

Abstract
Methylmalonic acidemia patients have complex rehabilitation needs that can be targeted to optimize societal independence and quality of life. Thirty-seven individuals with isolated MMA (28 mut, 5 cblA, 4 cblB), aged 2–33 years, were enrolled in a natural history study, and underwent age-appropriate clinical assessments to characterize impairments and disabilities. Neurological examination and brain imaging studies were used to document movement disorders and the presence of basal ganglia injury. A range of impairments and disabilities were identified by a team of physical medicine experts. Movement disorders, such as chorea and tremor, were common (n = 31, 83%), even among patients without evidence of basal ganglia injury. Joint hypermobility (n = 24,69%) and pes planus (n = 22,60%) were frequent and, in many cases, under-recognized. 23 (62%) patients required gastrostomy feedings. 18/31 patients >4 years old (58%) had difficulties with bathing and dressing. 16 of 23 school-aged patients received various forms of educational support. Five of the 10 adult patients were employed or in college; three lived independently. Unmet needs were identified in access to rehabilitation services, such as physical therapy (unavailable to 14/31), and orthotics (unavailable to 15/22). We conclude that patients with MMA are challenged by a number of functional limitations in essential activities of mobility, self-care, and learning, in great part caused by movement disorders and ligamentous laxity. Early assessment, referral, and implementation of age-appropriate rehabilitation services should significantly improve independence and quality of life.
Keywords: methylmalonic acidemia, rehabilitation, outcome, disabilities, basal ganglia
INTRODUCTION
The term isolated methylmalonic acidemia (MMA) refers to a heterogeneous group of autosomal recessive inborn errors of branched chain amino acid metabolism, affecting approximately 1:50,000–1:80,000 births [Coulombe et al., 1981; Shigematsu et al., 2010]. The disorders result from deficient activity of methylmalonyl-CoA mutase, encoded by the MUT gene (designated mut0 or mut− for complete or partial deficiency), which catalyzes the isomerization of L-methylmalonyl-CoA into succinyl-CoA at the terminal step of propionyl-CoA metabolism in the mitochondrial matrix. Defects in the synthesis of the enzymatic co-factor, 5′-deoxyadenosylcobalamin, corresponding to the cblA, cblB, or cblD-variant 2 subtypes, are caused by mutations in the MMAA, MMAB, or MMADHC genes, respectively [Manoli and Venditti, 2005]. The clinical spectrum of MMA is wide and varies from the severe infantile, vitamin B12 non-responsive types (mut0, mut−, many cblB), which present within the first days-weeks of life with metabolic ketoacidosis, hyperammonemia, and encephalopathy, to milder B12-responsive forms (cblA, some cblB) that manifest in the first months or years of life with failure to thrive, developmental delay, neurological syndromes, and renal tubular acidosis [Horster et al., 2007; Horster et al., 2009]. The patients are prone to recurrent episodes of acute metabolic decompensation and basal ganglia (globus pallidus) injury (BGI). The long-term outcome of MMA has not been fully explored but can be complicated by growth failure, intellectual impairment, end-stage renal disease, and high mortality [Roodhooft et al., 1990; Manoli and Venditti, 2005]. The implementation of expanded newborn screening (NBS) to diagnose MMA has magnified the importance of defining the long-term outcomes and clinical phenotypes in these disorders.
The combination of intensive treatment protocols for MMA, including specialized formulas, gastrostomy feeding, central catheters for parenteral nutrition, medication to augment ureagenesis, and increasingly, organ transplantation, has improved survival rates [Horster et al., 2009; Hauser et al., 2011; Baumgartner et al., 2014], perhaps at the expense of increased morbidity. Moreover, the demanding medical management of MMA patients often dominates the efforts of their metabolic teams, impacting on time dedicated toward addressing aspects of care that could enhance functional independence and improve quality of life. While most published data on long-term MMA outcomes have focused on medical endpoints such as survival and neurocognition, there have been fewer efforts to document functionality, independence and social integration or the need for rehabilitation interventions that could maintain and maximize function. [van der Meer et al., 1994; Leonard, 1995; Sniderman et al., 1999; de Baulny et al., 2005; Cosson et al., 2009].
Using a retrospective, cross-sectional, single-center study, we analyzed the spectrum of functional impairments and disabilities seen in a cohort of well-characterized MMA patients. In addition to delineating the functional phenotype of MMA for the first time, our results could provide guidance in addressing the rehabilitation needs and therapeutic services required to optimize long-term outcomes in this patient population and provide a metric for future studies.
MATERIALS AND METHODS
The patient population was evaluated between 2005 and 2012, through NIH protocol “Clinical and Basic Investigations of Methylmalonic Acidemia and Related Disorders” (ClinicalTrials.gov identifier: NCT00078078). All subjects and/or guardians provided consent/assent and the study was conducted in compliance with the Helsinki Declaration. The subtype of MMA was assigned by cellular biochemistry and complementation analysis (Dr. David S. Rosenblatt, Division of Medical Genetics, McGill University), and/or molecular genetic testing (GeneDx, Gaithersburg, MD). Participants were evaluated by a rehabilitation medicine team that included a physiatrist (physical medicine and rehabilitation physician/PM&R), physical therapist (PT), and occupational therapist (OT). The reports of these evaluations and their recommendations were reviewed to record functional impairments in various organ systems, and disabilities across the scope of function, including in mobility, self-care, and feeding. Based on accepted milestones [Hagan et al., 2008], four years of age was set as the threshold when age-appropriate independence in basic activities of daily living (eating, grooming, bathing, dressing, and toileting) would be anticipated. Joint range of motion was assessed in the clinical rehabilitation evaluations and hypermobility was defined as endpoint of range of motion well beyond expected norms; however, Beighton hypermobility scores were not recorded. Coordination and balance were assessed in an age-appropriate manner, which included examining eye/hand coordination, dexterity while performing specific tasks, posture control, quality of sitting and standing balance, and quality of gait. The presence of movement disorders and peripheral sensory deficits was ascertained through neurology consultation. Visual impairment and hearing loss were based on ophthalmology and audiology assessments. The diagnosis of a “metabolic stroke” (basal ganglia injury/BGI) was based on neuroimaging findings, or, in selected cases, a clear history of prior hospitalization consistent with metabolic decompensation and subsequent permanent neurologic sequelae. The MRI methodology on describing globus pallidus infarcts has been previously described [Baker et al., 2014].
Findings were classified as “impairments” and “functional limitations/disabilities.” According to accepted guidelines by the National Center for Medical Rehabilitation Research (NCMRR), an “impairment” is defined as loss of function/structure at the organ/organ system level; a “functional limitation” is a restriction to perform an action within the range consistent with the purpose of an organ/organ system; and a “disability” is the limitation in performing tasks, activities and social roles as expected for nondisabled peers (Fig. 1) [National Institute of Child Health & Human Development, 1993].
FIG. 1.
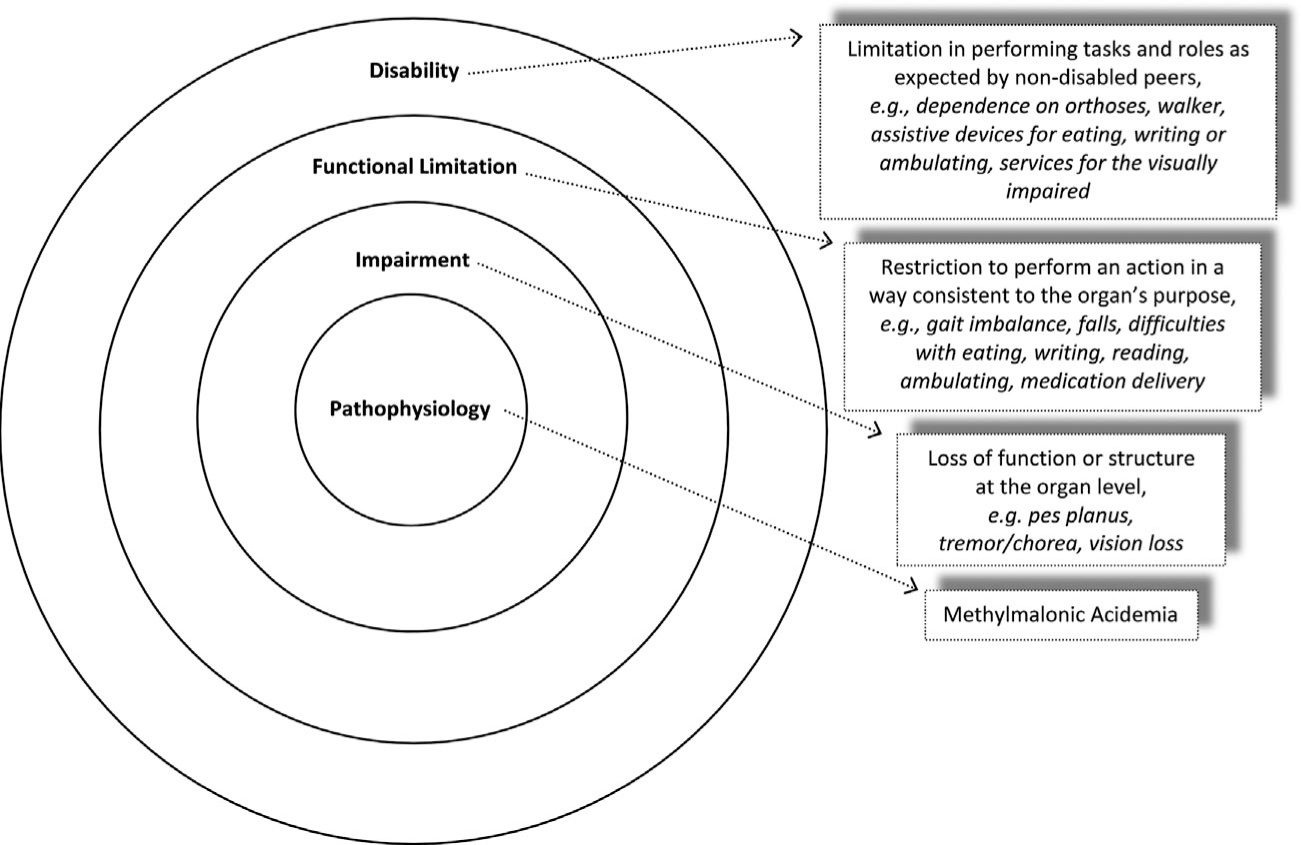
Definitions and MMA-specific examples of impairment, functional limitation, and disability, based on the NCMRR model of disablement.
Descriptive statistics were used to outline the prevalence of impairments and disabilities. To compare the prevalence of impairments between patients with and without metabolic strokes, Fisher’s exact tests and unpaired t-tests were performed, using SPSS v.19 (IBM Corporation, Armonk, NY). Statistical significance was defined as P < 0.05.
RESULTS
Demographics and Clinical Presentation
Thirty-seven patients (21 males, 16 females) with isolated MMA (28 mut, 5 cblA, 4 cblB) were evaluated by PM&R, PT, and OT; Table I presents selected clinical characteristics per subtype. In all patients, cellular enzymology and/or mutation analysis confirmed the biochemical diagnosis (Table II). Mean age at evaluation was 12.5 years (range: 2–33). In the mut group, two patients were diagnosed prenatally because of a previously affected offspring and two by NBS. One patient in the cblB group was asymptomatic and diagnosed due to an affected sibling. Mean age at diagnosis was 4.5 months (range: 0–24). Twenty-three patients had gastrostomies and six had central venous access devices. Eighteen patients had clear evidence of BGI, 14 confirmed by MRI, and four through past medical history, as previously described.
TABLE I.
Summary of the Patient Cohort
| Subtype | mut (n = 28) | cblA (n = 5) | cblB (n = 4) | Total (n = 37) |
|---|---|---|---|---|
| Age at Evaluation in years, Range (Mean) | 2–27 (10.6) | 2–33 (16.4) | 8–24 (18.3) | 2–33 (12.5) |
| Age at Diagnosis in months, Range (Mean) | 0–10 (1.1) | 10–24 (16.2) | 8–24 (12.5) | 0–24 (4.5) |
| Males : Females (n) | 14:14 | 5:0 | 2:2 | 21:16 |
| Basal Ganglia Injury (n) | 11 | 4 | 3 | 18 |
TABLE II.
Demographic, Cellular Enzymology. Molecular Genetic Testing, and Clinical Characteristics
| Pt | Age at Evaluation (years) | Age at Diagnosis (months) | Sex | Subtype | Gene | Mutation 1 | Mutation 2 | Presentation | Major Co-morbidities |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 0.2 | f | mut0, early onset | MUT | c.1106G>A p.R369H | c.682C>T p.R228X | HA, Hypotonia, +NBS, Leukopenia | |
| 2 | 2 | 10 | m | cblA | MMAA | c.433C>T p.R145X | c.1076G>A p.R359Q | BGI, MA | BGI |
| 3 | 2 | 23 | m | cblA | MMAA | c.592_595delACTG p.T198SfsX6 | c.387C>A p.Y129X | BGI, MA, Lethargy | BGI |
| 4 | 3 | 0, NBS | m | mut−, P/NBS | MUT | c.1942G>C p.G642R | c.1181T>A p.L394X | +NBS, Asymptomatic | |
| 5 | 3 | 0.1 | f | mut−, early onset | MUT | c.281G>T p.G94V | c.2150G>T p.G717V | HA, MA, Hypotonia | BGI |
| 6 | 3 | 0.1 | f | mut0, early onset | MUT | c.607G>A p.G203R | c.682C>T p.R228X | HA, MA, Lethargy, Hypothermia | |
| 7 | 4 | 0.1 | f | mut0, early onset | MUT | c.1207C>T p.R403X | c.2008G>C p.G670R | Lethargy, FTT | |
| 8 | 5 | 0, prenatal | f | mut0, P/NBS | MUT | c.2053dupCTC p.685insL | c.91C>T p.R31X | Affected sibling, Asymptomatic | BGI |
| 9 | 6 | 0.1 | f | mut0, early onset | MUT | c.927G>A p.W309X | c.983T>C p.L328P | HA, MA, Lethargy | |
| 10 | 6 | 0.1 | m | mut0, early onset | MUT | c.670G>T p.E224X | c.682C>T p.R228X | HA, MA, Hypothermia, Hypoglycemia | |
| 11 | 6 | 0.1 | m | mut0, early onset | MUT | c.17241C>T p.R581X | c.753+2T>A p.splice | HA, MA, Hypothermia | |
| 12 | 7 | 0, NBS | f | mut0, P/NBS | MUT | c.1942G>C p.G642R | c.1942G>C p.G642R | +NBS, Asymptomatic | |
| 13 | 7 | 0.2 | m | mut0, early onset | MUT | c.1106G>A p.R369H | |||
| c.1778_1782delAAAGT p.S594RfsX11 | Recurrent emesis, FTT | ||||||||
| 14 | 7 | 7 | m | mut0, late onset | MUT | c.1867G>C p.G623R | c.281G>T p.G94V | HA, MA, Lethargy, Hypoglycemia | BGI |
| 15 | 7 | 3 | m | mut0, early onset | MUT | c.1301_1302delTC p.L434HfsX3 | c.1844C>G p.P615R | Recurrent emesis, FTT | |
| 16 | 8 | 11 | f | cblB | MMAB | c.556C>T p.R186W | c.556C>T p.R186W | MA, FTT | BGI |
| 17 | 9 | 0.75 | m | mut0, early onset | MUT | c.1105C>T p.R369C | c.1207C>T p.R403X | Recurrent emesis, FTT | |
| 18 | 10 | 0.1 | f | mut0, early onset | MUT | c.1867G>C p.G623R | c.323G>A p.R108H | MA | BGI |
| 19 | 10 | 0.1 | f | mut0, early onset | MUT | c.572C>A p.A191E | unknown | Recurrent emesis, FTT | BGI |
| 20 | 10 | 0, prenatal | f | mut0, P/NBS | MUT | c.2053dupCTC p.685insL | c.91C>T p.R31X | Affected sibling, Asymptomatic | |
| 21 | 10 | 0.2 | m | mut0, early onset | MUT | c.682C>T p.R228X | c.1332+1delG p.splice | MA, Lethargy | |
| 22 | 11 | 0.1 | f | mut0, early onset | MUT | c.1658delT p.V553GfsX17 | c.29dupT p.L11TfsX38 | MA, FTT | Vision |
| 23 | 11 | 0.1 | m | mut0, early onset | MUT | c.682C>T p.R228X | c.682C>T p.R228X | HA, MA, Lethargy | |
| 24 | 12 | 0.1 | m | mut0, early onset | MUT | c.1106G>A p.R369H | c.1106G>A p.R369H | MA, Recurrent emesis | Vision |
| 25 | 13 | 16 | m | cblA | MMAA | c.433C>T p.R145X | c.433C>T p.R145X | MA, Lethargy, Recurrent emesis | |
| 26 | 18 | 8 | f | cblB | MMAB | c.556C>T p.R186W | c.556C>T p.R186W | MA, Hypotonia, FTT | BGI |
| 27 | 18 | 10 | m | mut0, early onset | MUT | c.572C>A p.A191E | c.655A>T p.N219Y | Recurrent emesis, FTT, Hypotonia | BGI |
| 28 | 19 | 0.1 | f | mut0, early onset | MUT | c.1106G>A p.R369H | c.1106G>A p.R369H | HA, MA, Lethargy, Hypothermia | BGI |
| 29 | 22 | 0.1 | m | mut0, early onset | MUT | c.572C>A p.A191E | c.682C>T p.R228X | MA, Recurrent emesis, Hypoglycemia | BGI, Hearing, Vision |
| 30 | 22 | 7 | m | mut0, early onset | MUT | c.1741C>T p.R581X | c.1741C>T p.R581X | HA, MA, Lethargy, Recurrent emesis | BGI, Vision |
| 31 | 23 | 9 | m | cblB, asympto-matic | MMAB | c.556C>T p.R186W | c.700C>T p.Q234X | Affected sibling, Asymptomatic | |
| 32 | 24 | 30 | m | cblB | MMAB | c.556C>T p.R186W | c.700C>T p.Q234X | MA, FTT | BGI |
| 33 | 24 | 3 | f | mut−, early onset | MUT | c.2150G>T p.G717V | c.2150G>T p.G717V | Recurrent emesis | BGI, Vision |
| 34 | 24 | 0.1 | m | mut0, early onset | MUT | c.655A>T p.N219Y | c.1048C>T p.H350Y | HA, MA, Lethargy | BGI, Hearing |
| 35 | 27 | 5 | f | mut0, early onset | MUT | c.1106G>A p.R369H | c.826G>T p.E276X | MA, Hypotonia, Recurrent emesis, FTT | Hearing, Vision |
| 36 | 32 | 13 | m | cblA | MMAA | c.433C>T p.R145X | c.433C>T p.R145X | MA, BGI | BGI |
| 37 | 33 | 18 | m | cblA | MMAA | c.450dupG p.P151AfsX19 | c.450dupG p.P151AfsX19 | MA, BGI | BGI |
BGI, basal ganglia injury; FTT, Failure to thrive; HA, hyperammonemia; MA, metabolic acidosis; NBS, newborn screening; Pt=patient. Early onset was defined as presentation of first symptoms prior to 30 days of life. Presenting symptoms were obtained through patient record review and/or parental interview.
Impairments
All 37 patients demonstrated evidence of one or more impairments (Fig. 2). Movement disorders and other neurological impairments that could affect function, including chorea, tremor, dystonia, dysmetria, asterixis, and ataxia, were the most prevalent impairment (n = 31). Choreiform movements were the most common finding (n = 13) and ranged from very mild, elicited only with the patient focused on a task such as tandem gait, to more obvious and extensive choreoathetoid movements at rest. Although movement disorders were more prevalent and more severe in patients who had BGI by imaging and/or history compared to those who did not (18/18 vs. 13/19, Fisher’s exact P = 0.02), most patients without clear evidence of BGI also had a detectable movement disorder (13/19) suggesting that subclinical BGI, even in the absence of a defined stroke, is common. The six (5 mut and 1 cblB) patients with a normal neurological exam had neither historical nor imaging evidence of BGI.
FIG. 2.
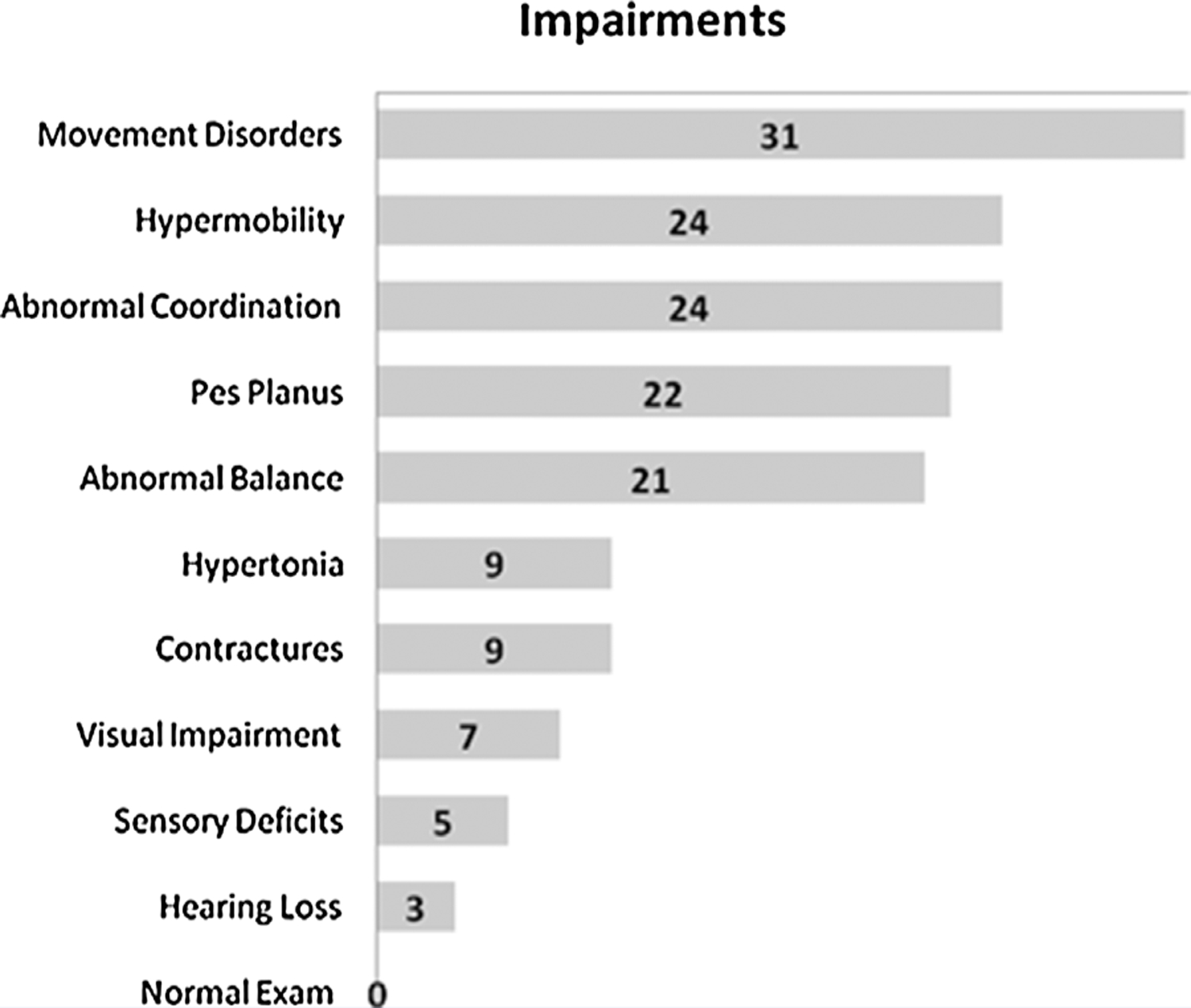
Type and frequency of impairments.
The most common musculoskeletal impairment identified was joint hypermobility (n = 24), and pes planus (n = 22) (Fig. 3), which has not been reported previously in patients with MMA. In addition, difficulties in coordination were noted in 24 and balance difficulties in 21 patients. A number of patients had hypertonia and contractures (n = 9), out of which seven had BGI, and the other two had negative MRIs but movement disorders suggestive of subclinical BGI. A smaller group (n = 5) had neurosensory deficits (decreased sense of touch, vibration, or proprioception). Lastly, visual pathology (optic nerve atrophy n = 6, retinal pathology n = 1) was present in seven patients and hearing loss was identified in three.
FIG. 3.
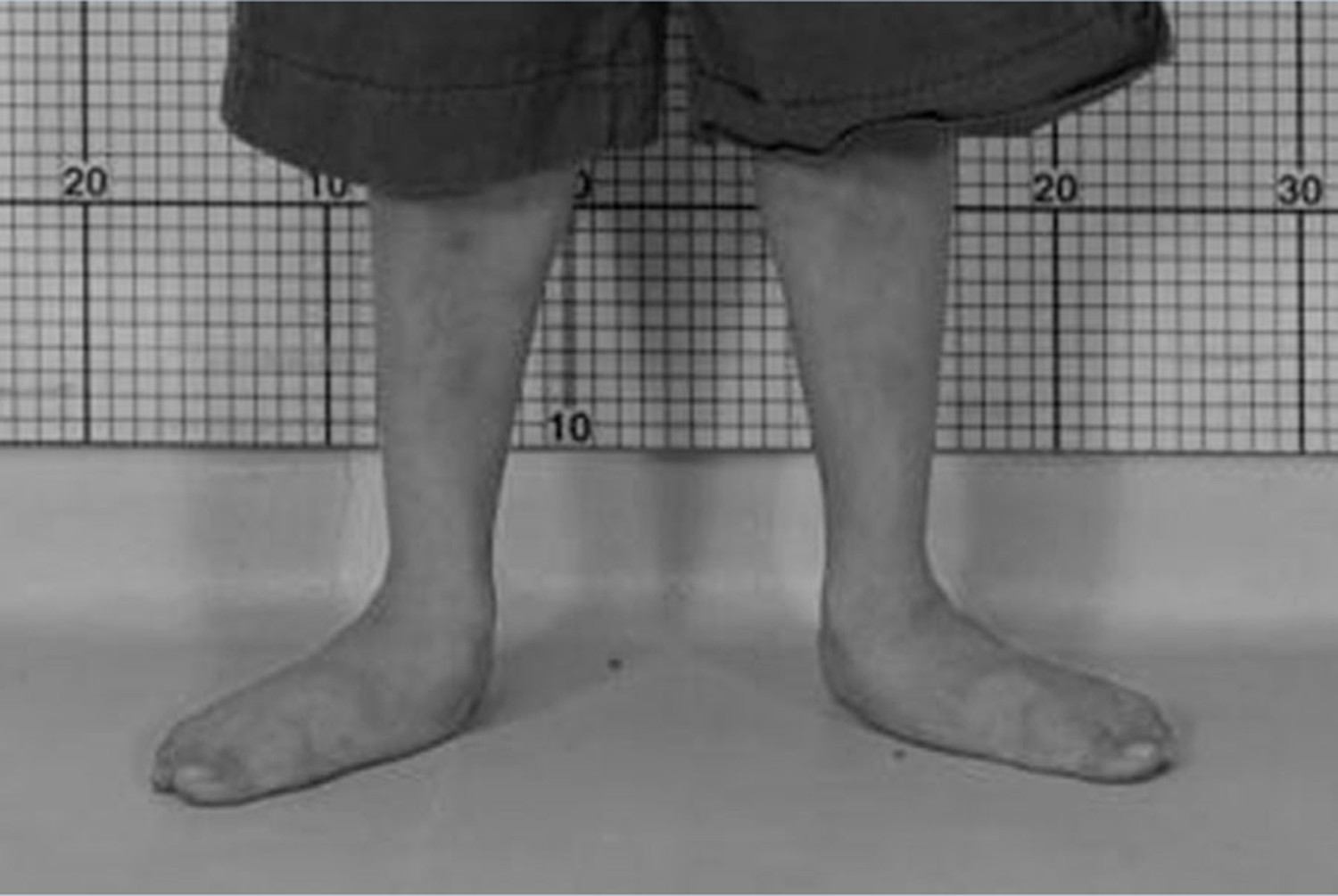
Pes Planus in an MMA patient.
Although the number of patients in the cblA (n = 5) and cblB (n = 4) groups was too small to allow for reliable statistical analysis, most patients in these groups had multiple impairments. Nearly all had a movement disorder: 4/5 cblA patients (with the exception of the patient that had no history of BGI) and all four cblB patients (including Patient 31, who was diagnosed due to an affected sibling and had no evidence of BGI on MRI, however had dysmetria and tremor elicited on neurological exam). In addition, most of these patients had additional impairments, with the most common ones being hypertonia (4/9), joint hypermobility (4/9), and contractures (3/9), however none had visual or hearing impairment at the time of evaluation.
Of the patients who were diagnosed via newborn screening or prenatally due to an affected sibling (Patients 4, 8, 12, 20), all of which were mut, one had severe movement disorder secondary to BGI confirmed by MRI and a wheelchair user, one had milder findings elicited on neurological exam, and all had hypermobility and pes planus. Only one mut patient in our cohort was late onset (presentation after 30 days of life), and had a known metabolic stroke with associated sequelae and multiple impairments. Three patients were classified as mut− based on enzymology, and all were found to have a movement disorder, which was more prominent and accompanied by hypertonia in two of the three with BGI identified on MRI.
Functional Limitations/Disabilities
Functional evaluations were performed in 31/37 patients that were >4 years old (Fig. 4). Eighteen patients had problems with dressing, where the most frequently reported challenge was the inability to fasten closures (e.g., buttons, zippers, snaps). Twelve of 26 of the patients >9 years of age had difficulties with bathing, including the inability to wash hair independently. Twenty-seven of 31 patients were able to eat independently, in an age-appropriate manner. Twenty-three patients had gastrostomies, and three were able to prepare and administer the tube feedings independently (ages 13, 19, and 22).
FIG. 4.
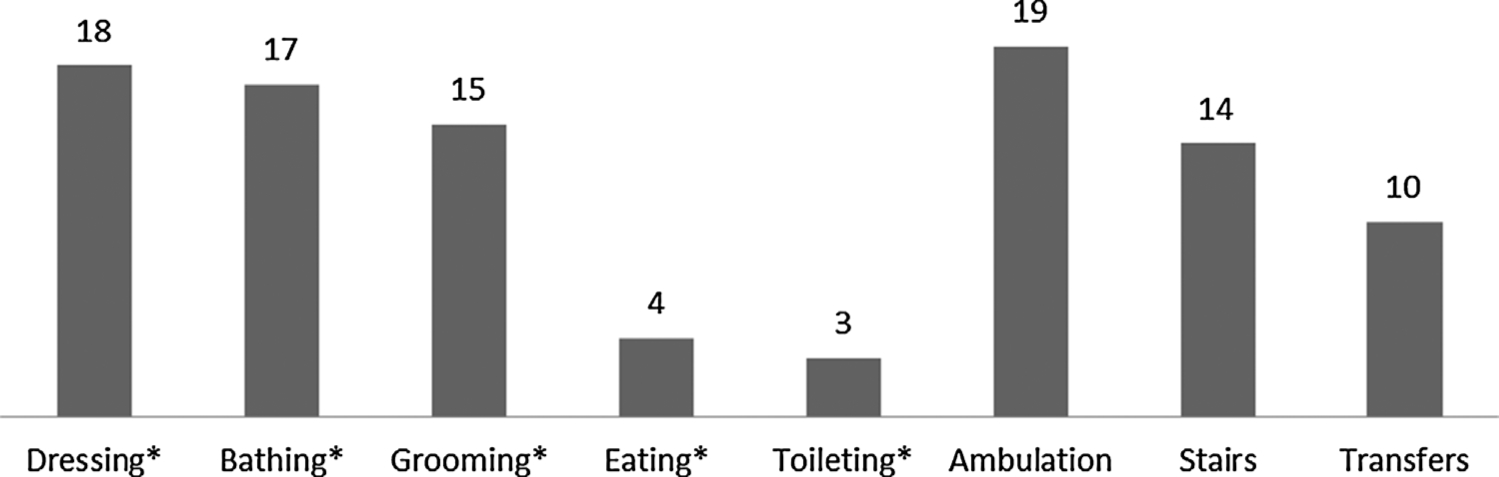
Prevalence of functional limitations/disabilities.
All 37 patients were >2 years old at the time of evaluation and developmentally expected to be ambulatory. However, only 18 ambulated independently on all surfaces without any devices. Seven patients wore orthoses, and 12 required significant assistive devices for functional mobility (walker, n = 4; wheelchair, n = 6; motorized scooter, n = 2). The most commonly encountered challenge was negotiating stairs (n = 14).
Difficulty with handwriting was a common problem (10/23 school-aged patients) and always associated with a movement disorder, even without clear radiographic/historical evidence of BGI (n = 5).
Rehabilitation Needs
The rehabilitation services that the patients were receiving at the time of evaluation were compared to the services recommended after assessment by the NIH Rehabilitation Medicine team (Fig. 5). The discrepancy between currently utilized and recommended services showed that the greatest areas of unmet needs were PM&R and PT, where 14/22 and 14/31 of patients, respectively, were not receiving any services despite clinical evidence of impairments and disabilities felt to require the coordination of a physiatrist and treatment from rehabilitation therapists. Only 8/22 patients that were in need of coordinated rehabilitation services had ever been evaluated by PM&R before their visit. Although all patients had some form of musculoskeletal or neuromotor impairment, only 17 were receiving physical therapy regularly. More than half (22/37) of the patients were in need of devices to improve ambulation ability, such as shoe orthoses, but most had never received them (15/22). Handwriting was also affected, mostly due to movement disorders, and 10/37 were thought to be able to benefit from adaptive writing utensils, but less than half of those had previously been provided with such equipment.
FIG. 5.
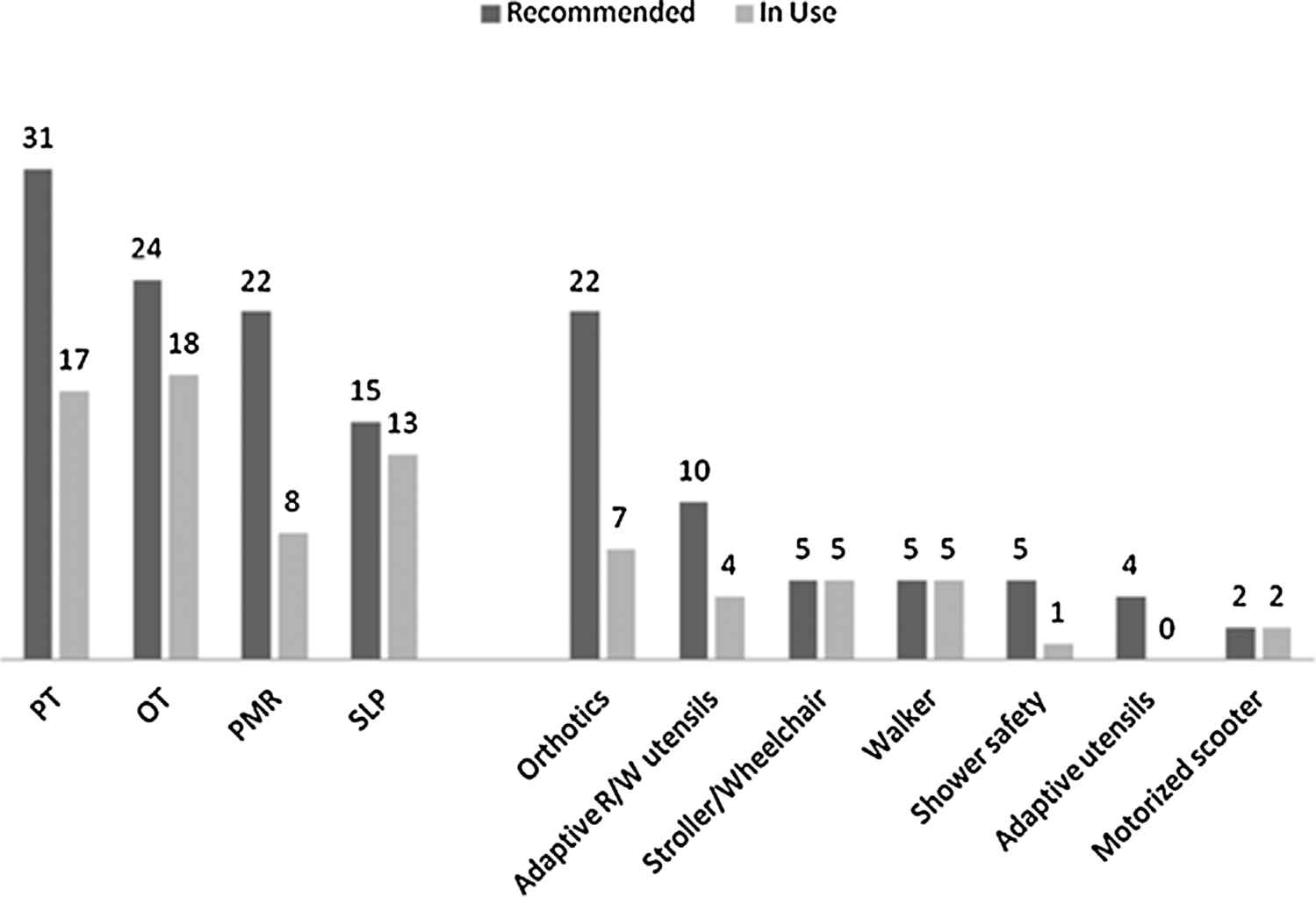
Needs in rehabilitation services and equipment
When patients were stratified by age, it was discovered that in those 2–9 years (n = 17), the greatest areas of unmet needs were PM&R (8/12), PT (6/15), orthotics (6/10) and adaptive reading/writing utensils (4/5). Between the ages of 10–18 (n = 10), the unmet needs were still apparent: PM&R (2/4), PT (5/9), OT (2/6), orthotics (9/10), and adaptive reading/writing utensils (2/4). In patients >18 years (n = 10) the greatest areas of unmet needs were PM&R (4/6), PT (3/7), OT (2/6), adaptive utensils (0/2), and shower safety equipment (0/2). Despite the fact that this older age group carried the highest prevalence of contractures (5/9), only two of the adult patients had been previously evaluated by PM&R, and four by PT and OT; the rest did not have access to rehabilitative services. Out of the all the patients with contractures (n = 9), only one had regular access to PM&R, and three to PT.
Education, Independent Living & Vocation
Twenty-four patients were school-aged (3–18 years) and the majority (16/23; Patient 5 had not started school yet) had individualized educational plans, including self-contained classes, resource classes, personal aide, special schools or home tutors (Fig. 6). At the time of evaluation, 10 patients were >18 years: four were employed at least part-time, one was in college and five were unemployed, out of whom one was previously employed and in search of new employment with the help of a vocational rehabilitation counselor; one patient lived alone and two patients with a partner; seven lived with parents or other family (ages 18–37, mean = 23).
FIG. 6.

Individualized educational plans 70% (16/23) of school-aged patients were receiving educational support. Two also required services for the visually impaired. APE is specifically designed to address individual needs, e.g., with activities to strengthen an atrophic limb. Self-contained classrooms are classrooms dedicated to students with learning difficulties; resource class is the part-time alternative.
DISCUSSION
While the mortality associated with MMA has been well documented since the 1980s [Matsui et al., 1983; Leonard et al., 1984; Rousson and Guibaud, 1984; Horster et al., 2007], the morbidity associated with the diagnosis requires further definition, especially because newborn screening for MMA and related disorders has been recently implemented. Our study is the first attempt to elucidate the functional phenotype of MMA across a broad age spectrum, in a well-characterized patient population. In addition to drawing attention to the functional challenges that MMA patients face, we document disease associations not widely recognizable, such as joint hypermobility, describe specific rehabilitation needs, and provide evidence on the chasm that exists between the functional deficits of these patients and the care that they receive. The NCMRR model (Fig. 1) serves as the framework for interpreting the results of this study; this rehabilitation medicine paradigm uses a classification which recognizes that disabilities stem from system impairments created by medical conditions, and provides a platform for the strategic identification of interventions to optimize patient functionality [National Institute of Child Health & Human Development, 1993].
Among the previously undescribed impairments was the documentation of generalized joint hypermobility/ligamentous laxity and associated pes planus (Figs. 2 and 3), suggesting connective tissue involvement as a novel musculoskeletal manifestation seen in patients with MMA. Increased joint angles, such as the ones noted in our patient cohort, can be caused by both low muscle tone and abnormal elasticity of the surrounding tissues. Interestingly, many of our patients who had increased muscle tone secondary to globus pallidus injury were also found to have joint hypermobility, which further underlines the involvement of the ligaments in the pathophysiology of the disease. Because hypermobility leads to diminished ambulation and can be improved with orthoses and PT [Adib et al., 2005; Falkerslev et al., 2013], our results suggest that the screening and treatment of pes planus should become part of the routine care for MMA patients. However, we should note that the retrospective data of this study were descriptive in nature and based on physical exam findings; future research with hypermobility scoring systems will provide further information.
The movement disorders in this patient cohort displayed a recognizable pattern, ranging from fine, distal, involuntary movements, to generalized choreoathetosis and/or dystonia in patients with extensive basal ganglial injuries. The resulting impairments ranged from subtle limitations in fine motor abilities (impacting writing, dressing, etc), to severe movement disorders that lead to global disability and a limited ability to participate in any purposeful physical activity. The well-recognizable sequelae of severe CNS injury, such as hypertonia, contractures, and muscle atrophy, were also seen. There is concern that these complications may have arisen or worsened as a consequence of the limited amount and scope of rehabilitation treatment these patients received. Neurocognitive outcomes in a subset of this cohort have been previously reported, where processing speed was identified as a distinct impairment and an association between hyperammonemia at diagnosis and cognitive impairment was recognized [O’Shea et al., 2012].
In our cohort, the largest areas of need included self-care, ambulation and fine motor motor skills, as well as educational support. The majority of patients had one or more difficulties with basic activities of daily living, and most required individualized education plans (Figs. 4 and 6). Although very few patients had been using assistive equipment prior to their visit, it was found that most patients required orthoses (60%), and one third needed adaptive reading or writing utensils (Fig. 5). Thus, in many cases, especially in the adult population, impairments had not been recognized prior to the evaluations under our protocol and functional needs had frequently gone unmet. The high prevalence of unmet needs, both in patients with and without metabolic strokes, emphasizes the importance for care providers to seek consultation from physiatry for the assessment and coordination of rehabilitation needs and services. Vigilance in addressing such needs is important, especially because simple interventions, such as the provision of arch support for pes planus and use of orthoses, may significantly improve the ability for patients to ambulate effectively and safely. In our study, information was not available as to the etiology of the gap between patient needs and the services actually provided. We hypothesize that the answer is multifactorial; a combination of under-recognition of needs, lack of financial resources, medical insurance coverage, or non-compliance.
Several other issues that might have an impact on the quality of life of MMA patients should also be taken into account when designing treatment plans. Most patients need gastrostomy tubes for part of their nutrition; however the majority of patients were unable to administer their feeds independently, either due to a movement disorder or cognitive impairment. Central venous access devices were implanted in the most severe cases, as they are critical for the prompt management of acute metabolic decompensation, which adds another dimension of care to their complex management. Also, long-term complications, even in recipients of organ transplantation, can further hinder functional outcomes. For example, optic nerve atrophy and sensorineural hearing loss, seen in a small number of patients in our cohort, immediately change quality of life and independence. Furthermore, education is often challenging not only because of the neurocognitive or sensory impairments, but, also because of behavioral problems and communication barriers. All of the above highlight the need for MMA-related anticipatory guidance in order to implement accommodations in educational programs and other services.
The data obtained in this study come from a cohort of patients referred from independent USA and international centers. A limitation of our study is that more severe patients may have been over-represented in our cohort. For example, 4/5 cblA patients born prior to NBS had a metabolic stroke, but the majority of cblA patients, if diagnosed and treated early, might be expected to have milder phenotypes due to the B12-responsive nature of the defect. Also, very few of our patients were diagnosed through NBS and therefore many suffer the long-term sequelae of symptomatic presentation. Our cohort only included one late onset mut patient, and three mut− patients. Therefore, extension of our observations to other cohorts needs to be accompanied by both enzymatic and genetic stratification as well as the timing of diagnosis (Table II). However, because severely affected patients can experience symptoms before the return of NBS results, and tend to experience recurrent symptoms and evolution of disease despite NBS, the functional phenotype of isolated MMA we have delineated should help guide practitioners in developing better rehabilitative care plans and provides an important historical metric for the impact of NBS on functional outcomes in MMA.
One goal of this study was to identify the areas of functional impairments and disability that are involved in MMA, as a first step towards choosing the best-suited standardized tools for future studies. More precise quantification of disabilities will aid in describing the functional phenotype of MMA in more depth, identifying rehabilitation treatment plans and assessing their efficacy. Among the global disability measures that should be considered for prospective monitoring are the WeeFIM (Functional Independence Measure, adapted for children) and the Pediatric Disability Inventory (PEDI). These tools take into account the amount of caregiver assistance required and the use of assistive equipment to perform a task, and evaluate performance in domains such as self-care, mobility, communication, and social cognition [Msall et al., 1994; Batshaw, 2007]. Future studies should concentrate on the collection of longitudinal functional data that will provide insight on the development and progression of impairments and disabilities in MMA. If generic scales, such as those mentioned above, are found insufficient, the development of an MMA-specific disability scale to monitor patients, measure outcomes, and assess therapeutic interventions, in MMA and other organic acidurias with overlapping phenotypes, will represent an important avenue of future research.
Limited data exist on the long-term outcome of organic acidurias [Baumgartner and Viardot, 1995; van der Meer et al., 1996]. The course of life and limited aspects of the functional phenotype in affected adults, including some with MMA, were discussed by Martin-Hernandez et al., who noted the importance of multidisciplinary support, especially as more patients survive into adulthood and desire independence [Martin-Hernandez et al., 2009]. In other organic acidurias, such as glutaric aciduria type I, and in conditions such as cerebral palsy, mitochondrial myopathies, and muscular dystrophies, the impact of carefully designed rehabilitation plans to maximize functionality is well-documented [Gitiaux et al., 2008,Abresch et al., 2012; Mazzone et al., 2012; Moll and Cott, 2012; Msall and Tremont, 1999; Prats Vinas, 2001]. Our results suggest that MMA should be addressed in a similar fashion, with thoughtful consideration of short and long-term patient needs and comprehensive treatment plans to address both.
ACKNOWLEGMENTS
The authors would like to thank the nurses, occupational therapists, and physical therapists of the NIH Clinical Research Center for their assistance with protocol studies; the efforts of Dr. Glenn Hanna for his assistance with data collection in the early phase of this project, and the patients and families for participating in the protocol. The authors have no conflict of interest to disclose.
REFERENCES
- Abresch RT, Carter GT, Han JJ, McDonald CM. 2012. Exercise in neuromuscular diseases. Phys Med Rehabil Clin N Am 23:653–673. [DOI] [PubMed] [Google Scholar]
- Adib N, Davies K, Grahame R, Woo P, Murray KJ. 2005. Joint hypermobility syndrome in childhood. A not so benign multisystem disorder? Rheumatology 44:744–750. [DOI] [PubMed] [Google Scholar]
- Baker EH, Sloan JL, Hauser NS, Gropman AL, Adams DR, Toro C, Manoli I, Venditti CP. 2015. MRI characteristics of globus pallidus infarcts in isolated methylmalonic acidemia. AJNR Am J Neuroradiol. 36(1): 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batshaw ML. 2007. Children with disabilities, Sixth Edition. Baltimore, MD: Paul H. Brookes. [Google Scholar]
- Baumgartner ER, Viardot C. 1995. Long-term follow-up of 77 patients with isolated methylmalonic acidaemia. J Inherit Metab Dis 18: 138–142. [DOI] [PubMed] [Google Scholar]
- Baumgartner MR, Hörster F, Assoun M, Ballhausen D, Burlina A, Chapman KA, Dionisi-Vici C, Fowler B, Grünert S, Grünewald S, Haliloglu G, Hochuli M, Honzik T, Huemer M, Karall D, MacDonald A, Martinelli D, Merinero B, Pérez-Cerdá C, Sass J, Scholl-Burgi S, Skovby F, Valayannopoulos V, Wijburg F, Chakrapani A. 2014. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 9(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson MA, Benoist JF, Touati G, Dechaux M, Royer N, Grandin L, Jais JP, Boddaert N, Barbier V, Desguerre I, Campeau PM, Rabier D, Valayannopoulos V, Niaudet P, de Lonlay P. 2009. Long-term outcome in methylmalonic aciduria: A series of 30 French patients. Mol Genet Metab 97:172–178. [DOI] [PubMed] [Google Scholar]
- Coulombe JT, Shih VE, Levy HL. 1981. Massachusetts Metabolic disorders screening program. II. methylmalonic aciduria. Pediatrics 67:26–31. [PubMed] [Google Scholar]
- de Baulny HO, Benoist JF, Rigal O, Touati G, Rabier D, Saudubray JM. 2005. Methylmalonic and propionic acidaemias: Management and outcome. J Inherit Metab Dis 28:415–423. [DOI] [PubMed] [Google Scholar]
- Falkerslev S, Baago C, Alkjaer T, Remvig L, Halkjaer-Kristensen J, Larsen PK, Juul-Kristensen B, Simonsen EB. 2013. Dynamic balance during gait in children and adults with Generalized Joint Hypermobility. Clin Biomech 28:318–324. [DOI] [PubMed] [Google Scholar]
- Gitiaux C, Roze E, Kinugawa K, Flamand-Rouviere C, Boddaert N, Apartis E, Valayannopoulos V, Touati G, Motte J, Devos D, Mention K, Dobbelaere D, Rodriguez D, Roubertie A, Chabrol B, Feillet F, Vidailhet M, Bahi-Buisson N. 2008. Spectrum of movement disorders associated with glutaric aciduria type 1: A study of 16 patients. Mov Disord 23:2392–2397. [DOI] [PubMed] [Google Scholar]
- Hagan JF, Shaw JS, Duncan P, eds. 2008. Bright futures: Guidelines for health supervision of infants, children, and adolescents. Elk Grove Village, IL: American Academy of Pediatrics. [Google Scholar]
- Hauser NS, Manoli I, Graf JC, Sloan J, Venditti CP. 2011. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. Am J Clin Nutr 93:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horster F, Baumgartner MR, Viardot C, Suormala T, Burgard P, Fowler B, Hoffmann GF, Garbade SF, Kolker S, Baumgartner ER. 2007. Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut−, cblA, cblB). Pediatr Res 62:225–230. [DOI] [PubMed] [Google Scholar]
- Horster F, Garbade SF, Zwickler T, Aydin HI, Bodamer OA, Burlina AB, Das AM, De Klerk JB, Dionisi-Vici C, Geb S, Gokcay G, Guffon N, Maier EM, Morava E, Walter JH, Schwahn B, Wijburg FA, Lindner M, Grunewald S, Baumgartner MR, Kolker S. 2009. Prediction of outcome in isolated methylmalonic acidurias: combined use of clinical and biochemical parameters. J Inherit Metab Dis 32:630–639. [DOI] [PubMed] [Google Scholar]
- Leonard JV, Daish P, Naughten ER, Bartlett K. 1984. The management and long term outcome of organic acidaemias. J Inherit Metab Dis 7:13–17. [DOI] [PubMed] [Google Scholar]
- Leonard JV. 1995. The management and outcome of propionic and methylmalonic acidaemia. J Inherit Metab Dis 18:430–434. [DOI] [PubMed] [Google Scholar]
- Manoli I, Venditti CP. 2005. Methylmalonic Acidemia. In: Pagon RA, Adam MP, Ardinger HH, et al. , editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1231/ [Google Scholar]
- Martin-Hernandez E, Lee PJ, Micciche A, Grunewald S, Lachmann RH. 2009. Long-term needs of adult patients with organic acidaemias: Outcome and prognostic factors. J Inherit Metab Dis 32:523–533. [DOI] [PubMed] [Google Scholar]
- Matsui SM, Mahoney MJ, Rosenberg LE. 1983. The natural history of the inherited methylmalonic acidemias. N Engl J Med 308:857–861. [DOI] [PubMed] [Google Scholar]
- Mazzone ES, Vasco G, Palermo C, Bianco F, Galluccio C, Ricotti V, Castronovo AD, Mauro MS, Pane M, Mayhew A, Mercuri E. 2012. A critical review of functional assessment tools for upper limbs in Duchenne muscular dystrophy. Dev Med Child Neurol 54:879–885. [DOI] [PubMed] [Google Scholar]
- Moll LR, Cott CA. 2012. The paradox of normalization through rehabilitation: growing up and growing older with cerebral palsy. Disabil Rehabil 35:1276–1283. [DOI] [PubMed] [Google Scholar]
- Msall ME, Tremont MR. 1999. Measuring functional status in children with genetic impairments. Am J Med Genet 89:62–74. [DOI] [PubMed] [Google Scholar]
- Msall ME, DiGaudio K, Rogers BT, LaForest S, Catanzaro NL, Campbell J, Wilczenski F, Duffy LC. 1994. The Functional Independence Measure for Children (WeeFIM). Conceptual basis and pilot use in children with developmental disabilities. Clin Pediatr 33:421–430. [DOI] [PubMed] [Google Scholar]
- Research Plan for the National Center for Medical Rehabilitation Research. 1993. National Institute of Child Health & Human Development, National Institutes of Health, editor: U.S. Department of Health and Human Services, Public Health Service, NIH Publication No. 93–3509. [Google Scholar]
- O’Shea CJ, Sloan JL, Wiggs EA, Pao M, Gropman A, Baker EH, Manoli I, Venditti CP, Snow J. 2012. Neurocognitive phenotype of isolated methylmalonic acidemia. Pediatrics 129:e1541–e1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prats J 2001. Glutaric aciduria type I: An organic acidemia without acidosis with severe movement disorders. Neurologia 16:337–341. [PubMed] [Google Scholar]
- Roodhooft AM, Baumgartner ER, Martin JJ, Blom W, Van Acker KJ. 1990. Symmetrical necrosis of the basal ganglia in methylmalonic acidaemia. Eur J Pediatr 149:582–584. [DOI] [PubMed] [Google Scholar]
- Rousson R, Guibaud P. 1984. 1984. Long term outcome of organic acidurias: survey of 105 French cases (1967–1983). J Inherit Metab Dis 7:10–12. [DOI] [PubMed] [Google Scholar]
- Shigematsu Y, Hata I, Tajima G. 2010. Useful second-tier tests in expanded newborn screening of isovaleric acidemia and methylmalonic aciduria. J Inherit Metab Dis 33:S283–S288. [DOI] [PubMed] [Google Scholar]
- Sniderman LC, Lambert M, Giguere R, Auray-Blais C, Lemieux B, Laframboise R, Rosenblatt DS, Treacy EP. 1999. Outcome of individuals with low-moderate methylmalonic aciduria detected through a neonatal screening program. J Pediatr 134:675–680. [DOI] [PubMed] [Google Scholar]
- van der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, Hubert P, Depondt E, Rapoport D, Rabier D, Charpentier C. 1994. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr 125:903–908. [DOI] [PubMed] [Google Scholar]
- van der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, Hubert P, Depondt E, Rapoport D, Rabier D, Charpentier C, Parvy P, Bardet J, Kamoun P, Saudubray JM. 1996. Clinical outcome and long-term management of 17 patients with propionic acidaemia. Eur J Pediatr 155:205–210. [DOI] [PubMed] [Google Scholar]