

ABSTRACT
In Plasmodium, the first two and rate-limiting enzymes of the pentose phosphate pathway, glucose 6-phosphate dehydrogenase (G6PD) and the 6-phosphogluconolactonase, are bifunctionally fused to a unique enzyme named GluPho, differing structurally and mechanistically from the respective human orthologs. Consistent with the enzyme’s essentiality for malaria parasite proliferation and propagation, human G6PD deficiency has immense impact on protection against severe malaria, making PfGluPho an attractive antimalarial drug target. Herein we report on the optimized lead compound N-(((2R,4S)-1-cyclobutyl-4-hydroxypyrrolidin-2-yl)methyl)-6-fluoro-4-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]thiazepine-8-carboxamide (SBI-0797750), a potent and fully selective PfGluPho inhibitor with robust nanomolar activity against recombinant PfGluPho, PvG6PD, and P. falciparum blood-stage parasites. Mode-of-action studies have confirmed that SBI-0797750 disturbs the cytosolic glutathione-dependent redox potential, as well as the cytosolic and mitochondrial H2O2 homeostasis of P. falciparum blood stages, at low nanomolar concentrations. Moreover, SBI-0797750 does not harm red blood cell (RBC) integrity and phagocytosis and thus does not promote anemia. SBI-0797750 is therefore a very promising antimalarial lead compound.
KEYWORDS: G6PDH, inhibitors, malaria, Plasmodium, Plasmodium falciparum, Plasmodium vivax
INTRODUCTION
Malaria is one of the world’s most devastating infectious diseases, transmitted to the human host by female Anopheles mosquitoes. In 2019, an estimated 229 million cases of malaria resulting in 409,000 deaths occurred worldwide (1). Plasmodium falciparum is responsible for 90% of all malaria-related deaths and is therefore the most important Plasmodium species; however, infections with Plasmodium vivax can also manifest as severe malaria and thereby threaten the health and welfare of people in countries where the organism is endemic (2, 3). Currently, WHO recommends an artemisinin-based combination therapy (ACT) for treating uncomplicated falciparum malaria and chloroquine or an ACT to treat uncomplicated vivax malaria (4). However, increasing resistance of both P. falciparum and P. vivax against these commonly used drugs is hampering the fight against the disease (4–6). Therefore, ambitious efforts to develop new antimalarial drugs are needed.
Naturally occurring evidence for a connection of the pentose phosphate pathway (PPP) and malaria is demonstrated by the fact that deficiency of the enzyme glucose 6-phosphate dehydrogenase in humans (Homo sapiens G6PD [HsG6PD]) leads to partial resistance to the disease. The very similar distribution patterns of malaria and HsG6PD deficiency further underscore this correlation (7, 8). Currently, around 400 million people are affected by G6PD deficiency, which can be caused by over 200 known mutations in the HsG6PD-encoding gene (8). The mild and most common mutations result in reduced G6PD activity. It is assumed that their protective effect against malaria is the result of increased phagocytosis of parasitized G6PD-deficient erythrocytes (9–11).
Plasmodium parasites have a complex life cycle with changing environments in the Anopheles mosquito vector and the human host (12). During their life, Plasmodium parasites are continuously exposed to reactive oxygen species (ROS) and reactive nitrogen species (RNS) of different sources, mainly detoxified by the thioredoxin and glutathione systems. These systems are highly dependent on the supply of NADPH as a reducing equivalent (13), underscoring the importance of the oxidative PPP as the major source of NADPH for the parasites (14). A noticeable structural difference between the plasmodial and human PPP is that in Plasmodium the first two enzymes, G6PD and 6-phosphogluconolactonase, are fused to form a unique bifunctional enzyme named GluPho, which differs structurally and functionally from the human orthologs (15). Using double-crossover disruption, we were able to show that PfGluPho is essential for the growth of asexual blood-stage parasites, validating this enzyme as an excellent target for the development of new, specific antimalarial drugs (16). To follow up on this hypothesis, we performed a high-throughput screen of 348,911 compounds in the NIH’s Molecular Libraries Small Molecule Repository (MLSMR) collection (17) and identified two promising compounds with selective inhibition of PfGluPho in the nanomolar range, ML276 (18) and ML304 (19). In this study, we now present the characterization of an optimized lead from the dibenzothiazepine-derived second probe ML304 (19), SBI-0797750 (Fig. 1), with low nanomolar activity against recombinant PfGluPho, PvG6PD, and P. falciparum blood-stage parasites and negligible effects on host red blood cell (RBC) integrity and phagocytosis.
FIG 1.

Previously described PfGluPho inhibitors ML276 and ML304 and optimized lead compound SBI-0797750.
RESULTS
SBI-0797750 inhibits PfGluPho and PvG6PD in a highly selective manner at low nanomolar concentrations.
The 50% inhibitory concentration (IC50) of SBI-0797750 was first determined using the diaphorase-coupled assay and confirmed with the orthogonal assay without diaphorase. We determined an IC50 of 6.7 ± 1.8 nM for PfGluPho and of 31.0 ± 3.1 nM for PvG6PD. The human homologue HsG6PD was tested up to 99,000 nM with inhibition below 50% (20), indicating high selectivity of SBI-0797750 for the plasmodial enzymes (Table 1).
TABLE 1.
Biochemical and cellular activities of ML304 and SBI-0797750a
| Compound | IC50 (nM) for biochemical/recombinant protein |
EC50 (nM) for cellular/in vitro
P. falciparum |
||||
|---|---|---|---|---|---|---|
| PfGluPho | PvG6PD | HsG6PD | 3D7, asexual | NF54-attB, asexual | NF54, sexual | |
| ML304 | 190* | 2,600 ± 800** | >79,000* | 471 ± 40†** | 1,700‡ | ND |
| SBI-0797750 | 6.7 ± 1.8 | 31.0 ± 3.1 | >99,000 | 22.5 ± 2.2,† 64.6 ± 9.6‡ | 83.8 ± 17.3‡ | 73.9 ± 22 |
Mode-of-inhibition studies indicated that SBI-0797750 competes with G6P for the substrate binding site, which was evident by increasing Km for G6P and constant Vmax with increasing drug concentration (Fig. 2A and B). Similarly, the Km for NADP+ increased with increasing compound concentrations, but Vmax decreased (Fig. 2C and D), suggesting a mixed-type inhibition of SBI-0797750 against NADP+.
FIG 2.

Mechanism of inhibition of SBI-0797750 against PfGluPho. (A and B) Various compound concentrations were titrated against G6P. SBI-0797750 acts as a competitive inhibitor against PfGluPho, since the Km value for G6P increases with increasing compound concentrations, while Vmax stays constant. (C and D) Titration of NADP+ against different compound concentrations. The Km value for NADP+ increases, while Vmax decreases, indicating a mixed-type inhibition. PfGluPho activity is in relative fluorescence units per second (modified from reference 20).
To test for reversibility of PfGluPho inhibition by SBI-0797750, the enzyme was first incubated with a high compound concentration (1.65 μM), following dilution below the IC50 (3.3 nM), and again incubated to allow compound dissociation. Activities were compared to controls containing either no SBI-0797750 (CTL, 0 nM; 100% activity) or 3.3 nM SBI-0797750 (CTL, 3.3 nM), which corresponds to the concentration after dilution. The diluted sample (postdilution, 3.3 nM) showed a residual activity of 79.2% ± 1.5%, almost identical to the control with 77.2% ± 1.6% (CTL, 3.3 nM). In contrast, incubation of the enzyme with 1.65 μM SBI-0797750 (predilution, 1.65 μM) revealed a residual activity of 2.4% ± 0.3%, proving complete inhibition of the enzyme before dilution. Since the small remaining activity could be due to the competition of SBI-0797750 with G6P at saturation (Fig. 2A and B), we performed the same experiment under Km conditions, and indeed, enzyme activity was reduced to 0.3% ± 0.2%. Finally, the data clearly indicate a fully reversible inhibition of PfGluPho by SBI-0797750 (Fig. 3).
FIG 3.

Reversibility of the PfGluPho inhibition by SBI-0797750. Reversibility of PfGluPho inhibition by SBI-0797750 (SBI-750) was determined by incubating the enzyme with a high compound concentration of 1.65 μM (predilution), followed by dilution to 3.3 nM (postdilution). The diluted sample had the same activity as a control containing the same final compound concentration (CTL, 3.3 nM), while the undiluted sample was completely inhibited. Treatment without SBI-750 was defined as 100% activity (CTL, 0 nM). Values are means and SD from three independent determinations, each including two measurements.
Activity against P. falciparum 3D7 and NF54-attB asexual parasites and NF54 gametocytes in culture.
Moreover, we tested the activity of SBI-0797750 against P. falciparum blood stages in vitro by using different assays, strains, and parasite stages (Table 1). For asexual stages of 3D7 strains, 50% effective concentrations (EC50s) of 22.5 ± 2.2 nM were determined using the [3H]hypoxanthine incorporation assay, and EC50s of 64.6 ± 9.6 nM were obtained using the SYBR green assay. For the NF54-attB parasites, the SYBR green assay resulted in EC50s of 83.8 ± 17.3 nM. In addition, initial experiments with P. falciparum NF54 gametocytes revealed comparable inhibitory effects with EC50s of 73.9 nM ± 22 nM.
Effects on the glutathione redox state of NF54-attB[hGrx1-roGFP2] parasites.
The oxidative effect of SBI-0797750 and ML304 on the glutathione redox system was determined using the cytosolic glutathione redox sensor hGrx1-roGFP2, stably integrated into NF54-attB parasites. Mid- and long-term exposures were carried out via confocal laser scanning microscopy (CLSM) as described in Materials and Methods. Control studies excluded a direct interaction of SBI-0797750, ML304, and the control drugs artesunate (ATS) and chloroquine (CQ) with the recombinant hGrx1-roGFP2 probe at the concentrations used in this study. Four-hour incubations with 1× EC50 SBI-0797750 and ML304 showed a highly significant increase in redox ratio (Fig. 4A) as well as in the degree of oxidation (OxD), which was 75.6% ± 1.6% for SBI-0797750 and 83.3% ± 2.8% for ML304. Thus, the improved drug SBI-07977504 disrupts the redox homeostasis to the same extent as ML304, even at lower concentrations. In contrast, the parasites could compensate for the oxidizing effect of 1× EC50 SBI-0797750 and ML304 after a long incubation period of 24 h (Fig. 4B), and no significant change in the redox ratio could be measured. For comparison, we tested antimalarials such as ATS and CQ on the redox sensors. However, in agreement with previous data (21), no effect was observed after 4 h or 24 h, even at concentrations of 100× and 10× EC50, respectively (see Fig. S1 in the supplemental material).
FIG 4.

Mid- and long-term effects of ML304 and its derivative SBI-0797750 on the redox ratio of P. falciparum NF54-attB[hGrx1-roGFP2] parasites. In 4-h and 24-h experiments, P. falciparum NF54-attB[hGrx1-roGFP2] parasites were incubated with 1× EC50 ML304 and SBI-0797750 (SBI-750). Via CLSM, a significant increase of the 405/488-nm fluorescence ratio of the redox sensor could be observed in the 4-h-incubation experiment (A). In the 24-h-incubation experiment, neither ML304 nor SBI-0797750 significantly changed the redox ratio (B). Nontreated parasites served as controls. All experiments included fully oxidized (1 mM DIA) and fully reduced (10 mM DTT) parasites. CLSM data were obtained from 10 to 20 trophozoites for each experiment and each incubation time. Values are means and SD from three independent experiments. A one-way ANOVA with 95% confidence intervals with the Dunnett’s multiple-comparison test was applied for statistical analysis of significance (***, P < 0.001; ****, P < 0.0001).
Effects on H2O2 homeostasis in NF54-attB[roGFP2-Orp1] and NF54-attB[Mito-roGFP2-Orp1] parasites.
To determine whether SBI-0797750 can directly affect H2O2 levels in the cytosol and mitochondrion of P. falciparum N54-attB parasites, mid- and long-term experiments were carried out using NF54-attB[roGFP2-Orp1] and NF54-attB[Mito-roGFP2-Orp1] parasites via CLSM (22). Control studies excluded a direct interaction of SBI-0797750 with the recombinant H2O2 probes. Studies using 4 h of incubation with approximately 1× EC50 ML304 and SBI-0797750 increased significantly the 405/488-nm fluorescence ratio (Fig. 5A). With a time delay, this increase could also be determined in the mitochondrion (Fig. 5B). In comparison, the control compound CQ increased the redox ratio after 4 h (100× EC50) in the cytosol (Fig. S2). However, in contrast to our previous publication (22), this did not reach significance, likely due to biological variations. No increase in redox ratio could be measured in the mitochondrion. Studies of 4 h and 24 h of incubation with the control compound ATS (100× and 10× EC50) showed hardly any effect on the 405/488-nm fluorescence ratio in the cytosol and the mitochondrion (Fig. S2).
FIG 5.

Mid- and long-term effects of ML304 and its derivative SBI-0797750 on the redox ratio of P. falciparum NF54-attB[roGFP2-Orp1] and NF54-attB[Mito-roGFP2-Orp1]-transfected parasites. Four and 24 h of incubation of NF54-attB[roGFP2-Orp1] transfectants with 1× EC50 SBI-0797750 (SBI-750) and ML304 significantly increased the fluorescence ratio of the cytosolic sensor, as determined using CLSM. In NF54-attB[Mito-roGFP2-Orp1] transfectants, ML304 and SBI-0797750 increased the redox ratio after 4 h (A), both of which reached significance after 24 h of incubation (B). Nontreated parasites served as controls. All experiments included fully oxidized (1 mM DIA) and fully reduced (10 mM DTT) parasites. CLSM data comprised 10 to 20 trophozoites analyzed per experiment for each incubation. Values are means and SD (error bars) for three independent experiments. A one-way ANOVA with 95% confidence intervals with the Dunnett’s multiple-comparison test was applied for statistical analysis of significance (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Absence of SBI-0797750 adverse effects on RBCs from G6PD-normal subjects and G6PD-deficient hemizygous males.
Since a very good therapeutic index of SBI-0797750 (Table 1) is a promising prerequisite for therapeutic application, we analyzed potential adverse effects of the substance on the parasites’ host cells, the RBCs, whose integrity depends on HsG6PD activity. The lack of effect of SBI-0797750 was tested in RBCs from G6PD-normal and -deficient donors ex vivo.
First, the potential oxidative effect of the PfGluPho inhibitor SBI-0797750 on RBCs was determined by measuring reduced glutathione (GSH). GSH levels of G6PD-normal RBCs did not change during 1 h and 24 h of incubation with any of the tested inhibitor concentrations up to 20 μM (4,000× IC50 against PfGluPho) (Table 1; Fig. 6A). The same results were obtained for G6PD-deficient RBCs, with the exception of the highest inhibitor concentration (20 μM), which provoked a significant loss (−20%) of GSH (Fig. 6A) after 24 h inhibitor exposure. Under these conditions, GSH dropped to approximately 1 mM within G6PD-deficient RBCs (which is half the normal GSH level in G6PD-normal RBCs). In contrast, treating G6PD-deficient and G6PD-normal RBCs with the HsG6PD inhibitor CB83 (23) for 24 h resulted in a significant GSH decrease at concentrations of 0.8 μM (2× IC50 against HsG6PD) (23) and 4 μM (10× IC50), respectively (Fig. 6B). G6PD-normal or -deficient RBCs pretreated with 2 μM (400× IC50) of SBI-0797750 did not potentiate the 70% GSH drop induced by the oxidative stress of diamide and did not interfere with the GSH recovery rate after exposure (Fig. 6C). These findings confirm the lack of HsG6PD inhibition by SBI-0797750 in intact RBCs at a biologically plausible concentration for parasite growth inhibition.
FIG 6.
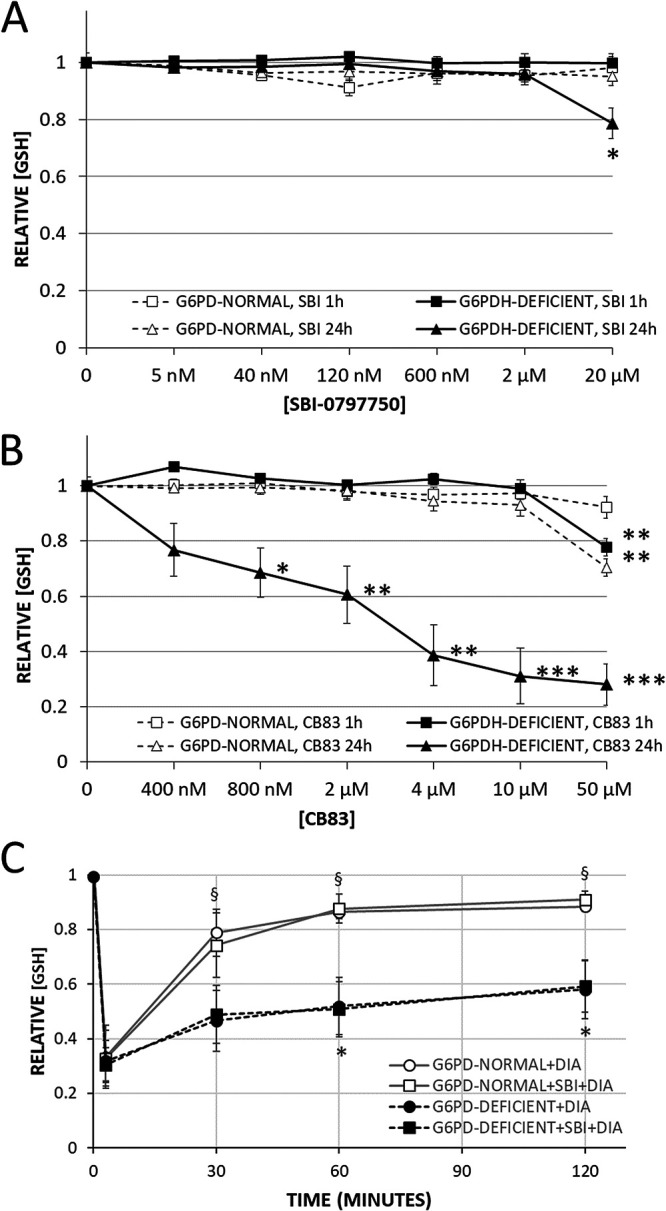
Effect of SBI-0797750 and CB83 on steady-state concentration and recovery of GSH after oxidative challenge in G6PD-normal and G6PD-deficient RBCs. G6PD-normal and -deficient RBCs were incubated with SBI-0797750 (A) or CB83 (B) at indicated concentrations or kept under the same conditions without inhibitor treatment (0 nM). GSH concentrations in RBCs were measured after 1 and 24 h of incubation as indicated. GSH concentrations were normalized by referring them to the hemolysis of respective untreated RBCs (0 nM). Mean GSH concentrations and SE in untreated RBCs (0 nM) were 2.86 ± 0.13 mM and 2.65 ± 0.13 mM for G6PD-normal RBCs (n = 5) and 1.89 ± 0.13 mM and 1.24 ± 0.23 mM for G6PD-deficient ones (n = 8) after 1 and 24 h of incubation, respectively. Normalized GSH concentrations in RBCs (relative [GSH]) of G6PD-normal (n = 5) and G6PD-deficient (n = 8) donors are shown as means and SE. Significant differences relative to untreated RBCs are indicated: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) GSH was measured in RBCs after preincubation with or without 2 μM SBI-0797750, and G6PD-normal and -deficient RBCs were subsequently challenged with 0.5 and 1.0 mM DIA (final concentrations), respectively, at time zero. RBC suspensions were incubated for 3 min at 4°C and afterward at 37°C. GSH values of RBCs measured at 3, 30, 60, and 120 min after DIA supplementation were referred to the corresponding starting GSH value (t = 0) of the same RBC suspension measured immediately before DIA supplementation (relative [GSH]). Mean GSH concentrations and SE of G6PD-normal and -deficient RBCs at time zero were 2.71 ± 0.15 mM and 1.66 ± 0.15 mM, respectively, without SBI-0797750 and 2.82 ± 0.17 mM and 1.68 ± 0.14 mM, respectively, with SBI-0797750. Relative GSH concentrations in RBCs of G6PD-normal (n = 5) and G6PD-deficient (n = 8) donors are shown as means and SE. Significant differences relative to respective values at 3 min (*, P < 0.05) and differences between G6PD-normal and -deficient RBCs (§, P < 0.05) are indicated.
Furthermore, SBI-0797750 was shown to have no major effect on RBC phagocytosis. Oxidatively modified RBCs, vulnerable to phagocytosis, are recognized by spleen phagocytes and removed from circulation in vivo, leading to anemia (24, 25). To exclude adverse effects of the PfGluPho inhibitor leading to oxidation-induced phagocytosis of RBCs, SBI-0797750 was tested in an in vitro phagocytosis assay. For this purpose, RBCs were treated with increasing concentrations of SBI-0797750 for 1 h and 24 h and subsequently exposed to human monocytic phagocytes.
Both parameters quantifying the phagocytosis of RBCs—the amount of phagocytes that phagocytosed (Fig. 7A, C, E, and G) and the mean number of RBCs taken up by a single phagocytosis-positive phagocyte (Fig. 7B, D, F, and H)—consistently show that the PfGluPho inhibitor does not significantly increase the phagocytosis rate of RBCs in G6PD-normal RBCs (up to 20 μM; 4,000× IC50) and G6PD-deficient RBCs (up to 2 μM; 400× IC50) compared to nontreated controls. Only in G6PD-deficient RBCs treated with 20 μM SBI-0797750 was the basal phagocytosis rate (5%) increased (10 to 20%), but it remained low compared to that of the positive control (IgG anti-D-opsonized RBCs), where 30 to 40% phagocytosing THP-1 was detected (Fig. 7). The compound did not harm RBCs during short- or long-term incubations at either low or high hematocrit. These different incubation conditions were used to ensure cytosolic concentrations in RBCs, which might be plausible for an in vivo supplementation of the substance.
FIG 7.

Effect of SBI-0797750 and CB83 on the phagocytosis of G6PD-normal and G6PD-deficient RBCs by human phagocytes. G6PD-normal and -deficient RBCs were incubated at a hematocrit of either 40% (A to D) or 2% (E to H) with SBI-0797750 (SBI) or CB83 at the indicated concentrations for 1 h and 24 h or kept under the same conditions without inhibitor treatment (0 nm). RBCs were fluorescence stained with carboxyfluorescein diacetate succinimidyl ester (CFDA-SE). Erythrophagocytosis by THP-1 phagocytes expressed as a proportion of phagocytosis-positive phagocytes (phTHP-1 [%]) and the number of RBCs phagocytosed per phagocyte (RBCs/phTHP-1) were assessed via FACS. IgG anti-D-opsonized RBCs were included as a positive phagocytosis control. For details, see Materials and Methods. Columns represent means and SE of RBC phagocytosis from G6PD-normal (n = 5) and G6PD-deficient (n = 8) donors. Significant differences compared to untreated RBCs are indicated: *, P < 0.05.
In contrast, 10 μM (25× IC50) of the HsG6PD inhibitor CB83 significantly increased phagocytosis in G6PD-deficient RBCs incubated at low hematocrit, allowing the substance to accumulate in the cytosol over time (Fig. 7). Here, the phagocytosis rate was similar to that of the positive phagocytosis control, IgG anti-D-opsonized RBCs. The highest concentration (125× IC50; 50 μM) of CB83 resulted in significantly increased phagocytosis of G6PD-deficient RBCs, even at high hematocrit, and of G6PD-normal RBCs at low hematocrit.
Finally, a loss of RBC integrity was quantified via hemoglobin release from RBCs, which occurs when the membrane becomes damaged and leaky from the inhibitor, and was expressed as the hemolysis rate. After 24 h of incubation with biologically relevant concentrations of SBI-0797750 (up to 2 μM; 400× IC50), the inhibitor did not induce significant hemolysis in either G6PD-normal or G6PD-deficient cells independent of hematocrit (Fig. 8). Only the highest concentration of the PfGluPho inhibitor tested (20 μM; 4,000× IC50) caused a moderate hemoglobin release. Thus, the basal hemolysis rates in G6PD-normal RBCs, with 2% and 40% hematocrit, were increased by 30% and 80%, respectively, after 1 h of incubation and by 80% and 40% after 24 h. Similarly, G6PD-deficient RBCs were not damaged by SBI-0797750, and only the highest concentration of 20 μM caused hemolysis after 1 h and 24 h of incubation (+80% and +100%, respectively, at 2% hematocrit and +60% and +65% at 40% hematocrit compared to untreated RBCs) (Fig. 8).
FIG 8.

Effect of SBI-0797750 and CB83 on hemolysis in G6PD-normal and -deficient RBCs. G6PD-normal and -deficient RBCs were incubated at a hematocrit of either 40% (A and B) or 2% (C and D) with SBI-0797750 (SBI) (A and C) or CB83 (B and D) at indicated concentrations or kept under the same conditions without inhibitor treatment (0 nM). The hemolysis rate was assessed in terms of hemoglobin release from RBCs and was measured in the supernatant after 1 h and 24 h of incubation. Supernatant hemoglobin was referred to total hemoglobin in the RBC suspension to obtain the hemolysis rate. These rates were normalized by referring any measured value to the hemolysis rate assessed in respective untreated RBCs (0 nM). Mean hemolysis rates and SE in untreated RBCs (0 nM) incubated at a hematocrit of 40% were 0.9% ± 0.09% and 1.47% ± 0.16% for G6PD-normal (n = 5) and 1.17% ± 0.23% and 1.56% ± 0.36% for G6PD-deficient RBCs (n = 8) after 1 h and 24 h of incubation, respectively (A and B). Mean hemolysis rates and SE in untreated RBCs (0 nM) incubated at a hematocrit of 2% were 1.95% ± 0.28% and 2.94% ± 0.48% for G6PD-normal (n = 5) and 2.40% ± 0.26% and 3.56% ± 0.59% for G6PD-deficient RBCs (n = 8) after 1 h and 24 h of incubation, respectively (C and D). Normalized hemolysis rates of RBCs are shown as means and SE of G6PD-normal (n = 5) and G6PD-deficient (n = 8) donors. Significant differences to untreated RBCs are indicated: *, P < 0.05; **, P < 0.01; ***, P ≤ 0.001.
We also showed that the HsG6PD inhibitor CB83 is safe for RBCs up to a concentration of 10 μM (25× IC50) in G6PD-normal and -deficient cells, incubated at 2 or 40% hematocrit. However, inhibitor concentrations of 50 μM (125× IC50) resulted in significantly higher hemolysis rates than in all experiments with SBI-0797750. In G6PD-normal and -deficient RBCs with 2% hematocrit, 2.0- and 3.7-fold-higher values were measured after 1 h and 2.8- and 4.8-fold higher values after 24 h than in untreated RBCs. During RBC incubation at 40% hematocrit with CB83, we observed a dark-brownish discoloration of the RBC suspension (not shown) which is typical of methemoglobin (MetHb) formation. Hemoglobin aggregation and Heinz body formation at the membrane accompany MetHb formation, which might explain the lower hemoglobin release (hemolysis rate) at high hematocrit than at 2% hematocrit (Fig. 8).
DISCUSSION
Bifunctional plasmodial GluPho, including the enzymes G6PD and 6PGL of the oxidative PPP, is a promising target for developing new antimalarial drugs against P. falciparum, since the enzyme differs structurally and functionally from the human orthologs and is essential for blood-stage parasites (15, 16). The enzyme shares high sequence identity to its orthologs in other Plasmodium species such as P. vivax (PvGluPho) (71.2% identity and 79.8% similarity at the amino acid level [26]), increasing the probability that potential drugs may also inhibit the G6PD of other Plasmodium species. Previously we identified two promising compounds, ML276 (18) and ML304 (19), which selectively inhibited PfGluPho in the nanomolar range via a high-throughput screening of about 400,000 molecules (17). In this study, we present the optimization of the dibenzothiazepine-derived second probe, ML304, to a single-digit-nanomolar lead compound, SBI-0797750.
ML304 inhibits recombinant PfGluPho with an IC50 of 190 nM (19) and P. falciparum 3D7 asexual blood stages with an EC50 of 471.3 ± 39.5 nM (26). Medicinal chemistry optimization around the ML304 scaffold led to SBI-0797750, which is approximately 25-fold more active against the recombinant enzyme (IC50 = 6.7 ± 1.8 nM) and in vitro (EC50 = 22.5 ± 2.2 nM). Consistent with previous results on ML304 (EC50 = 1.25 μM [27]), initial experiments with SBI-0797750 showed that it is also active against sexual P. falciparum gametocytes (EC50 = 73.9 ± 22 nM). These preliminary results suggest that SBI-0797750 could potentially also be used as a transmission-blocking agent and is currently being investigated in more detail.
Furthermore, the optimization did not change the nature of inhibition since we confirmed a competition with the substrate G6P and a mixed type of inhibition against NADP+ (Fig. 2). In line with that, we could also show that the inhibitor is fully reversible on increasing the substrate to compound ratio (Fig. 3).
As shown for ML304 (20), SBI-0797750 also inhibits the corresponding enzyme of P. vivax—PvG6PD—with an IC50 of 31.0 ± 3.1 nM which is comparable to the inhibitory activity on the P. falciparum enzyme. This value is about 2 orders of magnitude lower than ML304 (IC50 = 2.6 ± 0.8 μM [26]). Since the G6P and NADP+ binding sites are highly conserved in Plasmodium, the higher IC50 of PvG6PD compared to the full-length enzyme PfGluPho might be explained by the structural disadvantage of the truncated enzyme, also showing higher Km values (26). Consequently, higher substrate concentrations were used in the PvG6PD inhibition studies, providing another possible explanation for the higher IC50 against PvG6PD, as SBI-0797750 competes with the G6P binding pocket. Since the human enzyme was hardly inhibited up to a concentration of 99 μM, SBI-0797750 represents a highly selective and robust inhibitor of plasmodial GluPhos with in vitro activity comparable to those of FDA-approved antimalarials such as CQ and ATS (28).
Investigations of SBI-0797750 and ML304 with the cytosolically expressed glutathione redox sensor hGrx1-roGFP2 and with the cytosolically and mitochondrially expressed H2O2 biosensors roGFP2-Orp1 and Mito-roGFP2-Orp1, respectively, revealed that the mode of action of these drugs is likely linked to a disruption of glutathione and H2O2 homeostasis. The cytosolic probes demonstrated that oxidative effects were already occurring at 4 h of incubation with low nanomolar concentrations (1× EC50) of SBI-0797750, comparable to ML304. These effects were even increased in the cytosolic roGFP2-Orp1 sensor after 24 h, at which time the oxidation of the mitochondrial probes reached significance as well. In contrast, the clinically used drug CQ only increased the 405/488-nm ratio of the cytosolic roGFP2-Orp1 sensor in the 4-h experiment (100× EC50), but not significantly. The antimalarial ATS hardly affected the 405/488-nm ratio of all tested sensors and under all conditions. These results clearly indicate that the mode of action of ML304 and SBI-0797750 in P. falciparum blood stages is likely linked to GSH and H2O2 homeostasis, which perfectly fits the role of G6PD as a central and essential enzyme for redox balance.
Similarly to Plasmodium parasites, host RBCs are dependent on NADPH generation by its own G6PD. It was therefore important to demonstrate that SBI-0797750 did not harm RBCs from either G6PD-normal or G6PD-deficient individuals when the inhibitor was supplemented to cells ex vivo. Both mechanisms of RBC damage in vivo, hemolysis and phagocytosis, which result in anemia, were not increased by SBI-0797750 when added in a relevant concentration.
In conclusion, with the optimization of ML304 to SBI-0797750, we have developed a very potent and selective PfGluPho and PvG6PD inhibitor that gives robust inhibition in P. falciparum cellular assays. The excellent selectivity of SBI-0797750 over the human enzyme should result in a significant therapeutic window. Indeed, SBI-0797750 has great promise as a first-in-class therapeutic, and in vivo studies on humanized mouse models are under way to explore this utility. A cross-species effect of our G6PD inhibitor against a range of other parasites will be investigated to determine if this compound may have other applications.
MATERIALS AND METHODS
Drugs and chemicals.
All chemicals used were of the highest available purity and were obtained from Roth (Karlsruhe, Germany), Sigma-Aldrich (Steinheim, Germany), or Merck (Darmstadt, Germany). RPMI 1640 medium was purchased from Gibco (Paisley, United Kingdom), chloroquine (CQ) and artesunate (ATS) from Sigma-Aldrich (Steinheim, Germany), methylene blue (MB) and artemisinin (ART) from Roth (Karlsruhe, Germany), and SYBR green I from Thermo Scientific (Schwerte, Germany). SBI-0797750 was prepared in a fashion similar to that described for ML304 (19). Stock solutions of SBI-0797750, MB, and ART were prepared in dimethyl sulfoxide (DMSO); CQ was dissolved in double-distilled water (ddH2O).
Production and purification of recombinant PfGluPho, PvG6PD, and HsG6PD.
The recombinant enzymes used for kinetic characterization of SBI-0797750 were produced as described by Jortzik et al. (15). Since the recombinant production of full-length PvGluPho remained challenging, we used the isolated PvG6PD domain, representing the C-terminal part of the bifunctional enzyme (26), to characterize the inhibitory action of SBI-0797750.
Resazurin-/diaphorase-coupled assay.
Dose-response curves of SBI-0797750 for PfGluPho, PvG6PD, and HsG6PD were determined as described previously with slight modifications (18). Briefly, various volumes of compound in 100% DMSO (highest final compound concentration of 4 μM for PfGluPho and PvG6PD and 79 μM for HsG6PD) were transferred to 1,536-well plates using an Echo 550 (Labcyte) acoustic dispenser. One volume of enzyme mix (final concentrations, 50 mM Tris [pH 7.5], 0.005% Tween 20, 1 mg mL−1 BSA, 0.075 μg mL−1 PfGluPho/0.15 μg mL−1 PvG6PD/0.15 μg mL−1 HsG6PD) was added to all wells using the Multidrop Combi reagent dispenser (Thermo Fisher). To start the reaction, 1 volume substrate mix (final concentrations, 50 mM Tris [pH 7.5], 0.005% Tween 20, 1 mg mL−1 bovine serum albumin [BSA], 3.3 mM MgCl2, 1 U mL−1 diaphorase, 25/60/140 μM resazurin, 20/30/125 μM G6P, 4/12/6 μM NADP+ for PfGluPho/PvG6PD/HsG6PD) was added to the wells. Substrate mix without G6P served as a positive control (100% inhibition), and a mix without compound but with additional DMSO served as a negative control (100% activity). The plates were centrifuged at 1,500 rpm for 1 min and incubated in the dark for 2 h. Increasing fluorescence of resorufin was measured at an excitation wavelength of 530 nm and an emission wavelength of 580 nm (ex530/em580) using a ViewLux (PerkinElmer) plate reader. The reaction rate was calculated by dividing the relative fluorescence units (RFU) by time, and IC50 values were calculated using CBIS (Chemical and Biological Information Systems [www.cheminnovation.com]).
Orthogonal assay.
In order to exclude compounds that interfere with resazurin or diaphorase, IC50s were determined using the orthogonal enzyme assay without coupled resazurin and diaphorase. Plates were prepared as described above. For PfGluPho and PvG6PD, a 4 μM concentration of the highest compound concentration was used; for HsG6PD, 99 μM was used. The enzyme mix contained 0.05 μg mL−1 PfGluPho, 0.025 μg mL−1 PvG6PD, and 0.0045 μg mL−1 HsG6PD. The reaction was started by adding the substrate mix described above but without resazurin and diaphorase. After centrifuging the plates, NADPH production was monitored at ex350/em460 over 45 min with a PHERAstar multiwell plate reader. IC50s were calculated as described above.
Mechanism of inhibition.
SBI-0797750 was mechanistically characterized as described previously with slight modifications (17). Plates and PfGluPho concentrations were prepared as described above using the orthogonal assay with substrate and enzyme mix. To determine the mode of inhibition, either G6P or NADP+ were titrated at various constant concentrations against different compound concentrations, keeping the second substrate in saturation (200 μM). Measurement without adding substrate served as a positive control. Increasing NADPH fluorescence was monitored as described for the orthogonal assay. The relationship between initial velocity (v0) and substrate concentrations ([S]) at various constant compound concentrations was analyzed by replotting the data in a Lineweaver-Burk plot and calculating 1/Km and 1/Vmax values with the GraphPad Prism 8 software (GraphPad, San Diego, CA, USA) to determine the compound mode of inhibition (competitive or mixed type).
Reversibility of the inhibition.
The reversibility of the PfGluPho inhibition by SBI-0797750 was tested by incubating the enzyme with high compound concentrations, followed by dilution to a compound concentration below the IC50 and subsequent determination of the enzyme activity. In detail, 1.65 μM PfGluPho was incubated with 1.65 μM SBI-0797750 for 10 min at 22°C. Afterward, the enzyme-inhibitor mixture was diluted to a compound concentration below IC50 (3.3 nM) and again incubated for 10 min at 22°C to allow dissociation. Afterward, NADP+ and G6P were added to start the reaction, either at substrate saturation (200 μM) or at concentrations close to the Km (6.5 μM NADP+, 19 μM G6P). For comparison, controls containing either no inhibitor (100% activity) or inhibitor at the concentration remaining after dilution were prepared. NADPH production was monitored at ex340 using the Tecan Infinite M200 plate reader (Tecan, Maennedorf, Switzerland) in 96-well plates. Specific activities were calculated from reaction rates. Inhibition was considered irreversible if the activity of the sample after dilution was significantly below the activity of the controls. Inhibition was considered reversible if the activity of the sample was equal to that of the controls.
Cloning and heterologous overexpression of the hGrx1-roGFP2, roGFP2-Orp1, and Mito-roGFP2-Orp1 constructs.
The genetically encoded ratiometric sensor of the intracellular redox potential, hGrx1-roGFP2, was previously genomically integrated and stably expressed in NF54-attB parasites by using the pDC2-CAM-attP expression vector (21). Heterologous overexpression of hGrx1-roGFP2 to evaluate the in vitro interactions of SBI-0797750 and antimalarial drugs with the recombinant redox probe was performed according to Kasozi et al. (28). The genetically encoded ratiometric sensor of cytosolic H2O2 metabolism roGFP2-Orp1 and the mitochondrion-targeted sensor Mito-roGFP2-Orp1 were recently stably expressed in NF54-attB parasites by using the pDC2-CAM-attP expression vector (22). Heterologous overexpression of roGFP2-Orp1 to evaluate the in vitro interactions of SBI-0797750 and antimalarial drugs with the recombinant redox probe was performed according to Rahbari et al. (29).
Cultivation and transfection of P. falciparum.
The CQ-sensitive P. falciparum 3D7 and NF54-attB strains were cultivated and transfected according to Kasozi et al. (28) and Rahbari et al. (22), respectively. Briefly, the strains were propagated in A+ RBCs in RPMI 1640 medium supplemented with 0.5% (wt/vol) Albumax, 9 mM glucose, 0.2 mM hypoxanthine, 2.1 mM l-glutamine, 25 mM HEPES, and 22 μg mL−1 gentamicin at 3.3% hematocrit and 37°C in a gaseous mixture consisting of 3% O2, 3% CO2, and 94% N2. Deviating from that, NF54 parasites for the gametocyte assays were cultured in O+ RBCs at 10% hematocrit with 10% human serum instead of Albumax. Plasmodium falciparum parasites were synchronized with sorbitol and Percoll treatment (30, 31). To generate pure gametocyte cultures, prior to induction of sexual commitment, highly synchronous asexual parasites were seeded into multiple T75 flasks at a starting parasitemia of 0.06%, followed by daily medium changes of half the culture volume. Sexual commitment was induced on day 5 postsetup by doubling the medium volume (32, 33). Subsequently, medium was changed daily, with fresh medium containing 50 mM N-acetyl-d-glucosamine (GluNAc) to prevent asexual parasite development (34). The cultures were maintained for 8 to 10 days until pure gametocyte cultures were observed and were again enriched with Percoll for follow-up experiments. P. falciparum trophozoites were enriched via magnetic separation (35). Cell lysates were obtained via saponin lysis (36). Parasitemia was counted on Giemsa-stained blood smears.
In vitro activity of SBI-0797750 against P. falciparum 3D7 and NF54-attB asexual parasites.
Parasite growth inhibition was studied, and the half maximal effective concentration (EC50) of antimalarial drugs or compounds against P. falciparum was calculated using either the [3H]hypoxanthine incorporation assay according to Kasozi et al. (28) or the SYBR green I-based fluorescence assay for parasite nucleic acids according to Ekland et al. (37) in 96-well format with modifications. For the [3H]hypoxanthine assay, the DMSO-diluted compounds were serially double diluted in hypoxanthine-free medium in 96-well microtiter plates. Synchronized ring-stage parasites in hypoxanthine-free complete medium were added to each well (0.15% parasitemia, 1.25% final hematocrit) and incubated for 48 h under cell culture conditions. Negative-control wells contained only RBCs, and positive-control wells contained only infected RBCs (iRBCs). Afterward, [3H]hypoxanthine was added at a final concentration of 0.5 μCi/well, and the plate further incubated for 24 h. Plates were then frozen at −80°C for at least 1 h and thawed, and cells were harvested on glass fiber filters. After drying, the radioactivity in counts per minute from each well was measured and calculated proportional to the growth of P. falciparum in comparison to RBCs and iRBCs.
For SYBR green I assays, the DMSO-diluted compounds were serially double diluted in complete medium in 96-well microtiter plates. Synchronized ring-stage parasites in complete medium were added to each well (0.15% parasitemia, 1.25% final hematocrit) and incubated for 48 h (NF54-attB) or 44 h (3D7) at 37°C. Then, 20 μL of 5× SYBR green (10,000× stock solution) in lysis buffer (20 mM Tris-HCl, 5 mM EDTA, 0.16% [wt/vol] saponin, and 1.6% [vol/vol] Triton X-100) were added to each well and incubated in the dark for 24 h at room temperature (RT). Fluorescence was measured in the Clariostar plate reader at ex494/em530. For EC50 calculation, the percent growth inhibition was plotted against the log drug concentration using GraphPad Prism 8 software (GraphPad, San Diego, CA, USA).
In vitro activity of SBI-0797750 against P. falciparum NF54 gametocytes.
To test SBI-0797750 on P. falciparum NF54 gametocytes, sexual commitment was first induced as described above. At 9 to 11 days after induction of sexual commitment, when pure gametocyte cultures were observed, similar numbers of the mature stage V gametocytes were plated in 96-well plates. DMSO-diluted SBI-0797750 stock was serially double diluted in medium and added to each well. Each 96-well plate per experiment contained at least four wells that were cultured without drugs (positive control), three wells of DMSO-only controls, and a varying number of duplicate wells with decreasing concentrations of the inhibitor. Upon addition of the inhibitor, the 96-well plate was left for 3 days without medium change. Subsequently, smears of each well were prepared, Giemsa stained, and counted. The gametocyte numbers of the drug-cultured wells were related to the number of gametocytes observed in the positive control. GraphPad Prism 8 software was used to prepare graphs and calculate EC50s.
In vitro characterization of SBI-0797750 using recombinant hGrx1-roGFP2 and roGFP2-Orp1.
SBI-0797750 was used at 10 nM to 1 mM in degassed standard reaction buffer (100 mM potassium phosphate, 1 mM EDTA; pH 7.0). The in vitro interaction of the pharmacologically used antimalarial drugs ATS and CQ with the purified hGrx1-roGFP2 protein was previously determined and did not have an effect at the concentrations used in this study (28). For roGFP2-Orp1, ATS and CQ did not affect the fluorescence ratio of the probe either (29). ML304 was measured as described by Haeussler et al. (26). The in vitro interactions of hGrx1-roGFP2 and roGFP2-Orp1 with SBI-0797750 were characterized as described by Schuh et al. (21) and Rahbari et al. (22), respectively.
Confocal imaging and image processing.
A Leica confocal system TCS SP5 inverted microscope equipped with the objective HCX PL APO 63.0× 1.30 GLYC 37°C UV connected to a 37°C temperature chamber was used. The argon laser power was set to 20%; scanning was performed at a frequency of 400 Hz and a resolution of 512 by 512 pixels. The smart gain and smart offset were 950 V and −0.9%, respectively. With a sequential scan, we excited the hGrx1-roGFP2 and roGFP2-Orp1/Mito-roGFP2-Orp1 probes at 405 nm and 488 nm and detected emission at 500 to 550 nm. Laser intensity was adjusted to match the full dynamic range of the probe to the dynamic range of the detector (hGrx1-roGFP2: 405 nm, 10%; 488 nm, 4%; roGFP2-Orp1: 405 nm, 10%; 488 nm, 4%; Mito-roGFP2-Orp1: 405 nm, 10%; 488 nm, 5%). Autofluorescence images were simultaneously taken at ex405/em430 to 450 and were individually defined together with the background for every image, but no fluorescence signal could be detected. Leica LAS AF Lite software for fluorescence analysis was used. The 405/488-nm ratio was calculated. The graphs were plotted using GraphPad Prism 8 software (GraphPad, San Diego, CA, USA). For imaging, only parasites showing fluorescent signals at both 405 and 488 nm excitation and an intact host cell were chosen.
Effect of SBI-0797750 and other antimalarials on glutathione and H2O2 redox homeostasis.
The effect of ML304, its derivative SBI-0797750, and the antimalarial drugs ATS and CQ as reference drugs on P. falciparum were investigated in 4 h and 24 h of incubation experiments. The EC50 of ATS (4.2 nM), CQ (6.9 nM), and ML304 (1.7 μM) on NF54-attB parasites were determined previously (22). Trophozoite stage parasites (26 to 30 h post invasion) of NF54-attB[hGrx1-roGFP2], NF54-attB[roGFP2-Orp1], and NF54-attB[Mito-roGFP2-Orp1] (6 to 8% parasitemia) were treated with different SBI-0797750, ML304, ATS, and CQ concentrations (1× to 100× EC50) for 4 h. Subsequently, free thiol groups were blocked with 2 mM N-ethylmaleimide (NEM) for 15 min at 37°C. The parasites were magnetically enriched (Miltenyi Biotec, Germany) and returned to cell culture for at least 1 h to recover. For 24 h experiments, a 5-mL culture (5% hematocrit, 6 to 8% parasitemia) of ring-stage parasites (6 to 10 h postinvasion) was treated with different concentrations of SBI-0797750, ML304, ATS, and CQ (1× to 100× EC50). Prior to enrichment, cysteines were blocked with 2 mM NEM for 15 min at 37°C. Enriched parasites were returned to cell culture to recover for at least 1 h. Thereafter, cells were washed and resuspended in Ringer’s solution (122.5 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 0.8 mM MgCl2, 11 mM d-glucose, 25 mM HEPES, 1 mM NaH2PO4; pH 7.4). Fifty microliters of cells were seeded onto poly-l-lysine-coated μ-slides with 18 wells (flat) (Ibidi, Martinsried, Germany) and measured using CLSM (confocal laser scanning microscopy) with excitation wavelengths of 405 nm and 488 nm. All experiments included nontreated parasites as controls and both fully reduced and fully oxidized parasites with 10 mM dithiothreitol (DTT) and 1 mM diamide (DIA), respectively (2 min incubation), prior to blocking with NEM. Each experiment was carried out three times. Between 10 and 20 microscopy images were taken each time, resulting in at least 30 experimental values per concentration and incubation time. Means and errors (indicated as standard deviations [SD]) are shown. A one-way analysis of variance (ANOVA) with 95% confidence intervals with Dunnett’s multiple-comparison test (GraphPad Prism 8 software; GraphPad, San Diego, CA, USA) was applied for statistical analysis of significance. OxD of the glutathione redox sensor was calculated as follows:
R represents the 405/488 nm fluorescence ratio, Rred and Rox are the 405/488-nm fluorescence ratios of fully reduced or fully oxidized parasites, I485ox represents the fluorescence intensity (FI) at 488 nm for fully oxidized parasites, and I485red is the FI at 488 nm for fully reduced parasites.
Treatment of RBCs with PfGluPho inhibitor SBI-0797750 and HsG6PD inhibitor CB83.
This study was carried out in accordance with the Declaration of Helsinki and authorized by the local ethical committee. Blood samples were collected in heparin-containing vacutainers from G6PD-normal and -deficient adult donors after confirmed consent. RBCs were washed three times with phosphate-buffered saline containing 10 mM glucose (PBS-G) to deplete white blood cells and platelets. Cells were sedimented at 1,200 × g for 5 min after each washing step. A final washing step was performed in incubation buffer (50 mM HEPES, pH 7.4, supplemented with 110 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM NaH2PO4, 20 mM glucose). RBCs were sedimented at 2,700 × g for 5 min. The cell pellet was resuspended in incubation buffer at a hematocrit of 40% and incubated in a humidified CO2-air incubator at 37°C for recovery. After 1 h, an aliquot of RBC suspension was further diluted with incubation buffer to 2% hematocrit. Cell suspensions of 2 and 40% hematocrit were aliquoted and incubated with increasing concentrations of SBI-0797750 (between 5 nM and 20 μM; 1× to 4,000× IC50 against PfGluPho) and CB83 (between 400 nM and 50 μM; 1× to 125× IC50 against HsG6PD) (23). Inhibitor substances were first dissolved in DMSO to obtain 10 mM stock solutions. Before addition to the RBC suspension, the inhibitors were further diluted in PBS to obtain suitable working concentrations. Nontreated controls without inhibitor (0 nM) were instead supplemented with equivalent amounts of DMSO. The incubation of RBC suspension was performed under gentle movement in a CO2-air cell incubator at 37°C for 24 h. Aliquots of incubated suspensions were taken at 1 and 24 h for analysis of GSH, hemolysis, and phagocytosis and at 2 h for diamide challenge.
Effect of SBI-0797750 and the HsG6PD inhibitor CB83 on reduced GSH and recovery from oxidative stress with diamide in RBCs.
Aliquots were taken from RBCs suspended at a hematocrit of 40% and incubated with different inhibitor concentrations for 1 and 24 h to determine the effect of the G6PD inhibitors on GSH oxidation. A colorimetric assay of GSH in RBCs was run with Ellman’s reagent after precipitation of high-molecular-weight thiols with metaphosphoric acid as described by Beutler (38). Optical density (OD) was determined at 412 nm. GSH was measured in duplicate, and values were calculated as μmol/mL packed RBCs. These values were normalized to the GSH concentration in RBCs of the respective donor incubated in parallel without inhibitor supplementation (0 nM). Additionally, the effect of SBI-0797750 on GSH recovery was tested in RBCs, which were oxidatively challenged with diamide. For this, RBCs were preincubated with or without 2 μM SBI-0797750 (400× IC50 against PfGluPho) at 40% hematocrit in a CO2-air cell incubator at 37°C for 2 h. Thereafter, cells were placed on ice and supplemented with 0.5 and 1 mM diamide (final concentrations in G6PD-deficient and G6PD-normal RBC suspensions, respectively) at time zero (0 min). RBC suspensions were kept for 3 min on ice and then transferred to 37°C for another 2 h to monitor the recovery of reduced glutathione. Aliquots of the suspension were taken at 0, 3, 30, 60, and 120 min for GSH quantification. The values from RBCs of 5 G6PD-normal and 8 G6PD-deficient donors are presented as means and standard errors (SE). Statistical significance was calculated with a nonparametric Mann-Whitney test.
Effect of SBI-0797750 and the HsG6PD inhibitor CB83 on the hemolysis of nonparasitized G6PD-deficient and G6PD-normal RBCs.
Hemolysis was determined by measuring hemoglobin release from incubated RBCs into the supernatant. For this, aliquots from RBC suspensions were taken at 1 and 24 h of incubation and centrifuged at 1,200 × g for 5 min. The collected supernatant was further centrifuged at 16,000 × g for 15 s to sediment cell debris. Aliquots of the clear RBC-free supernatants and of the entire RBC suspension were solubilized in duplicate with 0.1 M NaOH containing 3 mM EDTA and 0.05% (vol/vol). Triton X-100, and hemoglobin content was quantified with heme-dependent luminol-enhanced luminescence (39). The hemolysis rate was calculated by referring the heme content of the RBC-free supernatant to that of respective RBC suspension. Statistical values and analysis were carried out as described above.
Effect of SBI-0797750 and HsG6PD inhibitor CB83 on the phagocytosis of nonparasitized G6PD-deficient and G6PD-normal RBCs by human phagocytes.
Treated or nontreated RBCs with increasing concentrations of G6PD inhibitors were used in an in vitro phagocytosis assay with human monocytes of the immortalized cell line THP-1 to verify potential damage to RBCs that leads to phagocytosis. Fluorescence-labeled and opsonized RBCs were added to suspend and preactivate THP-1 cells as described by Gallo et al. (40). The occurrence of phagocytosis was assessed via flow cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA), quantifying fluorescence-positive THP-1 cells and the intensity of fluorescence per phagocyte. Both parameters mirror the vulnerability of RBCs for phagocytosis. To standardize the phagocyte activity between different experiments, IgG-anti-D-opsonized RBCs were included as a positive phagocytosis control, and phagocytosis values can be referred to this internal cellular standard (40). Statistical values and analysis were carried out as described above.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01AI104916 to L.B. and A.B.P. and in part by an NIH Molecular Libraries grant (U54HG005033) to the Conrad Prebys Center for Chemical Genomics at the Sanford Burnham Medical Research Institute, one of the comprehensive centers of the NIH Molecular Libraries Probe Production Centers Network (MLPCN) during the 2008 to 2014 term of the NIH Molecular Libraries Program production phase. The Hessian LOEWE Center DRUID (project B3) and the German Research Foundation (grant BE1540/23-2 within the DFG Priority Program 1710 on Thiol Switches) also supported this work.
We thank Siegrid Franke for her excellent technical assistance. We thank David A. Fidock for sharing the NF54-attB strain with us.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.WHO. 2020. World Malaria Report 2020. https://www.who.int/publications/i/item/9789240015791.
- 2.Cowman AF, Healer J, Marapana D, Marsh K. 2016. Malaria: biology and disease. Cell 167:610–624. 10.1016/j.cell.2016.07.055. [DOI] [PubMed] [Google Scholar]
- 3.Naing C, Whittaker MA, Nyunt Wai V, Mak JW. 2014. Is Plasmodium vivax malaria a severe malaria?: a systematic review and meta-analysis. PLoS Negl Trop Dis 8:e3071. 10.1371/journal.pntd.0003071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO. 2017. World malaria report 2017. http://apps.who.int/iris/bitstream/handle/10665/259492/9789241565523-eng.pdf;jsessionid=5AC595247B17EBAC2AA1EE5F4BD070CB?sequence=1.
- 5.Mbengue A, Bhattacharjee S, Pandharkar T, Liu H, Estiu G, Stahelin RV, Rizk SS, Njimoh DL, Ryan Y, Chotivanich K, Nguon C, Ghorbal M, Lopez-Rubio J-J, Pfrender M, Emrich S, Mohandas N, Dondorp AM, Wiest O, Haldar K. 2015. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520:683–687. 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, Chotivanich K, Imwong M, Pukrittayakamee S, Dhorda M, Nguon C, Lim P, Amaratunga C, Suon S, Hien TT, Htut Y, Faiz MA, Onyamboko MA, Mayxay M, Newton PN, Tripura R, Woodrow CJ, Miotto O, Kwiatkowski DP, Nosten F, Day NPJ, Preiser PR, White NJ, Dondorp AM, Fairhurst RM, Bozdech Z. 2015. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347:431–435. 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cappellini MD, Fiorelli G. 2008. Glucose-6-phosphate dehydrogenase deficiency. Lancet 371:64–74. 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 8.Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Serrano-Posada H, Ortega-Cuellar D, González-Valdez A, Castillo-Rodríguez RA, Hernández-Ochoa B, Sierra-Palacios E, Rodríguez-Bustamante E, Arreguin-Espinosa R. 2016. Glucose-6-phosphate dehydrogenase: update and analysis of new mutations around the world. Int J Mol Sci 17:2069. 10.3390/ijms17122069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arese P, Turrini F, Schwarzer E. 2005. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell Physiol Biochem 16:133–146. 10.1159/000089839. [DOI] [PubMed] [Google Scholar]
- 10.Cappadoro M, Giribaldi G, O'Brien E, Turrini F, Mannu F, Ulliers D, Simula G, Luzzatto L, Arese P. 1998. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 92:2527–2534. 10.1182/blood.V92.7.2527.2527_2527_2534. [DOI] [PubMed] [Google Scholar]
- 11.Howes RE, Battle KE, Satyagraha AW, Baird JK, Hay SI. 2013. G6PD deficiency: global distribution, genetic variants and primaquine therapy. Adv Parasitol 81:133–201. 10.1016/B978-0-12-407826-0.00004-7. [DOI] [PubMed] [Google Scholar]
- 12.Ashley EA, Pyae Phyo A, Woodrow CJ. 2018. Malaria. Lancet 391:1608–1621. 10.1016/S0140-6736(18)30324-6. [DOI] [PubMed] [Google Scholar]
- 13.Jortzik E, Becker K. 2012. Thioredoxin and glutathione systems in Plasmodium falciparum. Int J Med Microbiol 302:187–194. 10.1016/j.ijmm.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Preuss J, Jortzik E, Becker K. 2012. Glucose-6-phosphate metabolism in Plasmodium falciparum. IUBMB Life 64:603–611. 10.1002/iub.1047. [DOI] [PubMed] [Google Scholar]
- 15.Jortzik E, Mailu BM, Preuss J, Fischer M, Bode L, Rahlfs S, Becker K. 2011. Glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase: a unique bifunctional enzyme from Plasmodium falciparum. Biochem J 436:641–650. 10.1042/BJ20110170. [DOI] [PubMed] [Google Scholar]
- 16.Allen SM, Lim EE, Jortzik E, Preuss J, Chua HH, MacRae JI, Rahlfs S, Haeussler K, Downton MT, McConville MJ, Becker K, Ralph SA. 2015. Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase is a potential drug target. FEBS J 282:3808–3823. 10.1111/febs.13380. [DOI] [PubMed] [Google Scholar]
- 17.Preuss J, Hedrick M, Sergienko E, Pinkerton A, Mangravita-Novo A, Smith L, Marx C, Fischer E, Jortzik E, Rahlfs S, Becker K, Bode L. 2012. High-throughput screening for small-molecule inhibitors of Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase. J Biomol Screen 17:738–751. 10.1177/1087057112442382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preuss J, Maloney P, Peddibhotla S, Hedrick MP, Hershberger P, Gosalia P, Milewski M, Li YL, Sugarman E, Hood B, Suyama E, Nguyen K, Vasile S, Sergienko E, Mangravita-Novo A, Vicchiarelli M, McAnally D, Smith LH, Roth GP, Diwan J, Chung TDY, Jortzik E, Rahlfs S, Becker K, Pinkerton AB, Bode L. 2012. Discovery of a Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase inhibitor (R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzob1,4thiazine-6-carboxamide (ML276) that reduces parasite growth in vitro. J Med Chem 55:7262–7272. 10.1021/jm300833h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maloney P, Hedrick M, Peddibhotla S, Hershberger P, Milewski M, Gosalia P, Li L, Preuss J, Sugarman E, Hood B, Suyama E, Nguyen K, Vasile S, Sergienko E, Salanawil S, Stonich D, Su Y, Dahl R, Mangravita-Novo A, Vicchiarelli M, McAnally D, Smith LH, Roth G, Diwan J, Chung TD, Pinkerton Ab Bode L, Becker K. 2010. Probe Reports from the NIH Molecular Libraries Program. A 2nd selective inhibitor of Plasmodium falciparum glucose-6-phosphate dehydrogenase (PfG6PDH)—probe 2. NIH, Bethesda, MD. [PubMed] [Google Scholar]
- 20.Haeussler K. 2019. Characterization and inhibition of NADPH-producing enzymes from the pentose phosphate pathway of Plasmodium parasites. Doctoral thesis. Justus-Liebig-Universität Gießen, Giessen, Germany. [Google Scholar]
- 21.Schuh AK, Rahbari M, Heimsch KC, Mohring F, Gabryszewski SJ, Weder S, Buchholz K, Rahlfs S, Fidock DA, Becker K. 2018. Stable integration and comparison of hGrx1-roGFP2 and sfroGFP2 redox probes in the malaria parasite Plasmodium falciparum. ACS Infect Dis 4:1601–1612. 10.1021/acsinfecdis.8b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahbari M, Rahlfs S, Przyborski JM, Schuh AK, Hunt NH, Fidock DA, Grau GE, Becker K. 2017. Hydrogen peroxide dynamics in subcellular compartments of malaria parasites using genetically encoded redox probes. Sci Rep 7:10449. 10.1038/s41598-017-10093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Preuss J, Richardson AD, Pinkerton A, Hedrick M, Sergienko E, Rahlfs S, Becker K, Bode L. 2013. Identification and characterization of novel human glucose-6-phosphate dehydrogenase inhibitors. J Biomol Screen 18:286–297. 10.1177/1087057112462131. [DOI] [PubMed] [Google Scholar]
- 24.Becker K, Tilley L, Vennerstrom JL, Roberts D, Rogerson S, Ginsburg H. 2004. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int J Parasitol 34:163–189. 10.1016/j.ijpara.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Uyoga S, Skorokhod OA, Opiyo M, Orori EN, Williams TN, Arese P, Schwarzer E. 2012. Transfer of 4-hydroxynonenal from parasitized to non-parasitized erythrocytes in rosettes. Proposed role in severe malaria anemia. Br J Haematol 157:116–124. 10.1111/j.1365-2141.2011.09015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haeussler K, Berneburg I, Jortzik E, Hahn J, Rahbari M, Schulz N, Preuss J, Zapol'skii VA, Bode L, Pinkerton AB, Kaufmann DE, Rahlfs S, Becker K. 2019. Glucose 6-phosphate dehydrogenase 6-phosphogluconolactonase: characterization of the Plasmodium vivax enzyme and inhibitor studies. Malar J 18:22. 10.1186/s12936-019-2651-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siciliano G, Santha Kumar TR, Bona R, Camarda G, Calabretta MM, Cevenini L, Davioud-Charvet E, Becker K, Cara A, Fidock DA, Alano P. 2017. A high susceptibility to redox imbalance of the transmissible stages of Plasmodium falciparum revealed with a luciferase-based mature gametocyte assay. Mol Microbiol 104:306–318. 10.1111/mmi.13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasozi D, Mohring F, Rahlfs S, Meyer AJ, Becker K. 2013. Real-time imaging of the intracellular glutathione redox potential in the malaria parasite Plasmodium falciparum. PLoS Pathog 9:e1003782. 10.1371/journal.ppat.1003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahbari M, Rahlfs S, Jortzik E, Bogeski I, Becker K. 2017. H2O2 dynamics in the malaria parasite Plasmodium falciparum. PLoS One 12:e0174837. 10.1371/journal.pone.0174837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 31.Wahlgren M, Berzins K, Perlmann P, Björkman A. 1983. Characterization of the humoral immune response in Plasmodium falciparum malaria. I. Estimation of antibodies to P falciparum or human erythrocytes by means of microELISA. Clin Exp Immunol 54:127–134. [PMC free article] [PubMed] [Google Scholar]
- 32.Buchholz K, Burke TA, Williamson KC, Wiegand RC, Wirth DF, Marti M. 2011. A high-throughput screen targeting malaria transmission stages opens new avenues for drug development. J Infect Dis 203:1445–1453. 10.1093/infdis/jir037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fivelman QL, McRobert L, Sharp S, Taylor CJ, Saeed M, Swales CA, Sutherland CJ, Baker DA. 2007. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol Biochem Parasitol 154:119–123. 10.1016/j.molbiopara.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Gupta SK, Schulman S, Vanderberg JP. 1985. Stage-dependent toxicity of N-acetyl-glucosamine to Plasmodium falciparum. J Protozool 32:91–95. 10.1111/j.1550-7408.1985.tb03020.x. [DOI] [PubMed] [Google Scholar]
- 35.Paul F, Roath S, Melville D, Warhurst DC, Osisanya JO. 1981. Separation of malaria-infected erythrocytes from whole blood: use of a selective high-gradient magnetic separation technique. Lancet ii:70–71. 10.1016/s0140-6736(81)90414-1. [DOI] [PubMed] [Google Scholar]
- 36.Orjih AU. 1994. Saponin haemolysis for increasing concentration of Plasmodium falciparum infected erythrocytes. Lancet 343:295. 10.1016/s0140-6736(94)91142-8. [DOI] [PubMed] [Google Scholar]
- 37.Ekland EH, Schneider J, Fidock DA. 2011. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J 25:3583–3593. 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beutler E. 1975. Red cell metabolism. A manual of biochemical methods, 2nd ed. Grune & Stratton, New York, NY. [Google Scholar]
- 39.Schwarzer E, Turrini F, Arese P. 1994. A luminescence method for the quantitative determination of phagocytosis of erythrocytes, of malaria-parasitized erythrocytes and of malarial pigment. Br J Haematol 88:740–745. 10.1111/j.1365-2141.1994.tb05112.x. [DOI] [PubMed] [Google Scholar]
- 40.Gallo V, Skorokhod OA, Schwarzer E, Arese P. 2012. Simultaneous determination of phagocytosis of Plasmodium falciparum-parasitized and non-parasitized red blood cells by flow cytometry. Malar J 11:428. 10.1186/1475-2875-11-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2. Download aac.02109-21-s0001.pdf, PDF file, 0.3 MB (262.4KB, pdf)