

Abstract
The global COVID-19 pandemic underscores the dire need of effective antivirals. Encouraging progress has been made in developing small molecule inhibitors targeting the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) and main protease (Mpro). However, the development of papain-like protease (PLpro) inhibitors faces several obstacles. Nevertheless, PLpro represents a high-profile drug target given its multifaceted roles in viral replication. PLpro is involved in not only the cleavage of viral polyprotein but also modulation of host immune response. In this study, we conducted a drug-repurposing screening of PLpro against the MedChemExpress bioactive compound library and identified three hits, EACC, KY-226, and tropifexor, as potent PLpro inhibitors with IC50 values ranging from 3.39 to 8.28 μM. The three hits showed dose-dependent binding to PLpro in the thermal shift assay. In addition, tropifexor inhibited the cellular PLpro activity in the FlipGFP assay with an IC50 of 10.6 μM. Gratifyingly, tropifexor showed antiviral activity against SARS-CoV-2 in Calu-3 cells at non-cytotoxic concentrations. Overall, tropifexor represents a novel PLpro inhibitor that can be further developed as SARS-CoV-2 antivirals.
Keywords: SARS-CoV-2, papain-like protease, PLpro, antiviral, tropifexor, GRL0617
Graphical Abstarct
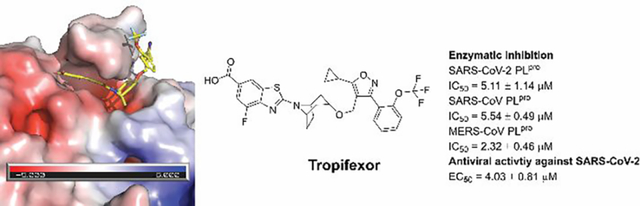
Drug repurposing screening identified tropifexor as a potent SARS-CoV-2 papain-like protease inhibitor with antiviral activity.
The etiological agent of COVID-19 is SARS-CoV-2, a single-stranded, positive-sense RNA virus that belong to the β-coronavirus genera. Given the catastrophic impact of COVID-19 on public health and global economy, researchers around of the globe are working relentlessly to develop vaccines and antiviral drugs. This effort led to the approval of vaccines and antiviral drugs in record breaking speed. Two mRNA vaccines from Moderna and Pfizer, and one adenovirus-based vaccine from Johnson & Johnson were approved by FDA.1
Although vaccines are the mainstay in combating the pandemic, antiviral drugs are nevertheless needed as complementary strategies. Vaccines are preventative, while antiviral drugs can be used for the treatment of COVID patients. In addition, the mRNA vaccines target the viral spike protein, which is prone to mutation as shown by the variants of concerns including the Delta variant and the most recent Omicron variant.2 As a result, vaccines might need to be frequently updated to match the circulating strains. In comparison, small molecule antiviral drugs targeting the conserved viral proteins are expected to have broad-spectrum antiviral activity and a high genetic barrier to drug resistance. The viral RNA-dependent RNA polymerase (RdRp) inhibitor remdesivir is the first FDA-approved COVID drug.3 In addition, the second RdRp inhibitor molnupiravir4–6 and the main protease (Mpro) inhibitor PF-07321332 (Paxlovid)7 are FDA-approved specific oral COVID drugs.
Despite the encouraging progress, additional antiviral drugs with a novel mechanism of action are still in dire need to override the emergence of new mutations. They can be used either alone or in combination with existing RdRp inhibitors or Mpro inhibitors to combat not only current COVID-19 pandemic, but also future coronavirus outbreaks. SARS-CoV-2 expresses two viral proteases, the Mpro and papain-like protease (PLpro), during viral replication. Both Mpro and PLpro are cysteine proteases that mediate the cleavage of viral polyprotein during viral replication.8 In addition, PLpro desregulates the host immune responses by cleaving ubiquitin and interferon-stimulated gene 15 protein (ISG15) from host proteins.9 Therefore, inhibiting PLpro is a two-pronged approach in protecting host cells from viral infection.
PLpro is a 35-kDa domain of Nsp3, a 215-kDa multidomain protein that is a key component of the viral replication complex.10 Compared to PLpro from SARS-CoV, SARS-CoV-2 PLpro displays decreased deubiquitination activity and enhanced deISGlyation activity.9, 11
In contrast to Mpro, PLpro is a more challenging drug target mainly for two reasons. First, the protein substrate of PLpro consists of LXGG.12 Accordingly, there is a lack of drug binding pockets in the S1 and S2 subsites. As such, majority of reported PLpro inhibitors are non-covalent inhibitors that bind to the S3 and S4 subsites that are located more than 10 Å away from the catalytic cysteine C111.13–15 Second, PLpro cleaves the same substrate sequence LXGG as the human deubiquitinase,16 which presents a challenge in developing selective PLpro inhibitors. Despite extensive high-throughput screening and lead optimization,11, 13–15, 17, 18 GRL0617 and its analogs remain the most potent PLpro inhibitors reported so far. To identify structurally novel PLpro inhibitors, we conducted a drug repurposing screening and identified EACC, KY-226, and tropifexor, as potent PLpro inhibitors with IC50 values ranging from 3.39 to 8.28 μM. EACC is a reversible autophagy inhibitor.19 KY-226 is a potent, selectivity, and orally bioavailable allosteric protein tyrosine phosphatase 1B (PTP1B) with an IC50 of 0.25 μM.20 Tropifexor is a highly potent agonist of the farnesoid X receptor and is currently undergoing phase II clinical trial for nonalcoholic steatohepatitis (NASH) and liver fibrosis.21 Their mechanism of action was further characterized in the thermal shift assay and the FlipGFP protease assay. Gratifyingly, tropifexor also had potent antiviral activity against SARS-CoV-2 in Calu-3 cells with an EC50 of 4.03 μM. Overall, tropifexor represents a potent PLpro inhibitor with a novel scaffold that can be further developed as SARS-CoV-2 antivirals.
RESULTS AND DISCUSSION
High-throughput screening of SARS-CoV-2 PLpro inhibitors
Using the previously optimized FRET assay condition,15 we performed a high-throughput screening of SARS-CoV-2 PLpro against the MedChemExpress bioactive compound library which consists of 9,791 compounds including FDA-approved drugs, clinical candidates, and natural products. The assay was performed in 384-well plate with a Z’ of 0.688 and GRL0617 was included as the positive control. All compounds were originally screened at 40 μM, and hits showing more than 50% inhibition were further titrated to determine the IC50 values. GRL0617 was included as a positive control. In total, three compounds, EACC, KY-226, and tropifexor (Figure 1A), were identified as positive hits with IC50 values of 8.28, 3.39, and 5.11 μM, respectively (Figure 1B). In comparison, the IC50 value for the positive control GRL0617 was 1.66 μM (Figure 1B). Next, the broad-spectrum activity of the three hits was tested against SARS-CoV PLpro (Figure 1C) and MERS-CoV PLpro (Figure 1D). It was found that EACC, KY-226, and tropifexor retained potent inhibition against SARS-CoV PLpro with IC50 values of 6.28, 3.53, and 5.54 μM, respectively (Figure 1C). In contrast, EACC and KY-226 were weak inhibitors of MERS-CoV PLpro with IC50 values of 27.8 and 30.6 μM, while GRL0617 was inactive (IC50 > 60 μM) (Figure 1D). Nevertheless, tropifexor showed higher potency against MERS-CoV PLpro with an IC50 of 2.32 μM (Figure 1D). The hits were further counter screened against the SARS-CoV-2 Mpro to rule out promiscuous cysteine protease inhibitors.22–25 It was found that EACC and KY-226 were not active (IC50 ≥ 60 μM), while tropifexor had weak inhibition with an IC50 of 43.65 μM, which corresponds to a selectivity index (SI) of 8.5 (Figure 1E). These results suggest the inhibition of SARS-CoV-2 PLpro by tropifexor is specific. The inhibition of PLpro’s deubiquitination and deISGlyation activities were characterized using the Ub-AMC and ISG15-AMC substrates, respectively.14, 15 While EACC and KY-226 were inactive in inhibiting the deubiquitinase activity of PLpro (IC50 > 100 μM), tropifexor showed moderate activity with an IC50 of 18.85 μM (Figure 1F). Similarly, EACC and KY-226 were not active in inhibiting the deISGlyation activity of PLpro (IC50 > 80 μM), tropifexor showed does-dependent inhibition with an IC50 of 27.22 μM (Figure 1G). Tropifexor is a hydrophobic compound with a CLogP of 5.69. To rule out the possibility that the observed PLpro inhibition was due to non-specific binding, we repeated the FRET assay against SARS-CoV-2 PLpro in the presence of 0.01% BSA, and it was found that tropifexor retained potent inhibition with an IC50 of 10.36 μM (Figure 1H), suggesting the inhibition of PLpro by tropifexor is unlikely due to non-specific hydrophobic interactions. Tropifexor had similar IC50 values against SARS-CoV-2 PLpro with and without a 30 mins pre-incubation (Figure 1I), suggesting a reversible binding. The mechanism of inhibition of tropifexor was further studied in enzymatic kinetic experiment and GRL-0617 was included as a control. The Lineweaver–Burk plots showed that both compounds are competitive inhibitors of SARS-CoV-2 PLpro (Figures 1J–K).
Figure 1.
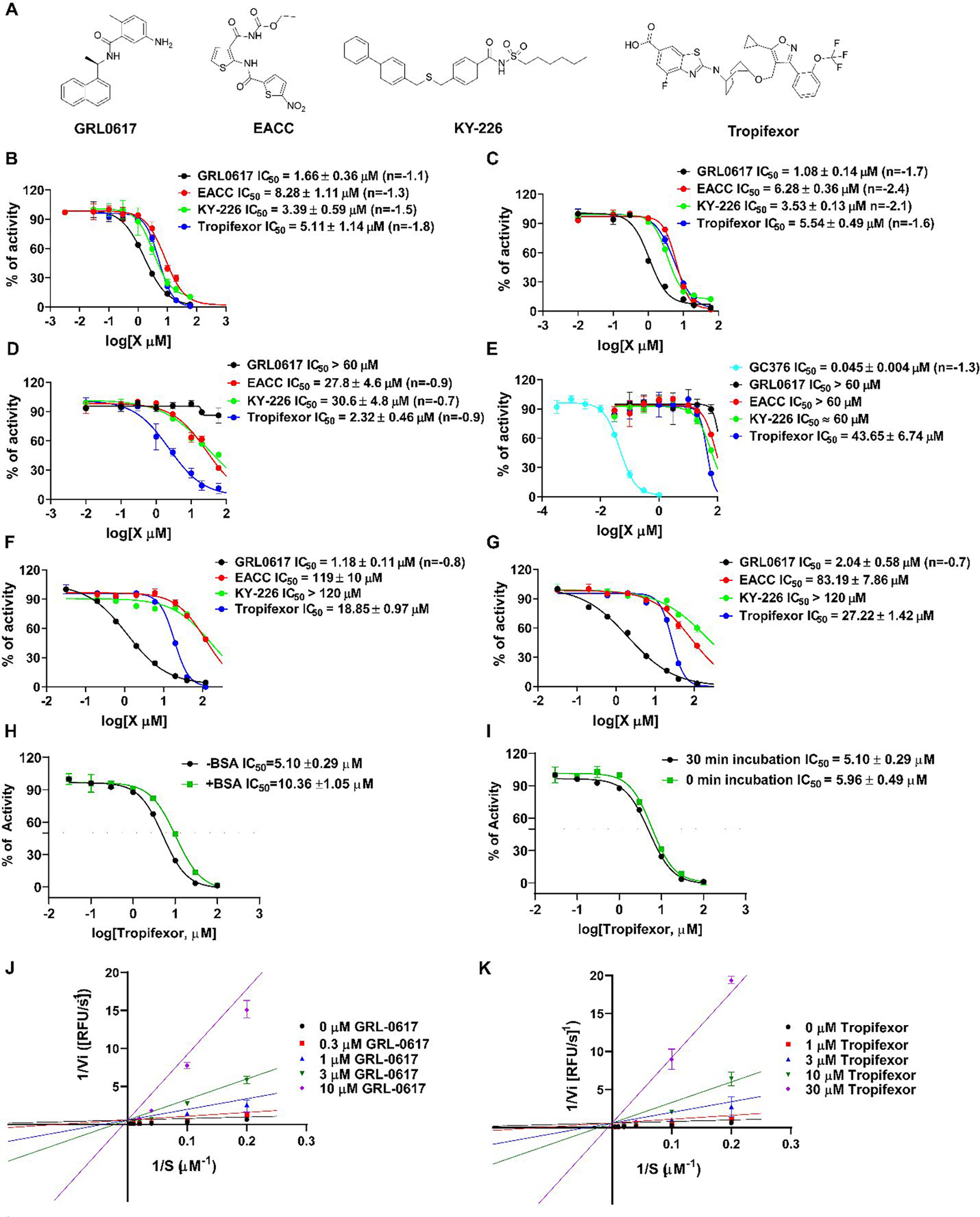
Characterization of SARS-CoV-2 PLpro inhibitors identified from the high-throughput screening. (A) Chemical structures of the positive control GRL0617 and the three hits EACC, KY-226, and tropifexor. (B) IC50 curves of the hits in inhibiting SARS CoV-2 PLpro with the FRET peptide substrate 1. (C) IC50 curves of the hits in inhibiting SARS CoV PLpro with the FRET peptide substrate 1. (D) IC50 curves of the hits in inhibiting MERS-CoV PLpro with the FRET peptide substrate 1. (E) IC50 curves of the hits in inhibiting SARS CoV-2 Mpro with the FRET peptide substrate 2. (F) IC50 curves of the hits in inhibiting SARS CoV-2 PLpro with the Ub-AMC substrate. (G) IC50 curves of the hits in inhibiting SARS CoV-2 PLpro with the ISG15-AMC substrate. Please refer to the methods and materials section for assay conditions. Values represent the average ± standard deviation of three replicates. (H) IC50 curves of tropifexor in inhibiting SARS CoV-2 PLpro with and without the addition of 0.01% BSA. (I) IC50 curves of tropifexor in inhibiting SARS CoV-2 PLpro with or without a 30 mins pre-incubation. (J) Lineweaver-Burk curves of GRL-0617 in inhibiting SARS-CoV-2 PLpro. (K) Lineweaver-Burk curves of tropifexor in inhibiting SARS-CoV-2 PLpro.
Overall, tropifexor appears to be the most promising hit with consistent inhibition against SARS-CoV-2, SARS-CoV, and MERS-CoV PLpros. In addition, tropifexor also inhibited the deubiquitination and deISGlyation activities of SARS-CoV-2 PLpro, albeit at lower potency.
Pharmacological characterization of the hits in the thermal shift assay and the cell-based FlipGFP PLpro assay
The mechanism of action of EACC, KY-226, and tropifexor in inhibiting SARS-CoV-2 PLpro was further characterized in the thermal shift assay and the cell-based FlipGFP PLpro assay.15, 22, 23, 26 Thermal shift assay measures the direct binding between the compound and the protein, therefore it can rule out hits that might bind to the FRET substrate in the enzymatic assay. Similar to the positive control GRL0617, all three hits displayed dose-dependent binding to PLpro as revealed by the enhanced melting temperatures with increasing drug concentration (Figure 2).
Figure 2.
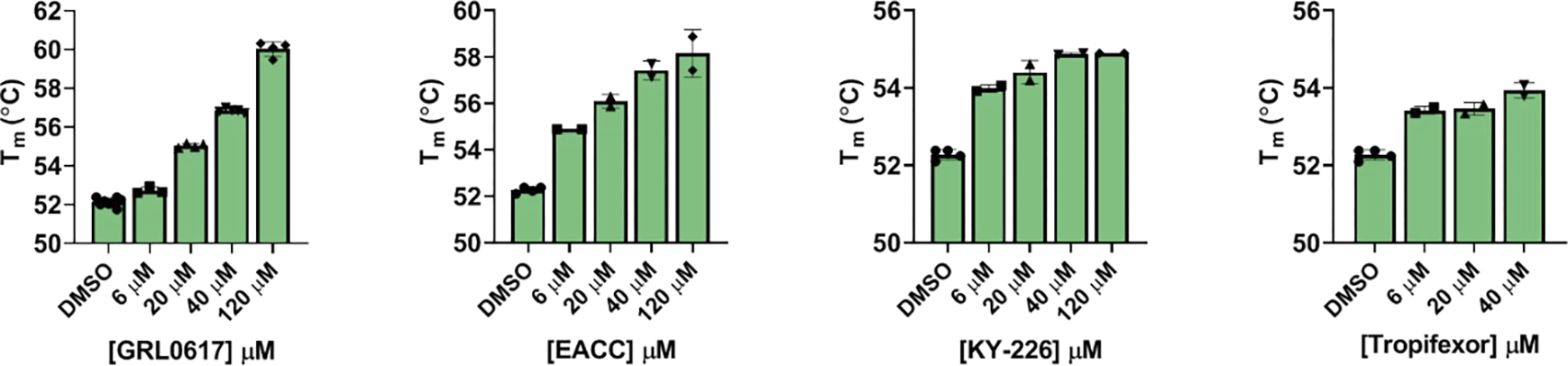
Thermal shift assay of SARS-CoV-2 PLpro protease against identified inhibitors. All inhibitors display dose-dependent melting temperature (Tm) shift. Values represent the average ± standard deviation of three replicates.
Next, we tested the three hits in the FlipGFP PLpro assay.15, 22, 23 The FlipGFP PLpro was recently developed by us as a surrogate assay to quantify the cellular activity of PLpro inhibitors in the biological safety level 2 facility, and we have shown that there is positive correlation between the FlipGFP IC50 values with the SARS-CoV-2 antiviral EC50 values.15 The FlipGFP assay is a virus free cell-based protease assay in which the 293T cells were transfected with PLpro and the GFP reporter. The GFP reporter consists of two fragments,27, 28 the β1–9 template, and the β10–11 strands that are constrained in the parallel inactive conformation through a PLpro substrate linker. Upon cleavage of the substrate linker, the β10 and β11 strands become parallel and can associate with the β1–9 template, leading to increased GFP signal. mCherry is included as an internal control to normalize transfection efficacy and compound cytotoxicity. In principle, the normalized GFP/mCherry ratio is proportional to the enzymatic activity of PLpro. The advantages of FlipGFP assay compared to the FRET assay is that it can rule out compounds that are cytotoxic, membrane impermeable, and having off-target effects that prevent cellular on-target engagement.22, 23
In the FlipGFP assay, the positive control GRL0617 showed dose-dependent inhibition with an IC50 of 14.67 μM, while the negative control GC376 was not active (IC50 > 60 μM) (Figure 3A, B). The results from EACC and KY-226 were not conclusive due to the cytotoxicity of the compounds. Tropifexor had an IC50 of 10.60 μM, but a low selectivity index (CC50 = 29.77 μM, SI = 2.8) (Figure 3A, B). Given the low selectivity, the results from the FlipGFP are not stringently conclusive. Nevertheless, tropifexor reduced the GFP/mCherry ratio by 50% at 10 μM, which was not cytotoxic. In summary, the FlipGFP assay results suggest tropifexor might have antiviral activity against SARS-CoV-2.
Figure 3.
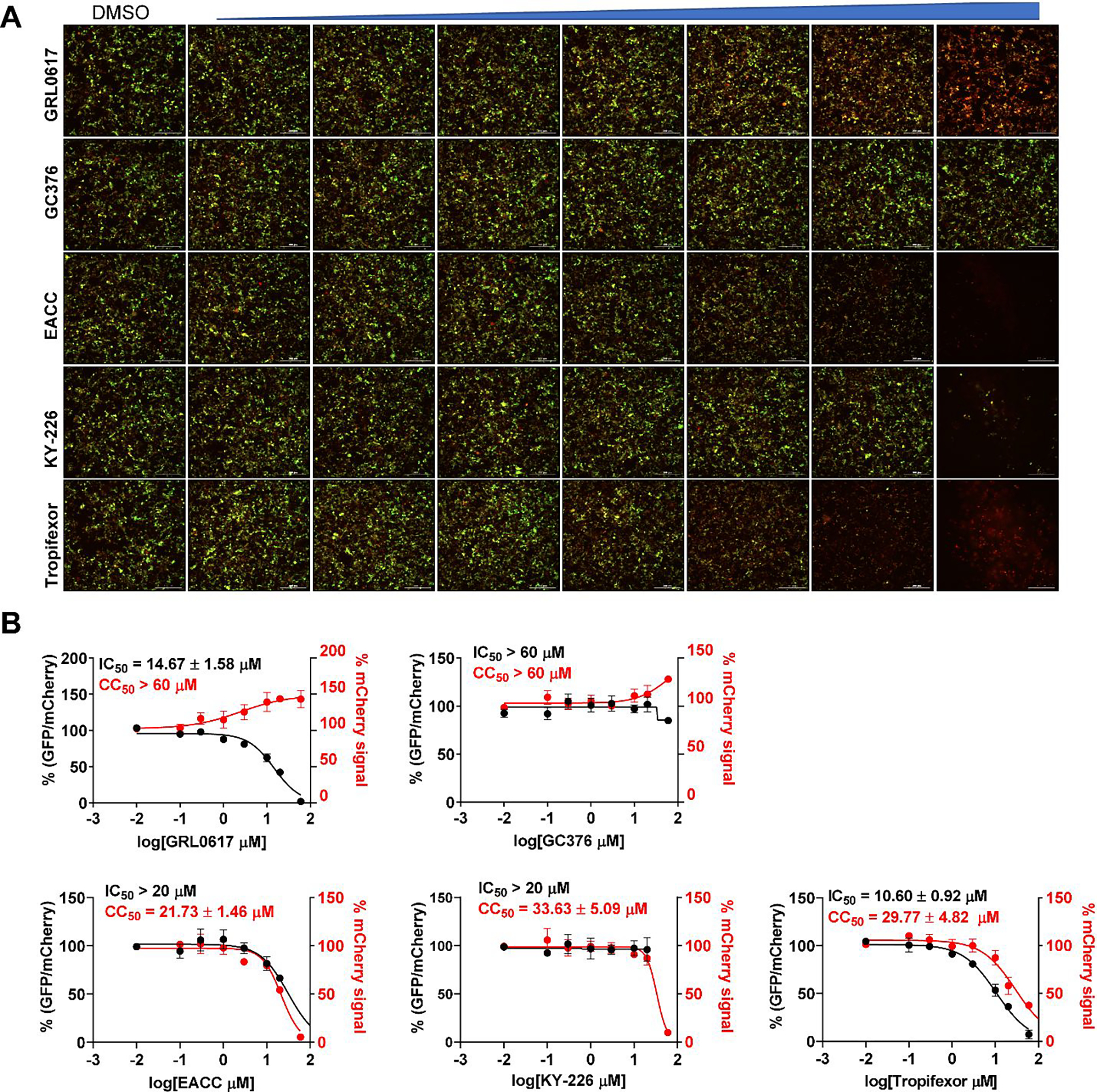
FlipGFP SARS CoV-2 PLpro assay to determine cellular protease inhibitory activity of identified inhibitors. (A) Representative images of FlipGFP-PLpro assay with increasing concentrations of GRL0617 (positive control), GC376 (negative control), EACC, KY-226, and tropifexor. GRL0617 showed does-dependent decrease of GFP signal with the increasing drug concentration, while almost no GFP signal change was observed with the increasing concentration of negative control compound GC376. (B) Dose−response curves of the GFP/mCherry ratio with increasing drug concentrations. mCherry signal alone was used to calculate transfection efficiency and compound cytotoxicity. All three hits displayed significant cytotoxicity at high drug concentrations. Values represent the average ± standard deviation of three replicates.
Antiviral activity of hits against SARS-CoV-2 in Calu-3 cells
The antiviral activity of EACC, KY-226, and tropifexor in inhibiting SARS-CoV-2 replication in Calu-3 cells was tested using the immunofluorescence assay (Figure 4). Calu-3 is TMPRSS2-positive and is a close mimetic of the human respiratory epithelial cells,29 enabling it a widely accepted cell line for SARS-CoV-2 studies.22, 30 The positive control GRL0617 had an EC50 of 31.4 μM (Figure 4A). EACC did not show antiviral activity at non-toxic drug concentration (EC50 > 35 μM, CC50 = 35.29 μM) (Figure 4B). Gratifyingly, both KY-226 and tropifexor had improved antiviral activity against SARS-CoV-2 with EC50 values of 25.0 (Figure 4C) and 4.03 μM (Figure 4D), respectively. While KY-226 had a low selectivity index (SI = 1.65), tropifexor had a moderate selectivity window (SI = 6.97) and the observed antiviral activity was likely not caused by the cytotoxicity of the compound.
Figure 4.
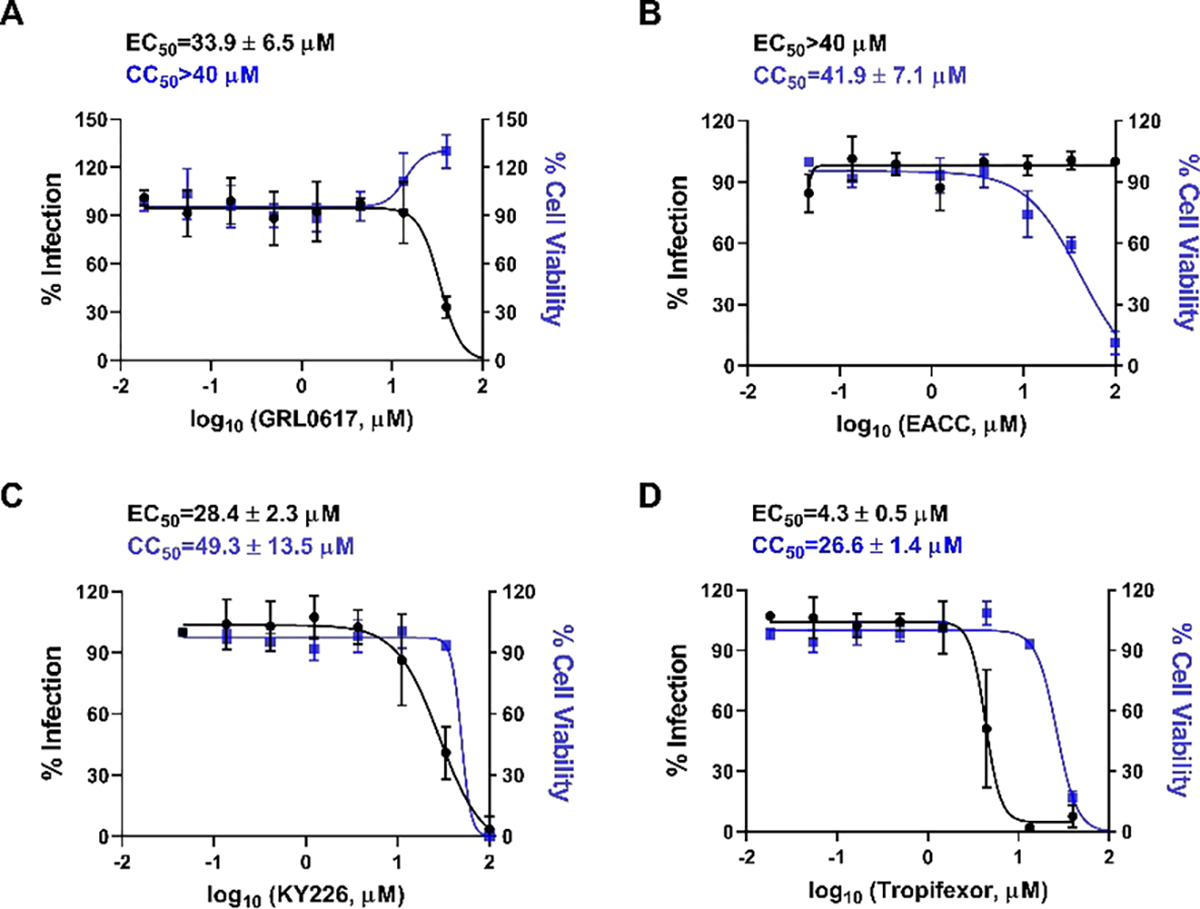
Antiviral activity of SARS-CoV-2 PLpro inhibitors GRL0617 (A), EACC(B), KY-226 (C), and tropifexor (D) against SARS-CoV-2 in Calu-3 cells. The results were quantified by immunofluorescence assay. Values represent the average ± standard deviation of three replicates.
Molecular docking of EACC, KY-226, and tropifexor in SARS-CoV-2 PLpro
To gain insights of the binding mode of the three hits, we performed molecular docking with Schrödinger Glide XP (extra precision) using the wild-type SARS-CoV-2 PLpro structure we recently solved (PDB: 7JRN).15 The binding sites were calculated by sitemap and the GRL0617 binding site was identified as the top-ranked binding site, therefore, it was selected for docking. GRL0617 was included as a positive control. The docking pose of GRL0617 was superimposable with binding mode in the X-ray crystal structure (Figure 5A). Tropifexor, EACC, and KY-226 all fit snuggly into the U-shape binding pocket that is covered by the BL2 loop where GRL0617 binds (Figures 5B–D). Among the three hits, tropifexor showed the most favorable binding pose with a Glide score of −4.085 (Figure 5B). The docking poses might provide a guidance for the following lead optimization.
Figure 5.
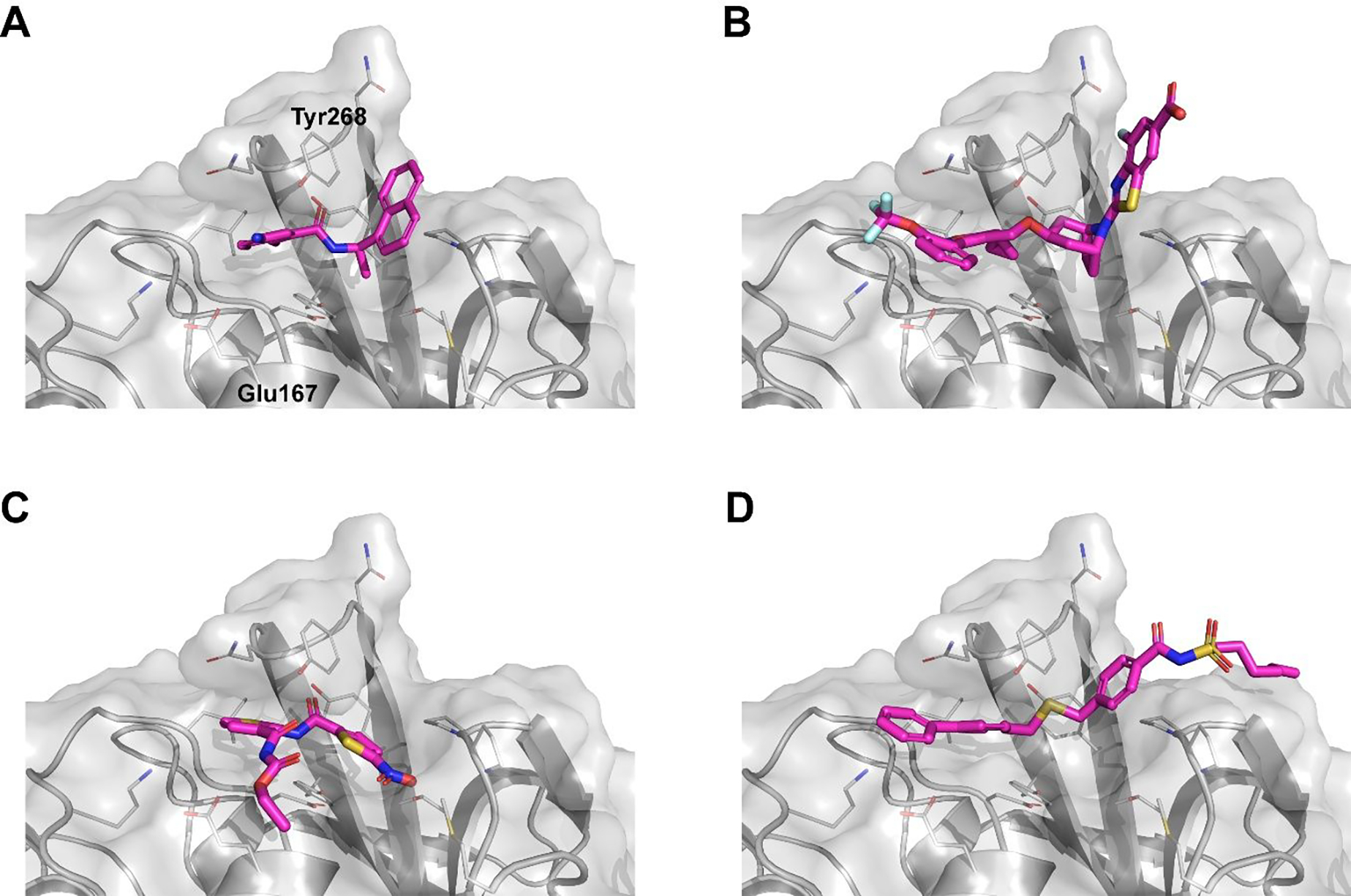
Molecular docking of SARS-CoV-2 PLpro inhibitors GRL0617 (A), tropifexor(B), EACC (C), and KY-226 (D) in PLpro (PDB: 7JRN). The Glide scores are −7.161 (GRL0617), −4.085 (Tropifexor), −3.794 (EACC), and −3.332 (KY-226).
CONCLUSION
Although PLpro is a validated antiviral drug target, the development of PLpro inhibitors falls behind Mpro and RdRp inhibitors. As of date, no PLpro inhibitors have been advanced to the in vivo animal model studies yet. The naphthalene compounds such as GRL0617 and its analogs are the only class of validated PLpro inhibitors with antiviral activity against SARS-CoV-2. However, the low metabolic stability of this series of compounds might prevent its further development.14, 31 In this study, we aimed to identify structurally novel PLpro inhibitors that can serve as starting points for further optimization. Through screening the MedChemExpress bioactive compound library, three hits EACC, KY-226, and tropifexor were identified as SARS-CoV-2 PLpro inhibitors with IC50 values in the single digit micromolar range. Among the three hits, tropifexor appears to be the most promising hit as it also showed potent inhibition against SARS-CoV PLpro (IC50 = 5.54 μM) and MERS-CoV PLpro (IC50 = 2.32 μM). In addition to the inhibition of PLpro mediated cleavage of viral polyprotein substrate, tropifexor also inhibited the deubiquitination and deISGlation activities of SARS-CoV-2 PLpro. Consistent with the enzymatic inhibition, tropifexor showed dose-dependent stabilization of SARS-CoV-2 PLpro in the thermal shift assay. Importantly, tropifexor displayed cellular PLpro inhibitory activity in the FlipGFP assay and the antiviral activity against SARS-CoV-2 in Calu-3 cells. Although the low selectivity index (SI = 6.2) of tropifexor in the antiviral assay prevents its direct repurposing as a SARS-CoV-2 antiviral, the discovery of tropifexor as a novel PLpro inhibitor provides an additional scaffold for further medicinal chemistry optimization. Follow up studies will focus on improving the target and cellular selectivity. Furthermore, tropifexor is a fairly large molecule (MW: 603.59), efforts will be made to reduce the size as well as the hydrophobicity of the compound to optimize ligand efficiency and drug-likeness properties.
MATERIALS AND METHODS
Protein Expression and Purification.
Detailed expression and purification procedures untagged SARS-CoV-2 PLpro and SARS-CoV-2 Mpro were described in our previous publications.15, 32 SARS-CoV papain-like protease gene (ORF 1ab 1541−1855) (accession # AEA10621.1) from strain SARS coronavirus MA15 with E. coli codon optimization in the pET28b-(+) vector was ordered from GenScript. Then the SARS-CoV PLpro gene (ORF 1ab 1541−1855) was subcloned from the pET28b-(+) to pE-SUMO vector according to the manufacturer’s protocol (LifeSensors Inc., Malvern, PA). The forward primer with the Bsa I site is GCGGTCTCAAGGTGAGGTGAAGACCATCAAAGTGTTCACCACC; the reverse primer with a Bsa I site is GCGGTCTCTCTAGATTATTTAATGGTGGTGGTATAGCTGGTTTCCTTGTAG. The expression and purification protocol of SARS-CoV PLpro is identical to SARS CoV-2 PLpro.15 MERS-CoV PLpro gene (ORF 1ab 1482–1803) (accession # KY581684) from strain MERS coronavirus Hu/UAE_002_2013 with E. coli codon optimization in the pET28b-(+) vector was ordered from GenScript. Then MERS-CoV PLpro gene (ORF 1ab 1482–1803) was subclone into pE-SUMO vector with the pair primers: GCGGTCTCAAGGTCAGCTGACCATCGAGGTGCTGGTTACCGTGG and GCGGTCTCTCTAGATTAGTTGCAATCGCTGCTATATTTTTGACCCGGGAAC. The expression and purification protocol of MERS-CoV papain-like protease is identical to SARS CoV-2 PLpro.15
FRET substrate synthesis:
The SARS-CoV-2 PLpro FRET substrate 1 is Dabcyl-FTLRGG/APTKV(Edans); this substrate was also used as SARS-CoV PLpro and MERS-CoV PLpro substrates. SARS-CoV-2 Mpro FRET substrate 2 is Dabcyl-KTSAVLQ/SGFRKME- (Edans). These FRET substrates were synthesized by solidphase synthesis through iterative cycles of coupling and deprotection using the previously optimized procedure.33 Ub-AMC and ISG15-AMC were purchased from BostonBiochem (catalog no. U-550–050 and UL-553–050, respectively).
Enzymatic Assays.
The high-throughput screening was carried out in 384-well format as described previously.15 The bioactive compound library consisting of 9,791 compounds was purchased from MedChemExpress (catalog no. HY-L001). The enzymatic reactions for SARS-CoV-2, SARS-CoV, MERS-CoV PLpros were carried out in reaction buffer consisting of 50 mM HEPES pH 7.5, 5 mM DTT and 0.01% Triton X-100. For the IC50 measurement with FRET peptide-Edans substrate, the reaction was carried out in 96-well format with 100 μl reaction volume. SARS-CoV-2 PLpro (200 nM) SARS-CoV PLpro (200 nM) or MERS-CoV PLpro (2 μM) was pre-incubated with various concentrations of testing compounds at 30 °C for 30 min before the addition of FRET-peptide substrate to initiate the reaction. The reaction was monitored in a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30 °C for 1 h. The initial enzymatic reaction velocity was calculated from the initial 10 min enzymatic reaction via a linear regression function and was plotted against the substrate concentrations in Prism 8 with a four-parameter dose-response function. For the IC50 measurements with Ub-AMC or ISG15-AMC substrate, the reaction was carried out in 384-well format in 50 μl reaction volume. In the Ub-AMC cleavage assay, the final SARS-CoV-2 PLpro concentration is 50 nM, and substrate Ub-AMC concentration is 2.5 μM. IN the ISG15-AMC assay, the final SARS-CoV-2 PLpro concentration is 2 nM, and substrate ISG15-AMC concentration is 0.5 μM. The SARS-CoV-2 Mpro enzymatic assays were carried out in the reaction buffer containing 20 mM HEPES pH 6.5, 120 mM NaCl, 0.4 mM EDTA, 20% glycerol, and 4 mM DTT as described previously.32, 34 To rule out that the inhibition of Tropifexor on PLPro is due to aggregation, 200 nM PLPro was incubated with serial concentrations of Tropifexor (0, 0.1, 0.3, 1, 3, 10, 30, 100 μM) in the reaction buffer in the presence or absence of 0.01% BSA (0.1 mg/ml) at 30 °C for 30 min. The reaction was initiated by adding 10 μM FRET substrate and monitored every 90 seconds for 1 h at 30 °C. The initial velocity was determined in the first 15 min by linear regression. The IC50 values were determined by fitting the curves with nonlinear regression using log (concentration of inhibitor) vs response with variable slopes in Prism 8.
To determine whether preincubation affect the IC50 value of tropifexor, 200 nM PLPro was mixed with serial concentrations of Tropifexor (0, 0.1, 0.3, 1, 3, 10, 30, 100 μM) in the reaction buffer with or without preincubation at 30 °C for 30 min, and the reaction was initiated by adding 10 μM FRET substrate. IC50 values were determined as previously described.
To determine the binding mode of Tropifexor, KM and Vmax were determined at different concentrations of GRL-0617 (0, 0.3, 1, 3, 10 μM) or Tropifexor (0, 1, 3, 10, 30 μM). 200 nM SARS-CoV-2 PLPro was mixed with the indicated concentrations of GRL-0617 or Tropifexor in the reaction buffer and incubated at 30 °C for 30 min. The reaction was initiated by adding different concentrations of FRET-peptide (5, 10, 25, 50, 100, 200 μM). Michaelis-Menten and Lineweaver-Burk curves were plotted in Prism 8.
Differential Scanning Fluorimetry (DSF).
The thermal shift assay (TSA) was carried out using a Thermo Fisher QuantStudio 5 Real-Time PCR system as described previously.15, 32 Briefly, 4 μM SARS-CoV-2 PLpro protein in PLpro reaction buffer (50 mM HEPES pH 7.5, 5 mM DTT and 0.01% Triton X-100) was incubated with various concentrations of testing compounds at 30 °C for 30 min. 1× SYPRO orange dye was added, and the fluorescence of each well was monitored under a temperature gradient range from 20 to 90 °C with 0.05 °C/s incremental step. The melting temperature (Tm) was calculated as the mid-log of the transition phase from the native to the denatured protein using a Boltzmann model in Protein Thermal Shift Software v1.3.
Cell-Based FlipGFP PLpro Assay.
Plasmid pcDNA3-PLpro-flipGFP-T2A-mCherry was constructed from pcDNA3-TEV-flipGFP-T2A-mCherry.15 SARS-CoV-2 PLpro expression plasmid pcDNA3.1-SARS2 PLpro was ordered from Genscript (Piscataway NJ) with codon optimization. For transfection, 293T cells were seeded into 96-well Greiner plate (catalog no. 655090) to overnight with 70−90% confluency. 50 ng of pcDNA3-PLPro-flipGFP-T2A-mCherry plasmid and 50 ng of protease expression plasmid pcDNA3.1-PLpro were added to each well in the presence of transfection reagent TransIT-293 (Mirus) according to manufacturer’s protocol. Three hours after transfection, 1 μL of testing compound was added to each well at 100-fold dilution. Images were acquired 2 days after transfection with a Cytation 5 imaging reader (Biotek) GFP and mCherry channels and were analyzed with Gen5 3.10 software (Biotek). SARS-CoV-2 PLpro protease activity was calculated by the ratio of GFP signal over the mCherry signal. The FlipGFP PLpro assay IC50 value was determined by plotting the GFP/ mCherry signal over the compound concentration with a four-parameter dose–response function in Prism 8. The mCherry signal alone was utilized to evaluate the transfection efficiency and compound cytotoxicity.
Antiviral Assay in Calu-3 Cells.
Calu-3 cells (ATCC, HTB-55) grown in Minimal Eagles Medium supplemented with 1% nonessential amino acids, 1% penicillin/streptomycin, and 10% FBS are plated in 384 well plates. The next day, 50 nL of drug suspended in DMSO is added as an 8-pt dose response with 3-fold dilutions between test concentrations in triplicate, starting at 40 μM final concentration. The negative control (DMSO, n = 32) and positive control (10 μM Remdesivir, n = 32) are included on each assay plate. Calu3 cells are pretreated with controls and test drugs (in triplicate) for 2 h prior to infection. In BSL3 containment, SARS-CoV-2 (isolate USA-WA1/2020) diluted in serum free growth medium is added to plates to achieve an MOI = 0.5. Cells are incubated continuously with drugs and SARS-CoV-2 for 48 h. Cells are fixed and then immunstained with anti-dsRNA (J2), and nuclei are counterstained with Hoechst 33342 for automated microscopy. Automated image analysis quantifies the number of cells per well (toxicity) and the percentage of infected cells (dsRNA+ cells/cell number) per well. SARS-CoV-2 infection at each drug concentration was normalized to aggregated DMSO plate control wells and expressed as percentage-ofcontrol (POC = % Infection sample/Avg % Infection DMSO cont). A nonlinear regression curve fit analysis (GraphPad Prism 8) of POC infection and cell viability versus the log10 transformed concentration values to calculate EC50 values for infection and CC50 values for cell viability. Selectivity index (SI) was calculated as a ratio of drug’s CC50 and EC50 values (SI = CC50/IC50).
Molecular modeling of the binding of EACC, KY-226, and tropifexor to SARS-CoV-2 PLpro.
Docking was performed using Schrödinger Glide extra precision (XP). The SARS-CoV-2 PLpro structure was downloaded from PDB code 7JRN. The binding sites were calculated by the sitemap and the GRL0617 binding site is the highest scored binding site, and therefore it was chosen for docking. The docking grid was centered around GRL0617 with the coordinates of X = 9.88, Y = −11.74, and Z = 32.55. GRL0617 was added as a positive control for the docking. The final docking poses were generated in PyMOL.
ACKNOWLEDGMENTS
This research was partially supported by the National Institute of Allergy and Infectious Diseases of Health (NIH) (grants AI147325, AI157046, and AI158775) and the Arizona Biomedical Research Commission Centre Young Investigator grant (ADHS18–198859) to J. W. The SARS-CoV-2 antiviral assay in Calu-3 cells was conducted by Drs. David Schultz and Sara Cherry at the University of Pennsylvania (USA) through the NIAID preclinical service under a non-clinical evaluation agreement.
REFERENCES
- (1).Tregoning JS; Flight KE; Higham SL; Wang Z; Pierce BF, Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021, 21, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Harvey WT; Carabelli AM; Jackson B; Gupta RK; Thomson EC; Harrison EM; Ludden C; Reeve R; Rambaut A; Peacock SJ; Robertson DL; Consortium C-GU, SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Beigel JH; Tomashek KM; Dodd LE; Mehta AK; Zingman BS; Kalil AC; Hohmann E; Chu HY; Luetkemeyer A; Kline S; Lopez de Castilla D; Finberg RW; Dierberg K; Tapson V; Hsieh L; Patterson TF; Paredes R; Sweeney DA; Short WR; Touloumi G; Lye DC; Ohmagari N; Oh M.; Ruiz-Palacios GM; Benfield T; Fätkenheuer G; Kortepeter MG; Atmar RL; Creech CB; Lundgren J; Babiker AG; Pett S; Neaton JD; Burgess TH; Bonnett T; Green M; Makowski M; Osinusi A; Nayak S; Lane HC, Remdesivir for the treatment of covid-19 — final report. N. Engl. J. Med. 2020, 383, 1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wahl A; Gralinski LE; Johnson CE; Yao W; Kovarova M; Dinnon KH; Liu H; Madden VJ; Krzystek HM; De C; White KK; Gully K; Schäfer A; Zaman T; Leist SR; Grant PO; Bluemling GR; Kolykhalov AA; Natchus MG; Askin FB; Painter G; Browne EP; Jones CD; Pickles RJ; Baric RS; Garcia JV, SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 2021, 591, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cox RM; Wolf JD; Plemper RK, Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sheahan TP; Sims AC; Zhou S; Graham RL; Pruijssers AJ; Agostini ML; Leist SR; Schafer A; Dinnon KH 3rd; Stevens LJ; Chappell JD; Lu X; Hughes TM; George AS; Hill CS; Montgomery SA; Brown AJ; Bluemling GR; Natchus MG; Saindane M; Kolykhalov AA; Painter G; Harcourt J; Tamin A; Thornburg NJ; Swanstrom R; Denison MR; Baric RS, An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y, An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [DOI] [PubMed] [Google Scholar]
- (8).Meyer B; Chiaravalli J; Gellenoncourt S; Brownridge P; Bryne DP; Daly LA; Grauslys A; Walter M; Agou F; Chakrabarti LA; Craik CS; Eyers CE; Eyers PA; Gambin Y; Jones AR; Sierecki E; Verdin E; Vignuzzi M; Emmott E, Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shin D; Mukherjee R; Grewe D; Bojkova D; Baek K; Bhattacharya A; Schulz L; Widera M; Mehdipour AR; Tascher G; Geurink PP; Wilhelm A; van der Heden van Noort GJ; Ovaa H; Müller S; Knobeloch KP; Rajalingam K; Schulman BA; Cinatl J; Hummer G; Ciesek S; Dikic I, Papain-like protease regulates sars-cov-2 viral spread and innate immunity. Nature 2020, 587, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Baez-Santos YM; St John SE; Mesecar AD, The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Klemm T; Ebert G; Calleja DJ; Allison CC; Richardson LW; Bernardini JP; Lu BG; Kuchel NW; Grohmann C; Shibata Y; Gan ZY; Cooney JP; Doerflinger M; Au AE; Blackmore TR; van der Heden van Noort GJ; Geurink PP; Ovaa H; Newman J; A. Riboldi-Tunnicliffe; Czabotar PE; Mitchell JP; Feltham R; Lechtenberg BC; Lowes KN; Dewson G; Pellegrini M; Lessene G; Komander D, Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J 2020, 39, e106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rut W; Lv Z; Zmudzinski M; Patchett S; Nayak D; Snipas SJ; El Oualid F; Huang TT; Bekes M; Drag M; Olsen SK, Activity profiling and crystal structures of inhibitor-bound sars-cov-2 papain-like protease: A framework for anti-covid-19 drug design. Sci. Adv. 2020, 6, eabd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shan H; Liu J; Shen J; Dai J; Xu G; Lu K; Han C; Wang Y; Xu X; Tong Y; Xiang H; Ai Z; Zhuang G; Hu J; Zhang Z; Li Y; Pan L; Tan L, Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021, 28, 855–865.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shen Z; Ratia K; Cooper L; Kong D; Lee H; Kwon Y; Li Y; Alqarni S; Huang F; Dubrovskyi O; Rong L; Thatcher GRJ; Xiong R, Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ma C; Sacco MD; Xia Z; Lambrinidis G; Townsend JA; Hu Y; Meng X; Szeto T; Ba M; Zhang X; Gongora M; Zhang F; Marty MT; Xiang Y; Kolocouris A; Chen Y; Wang J, Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ratia K; Kilianski A; Baez-Santos YM; Baker SC; Mesecar A, Structural basis for the ubiquitin-linkage specificity and deisgylating activity of sars-cov papain-like protease. PLoS Pathog. 2014, 10, e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fu Z; Huang B; Tang J; Liu S; Liu M; Ye Y; Liu Z; Xiong Y; Zhu W; Cao D; Li J; Niu X; Zhou H; Zhao YJ; Zhang G; Huang H, The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Osipiuk J; Azizi SA; Dvorkin S; Endres M; Jedrzejczak R; Jones KA; Kang S; Kathayat RS; Kim Y; Lisnyak VG; Maki SL; Nicolaescu V; Taylor CA; Tesar C; Zhang YA; Zhou Z; Randall G; Michalska K; Snyder SA; Dickinson BC; Joachimiak A, Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vats S; Manjithaya R, A reversible autophagy inhibitor blocks autophagosome-lysosome fusion by preventing Stx17 loading onto autophagosomes. Mol. Biol. Cell 2019, 30, 2283–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ito Y; Fukui M; Kanda M; Morishita K; Shoji Y; Kitao T; Hinoi E; Shirahase H, Therapeutic effects of the allosteric protein tyrosine phosphatase 1B inhibitor KY-226 on experimental diabetes and obesity via enhancements in insulin and leptin signaling in mice. J. Pharmacol. Sci. 2018, 137, 38–46. [DOI] [PubMed] [Google Scholar]
- (21).Tully DC; Rucker PV; Chianelli D; Williams J; Vidal A; Alper PB; Mutnick D; Bursulaya B; Schmeits J; Wu X; Bao D; Zoll J; Kim Y; Groessl T; McNamara P; Seidel HM; Molteni V; Liu B; Phimister A; Joseph SB; Laffitte B, Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 60, 9960–9973. [DOI] [PubMed] [Google Scholar]
- (22).Ma C; Tan H; Choza J; Wang Y; Wang J, Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and Protease-Glo luciferase assays. Acta Pharm. Sin. B 2021, DOI: 10.1016/j.apsb.2021.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ma C; Wang J, Validation and invalidation of SARS-CoV-2 papain-like protease inhibitors. ACS Pharmacol. Transl. Sci. 2022, 5, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ma C; Hu Y; Townsend JA; Lagarias PI; Marty MT; Kolocouris A; Wang J, Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ma C; Wang J, Dipyridamole, chloroquine, montelukast sodium, candesartan, oxytetracycline, and atazanavir are not SARS-CoV-2 main protease inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2024420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xia Z; Sacco M; Hu Y; Ma C; Meng X; Zhang F; Szeto T; Xiang Y; Chen Y; Wang J, Rational Design of Hybrid SARS-CoV-2 Main Protease Inhibitors Guided by the Superimposed Cocrystal Structures with the Peptidomimetic Inhibitors GC-376, Telaprevir, and Boceprevir. ACS Pharmacol. Transl. Sci. 2021, 4, 1408–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li X; Lidsky PV; Xiao Y; Wu C-T; Garcia-Knight M; Yang J; Nakayama T; Nayak JV; Jackson PK; Andino R; Shu X, Ethacridine inhibits SARS-CoV-2 by inactivating viral particles. PLOS Pathog. 2021, 17, e1009898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Froggatt HM; Heaton BE; Heaton NS, Development of a Fluorescence-Based, High-Throughput SARS-CoV-2 3CL(pro) Reporter Assay. J. Virol. 2020, 94, e01265–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hoffmann M; Kleine-Weber H; Schroeder S; Kruger N; Herrler T; Erichsen S; Schiergens TS; Herrler G; Wu NH; Nitsche A; Muller MA; Drosten C; Pohlmann S, SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kitamura N; Sacco MD; Ma C; Hu Y; Townsend JA; Meng X; Zhang F; Zhang X; Ba M; Szeto T; Kukuljac A; Marty MT; Schultz D; Cherry S; Xiang Y; Chen Y; Wang J, Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Báez-Santos YM; Barraza SJ; Wilson MW; Agius MP; Mielech AM; Davis NM; Baker SC; Larsen SD; Mesecar AD, X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014, 57, 2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J, Boceprevir GC -376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cady SD; Wang J; Wu Y; DeGrado WF; Hong M, Specific Binding of Adamantane Drugs and Direction of Their Polar Amines in the Pore of the Influenza M2 Transmembrane Domain in Lipid Bilayers and Dodecylphosphocholine Micelles Determined by NMR Spectroscopy. J. Am. Chem. Soc. 2011, 133, 4274–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sacco MD; Ma C; Lagarias P; Gao A; Townsend JA; Meng X; Dube P; Zhang X; Hu Y; Kitamura N; Hurst B; Tarbet B; Marty MT; Kolocouris A; Xiang Y; Chen Y; Wang J, Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci Adv 2020, 6, eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]