

ABSTRACT
How proteins move through space and time is a fundamental question in biology. While great strides have been made toward a mechanistic understanding of protein movement, many questions remain. We discuss the biological implications of motion in the context of the peptidoglycan (PG) synthesis machines. We reviewed systems in several bacteria, including Escherichia coli, Bacillus subtilis, and Streptococcus pneumoniae, and present a comprehensive view of our current knowledge regarding movement dynamics. Discrepancies are also addressed because “one size does not fit all”. For bacteria to divide, new PG is synthesized and incorporated into the growing cell wall by complex multiprotein nanomachines consisting of PG synthases (transglycosylases [TG] and/or transpeptidases [TP]) as well as a variety of regulators and cytoskeletal factors. Advances in imaging capabilities and labeling methods have revealed that these machines are not static but rather circumferentially transit the cell via directed motion perpendicular to the long axis of model rod-shaped bacteria such as E. coli and B. subtilis. The enzymatic activity of the TG:TPs drives motion in some species while motion is mediated by FtsZ treadmilling in others. In addition, both directed and diffusive motion of the PG synthases have been observed using single-particle tracking technology. Here, we examined the biological role of diffusion regarding transit. Lastly, findings regarding the monofunctional transglycosylases (RodA and FtsW) as well as the Class A PG synthases are discussed. This minireview serves to showcase recent advances, broach mechanistic unknowns, and stimulate future areas of study.
KEYWORDS: cell wall, peptidoglycan, PG synthases, circumferential motion, SEDs enzymes, movement dynamics, morphogenic complex, cell wall, circumferential movement, diffusion, directed motion, peptidoglycan synthases, septal machine, treadmilling
INTRODUCTION
The intracellular environment of a bacterial cell is dynamic, with various events occurring on timescales ranging from fractions of milliseconds (transient interactions) to several minutes (chromosomal replication). While some processes, e.g., spore formation, occur at distinct stages of the life cycle, many occur simultaneously with minimal physical or temporal separation. Moreover, the typical bacterial cell possesses millions of copies of diverse proteins, tens of thousands of ribosomes, and an ∼5.0 Mbp chromosome, all contained within an ∼3.0 μm by 0.8 μm cell body (1). A large amount of biological material and relatively small size of the cell results in macromolecular crowding, where 20 to 30% of the total intracellular volume is occupied (1–3). Confinement and compaction of these contents exert considerable outward force (4–7). The main “load-bearing” structure of the bacterial cell is peptidoglycan (PG) or cell wall, a mesh-like macromolecular structure that encases the cell, protecting from turgor pressure and lysis (8–11). For most bacteria, growth requires that new PG be inserted into the existing cell wall (12–16). This process is carried out by tightly regulated, complex multiprotein machines, mainly consisting of PG synthases (transglycosylases [TG] and/or transpeptidases [TP]) as well as regulators and cytoskeletal proteins (12–14). Importantly, these nanomachines are not static, but rather dynamically transit the crowded, cellular environment during PG synthesis and cell division.
Here, we reviewed PG synthesis in the context of motion. We discuss advances in microscopy and single-molecule tracking (SMT) that have greatly increased our ability to capture, quantify, and understand movement (directed versus diffusive) of single molecules as well as large treadmilling polymeric filaments. We address the biological implications of directed motion and dissect the role of diffusion within the cell. Lastly, we discuss pivotal findings concerning the bifunctional class A PG synthases and newly discovered monofunctional transglycosylases, RodA and FtsW. We drew from several model organisms (Escherichia coli, Bacillus subtilis, Streptococcus pneumoniae, and Staphylococcus aureus) to establish a comprehensive understanding of motion regarding the complex processes of PG synthesis and cell division. In addition, we address techniques to detect and quantify molecules in motion.
UNDERSTANDING THE SYSTEM: MECHANISMS OF PG SYNTHESIS AND CELL DIVISION
Different species of bacteria display diverse morphologies ranging from simple cocci to six-pointed stars. Diversity in cell shape is thought to aid bacteria in occupying specific environmental niches (8, 9, 17). Cell shape is an inherited trait, imparted by PG, with distinct morphologies being faithfully reproduced through generations (8, 9, 17). PG is comprised of long glycan chains cross-linked by flexible peptide bonds (12, 18, 19). Alternating units of β-1-4-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) form the glycan chains with slight variations (chain length and special modifications) existing between species (12, 20). Attached to MurNAc is the stem peptide, which is synthesized as a pentapeptide containing l- and d-amino acids and a dibasic amino acid that facilitates cross-linking (12, 20). Common cross-linkers include mesodiaminopimelic acid (Gram-negative bacteria) and l-lysine (Gram-positive bacteria) (12). PG layers likely align anti-parallel to the cell body and range from 1 to 3 nm (Gram-negative) to 60 nm (Gram-positive) (12, 21). These interconnected layers of glycan chains and peptide bonds encase the full cell body forming the sacculus, imparting shape as well as protection from lysis due to turgor pressure (8, 9, 12, 21). The pore size of PG is estimated to be around 2 nm (22). Because bacteria of diverse shapes share similar PG scaffolds, the building blocks (disaccharide-pentapeptides) are unlikely determinants of cell shape (8, 9). Rather, variations in mechanisms of PG synthesis such as accurately determining the correct location to insert new PG along the cell body, likely give rise to morphological diversity (8, 9).
PG synthesis is divided into three stages: (i) precursor biosynthesis, (ii) export of lipid II, and (iii) insertion via transglycosylation and transpeptidation (Fig. 1) (12, 20). Briefly, biosynthesis (step 1) of lipid II (substrate of the machines) occurs in the cytoplasm by way of an enzymatic reaction cascade (12, 20, 23). Disruption of the cascade alters PG precursor pools within the cell and can be exploited to investigate downstream PG synthesis mechanisms (24). Lipid II must be exported out of the cytoplasm because the machine’s membrane-bound PG synthases function outside the cell (Gram-positive) or within the periplasmic space (Gram-negative) (12, 14, 16). In step 2, lipid II is transported across the membrane by a “flippase,” typically MurJ. Alternative flippases, such as Amj of B. subtilis, also exist (25–30). Export is essential as MurJ-deficient cells accrue a surplus of PG precursors in the cytoplasm, fail to divide, and subsequently lyse (27, 29, 31, 32). Once exported, the disaccharide-pentapeptide of lipid II is incorporated into the existing sacculus by the PG synthesis machines (step 3) (Fig. 1) (12). Typically, bacteria that divide by binary fission possess two distinct PG synthesis machines which are highly conserved and distributed across bacterial species (Table 1 and Fig. 1) (6, 12–16). For continuity and simplicity, the divisome and elongasome (elongase, rod-system, peripheral machine, or PGEM) will be referred to as the septal and elongation machines, respectively.
FIG 1.

Peptidoglycan is inserted in the existing sacculus by distinct multiprotein machines. This process is divided into three steps: precursor biosynthesis, export of lipid II, and insertion into the existing sacculus via transglycosylation (TG) and transpeptidation (TP). (i) Biosynthesis of lipid II occurs in the cytoplasm and is facilitated by a sequence of biochemical reactions discussed elsewhere (1, 12, 114). (ii) Export of lipid II is mediated by MurJ, known as a “flippase,” which translocates lipid II across the cytoplasmic membrane. (iii) Incorporation of lipid II’s disaccharide-pentapeptide into the existing sacculus is carried out by transglycosylation (TG) and transpeptidation (TP) reactions of the PG synthesis machines. The septal machine accomplishes cell division, and the elongation machine mediates lateral growth. While insertion is carried out by the TG:TP reactions of the machine’s PG synthases, incorporation does not occur in the absence of other key machine components. The septal machine is comprised of >30 proteins while the elongation machine consists of approximately 5 to 8 proteins, to date. Division and elongation can be temporally separated as in the “two-competing-sites” model or occur simultaneously as in S. pneumoniae.
TABLE 1.
Conserved components of the peptidoglycan synthesis machines of key model organismsj
| Machine | Protein | Canonical functiona |
Ecob rod, G − |
Bsu rod, G + |
Cau crescent-rod, G − |
Spn ovococcal, G + |
Sau cocci, G + |
Ct cocci, G − |
|---|---|---|---|---|---|---|---|---|
| Septal machine components | Class B PBP | Monofunctional transpeptidase (TP) | + | + | + | + | + | + |
| FtsW | Monofunctional transglycosylase (TG) | + | + | + | + | + | + | |
| FtsA | Actin-like, Z-ring tether | + | + | + | + | + | − | |
| FtsZ | Z-ring, division orchestrator | + | + | + | + | + | − | |
| FtsK | Division regulator, DNA pump | + | + | + | + | + | + | |
| EzrA | Z-ring regulator | − | + | − | + | + | − | |
| FtsQLB | Forms FtsQ, FtsL, and FtsB complex and division regulatorb | + | +c | + | + | ?d | + | |
| FtsN | Division regulator | + | − | + | − | − | − | |
| DivIVA | Division regulator | − | + | − | + | + | − | |
| FtsX | Regulates cell wall hydrolases | + | +e | + | + | ? | − | |
| FtsE | Regulates cell wall hydrolases | + | +e | + | + | ?f | − | |
| ZapA | Z-ring stabilizer | + | + | + | + | + | − | |
| Elongation machine | Class B PBP | Monofunctional TP | + | + | ? | + | ? | + |
| RodA | Monofunctional TG | + | + | + | + | + | + | |
| MreB | Morphogenic, elongation orchestrator | + | + | + | − | − | + | |
| MreC | Morphogenic protein | + | + | + | + | + | + | |
| MreD | Morphogenic protein | + | + | + | + | + | ?g | |
| RodZ | Morphogenic protein, MreB-filament stabilizer |
+ | + | + | + | +h | + | |
| Machine independent and/or unknown | Class A PBPs | Bifunctional TP and TG | + | + | + | + | + | ? |
| GpsB | Cell wall regulator, associated with Class A PBPs | − | + | − | + | + | − | |
| StkP | PG synthesis associated kinase | − | + | − | + | + | − | |
| PhpP | Cell wall-associated phosphatase | − | ? | ? | + | + | − | |
| MurJ | “Flippase” that translocates lipid II across the cytoplasmic membrane | + | + | + | + | +i | + |
The most predominant function of the various homologs.
Eco, E. coli; Bsu, B. subtilis; Cau, C. crescentus; Spn, S. pneumoniae; Sau, S. aureus; Ct, C. trachomatis; G +, Gram positive; G −, Gram negative.
Known as FtsB (DivIB), FtsL, and DivIC in B. subtilis.
To date, FORC090_RS06470 (FtsQ), and FORC090_RS06450 (FtsL) were detected bioinformatically in S. aureus, but FtsB was not (112). Protein detected that possesses serine/threonine kinase domain; however, the PASTA (penicillin-binding protein and serine/threonine kinase-associated) domain is not conserved (13).
FtsEX is involved in cell elongation in B. subtilis (113).
FORC090_RS03700 encodes FtsX-like permease family protein in S. aureus; however, FtsE-like protein is not detected bioinformatically (112).
MreD homolog annotated in C. muridarum, while an unannotated MreD homolog is detected in C. trachomatis in Chlambase (108).
Present in Staphylococcus but species not specified (111).
Evidence suggests SAV1754 is the MurJ homolog in S. aureus (110).
For over 30 years, researchers have exploited genetics and chemical perturbations to uncover the individual components and functions of the PG synthesis machines (12–16, 33). The septal machine, comprised of >30 proteins, mediates septation and is largely directed by the tubulin homolog and cytoskeletal protein, FtsZ (Table 1 and Fig. 1). Elongation or lateral growth is accomplished by the elongation machine, which is largely coordinated by the actin homolog and cytoskeletal element MreB (Table 1). Within the respective machines, glycan polymerization is carried out by specific PG transglycosylases (TG) such as FtsW (septal) or RodA (elongation), while cross-linking occurs via the transpeptidases (TP) including FtsI-/PBP3 (septal) and PBP2 (elongation) (24, 34–36). Components of the separate machines exhibit distinct movement trajectories that correlate with their respective PG insertion patterns (24, 37–40). In cell division, the septal machine is restricted to midcell, while during growth the elongation machine transits the length of the cell perpendicular to the long axis of rod-shaped bacteria (24, 39–43). Substrate (lipid II) availability is a key determinant of machine processivity. At present, the length of the glycan chain (derived from lipid II subunits) inserted during one processive run of the machine remains elusive. Estimations of E. coli’s septal TG:TP (FtsW:FtsI) calculate that ∼6 to 14 disaccharides can be inserted per second because one disaccharide measures ∼1 nm (41, 44).
In considering biosynthesis and insertion, a spatiotemporal conundrum arises in that nucleotide-PG precursors reside in the cytoplasm while the machine’s TG:TP enzymes work in the periplasmic space (Gram negative) or extracellularly (Gram positive). How do lipid II and machine arrive at the precise location of insertion in time and space? If lipid II is not present, hydrolyzed gaps within the sacculus will remain unfilled, and the crowded cytoplasm will exert force onto the compromised sacculus, threatening structural collapse (1, 3). Furthermore, unequal osmotic gradients between the cell body and the external environment stress the sacculus inward from the outside. The side walls of a rod-shaped bacterium experience twice the mechanical stress than that of the poles (4–6). Gaps must be filled promptly to avoid structural collapse. Thus, by what mechanism(s) might exported lipid II and machine encounter one another? This spatiotemporal conundrum is discussed in greater detail later. At present, we will explore molecular motion and the machines’ movement patterns.
TRACKING AND QUANTIFYING MOLECULAR MOVEMENT PATTERNS
Here, we discuss the molecular movement patterns of directed motion, diffusive motion, and treadmilling, as well as means to represent and quantify movement. Motion is defined as an occurrence in which an object changes its position over time. It can be mathematically described in terms of time, velocity, and displacement. Movement patterns of an object can be clear and distinct (directed and treadmilling), as well as rapid and stochastic (diffusive) (1–3, 7). For this review, directed motion is defined as a clear linear path of an object moving from point A to point B over time. Energy is required for directed motion and directionality can be attributed to an object’s path (1, 2, 7). As illustrated in Fig. 2A, the green dot (representing a fluorescent protein) moves from the middle to the bottom of the cell during a 180 s time frame. When ascertaining the best approach to capture a novel object’s motion, (e.g., MurJ), time is a problematic variable. On one hand, choosing an imaging time frame that is too short results in an object appearing static because its movement occurs outside the applied time frame. In Fig. 2A, the green dot appears static if tracked using a 10 s time frame. On the other hand, long imaging frames are incapable of capturing proteins displaying rapid, stochastic movement such as diffusive motion. Therefore, it is best to empirically apply various imaging and video run times.
FIG 2.

Methods of representing and quantifying molecular movement patterns. Illustration of the cell body (gray with black outline) and single fluorescent foci (green dots) imaged using high-resolution microscopy. (A) Directed motion over 180 s and resulting kymograph showing focus trajectory (green) against cell body background (black for contrast). Kymographs can be generated using ImageJ and represent motion in 2D by stacking video pixels chronologically. For quantification, the velocity of the molecule displaying directed motion can be delineated by using the equation shown (nm = nanometer). (B) Diffusive motion captured using “fast” acquisition rate and resulting kymograph. Observe that kymographs are insufficient in representing diffusion because diffusive molecules do not form linear trajectories and often leave the plane of vision. Loss of visualization is illustrated as a blank cell in T = 60 and black gaps in kymographs. For quantification, the mean squared displacement (MSD) can be determined and plotted from raw image files using ImageJ. Analyzing MSDs reveals diffusion coefficients (diffusive molecules) as well as velocities (directed motion). (C) Single molecule within a treadmilling filament. Here, the molecule’s zero change in distance over time results in a straight-line trajectory. As expected, calculating the velocity using tanΦ of zero, results in V = zero, thereby adding mathematical confidence to the observed stagnation. (D) Directional movement of a treadmilling polymeric cytoskeletal filament (green arrow). A filament’s treadmilling rate is easily extrapolated by solving for V (velocity) or plotting the MSD.
Widefield, confocal, and total internal reflection microscopy (TIRm) can detect single molecules within a bacterial cell (Fig. 3A and B) (2, 7, 45–48). For SMT experiments, the field of view must not be saturated with multiple copies of the molecule (1, 2). Fluorochromes, such as GFP, emit visible light when excited by a specific wavelength of light. As a result, all GFP molecules within a field fluoresce when excited, thereby saturating the system with multiple copies of fluorescent molecules (49, 50). Classically, to achieve single-molecule imaging conditions, exogenous plasmids are used to titrate expression levels of fluorochrome fusion proteins (1, 2). The development of Halo-tag technology has revolutionized SMT (51, 52). Unlike GFP, Halo-tag proteins only fluoresce if appropriately excited and covalently bound by an exogenously added Halo-tag specific ligand (51, 52). Interaction between the Halo-tag and ligand is covalent and occurs in a 1:1 ratio with ligand titration allowing for SMT (51, 52). With either approach, plasmid expression or ligand titration, SMT experiments require sparse conditions where 1 to 10 fluorescent molecules occupy the cell (1, 2). Otherwise, it is difficult to accurately track movement patterns as fluorescent signals of multiple molecules overlap, making one molecule’s path indistinguishable from another (1, 2). Fluorescence microscopy coupled with saturating conditions, where as many molecules as possible fluoresce, is a powerful tool to visualize global protein localization patterns as well as large polymeric structures such as the FtsZ ring (Fig. 3C and D). Speaking to the versatility of Halotags, both sparse and saturating labeling is achieved by modulating ligand amount before imaging (51, 52).
FIG 3.

Achieving single-molecule imaging. Representative illustrations of bacterial cells that were imaged using confocal, widefield, total internal reflection microscopy (TIRFm), and fluorescence microscopy. Cell symbolized as in Fig. 2 with green dots representing monomeric units of FtsZ-Halo-tag protein. (A) Illustration showing imaging power of pairing confocal or widefield fluorescence microscopy with sparse labeling conditions. Theoretically, sparse labeling aims to label one molecule per cell to achieve single-molecule imaging. In practice, less than 10 molecules per cell are required to accurately track the movement dynamics of single molecules. This is achieved by modulating the amount of dye/ligand added to the system (halo-tag fusions) or titrating expression levels (GFP fusions) via plasmids with inducible promoters. (B) Illustration of TIRFm paired with sparse labeling conditions. With TIRFm, only molecules within the evanescent field (200 nm at the cell’s surface) are excited and therefore fluoresce (symbolized as a green dot within the evanescent field [blue box] and white nonfluorescent dot outside the field) (115). (C) Illustration of Z-ring captured using fluorescence microscopy and saturated labeling conditions. Contrary to sparse conditions, saturating aims to label all molecules. Here, the Z-ring appears as a distinct band at midcell as all molecules within the cell are excited and fluoresce. (C and D) While it is theoretically improbable that “all” fluorochrome-fusions are labeled with an exogenous dye (halo-tag ligand) and fluoresce, saturating conditions aim to flood the system and label as many molecules as possible. If fluorescent fluorochrome-fusions (GFP) are used, then expression levels are not lessened, again aiming to visualize as many fluorescent molecules as possible. Saturating conditions allow for the detection of polymeric structures (Z-ring) and/or visualization of a protein’s global localization pattern(s). (D) Coupling saturating conditions with confocal or widefield fluorescence microscopy increases the signal to background ratio, thereby greatly increasing an image’s contrast. As a result, the Z-ring appears as a distinct ring when imaged with confocal or widefield under saturating conditions.
Once captured, pixel data contained in the movie are stacked through time to construct a kymograph, allowing one to visualize and represent motion two-dimensionally (Fig. 2). Kymographs are sound, reproducible data formats from which concrete numerical values such as velocity can be extrapolated. Consider a bound flipbook that, when flipped, displays a stick figure walking from the left to the right side of the page, transiting from point A to point B. In a kymograph, the individual pages of the flipbook are torn out and stacked linearly in chronological order, forming a 2D trajectory of the stick figure’s movement. Mathematically, linear change in an object’s position in time results in a slope, which can be fitted to a right angle. Thereby, the velocity of an object displaying directed motion can be extrapolated using tanΦ of the right angle delineated from the kymograph if the pixel size is known. Kymographs can be generated using ImageJ plugins (53). Another way to represent directed motion is by constructing a montage where individually captured images (still frames) are positioned in chronological order next to one another (Fig. 2). Montages do not require ImageJ and can be constructed in pixel-preserving applications such as Photoshop or Illustrator. Kymographs and montages are powerful approaches to illustrate directed motion two-dimensionally (24, 35, 39, 40, 42). However, kymographs are insufficient in representing diffusion as its motion is rapid and stochastic as observed in live-cell imaging (1, 2, 7).
The environment of the bacterial cell serves as a medium for diffusion. By harnessing the thermal energy of the environment (random collisions with other molecules) proteins can transit from one end of the cell to the other (∼1 μm) without the use of molecular motors or ATP consumption (1, 7). Protein encounters, which determine the kinetics of machine formation and subsequent applications, rely on diffusion (1, 7). Because molecule size, environmental viscosity, and macromolecular crowding directly influence diffusion, the rate of diffusion can be the limiting step of certain reactions (1, 3, 7). Osmotic upshifts, which drastically increase the crowding of cellular components, decrease the rate of diffusion (1, 3). Protein concentrations in the cytoplasm of E. coli are typically ∼200 to 300 g/liter, whereas osmotic upshifts resulting in water loss can lead to concentrations upwards of ∼400 g/liter (54). Within the cytoplasm, the diffusion coefficient of free GFP molecules is ∼7.7 μm2/s, whereas in vitro GFP diffuses at 87 μm2/s. Thus, GFP transits 11-fold slower in vivo than in vitro due to macromolecular crowding (55). Nonetheless, the rate of diffusive motion is well above that of energy-dependent, enzymatic-driven processes. During PG synthesis, the enzymatically active septal TG FtsW transits the septum via directed motion 15 to 20-fold slower than inactive, diffusive FtsW molecules transiting outside the PG synthesis zone in S. pneumoniae cells (24, 43). Thus, diffusion is a rapid, net zero-cost mechanism of protein movement through the intracellular environment (1, 7).
Because the cell body geometrically confines a molecule’s diffusion to the cell’s volume, molecules exhibit confined diffusion when tracked via live-cell imaging (1). Due to the intrinsic properties of diffusion, it is best captured using fast image acquisition rates, such as ∼20 frames per second (FPS) (43). For comparison, an image acquisition rate of 1 FPS is sufficient to capture the directed motion of the PG synthases (24). Diffusion can be visualized using video compilations and image montages. Because diffusive motion does not take a linear path, measuring the total distance traveled is not accurate, rather its net displacement should be calculated (1, 7). Displacement is a vector quantity and is typically extrapolated by calculating the mean squared displacement (MSD), where the trajectory of the object is analyzed in terms of net displacement between lag times (56). MSDs can be used to delineate diffusion coefficients as well as velocities of directed motion (1, 56). Plotting the MSD versus time reveals whether molecules exhibit Brownian or non-Brownian directed motion (1, 7, 57). Minimal coding coupled with ImageJ can be used to delineate the MSD from raw imaging files (53). For plotted MSDs and montages generated from biological samples (43, 58, 59).
In addition to the directed and diffusive movement of single molecules, treadmilling of large polymeric filaments, such as FtsZ filaments, is another means of intracellular transit. Here, individual molecules within a filament are static, yet the net movement of a filament occurs about the cell due to polymerization (positive-end) and depolymerization (negative-end) events at the respective ends of the filament (Fig. 4) (14, 60–62). For instance, in SMT, individual FtsZ molecules are static and, thus, generate kymographs that display a straight line as no change in distance over time occurred (24). Conversely, zooming out and imaging the polymeric filament reveals distinct, directional motion from which the filament’s velocity can be extrapolated (24, 40–42). In sum, treadmilling is active movement, utilized by polymeric filaments to transit the cell.
FIG 4.
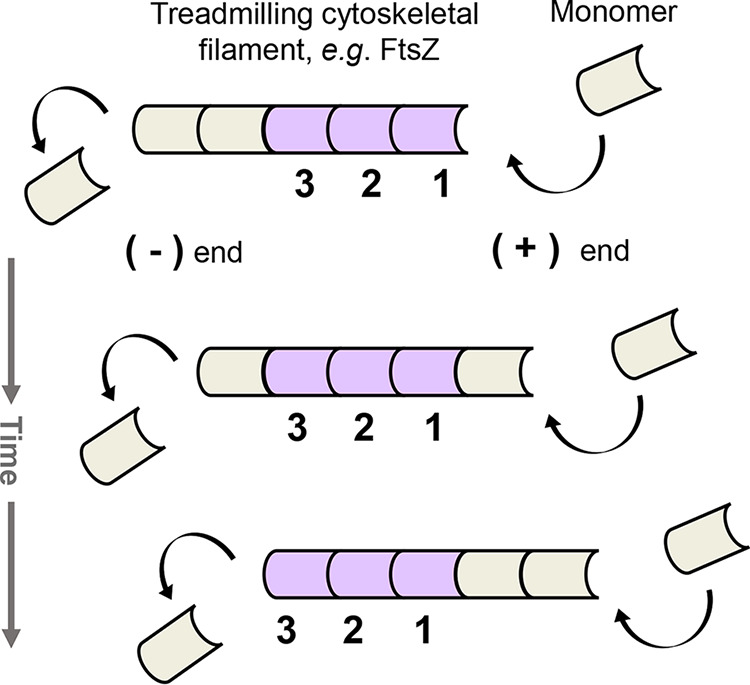
Treadmilling as a means of motion. The illustration shows the net movement of filament to the right. To keep track, single monomeric units are represented as either lilac or tan. Treadmilling is active motion and requires an energy source such as GTP hydrolysis. In treadmilling, individual cytoskeletal monomers within the filament are stationary. However, net filament movement occurs due to reoccurring polymerization and depolymerization events at the respective positive (+) and negative (−) ends of the filament. Filament growth requires unequal rates of polymerization and depolymerization and is dependent on the cytoplasmic concentrations of cytoskeletal monomer and substrate, such as GTP. It is unknown what triggers the initial polymerization event of monomer to filament within the cell.
DRIVING CONSTRICTION EVENTS
Although components of the PG synthesis machines are well-established, we are just beginning to understand mechanisms of transit and timing. During division >30 proteins localize to the midcell, site of the future septum (12–14). Diffusion is thought to facilitate this stepwise migration (Table 1). At some point in time, FtsZ monomers in the cytoplasm form polymeric filaments at midcell where interactions with various membrane anchors and regulators modulate Z-ring formation and stability (14, 37, 63–65). The Z-ring is a discontinuous subcellular structure comprised of FtsZ filaments that trace the circumference of the cell (37, 63, 64, 66, 67). Within the Z-ring, FtsZ filaments align with one another forming filament bundles (66). Z-ring formation is well documented in rods (E. coli and B. subtilis), crescent-rods (C. crescentus), ovococci (S. pneumoniae), and cocci (S. aureus) (14, 24, 65, 68–70). Intriguingly, the obligate intracellular pathogen Chlamydia trachomatis (coccus) accomplishes FtsZ-independent division with MreB likely serving as an FtsZ surrogate (71–73). Nonetheless, the formation of the Z-ring marks the onset of division and orchestrates constriction in most bacteria (14, 61, 62).
The Z-ring itself is dynamic in that its FtsZ filaments bi-directionally transit the septum perpendicular to the long axis of the cell via treadmilling (24, 40, 43, 60, 74). Moreover, cytoskeletal filaments exist in a constant state of flux, stochastically assembling and disassembling. High-resolution microscopy has captured treadmilling FtsZ filaments/bundling in E. coli, B. subtilis, and S. pneumoniae (24, 40, 42). Treadmilling of FtsZ filaments is facilitated by the intrinsic guanosine triphosphatase (GTPase) activity of FtsZ. Here, FtsZ-GTP monomers are added to the (+) end of the filament while hydrolyzed FtsZ-GDP monomers disassociate at the (−) end (61, 62, 75). In vitro, FtsZ treadmilling ranges from 10 to 100 nm/s, whereas, in vivo in Gram-positive bacteria (B. subtilis and S. pneumoniae), treadmilling FtsZ filaments exhibit an average velocity of ∼32 nm/s with motion following a Gaussian distribution (24, 37, 67). Disruption of the GTPase activity of FtsZ via mutagenesis or chemical perturbation halts motion (24, 40, 42). Thus, multiple bacterial species display a large structurally discontinuous, bidirectionally fluid ring comprised of FtsZ filaments that consumes energy. What purpose does this intrinsically transient and dynamic ring serve?
Work in B. subtilis demonstrates that treadmilling FtsZ filaments drive septal PG synthesis and thereby constriction (40). The use of high-resolution microscopy with sequential FDAA (fluorescent d-amino acids) pulse-labeling demonstrated that the septum gradually decreases in size inward from the cell surface as new PG is synthesized (40). Exploiting different pulse times showed that PG synthesis occurs at discrete sites at the midcell and that these sites increase in area over time. Tracking experiments show that FtsZ, FtsA, and the PG synthase PBP2B transit the cell at similar velocities, ∼32 nm/s (40). In B. subtilis, FtsZ and the membrane-associated actin homolog FtsA, form filaments on the cytoplasmic face of the inner membrane. FtsA is highly conserved (Table 1), possesses ATPase activity, and influences Z-ring formation (14, 37, 63, 68, 69, 76). Overexpression of GTPase-deficient FtsZ halts directed motion of PBP2b (PG synthase), implicating FtsZ treadmilling as the source of motion (40). Meanwhile, FtsZ treadmilling is unaltered by depletion of PBP2b as well as exposure to the PG synthase-inhibiting antibiotic, penicillin G (40). Thus, in B. subtilis FtsZ treadmilling directs the progressive insertion of PG at the septum by building increasingly smaller rings that ultimately divide the cell.
While it seems that B. subtilis provides an elegant model that can be superimposed onto binary-fission, FtsZ-dependent bacteria, “one-size-does-not-fit-all” and distinct processes power constriction across bacterial species. In S. pneumoniae constriction is independent of FtsZ treadmilling and is driven by the TG:TP reactions of the septal machine (24). The directed motion of the septal monofunctional PG synthases (FtsW and PBP2x) occur at velocities of ∼19 to 21 nm/s where treadmilling FtsZ filaments transit at 32 nm/s at S. pneumoniae’s midcell (24). Decreasing PG precursor pools reduces FtsW and PBP2x velocity to 14 nm/s whereas treadmilling FtsZ filaments remained constant (24). As in B. subtilis, PG synthesis occurs at distinct “nodal” sites with ring closure resulting from the outward to inward addition of PG (40, 77). Yet, constriction is powered by different means in B. subtilis and S. pneumoniae, despite each possessing a thick PG sacculus. Of note, the role of FtsZ in Gram-negative E. coli will be discussed later. Gram-positive S. aureus demonstrates that energetic drivers of constriction are not mutually exclusive. Chemical perturbation and microscopy revealed that S. aureus exhibits bi-phasic division in which early constriction rings are FtsZ-treadmilling-dependent and subsequently switch to FtsZ-independent constriction that is then driven by PG synthesis (TG:TP) reactions. Moreover, it seems that this energetic switch is triggered by the arrival of MurJ at midcell (74). Thus, the FtsZ ring drives constriction in B. subtilis and seems to partially power closure in S. aureus. Irrespective of generating a constrictive force, Z-rings are required to secure homogenous, symmetric constriction events across an array of bacterial species (61, 62).
In S. pneumoniae (TG:TP-dependent), FtsZ treadmilling is theorized to serve as a subcellular scaffold, ensuring assembly of the septal machine at the appropriate location in space and time. Observe that the scaffolding function of FtsZ is not restricted to S. pneumoniae, but is likely widely distributed (61, 62). In S. pneumoniae, Z-ring scaffolds serve to support PG synthesis at the septa as well as mark the future division site of daughter cells (24). It remains to be determined whether support is physical (load-bearing structure) or instead regulatory (a cascade of hierarchical interactions). Because the Z-ring is discontinuous and dynamic, it seems probable that the Z-ring serves to mark the future septa, rather than acting as a physical subcellular, stable-architectural structure (61). This is supported in that rings of FtsZ, FtsA, and ErzA are the first to migrate from the mature septal ring to the site of nascent PG synthesis in daughter S. pneumoniae cells, thereby marking the future division site (24, 78). The active PG synthases (PBP2x and FtsW) remain at the constricting septum and are among the last to leave (24, 62, 79). While it is fascinating to envision these nanomachines (regulatory proteins, cytoskeletal filaments, and PG synthases) transiting the circumference of the cell, it leads to the question: how do other components stay clear of these massive machines?
CLASSIFICATION AND FUNCTIONS OF THE PG SYNTHASES
High molecular weight PG synthases are either bifunctional TP and TG, classically termed Class A penicillin-binding proteins (PBPs), or monofunctional TP, termed Class B PBPs (12, 20). For many years the bifunctional PG synthases were classified as machine components (12–14, 80). However, present data warrant reclassification of the Class A PG synthases role(s). The discovery of the monofunctional TGs, FtsW, and RodA, eliminates the Class A PBPs as the sole source of the machine’s TG reactions (34–36, 41). FtsW and RodA belong to the highly distributed SEDS (shape, elongation, division, and sporulation) family of proteins (34, 35). Biochemical and in vivo work show that FtsW and its septal monofunctional TP (E. coli PBP3/FtsI or S. pneumoniae PB2x) form a cognate, codependent pair capable of fulfilling the required TG-TP of the septal machine (24, 36, 41). For elongation, RodA pairs with the homologous monofunctional TP, e.g., E. coli PBP2 (81, 82). Thus, both machines no longer require a transglycosylating Class A PBP. Moreover, mutational analysis reveals that the Class A and B PBPs do not phenocopy one another. Typically, single deletion of a Class B PBP is lethal whereas the Class A PBPs are synthetically lethal, requiring double deletions (83–90). Additionally, single perturbation of Class A PBPs mildly alters cell shape, e.g., smaller diameter, while perturbing other elongation machine components results in overt morphological defects, e.g., enlarged spheres (83, 84, 90). Double perturbation of the Class A’s (specifically PBP1ab) leads to abrupt lysis, not spheres (87, 91). These discrepancies may be due to the genetic redundancy of class A PBPs or hint at similar yet distinct roles regarding cell wall integrity and shape.
In support of the latter, SMT revealed that the Class A PBPs display two distant movement patterns: diffusive and static. These patterns contrast with those of Class B whose movement patterns correlate with the insertion patterns of the respective machines (39, 40, 77). The directed circumferential motion of a Class A PBP has not been observed in B. subtilis or E. coli (35, 84, 92). Tracking PBP1 in B. subtilis showed that PBP1 exhibits static, nonmotile behavior and diffusive motion with a diffusion coefficient of ∼0.004 to 0.007 μm2/s (35). To date, the only processive circumferential motion has been correlated with PG synthesis, be it mediated by treadmilling or TG:TP reactions. However, evidence suggests that these nondirectional Class A PBPs are functional. In the context of the elongation machinery, mutational analyses (TP) and chemical perturbations (TG) showed that inactivation of the Class A PG synthase (PBP1b:E. coli) reduces cell wall synthesis levels by 20% whereas evidence suggests that the Class A PBPs insert 50% of the new PG at the septum (35, 41). Because diffusive motion is passive, it is highly unlikely that diffusive Class A PBP molecules are enzymatically active and synthesizing PG. It is possible that static Class A PBPs, e.g., B. subtilis:PBP1, represent enzymatic activity. One can envisage a “one and done” scenario in which PBP1 freely diffuses, docks, accomplishes one cross-linking or polymerization event, and then remains stalled and inactive at the insertion site until conditions favor its subsequent diffusion, such as exported lipid II or detected gaps/weakness in the sacculus. In this scenario, the independently functioning septal or elongation machines may accomplish longer runs of TG/TP activity, hence the machine’s measurable trajectory of synthesis. Without question, evidence is needed to address these speculations and the enzymatic activity of these static Class A PBPs.
Functionality studies strongly indicate that the Class A PG synthases function autonomously to modify existing PG laid by the respective machines (35, 84, 93). In S. pneumoniae, the use of enzyme-specific antibiotics and mutant backgrounds revealed that PG synthesized by the septal machine is further modified by Class A PG synthases (93). Class A-mediated modification of nascent PG is inhibited by oxacillin and moenomycin, strongly suggesting that both TG:TP activities of the Class A synthases are required for PG remodeling (93). Here, PG remodeling likely involves the Class A PG synthases further processing the PG meshwork synthesized by the machines, be it increasing the structural integrity of the sacculus by removing imperfections and/or increasing cell wall density (93). This is supported by recent discoveries in E. coli where the Class A PBPs were found essential to maintain the mechanical plasticity and integrity of the cell wall, yet fully dispensable regarding overall cell shape (84).
Unequivocally, the Class A PG synthases serve an essential function across bacterial species. Mounting evidence strongly indicates that Class A’s function is connected to, but regulated separately from, the PG synthesis machines (35, 86, 93). Considering motion, logically Class A’s function likely lies outside the machine. Otherwise, their movement patterns would match the circumferential motion of other machine components. It appears that the Class B PBPs work within the confines of the PG synthesis machines to form and maintain cell shape while the Class A PBPs work autonomously to sustain the structural integrity (repair) and mechanical plasticity of the sacculus. Theoretically, the machine’s Class B PBPs may lay the structural foundation of the septum while the Class A PBPs subsequently strengthen the structural integrity of the septum – owing to related, yet distinct roles (35, 84, 86, 93). Alternatively, the Class A PBPs may partner with their respective outer membrane lipoprotein (LpoA/B) partner and solely function as repair enzymes, again separate from the machines (35, 84, 94–97). Pinpointing the specific roles of the various PG synthases is important as antibiotic resistance often arises in genes encoding these genetically redundant yet functionally disparate enzymes (98).
NAVIGATING THE TOPOLOGICAL LANDSCAPE: MOVEMENT STRATEGIES OF ACTIVE VERSUS INACTIVE PG SYNTHASES
While a multitude of proteins arrive at midcell during division, not all molecules in a population incorporate into the assembling machines. Septal Class B PG synthases of S. pneumoniae and B. subtilis display diffusive movement outside the septa (24, 40). Comparing the movement dynamics of PG synthases to FDAA labeling patterns, a surrogate of TP activity indicates that these diffusive PG synthases are likely inactive. To reiterate, the Class B PG synthases (FtsW and PBP2x) of S. pneumoniae display two distinct movement patterns and velocities (24, 43, 77). One active population is slow, showing directed motion at the septa with a velocity of ∼19 to 21 nm/s and the other is fast, displaying diffusive motion outside the septa with a diffusion coefficient of 0.028 ± 0.0004 μm2/s (24, 43). This results in active PG synthases transiting the septa 15- to 20-fold slower than inactive PG synthases diffusing freely about the membrane. Mechanisms that partition the population into directed (active, slow) versus diffusive (inactive, fast) remain to be determined. Quantitative work in S. pneumoniae indicates that low expression of the elongation TP (PBP2b) is sufficient for normal growth and morphology, suggesting that key proteins involved in PG synthesis likely exist in excess (77). Maintaining a reservoir of essential cell wall enzymes is likely beneficial because the loss of cell wall integrity results in death. Given the relatively small size of a bacterial cell and the high rate of diffusion, an inactive PG synthase has the potential to arrive at the septa within seconds via diffusion (1).
SMT revealed that three distinct populations of transiting PG synthases exist in E. coli, one population of diffusive PG synthases and two populations of directionally transiting PG synthases at the septum, a slow (∼8 nm/s) active population, and a fast (∼30 nm/s) inactive population (41, 43). A combination of theoretical modeling and SMT determined that FtsZ treadmilling (20 to 40 nm/s in vivo) in E. coli drives the directional movement of inactive monofunctional PG synthases (FtsI) via a Brownian rachet mechanism, ultimately aiding in the spatiotemporal distribution of the PG synthases about the septum (41, 43). A Brownian rachet mechanism occurs when mechanical force and/or motion is produced in an anisotropic closed environment by nonequilibrium fluctuations of an isothermal medium (99–101). The fluctuating crowded heterogenous environment of the cell is primed for such exchanges of energy (1, 3).
FtsZ treadmilling events in the cytoplasm introduce a disequilibrium that biases the system toward the random diffusion of PG synthases in the membrane plane, upon which FtsI persistently trails the shrinking end of a treadmilling FtsZ filament. This results in the directional movement of FtsI with the PG synthase ratcheting “forward” ∼5 nm per event (43). However, the stochastic nature of Brownian Ratcheting makes the directional movement of inactive FtsI molecules probabilistic, rather than deterministic because FtsI may uncouple from the treadmilling filament at any moment (43). Importantly, the slow-moving (8 nm/s), enzymatically active population remains independent of FtsZ treadmilling but dependent on PG precursor pools and the TG:TP reactions of the septal machine (42). In opposition, fast (inactive) PG synthase motion is independent of these resources but strictly reliant on the intrinsic binding potential of FtsI-FtsZ, random diffusion of FtsI, and FtsZ treadmilling speed (43).
Regarding diffusion, the rate of FtsI diffusion is critical to its ability to end track. For instance, if FtsI diffuses too rapidly it will not couple to the (–) end of the filament, showing very little end-tracking behavior. Conversely, diffusing too slowly hinders FtsI’s ability to keep up with a treadmilling FtsZ filament. Therefore, the processivity of the Brownian rachet hinges on the balance between FtsZ treadmilling speed and random diffusion of FtsI. Under normal conditions, the diffusion coefficient of E. coli FtsI measures ∼0.041 ± 0.0051 μm2/s. By comparison, B. subtilis PBP2b (septal TP) measures 0.038 ± 0.0019 μm2/s with FtsW (septal TG) of S. pneumoniae measuring 0.028 ± 0.0004 μm2/s (43). Experimentally, FtsZ end-tracking by inactive PG synthases has only been recorded in E. coli, however, theoretical modeling predicts that B. subtilis PG synthases have the propensity to display end-tracking behavior as well (40, 43). Moreover, the diffusion coefficients of these various PG synthases are similar, heralding diffusion as a primordial means of transit across species. A molecule’s diffusion rate is at the mercy of macromolecular crowding conditions of the cell. In sum, mechanisms that control the spatiotemporal distribution of active versus inactive PG synthases are emerging and appear to share overlapping but distinct trends between species.
LIPID II AND THE MACHINES: A SPATIOTEMPORAL CONUNDRUM
Reflecting on our current knowledge of cellular architecture, motion, and movement patterns the mechanism by which lipid II and PG machines find one another is quite perplexing, (Fig. 5). An alluring theory is that lipid II and PG machine encounters are driven by diffusion, relying on chance. One can envision that a large amount of lipid II is synthesized and exported at the forming septum and its disaccharide-pentapeptide subsequently inserted by the septal machine. Indeed, the septal machine’s trajectory and FDAA labeling show that a large mass of new PG is deposited at the septum. Yet, given the stochastic nature of diffusion how is exported lipid II corralled to midcell? Moreover, diffusion-mediated encounters seem unlikely in considering elongation. PG synthases are restricted to the membrane plane; however, a PG synthase can traverse the cell’s circumference, sidewalls, and poles as the membrane surrounds the full cell body. Thus, the likelihood of independently diffusing membrane-linked lipid II and PG synthase randomly colliding at the exact position of insertion in time and space seems slim. To increase the odds of collision perhaps lipid II levels exist in saturation. However, maintaining such membrane saturation seems costly and unproductive. In addition, lipid II levels can be increased artificially, thereby refuting that lipid II levels exist in saturation (41). Furthermore, data show that the machines circumferentially transit the cell in processive linear paths, inserting PG in distinct patterns (24, 39, 40, 42, 74). These observations do not suggest diffusion-mediated encounters (stochastic or random patterns), but rather point to unknown mechanisms of spatiotemporal coordination that tether precursor biosynthesis, lipid II export, and insertion together.
FIG 5.

Illustrating the machine’s motion and spatiotemporal organization. (A) The trajectory of elongation machine components. Directional movement, signifying active PG synthesis, is indicated by magenta arrows. Gray circles and dotted lines symbolize diffusive, nonactive machine components as well as other components occupying the cell. (B) The trajectory of septal machine components. The septal ring is illustrated as a light blue ring. (C) Illustration of forming septum, rotate cell body 90 degrees. The outer light blue ring symbolizes the edge of the sacculus at the constriction site. Gaps in sacculus are shown as blank squares accompanied by cell wall hydrolases (gray Pac-Man). Dark blue trapezoids represent new PG inserted in distinct patches/nodes. The tan ring represents the constriction zone that is occupied by a multitude of proteins contained in the septal machine, including septal PG synthases, cytoskeletal filaments, and various regulatory proteins (Table 1). Of note, the septal machine contains cytoplasmic, membrane-associated, and integral membrane proteins. For protein topology see Fig. 1. (A to C) Arrows signify bidirectional, circumferential movement patterns of the respective machine components observed using microscopy and tracking techniques. The septal PG synthases, MurJ, lipid II, and various PG precursors drawn in areas in Fig. 1 FtsA (the membrane anchor and cytoskeletal protein) are shown as an orange circle. Treadmilling FtsZ filaments and diffusive filament monomers are drawn as beveled squares and colored tan or lilac. See Fig. 4 for more details. Other various PG precursors are indicated in the Key. The inner membrane is illustrated as a gray ring. For simplicity, the outer membrane/periplasmic space is not drawn. Likewise, the individual amino acids that comprise the pentapeptide are not drawn, but also reside in the cytoplasm. Architectural organization and PG deposition pattern based on B. subtilis (rod-shaped) and S. pneumoniae (football-shaped) (40, 77).
One can theorize that each machine (septal and elongation) possesses its flippase, allowing for streamlined export and subsequent insertion when areas rich in membrane-linked lipid II located below the membrane are happened upon. Here, the MurJ-containing-machine transits its cytoskeletal track as it simultaneously exports and inserts lipid II’s disaccharide-pentapeptide, thereby physically connecting the cytoplasm to the periplasm/cell exterior. Accordingly, machine and lipid II do not collide by chance but instead are joined together by the machine’s flippase, MurJ, an integral membrane protein. The necessity of the cytoplasmic, cytoskeletal proteins (FtsZ:septation and MreB:elongation) for motion supports such a theory. Consider that MurJ specifically localizes to the midcell (septation) and side walls (elongation) (102). Moreover, the septal PG synthases FtsW:FtsI is required for localization of MurJ to midcell, suggesting that lipid II export is contingent on available and active machines (102). Temporal studies show that MurJ as well as the PG synthases are among the last to assemble into the building machine (14, 68, 69, 102). Lastly, cytoplasmic biosynthesis of lipid II is also required for MurJ localization at midcell (102). Thus, a coupling of biosynthesis, export, and insertion is evident and appears to be orchestrated through the machine’s flippase, – a key component, connected to elements below and above the cytoplasmic membrane. Pivotal to validating such a hypothesis is understanding the movement dynamics of MurJ.
Be it by simple diffusion or coordinated spatiotemporal regulation, it remains to be empirically determined how essential lipid II (substrate) and the PG machines (inserter) appear at the same location (gap in sacculus) within the respective periplasmic or extracellular space in time. This inquiry of substrate-machine timing and localization extends to the cell wall hydrolases. They prime the sacculus for growth by hydrolyzing gaps so that new material may be inserted as well as serving to separate daughter cells by cleaving new PG at the septum (94, 103–105). Resolving this spatiotemporal conundrum represents one of the last frontiers of cellular division.
CONCLUSIONS AND UNKNOWNS
The pairing of high-resolution microscopy with SMT has revealed that many components of the PG synthesis machines exhibit distinct and discernible movement patterns to transit the cell. Moreover, the circumferential motion of the machines is a by-product of PG synthesis, be it driven by TG:TP reactions (S. pneumoniae), FtsZ treadmilling (B. subtilis), or both (S. aureus and E. coli). Our current knowledge suggests that machine components utilize three main movement patterns to transit the cell: directed motion, diffusive motion, and treadmilling. Directed motion and treadmilling require energy while diffusion is passive and harnesses the thermal energy potential of the environment. Treadmilling involves polymeric filaments while single molecules have the propensity to display both directed and diffusive movement. If all the patterns are sufficient for transit, then why do proteins employ multiple movement patterns?
At first glance, directed motion appears superior to diffusive as the shortest path between two objects is a straight line. However, directed (linear) motion is not processive in the context of a bacterial cell exhibiting macromolecular crowding, be it in the cytoplasm or membrane plane. As illustrated in Fig. 6, transit events are stymied if an object travels a set linear path where only ∼30% of the area is occupied by obstacles, such as metabolites, proteins, and the chromosome. While topological rearrangements may clear paths, time is lost during the blockage. Conversely, an object exhibiting diffusive motion is capable of maneuvering through obstacles via a random walk. Granted, an object displaying diffusive motion travels greater distances than that required of linear transit. However, because biological systems impose oscillating environmental and spatial constraints on particle movement and velocity, it appears advantageous to possess both directed and diffusive motion. This is reflected in the ability of Class B PG synthases to transit both directionally and diffusively.
FIG 6.

Guaranteeing success: directed versus diffusive motion. Illustration of an object (green dot) transiting from point A to point B to interact with cup complex (concave green tower) within three different environmental conditions. Object trajectory is outlined by arrows. (A) In vitro as well as the minimally crowded cytoplasmic environment, i.e., <10% of the area occupied. The absence of environmental barriers leads to rapid and reproducible interaction with the cup complex. Highlights that the shortest path between two points is a straight line. (B) Molecular crowding of ∼30% halts the object’s ability to transit if alternative movement patterns/paths do not exist outside directed motion. Topological rearrangement may clear the path; however, time is lost. (C) Confined diffusion of the object leads to changes in the object’s direction via random collisions. While the distance traveled in panel C is greater than in panel A, diffusive motion overcomes and utilizes macromolecular crowding to increase its rate of transit, thereby connecting interactors more efficiently than in panel B. Yet, processivity is lost in that interaction relies on chance. Because the molecular environment is transient, it appears advantageous to possess both directed and diffusive movement patterns, like that of the PG synthases.
Without a doubt, developments in labeling techniques and tracking programs have led to major gains in understanding the elegant mechanisms of cell division. Among these discoveries, many questions remain, including what determines the ratio of active-directed to inactive-diffusive PG synthases within a given population of molecules? Are static Class A PG synthases enzymatically active? By extension, are the Class A PG synthases contained in and governed by the machines? Does MurJ transit with the machine, thereby coupling precursor biosynthesis to insertion? Future research will provide answers to these and other questions about molecular movement patterns of proteins within the bacterial cell.
Contributor Information
Anthony T. Maurelli, Email: amaurelli@phhp.ufl.edu.
Conrad W. Mullineaux, Queen Mary University of London
REFERENCES
- 1.Kapanidis AN, Uphoff S, Stracy M. 2018. Understanding protein mobility in bacteria by tracking single molecules. J Mol Biol 430:4443–4455. 10.1016/j.jmb.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie XS, Choi PJ, Li G-W, Lee NK, Lia G. 2008. Single-molecule approach to molecular biology in living bacterial cells. Annu Rev Biophys 37:417–444. 10.1146/annurev.biophys.37.092607.174640. [DOI] [PubMed] [Google Scholar]
- 3.Ellis RJ. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci 26:597–604. 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 4.Amir A, Nelson DR. 2012. Dislocation-mediated growth of bacterial cell walls. Proc Natl Acad Sci USA 109:9833–9838. 10.1073/pnas.1207105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hussain S, Wivagg CN, Szwedziak P, Wong F, Schaefer K, Izoré T, Renner LD, Holmes MJ, Sun Y, Bisson-Filho AW, Walker S, Amir A, Löwe J, Garner EC. 2018. MreB filaments align along greatest principal membrane curvature to orient cell wall synthesis. Elife 7:e32471. 10.7554/eLife.32471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang F, Huang KC. 2014. How and why cells grow as rods. BMC Biol 12:54. 10.1186/s12915-014-0054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian H, Sheetz MP, Elson EL. 1991. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys J 60:910–921. 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young KD. 2006. The selective value of bacterial shape. Microbiol Mol Biol Rev 70:660–703. 10.1128/MMBR.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young KD. 2010. Bacterial shape: two-dimensional questions and possibilities. Annu Rev Microbiol 64:223–240. 10.1146/annurev.micro.112408.134102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holtje JV. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev 62:181–203. 10.1128/MMBR.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rojas ER, Billings G, Odermatt PD, Auer GK, Zhu L, Miguel A, Chang F, Weibel DB, Theriot JA, Huang KC. 2018. The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature 559:617–621. 10.1038/s41586-018-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheffers DJ, Pinho MG. 2005. Bacterial cell wall synthesis: new Insights from localization studies. Microbiol Mol Biol Rev 69:585–607. 10.1128/MMBR.69.4.585-607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinho MG, Kjos M, Veening JW. 2013. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 14.den Blaauwen T, Hamoen LW, Levin PA. 2017. The divisome at 25: the road ahead. Curr Opin Microbiol 36:85–94. 10.1016/j.mib.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carballido-Lopez R. 2006. Orchestrating bacterial cell morphogenesis. Mol Microbiol 60:815–819. 10.1111/j.1365-2958.2006.05161.x. [DOI] [PubMed] [Google Scholar]
- 16.den Blaauwen T, de Pedro MA, Nguyen-Distèche M, Ayala JA. 2008. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev 32:321–344. 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 17.Randich AM, Brun YV. 2015. Molecular mechanisms for the evolution of bacterial morphologies and growth modes. Front Microbiol 6:580. 10.3389/fmicb.2015.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glauner B, Holtje JV, Schwarz U. 1988. The composition of the murein of Escherichia coli. J Biol Chem 263:10088–10095. 10.1016/S0021-9258(19)81481-3. [DOI] [PubMed] [Google Scholar]
- 19.Vollmer W, Holtje JV. 2004. The architecture of the murein (peptidoglycan) in gram-negative bacteria: vertical scaffold or horizontal layer(s)? J Bacteriol 186:5978–5987. 10.1128/JB.186.18.5978-5987.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajagopal M, Walker S. 2017. Envelope structures of gram-positive bacteria. Curr Top Microbiol Immunol 404:1–44. 10.1007/82_2015_5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demchick P, Koch AL. 1996. The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J Bacteriol 178:768–773. 10.1128/jb.178.3.768-773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barreteau H, Kovac A, Boniface A, Sova M, Gobec S, Blanot D. 2008. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev 32:168–207. 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 24.Perez AJ, Cesbron Y, Shaw SL, Bazan Villicana J, Tsui H-CT, Boersma MJ, Ye ZA, Tovpeko Y, Dekker C, Holden S, Winkler ME. 2019. Movement dynamics of divisome proteins and PBP2x:FtsW in cells of Streptococcus pneumoniae. Proc Natl Acad Sci USA 116:3211–3220. 10.1073/pnas.1816018116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar S, Rubino FA, Mendoza AG, Ruiz N. 2019. The bacterial lipid II flippase MurJ functions by an alternating-access mechanism. J Biol Chem 294:981–990. 10.1074/jbc.RA118.006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubino FA, Mollo A, Kumar S, Butler EK, Ruiz N, Walker S, Kahne DE. 2020. Detection of transport intermediates in the peptidoglycan flippase MurJ identifies residues essential for conformational cycling. J Am Chem Soc 142:5482–5486. 10.1021/jacs.9b12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sham L-T, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. 2014. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 345:220–222. 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pomorski T, Menon AK. 2006. Lipid flippases and their biological functions. Cell Mol Life Sci 63:2908–2921. 10.1007/s00018-006-6167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruiz N. 2015. Lipid flippases for bacterial peptidoglycan biosynthesis. Lipid Insights 8:21–31. 10.4137/LPI.S31783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meeske AJ, Sham L-T, Kimsey H, Koo B-M, Gross CA, Bernhardt TG, Rudner DZ. 2015. MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc Natl Acad Sci USA 112:6437–6442. 10.1073/pnas.1504967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue A, Murata Y, Takahashi H, Tsuji N, Fujisaki S, Kato J-i. 2008. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J Bacteriol 190:7298–7301. 10.1128/JB.00551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz N. 2008. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc Natl Acad Sci USA 105:15553–15557. 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pichoff S, Lutkenhaus J. 2007. Overview of cell shape: cytoskeletons shape bacterial cells. Curr Opin Microbiol 10:601–605. 10.1016/j.mib.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ. 2016. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537:634–638. 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho H, Wivagg CN, Kapoor M, Barry Z, Rohs PDA, Suh H, Marto JA, Garner EC, Bernhardt TG. 2016. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat Microbiol 1:16172. 10.1038/nmicrobiol.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taguchi A, Welsh MA, Marmont LS, Lee W, Sjodt M, Kruse AC, Kahne D, Bernhardt TG, Walker S. 2019. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat Microbiol 4:587–594. 10.1038/s41564-018-0345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loose M, Mitchison TJ. 2014. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat Cell Biol 16:38–46. 10.1038/ncb2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carballido-Lopez R, Errington J. 2003. A dynamic bacterial cytoskeleton. Trends Cell Biol 13:577–583. 10.1016/j.tcb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 39.Garner EC, Bernard R, Wang W, Zhuang X, Rudner DZ, Mitchison T. 2011. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333:222–225. 10.1126/science.1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bisson-Filho AW, Hsu Y-P, Squyres GR, Kuru E, Wu F, Jukes C, Sun Y, Dekker C, Holden S, VanNieuwenhze MS, Brun YV, Garner EC. 2017. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355:739–743. 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang X, McQuillen R, Lyu Z, Phillips-Mason P, De La Cruz A, McCausland JW, Liang H, DeMeester KE, Santiago CC, Grimes CL, de Boer P, Xiao J. 2021. A two-track model for the spatiotemporal coordination of bacterial septal cell wall synthesis revealed by single-molecule imaging of FtsW. Nat Microbiol 6:584–593. 10.1038/s41564-020-00853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, Xiao J. 2017. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355:744–747. 10.1126/science.aak9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCausland JW, Yang X, Squyres GR, Lyu Z, Bruce KE, Lamanna MM, SöDerströM B, Garner EC, Winkler ME, Xiao J, Liu J, 2021. Treadmilling FtsZ polymers drive the directional movement of sPG-synthesis enzymes via a Brownian ratchet mechanism. Nat Commun 12:609. 10.1038/s41467-020-20873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vollmer W, Seligman SJ. 2010. Architecture of peptidoglycan: more data and more models. Trends Microbiol 18:59–66. 10.1016/j.tim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Martin-Fernandez ML, Tynan CJ, Webb SED. 2013. A 'pocket guide' to total internal reflection fluorescence. J Microsc 252:16–22. 10.1111/jmi.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verdaasdonk JS, Stephens AD, Haase J, Bloom K. 2014. Bending the rules: widefield microscopy and the Abbe limit of resolution. J Cell Physiol 229:132–138. 10.1002/jcp.24439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.St Croix CM, Shand SH, Watkins SC. 2005. Confocal microscopy: comparisons, applications, and problems. Biotechniques 39:S2–S5. 10.2144/000112089. [DOI] [PubMed] [Google Scholar]
- 48.Jonkman J, Brown CM, Wright GD, Anderson KI, North AJ. 2020. Tutorial: guidance for quantitative confocal microscopy. Nat Protoc 15:1585–1611. 10.1038/s41596-020-0313-9. [DOI] [PubMed] [Google Scholar]
- 49.Remington SJ. 2011. Green fluorescent protein: a perspective. Protein Sci 20:1509–1519. 10.1002/pro.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lichtman JW, Conchello J-A. 2005. Fluorescence microscopy. Nat Methods 2:910–919. 10.1038/nmeth817. [DOI] [PubMed] [Google Scholar]
- 51.England CG, Luo H, Cai W. 2015. HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem 26:975–986. 10.1021/acs.bioconjchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. 2008. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3:373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 53.Collins TJ. 2007. ImageJ for microscopy. Biotechniques 43:25–S30. 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 54.Zimmerman SB, Trach SO. 1991. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J Mol Biol 222:599–620. 10.1016/0022-2836(91)90499-V. [DOI] [PubMed] [Google Scholar]
- 55.Swaminathan R, Hoang CP, Verkman AS. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J 72:1900–1907. 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gal N, Lechtman-Goldstein D, Weihs D. 2013. Particle tracking in living cells: a review of the mean square displacement method and beyond. Rheol Acta 52:425–443. 10.1007/s00397-013-0694-6. [DOI] [Google Scholar]
- 57.Shukron O, Seeber A, Amitai A, Holcman D. 2019. Advances using single-particle trajectories to reconstruct chromatin organization and dynamics. Trends Genet 35:685–705. 10.1016/j.tig.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Niu L, Yu J. 2008. Investigating intracellular dynamics of FtsZ cytoskeleton with photoactivation single-molecule tracking. Biophys J 95:2009–2016. 10.1529/biophysj.108.128751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.English BP, Hauryliuk V, Sanamrad A, Tankov S, Dekker NH, Elf J. 2011. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci USA 108:E365–E373. 10.1073/pnas.1102255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterman-Storer CM, Salmon ED. 1997. Microtubule dynamics: treadmilling comes around again. Curr Biol 7:R369–R372. 10.1016/S0960-9822(06)00177-1. [DOI] [PubMed] [Google Scholar]
- 61.McQuillen R, Xiao J. 2020. Insights into the structure, function, and dynamics of the bacterial cytokinetic FtsZ-ring. Annu Rev Biophys 49:309–341. 10.1146/annurev-biophys-121219-081703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haeusser DP, Margolin W. 2016. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol 14:305–319. 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lan G, Daniels BR, Dobrowsky TM, Wirtz D, Sun SX. 2009. Condensation of FtsZ filaments can drive bacterial cell division. Proc Natl Acad Sci USA 106:121–126. 10.1073/pnas.0807963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bi EF, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161–164. 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 65.Fujita J. 2021. Dynamic assembly/disassembly of Staphylococcus aureus FtsZ visualized by high-speed atomic force microscopy. Int J Mol Sci 22:1697. 10.3390/ijms22041697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lan G, Dajkovic A, Wirtz D, Sun SX. 2008. Polymerization and bundling kinetics of FtsZ filaments. Biophys J 95:4045–4056. 10.1529/biophysj.108.132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramirez-Diaz DA, García-Soriano DA, Raso A, Mücksch J, Feingold M, Rivas G, Schwille P. 2018. Treadmilling analysis reveals new insights into dynamic FtsZ ring architecture. PLoS Biol 16:e2004845. 10.1371/journal.pbio.2004845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aarsman MEG, Piette A, Fraipont C, Vinkenvleugel TMF, Nguyen-Distèche M, den Blaauwen T. 2005. Maturation of the Escherichia coli divisome occurs in two steps. Mol Microbiol 55:1631–1645. 10.1111/j.1365-2958.2005.04502.x. [DOI] [PubMed] [Google Scholar]
- 69.Gamba P, Veening J-W, Saunders NJ, Hamoen LW, Daniel RA. 2009. Two-step assembly dynamics of the Bacillus subtilis divisome. J Bacteriol 191:4186–4194. 10.1128/JB.01758-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hou S, Wieczorek SA, Kaminski TS, Ziebacz N, Tabaka M, Sorto NA, Foss MH, Shaw JT, Thanbichler M, Weibel DB, Nieznanski K, Holyst R, Garstecki P. 2012. Characterization of Caulobacter crescentus FtsZ protein using dynamic light scattering. J Biol Chem 287:23878–23886. 10.1074/jbc.M111.309492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ranjit DK, Liechti GW, Maurelli AT. 2020. Chlamydial MreB directs cell division and peptidoglycan synthesis in Escherichia coli in the absence of FtsZ activity. mBio 11:e03222-19. 10.1128/mBio.03222-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liechti GW, Kuru E, Hall E, Kalinda A, Brun YV, VanNieuwenhze M, Maurelli AT. 2014. A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature 506:507–510. 10.1038/nature12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ouellette SP, Karimova G, Subtil A, Ladant D. 2012. Chlamydia co-opts the rod shape-determining proteins MreB and Pbp2 for cell division. Mol Microbiol 85:164–178. 10.1111/j.1365-2958.2012.08100.x. [DOI] [PubMed] [Google Scholar]
- 74.Monteiro JM, Pereira AR, Reichmann NT, Saraiva BM, Fernandes PB, Veiga H, Tavares AC, Santos M, Ferreira MT, Macário V, VanNieuwenhze MS, Filipe SR, Pinho MG. 2018. Peptidoglycan synthesis drives an FtsZ-treadmilling-independent step of cytokinesis. Nature 554:528–532. 10.1038/nature25506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodionov VI, Borisy GG. 1997. Microtubule treadmilling in vivo. Science 275:215–218. 10.1126/science.275.5297.215. [DOI] [PubMed] [Google Scholar]
- 76.Mura A, Fadda D, Perez AJ, Danforth ML, Musu D, Rico AI, Krupka M, Denapaite D, Tsui H-CT, Winkler ME, Branny P, Vicente M, Margolin W, Massidda O. 2017. Roles of the essential protein FtsA in cell growth and division. J Bacteriol 199:e00608-16. 10.1128/JB.00608-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perez AJ, et al. 2020. Organization of peptidoglycan synthesis in nodes and separate rings at different stages of cell division of Streptococcus pneumoniae. Mol Microbiol 115:1152–1169. 10.1111/mmi.14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fleurie A, Lesterlin C, Manuse S, Zhao C, Cluzel C, Lavergne J-P, Franz-Wachtel M, Macek B, Combet C, Kuru E, VanNieuwenhze MS, Brun YV, Sherratt D, Grangeasse C. 2014. MapZ marks the division sites and positions FtsZ rings in Streptococcus pneumoniae. Nature 516:259–262. 10.1038/nature13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsui H-CT, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. 2014. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Mol Microbiol 94:21–40. 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pazos M, Peters K, Vollmer W. 2017. Robust peptidoglycan growth by dynamic and variable multi-protein complexes. Curr Opin Microbiol 36:55–61. 10.1016/j.mib.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 81.Rohs PDA, Buss J, Sim SI, Squyres GR, Srisuknimit V, Smith M, Cho H, Sjodt M, Kruse AC, Garner EC, Walker S, Kahne DE, Bernhardt TG. 2018. A central role for PBP2 in the activation of peptidoglycan polymerization by the bacterial cell elongation machinery. PLoS Genet 14:e1007726. 10.1371/journal.pgen.1007726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Emami K, Guyet A, Kawai Y, Devi J, Wu LJ, Allenby N, Daniel RA, Errington J. 2017. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat Microbiol 2:16253. 10.1038/nmicrobiol.2016.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Land AD, Winkler ME. 2011. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol 193:4166–4179. 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vigouroux A, Cordier B, Aristov A, Alvarez L, Özbaykal G, Chaze T, Oldewurtel ER, Matondo M, Cava F, Bikard D, van Teeffelen S. 2020. Class-A penicillin binding proteins do not contribute to cell shape but repair cell-wall defects. Elife 9:e51998. 10.7554/eLife.51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsui H-CT, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, Winkler ME. 2016. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Mol Microbiol 100:1039–1065. 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pazos M, Vollmer W. 2021. Regulation and function of class A penicillin-binding proteins. Curr Opin Microbiol 60:80–87. 10.1016/j.mib.2021.01.008. [DOI] [PubMed] [Google Scholar]
- 87.García del Portillo F, de Pedro MA, Joseleau-Petit D, D'Ari R. 1989. Lytic response of Escherichia coli cells to inhibitors of penicillin-binding proteins 1a and 1b as a timed event related to cell division. J Bacteriol 171:4217–4221. 10.1128/jb.171.8.4217-4221.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.García del Portillo F, de Pedro MA. 1991. Penicillin-binding protein 2 is essential for the integrity of growing cells of Escherichia coli ponB strains. J Bacteriol 173:4530–4532. 10.1128/jb.173.14.4530-4532.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yousif SY, Broome-Smith JK, Spratt BG. 1985. Lysis of Escherichia coli by beta-lactam antibiotics: deletion analysis of the role of penicillin-binding proteins 1A and 1B. J Gen Microbiol 131:2839–2845. 10.1099/00221287-131-10-2839. [DOI] [PubMed] [Google Scholar]
- 90.Dion MF, Kapoor M, Sun Y, Wilson S, Ryan J, Vigouroux A, van Teeffelen S, Oldenbourg R, Garner EC. 2019. Bacillus subtilis cell diameter is determined by the opposing actions of two distinct cell wall synthetic systems. Nat Microbiol 4:1294–1305. 10.1038/s41564-019-0439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wientjes FB, Nanninga N. 1991. On the role of the high molecular weight penicillin-binding proteins in the cell cycle of Escherichia coli. Res Microbiol 142:333–344. 10.1016/0923-2508(91)90049-G. [DOI] [PubMed] [Google Scholar]
- 92.Lee TK, Meng K, Shi H, Huang KC. 2016. Single-molecule imaging reveals modulation of cell wall synthesis dynamics in live bacterial cells. Nat Commun 7:13170. 10.1038/ncomms13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Straume D, et al. 2020. Class A PBPs have a distinct and unique role in the construction of the pneumococcal cell wall. Proc Natl Acad Sci USA 117:6129–6138. 10.1073/pnas.1917820117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Typas A, Banzhaf M, Gross CA, Vollmer W. 2011. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Egan AJF, Jean NL, Koumoutsi A, Bougault CM, Biboy J, Sassine J, Solovyova AS, Breukink E, Typas A, Vollmer W, Simorre J-P. 2014. Outer-membrane lipoprotein LpoB spans the periplasm to stimulate the peptidoglycan synthase PBP1B. Proc Natl Acad Sci USA 111:8197–8202. 10.1073/pnas.1400376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jean NL, Bougault CM, Lodge A, Derouaux A, Callens G, Egan AJF, Ayala I, Lewis RJ, Vollmer W, Simorre J-P. 2014. Elongated structure of the outer-membrane activator of peptidoglycan synthesis LpoA: implications for PBP1A stimulation. Structure 22:1047–1054. 10.1016/j.str.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Markovski M, Bohrhunter JL, Lupoli TJ, Uehara T, Walker S, Kahne DE, Bernhardt TG. 2016. Cofactor bypass variants reveal a conformational control mechanism governing cell wall polymerase activity. Proc Natl Acad Sci USA 113:4788–4793. 10.1073/pnas.1524538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. 2006. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol Rev 30:673–691. 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 99.Ait-Haddou R, Herzog W. 2003. Brownian ratchet models of molecular motors. Cell Biochem Biophys 38:191–213. 10.1385/CBB:38:2:191. [DOI] [PubMed] [Google Scholar]
- 100.Moore A. 2019. Brownian ratchets of life: stochasticity combined with disequilibrium produces order. BioEssays 41:1900076. 10.1002/bies.201900076. [DOI] [PubMed] [Google Scholar]
- 101.Saffman PG, Delbruck M. 1975. Brownian motion in biological membranes. Proc Natl Acad Sci USA 72:3111–3113. 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu X, Meiresonne NY, Bouhss A, den Blaauwen T. 2018. FtsW activity and lipid II synthesis are required for recruitment of MurJ to midcell during cell division in Escherichia coli. Mol Microbiol 109:855–884. 10.1111/mmi.14104. [DOI] [PubMed] [Google Scholar]
- 103.Uehara T, Park JT. 2008. Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and septation. J Bacteriol 190:3914–3922. 10.1128/JB.00207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schaefer K, et al. 2020. Structure and reconstitution of a hydrolase complex that releases peptidoglycan from the membrane after polymerization. Nat Microbiol 6:34–43. 10.1038/s41564-020-00808-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Uehara T, Bernhardt TG. 2011. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr Opin Microbiol 14:698–703. 10.1016/j.mib.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 107.The UniProt C. 2019. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Putman T, Hybiske K, Jow D, Afrasiabi C, Lelong S, Cano MA, Stupp GS, Waagmeester A, Good BM, Wu C, Su AI. 2019. ChlamBase: a curated model organism database for the Chlamydia research community. Database 2019:baz041. 10.1093/database/baz041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huber J, Donald RGK, Lee SH, Jarantow LW, Salvatore MJ, Meng X, Painter R, Onishi RH, Occi J, Dorso K, Young K, Park YW, Skwish S, Szymonifka MJ, Waddell TS, Miesel L, Phillips JW, Roemer T. 2009. Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem Biol 16:837–848. 10.1016/j.chembiol.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 111.Alyahya SA, Alexander R, Costa T, Henriques AO, Emonet T, Jacobs-Wagner C. 2009. RodZ, a component of the bacterial core morphogenic apparatus. Proc Natl Acad Sci USA 106:1239–1244. 10.1073/pnas.0810794106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, Keseler IM, Krummenacker M, Midford PE, Ong Q, Ong WK, Paley SM, Subhraveti P. 2019. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform 20:1085–1093. 10.1093/bib/bbx085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Meisner J, Montero Llopis P, Sham L-T, Garner E, Bernhardt TG, Rudner DZ. 2013. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Mol Microbiol 89:1069–1083. 10.1111/mmi.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marquardt JL, Siegele DA, Kolter R, Walsh CT. 1992. Cloning and sequencing of Escherichia coli murZ and purification of its product, a UDP-N-acetylglucosamine enolpyruvyl transferase. J Bacteriol 174:5748–5752. 10.1128/jb.174.17.5748-5752.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mattheyses AL, Simon SM, Rappoport JZ. 2010. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci 123:3621–3628. 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]