not penalized even if other cells have significantly more fragments.
(D) Random corruption of the data at increasing rates leads to a small but steady increase in the mean squared error (measured from corrupted indices).
See also Figures S1A and S1B; Table S1.
PeakVI uses a variational autoencoder (Kingma and Welling, 2013) (VAE) and an auxiliary neural network to estimate these factors. The VAE consists of two major components: (1) the encoder network infers the distributional parameters of the d-dimensional (for ) latent representation (also known as the variational posterior) from the observed data: , and (2) the decoder network and the generative model, which takes a sample from the latent representation and the batch annotations and generates an estimate of the probability of each genomic region being accessible in the cell i: . The cell-specific scaling factor is inferred from the observed data using an additional neural net , and the region-specific scaling factor is optimized directly as a model parameter. Finally, the probability of observing a region in a cell (i.e., ) are computed as the product of the three probabilities: (Figure 1A). Formally:
Conditioning on batch annotations, or any other known sources of unwanted variation, encourages the encoder to capture batch-independent biological variation in the latent representation , which can then be used for normalized and batch-corrected visualization, clustering, and other downstream analyses. The inferred accessibility probabilities are an estimate of the true chromatin landscape in each cell, while technical effects that stem from either region-specific biases or cell-specific biases are captured by the r and scaling factors, respectively. We can then estimate the probability of observing a region in each cell as the product of these factors and compute the likelihood of the observations. During training, a lower bound of the marginal log likelihood is then maximized using auto-encoding variational Bayes (Kingma and Welling, 2013). Full model architecture and training parameters are provided in the STAR methods section.
Benchmark datasets
To evaluate the performance of PeakVI, we examined both simulated and real datasets. We found, however, that current simulation techniques (Chen et al., 2019) rely on independent sampling from distributions attained from bulk ATAC-seq data, which creates a highly sparse covariance structure that does not realistically reflect assayed datasets (Figure S1A). Our analysis therefore relies primarily on two publicly available datasets: (1) hematopoiesis data from Satpathy et al., (2019), which consists of bone marrow and blood samples that were flow-sorted for different cell subsets, as well as several batches of unsorted samples that consist of multiple cell types, and (2) a dataset released by 10X Genomics of joint RNA-seq and ATAC-seq from single human peripheral blood mono-nuclear cells (PBMC°s). The first dataset contains cell-type-specific labels that provide an established benchmark, as well as multiple batches that allow comparison of batch effect correction. The second dataset provides an orthogonal modality of data that can be used to validate scATAC-based analyses. Finally, the two datasets are generated using different protocols and are processed differently, allowing us to demonstrate that the PeakVI’s performance is protocol and processing independent.
PeakVI captures nuanced effects of technical confounders
Since the normalization factors included in the PeakVI model, r and , are optimized by the training process, we set out to confirm that they converge on values that correspond to the empirical, technical confounders. We used the 10× PBMC data for these analyses. For the region-specific factor r, we examined how it corresponds to the width of the genomic region, a known technical confounder. We found that PeakVI assigns the vast majority of regions with a value around 0.5, with higher values indeed being assigned to wider regions, which have a higher probability of being fragmented (Figure 1B). Notably, the overall distribution of this factor only reaches as high as roughly 0.75, well below the max value of 1. This translates to a global penalty imposed on all observations, which implicitly reflects the limited sensitivity of this assay and the resulting abundance of false-negative observations. For the cell-specific factor we examined how it corresponds to the number of reads captured in each cell. We find that the vast majority of cells have , and the dynamic values of indeed correspond to the empirical library size (Figure 1C). The saturation of this factor reflects an important consideration when normalizing library sizes for chromatin profiling: different cell types may have different levels of accessibility (e.g., unbalanced chromatin remodeling during differentiation [Sen et al., 2016]), therefore this factor should not penalize cells states with less accessible chromatin, but rather only weigh down cells in cases where the decrease in fragments is due to technical effects. Overall we see that the normalization factors used by the model have a clear but nuanced correspondence to empirical confounders.
PeakVI is robust to increased sparsity and stable across hyperparameters
Limited sensitivity, which results in an abundance of missing observations, is a major problem in single-cell assays and particularly scATAC-seq. We therefore examined how PeakVI handles increasing levels of sparsity. We corrupted the 10X PBMC data by randomly replacing non-zero observations with zeros at a range of probabilities (10%–90%) and trained PeakVI on each corrupted dataset. We then used PeakVI’s estimates of the probability of accessibility for these corrupted observations and compared the estimates from the models trained on corrupted data, in which these observations were 0, to the original estimates from the model trained on the full data, where these observations where non-zero. We computed the error: , where C is the set of corrupted observations, is the probability of accessibility estimated by PeakVI when trained on the corrupted data, and y is the probability of accessibility estimated from the original, uncorrupted, data. We found that PeakVI produces highly consistent results, even in highly sparse situations: with a mean squared error of 0.06 when 10% of the observations are removed, to 0.17 when 90% of the data are removed (Figures 1D and S2). We also observed that the corrupted estimates are generally lower than the original estimates, consistent with the corruption being one-directional (introducing false negatives, not false positives). These results demonstrate that PeakVI is robust to low-quality and highly sparse data.
Since training PeakVI involved stochastic optimization of a non-convex function, the model can produce different results in different runs. We examined how stable PeakVI is to changes in architecture and training hyperparameters by training PeakVI on a variety of configurations and comparing how the different models perform on held-out data. We varied the number of hidden layers in the neural networks, the size of the mini-batch used in training, the dropout rate and learning rate, and the weight decay. For each set of hyperparamters, we trained the model three times and measured the likelihood the model achieves on the held-out data in each run. We found that PeakVI is highly stable and that the default hyperparameters perform well without a need to fine-tune the model for each analysis (STAR Methods; Table S1). Finally, to see how PeakVI stability is impacted by the sparsity of the data, we artificially corrupted data to only retain 50% and 10% of observations and repeated the stability analysis. In both cases, while the model performance decreased compared with the full data, the model remained stable in terms of hyperparameters, indicating that the default hyperparameters perform well even in highly sparse situations (Table S1).
PeakVI learns an informative batch-corrected latent representation
PeakVI learns a low-dimensional representation of each cell that preserves biological heterogeneity while reducing noise, technical artifacts, and batch effects. We compared the latent space learned by PeakVI with representations from published methods. We compared these using four methods: (1) LSA, a natural language processing technique commonly used in scATAC-seq analysis pipelines, such as Signac (Stuart et al., 2021) and ArchR (Granja et al., 2021); (2) cisTopic (González-Blas et al., 2019), which uses latent Dirichlet allocation; (3) SCALE (Xiong et al., 2019), which also employs a VAE and incorporates Gaussian mixture modeling to create a clustered latent space; and (4) chromVAR (Schep et al., 2017), an algorithm that aggregates genomic regions by known binding motifs and normalizes these aggregates to motif accessibility scores. The first two methods, LSA and cisTopic, were chosen since a recent benchmark of computational analysis methods for scATAC-seq methods (Chen et al., 2019) found them to be the best performing methods. SCALE is included in our comparison due to the conceptual similarities with PeakVI. Finally, we included chromVAR since it is commonly used as both a dimensionality reduction method as well as an annotation technique.
First we used the 10× PBMC scATAC-seq data to measure how consistent each latent representation is with the gene expression profiles that are also measured from each cell. We ran all methods on the 10× PBMC data and extracted the latent representation computed by each. We then independently analyzed the paired scRNA-seq data and clustered the cells based on their gene expression profiles (STAR Methods). We then overlaid the scRNA-based cluster labels on the scATAC-based representations (Figure 2A), and measured for each cell the fraction of its chromatin-based K-nearest neighbors that are from the same RNA-based cluster for varying values of K (STAR Methods; Figure 2B). We found that PeakVI and cisTopic outperformed all other methods, with PeakVI doing marginally better than cisTopic. We also measured how robust each method is to library size effects, by computing for each latent space the correlation of the latent representation with the empirical library size (), using Geary’s C (Geary, 1954) (STAR Methods; Figure S2A). We found that LSA and SCALE are especially sensitive to library size effects, while PeakVI and cisTopic are more robust, and chromVAR is insensitive to library size effects.
Figure 2.

UMAP visualizations of latent representations from PeakVI, LSA, cisTopic, SCALE, and chromVAR
(A) The paired scRNA-scATAC sample PBMC dataset from 10× Genomics. Cells are colored based on the scRNA-based clustering; umaps are computed from the scATAC representations. All methods except for chromVAR are comparably consistent with the scRNA data.
(B) Quantitative consistency of the latent representation with the scRNA data; fraction of the K-nearest neighbors in the scATAC representation that are also among the K-nearest neighbors in the scRNA representation, for various values of K. PeakVI marginally outperforms cisTopic, followed by LSA, SCALE, and chromVAR.
(C) Data from Satpathy et al. (2019); cells are colored using the FACS-based cell-type-specific labels. Cells from unsorted samples or non-specific sorted samples are colored in light gray. PeakVI, LSA, and cisTopic all achieve good separation of cell types.
(D) Data from Satpathy et al. (2019); cells are colored using the unsorted PBMC replicates. Cells from all other samples are colored in light gray. Batch effects are reduced with PeakVI, chromVAR, and SCALE.
(E) Enrichment of labels among the K-nearest neighbors for each cell; the x axis is the enrichment of batch labels, where lower enrichment indicates better batch mixing. The y axis is the enrichment of cell-type labels, where higher enrichment indicates better separation. PeakVI reaches a better balance of the two tasks.
See also Figures S2A, S2B, and S3.
Next we looked into how each method handles a more complex experimental design, as in the hematopoiesis dataset, which consists of multiple samples of different sizes, some cell-type-specific and others general. We analyzed the data with all methods. For completeness, we also included a variation of LSA used by the ArchR pipeline (Granja et al., 2021) called Iterative LSA (STAR Methods), as well as three configurations of PeakVI: (1) “no batch,” without any batch annotation; (2) “full batch,” treating each sample as a separate batch; and (3) “replicate batch,” treating each replicate from multi-replicate conditions as a separate batch (STAR Methods). These configurations correspond to having no batch correction, strict batch correction, or an intermediate approach, respectively. We examined how well each method preserves biological heterogeneity by measuring how separated the sorted cell populations are, using the cell-type-specific fluorescence-based labels (Figures 2C, S2B, and S3). We also examined how well each method handles batch effects, which none of the examined methods explicitly corrects, by measuring how well mixed the four different batches of unsorted PBMC samples are (Figures 2D and S2B). For both analyses we computed an enrichment score by computing for every cell the number of neighbors out of its K-nearest neighbors that share its label, and comparing them with the random expectation (STAR Methods), for varying values of K (scores in the text are for ) (Figure 2E). Ideally, this enrichment score would be high for biological labels and low for batch labels. We find that LSA, cisTopic, and PeakVI with nobatch configuration all achieve high separation (enrichment scores 9.1, 9.13, and 9.42, respectively) but separate the different batches as well (enrichment scores 2.33, 2.28, and 2.39, respectively); conversely, chromVAR and SCALE outperform all methods in batch mixing (1.57 and 1.59, respectively) but do worse on cell-type separation (5.78 and 7.03, respectively). Iterative LSA seems to underperform on both tasks. In contrast, we find that PeakVI with replicate-batch strikes a desirable balance, preserving biological heterogeneity comparably well (enrichment score 9.04) while more effectively mixing the batches (enrichment score 1.85). Finally, PeakVI with full-batch configuration also achieves a good balance (8.37 for cell-type separation, 1.88 for batch mixing), but underperforms the replicate-batch configuration on both tasks. Overall, these results demonstrate that PeakVI is better able to correct batch effects while preserving biological heterogeneity, reaching an overall better latent representation than all examined methods.
PeakVI performs differential accessibility analysis at a single-region resolution
Among the main promises of scATAC-seq is the ability to better identify individual genomic elements that help regulate certain biological processes. Achieving this requires the ability to identify individual regions that are differentially accessible between different groups of cells. In practice this task is challenging due to the binary nature of each observation, batch effects, and the high levels of noise and sparsity. Most differential analyses thus choose to aggregate the differential signal across different regions, either by the binding motifs they harbor (i.e., the differential analysis chromVAR performs) or by aggregating the surrounding regions to each gene and creating a gene activity score (Pliner et al., 2018). While these analyses are useful, they do not enable identification of individual regions, thereby not fully unlocking the promise of scATAC-seq data. Some differential analyses are performed in single-region resolution: ArchR (Granja et al., 2021) uses Wilcoxon rank-sum test, and Signac (Stuart et al., 2021) uses a logistic regression model, which models the total number of fragments to account for library size effects. Both of these approaches offer partial solutions to the noise and sparsity issues presented by scATAC-seq.
PeakVI addresses this problem by leveraging the probabilistic nature of the latent space to produce denoised and normalized estimates of accessibility, which enable a robust and accurate estimate of differential accessibility at a single-region resolution. In brief, given a population of cells C and a region j, PeakVI samples from the area of the latent space that corresponds to C and estimates the probability of region j being accessible for each sample, then averages over the samples to get a stable estimate of accessibility: (STAR Methods). Importantly, the representation of the latent space using random variables means that each cell in the original data can be sampled multiple times, allowing PeakVI to sample beyond the available number of observed cells. In addition, this procedure can be conditioned on batch annotation, thereby correcting batch effects. When comparing two populations of cells, and , we use the absolute difference between estimates () as a measure for the extent of differential accessibility (effect size). Compared with ratio-based statistics (e.g., odds ratio), this estimate is more interpretable (representing absolute increase or decrease in binding propensity) and more stable to low-level signals. For instance, this means that an increase from 0.01 to 0.21 will be equivalent to an increase from 0.7 to 0.9 as opposed to the first being a 20-fold increase and the second being a 1.3-fold increase.
Using PeakVI estimates for differential accessibility is more sensitive and robust than using the observed data directly
To compare the estimated effect from PeakVI to the empirical effect calculated directly from the observations, we used the hematopoiesis data and the replicate-batch PeakVI model. We define the empirical accessibility as the proportion of cells in C in which j is observed as accessible: , and the empirical effect is defined equivalently to the estimated effect, as . We clustered the latent representations of the cells and ran a series of comparisons for each cluster. First, we ran two comparisons for each cluster: (1) a “biological” comparison, comparing all cells within the cluster to all other cells, and (2) an “artifactual” comparison, comparing within each cluster cells that originated from the two large PBMC batches (replicates 1 and 2; excluding clusters with less than five cells in either group) (Figure 3A). The biological comparisons are a common use for differential analyses where some real differences in accessibility are expected, whereas the artifactual comparisons are used as negative controls. We ran two additional comparisons for each cluster, comparing cells within that cluster that originated from a given PBMC batch (either replicate 1 or 2) to all cells in all other clusters, which essentially provided two technical replicates of the biological analysis (denoted “biological b1” and “biological b2”).
Figure 3.

Differential accessibility analysis with PeakVI
(A) Illustration of the different comparisons. “real,” compare cells between two population; “null,” compare cells from different batches within a single population; “real b1”/“real b2,” compare cells from a specific batch in a population to all cells in the other population.
(B) Pearson correlations between the estimated and empirical effects.
(C) Correlation of effect size in “real b1” and corresponding effect in “real b2” comparisons. PeakVI estimated effects are far less sensitive to batch effects.
(D) An example (using cluster 14) relationship between the PeakVI estimated effect to the empirical effect in real (top) and null (bottom) comparisons.
(E) The width (measured by the SD) of the effect distributions; PeakVI amplifies real differential effects, and silences nuisance ones.
(F) Level of amplification/silencing depends on level of noise in the empirical effect.
(G) Volcano plots for a GLM (top), Wilcoxon (middle), and PeakVI (bottom) when comparing between two batches of NK cells.
(H) Volcano plots for a GLM (top), Wilcoxon (middle), and PeakVI (bottom) when comparing between B cells and NK cells.
(I) PeakVI (bottom) effect is better correlated with a bulk ATAC-based ground truth comparison and more numerically stable than GLM (top) and Wilcoxon (middle).
We first measured the correlation between the PeakVI estimated effects and the raw data (empirical) effects. We found that the effects are highly correlated in biological comparisons (mean Pearson correlation 0.97), but less so in artifactual comparisons (mean correlation 0.52) (Figure 3B). We then used the results from “biological b1” and “biological b2” results, and found that the estimated effect is highly reproducible (mean correlation 0.95), while we see a marked decrease in reproducibility of the empirical effect (mean correlation 0.66) (Figure 3C). We also noticed that, while the results were highly correlated, there was a difference in the width of the distributions between the estimated and the empirical effects (Figures 3C and S4A). To investigate this effect more thoroughly, we calculated the SD of the distributions for each comparison, and found that, in all biological comparisons (including “biological b1” and “biological b2”), the estimated effect had a wider distribution than the empirical effect, whereas in artifactual comparisons the distributions were either similarly wide or the estimated effect had a narrower distribution (Figure 3D). We also found that this is related to the number of cells included in the compared groups, especially in comparisons that rely on small numbers of cells: in these cases we observed the least difference in standard deviations for the biological comparisons, and the most difference for the artifactual comparisons (Figure 3E).
Taken together, these results demonstrate that PeakVI is amplifying the empirical effect when the effect corresponds to real biological difference, but silences it when it is a product of noise. When the empirical effect is more susceptible to noise (e.g., a smaller number of cells included in the comparison), PeakVI is less able to amplify biological signal, but more efficient in silencing the noise. In contrast, when the empirical effect is calculated with a large number of cells, and is therefore less noisy, PeakVI has less silencing effect, but is able to amplify real differences better.
Statistical significance with PeakVI
To estimate the statistical significance of differential effects, PeakVI uses techniques described in previous methods from our group (Lopez et al., 2018, 2020). In brief, during the sampling procedure described above, PeakVI considers pairs of samples, one from each of the compared groups . PeakVI determines for each pair if the measured effect for each region j is greater than some minimal effect size δ: (for one-sided tests: ). We repeat this many times and define the probability of differential accessibility, , as the proportion of pairs for which (STAR Methods). We then use a conservative multiple hypothesis correction procedure described previously by Lopez et al., (2020) to identify differentially accessible regions with some nominal false discovery rate.
Established pipelines perform this analysis using generalized linear models (e.g., Signac [Stuart et al., 2021]) or standard statistical tests, such as the Wilcoxon rank-sum test or a two-sided t test (e.g., ArchR [Granja et al., 2021]). We therefore compared our differential accessibility analysis with a generalized linear model (GLM) equivalent to that used by Signac: a logistic regression with an additional covariate for the number of fragments in each cell to avoid library size effects dominating the analysis (STAR Methods), as well as to a Wilcoxon rank-sum test used by ArchR. We performed two comparisons using all methods: (1) an artifactual comparison, using the hematopoiesis data we compared between cells from the two PBMC replicates that mapped to cluster 1, corresponding to cells the NK cell label (Figure 3G), and (2) a biological comparison, comparing cells from the NK cell sample to cells from the B cell sample (using only cells that were FACS sorted) (Figure 3H). We found that all approaches show a clear relationship between effect size and statistical significance in both analyses. Both GLM and Wilcoxon results revealed two common issues: (1) some regions have a very large effect size but are not statistically significant, corresponding to regions that have very low detection rates in both populations, and (2) the p values were inflated due to the large sample size. In the artificial comparison, where no biological signal is expected, PeakVI correctly identified no regions as differentially accessible, compared with 910 regions identified by the GLM model and 6,761 regions identified by the Wilcoxon rank-sum test. In the biological comparison, PeakVI identified 11,362 (16.5%) regions as differentially accessible, compared with 33,679 (48.9%) identified by the GLM, and 26,410 (19.7%) identified by Wilcoxon test.
We then ran an equivalent comparison between B cells and NK cells using bulk ATAC-seq data from Calderon et al. (2019) with sorted immune cell populations, as a ground truth (STAR Methods), and compared the results with the scATAC-seq-based results from both analyses (Figure 3I). Overall results from all methods are consistent with the bulk results, but PeakVI achieves higher correlation between the effect sizes (0.74 compared with 0.48 and 0.52 for the GLM and Wilcoxon results, respectively). In terms of correctly identifying differential regions, for both PeakVI and Wilcoxon, 86% of the regions identified were also differential according to the bulk analysis, compared with 65.6% for the GLM. In terms of overlap between the regions found with bulk comparison versus single cell, all analyses resulted in sets of regions that are over-represented at the bulk results, with PeakVI reaching an odds ratio of 1.92, Wilcoxon reaching 1.93, and GLM reaching 1.47. Overall, these results demonstrate that PeakVI provides a well-calibrated statistical significance estimation and enables identification of differentially active regions at a single-region resolution, while minimizing false discovery and avoiding numerical issues due to low detection rates.
PeakVI supports multiple approaches for annotation and discovery of cell states
A major challenge in analyzing scATAC-seq data is the lack of region-based annotations of cell state, in contrast to the abundant resources for RNA-based annotation. Current methods therefore rely on annotations that were generated from gene expression profiles, which are useful but only provide a partial solution, since chromatin accessibility may carry information that is not discernible from gene expression alone. We therefore set out to demonstrate two different approaches for how PeakVI can be leveraged for annotation and downstream discovery. First, PeakVI’s integration capabilities can be used for transfer learning, projecting annotated reference data and un-annotated query data onto a joint space, and transferring insights from the former to the latter. Importantly, this approach relies solely on the regions, without associating regions to target genes or identifying harbored motifs. Second, in the lack of an annotated reference, PeakVI’s differential accessibility analysis can be leveraged for de novo annotation, associating marker regions with nearby genes and identifying enriched gene sets or known marker genes.
PeakVI can be used for transfer learning, by leveraging an annotated reference dataset to annotate a query dataset. First, the reference and query datasets need to be integrated into a joint space, which can be achieved using PeakVI in one of two ways: (1) naively, by analyzing both datasets together and conditioning on the dataset of origin, or (2) using a two-step procedure first presented in scArches (Lotfollahi et al., 2021), in which the reference data are processed in advance, and then incoming query data can be projected onto the reference-based space. The scArches procedure is particularly useful when creating a detailed atlas to be used as a reference resource. After the query and reference are in a shared space, transferring annotations from one to the other can be done using proximity-based classifiers, such as KNN or cluster majority vote (which we utilized here). We demonstrate this ability using the hematopoiesis data as the reference, and a dataset of human PBMCs provided by 10× as a query (note that this dataset is different from the multiomic dataset used in previous sections). Notably, the reference data cover both bone marrow and blood, and consists of samples that were sorted to specific cell types, as well as samples that consist of the entire PBMC compartment. We therefore expect the query data to align only to the parts covered by the reference PBMC samples, and not next to cell subsets that are more abundant in the bone marrow. Furthermore, we expect technical hurdles to complicate the integration of the datasets as they were generated by different experimental protocols and processed with different computational pipelines.
We began by creating a reference model, by analyzing the hematopoiesis data using PeakVI in a scArches-compatible configuration (STAR Methods). We then used PeakVI to project the query PBMC data onto the reference space. PeakVI was able to mix the datasets well, only mapping query cells onto areas of the space occupied by PBMCs, but not those corresponding to progenitor cells, which are absent from the query PBMC data (Figures 4A and S4B). We then clustered the cells and assigned each cluster with the most abundant cell-specific FACS-based label in that cluster from the reference data. Importantly, these annotations are based on similarity of chromatin landscapes between cells in the query and reference data, without any association to other biological features or aggregation, resulting in a straight-forward labeling of the query data (Figure S4C).
Figure 4.

PeakVI unlocks multiple paths for annotation and identification
(A–C) PeakVI supports transfer learning. (A) Mapping of query data (sample PBMC data from 10× Genomics) onto reference data (from Satpathy et al., 2019). PeakVI mixes the query data with the reference despite the data being generated by a different protocol and processed by a different pipeline. (B) The reference data, colored by FACS-based cell-type-specific labels. (C) The query data, colored by the transferred cell-type-specific labels.
(D–F) De novo annotation using PeakVI’s differential accessibility analysis. (D) Hematopoiesis data colored by clusters. (E) Regions that are preferentially accessible in each cluster were analyzed for enriched cell-type signatures from ARCHS (Lachmann et al., 2018) signatures, using enrichr (Chen et al., 2013; Kuleshov et al., 2016). Heatmap shows distribution of cell-type-specific labels for each cluster, normalized by row. (F) Volcano plot for a differential accessibility analysis between the two B cell clusters (clusters 13 and 17).
(G) Volcano plot for only significant regions, labeled by associated genes that are implicated in naive B cells (red) and memory B cells (blue).
See also Figures S4B and S4C.
However, this procedure requires an annotated atlas from a corresponding system, while many scenarios require de novo annotation, which PeakVI facilitates using the differential accessibility analysis. We demonstrate this using the hematopoiesis data, by de novo annotating the data and using the FACS-based labels as a ground truth. We first clustered the latent space (Figure 4D), and consistent with our previous findings we found that clusters tend to consist primarily of cells that have the same label. Next, using our differential accessibility analysis, we compared each cluster to all other clusters except for the three most similar clusters, to avoid highly similar clusters masking the differences (STAR Methods). For each cluster we used a one-sided test to only identify regions that are preferentially open in the target cluster. We then used enrichr (Chen et al., 2013; Kuleshov et al., 2016) to associate the regions to nearby genes and leveraged the ARCHS4 (Lachmann et al., 2018) collection to find over-represented cell-type-specific gene signatures. We were able to confidently identify many of the cell-type-specific clusters, which matched their FACS-based label (Figure 4E; STAR Methods). For instance, marker regions for clusters 13 and 17, in which labeled cells are overwhelmingly B cells, were indeed enriched for regions associated with B cell marker genes; cluster 1 marker regions were enriched for NK cell marker genes, and indeed, the labeled cells in that cluster are NK cells. Similarly signatures for CD4+ T cells, Regulatory T cells, and pDCs, were all highly enriched in the clusters with the corresponding FACS-based labels. Thus, using PeakVI and gene-based signatures, we are able to annotate the data and recapitulate many of the FACS-based labels.
These results are nonetheless limited by the availability of gene signatures, which may not be available for all cell types, or provide only a high-level annotation at a limited resolution. Specifically, most progenitor cells in the hematopoiesis data could not be annotated in a similar fashion for lack of corresponding signatures, and despite clustering separately, both CD4+ naive T cells and CD4+ memory T cells were annotated simply as CD4+ T cells, since higher-resolution signatures were not available. PeakVI can therefore be used in a two-step approaches whereby cells can be stratified into broad types, using reference-based annotation, and then assigned with more high-resolution labels of cell sub-types or states using de novo analysis. As a case in point, we focused on the set of cells which were annotated as B cells in our reference-based analysis. These cells can be divided into two clusters (clusters 13 and 17). To derive a higher-resolution annotation of the B cell compartment, we ran a two-sided comparison between the two clusters and identified 1,043 differentially accessible regions in total, 207 preferentially accessible in cluster 13 and 836 preferentially accessible in cluster 17 (STAR Methods; Figure 4F; Table S2). Among the genes associated with regions detected for cluster 13 we found TCL1A, known to be expressed throughout B cell differentiation up to naive B cells but silenced in memory B cells and plasma cells (Teitell, 2005; Virgilio et al., 1994), and YBX3, implicated in B cell differentiation as an immature B cell marker (Lee et al., 2020). We also found SATB1, TENT5A, and ZNF667-AS1, which along with TCL1A and YBX3, were previously found to be differentially expressed in naive B cells compared with memory B cells (Longo et al., 2009). Concordantly, genes associated with cluster 17 included known markers for memory B cells AIM2 (Svensson et al., 2017) and CD80 (Sahoo et al., 2002), and nine other genes previously found to be differentially expressed in memory B cells compared with naive B cells (Longo et al., 2009) (Figure 4G). Taken together, we concluded that cluster 13 consists of naive B cells and cluster 17 consists of memory B cells, therefore demonstrating that PeakVI’s differential accessibility analysis can be used in conjunction with a reference-based annotation to increase the resolution of annotations and identify new targets for further study.
Discussion
PeakVI is a deep generative model for analyzing single-cell chromatin accessibility data. The model is designed to explicitly account for various technical effects that mask and distort the biological signal. The latent representation learned by the model is probabilistic in nature, embedding the observed cells in a smooth variational space that preserves the biological heterogeneity, minimizes confounding effects, and can be used directly to explore the chromatin landscape of a population of cells. Importantly, PeakVI takes as input a region-by-cell count matrix, allowing the user to integrate PeakVI with current and future preprocessing and peak calling methods.
PeakVI improves upon previous attempts to use deep learning to analyze scATAC-seq data in several manners. First, the architecture used in the underlying neural networks scales with the size of the input data, increasing the expressiveness of the model to match with increasingly large and complex datasets (STAR Methods). Second, PeakVI accounts for technical confounders and enables correction of batch effects, with clear benefits to downstream results. Thirdly, since it is common for features (regions) to outnumber the samples (cells), and the observations are mostly binary and therefore contain little information, PeakVI also takes measures to successfully prevent the model from over-fitting, by holding out some of the data as a validation set, tracking the model’s performance on the validation data, and halting the training process when the performance on the validation data stops improving, thus ensuring that the model is learning generalizable features. Finally, PeakVI provides extensive methods to take advantage of the learned latent space for analysis tasks beyond dimensionality reduction, visualization, and clustering. Specifically, PeakVI enables high-resolution annotation of cell state, by allowing both reference-based analysis and de novo annotation analysis. In that capacity, PeakVI enables accurate differential accessibility analysis at a single-region resolution that reduces the effect of confounders and avoids common issues with the current practices for differential accessibility, namely numerical instability and inflation of significance scores.
Since PeakVI takes as input a region-by-cell matrix, it does not offer a full end-to-end solution to all of the challenges presented by scATAC-seq, instead relying on other methods and pipelines to perform upstream tasks, such as fragment alignment, peak calling, and cell calling. This allows users to match PeakVI with other methods; for instance, using specialized peak callers such as Lancetron (Hentges et al., 2021) or AtacWorks (Lal et al., 2021), and analyzing the resulting matrix with PeakVI to benefit from superior dimensionality reduction, batch correction, differential accessibility, and annotation. In addition, PeakVI is implemented in the scvi-tools suite (Gayoso et al., 2021), which provides interfaces with popular processing environments, such as scanpy (Wolf et al., 2018) and Signac (Stuart et al., 2021). Finally, PeakVI is robust to low-quality data and easy to configure, train, and use. It can be easily incorporated in existing analysis pipelines to enhance current analyses for dimensionality reduction, batch correction, differential accessibility, and annotation.
Limitations of the study
PeakVI relies on peaks that are called upstream of the model, which limits the model’s power in situations where informative peaks were discarded (i.e., in rare populations), unlike methods that perform their own peak calling or that use the fragments directly. This also means that, when integrating multiple datasets, the datasets need to be process jointly to get a shared set of peaks before PeakVI can be employed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| 10× sample PBMC dataset | 10× Genomics | https://www.10xgenomics.com/resources/datasets/10-k-peripheral-blood-mononuclear-cells-pbm-cs-from-a-healthy-donor-next-gem-v-1-1-1-1-standard-2-0-0 |
| Hematopoiesis dataset | Satpathy et al., 2019 | GSE129785 |
| Software and algorithms | ||
| analysis scripts | This paper | https://doi.org/10.5281/zenodo.4728534 |
| PeakVI implementation | This paper | https://github.com/YosefLab/scvi-tools/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Nir Yosef (niryosef@berkeley.edu)
Materials availability
This study did not generate new unique reagents.
Method details
The PeakVI model
Let be a scATAC-seq region-by-cell matrix with N cells and K regions, where is the number of fragments from cell i that map to region j. Since PeakVI models the probability of observing a region, regardless of the number of reads supporting that observation, the observations are treated as binary: , where . The observations are therefore generated from a Bernoulli distribution . PeakVI computes as a product of three probabilities: , where captures the true biological heterogeneity; captures region-specific biases (e.g width, sequence); captures cell-specific biases (e.g library size). The three probabilities are estimated jointly using deep neural networks.
The biological component is estimated using a VAE (Kingma and Welling, 2013), which is composed of two deep neural networks, the encoder and decoder . Briefly, the encoder , computes the distributional parameters of a D-dimensional multivariate normal random variable: . The sample is then concatenated to the batch annotation for cell i, and passed through the decoder , for S being the dimension of the one-hot batch annotation (the number of batches). The cell-specific factor computed from the input data for cell i via a deep neural network . Finally, the region-specific factor , since it is optimized across samples, is stored as a K-dimensional tensor, used and optimized directly.
Architecture
All PeakVI neural nets are fully connected networks, composed of repeated blocks that share a basic structure. For convenience, we define a fully connected block as having a fully connected layer with I input nodes and O output nodes, followed by a drop-out layer with a D probability of dropout, a layer-norm layer, and finally an A activation function.
The encoder is constructed as follows:
With being the default dimensionality of the latent representation. This ensures that the model architecture scales with the number of features in the data and the complexity of the representation.
The decoder is constructed as follows:
With S as the dimensionality of the batch annotations, concatenated to the latent representation.
The cell-specific factor network is constructed similarly:
Training procedure
By default, PeakVI is optimized using AdamW (Loshchilov and Hutter, 2017) with a learning rate of 0.0001, weight decay of 0.001, and minibatch size of 128. The model is trained on of the data, with the remaining used as a validation set. Training is performed for at most 500 epochs, with early stopping: if there is no improvement in terms of the reconstruction loss on the validation set for 50 epochs, the training stops. For epochs the KL divergence term is weighed done by a factor of . The best state throughout training, defined as the state that achieves the best reconstruction loss, is saved during the training and used as the final state. All training settings are configurable.
Differential accessibility analysis
For a differential accessibility analysis between two populations A and B, the analysis is performed as follows:
-
1)
N cells are sampled from each population, with replacement (default ). We denote the resulting cells for the i-th sample from population X, for and .
-
2)
for each cell C, we apply the inference model on the cell’s chromatin accessibility profile to get the variation distribution corresponding to that cell, , sample from that distribution to get an estimated profile of the probability of accessibility of all regions in that cell: . We then use the generative model to estimate the probability of accessibility of each region j in that cell: . Sampling from the variational space allows us to sample the same cell multiple times and get different estimates, thereby enabling statistical power beyond the original sample size.
-
3)
to calculate the effect size for each region, we simply take the average estimated probability of accessibility across all samples from each population, and compute the absolute difference between the averages: .
-
4)
to calculate the statistical significance, we randomly pair samples from each population into N pairs of estimates , then for each region we count for how many pairs the difference between estimates was greater than some minimal δ (default 0.05): . This procedure has been previously described by (Lopez et al., 2020).
-
5)
In addition to , we also compute the Bayes factor: , and perform multiple testing correction using the procedure previously described by Lopez et al. (Lopez et al., 2020) to get a qualitative, binary label for each region.
Quantification and statistical analysis
Stability analysis
To measure the stability of PeakVI to hyperparameter selection, we ran a full grid search using the 10× Genomics sample data. We held out 10% of the data as a test set and trained all models on the remaining set. We trained each model 3 times (with an independent train-validation split) and measured the likelihood on the held-out data. The full results are available in Table S1. The hyperparameters we varied and the values used are as follows: learning rate (1e-2, 1e-3, 1e-4); number of hidden layers (1,2,3,4); dropout rate (0.1, 0.3); minibatch size (64, 128, 256); weight decay (0.1, 0.01, 0.001).
Dataset processing
The hematopoiesis data was downloaded from GEO (Accession GSE129785); specifically the processed peak-by-cell matrix and metadata files: scATAC-Hematopoiesis-All.cell-barcodes.txt.gz, scATAC-Hematopoiesis-All.mtx.gz, scATAC-Hematopoiesis-All.peaks.txt.gz. We then filtered the genomic region to only those that are detected in at least 0.1% of the cells in the sample, reducing the data from 571400 regions to 133962 regions. The sample data from 10× genomics was also downloaded as preprocessed peak-by-cell matrices, without any additional filters.
Running published methods
For all methods, we followed the standard recommended procedure for analyzing data. For visualization, we computed the umap (McInnes et al., 2018) coordinates using the python implementation from the latent space computed by the respective method (except for SCALE, see below). cisTopic (v0.3.0): We used the WarpLDA model fitting procedure, and chose the best number of topics based on the second derivative, as recommended by the package documentation. For the hematopoiesis data the model used 100 topics, and 40 topics for the paired PBMC sample data from 10X Genomics. chromVAR (v1.12.0): We used the JASPAR2016 motif set, containing 386 motifs, and followed the standard analysis outlined in the package documentation. We used the unnormalized motif deviation scores. For dimensionality reduction, we found no clear difference between using the chromVAR scores directly and applying an additional linear procedure (i.e principle component analysis). Results described in the manuscript use the deviation scores directly. LSA: We used the python implementation from the Scikit-learn (Pedregosa et al., 2011). We first binarized the data, then computed the top 50 components used the TruncatedSVD method, on the tfidf-transformed data. SCALE (v1.0.4): we used the external script to run SCALE without a pre-determined number of clusters, using the default arguments. In all visualizations, we used the umap coordinates computed by SCALE.
Enrichment score calculation
Enrichment scores used to quantify cell type separation and batch mixing were computed in an identical way. Given a latent representation R, an integer k, and cell labels L, we first compute , the K-nearest neighbor graph from R with k neighbors. We then compute for each cell the proportion of neighbors that share the same label: . The overall score is the average score across all cells, , normalized by the expected score for a random sample from the distribution of labels: , for being the set of available labels, and being the proportion of each label . The enrichment score is then .
Differential expression with logistic regression
As a simple benchmark for differential accessibility, we constructed a standard logistic regression model to compare B-cells to NK-cells, using the design , where y is the binary detection of a genomic region. We fit the model using the function in R. Due to the runtime of this analysis, we limited the results to regions that are detected in at least 1% of the compared cells.
Analysis of bulk ATAC-seq data
The bulk ATAC-seq data used as a ground truth reference for differential accessibility analysis was downloaded from GEO (accession GSE118189). We used the unstimulated samples of all B-cell and NK-cell subtypes included in the study and used DESeq2 (Love et al., 2014), which was found to be among the ebst performing methods for differential accessibility from bulk ATAC-seq data (Gontarz et al., 2020) for differential accessibility between the two group. We then found regions in the hematopoiesis data that overlap with the regions in the bulk data, and used the differential signal found in the bulk data for the overlapping regions in the hematopoiesis data.
Projection of query data onto reference
Projection of query data onto a latent space learned from reference data is done using scArches (Lotfollahi et al., 2021). First, the 10× sample PBMC data was downloaded and processed (using CellRanger v3.1.0) using the hematopoiesis peaks. We then trained a PeakVI model on the hematopoiesis data using ell covariate injection, which adds one-hot encoded batch annotation to each layer in the VAE (as opposed to only the decoder layers, which is the default behavior). We then trained the resulting model on the query data, which involves adding batch annotations corresponding to the query data, and only training the nodes in the network that interact with these additional batches. This preserves the latent representation of the reference data while projecting the query data onto the same space, while correcting batch effects between the query and data.
Cluster annotation with differential accessibility
Differential accessibility to identify marker regions for each cluster was performed between each cluster and all other clusters except the three most similar clusters. This was in order to avoid sampling pairs of cells that are highly similar from the two groups, which would reduce the signal. We therefore calculated the centroid of each cluster (the average position in the latent space of all cells in the cluster), computed the Euclidean distance matrix between all centroids, and identified for each cluster the 3 most similar clusters. We then used the identified regions (using the Bayesian FDR method described by Lopez et al. (Lopez et al., 2020)), ran them through enrichr (Chen et al., 2013; Kuleshov et al., 2016), and downloaded the enrichment results for the ARCHS4 Tissues set. For associating regions with genes, we used the bioconductor package TxDb.Hsapiens.UCSC.hg19.knownGene (Carlson and Maintainer, 2015) and considered only strict overlaps between the region and the annotated gene body or promoter.
Acknowledgments
We thank Florian Wimmers for many helpful discussions and insightful feedback. We thank Christina Usher for assistance with visualizations. This work was funded by Chan Zuckerberg Foundation Network grant no. 2019-02452 and NIH–NIAID grant U19 AI090023.
Author contributions
T.A. and N.Y. conceived of the model and designed the analyses. T.A. implemented the model with input from A.G. T.A. and D.A.R. performed the analyses. N.Y. supervised the work. T.A. and N.Y. wrote the manuscript.
Declaration of interests
All authors declare no competing interests.
Published: March 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100182.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P., Chang H.Y., Greenleaf W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon D., Nguyen M.L.T., Mezger A., Kathiria A., Müller F., Nguyen V., Lescano N., Wu B., Trombetta J., Ribado J.V., et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet. 2019;51:1494–1505. doi: 10.1038/s41588-019-0505-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M., Maintainer B.P. 2015. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation package for TxDb object(s). (R package version 3.2.2.) https://bioconductor.org/packages/release/data/annotation/html/TxDb.Hsapiens.UCSC.hg19.knownGene.html. [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Lareau C., Andreani T., Vinyard M.E., Garcia S.P., Clement K., Andrade-Navarro M.A., Buenrostro J.D., Pinello L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019;20:241. doi: 10.1186/s13059-019-1854-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford G.E., Holt I.E., Whittle J., Webb B.D., Tai D., Davis S., Margulies E.H., Chen Y.D., Bernat J.A., Ginsburg D., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16:123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R., Preissl S., Li Y., Hou X., Lucero J., Wang X., Motamedi A., Shiau A.K., Zhou X., Xie F., et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021;12:1337. doi: 10.1038/s41467-021-21583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso A., Lopez R., Xing G., Boyeau P., Wu K., Jayasuriya M., Melhman E., Langevin M., Liu Y., Samaran J., et al. scvi-tools: a library for deep probabilistic analysis of single-cell omics data. bioRxiv. 2021 doi: 10.1101/2021.04.28.441833. [DOI] [PubMed] [Google Scholar]
- Geary R.C. Vol. 5. Royal Statistical Society; 1954. The contiguity ratio and statistical mapping; pp. 115–146. (The Incorporated Statistician). [Google Scholar]
- Gontarz P., Fu S., Xing X., Liu S., Miao B., Bazylianska V., Sharma A., Madden P., Cates K., Yoo A., et al. Comparison of differential accessibility analysis strategies for ATAC-seq data. Sci. Rep. 2020;10:10150. doi: 10.1038/s41598-020-66998-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Blas C.B., Minnoye L., Papasokrati D., Aibar S., Hulselmans G., Christiaens V., Davie K., Wouters J., Aerts S. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods. 2019;16:397–400. doi: 10.1038/s41592-019-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges L.D., Sergeant M.J., Downes D.J., Hughes J.R., Taylor S. LanceOtron: a deep learning peak caller for ATAC-seq, ChIP-seq, and DNase-seq. bioRxiv. 2021 doi: 10.1101/2021.01.25.428108. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Welling M. Auto-encoding variational Bayes. arXiv. 2013 https://arxiv.org/abs/1312.6114v10 Preprint at. [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A., Torre D., Keenan A.B., Jagodnik K.M., Lee H.J., Wang L., Silverstein M.C., Ma’ayan A. Massive mining of publicly available RNAseq data from human and mouse. Nat. Commun. 2018;9:1366. doi: 10.1038/s41467-018-03751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A., Chiang Z.D., Yakovenko N., Duarte F.M., Israeli J., Buenrostro J.D. Deep learning-based enhancement of epigenomics data with AtacWorks. Nat. Commun. 2021;12:1507. doi: 10.1038/s41467-021-21765-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R.D., Munro S.A., Knutson T.P., LaRue R.S., Heltemes-Harris L.M., Farrar M.A. Single-cell analysis of developing B cells reveals dynamic gene expression networks that govern B cell development and transformation. bioRxiv. 2020 doi: 10.1101/2020.06.30.178301. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo N.S., Lugar P.L., Yavuz S., Zhang W., Krijger P.H.L., Russ D.E., Jima D.D., Dave S.S., Grammer A.C., Lipsky P.E. Analysis of somatic hypermutation in X-linked hyper-IgM syndrome shows specific deficiencies in mutational targeting. Blood. 2009;113:3706–3715. doi: 10.1182/blood-2008-10-183632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R., Boyeau P., Yosef N., Jordan M.I., Regier J. Decision-making with auto-encoding variational Bayes. arXiv. 2020 doi: 10.48550/arXiv.2002.07217. Preprint at. [DOI] [Google Scholar]
- Loshchilov I., Hutter F. Decoupled weight decay regularization. arXiv. 2017 https://arxiv.org/abs/1711.05101 Preprint at. [Google Scholar]
- Lotfollahi M., Naghipourfar M., Luecken M.D., Khajavi M., Büttner M., Wagenstetter M., Avsec Ž., Gayoso A., Yosef N., Interlandi M., et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 2021;40:1–10. doi: 10.1038/s41587-021-01001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L., Healy J., James M. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv. 2018 https://arxiv.org/abs/1802.03426 Preprint at. [Google Scholar]
- Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Müller A., Nothman J., Louppe G., et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- Pliner H.A., Packer J.S., McFaline-Figueroa J.L., Cusanovich D.A., Daza R.M., Aghamirzaie D., Srivatsan S., Qiu X., Jackson D., Minkina A., et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol. Cell. 2018;71:858–871.e8. doi: 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissl S., Fang R., Huang H., Zhao Y., Raviram R., Gorkin D.U., Zhang Y., Sos B.C., Afzal V., Dickel D.E., et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci. 2018;21:432–439. doi: 10.1038/s41593-018-0079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo N.C., Rao K.V.S., Natarajan K. CD80 expression is induced on activated B cells following stimulation by CD86. Scand. J. Immunol. 2002;55:577–584. doi: 10.1046/j.1365-3083.2002.01093.x. [DOI] [PubMed] [Google Scholar]
- Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones D.E., Cui K., Cuddapah S., Roh T.-Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D.R., Kaminski J., Barnitz R.A., Kurachi M., Gerdemann U., Yates K.B., Tsao H.-W., Godec J., LaFleur M.W., Brown F.D., et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson A., Churqui M.P., Schlüter K., Lind L., Eriksson K. Maturation-dependent expression of AIM2 in human B-cells. PLoS One. 2017;12:e0183268. doi: 10.1371/journal.pone.0183268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitell M.A. The TCL1 family of oncoproteins: co-activators of transformation. Nat. Rev. Cancer. 2005;5:640–648. doi: 10.1038/nrc1672. [DOI] [PubMed] [Google Scholar]
- Virgilio L., Narducci M.G., Isobe M., Billips L.G., Cooper M.D., Croce C.M., Russo G. Identification of the TCL1 gene involved in T-cell malignancies. Proc. Natl. Acad. Sci. U S A. 1994;91:12530–12534. doi: 10.1073/pnas.91.26.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L., Xu K., Tian K., Shao Y., Tang L., Gao G., Zhang M., Jiang T., Zhang Q.C. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat. Commun. 2019;10:4576. doi: 10.1038/s41467-019-12630-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.