

Abstract
Background:
Epidemiological studies have reported an association between Parkinson’s disease (PD) and restless legs syndrome.
Objectives:
We aimed to use genetic data to study whether these 2 disorders are causally linked or share genetic architecture.
Methods:
We performed two-sample Mendelian randomization and linkage disequilibrium score regression using summary statistics from recent genome-wide meta-analyses of PD and restless legs syndrome.
Results:
We found no evidence for a causal relationship between restless legs syndrome (as the exposure) and PD (as the outcome, inverse variance-weighted; b = −0.003, SE = 0.031, P = 0.916; F statistic = 217.5). Reverse Mendelian randomization also did not demonstrate any causal effect of PD on restless legs syndrome (inverse variance-weighted; b = −0.012, SE = 0.023, P = 0.592; F statistic = 191.7). Linkage disequilibrium score regression analysis demonstrated lack of genetic correlation between restless legs syndrome and PD (rg = −0.028, SE = 0.042, P = 0.507).
Conclusions:
There was no evidence for a causal relationship or genetic correlation between restless legs syndrome and PD. The associations observed in epidemiological studies could be attributed, in part, to confounding or nongenetic determinants. © 2021 International Parkinson and Movement Disorder Society
Keywords: restless legs syndrome, Parkinson’s disease, Mendelian randomization, linkage disequilibrium score regression, genetic correlation
Restless legs syndrome (RLS) and Parkinson’s disease (PD) are common neurological disorders, with a prevalence of 1.9%–4.6% and 0.1%–2.9%, respectively, in Europeans.1,2 Epidemiological studies suggest that RLS is more common than expected in PD patients, and PD affects RLS patients more frequently than matched controls or the general population.3 Some studies suggest that RLS may be an early clinical manifestation of PD,4–6 whereas other studies found no association between RLS and PD.3 A recent meta-analyses showed higher odds ratios for RLS in PD patients compared with controls.3 In this study, the previous contradictory results were explained by different inclusion and diagnostic criteria and differences in sex distribution.3 However, there are major differences between RLS and PD, including clinical, ultrasonographic, functional, and neuroimaging aspects, which do not support an association between RLS and PD.7–10
Therefore, the true nature of the association between RLS and PD remains unclear. Mendelian randomization (MR) may help to mitigate some of the bias introduced by reverse causation and confounding in traditional observational studies.11 In addition, genetic correlation using linkage disequilibrium (LD) score regression (LDSC) may help to determine whether different traits have overlapping genetic background, which may explain some of the observed associations between traits.
Here, we used bidirectional MR and LDSC to seek evidence for a causal relationship and/or shared genetic architecture between RLS and PD.
Methods
Study Population and Genetic Data
To perform MR and LDSC, we used summary statistics from 2 recent genome-wide association study (GWAS) meta-analyses of RLS and PD.12,13 The RLS summary statistics included data from 15,126 patients and 95,725 controls,12 and the PD summary statistics included data from 33,674 cases (15,056 PD patients and 18,618 proxy cases), and 449,056 controls.13 A subset of data (23andMe data) was not included in the PD summary statistics to avoid potential overlap with the RLS data, which included 23andMe data. The 23andMe participants provided informed consent and participated in the research under a protocol approved by the external Association for the Accreditation of Human Research Protection Programs–accreted institutional review board Ethical & Independent Review Services. The full GWAS summary statistics for the 23andMe discovery data set will be made available through 23andMe to qualified researchers under an agreement with 23andMc that protects the privacy of the 23andMe participants. Please visit https://research.23andme.eom/collaborate/#dataset-access/ for more information and to apply to access the data. Information on recruitment procedures and diagnostic criteria is detailed in the original publications.12,13 All cases and controls in this study were of European ancestry.
Power Calculation
Power was calculated for detecting an effect size of an odds ratio of 1.2 on RLS and PD risk, using online sample size and power calculator for Mendelian randomization with a binary outcome (https://sb452.shinyapps.io/power/).14 For all analyses power was estimated at >80%.
Mendelian Randomization
We performed bidirectional MR, that is, examining whether RLS is a causal risk factor (exposure) for PD (outcome) and if PD is a causal risk factor for RLS. For each MR analysis, we constructed multivariant instruments from the independent (“index”) GWAS significant single-nucleotide polymorphisms (P ≤ 5 × 10−08) from the exposure GWAS. In brief, index single-nucleotide polymorphisms (SNPs) were obtained by clumping all GWAS significant SNPs within each linkage-disequilibrium block using an R2 threshold of 0.001 or a distance of 10,000 kb from the index SNP. This process increased the independence of each index SNP based on the above parameters. Additional details regarding the instrument construction and the code used for the analysis are available at https://github.com/gan-orlab/MR_LDSC_RLS-PD.
To calculate the proportion of variability in the exposure explained by the SNPs and to test the strength of the instrument variables (IVs), we used the statistical power for MR analyses (the coefficient of determination, R2) and F-statistic tests, as previously described.15 To perform MR, an estimate of the individual effect of SNPs on the exposure and outcome (RLS and PD, interchangeably) was used to calculate the Wald ratio. Then, the effect estimates were combined using the inverse-variance weighted (IVW) method, which is a weighted mean of the Wald ratio estimates obtained from each individual SNP separately.16
Sensitivity Analyses
To explore whether IVW results might be biased because of violations of MR assumptions and to evaluate the robustness of the results, we used weighted median and MR Egger16 estimators as sensitivity analyses. The weighted median estimate provides a reliable pooled estimate assuming that at least half the weight of the SNPs in the instrument are valid. MR Egger assesses directional pleiotropy similarly to the IVW approach except that the regression slope y intercept is not constrained to pass through the origin. For each approach, we constructed funnel plots to detect outliers. We evaluated the heterogeneity statistics Q for IVW and Q′ for MR-Egger. Mendelian Randomization Pleiotropy RESidual Sum, and Outlier (MR-PRESSO)17 was used to examine outlier SNPs that might occur in the presence of horizontal pleiotropy and correct pooled estimates. Steiger filtering was used to discard SNPs that explain more variance in the outcome than in the exposure. To find all the SNPs that are in LD with the index SNP, the LDmatrix module on the LDlink web tool was used.18
Genetic Correlation Analyses
To assess the genetic correlation between RLS and PD, we performed LDSC after computing z scores and formatting data from the two GWASs as previously described.19,20 In brief, LDSC calculates genetic correlation between two traits by incorporating LD scores (the more variants in LD with a SNP, the higher the LD score) and GWAS summary statistics (z scores) in a regression model.
Results
In total, 20 and 55 index SNPs for RLS and PD, respectively, were initially used as IVs for exposure. These IVs explain 3.5% and 2.1% of the risk in RLS and PD, respectively. All SNPs were strong instruments for MR analysis as measured by the F statistic (RLS F statistic = 217.5; PD F statistic = 191.7). There was no overlap between the genes where the clumped SNPs are in both meta-analyses (Table S1).
We then performed MR analyses to assess the bidirectional causal relationship between RLS and PD. RLS, as the exposure, was not causally associated with PD (IVW; b = −0.051, SE = 0.037, P = 0.172). However, the P values of IVW-Q and MR Egger-Q′ tests were 0.034 and 0.025, respectively, raising the possibility of pleiotropic SNPs in our data set, which violates MR assumptions. MR-PRESSO17 was applied, and a pleiotropic index SNP, rs11860769 (P = 0.02) was identified when RLS was used as exposure. This SNP has an opposite effect on risk of RLS and PD, as was previously shown.21 After removing the pleiotropic index SNP (rs11860769), 19 index SNPs were used as IVs for RLS. Again, there was no causal effect of RLS on PD (b = −0.003, SE = 0.031, P = 0.916) or of PD on RLS (b = −0.012, SE = 0.023, P = 0.592) with 55 IVs, and the results of sensitivity analyses suggested that there were no additional deviations from the MR assumptions (Table 1, Fig. 1; Figs. S1 and S2).
Table 1.
MR analysis between RLS and PD
| Inverse variance weighted |
MR Egger |
Weighted median |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exposure | Outcome | F | R 2 | MR-PRESSO | b | SE | P | Q test p | b | SE | P | Q test p | b | SE | P |
|
| |||||||||||||||
| RLS | PD | 217.558 | 0.035 | 0.832 | −0.003 | 0.031 | 0.916 | 0.780 | −0.019 | 0.064 | 0.767 | 0.729 | −0.020 | 0.043 | 0.632 |
| PD | RLS | 191.791 | 0.0315 | 0.662 | −0.012 | 0.023 | 0.592 | 0.300 | −0.002 | 0.050 | 0.958 | 0.269 | −0.011 | 0.036 | 0.749 |
RLS, restless legs syndrome; PD, Parkinson’s disease; F, “strength” of the genetic instrumental variable; R2, proportion of variance in exposure variable explained by SNPs; MR-PRESSO, Mendelian Randomization Pleiotropy RESidual Sum and Outlier; MR, Mendelian randomization; b, beta; SE, standard error; Q, Cochran’s Q test.
FIG. 1.
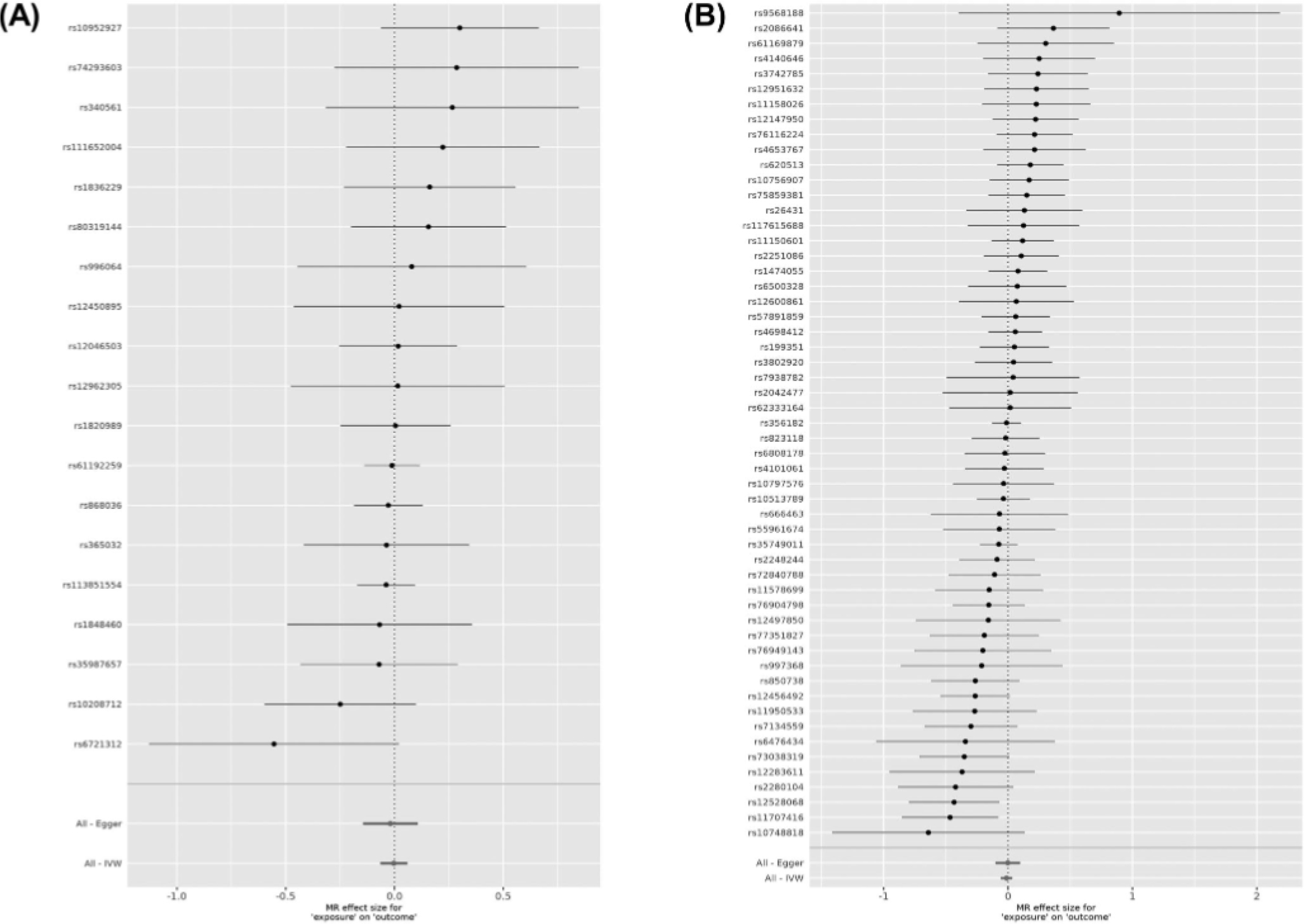
Forest plots showing results from the Mendelian randomization study to evaluate the potential causal relationships between RLS and PD. (A) Forest plot showing point estimates of RLS as an exposure on PD (outcome). In total, 19 index SNPs were left after excluding the pleiotropic SNPs to construct instrument variables. The black dots represent the causal estimate (b = log odds ratio) of each SNP on the risk of PD. Red dots represent the causal estimate when combining all SNPs together, using MR Egger and IVW methods. Horizontal lines denote 95% CI. (B) Forest plot showing point estimates of PD as an exposure on RLS (outcome). The instrument variables were constructed by 55 index SNPs. The black dots represent the causal estimate (b, the log odds ratio) of each SNP on the risk of RLS. Red dots represent the causal estimate when combining all SNPs together, using MR Egger and IVW methods. Horizontal lines denote 95% CI.
We then sought to examine whether there is genetic correlation between RLS and PD that may explain the overrepresentation of these disorders in one another. There was no genetic correlation between RLS and PD (rg = −0.028, SE = 0.042, P = 0.507).
Discussion
Our findings suggest lack of a causal relationship between RLS and PD and lack of a genetic correlation. One locus on chromosome 16, including the genes TOX3 and CASCA6, is pleiotropic, with opposite direction of effect, as SNPs associated with increased risk of RLS are associated with reduced risk of PD, as previously reported.21 Therefore, this locus also cannot explain the observed increased frequency of PD in RLS and of RLS in PD.
Although RLS and PD co-occur at a rate higher than expected and share several traits such as dopaminergic treatment response, multiple lines of evidence have shown differences between RLS and PD from a pathophysiological perspective. PD arises from the loss of dopaminergic neurons in the substantia nigra, whereas in RLS there is no loss of dopaminergic cells and no reduced dopamine22; instead, there is increased presynaptic dopaminergic activity.23 The neuronal loss may explain the elevated iron (seen as hyperechogenicity in transcranial sonography) and impairment in motor performance in PD versus reduced iron content (hypo echogenicity in transcranial sonography) and normal motor function in idiopathic RLS.3,24,25
Our LDSC analyses showed lack of genetic correlation between RLS and PD. Similarly, various genetic studies found no association between known RLS-associated variants and PD in the BTBD9,26,27 MAP2K5/SKOR1,26,27 MEIS1,26,27 and PTPRD26 loci. In a study of 2 Italian families, 10 of 20 RLS patients carried compound heterozygous or single heterozygous PRKN variants. It is not clear if these variants are pathogenic, and the clinical symptoms did not differ between RLS patients with and without PRKN variants in these 2 families, indicating that their presence was likely random.28 In an Asian cohort of 80 PD patients, 1 patient with a homozygous PINK1 mutation presented features of RLS, but 2 other unrelated PD patients with PINK1 mutations in the same cohort did not show RLS features.29 In a study of 258 RLS patients versus 235 healthy controls, the authors reported that the SNCA Rep1 allele was associated with reduced risk of RLS.30 However, this association was not replicated by the much larger RLS GW AS.12 Overall, genetic studies, including the current study, do not support a genetic overlap between RLS and PD.
Our study has some limitations. We could not exclude PD patients with RLS and RLS patients with PD in the data sets used for this analysis, which would have made the results cleaner, because these data were not available. In the RLS GWAS, the samples from the EU-RLS-GENE consortium included RLS patients who were diagnosed by expert neurologists, yet the 23andMe and INTERVAL RLS GWAS data sets included participants based on self-report, potentially diluting the GWAS accuracy. In addition, this study focused on individuals of European ancestry. Studies from multiple ethnicities are required to further study PD, RLS, and the association between them. Of note, the underlying genetics explains only a portion of the variance in PD and RLS, and it is possible that pathways not influenced by genetics may underlie some of the variance in these 2 conditions. It is possible that rare or structural variants outside what can be detected with current GWAS technologies are contributing to a shared genetic etiology.
In light of the current and previous findings, it is likely that confounding factors such as treatment, closer neurological follow-up, and others may have contributed to the observed epidemiological association between RLS and PD. Although additional studies are required to identify these potential confounders, the observed association between RLS and PD should not be considered causal on current evidence.
Supplementary Material
Acknowledgments:
We thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). We also thank the research participants and employees of 23andMe for making this work possible. For a complete overview of members, acknowledgements, and funding, please see http://pdgenetics.org/partners. M.A.E. is funded by the Fonds de Recherche du Québec-Santé (FRQS). G.A.R. holds a Canada Research Chair (Tier 1) in Genetics of the Nervous System and the Wilder Penfield Chair in Neurosciences. Z.G.O. is supported by the Fonds de Recherche du Québec-Santé Chercheur-Boursier award and is a Parkinson Canada New Investigator awardee. We thank the International EU-RLS-GENE consortium (https://www.helmholtz-muenchen.de/ing/rls-gene-consortium/memhers/index.html ) for providing RLS GWAS summary statistics. The International EU-RLS-GENE consortium includes data from the COR study, which was supported by unrestricted grants to the University of Münster from the German Restless Legs Patient Organisation (RLS Deutsche Restless Legs Vereinigung), the Swiss RLS Patient Association (Schweizerische Restless Legs Selbsthilfegruppe), and a consortium formed by Boeringer Ingelheim Pharma, Mundipharma Research, Neurobiotec, Roche Pharma, UCB (Germany + Switzerland), and Vifor Pharma. The clinical material and biospecimens of the Mayo Clinic Florida RLS collection were collected with the assistance of the Mayo Clinic internal funding through the Neuroscience Focused Research Team grant, Genotyping of the International EU-RLS-GENE consortium data set was supported by DFG grant 218143125 to Professor Juliane Winkelmann. Participants in the INTERVAL randomized, controlled trial were recruited with the active collaboration of NHS Blood and Transplant England (www.nhsbt.nhs.uk), which has supported fieldwork and other elements of the trial. DNA extraction and genotyping were cofunded by the National Institute for Health Research (NIHR), the NIHR BioResource (http://bioresource.nihr.ac.uk/), and the NIHR (Cambridge Biomedical Research (Centre at the Cambridge University Hospitals NHS Foundation Trust).* The academic coordinating center for INTERVAL was supported by core funding from NIHR Blood and Transplant Research Unit in Donor Health and Genomics (NIHR BTRU-2014–10024), UK Medical Research Council (MR/L003120/1), British Heart Foundation (SP/09/002; RG/13/13/30194; RG/18/13/33946), and the NIHR (Cambridge Biomedical Research (Centre at the Cambridge University Hospitals NHS Foundation Trust]).* A complete list of the investigators and contributors to the INTERVAL trial is provided in reference 31. The academic coordinating center thanks blood donor center staff and blood donors for participating in the INTERVAL trial and Dr Brendan Burchell (University of Cambridge) for advice on RLS phenotyping. *The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care. This work was supported in part by the Intramural Research Program of the National Institute on Aging and National Institute of Neurological Disorders and Stroke (project number Z01-AG000949–02).
Funding agencies:
M.A.E. is funded by the Fonds de Recherche du Québec-Santé (FRQS). This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund, awarded to McGill University tor the Healthy Brains for Healthy Lives initiative granted to M.A.E. This study was funded by a CIHR Foundation grant granted to G.A.R. G.A.R. holds a Canada Research Chair (Tier 1) in Genetics of the Nervous System and the Wilder Penfield Chair in Neurosciences. Z.G.O. is supported by the Fonds de recherche du Québec-Santé Chercheur-Boursier award and is a Parkinson Canada New Investigator awardee.
Footnotes
Relevant conflicts of interest/financial disclosures: There is no conflict of interest.
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord 2014;29(13):1583–1590. [DOI] [PubMed] [Google Scholar]
- 2.Ohayon MM, O’Hara R, Vitiello MV. Epidemiology of restless legs syndrome: a synthesis of the literature. Sleep Med Rev 2012;16(4):283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alonso-Navarro H, García-Martín E, Agúndez JA, Jiménez-Jiménez FJ. Association between restless legs syndrome and other movement disorders. Neurology 2019;92(20):948–964. [DOI] [PubMed] [Google Scholar]
- 4.Gao X, Schwarzschild MA, O’Reilly EJ, Wang H, Ascherio A. Restless legs syndrome and Parkinson’s disease in men. Mov Disord 2010;25(15):2654–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong JC, Li Y, Schwarzschild MA, Ascherio A, Gao X. Restless legs syndrome: an early clinical feature of Parkinson disease in men. Sleep 2014;37(2):369–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szatmari S Jr, Bereczki D, Fornadi K, Kalantar-Zadeh K, Kovesdy CP, Molnar MZ. Association of restless legs syndrome with incident Parkinson’s disease. Sleep 2017;40(2):zsw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agúndez JA. Neurochemical features of idiopathic restless legs syndrome. Sleep Med Rev 2019;45:70–87. [DOI] [PubMed] [Google Scholar]
- 8.Ryu JH, Lee MS, Baik JS. Sonographic abnormalities in idiopathic restless legs syndrome (RLS) and RLS in Parkinson’s disease. Parkinsonism Relat Disord 2011;17(3):201–203. [DOI] [PubMed] [Google Scholar]
- 9.Alberts J, Adler CH, Saling M, Stclmach G. Prehension patterns in restless legs syndrome patients. Parkinsonism Relat Disord 2001;7(2):143–148. [DOI] [PubMed] [Google Scholar]
- 10.Ferini-Stramhi L, Carli G, Casoni F, Galbiati A. Restless legs syndrome and Parkinson disease: a causal relationship between the two disorders? Front Neurol 2018;9:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kia DA, Noyce AJ, White J, et al. Mendelian randomization study shows no causal relationship between circulating urate levels and Parkinson’s disease. Ann Neurol 2018;84(2):191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schormair B, Zhao C, Bell S, et al. Identification of novel risk loci for restless legs syndrome in genome-wide association studies in individuals of European ancestry: a meta-analysis. Lancet Neurol 2017;16(11):898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18(12):1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgess S Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol 2014;43(3):922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011;40(3):755–764. [DOI] [PubMed] [Google Scholar]
- 16.Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods 2019;10(4):486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verbanck M, Chen C-y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50(5):693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Machiela MJ, Chanock SJ. LDIink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015;31(21)3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulik-Sullivan BK, Loh P-R, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015;47(3):291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47(11):1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohtashami S, He Q, Ruskey JA, et al. TOX3 variants are involved in restless legs syndrome and Parkinson’s disease with opposite effects. J Mol Neurosci 2018;64(3):341–345. [DOI] [PubMed] [Google Scholar]
- 22.Pittock SJ, Parrett T, Adler CH, Parisi JE, Dickson DW, Ahlskog JE. Neuropathology of primary restless leg syndrome: absence of specific τ-and α-synuclein pathology. Mov Disord 2004;19(6):695–699. [DOI] [PubMed] [Google Scholar]
- 23.Connor JR, Wang X-S, Allen RP, et al. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain 2009;132(9):2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Connor JR, Boyer P, Menzies S, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 2003;61(3):304–309. [DOI] [PubMed] [Google Scholar]
- 25.Connor JR, Wang X, Patton S, et al. Decreased transferrin receptor expression by neuromelanin cells in restless legs syndrome. Neurology 2004;62(9):1563–1567. [DOI] [PubMed] [Google Scholar]
- 26.Gan-Or Z, Alcalay RN, Bar-Shira A, et al. Genetic markers of restless legs syndrome in Parkinson disease. Parkinsonism Relat Disord 2015;21(6):582–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilariño-Güell C, Soto A, Young J, et al. Susceptibility genes for restless legs syndrome are not associated with Parkinson disease. Neurology 2008;71(3):222–223. [DOI] [PubMed] [Google Scholar]
- 28.Adel S, Djarmati A, Kabakci K, et al. Co-occurrence of restless legs syndrome and Parkin mutations in two families. Mov Disord 2006; 21(2)358–263. [DOI] [PubMed] [Google Scholar]
- 29.Tan EK, Yew K, Chua E, et al. PINK1 mutations in sporadic early-onset Parkinson’s disease. Mov Disord 2006;21(6):789–793. [DOI] [PubMed] [Google Scholar]
- 30.Lahut S, Vadasz D, Depboylu C, et al. The PD-associated alpha-synuclein promoter Rep1 allele 2 shows diminished frequency in restless legs syndrome. Neurogenetics 2014;15(3):189–192. [DOI] [PubMed] [Google Scholar]
- 31.Di Angelantonio E, Thompson SG, Kaptoge S, et al. Efficiency and safety of varying the frequency of whole blood donation (INTERVAL): a randomised trial of 45 000 donors. Lancet 2017;390(10110):2360–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.