

ABSTRACT
Cephalosporins are commonly prescribed antibiotics that impair cross-linking of the bacterial cell wall. The Gram-positive opportunistic pathogen, Enterococcus faecalis, is intrinsically resistant to these antibiotics and proliferates substantially during cephalosporin therapy. As a result, the usage of cephalosporins has the potential to lead to life-threatening enterococcal infections. Yet, the molecular mechanisms that drive cephalosporin resistance (CR) are incompletely understood. Previously, we demonstrated that MurAA, an enzyme that catalyzes the first committed step in peptidoglycan (PG) synthesis, is required for CR. However, the mechanism by which MurAA contributes to CR remained unknown. Here, we tested the hypothesis that MurAA drives CR by controlling metabolic flux through the PG synthesis pathway. To do so, we developed and exploited an inducible gene expression system for E. faecalis based on an interspecies chimeric receptor that responds to exogenous nitrate for control of expression from a NisR-regulated promoter (PnisA). We used this tool to demonstrate synthetic lethality of MurAA with its homolog MurAB, to titrate expression of MurAA, and to conditionally deplete multiple PG synthesis enzymes downstream of MurAA that are predicted to be essential. These genetic manipulations, in addition to pharmacological inhibition of the PG synthesis pathway, all led to reductions in PG synthesis that correlated with reductions in CR. Our findings are consistent with a model in which control of metabolic flux through the PG synthesis pathway is a major driver of CR.
IMPORTANCE Enterococci are dangerous opportunistic pathogens with the potential to cause life-threatening infections due in part to their intrinsic resistance to cephalosporin antibiotics. Elucidating the molecular mechanisms that provide this resistance is critical for the development of strategies to both prevent and treat enterococcal infections. Here, we report that the cell wall synthesis enzyme, MurAA, drives cephalosporin resistance at least in part by controlling metabolic flux through the peptidoglycan synthesis pathway. To demonstrate this, we designed and validated an inducible gene expression system based on a chimeric receptor that is functional in multiple lineages of E. faecalis. In doing so, we provided a new tool for inducible gene expression with broad applications beyond our studies, including studies of essential genes.
KEYWORDS: Enterococcus, cephalosporin resistance, peptidoglycan synthesis
INTRODUCTION
Enterococci are Gram-positive, commensal bacteria of the gastrointestinal tract of healthy individuals. However, they possess both intrinsic resistance to many commonly prescribed antibiotics and the ability to rapidly acquire resistance to additional antimicrobial agents (1, 2). These features make enterococci dangerous opportunistic pathogens. Enterococcal species are the third leading cause of infectious endocarditis (IE), a life-threatening disease. Most cases of enterococcal IE are caused by a single species, Enterococcus faecalis, while a smaller proportion is caused by Enterococcus faecium (3, 4).
One of the major drivers of enterococcal infection is treatment with cephalosporin antibiotics (5, 6). Cephalosporins, which belong to the β-lactam family, impair peptidoglycan (PG) cross-linking in susceptible organisms by inhibiting penicillin-binding proteins (PBPs) (7). Disruption of cross-linking destabilizes the cell wall, causing cell lysis and death. However, enterococci are intrinsically resistant to cephalosporins (1, 2). Therefore, therapy with this class of antibiotics provides an opportunity for enterococci to proliferate, disseminate from the gut, and cause infection.
Enterococcal cephalosporin resistance (CR) is provided, in part, by two class B PBPs (PbpA/2b and Pbp5/4) that exhibit low reactivity toward cephalosporins (8, 9). However, these low-reactivity PBPs are not sufficient for cephalosporin resistance. Several additional factors are required for CR, including some involved in sensing of cell wall stress (e.g., CroS/R) (10–12) and in the synthesis of PG (e.g., MurAA) (13). The CroS sensor-histidine kinase is activated by antibiotic-mediated cell wall stress to phosphorylate its cognate response regulator, CroR, which regulates genes to help combat the stress (10–12).
MurAA is a UDP-N-acetylglucosamine 1-carboxyvinyl transferase, which catalyzes the first committed step in the PG biosynthesis pathway, the conversion of UDP-N-acetylglucosamine (UDP-GlcNAc) to enolpyruvyl UDP-N-acetylglucosamine (Fig. 1). Like most low-GC Gram-positive bacteria (14), E. faecalis encodes a homolog of MurAA called MurAB which exhibits ∼45% identity with MurAA. We previously showed that either murAA or murAB can be individually deleted from the E. faecalis genome (13), consistent with the hypothesis that both can provide UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity to promote PG synthesis. Because MurAA and MurAB represent the only UDP-N-acetylglucosamine 1-carboxyvinyl transferase homologs in the E. faecalis genome, we predicted murAA and murAB are a synthetically lethal pair (i.e., both genes cannot be simultaneously deleted), although this has previously not been experimentally tested.
FIG 1.

Schematic of the E. faecalis PG synthesis pathway.
Our previous work demonstrated that murAA, but not murAB, is required for CR (13). Genetic deletion of murAA led to a substantial loss of CR, while overexpression of murAA from a plasmid with a constitutively active promoter led to a cephalosporin hyper-resistance phenotype, suggesting that the amount of MurAA is a primary driver of CR. Moreover, the catalytic activity of MurAA is essential to promote CR, because a catalytically inactive MurAA variant was unable to do so. However, in those studies, we were unable to titrate the amount of MurAA in cells to determine if there is a dose-dependent relationship between MurAA and CR.
Collectively, these findings led to the hypothesis that MurAA influences CR, at least in part, by regulating the flux of metabolites through the PG synthesis pathway. As one test of this, we sought to control the abundance of MurAA protein in E. faecalis cells using an inducible promoter to regulate murAA gene expression. The two best-characterized tools for inducible gene expression in E. faecalis are the nisin-regulated NisK/R two-component signaling system (15) and the cCF10 peptide pheromone-inducible promoter (16). Although widely used, both tools possess some inherent limitations that rendered them incompatible with our experiments. For example, the inducer of NisK/R (nisin) is itself an antimicrobial peptide that is known to inhibit peptidoglycan synthesis by targeting the lipid II intermediate in the PG synthesis pathway (17). Thus, the use of nisin to induce gene expression has significant potential to confound any studies designed to examine PG synthesis. Similarly, the cCF10 peptide pheromone is a highly hydrophobic peptide (i) whose activity is sensitive to the composition of the growth medium, exhibiting reduced activity in medium containing competing peptides such as Mueller-Hinton broth, commonly used to measure antimicrobial sensitivity, (ii) that, from a practical perspective, would be difficult to include as an inducer in agar plates given its hydrophobicity, and (iii) that we found to itself influence enterococcal CR (through unknown mechanisms) at high concentrations needed to achieve maximal induction (unpublished data).
To overcome these limitations, we developed an alternative tool for inducible gene expression in E. faecalis. To do so, we exploited the modular nature of two-component signaling system kinases to engineer a chimeric receptor that controls gene expression in E. faecalis in response to the nitrate concentration in the environment, enabling the study of MurAA and other essential PG synthesis proteins at various levels of expression. With this new tool, we analyzed CR in strains exhibiting various abundances of multiple PG synthesis proteins, including some that are essential for viability. Collectively, our data are consistent with a model in which MurAA drives CR in E. faecalis at least in part by modulating the flux of metabolites through the PG synthesis pathway.
RESULTS
Chimeric receptor for inducible gene expression in E. faecalis.
We sought to develop a system for inducible gene expression in E. faecalis that relied on heterologous components not normally found in enterococci, to reduce the potential for unintended regulation of other enterococcal genes. Thus, we engineered a chimeric receptor that exploited the modular nature of sensor kinases that comprise two-component signaling systems. Prior work demonstrated that the extracellular nitrate-responsive sensing module of the NarQ sensor kinase from Escherichia coli could be fused to intracellular signaling modules from other transmembrane signaling proteins to create a functional nitrate-responsive chimeric receptor (18). In addition, the lactococcal NisK/R two-component system comprised of the NisK sensor kinase and NisR DNA-binding response regulator has been widely used for inducible gene expression in E. faecalis, most commonly by coexpression of NisK/R from a plasmid that also contains a NisR-regulated promoter, PnisA (such as pMSP3535; (15)). We, therefore, engineered an interspecies chimeric receptor by replacing the extracellular and transmembrane domains of the nisin-responsive sensor kinase, NisK, with those of E. coli NarQ (Fig. 2), creating plasmid pJLL286 (Fig. S1). We hypothesized that the resulting chimeric NarQ169-NisK168 kinase would no longer be responsive to nisin, but instead would respond to exogenous nitrate in the environment to drive the phosphorylation of the NisR response regulator, ultimately leading to enhanced transcription from the PnisA promoter via the canonical two-component signaling mechanism of the NisK/R system that remained otherwise intact.
FIG 2.

(A) Schematic of the NarQ169-NisK168 receptor and signaling pathway. (B) The fusion point for chimeric receptor construction was at the C terminus of the predicted second transmembrane segment. The chimeric receptor NarQ169-NisK168 contains residues 2 to 169 of E. coli NarQ and 168 to 447 of L. lactis NisK (also contains residues 1 to 4 of L. lactis NisK, included to avoid perturbing NisR which overlaps slightly with nisK). The predicted transmembrane sequences are highlighted in bold.
To test this, we cloned E. faecalis murAA downstream of PnisA in pJLL286 and introduced the resulting plasmid into the E. faecalis ΔmurAA mutant. Immunoblotting for MurAA revealed that in the absence of exogenous nitrate very little MurAA was produced (Fig. 3). The small amount of MurAA that could be detected suggested low-level leakiness of the PnisA promoter. Increasing concentrations of nitrate resulted in an increased abundance of MurAA in a dose-dependent manner (Fig. 3A). As expected, exposure to nisin at a concentration known from previous work to result in robust expression via NisK/R (15) did not lead to enhanced production of MurAA (Fig. 3B), indicating that the ability of NisK to sense nisin has been eliminated in the NarQ169-NisK168 chimera, and replaced by the ability to sense nitrate. We also found that induction of MurAA could restore ceftriaxone resistance to the ΔmurAA mutant on agar plates (Fig. S2), demonstrating that the nitrate-inducible promoter would function on solid media as well as in liquid culture.
FIG 3.
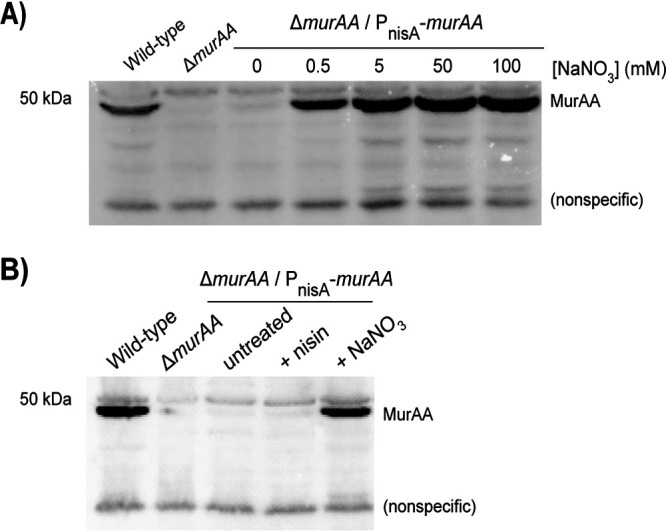
NarQ169-NisK168 mediated nitrate-responsive expression in E. faecalis. Immunoblot analysis for MurAA of total protein lysates from exponentially growing E. faecalis cells. (A) E. faecalis grown with various concentrations of NaNO3 as indicated. (B) E. faecalis cells were untreated or grown with 25 ng/mL nisin or 0.5 mM NaNO3. The anti-MurAA antisera cross-reacts with a nonspecific lower band that serves as a loading control. Results are representative of a minimum of 2 independent biological replicates. Strains were wild-type, OG1/pJLL286; ΔmurAA, JL626/pJLL286; and ΔmurAA/PnisA-murAA, JL626/pJLL288.
To test the NarQ169-NisK168 chimera more broadly, we cloned murC (another enzyme in the PG synthesis pathway; Fig. 1) under the control of PnisA. An HA tag was fused to murC to allow detection of MurC-HA via immunoblot because no antisera recognizing MurC was available. Immunoblot analyses revealed that, similar to MurAA, a small amount of MurC-HA was detectable in the absence of nitrate. The addition of nitrate led to substantial increases in MurC-HA abundance in a dose-dependent manner (Fig. S3). To ensure that the functionality of the NarQ169-NisK168 chimera was not restricted to only the OG1 lineage of E. faecalis, we introduced the MurC-HA encoding plasmid into 4 additional evolutionarily divergent lineages of E. faecalis. In each case, expression of MurC-HA was nearly off in the absence of nitrate and was induced in the presence of nitrate (Fig. S4). However, expression of MurC-HA was constitutively on and insensitive to exogenous nitrate concentrations when the MurC-HA plasmid was introduced into 3 divergent lineages of Enterococcus faecium (unpublished data). Although the lack of nitrate-inducible regulation in E. faecium remains unexplained, our results indicate that the engineered NarQ169-NisK168 chimeric receptor enables robust nitrate-responsive control of gene expression in E. faecalis.
MurAA is the main driver of PG synthesis in E. faecalis.
In previous work, we demonstrated that either MurAA or MurAB (the 2 UDP-GlcNAc carboxyvinyl transferase homologs encoded in the enterococcal genome) could be individually deleted, but we were unable to isolate a mutant simultaneously lacking both MurAA and MurAB. Given that the reaction catalyzed by MurAA and MurAB is the first committed step in the synthesis of PG, we hypothesized that mutations in murAA and murAB were synthetically lethal. In other words, E. faecalis cells must possess one or the other to make PG and maintain viability. To test this, we used our nitrate inducible MurAA expression plasmid to control the synthesis of MurAA. In the presence of nitrate and the MurAA expression plasmid, we were able to successfully introduce simultaneous deletions of both murAA and murAB into the E. faecalis genome. The growth of the resulting strain was analyzed in liquid culture. While nitrate did not affect the growth of wild-type E. faecalis, growth of the ΔmurAA murAB double mutant was dependent on the presence of nitrate in the medium (Fig. 4), confirming that murAA and murAB are synthetically lethal and that they together represent the only sources of UDP-GlcNAc carboxyvinyl transferase activity to support PG biosynthesis in E. faecalis cells.
FIG 4.
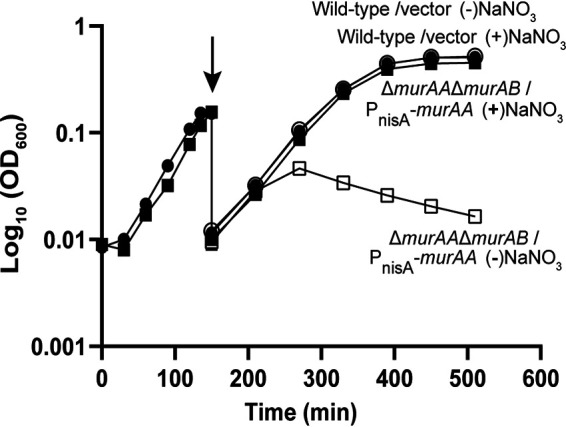
MurAA and MurAB are a synthetically lethal pair. Cultures were grown to exponential phase in the presence of 5 mM NaNO3, then washed (arrow) and suspended in media with or without 5 mM NaNO3. Data represent two independent biological replicates for each strain. Error bars are present but too small to be seen. Strains were wild-type/vector (circles), OG1/pJLL286; ΔmurAA ΔmurAB/PnisA-murAA (squares), DDJ343/pJLL288. Open symbols, no NaNO3, closed symbols, 5 mM NaNO3.
In previous work, we found that the ΔmurAA mutant, but not the ΔmurAB mutant, exhibited a growth defect in liquid cultures in the absence of antimicrobial stress (13). To determine if differences in PG synthesis contributed to the growth defect, we monitored [14C] GlcNAc incorporation into SDS-insoluble PG using a method we and others described previously (9, 19, 20). Consistent with the prior growth studies, we did not detect any change in PG synthesis for the ΔmurAB mutant but observed an ∼42% reduction relative to wild-type for the ΔmurAA mutant (Fig. 5). Together, the growth and PG synthesis data indicate that although MurAB can provide enough UDP-GlcNAc carboxyvinyl transferase activity to sustain viability in the absence of MurAA, MurAA appears to be the main driver of PG synthesis in E. faecalis, at least in unstressed cells.
FIG 5.

The rate of PG synthesis is reduced in the absence of MurAA. Incorporation of [14C] GlcNAc into SDS-insoluble peptidoglycan of exponentially growing E. faecalis was monitored following pulse-labeling for the ΔmurAA mutant (A) or the ΔmurAB mutant (B). The label incorporation rates calculated from data points between 10 and 25 min are shown in the tables below the graphs. Data represent mean ± standard deviation for two independent replicates. Strains were wild-type, OG1; ΔmurAA, JL626; ΔmurAB, CM1.
Cephalosporin resistance correlated with MurAA expression levels.
In previous work, we found that genetic deletion of MurAA led to a dramatic reduction in CR, and overexpression of MurAA led to elevated CR (13), suggesting that the amount of MurAA activity in cells was an important driver of CR. To test this, we used our nitrate inducible promoter to express MurAA at different levels and determined MICs for ceftriaxone, a widely used representative cephalosporin. Increasing concentrations of nitrate did not affect CR for cells carrying an empty vector, but an inducible expression of MurAA led to dose-dependent increases in CR (Table 1). Because MurAA expression correlates with both PG synthesis and CR, we hypothesized that MurAA-driven flux of metabolites through the PG synthesis pathway was an important driver of CR.
TABLE 1.
Inducible expression of MurAA yields dose-dependent increases in ceftriaxone resistance
| Strainb | Ceftriaxone MIC (μg/mL)a |
||||||
|---|---|---|---|---|---|---|---|
| NaNO3 (mM) | |||||||
| 0 | 0.10 | 0.15 | 0.20 | 0.25 | 0.30 | 0.35 | |
| Wild-type/vector | 128 | ndc | ndc | ndc | ndc | ndc | 128 |
| ΔmurAA/vector | 1 | ndc | ndc | ndc | ndc | ndc | ≤0.5 |
| ΔmurAA/PnisA-murAA | 1 | 2 | 4 | 8 | 32 | 64 | 128 |
Minimal inhibitory concentration for ceftriaxone was determined at various levels of MurAA induction. Data represent the median MIC from at least 2 independent biological replicates.
Strains analyzed were wild-type/vector, OG1/pJLL286; ΔmurAA/vector, JL626/pJLL286; and ΔmurAA/PnisA-murAA, JL626/pJLL288.
nd, not determined.
Depletion of PG synthesis enzymes downstream of MurAA impairs PG synthesis and CR.
If MurAA-driven flux of metabolites through the PG synthesis pathway enhances CR, we hypothesized that restricting the pathway at points downstream of MurAA would impair CR even in the presence of MurAA. To test this, we chose two enzymes downstream of MurAA in the PG synthesis pathway (MurC and MurJ; Fig. 1) and constructed strains in which the expression of each was under the control of the nitrate-inducible promoter in pJLL286 (while the chromosomal copy of the gene was deleted) to enable depletion of the corresponding enzyme as a function of nitrate concentration. The enzymes chosen represented both a cytoplasmic step (MurC; UDP-N-acetylmuramic acid:l-alanine ligase) and a membrane-bound step (MurJ; predicted Lipid II flippase responsible for translocation of cytoplasmic lipid-linked PG precursors to the external surface of the membrane, previously referred to as YtgP in Bacillus subtilis) of the PG synthesis pathway. Note that neither MurC nor MurJ possess homologs in the E. faecalis OG1 genome that were identifiable by BLAST searches; hence both are expected to be uniquely responsible for their respective step in the PG synthesis pathway, and depletion of the respective gene products is expected to create a bottleneck in the PG synthesis pathway that restricts the flow of PG precursors through the pathway. Genetic manipulations to delete the chromosomal copy of murC or murJ, as well as subsequent propagation of the resulting strains, were performed in the presence of nitrate and a nitrate-inducible copy of the corresponding gene. Note, for these experiments, the nitrate-inducible clones encoded wild-type forms of MurC or MurJ to avoid the possibility that the addition of an epitope tag, such as the HA tag noted above, would interfere with the function of the resulting proteins.
As expected, the ΔmurC PnisA::murC strain required nitrate for growth (Fig. S5), confirming that MurC is essential for viability and indicating that MurC was successfully depleted at low nitrate concentrations. Depletion of MurC at low nitrate concentration led to an ∼47% reduction in the rate of PG synthesis (Fig. 6A), consistent with its requirement for viability, that was rescued by boosting MurC expression with nitrate induction. Depletion of MurC also triggered activation of the CroS/R signaling system, which was rescued by boosting MurC expression with nitrate. Similar activation of the CroS/R signaling system is seen upon deletion of MurAA (Fig. S6A). These phenotypes in the MurC depletion strain are due specifically to depletion of MurC because inducible expression of MurC rescued the defects, and because immunoblotting confirmed that MurAA was still expressed at similar levels despite depletion of MurC (Fig. S6B). These results are consistent with the expectation that impairment of the PG synthesis pathway – by either MurAA deletion or MurC depletion – leads to cell wall stress that is sensed by CroS/R and results in activation of CroS/R signaling, thereby validating the depletion of MurC by an orthogonal approach. To assess the impact of MurC depletion on CR, we determined MICs for ceftriaxone at a variety of nitrate concentrations. At concentrations of nitrate that led to minimal change in growth rate (Fig. S7A) due to presumably modest depletion of MurC, the MurC-depleted cells exhibited a substantial reduction in CR (Table 2). Increases in nitrate concentration led to enhanced CR in a dose-dependent manner, although we were unable to fully restore CR to wild-type levels at any nitrate concentration tested, possibly because MurC expression levels did not achieve those of wild-type cells even at the highest nitrate concentrations tested.
FIG 6.

The rate of PG synthesis is reduced upon depletion of MurC (A) and MurJ (B). Incorporation of [14C] GlcNAc into SDS-insoluble peptidoglycan of exponentially growing E. faecalis was monitored following pulse-labeling for the MurC depletion mutant (A) and the MurJ depletion mutant (B). The label incorporation rate calculated from data points between 10 and 25 min is shown in the tables below the graphs. Data represent mean ± standard deviation for two independent replicates. Strains were wild-type/vector, OG1/pJLL286 + pJRG9; ΔmurC/PnisA-murC, JL657/pJLL297 + pJRG9; ΔmurJ/PnisA-murJ, JL656/pJLL296 + pJRG9.
TABLE 2.
Depletion of MurC impairs ceftriaxone resistance
| Strainb | Ceftriaxone MIC (μg/mL)a |
||
|---|---|---|---|
| NaNO3 (mM) | |||
| 2 | 5 | 12 | |
| Wild-type/vector | 128 | 128 | 128 |
| ΔmurC/PnisA-murC | 2 | 4 | 8 |
Minimal inhibitory concentration for ceftriaxone was determined at various levels of MurC induction. Data represent the median MIC from four independent biological replicates.
Strains analyzed were wild-type/vector, OG1/pJLL286; and ΔmurC/PnisA-murC, JL657/pJLL297.
Growth of the ΔmurJ PnisA::murJ strain was impaired in the absence of nitrate induction (Fig. S7B), but not eliminated. The simplest explanation for this is that residual murJ expression in the absence of inducer (as observed for MurAA and MurC-HA in Fig. 3 and Fig. S3) led to low levels of MurJ that were sufficient to sustain some growth (no antibody is available to assess MurJ abundance), although we cannot rule out the possibility of an alternative enzyme that can functionally compensate for the absence of MurJ at some low level. Regardless, depletion of MurJ led to an ∼44% reduction in the rate of PG synthesis (Fig. 6B) and, like depletion of MurC, triggered activation of the CroS/R signaling system without changing MurAA expression (Fig. S8), consistent with the prediction that enterococcal MurJ functions in the PG synthesis pathway as a PG flippase. To assess the impact of MurJ depletion on CR, we determined MICs for ceftriaxone at a variety of nitrate concentrations. MurJ-depleted cells exhibited a substantial reduction in CR and increases in nitrate concentration led to enhanced CR in a dose-dependent manner (Table 3). Together with the MurC depletion data, these results are consistent with a model in which restricting the PG synthesis pathway downstream of MurAA compromises CR in E. faecalis.
TABLE 3.
Depletion of MurJ impairs ceftriaxone resistance
| Strainb | Ceftriaxone MIC (μg/mL)a |
||
|---|---|---|---|
| NaNO3 (mM) | |||
| 0 | 0.1 | 5 | |
| Wild-type/vector | 64 | 64 | 64 |
| ΔmurJ/PnisA-murJ | 2 | 16 | 64 |
Minimal inhibitory concentration for ceftriaxone was determined at various levels of MurJ induction. Data represent the median MIC from four independent biological replicates.
Strains analyzed were wild-type/vector, OG1/pJLL286; and ΔmurJ/PnisA-murJ, JL656/pJLL296.
Pharmacologic block of the PG synthesis pathway downstream of MurAA impairs PG synthesis and CR.
To test that model pharmacologically, we used the antibiotic d-cycloserine (d-cyc) to inhibit the PG synthesis pathway downstream of MurAA. d-cyc inhibits two sequential enzymes in the PG synthesis pathway, alanine racemase (Alr) and d-alanine:d-alanine ligase (Ddl). Alr and Ddl together are responsible for the generation of the d-alanyl-d-alanine dipeptide, which is essential for the synthesis of complete PG precursors (Fig. 1). Hence, d-cyc blocks the PG synthesis pathway at a cytoplasmic step, which is distinct from the steps catalyzed by MurC or MurJ.
As expected, treatment of wild-type E. faecalis with d-cyc reduced the rate of PG synthesis in a dose-dependent manner (Fig. 7), with reductions of ∼36% and ∼77% observed at 100 μg/mL and 250 μg/mL d-cyc, respectively. Moreover, treatment of E. faecalis with d-cyc has been previously demonstrated to trigger activation of the CroS/R signaling system (10), in response to the cell wall stress imposed by inhibition of the PG synthesis pathway. To assess the impact of d-cyc on CR, we determined MICs for ceftriaxone in the presence of various d-cyc concentrations. d-cyc treatment resulted in a dose-dependent reduction in CR (Table 4), with higher d-cyc concentrations leading to more substantial reductions in CR. Moreover, d-cyc also impaired CR of E. faecium, the other most clinically relevant species of Enterococcus (Table 4), suggesting a common mechanism among enterococci. Collectively, our genetic and pharmacological studies all support a model in which MurAA-driven flux of metabolites through the PG synthesis pathway is a critical determinant of CR in enterococci.
FIG 7.
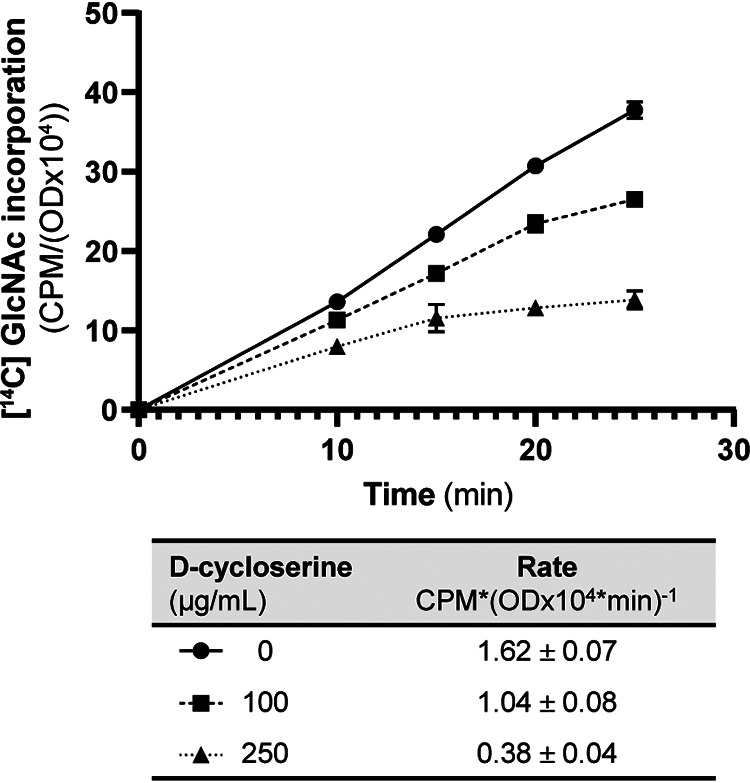
The rate of PG synthesis is reduced upon treatment with d-cyc. Incorporation of [14C] GlcNAc into SDS-insoluble peptidoglycan of exponentially growing E. faecalis OG1 was monitored following pulse-labeling. The label incorporation rate calculated from data points between 10 and 25 min is shown in the table below the graph. Data represent mean ± standard deviation for two independent replicates.
TABLE 4.
Pharmacological inhibition of d-ala-d-ala production by d-cycloserine impairs ceftriaxone resistance
| Strain | Ceftriaxone MIC (μg/mL)a |
|||
|---|---|---|---|---|
| d-cycloserine (μg/mL) | ||||
| 0 | 20 | 40 | 50 | |
| E. faecalis OG1 | 64 | ndb | 8 | 4 |
| E. faecium 1141733 | 32 | 4 | 0.5 | ndb |
Minimal inhibitory concentration for ceftriaxone was determined at various levels of d-cycloserine. Data represent the median MIC from three independent biological replicates.
nd, not determined.
DISCUSSION
Our previous work revealed that MurAA, one of two UDP-GlcNAc carboxyvinyl transferase homologs encoded in the E. faecalis genome, was specifically required for enterococcal CR (13). However, the molecular mechanism(s) by which MurAA drives CR remained unknown. Given that MurAA catalyzes the first committed step in the PG synthesis pathway and MurAA catalytic activity is required for CR, we hypothesized that MurAA-driven flux of metabolites through the PG synthesis pathway enhanced CR.
To test this, we sought to restrict the flow of metabolites through the PG synthesis pathway by depleting the expression of PG synthesis genes downstream of MurAA, most of which do not possess any obvious homologs in the enterococcal genome and are therefore expected to be essential for viability. However, such experiments presented a technical challenge, because the two most used tools for inducible gene expression in E. faecalis each suffer from limitations that rendered them incompatible with our studies. As a result, we developed a new tool for inducible gene expression in E. faecalis based on an interspecies chimeric receptor that senses external nitrate concentrations.
Transmembrane sensor kinases from canonical two-component signaling systems are well-known to exhibit a modular domain architecture in which the N-terminal domain comprises a sensing module, tailored to respond to specific cognate signal(s). The sensing module transmits information about ligand occupancy to the intracellular signaling module, typically resulting in enhanced phosphorylation of the response regulator partner that is then able to stimulate transcription of genes in the regulon. The modular nature of these signaling proteins has enabled the engineering of functional chimeras that exhibit altered ligand specificity defined by the identity of the N-terminal sensing domain. For example, the extracellular nitrate-responsive sensing module of the NarQ sensor kinase from E. coli could be fused to an intracellular signaling module from other transmembrane signaling proteins to create a functional nitrate-responsive chimeric receptor (18). We exploited this strategy to engineer an interspecies chimeric receptor in which the nitrate-responsive sensing module of E. coli NarQ replaced the sensing module of the lactococcal NisK nisin-sensing receptor that has been widely used as a tool for nisin-inducible gene expression in E. faecalis and other Gram-positive bacteria. Our engineered NarQ169-NisK168 chimeric receptor was no longer responsive to nisin but is functional in 5 diverse lineages of E. faecalis because it modulated expression from the PnisA promoter in a manner dependent on the extracellular concentration of nitrate.
We exploited our nitrate-inducible expression system in multiple ways in this work. By controlling the expression of MurAA, we were able to demonstrate the synthetic lethality of the 2 UDP-GlcNAc carboxyvinyl transferase homologs in E. faecalis, murAA, and murAB, confirming that at least one of them is essential for viability. We also depleted the expression of 2 additional genes in the PG synthesis pathway, that are each expected to be essential for viability (and therefore could not be deleted from the genome). In the case of MurC, subsequent studies demonstrated its essentiality because growth was strictly dependent on the induction of MurC from the nitrate-responsive promoter. Furthermore, in this work, we successfully used our nitrate-inducible expression system to regulate gene expression in both liquid cultures and on agar plates, a necessity for genetic manipulations of essential genes. A small amount of residual expression was observed in the uninduced state with our system, which might be a problem in some circumstances. We speculate that changes to the ribosome binding site of cloned genes to reduce the efficiency of translation could diminish such uninduced expression and mitigate any phenotypic consequences, although such modifications would likely also reduce the maximal level of expression that can be obtained in the induced state.
To test our hypothesis that the MurAA-driven flux of metabolites through the PG synthesis pathway enhanced CR, we used our nitrate-inducible expression system to regulate the expression of multiple genes in the PG synthesis pathway. Although MurAB is capable of sustaining (poor) growth in the absence of MurAA (13), MurAA appears to be the primary driver of PG synthesis in E. faecalis (Fig. 5) and increased expression of MurAA as a function of nitrate concentration correlated with MurAA-dependent increases in CR (Table 1). Given that MurAA catalyzes the first committed step in the PG synthesis pathway, these results are consistent with the hypothesis that MurAA-driven flux through the pathway is a key determinant of CR.
If so, restriction of metabolic flow through the PG synthesis pathway downstream of MurAA would be expected to compromise CR, despite the presence of MurAA. To test this, we used our nitrate-inducible system to deplete the expression of two downstream enzymes from distinct points in the PG synthesis pathway, the cytoplasmic MurC ligase, and the membrane-bound MurJ flippase. MurC catalyzes the addition of an l-alanine residue to the cytoplasmic peptidoglycan precursor, UDP-MurNAc, the first step in the formation of the peptide stem. Following the cytoplasmic steps of peptidoglycan biosynthesis, the PG precursors (known as Lipid II) must be flipped to the outside the membrane, where they are used for polymerization into linear glycan chains that are subsequently cross-linked to form mature peptidoglycan (Fig. 1). MurJ (previously referred to as YtgP in other Gram-positive organisms) is the predicted Lipid II flippase (21, 22). Both MurC and MurJ were predicted to be essential for viability given the lack of any known homologs encoded in the genome of E. faecalis. We confirmed this prediction for MurC because the ΔmurC PnisA::murC strain required nitrate for growth. The MurJ depletion strain was, however, able to grow in the absence of inducer, albeit at a much lower rate than wild type. We hypothesize that this is due to the leakiness of the PnisA promoter and residual expression of MurJ. However, it remains possible that another enzyme is capable of supporting Lipid II flipping in the absence of MurJ. Studies in Bacillus subtilis demonstrated that MurJ (referred to as YtgP) was not essential for growth in this species, nor were three identified putative homologs (23), and subsequent work supported the existence of secondary flippase (24). However, in addition to lacking any homologs of MurJ, the genome of E. faecalis does not appear to encode a homolog of this alternative flippase.
Using the MurC and MurJ depletion strains, we successfully created bottlenecks in the PG synthesis pathway as evidenced by the significant reduction observed in the rate of PG synthesis, which was rescued upon induction of the respective genes. The finding that depletion of MurC or MurJ activates the CroS/R signaling system (as is also observed with deletion of murAA), further demonstrates that MurC (or MurJ) depletion in our experiments impaired synthesis of PG. Concomitant with the restriction of peptidoglycan synthesis, we observed a substantial loss of CR in our MurC and MurJ depletion strains. Upon addition of increasing concentrations of inducer, we saw dose-dependent increases in the CR, indicating that CR is dependent on flux through the peptidoglycan synthesis pathway.
To augment the genetic depletion studies, we used a pharmacological approach to restrict PG synthesis downstream of MurAA with the antibiotic d-cycloserine. d-cycloserine is an antibiotic that inhibits the generation of the essential d-alanyl-d-alanine dipeptide moiety during the cytoplasmic phase of PG precursor synthesis, preventing the synthesis of complete PG precursors. Upon inhibition of PG synthesis with d-cycloserine, we observed dose-dependent defects in both PG synthesis and CR. Thus, we have shown that restriction of PG synthesis, using two unique methods (genetic and pharmacological) targeting four distinct steps in the PG synthesis pathway, correlates with impairment of CR in E. faecalis. Moreover, the reduction of CR occurs even under conditions in which overall growth is not substantially impaired, suggesting that the CR machinery is carefully regulated by some aspect of the PG synthesis pathway.
Taken together, our data indicate that control of flux through the peptidoglycan synthesis pathway is an important driver of CR in E. faecalis. Moreover, the PG synthesis enzyme, MurAA, likely contributes to CR by modulating this flux. A similar model has recently been proposed in Listeria monocytogenes. Elevated accumulation of the MurAA homolog, MurA, in L. monocytogenes was associated with enhanced cephalosporin resistance co-occuring with thicker peptidoglycan at the cell poles, suggestive of increased PG synthesis (25). Another group reported that PG production is increased in an L. monocytogenes mutant that accumulates elevated levels of MurA using a click-chemistry-based PG-labeling approach (26). However, these prior studies did not distinguish whether the proposed increases in PG synthesis or the increase in MurA protein abundance per se, was responsible for the enhanced CR phenotype. Given that MurC and MurJ depletion impair CR without affecting MurAA protein levels, our results suggest that flux through the PG synthesis pathway is itself an important driver of CR in E. faecalis.
It is not necessarily obvious how modulating metabolite flow through the PG synthesis pathway could alter resistance to cephalosporins antibiotics, which impair cross-linking of PG polymers by PBPs. One possibility is that the availability of linear PG glycan polymers may dictate the relative importance of PBP activity. When PG synthesis is limited and fewer linear PG polymers are being produced, each potential cross-linking reaction may be more critical for cell wall stability. In contrast, when linear PG polymers are plentiful, the cell may tolerate greater inhibition of PBPs. Alternatively, the cell may modulate PBP expression, stability, or activity concurrently with the regulation of metabolite flux through the PG synthesis pathway. In Listeria monocytogenes, MurA stability (and thus metabolite flux into the PG synthesis pathway) is regulated by the PASTA domain-containing eukaryotic-like serine/threonine kinase, PrkA (25). It seems likely that a similar mechanism exists in enterococci involving the enterococcal PrkA homolog, IreK. Thus, it is easy to imagine that the regulation of PBPs could be coordinated with metabolic flow through the PG synthesis pathway or with the regulation of MurAA via IreK signaling.
In any case, we suspect that these mechanisms apply more broadly to related species given that a dose-dependent effect of d-cycloserine treatment on cephalosporin resistance was also observed in E. faecium, the other clinically relevant species of Enterococcus. Finally, the fact the MurAA homolog, MurAB, is not required for CR and does not seem to contribute significantly to PG synthesis suggests a unique function or level of regulation for MurAA that allows it to contribute to these processes but is yet to be fully explored.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 5. Enterococci were propagated on Mueller-Hinton broth (MHB) or agar (prepared according to the manufacturer’s instructions; Difco) at 30°C unless indicated otherwise. Erythromycin and/or chloramphenicol were included at 10 μg/mL when required for maintenance of plasmids.
TABLE 5.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Routine cloning host | Lab stock |
| E. faecalis | ||
| OG1 | Wild-type reference strain (MLST 1) | (29) |
| T1 | Clinical isolate (MLST 21) | (30) |
| E1 Sol | Fecal isolate from Solomon Islands (MLST 93) | (31) |
| T3 | Clinical isolate from Japan (MLST 67) | (31) |
| X98 | Fecal isolate (MLST 19) | (31) |
| CM1 | OG1 ΔmurAB (ΔK22-E418) | This work |
| JL626 | OG1 ΔmurAA (ΔV17-K411) | This work |
| DDJ343 | OG1 ΔmurAA murAB | This work |
| JL657 | OG1 ΔmurC (ΔK7-T440) | This work |
| JL656 | OG1 ΔmurJ (ΔS7-R543) | This work |
| E. faecium | ||
| 1,141,733 | Wild-type reference strain, clinical isolate (MLST 327) | (31) |
| Plasmids | ||
| pJH086 | E. faecalis allelic exchange vector (CmR); pheS* counterselection | (11) |
| pJLL286 | Nitrate-inducible expression vector (EmR) | This work |
| pJLL288 | pJLL286 carrying PnisA::murAA | This work |
| pJLL294 | pJLL286 carrying PnisA::murC-HA | This work |
| pJLL297 | pJLL286 carrying PnisA::murC | This work |
| pJLL296 | pJLL286 carrying PnisA::murJ | This work |
MLST, multilocus sequence type.
Construction of pJLL286.
Plasmid pMSP3535 (15) was used as the starting point for modifications to create pJLL286. pMSP3535 carries the PnisA promoter that is regulated by the NisK/R two-component signaling system (also encoded on pMSP3535) in response to exogenously provided nisin. To construct pJLL286, we first performed site-directed mutagenesis via inverse PCR to delete segments containing the ribosome binding site of nisA and initial 40 residues of NisA found in pMSP3535, as previously described (27). The resulting shortened version of the PnisA promoter was previously referred to as PnisA-m to distinguish it from the originally cloned PnisA (27), but here we will refer to it simply as PnisA because none of the actual promoter elements were altered. Subsequently, we deleted the segment of the nisK open reading frame encoding residues 5 to 167 of NisK and simultaneously replaced it with a synthetic DNA fragment (IDT) encoding residues 2 to 169 of E. coli NarQ that had been manually codon-optimized to align with enterococcal codon usage. Note that the first 4 codons of nisK were retained in the chimeric receptor to avoid perturbing translation of nisR, which is encoded immediately upstream of nisK and overlaps slightly with nisK.
Genes of interest were amplified by PCR and introduced into BamHI/XhoI-digested pJLL286 via isothermal assembly, incorporating a synthetic ribosome-binding site (AGGAGGAACTCAATATG, where “ATG” is the initiation codon) upstream of the gene open reading frame.
Genetic manipulation of enterococci.
Mutant strains were constructed using the temperature-sensitive, counter selectable allelic exchange plasmid pJH086 as previously described (11). In addition to p-Cl-Phe, counterselection plates contained 20% sucrose. Fragments of genomic DNA from upstream and downstream of the gene of interest were amplified by PCR and introduced into pJH086 via isothermal assembly (28). Deletion alleles were in-frame and retained a variable number of codons at the beginning and end of each gene, to avoid perturbing the expression of adjacent genes. Table 5 contains the specific details of each deletion allele. All mutant or complemented strains were constructed independently at least twice and analyzed to ensure that their phenotypes were concordant. The inserts of all recombinant plasmids were sequenced in their entirety to ensure the absence of any undesired mutations.
Construction of ΔmurAA murAB, ΔmurC, and ΔmurJ strains was carried out with cells carrying an inducible copy of the corresponding gene expressed from pJLL286 (murAA, murC, or murJ, respectively) in the presence of erythromycin. In these cases, 5 mM NaNO3 was included in the counterselection plates and the growth media for all subsequent propagation, except as noted for specific experiments.
Antimicrobial susceptibility assays.
MICs were determined using a broth microdilution method. Briefly, bacteria from stationary-phase cultures were inoculated into wells containing 2-fold serial dilutions of antibiotics in MHB (including erythromycin when required for maintenance of plasmids and indicated concentrations of NaNO3) at a normalized optical density at 600 nm (OD600) of 4 × 10−5 (∼1 × 105 CFU). If NaNO3 was required for the growth of overnight cultures (e.g., for the ΔmurC mutant), cultures were washed twice with MHB before dilution. Plates were incubated in a Bioscreen C plate reader at 37°C for 24 h with brief shaking before each measurement. The optical density at 600 nm (OD600) was determined every 15 min, and the lowest concentration of antibiotic that prevented growth was recorded as the MIC.
Immunoblot analysis.
Stationary-phase cultures were diluted to OD600 = 0.01 in MHB supplemented with erythromycin for plasmid maintenance and NaNO3. Cultures were incubated at 37°C with shaking until the exponential phase (OD600 ∼0.2), at which time they were harvested by mixing with an equal volume of cold EtOH/acetone (1:1), collected by centrifugation, and washed once with water. To prepare total cell lysates, bacteria were suspended in lysozyme buffer (10 mM Tris [pH 8], 50 mM NaCl, 20% sucrose), normalized to equivalent OD600, treated with lysozyme (5 mg/mL) for 30 min at 37°C, and mixed with Laemmli SDS sample buffer. After SDS-PAGE, proteins were transferred to polyvinylidene difluoride (PVDF; Bio-Rad) and probed with rabbit anti-MurAA, rabbit anti-CroR, rabbit anti-RpoA (all custom polyclonal antisera), or rabbit anti-HA antisera (Abcam) as indicated. Detection was performed with goat anti-rabbit HRP-conjugated secondary antibody (Invitrogen) on a Bio-Rad Chemidoc. Phos-tag SDS-PAGE for CroR was performed as described previously (12).
Peptidoglycan synthesis assays.
Peptidoglycan synthesis was monitored by uptake of [14C] GlcNAc into SDS-insoluble peptidoglycan as described previously (9, 19, 20) with minor modifications. Bacteria were grown to exponential phase in MHB at 37°C as described above. Cultures were diluted 6-fold into prewarmed MHB supplemented with a final concentration of 0.33 μCi/mL [14C] GlcNAc. At intervals, aliquots were mixed with equal volumes of 0.2% SDS. The cells were collected by centrifugation and suspended in 150 μL water, then transferred into 5 mL Econo-Safe scintillation fluid for scintillation counting. Optical density measurements were done in triplicate by transferring 200 μL from parallel label-free cultures to a 96-well plate and measuring OD600 using a SpectraMax5 plate reader (Molecular Devices). Data were expressed as CPM/(OD600 x 104). We previously demonstrated that this method measures incorporation of [14C] GlcNAc into peptidoglycan specifically because treatment with fosfomycin, an antibiotic that inhibits the first committed step in the cytoplasmic PG synthesis pathway, largely prevented the incorporation of the labeled substrate (9). Therefore, this method allowed us to monitor peptidoglycan synthesis by measuring [14C] GlcNAc incorporation over time and normalizing to total biomass.
For the MurC and MurJ depletion experiments cultures were grown to exponential phase in MHB containing NaNO3 (at the indicated concentration) as well as chloramphenicol and erythromycin for retention of plasmids. For these experiments, all strains also carried pJRG9, a chloramphenicol-resistant plasmid unrelated to the specific goals of these experiments. For experiments using d-cycloserine, treatment with d-cycloserine was initiated at time zero, when exponential-phase cultures were diluted into prewarmed labeled media (and unlabeled media for parallel cultures) containing the indicated concentration of d-cycloserine.
Nitrate-dependent growth to establish gene essentiality.
Stationary-phase cultures in MHB supplemented with erythromycin and 5 mM NaNO3 were diluted to OD600 = 0.01 in fresh media of the same composition and incubated at 37°C with shaking until exponential phase (OD600 ∼0.14 to 0.15), at which time cultures were collected by centrifugation (4,000 rpm, 10 min, 4°C) in prechilled 50 mL conical tubes. Cells were washed twice with MHB to remove NaNO3 then resuspended at OD600 = 0.01 in MHB supplemented with erythromycin in the presence or absence of 5 mM NaNO3. OD600 was monitored during incubation at 37°C with shaking.
ACKNOWLEDGMENTS
This study was supported in part by grants AI134660 and AI150895 from the National Institutes of Health (NIH). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection, interpretation, or the decision to submit the work for publication.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Christopher J. Kristich, Email: ckristich@mcw.edu.
Michael J. Federle, University of Illinois at Chicago
REFERENCES
- 1.Kristich CJ, Rice LB, Arias CA. 2014. Enterococcal infection-treatment and antibiotic resistance. In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection. Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 2.Hollenbeck BL, Rice LB. 2012. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 3:421–433. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baddour LM, Wilson WR, Bayer AS, Fowler VG, Tleyjeh IM, Rybak MJ, Barsic B, Lockhart PB, Gewitz MH, Levison ME, Bolger AF, Steckelberg JM, Baltimore RS, Fink AM, O'Gara P, Taubert KA, American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. 2015. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications. Circulation 132:1435–1486. doi: 10.1161/CIR.0000000000000296. [DOI] [PubMed] [Google Scholar]
- 4.Holland TL, Baddour LM, Bayer AS, Hoen B, Miro JM, Fowler VG. 2016. Infective endocarditis. Nat Rev Dis Prim 2:16059. doi: 10.1038/nrdp.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmeli Y, Eliopoulos GM, Samore MH. 2002. Antecedent treatment with different antibiotic agents as a risk factor for vancomycin-resistant Enterococcus. Emerg Infect Dis 8:802–807. doi: 10.3201/eid0808.010418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shepard BD, Gilmore MS. 2002. Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect 4:215–224. doi: 10.1016/s1286-4579(01)01530-1. [DOI] [PubMed] [Google Scholar]
- 7.Bush K, Bradford PA. 2016. β-lactams and β-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med 6:a025247. doi: 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Signoretto C, Boaretti M, Canepari P. 1994. Cloning, sequencing and expression in Escherichia coli of the low-affinity penicillin binding protein of Enterococcus faecalis. FEMS Microbiol Lett 123:99–106. doi: 10.1111/j.1574-6968.1994.tb07207.x. [DOI] [PubMed] [Google Scholar]
- 9.Djorić D, Little JL, Kristich CJ. 2020. Multiple low-reactivity class B penicillin-binding proteins are required for cephalosporin resistance in enterococci. Antimicrob Agents Chemother 64:1–13. doi: 10.1128/AAC.02273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comenge Y, Quintiliani R, Li L, Dubost L, Brouard JP, Hugonnet JE, Arthur M. 2003. The CroRS two-component regulatory system is required for intrinsic β-lactam resistance in Enterococcus faecalis. J Bacteriol 185:7184–7192. doi: 10.1128/JB.185.24.7184-7192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kellogg SL, Little JL, Hoff JS, Kristich CJ. 2017. Requirement of the CroRS two-component system for resistance to cell wall-targeting antimicrobials in Enterococcus faecium. Antimicrob Agents Chemother 61:e02461-16. doi: 10.1128/AAC.02461-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellogg SL, Kristich CJ. 2016. Functional dissection of the CroRS two-component system required for resistance to cell wall stressors in Enterococcus faecalis. J Bacteriol 198:1326–1336. doi: 10.1128/JB.00995-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vesić D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob Agents Chemother 56:2443–2451. doi: 10.1128/AAC.05984-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du W, Brown JR, Sylvester DR, Huang J, Chalker AF, So CY, Holmes DJ, Payne DJ, Wallis NG. 2000. Two active forms of UDP-N-acetylglucosamine enolpyruvyl transferase in gram-positive bacteria. J Bacteriol 182:4146–4152. doi: 10.1128/JB.182.15.4146-4152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryan EM, Bae T, Kleerebezem M, Dunny GM. 2000. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44:183–190. doi: 10.1006/plas.2000.1484. [DOI] [PubMed] [Google Scholar]
- 16.Weaver KE, Chen Y, Miiller EM, Johnson JN, Dangler AA, Manias DA, Clem AM, Schjodt DJ, Dunny GM. 2017. Examination of Enterococcus faecalis toxin-antitoxin system toxin Fst function utilizing a pheromoneinducible expression vector with tight repression and broad dynamic range. J Bacteriol 199:e00065-17. doi: 10.1128/JB.00065-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panina I, Krylov N, Nolde D, Efremov R, Chugunov A. 2020. Environmental and dynamic effects explain how nisin captures membrane-bound lipid II. Sci Rep 10:8821. doi: 10.1038/s41598-020-65522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi S, Pollard AM, Yang Y, Jin F, Sourjik V. 2016. Engineering hybrid chemotaxis receptors in bacteria. ACS Synth Biol 5:989–1001. doi: 10.1021/acssynbio.6b00053. [DOI] [PubMed] [Google Scholar]
- 19.Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, Jarmer H, Devine KM. 2007. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol Microbiol 65:180–200. doi: 10.1111/j.1365-2958.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- 20.Mesnage S, Chau F, Dubost L, Arthur M. 2008. Role of N-acetylglucosaminidase and N-acetylmuramidase activities in Enterococcus faecalis peptidoglycan metabolism. J Biol Chem 283:19845–19853. doi: 10.1074/jbc.M802323200. [DOI] [PubMed] [Google Scholar]
- 21.Sham L-T, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. 2014. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz N. 2009. Streptococcus pyogenes YtgP (Spy-0390) complements Escherichia coli strains depleted of the putative peptidoglycan flippase MurJ. Antimicrob Agents Chemother 53:3604–3605. doi: 10.1128/AAC.00578-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fay A, Dworkin J. 2009. Bacillus subtilis homologs of MviN (MurJ), the putative Escherichia coli lipid II flippase, are not essential for growth. J Bacteriol 191:6020–6028. doi: 10.1128/JB.00605-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meeske AJ, Sham LT, Kimsey H, Koo BM, Gross CA, Bernhardt TG, Rudner DZ. 2015. MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc Natl Acad Sci USA 112:6437–6442. doi: 10.1073/pnas.1504967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wamp S, Rutter ZJ, Rismondo J, Jennings CE, Möller L, Lewis RJ, Halbedel S. 2020. PrkA controls peptidoglycan biosynthesis through the essential phosphorylation of ReoM. Elife 9:e56048. doi: 10.7554/eLife.56048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelliher JL, Grunenwald CM, Abrahams RR, Daanen ME, Lew CI, Rose WE, Sauer J-D. 2021. PASTA kinase-dependent control of peptidoglycan synthesis via ReoM is required for cell wall stress responses, cytosolic survival, and virulence in Listeria monocytogenes. PLoS Pathog 17:e1009881. doi: 10.1371/journal.ppat.1009881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J-H, Mills DA. 2007. Improvement of a nisin-inducible expression vector for use in lactic acid bacteria. Plasmid 58:275–283. doi: 10.1016/j.plasmid.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 29.Gold O, Jordan H, van Houte J. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch Oral Biol 20:473–477. doi: 10.1016/0003-9969(75)90236-8. [DOI] [PubMed] [Google Scholar]
- 30.Maekawa S, Yoshioka M, Kumamoto Y. 1992. Proposal of a new scheme for the serological typing of Enterococcus faecalis strains. Microbiol Immunol 36:671–681. doi: 10.1111/j.1348-0421.1992.tb02070.x. [DOI] [PubMed] [Google Scholar]
- 31.Palmer KL, Carniol K, Manson JM, Heiman D, Shea T, Young S, Zeng Q, Gevers D, Feldgarden M, Birren B, Gilmore MS. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J Bacteriol 192:2469–2470. doi: 10.1128/JB.00153-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S8. Download jb.00602-21-s0001.pdf, PDF file, 0.4 MB (418.2KB, pdf)