

Abstract
Ischemia-reperfusion (I-R) injury is damage caused by restoring blood flow into ischemic tissues or organs. This complex and characteristic lesion accelerates cell death induced by signaling pathways such as apoptosis, necrosis, and even ferroptosis. In addition to the direct association between I-R and the release of reactive oxygen species and reactive nitrogen species, it is involved in developing mitochondrial oxidative damage. Thus, its mechanism plays a critical role via reactive species scavenging, calcium overload modulation, electron transport chain blocking, mitochondrial permeability transition pore activation, or noncoding RNA transcription. Other receptors and molecules reduce tissue and organ damage caused by this pathology and other related diseases. These molecular targets have been gradually discovered and have essential roles in I-R resolution. Therefore, the current study is aimed at highlighting the importance of these discoveries. In this review, we inquire about the oxidative damage receptors that are relevant to reducing the damage induced by oxidative stress associated with I-R. Several complications on surgical techniques and pathology interventions do not mitigate the damage caused by I-R. Nevertheless, these therapies developed using alternative targets could work as coadjuvants in tissue transplants or I-R-related pathologies
1. Introduction
Ischemia-reperfusion (I-R) injury is a cellular phenomenon caused by the interruption of oxygen flow and the consecutive restoration of oxygen concentration, which is known as reperfusion [1]. The reperfusion of ischemic tissues subjected to arterial occlusion causes the formation of a characteristic lesion that accelerates apoptosis and necrosis development [2]. I-R occurs in individuals with multiple pathologies and those receiving an intervention. Thus, it is inevitable in different conditions, such as cardiac, thoracic, and peripheral vascular diseases, and interventions, including major vascular surgery and solid organ transplantation [3–5]. Although the prevalence of ischemia is high, the treatment and preventive strategies for this lesion are not standardized or, simply, not effective enough to resolve damage [6]. Due to the impact of this condition on health systems and its epidemiological distribution, preventive pharmacological strategy is needed urgently. Even when effective therapy is necessary, I-R injury is still poorly understood, and researchers are looking for alternatives or relevant molecular targets that can modulate damage induced by this injury [7]. To date, I-R is characterized by an augmented inflammatory reaction that increases the expression of reactive oxygen species and reactive nitrogen species, which exacerbate tissue damage [8]. Hypoxia-inducible factors (HIF) are oxygen-regulated transcription factors that play important roles in the detection and adaptation of hypoxia [9]. Besides, they act as critical effectors in response to reduced oxygen levels and have a large number of genes under their control [10]. The expression of HIF-1α together with the generation of mitochondrial reactive oxygen species (ROS) is reinforced in response to ischemic oxidative stress [11]. In hypoxia, HIF-1α stabilizes by the accumulation of elevated levels of ROS generated from complex III in the mitochondria [11]. The mechanism behind this is oxidative inactivation of nonheme iron at the catalytic site of the enzyme prolyl hydroxylase [12]. Notably, ROS driven by hypoxia activates NF-kB and other transcription factors such as nuclear factor erythroid 2-related factor 2 (NrF2), which plays a vital role in the regulation of protein transcription involved in antioxidant defense [13]. The mitogen-activated protein kinase (MAPK) pathway has important implications as it interacts with ROS, which leads to a higher expression of vascular endothelial growth factor (VEFG) [14, 15]. The increased expressions of vascular endothelial growth factor (VEGF) and its receptors VEGF-R1 and R2 play a part in the activation of HIF-1α by ROS, and they have fundamental roles in maximizing cell survival [16]. Moreover, ROS activates other intracellular signaling pathways including MAPK, NF-kB, and upstream of MMP [17]. In addition, mitochondrial ROS can enhance damage via different mechanisms, such as mitochondrial permeability induction, ROS-mediated inflammatory and proapoptotic signaling, extracellular remodeling, and primarily oxidative damage in structures and intramitochondrial molecules, which contribute to the development of I-R lesion [18]. The therapeutic value of mitochondrial ROS attenuation in modulating I-R damage to the cell must be emphasized. Hence, effective therapeutic alternatives for ischemic reconditioning and tissue preparation for a possible ischemic event can be developed [19]. Research continues its course. However, certain points must be clarified, and the active principles and crucial receptors that can be an alternative for modulating this phenomenon should be determined [20].
The I-R phenomenon is poorly understood and highly variable between tissues. Although multiple mechanisms are known day by day, there is no effective therapy in clinical phases to date [21]. However, the design of effective therapy is necessary due to the significant relationship between this phenomenon and multiple cardio-obstructive pathologies and surgical procedures [22]. Two main approaches come to light: inflammation and oxidative stress induced by I-R damage [23]. Oxidative stress is very relevant due to the multiple opportunities for damage control. Unfortunately, the mechanisms are not applied. That is the reason for doing this work. Specify and conceptualize the main therapeutic targets towards which the pharmacological designs that allow a resolution of ischemic pathologies should be oriented. Therefore, the current study is aimed at providing ideas and research objectives for resolving oxidative and mitochondrial damage to modulate I-R injury in different tissues.
2. Ischemia-Reperfusion
Over the years, the concept of I-R has been changing and developing, thereby making us closer to discovering or establishing effective therapeutic interventions [7]. In I-R injury, triggering mechanisms begin at the time of arterial blood flow interruption in a tissue or organ, which produces an imbalance of metabolic substrates, leading to hypoxia [18]. I-R is a critical clinical condition, and physicians find it challenging to manage as it requires the preservation of tissue or organ function among individuals with different pathologies or those undergoing surgical procedures [6]. However, in clinical practice, the outcomes after reperfusion in ischemic tissues are far from optimal, and numerous damages are induced to the tissues [24]. As a consequence, reoxygenation is correlated with the exacerbation of local tissue injury and severe local or systemic inflammatory response. This was observed in tissues subjected to I-R, which are comparable with the degree of necrosis observed 24 h after permanent ischemia [25]. Cell dysfunction, damage, and death are associated with the magnitude and duration of ischemia. Therefore, blood flow restoration is still based on injury resolution. However, not all tissues or organs respond similarly to ischemic insult; thus, reperfusion is important to improve cell necrosis [26, 27].
3. Mitochondrial Oxidative Damage in Ischemia-Reperfusion Injury
Mitochondrial oxidative damage is important for the development of I-R, which is directly correlated with mitochondrial ROS and reactive nitrogen species (RNS) formation [28]. In myocardial infarction, the heart requires substantial amounts of energy from phosphates to maintain function and transport [29]. Nevertheless, ATP must be continually synthesized by the oxidative substrate in the mitochondria, thereby increasing the demand for reactive species formation [22]. The inhibition of electron flow along the respiratory chain leads to energy conservation. In addition, limited oxygen supply can inhibit mitochondrial complex IV, which blocks electron transfer to molecular oxygen and reduces ATP concentrations [30]. During the ischemic phase, ATP concentrations are unsuccessfully maintained by glycolysis; hence, the condition further exacerbates, which leads to lactic acid accumulation. Next, intracellular pH decreases. Simultaneously, the Na+/H+ antiporter is activated in response to decreased cytosolic hydrogen potential. The cell is overloaded with Na+, which cannot be pumped out of the cell by Na/K-ATPase. If the ATP concentrations are low due to decreased inner mitochondrial membrane gradient, FOF1-ATPase hydrolyzes ATP to regulate the condition [31]. Due to the inability of the mitochondria to produce significant amounts of ATP, compensatory anaerobic glycolysis occurs as a resolution mechanism. However, paradoxically, a considerable amount of this ATP will be hydrolyzed by FOF1-ATPase [32]. Concurrently, Na+ can prevent the release of Ca2+ by the Na+ /Ca2+ antiporter, thereby attempting to reverse the process. Calcium could enter the cytosol or even the mitochondria via the reversal of the Na+/Ca2+ antiporter mechanism [18]. However, the mitochondrial matrix absorbs Ca2+ after the reperfusion process via the uniporter, which then overloads the matrix with this ion. The opening of the mitochondrial permeability transition pore (mPTP) is one of the essential mitochondrial mechanisms in I-R. These pores are strictly linked with mitochondrial ROS and RNS release. Mitochondrial permeability allows ions and solutes with a low weight to freely move between the mitochondrial matrixes [33]. The main concept of mPTP was considered as an in vitro artifact without any pathophysiological significance. A previous study has later supported this notion and confirmed their role in the development of some diseases [34]. ROS are some of the main triggers of mPTP opening by overloading the matrix with high Ca2+ concentrations. However, there are other factors and molecules implicated in reperfusion. One of them is the influx of oxygen in anoxic cells, which leads to the formation of free radicals, a consequence of respiratory chain inhibition [34]. Almost all free radicals may be produced via the activation of xanthine oxidase. This enzyme is activated in hypoxia during ischemia [35].
In addition to cellular phosphate and depleted adenine nucleotide levels, which are commonly correlated with the ischemia process, high Ca2+ concentrations and oxidative stress conditions can activate mPTP [35]. During the reperfusion phase, the pH returns to preischemic insult values. This phenomenon is attributed to the activity of Na+/H+ antiporter that grants the release of lactic acid, which makes mPTP relevant and facilitates its full ability to exhibit its effect [36].
4. Oxidative Molecular Mechanisms Involved in Ischemia-Reperfusion Injury
There are complementary processes that are directly or indirectly correlated with mitochondrial oxidative stress and that play an essential role in the development of I-R injury [37]. Events, such as increased cations at the cytosolic level, mitochondrial injury, formation of oxidative and nitrosative species, transcriptional reprogramming, apoptosis activation processes, autophagy, necrosis, inflammation, immunity-mediated injury, endothelial injury, activation of ferroptosis, and the nonreflux phenomenon, are triggered or enhanced by blood flow obstruction and restoration [38].
4.1. Calcium Overload
Calcium overload and cytosolic cation increment are the initial mechanisms activated after the start of ischemia. All tissues and cells affected by this condition become dependent on anaerobic glycolysis ATP supply [39]. However, as an alternative for restoring pH to normal levels, some anticarriers including Na+/H+ are activated to address the accumulation of cytosolic Ca2+. Nevertheless, the expression of cytosolic Ca2+ is even higher during reperfusion, when the removal of H+ ions of extracellular origin paradoxically raises the proton gradient, thereby accelerating the proton exchange function [40]. All these events and alterations as well as high Ca2+ concentrations activate different pathways involved in I-R-induced cell death. Pumping up Ca+ directly to the mitochondria via Ca2+ uniporters is a mechanism that can help cells manage Ca+ overload [41].
4.2. Formation of Reactive Oxygen and Nitrogen Species in Mitochondria
ROSs are normally produced in the mitochondria, endoplasmic reticulum, plasma membrane, and cytoplasm during physiological metabolic processes [42]. ROS and RNS production in the cell starts with the reduction of oxygen and nitrogen levels, which are extremely basic and simple reactions. However, they are extremely important in cell function [43]. Generally, the mitochondria are the main source of cellular oxidative and nitrosative stress. Nonetheless, study results that reinforce this argument, particularly in nitrosative stress and heart disease, must be further validated [44, 45]. The mitochondria are involved in reactive species formation, with production directly involved with cytosol reactive species concentrations [46]. Moreover, complexes I and III of the electron transport chain are involved in ROS formation along with NAD+-linked oxidoreductases in the mitochondrial matrix. This notion has been reviewed and presented in several studies [42, 47, 48]. The reactive species correlated with the mitochondria and oxidative and nitrosative damage are superoxide molecules, hydrogen peroxide, hydroxyl radicals (-OH), nitric oxide (NO), nitroxyl anion, nitrosonium cation (NO+), and peroxynitrite (ONOO-) [49, 50].
4.3. Ferroptosis in I-R Phenomenon
Ferroptosis is a type of cell death that is an alternative to apoptosis. It is characterized by the accumulation of iron-dependent lipid hydroperoxides at alarming levels. Moreover, it cannot be inhibited by factors associated with other known types of cell death [51]. Hence, it is morphologically, biochemically, and genetically different from other types of cell death, and it is involved in various pathological events in which I-R is not an exemption [52]. Further, it is one of the relevant oxidative pathways that could modulate I-R damage due to its close association with some oxidative components in this pathology [53]. Lipid ROS accumulation, which leads to oxidation and antioxidation activity mechanism via toxic lipid peroxidation, is a principal ferroptosis pathway that could be correlated with I-R [54, 55]. As an initiation mechanism, glutathione peroxidase (GPx) has an important antioxidant role in ferroptosis during reperfusion. Oxygenated blood promotes the stimulation of enzyme activity, primarily its isoform 4 (GPX4), which has the capabilities of a cytosolic antioxidant enzyme. This phenomenon then modulates the substrates of the lipoperoxide pathways such as H2O2, small hydroperoxides, and phospholipids that are inserted in the biomembranes [56, 57]. By contrast, arachidonic acid contains phosphatidylethanolamine, which plays a key role in the ferroptosis cell death signaling. Hence, it is a crucial target for modulating this oxidizing process [57–59].
5. Antioxidant Enzymes in I-R
Researchers are examining the safest and most effective therapies for regulating I-R damage. However, this tissue pathology is characterized by excessive oxidative damage that is challenging to resolve because free radicals can drive cells via different routes of cell death and subsequent necrosis [20]. Nevertheless, in recent years, a previous study about ROS has shown that these molecules are involved in different pathological processes closely correlated with I-R [60]. Therefore, antioxidant activity is the main therapeutic target of most pharmacological therapies to address this phenomenon [60]. However, not all therapeutic approaches have shown conclusive or favorable results. Thus, it is constantly necessary to make updates on antioxidant therapy in this event to improve the resolution of this pathophysiological condition [61]. The scientific community accepts the role of ROS and other important free radicals, such as superoxide radical (O2), formed by adding extra electrons in an oxygen molecule (OH), which is created from O2 via the interaction of H2O catalyzed by transition metals including iron in I-R [62]. Oxygen radicals can be formed by the action of singlet oxygen, which commonly occurs in ischemic tissues [63]. The eukaryotic cell has a defense system similar to that of enzymes, such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), and glutathione reductase, which inhibit reactive species formation [64, 65] For example, the concentrations of SOD1, SOD2, CAT, and GSH-Px decrease in I-R [66, 67]. Another important antioxidant complication is the reduction of GSH in the ischemic myocardium by buthionine sulfoximine, a cellular inhibitor of GSH, thereby making the tissue more susceptible to reperfusion damage [68]. One of the main concerns when choosing antioxidant enzymes as possible therapeutic targets is that the enzyme activity and the average concentration in tissue and the relationship they have in ischemia-reperfusion damage have not yet been correctly described or have not been conclusive [69].
6. Therapeutic Targets in Oxidative Damage
A detailed review of the therapeutic targets that can be a receptor for some antioxidant drugs should be performed. This allows redirecting research to the establishment of therapeutic alternatives that can have more interesting effects [70]. Calcium overload, which triggers the formation of reactive species in the mitochondria, and MPTP opening, which is involved in the release of Ca2+ from the mitochondrial matrix to the cytosol, are relevant [71]. Regulating the ferroptosis process, one of the key pathways in nonapoptotic death that is strictly correlated with cellular antioxidant capacity could be an interesting alternative for different pathologies involving I-R [64]. Undoubtedly, the modulation of ROS/RNS in the cytosol, which can prevent all types of cell damage, is a strategy that remains undiscovered due to a large number of possibilities [72]. Notably, future research must focus on the different types of tissues, variations in I-R injury, and modulating strategies.
6.1. Mitochondrial Receptors
Alternatives for modulating I-R damage should be identified. Oxygen depletion after ischemia is correlated with the inhibition of mitochondrial respiratory chain electron transport and the consecutive decrease in ATP levels, which leads to failure in Na+/K+ pump and Ca2+ accumulation [73, 74]. The electron transport chain can be the primary target of the mitochondria. This is confirmed by five main types of enzyme complexes, which are as follows: NADH-CoQ reductase, succinate-COQ reductase, CO-Q-cytochrome c reductase, cytochrome c oxidase, and ATP synthase, which are known in that order as complexes I–V and are all integrated into the inner mitochondrial membrane [75, 76]. Concurrently, ubiquinone and cytochrome c are the two freely diffusible molecules implicated in electron transfer between complexes previously mentioned [77]. Various signaling pathways, which are essential to normal cell function, require ROS activity from hydrogen peroxide, hydroxyl radicals, and superoxide anions. The main keys for triggering the formation of ROS are found in complexes I and III [78]. NADH commonly binds to the complex and promotes electron transfer flavin mononucleotide (FMN). Reduced levels of flavin decrease O2 superoxide concentrations and promote proton transfer that conducts ATP synthesis [78]. Mitochondrial complex II is the only enzyme that is part of both the Krebs cycle and electron transport chain. Succinate, which is oxidized to fumarate via the action of adenine flavin, mediated by the dinucleotide cofactor (FAD), is involved in this reaction [79]. Undoubtedly, mitochondrial complex II is a central modulator in metabolic and respiratory adaptation in response to different stimuli and abnormalities. Thus, it is a key receptor in the modulation of oxidative damage in I-R [80]. Previous reports have shown an overlap in respiratory complex II and mKATP channel agonists that can activate it. The association between mKATP and respiratory chain complexes has shown a correlation between complex II and decreased ROS production [81].
The complex III Q-cytochrome c reductase molecule, which is implicated in the addition of four protons to the intermembrane space, is a significant site for ROS productions. The free radical ubisemiquinone leads electrons to oxygen, and this reaction results in a superoxide ion formation process that is enhanced by complex III inhibition [82]. Cytochrome c oxidase, better known as complex IV, mediates O2 reduction from H2O molecules by transferring four protons from the matrix into the intermembrane space, thereby increasing the electrochemical gradient and then entering as part of the intermediaries to this reaction [83, 84]. ATP synthase complex V promotes oxidative phosphorylation and induces ATP synthesis resulting in ATP formation [85]. Therefore, failure in the activity of this complex leads to inefficiency and dysregulation of mitochondrial function [86]. Complexes I and III are the principal targets because small amounts of free radicals are correlated with oxidative damage induction, mainly during hypoxia or ischemia. The inhibitors or modulators of these two molecules could manage hypoxic or ischemic conditions [87]. Interestingly, modulating mPTP opening is another interesting point for preventing ROS damage or even necrosis. This complex is a crucial effector in the cell death pathway. In addition, the activation of the mPTP function is the first step in the mitochondrial intrinsic necrosis pathway, leading to mitochondrial permeability transition and loss of inner mitochondrial potential [88]. Several pathways lead to the opening of mPTP. As a protein complex, they must interfere in one of its subunits, cyclophilin D (CyD), an essential modulator of mPTP. This makes it a key target for preventing cell death due to necrosis [89, 90]. A recent study focuses on the identification of novel compounds that can inhibit mPTP opening without any modulation of the CyD [91]. Mitochondrial ATP-sensitive potassium channels (mKATP) are opened after ischemia as a resolution measure, thereby modifying the activation of mPTP and delaying apoptosis. In addition, nonmitochondrial KATP can provide protective effects by promoting blood flow and excessive production of substrates [92]. These complexes promote the blocking of mitochondrial respiration and membrane disruption during diseases. Further, they are considered the primary cause of cell death in myocardial infarction I-R [93–95]. The mitochondrial antioxidant manganese SOD (MnSOD) expression is one of the objectives for modulating its dismutase scavenging function in superoxide radical O2 affecting several cell compartments. These are correlated with the pathophysiology of I-R, with the endoplasmic reticulum being sensitive to ROS, thereby making it responsible for maintaining calcium homeostasis [96, 97]. By contrast, autophagy is a crucial modulation target, which is responsible for cell recycling [98]. Previous studies have shown that this event contributes to the processes of cellular damage, and the key molecules are Beclin 1, mTOR, and PI3K [99, 100]. The mitochondria are important in pathological processes. To date, there is sufficient evidence about the morphological differences between the mitochondria, and that they are structurally and physiologically distinguished even in the same tissue [101] (Figure 1).
Figure 1.
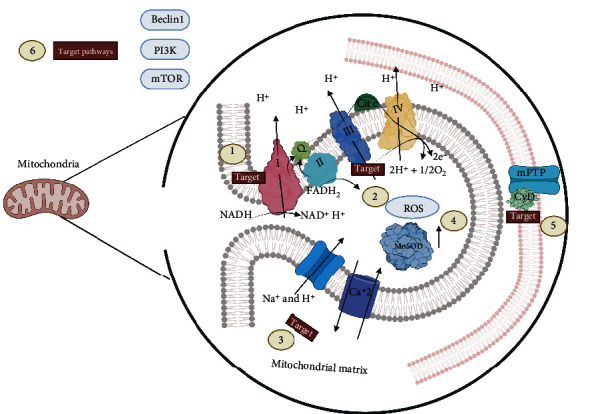
Mitochondrial targets for modulating oxidative damage induced by ischemia-reperfusion. The electron transport chain mainly in complexes I (1) and III (2) is the target of interest for reducing the formation of reactive oxygen species in the mitochondrial matrix as well as the activation of Na+/ H+ antiporters and calcium saturation (3) in the matrix along with the increased expression of antioxidant enzymes such as MnSOD (4) that carry out free radicals. In the mitochondria membrane, the blocking of MPTP by cyclophilin (CyD) is also essential (5), thereby preventing ROS from leaving the mitochondria to the cytosol. Beclin 1, PI3K, and mTOR (6), which are alternatives of interest for blocking mitochondrial damage, are some pathways that have good outcomes for reducing such damage.
7. Molecular Targets for Ischemia in Different Tissues
Although the I-R phenomenon has many similarities in different tissues, the lack of oxygen is the leading cause of cellular imbalance. On the other hand, it is necessary to highlight the differential characteristics between tissues, mainly the critical therapeutic targets that will elucidate pathophysiological mechanisms and the design or implementation of new therapeutics or interventions. In this work, we selected some groups of tissues most affected by this phenomenon and try to highlight the molecular targets.
7.1. Myocardium
The myocardium is an I-R susceptible tissue after epicardial coronary artery occlusion. The hypoperfused myocardial zone during myocardial infarction is a risk zone for oxidative damage and inflammation [102]. Clinical and preclinical research has shown a large number of cardioprotective agents, with mechanisms ranging from calcium overload to oxidative stress modulation. However, targeted therapy remains a challenge that has not been addressed altogether [103]. Calcium (Ca2+) released from the sarcoplasmic reticulum (SR) is important for excitation-contraction (E-C) coupling. The mitochondria, the major source of energy in the form of ATP, which is required for cardiac contractility, are closely interconnected with the SR, and Ca2+ is essential for the optimal function of these organelles. However, Ca2+ accumulation can impair mitochondrial function, leading to reduced ATP production and increased release of ROS. The calcium (Ca2 +) released by the SR is essential for cardiac excitation and contraction. ATP from the mitochondria is the main source of energy for the myocardial contraction process. However, the accumulation of mitochondrial Ca2+ affects the functioning of this organelle, which significantly decreases ATP and increases the formation of ROS [104]. Oxidative stress is directly associated with heart failure. Some studies have validated the role of Ca2+ in the development of this event, and it was found to be closely related to mitochondrial dysfunction [105]. Notably, there are two ways of releasing Ca2+ accumulating in the mitochondria of the cardiac cell, which are as follows: via type 2 ryanodine receptors RyR2 and type 2 inositol 1,4,5-triphosphate (IP3R2) receptors [106, 107] (Figure 2).
Figure 2.
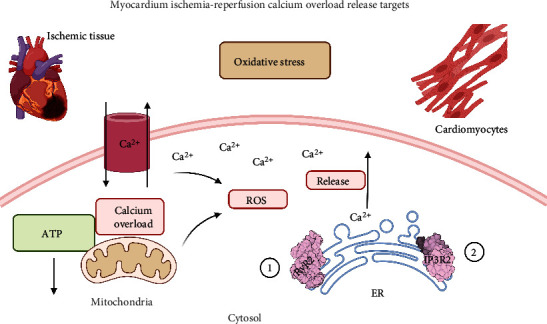
Calcium overload release targets. There are two principal ways of releasing Ca2+ accumulating in the mitochondria of the cardiomyocytes due to mitochondrial ROS formation. The first one is via type 2 ryanodine receptors RyR2 (1) and type 2 inositol 1,4,5-triphosphate (IP3R2) receptors (2). A mechanism that reduces ROS and Ca2+ cell damage.
7.2. Hepatic I-R
The liver is extremely sensitive to oxidative damage caused by I-R. Therefore, blood flow must be restored to prevent or slow down cell death [108]. Some studies have reported the importance of ischemic preconditioning for the management of this pathology and the role of lipoperoxidation modulation in this mechanism. This explains why peroxidation signaling pathways are relevant in reducing this condition [109]. By contrast, it is important to identify the role of peroxisome proliferator-activated receptor-gamma (PPAR-γ). That is, it inhibits the production of ROS in a pre- and posttransductional method, via the FAM3A complex and noncoding RNA axis, as reported by several workgroups [110]. In addition, the other important targets for modulating oxidative damage are metalloproteinases and malondialdehyde, which are the enzyme complexes involved in the cellular oxidative process [111, 112] (Figure 3).
Figure 3.
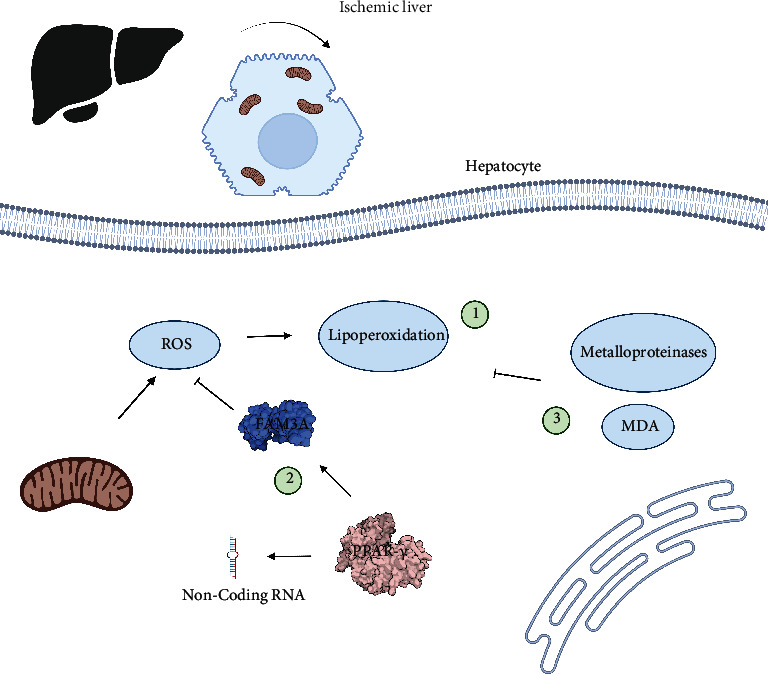
Main targets to regulate oxidative damage during hepatic ischemia. There are three principal points in hepatic tissue when oxidative damage is modulated. (1) Lipoperoxidation interferes with normal cellular functions and is the principal objective during hepatic I-R. That is why the peroxisome proliferator-activated receptor-gamma (PPAR-γ) is considered a good target. (2) It inhibits ROS production via the FAM3A complex and noncoding RNA. Besides, ROS formation of release blockade is crucial for diminishing lipoperoxidation, in addition, with the activity of metalloproteinases and malondialdehyde antioxidative complex.
7.3. Renal Tissue
The kidney is a specific organ that can be affected by I-R, which could lead to irreversible kidney injury. However, renal occlusion is inevitable during transplantation. That is why there are countless models for this phenomenon [113]. These advancements are crucial in understanding the pathophysiology of renal I-R, and they propose some therapeutic targets that can improve management. It is important to provide an overview of the possible therapies and receptors that can reduce oxidative damage in the kidneys [113, 114]. Similar to other organs, ROS plays a fundamental role in oxidative stress, which changes mitochondrial oxidative phosphorylation, ATP depletion, an increase of intracellular calcium, and activation of membrane phospholipid proteases, processes that could have results as a therapeutic alternative [115]. The interesting molecules to modulate the damage induced by renal I-R are more aimed at increasing the expression of antioxidant enzymes or their activity to offer a resolution of oxidative damage [116, 117]. Therefore, some interesting targets are the SOD, CAT, and GPX receptors, since, without a doubt, the neutralization of ROS and hydroperoxides mediated by these enzymes is a probable target for the treatment of renal I-R [118]. Notably, although antioxidant therapy is a viable alternative, damage caused by I-R cannot be fully treated. However, its efficiency is sufficient to considerably reduce oxidative damage [62, 118]. Some studies have shown the beneficial effects of free radical scavenging molecules on renal I-R. Molecules including melatonin can modulate the damage induced by renal reperfusion in ischemic kidneys due to its antioxidant activity [119]. Moreover, lipoperoxidation is a proven mechanism with good outcomes in renal tissues, where we could highlight the increase in SOD activity as the main target [120]. Furthermore, Diao et al. showed that the inhibition of protein arginine methylation transferase 5 (PRMT5) blocked ROS-mediated pyroptosis via the Nrf2/HO-1 signaling pathway. Therefore, PRMT5 is an interesting management target in renal I-R injury [121, 122]. The mitochondrial receptor MnSOD, an antioxidant enzyme capable of scavenging O2 free radicals, while controlling peroxynitrite radical (ONOO-), can successfully modulate I-R in renal tissues [123]. Although the role of ferroptosis in the renal I-R phenomenon has not been completely elucidated, several molecules can be targeted and provided interesting possibilities for therapeutics [124]. Pannexin 1 is an ATP-releasing protein that exhibits proapoptotic properties in renal I-R [125]. With consideration of molecular targets for modulating ferroptosis, the GPX4 enzyme can be a key regulator of lipoperoxidation [126]. Therefore, its activation is strictly correlated with the process of cell death and, consequently, the accumulation of ROS. This mechanism was found to be successful in pharmacological alternatives including irisin [127] (Figure 4).
Figure 4.
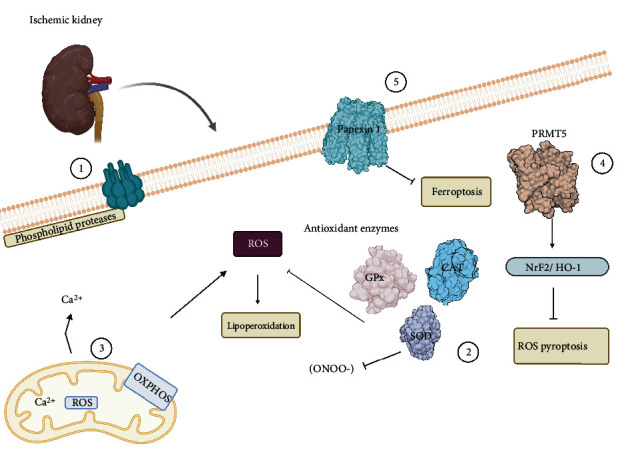
Relevant targets during ischemia-reperfusion in renal tissue. The main ways to decrease renal oxidative damage during I-R are (1) activation of phospholipid proteases, (2) the increment of antioxidant enzyme complex reducing free radicals and blocking lipoperoxidation, (3) modulation of Ca2+ and ROS mitochondrial release as well as controlling oxidative phosphorylation, (4) PRMT5 protein arginine methylation transferase is a regulation mechanism to stop ferroptosis, and (5) the membrane receptor Pannexin 1 through that blockaded ROS pyroptosis through Nrf2/HO-1 signaling pathway.
7.4. Brain Tissue
The brain is the most sensitive organ to blood supply interruption without the possibility of repair in I-R. That is, 20 minutes of ischemia is enough to exceed the threshold of damage it can withhold [18]. That can potentially cause or lead to oxidative stress-induced behavioral and cognitive decline. Oxidative stress in the brain caused by I-R leads to the primary etiologies of brain damage and significant neuronal effects, resulting in tissue destruction and cell death. These include lipid peroxidation, protein denaturation, inactivation of enzymes, nucleic acid, and DNA damage, the release of Ca2+ from intracellular stores, damage to the cytoskeletal structure, and chemotaxis [128]. Phospholipids in the brain are vulnerable to ROS-mediated peroxidation. However, proteins and DNA are targeted by ROS, and they become problematic with aging as aging brains exhibit high oxidative stress-induced mutation levels in the mitochondrial DNA [129, 130].
Perhaps, we cannot find an effective therapeutic target for reducing this damage. That is why ischemic preconditioning combined with antioxidant therapies will most likely be critical regulators for ischemic stroke [131]. Current studies have focused on the pathways of oxidative stress that involve a variety of cellular pathways, receptors, and processes that can be used on focused therapy for oxidative damage, such as autophagy, mitophagy, and necrosis, which are involved in eliminating excess ROS and subsequent cell death triggered by these free radicals [132, 133]. The endogenous protective mechanisms in the brain included the antioxidant enzyme systems and the low-molecular-weight antioxidants [134]. In response to stress, cells increase their antioxidant defenses with nuclear factor erythroid 2-related factor (Nrf2), an important transcription factor [135]. Therefore, Nrf2 has been proposed as a pharmacological target in pathologies with oxidative features since it modulates several genes encoding antioxidants and detoxification enzymes such as heme oxygenase 1 (HO-1), NAD(P)H dehydrogenase quinone 1, superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1), and catalase (CAT) [136]. By contrast, mitochondrial dysfunction suggests several diseases, including neurodegeneration [137].
The mitochondrial role in ischemic shock and its pathogenesis mainly involves the formation of free radicals [138]. mtDNA is particularly susceptible to oxidative damage because of its proximity to high levels of mitochondrial ROS production and its relatively poor defense against damage. Healthy mitochondria contribute to oxidative stress resistance by increasing respiratory capacity [139]
Taken together, ATP synthase and the electron transport chain make up the OxPhos system, which is the leading promoter of the mitochondrial electrochemical gradient [140, 141]. Crucial key points for electrons to enter are complex I and II of the electron transport chain, which through ubiquinone transfer electrons to complex III and this in turn to complex IV. Proton pumping via the mitochondrial membrane is the primary mechanism for maintaining the membrane potential. These protons are then used by complex V ATP synthase to form ATP and complete the oxidative phosphorylation process [142]. Reversible phosphorylation mechanisms are relevant targets of this whole process. Preserving these phosphorylation epitopes could offer a regulatory control for reducing oxidative damage since it could allow the regulation of OxPhos mediated by calcium and the ADP-shuttle mechanism [143]. The OxPhos complexes are phosphorylated in vivo by the second messenger Ca2+, thereby triggering the phosphorylation of most mitochondrial proteins, a process mediated by calcium-dependent phosphatases during ischemic stress [144]. This phosphorylation alters the electron transfer kinetics, which affects the allosteric regulation of ATP and ADP [145]. The mitochondrial membrane potential (ΔΨm) becomes positive in the inner chamber during oxidation, and cytochrome c (CytC) is released into the cytoplasm. The release of CytC from the mitochondria is an important pathway for the cascades of apoptotic events [146]. These proapoptotic proteins, such as Bid, Bad, Bax, Bak, Bok, and Bim, in the outer mitochondrial membrane, increase the permeability of membranes, thereby forming specific pores and stimulating free CytC release. Third, CytC binds to apoptosis protein-associated factor 1 (Apaf-1) and forms the Apaf-1/caspase-9/CytC complex. Finally, caspase-3 is activated, which triggers apoptosis and delays neuronal death [147]. Lipid peroxidation is one of the significant consequences of ROS-mediated injury to the brain. This ultimately leads to the production of conjugated diene hydroperoxides that attack lipids containing carbon-carbon double bond(s) in specific polyunsaturated fatty acids (PUFAs). Among these compounds, malondialdehyde (MDA) and HNE are the breakdown products of lipid peroxidation, and they are elevated in patients with ischemic stroke [148], with infarct size, stroke severity, and patient outcome. MDA can be the most mutagenic lipid peroxidation product, and HNE is the most toxic [149]. MDA is widely used as a biomarker for lipid peroxidation of omega-fatty acids, HNE is a cytotoxic product originating from peroxidation, and it is considered as one of the significant toxic products generated from lipid peroxides. The highly toxic characteristic of HNE can be explained by its rapid reactions with thiols and amino groups [150]. HNE is a bioactive marker of lipid peroxidation and is a signaling molecule involved in the regulation of several transcription factors, such as nuclear factor erythroid 2-related factor 2 (Nrf2), activating protein-1 (AP-1), NF-κB, and peroxisome proliferator-activated receptors (PPAR), cell proliferation and differentiation, cell survival, autophagy, senescence, apoptosis, and necrosis [151]. Hemoglobin (Hb)/haem is a putative neurotoxin. Hb is the most abundant protein in the blood and is released from lysed red blood cells after stroke. It can be engulfed by the microglia in the perihematomal zone and metabolized into ferrous/ferric iron, which induces ROS formation and lipid peroxidation [152]. The excess ferrous iron accumulates in the neurons via the transferrin (Tf)–Tf receptor system that forms highly toxic hydroxyl radicals (∙OH). These hydroxyl radicals attack DNA, proteins, and lipid membranes, leading to the disruption of cellular function. Ferroptosis was found in organotypic hippocampal slice cultures exposed to glutamate [153]. It can be distinguished from other types of regulated cell death because it does not require caspases ATP depletion or mitochondrial ROS generation (Bax/Bak) or elevations in intracellular Ca2+ levels [154]. Ferroptosis is triggered by glutathione biosynthesis or glutathione peroxidase 4 (GPX4) activity inhibition and is associated with shrunken and electron-dense mitochondria morphologically [155]. Tryptophan (TRP) is an aromatic essential amino acid whose route of TRP metabolism is the kynurenine (KYN) pathway, and the primary end products are nicotinic acid and its derivatives and NAD+ and NADP, which are two ubiquitous coenzymes In this catabolic process, starting from the central compound, kynurenine (KYN) forms kynurenic acid (KYNA), xanthurenic acid (XA), and picolinic acid [156] (Figure 5). KYNA is produced mainly in astrocytes, and quinolinic acid (QUIN) degradation occurs in microglial cells in the central nervous system. More recently, tryptophan oxidation via the kynurenine pathway has been implicated in inflammation and oxidative stress in the brain that occurs after stroke [157]. Elevated QUIN levels can cause excitotoxic cell death. The hippocampus and striatum are most sensitive to QUIN neurotoxicity. QUIN can directly interact with free iron ions to form toxic complexes that exacerbate ROS formation, oxidative stress, and excitotoxicity [158]. Moreover, it induces lipid peroxidation, produces ROS increases iNOS expression, decreases SOD activity, and causes mitochondrial dysfunction QUIN which stimulates mitochondrial dysfunction and apoptosis [159]. By contrast, the advantage of KYNA is that it cannot be metabolized to excitotoxic agents and scavenges oxygen radicals, thereby decreasing cellular damage. The application of KYNA in high concentrations or for a prolonged time causes neuronal cell damage [160]. The multiple effects of the kynurenine pathway and its changes during stroke have increased in recent years, thereby allowing interference with therapeutic targets. DNA damage includes oxidative modification and endonuclease-mediated DNA fragmentation. DNA oxidation may activate repair enzymes, such as poly (ADP-ribose) polymerase (PARP). PARP activation progresses from neuronal elements and localization of infiltrating inflammatory cells 3–4 days after stroke. The activation of PARP leads to DNA injury in the brain [161]. Indeed, there is a strong association between oxidative stress and PARP activation in the brain, and oxidative stress in the neurons can induce PARP activation [162]. PAR can directly affect mitochondrial membrane potential collapse [163]. Thus, PARP-1 activation may inhibit glycolysis and cause energy depletion, thereby leading to altered cellular metabolism.
Figure 5.
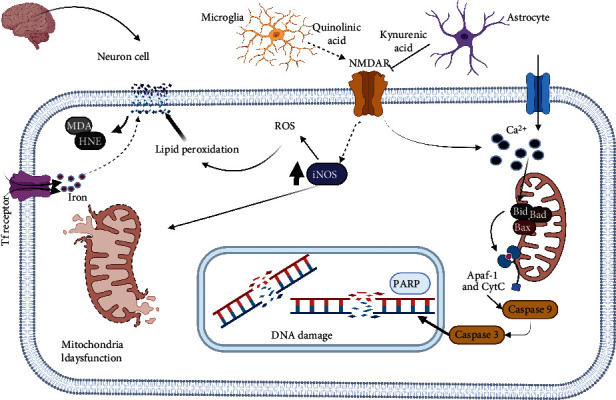
Oxidative stress in neuron cells. Several mechanisms activate oxidative stress during ischemia in neuron cells, principally leading by the quinolinic acid released by microglia and mitochondrial Ca2+ overload and ROS formation after BLD, BAD, BAX complex, and APAF-1 and CytC activate caspases pathways that result in DNA damage. Nevertheless, astrocytes intend to block that oxidative stress and lipoperoxidation that could damage plasmatic membrane releases kynurenic acid. Unfortunately, Tf receptors promote cell damage through ferroptosis. Therefore, all of these would function as excellent regulatory points for I-R oxidative injury in nervous tissue.
7.5. Lung Tissue
The lungs are affected by I-R indirectly. Several signaling pathways such as Nrf2/HO-1 and HIF 1α/VEGF have protective effects on this organ [164]. NrF2 is a transcriptional factor that protects cells from stress, and oxidative processes activate NrF2 to initiate such an effect. In turn, HO-1 becomes a rate-limiting enzyme that can reduce oxidative stress by increasing its expression in the lung either via local or peripheral ischemia [165, 166]. Hypoxia-inducible factor 1-α (HIF-1α) and cell repair mechanisms mediated by VEGF are implicated in the regulation of angiogenesis in ischemic events [167, 168]. After ischemia damage to the lungs, there is a significant loss of plasma proteins and inflammatory cells, and there are high amounts of HIF-α and its regulatory target VEGF during I-R in local tissues [169]. Moreover, recently, the close association between these two molecules has been correlated with repair mechanisms independent of angiogenic activity [164]. The mitochondrial approach may also be a good alternative to modulating oxidative damage. Some reports have shown interesting results regarding protecting the integrity of the mitochondrial DNA using the oxidative approach. That is, mtDNA could serve as a sentinel of ROS-mediated functions, as observed primarily in the lung tissues [170]. Using conventional oxidative stress as a therapeutic target in ischemia could be complicated. Therefore, preconditioning alternatives can be another therapeutic option. Researchers have designed in vitro and ex vivo experimental data in which the tissues and cells are exposed to high concentrations of polyethylene glycol-catalase (PEG-CAT) to protect against cytotoxicity caused by oxidative stress. This mechanism then preserves cellular metabolism and mitigates pulmonary I-R. Therefore, PEG-CAT can be an important therapeutic target [171]. Anti-inflammatory approaches for decreasing pulmonary ischemia remain unclear. It was proposed that the establishment of novel therapeutic strategies should involve the inhibition of transcriptional factors that activate oxidative stress with better techniques. For example, MAPKs that are activated after oxidative stress in the inflammatory models of pulmonary ischemia and different signaling pathways, such as p38, c-jun N-terminal kinase, p38 inhibition, or JNK, have protective effects in this organ [172]. In addition, ROS and RNS mediate inflammatory reactions by activating alveolar macrophages. With the activation of the inflammatory cascade, multiple potential ROS generators such as the mitochondria, xanthine oxidase, NOX, NOS uncoupling, and neutrophils must be considered as a therapeutic target for oxidative damage [173]. By contrast, the expression of DPP4 is directly correlated with decreased oxidative damage. That is, the capillaries are the main concentration regions of this expression, and DPP4, a serine protease, commonly cleaves the substrates with proline and alanine in the latter position [174] (Figure 6).
Figure 6.
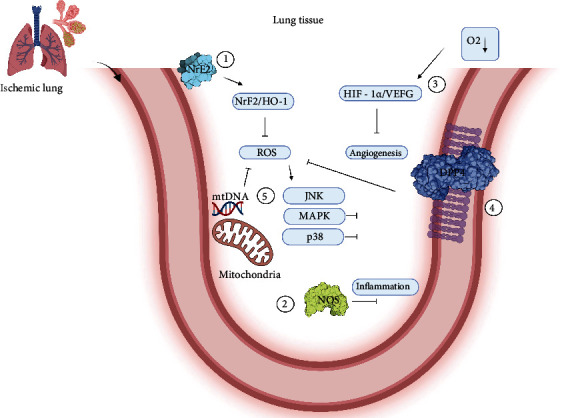
Molecular targets to decrease oxidative damage during I-R. (1) One of the main points to reduce oxidative damage is the NrF2 factor, the starting point of the NrF2/HO-1 signaling pathway that triggers ROS blockade. (2) Nitric oxide synthase, being blocked, can prevent the activation of the inflammatory cascade. (3) Low oxygen concentrations in the cell lead to the activation of HIF1 and HIF 1α/VEGF, one of the primary mechanisms for generating new vasculature. (4) DPP4s decrease oxidative stress when their expression increases. (5) In addition, mitochondrial DNA can function as a signaling mechanism that allows the activation of programs to block the production of reactive oxygen species.
7.6. Skeletal Muscle
In the limbs, skeletal muscle is the predominant tissue, and pathophysiological literature indicates that the damage threshold of this tissue is exceeded after 3 h of ischemia and is irreversible at 6 h [175]. Some studies about I-R showed that the main mechanisms of cell damage and death are mitochondrial dysfunction and mitochondrial proapoptotic protein release [176]. Similar to other cells, I-R in the myocyte is mediated by the mitochondrial membrane potential and the proton gradient that promotes ATP synthesis via oxidative phosphorylation [177]. This reduction during the ischemic process promotes ATP synthesis and inhibits Na+/K+ ATPase, thereby increasing intracellular Na+ and Ca2+ and anaerobic glycolysis. Further, the mitochondria play an important role in the pathophysiology of I-R in this tissue, and the free radicals generated by the skeletal muscle during rest and activity are NO and superoxide, which is dismuted into H2O2. However, there are still several limitations, and few studies have identified the nature of ROS or RNS present in the muscle fibers. Most reports have only examined cell surface free radicals [152]. Consequently, there are only a few reports about NO or H2O2 or substances that can cross mitochondrial barriers, but there are a large number of reactive species that have not been confirmed to be involved in skeletal muscle physiopathology [152]. To avoid deleterious effects on tissues, there are several cellular mechanisms to modulate free radicals such as the mitochondrial and cytosolic isoforms of superoxide dismutase (MnSOD and CuZnSOD) in addition to CAT and GPX modulation of their expression [178]. Any cellular processes are regulated by ROS and RNS, such as the activity of transcriptional factors, ionic transportation, apoptosis, and metabolism [179]. Proteins in skeletal muscle are susceptible to oxidation of their sulfhydryl groups or the formation of disulfide bonds. These processes are involved in the modulation of protein functions [180]. In the same way, ROS also functions as excellent second messengers in the activation of apoptosis programs such as the NF-kappaB pathway, which is involved in muscle degeneration and atrophy [181]. Several alternatives of regulation, such as the PAR-gamma coactivator-1 alpha (PGC1-α) pathway, are redox-sensitive, in which ROS would play a regulatory role [182]. In addition, ROS dependent on Nox2 is involved in the regulation of histones such as histone deacetylase 4 (HDAC4), this happens during vigorous muscle activity, as a regulation mechanism [183]. Not less important is the already described NRF2 transcriptional factor involved in multiple regulations of antioxidant defense [184]. Under conditions of oxidative stress, NRF2 is found in the cytoplasm thanks to the activity of degrading proteins; after it is released, it translocates into the nucleus where it activates the transcription of antioxidant gene programs and their respective protein [185]. Although it well tolerates oxidative damage, these modulation strategies are essential for the resolution of countless pathologies correlated with ischemia [186] (Figure 7)
Figure 7.
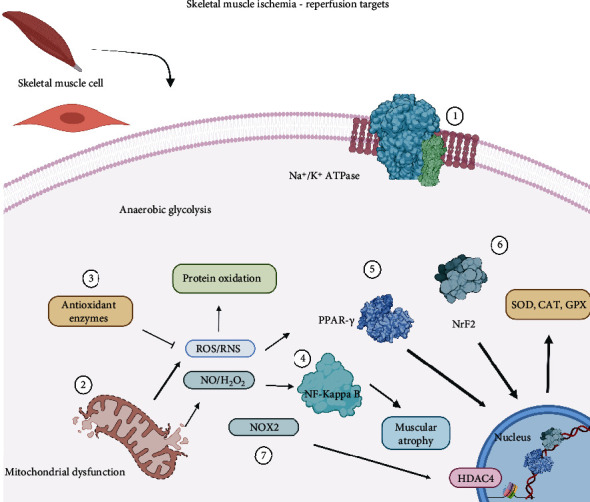
Skeletal muscle I-R targets. (1) Na+/K+ ATPase activation promotes the saturation on Na+ and K+. That is why modulation of these enzymes could be an excellent alternative to reduce damage. (2) Blocking mitochondrial release of ROS and RNS during dysfunction could improve cellular damage. (3) Scavenger activity of antioxidant enzymes is one of the main alternatives to modulate this condition. (4) Activation NF-κB leads to muscular atrophy for several inflammatory mechanisms. (5) PPAR-γ translocations, as well as NrF2 (6), result in the expression of the antioxidant complex. (7) NOX2-depending pathways conduce to activation of HDAC4 that facilitates gene expression.
8. Ischemia-Reperfusion Antioxidant Pharmacodynamics
Antioxidant therapy has been used to modulate oxidative stress in different experimental models. Generally, some proven strategies are used as antioxidant preconditioning without completely effective outcomes [7, 187, 188]. This can be explained by the nonselective characteristic of ROS modulation, which directly interferes with cell signaling pathways [189]. This has led to alternative approaches such as activation of the Nrf2 pathway by fumaric acid derivatives, resulting in a proven antioxidant activity [190]. Another relevant strategy is using ROS-producing enzymes such as Nox and MPO, which induce a more specific response by modulating pathological conditions [191, 192]. However, the most promising approach involves enzyme activity, particularly via drugs with a potential to reverse eNOS activity in pathologies correlated with oxidative stress [193]. However, despite advancements, almost all innovative cardiovascular therapies have been inadequate in the management of these pathologies.
8.1. Free Radical Scavengers
Notably, reactive species at low concentrations fulfill cellular functions as metabolic bioproducts or second messengers. At high concentration, they have deleterious effects, mainly in pathological events such I-R [194]. These effects conceptualize as oxidative stress and lead to the opening of mPTP, resulting in protein and DNA damage [195]. ROS signaling can be interfered with via the inhibition of complex I, using drugs such as metformin. This then reduces the amount of ROS in the cytosol [196].
8.2. Mitochondrial Respiration Chain Blockers
The electron transport chain stands out during the reperfusion process. This explains why direct modulation can be an alternative for reducing ROS production and the consequent activation of mPTP with cell death as an outcome [197]. The deregulated production of ROS in the mitochondrial respiratory complexes is associated with the I-R process. Regulating these processes modifies the harmful nature of ROS to a protective one, and these respiratory complexes are the main targets to carry this out [198]. Complexes I and III are major superoxide production sites. Electrons are transferred along the chain and back to complex I where NAD+/NADH is reversed and ROS production increases [199]. Using this approach, several drugs have been tested under I-R conditions, thereby providing varying but important results for understanding the phenomenon [200–203]. Some reports used the reversible inhibition of transiently inactivated complex I to diminish the generation of ROS without losing its function [59, 204, 205]. In these categories, some compounds such as biguanides, amobarbital, nicorandil, rotenone, and S-nitroso-2-mercaptopropionyl glycine have shown interesting results [205–210]. Highlighting metformin has exhibited cardioprotective properties by modulating complex I at high doses [211, 212]. Some strategies use acidic citric intermediates, malate, and oxaloacetate to inhibit complex II. Although it is not a specific site for the formation of ROS during the reperfusion process, it is correlated with complex I and III modulations, which allows cardioprotection regardless of K+ concentrations [213, 214]. Notably, some important mechanisms are not directly involved in the production of ROS nor as second messengers in the adaptation mechanism to hypoxia [215]. These signaling pathways are directly linked to ischemic preconditioning in different pathological conditions [216]. A consecutive modulation of complex III via the ubiquinol oxidation center (Qo site) has shown cardioprotection [217]. Reduced cytochrome c activity has a similar effect in electron transport from complex III to IV, an event that reduces superoxide-free radical production, which is a poorly understood mechanism [218] (Figure 8).
Figure 8.
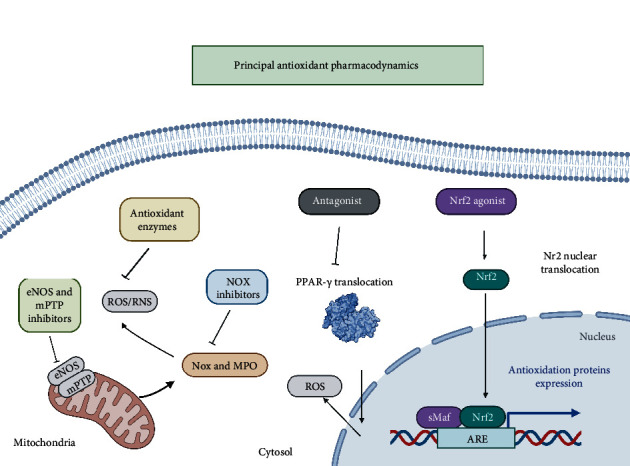
Principal antioxidant pharmacodynamics. The enlisted mechanisms are promising targets that are part of the pharmacodynamics of several drugs used in clinical practice. Antioxidant enzymes as a scavenger to reduce ROS and RNS, NOX inhibitors blocking free radical production, eNOS and MTTP inhibitors leading to reduction of reactive species and avoiding is release. Nuclear factors agonism and antagonism regulate the antioxidant expression and ROS production.
8.3. MPTP Inhibitors
The importance of MPTP for the development of I-R-mediated oxidative damage has been discussed. They represent a key point for the release of ROS/RNS from the mitochondria [36]. However, the therapeutic target approach of this protein complex is via its subunits, with CyD as one of the main ones, to which countless drug prototypes have been designed [219]. Nevertheless, in vivo and clinical data are not favorable enough to establish a therapy [220, 221]. Some analog drugs of CyA have shown favorable outcomes in myocardial, hepatic, and cerebral animal models [222]. Even some drugs that are not CyA analogs have interesting outcomes via this pathway in vivo models [223]. Other than this therapeutic target, other possible alternative therapies such as N-phenylbenzamides and cinnamic anilides can inhibit mPTP activity, thereby providing protective effects against oxidative damage [224, 225]. Nrf2 and NF-κB regulators are also good alternatives to ameliorate oxidative damage via this pathway [226–228] (Figure 1).
8.4. PPR's Gamma Inhibitors
Peroxisome proliferator-activated receptor gamma (PPAR-γ) is the target of multiple studies about cardiovascular pathologies [229]. Several isoforms have been described. However, the γ isotype is the most relevant in these I-R-related diseases [230]. After binding to endogenous ligands, the retinal X receptor is heterodimerized with a nuclear receptor, thereby inducing or repressing gene expression [231]. Therefore, PPAR has relevant roles in hepatic IR injury [232]. Some angiotensin II drugs are associated with this receptor, which exhibits an inhibitory effect [233, 234]. Several drugs attenuate PPAR I-R via antagonism, thereby reducing ROS production.
8.5. RNAS Transcripts in I-R
RNA and DNA are targets for modulating gene expression. Currently, advancements in molecular biology can allow them to be used as excellent therapeutic targets in multiple pathologies [235]. For example, the modulation of antioxidant enzymes using gene therapy has been useful in oxidative stress if a specific target receptor is already known [236]. Redox homeostasis has a direct correlation with cell function. Redox imbalance leads to oxidative stress production, which inhibits the development of vascular diseases and I-R injury, as well as triggers transcriptional and posttranscriptional modulation in gene expression [237, 238]. In addition, hypoxia is considered an important stimulus to regulate microRNA (miRNAs) expression [239]. Some miRNAs, known as hypoxia MIR, are even associated with hypoxia, and some of these transcripts are involved in the pathophysiology of ischemic and cardiovascular diseases [240]. An example is miR-210, a hypoxia-inducible transcript that promotes cell survival and improves cardiac function via antiapoptotic and angiogenic mechanisms [241]. Moreover, HIF is regulated by these transcripts with an important role in these pathologies [242]. Other examples of these nuclei of response to oxidative damage are NRF2, FOXO1, and NF-kB in the inflammatory part [243–247]. And the widely studied transcriptional factor p53, among the multiple stimuli that activate it, is ROS one of them, which consecutively triggers proapoptotic and antiproliferative mechanisms [248]. Even a single oxidative stress stimulus, such as H2O2 and O2, could be correlated with complex redox imbalance mechanisms in pathologies such as aging and limb ischemia [249, 250]. However, even though the role in oxidative stress of noncoding transcripts (miRNA and lncRNA) is widely known, their use as a therapeutic strategy remains premature [251, 252]. miRNAs are RNAs that regulate gene expression by forming hybrids with mRNAs, altering their translation [253]. Moreover, they have a regulatory role in oxidative stress via their interactions with SIRT1, FOXO1, and eNOS [254]. Furthermore, in different pathologies correlated with oxidative stress, these regulatory mechanisms have begun to take center stage [224] In I-R, miRNAs have great advantages when used as a therapeutic target. In the future, they could be considered pharmacological approaches in clinical practice [255]. Noncoding RNA similar to miR-92a has a proangiogenic effect, and miR-499 can decrease damage induced by hypoxia-reoxygenation. miR-24 had a similar mechanism correlated to the attenuation of infarct size in animal models [256–258]. Recently, the use of novel noncoding RNAs as therapeutic alternatives is emerging. For example, miR-181 is a drug target with an effect on experimental myocardial I-R [259]. miR-148a alleviates hepatic I-R and is implicated in resolution pathways [260, 261]. Another alternative is miR-374a-5p. This decreased myocardial cell damage in an I-R model [262]. Small RNAs are not the only noncoding transcripts relevant to disease-related I-R. Long RNAs (lncRNAs) are expressed by an opposite strand of mRNA, and they are located in the introns of annotated genes and transcribed from enhancer regulatory elements (eRNA) [263]. Moreover, this transcript mechanism varies, ranging from repressors to activators of gene expression, or even posttranductional regulators. They are also mRNA splicing and stability modulators; lncRNAs play an important role in regulating the response to oxidative stress [264]. For example, lncRNA Gpr19 inhibits and attenuates (I-R) injury after acute myocardial infarction, and lncRNA NEAT1 alleviates sepsis-induced myocardial injury by modulating oxidative stress [265, 266]. Some drugs including propofol have shown a beneficial effect against I-R oxidative stress under different conditions via the lncRNA-TUG1/Brg1 pathway in liver cells in an IR model [267]. Drugs such as metformin regulate oxidative stress via transcripts such as lncRNA-H19 [268]. In a recent study, ZFAS 1 lncRNA was found to reduce ischemic stroke via the regulation of some oxidative stress mechanisms [269]. (Figure 9).
Figure 9.
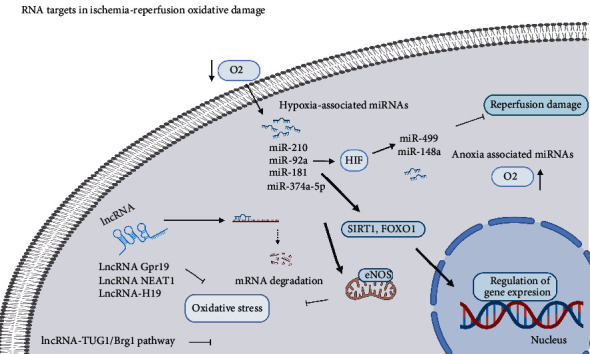
RNA targets in ischemia-reperfusion oxidative damage. We can make a group of the different miRNAs among those directly associated with the decrease in oxygen or hypoxia that directly activate the expression of HIF or are sensitive to it. Noncoding RNA activates various transcription factors to regulate the gene expression of antioxidant enzymes and prevent the formation of reactive species that exacerbate oxidative stress. Furthermore, those related to reperfusion or anoxia mechanisms are responsible for the resolution or inflammatory mechanism. Similarly, various lncRNAs block signaling pathways to reduce oxidative stress.
9. Alternative Interventions in Clinical Trials
Regardless of the several researches carried out to decrease damage caused by I-R injury, there is still no relevant pharmacological therapy that reduces damage caused by this condition in clinical practice. However, some studies have shown important data, particularly about those regarding Cyst A, in advanced pharmacological phases or pilot studies [270]. By contrast, metoprolol, a β-blocker, exhibited interesting activity in patients with I-R induced by myocardial infarction [271, 272]. Moreover, some glucose modulators have inhibited I-R damage via several mechanisms, with glucose/insulin/potassium (GIK) as the primary on [273]. In terms of clinical results, patients with acute coronary syndrome have presented with a significant reduction in infarct size [274]. In addition, intracoronary administration combined with thrombectomy significantly reduced infarct size [275]. The application of novel therapies in clinical practice is challenging, and there is an extensive list of not favorable outcomes in I-R treatments in clinical trials. One of the main reasons for these failures is the lack of clarity and significance in preclinical trials, thereby making it impossible to obtain relevant clinical information [276]. The most relevant examples are studies about inhaled nitric oxide and sodium nitrite for myocardial infarction. These strategies were tested in clinical practice. However, they were not effective in reducing infarct size [277]. The use of TRO40303, an MPTP inhibitor, had unfavorable outcomes in the MITOCARE study [253]. In the TREAT study, the outcomes of ticagrelor and clopidogrel treatment in similar pathological conditions were not significant [278]. However, some reports highlight interesting epitopes or therapeutic approaches, thereby allowing the appropriate incorporation of new clinical evidence into the practice guidelines of phase III trials, which are required to assure a solid preclinical background (Table 1).
Table 1.
Pharmacological approaches against I-R pathologies.
| Family | Pharmacodynamic | Results |
|---|---|---|
| β-Blockers | Reduce the cardiac frequency and calcium overload blockade | Regulate myocardial infarction |
| Glucose modulators | Regulate glucose/insulin/potassium concentration | Reduce myocardial infraction and infarct size |
| Immunomodulators (abciximab) | Reduce Inflammation and oxidative stress activation | Reduce infarct size in acute coronary syndrome |
| Inhaled NO and NaNO2 | Regulates oxidative stress | Failure in reducing myocardial infarction |
| MPTP inhibitors | Blockade of mitochondrial ROS release | Adverse effects and not significant data |
| Statins | Oxidative scavengers and IL10 expression | No significant data in acute coronary infarction |
| ARA II | PPAR-γ expression and antioxidant activity, SOD2 expression | Significant data were preventing I-R |
10. Perspective
The therapeutic targets most likely to be extrapolated in clinics settings are not yet fully elucidated. Hence, further studies should be performed to assess relevant epitopes. Most approaches used to reduce I-R damage are inflammatory. Therefore, many pharmacological effects of the peroxidative type have not been considered, thereby establishing the grouping of the said markers and targets of oxidative damage. After elucidating the molecular targets implicated in their physiopathology, a realistic approach in all pathology therapies is a pragmatic approach in all pathology therapies. The direct modulation of calcium overload in the cytosol can be a common strategy and, undoubtedly, a key element in stabilizing cellular pH. By contrast, the uptake of ROS mainly via scavenger molecules that may reduce damage can also be an exciting alternative.
11. Conclusion
I-R injury caused by the interruption of oxygen flow and the consecutive restoration of oxygen concentration, which is known as reperfusion, is still poorly understood [279]. These therapeutic targets can be a great alternative for modulating I-R injury. Reducing oxidative damage in cells could address several pathologies associated with I-R. Redox signaling is one of the processes with several therapeutic targets. However, most interventions present extremely ambiguous pharmacodynamics [280]. Thus, it is essential to elucidate the specific molecular mechanisms by which pharmacological interventions work. The mitochondria play a key role in the development of these cellular signaling [281]. Therefore, researchers are interested in approaches with correlated epitopes. Although the preclinical outcomes of targeted therapies are favorable, they have not yet been applied in clinical practice, and their adverse effects were not evaluated. Hence, pharmacological repositioning is a good alternative at present [282]. However, it is necessary to develop new active principles with a specific activity to resolve this pathology or to consider therapeutic combinations via in vitro and even in vivo tests [283]. Nevertheless, new active compounds with specific activity must dress to resolve pathologies or take advantage of the drug's antioxidant properties that can be considered adjuvant therapies in clinical settings. Proposing new therapy is necessary. This phenomenon is related to multiple pathologies and surgical procedures in oxidative stress and alterative to modulate ischemia-reperfusion.
Acknowledgment
We want to thank the Health Division of the Centro Universitario de Tonalá for the support provided in carrying out this work. The present work was performed with finance from the Department of Biomedical Sciences of the Centro Universitario de Tonalá, belonging to Universidad de Guadalajara.
Data Availability
We provide editable versions of our figures and editing platform publishing license.
Conflicts of Interest
The authors declare that they have no conflict of interest during the performance of this review, and it was conducted for academic purposes.
References
- 1.Anaya-Prado R., Toledo-Pereyra L. H., Lentsch A. B., Ward P. A. Ischemia/reperfusion injury. The Journal of surgical Research . 2002;105(2):248–258. doi: 10.1006/jsre.2002.6385. [DOI] [PubMed] [Google Scholar]
- 2.Eltzschig H. K., Eckle T. Ischemia and reperfusion--from mechanism to translation. Nature Medicine . 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhai Y., Petrowsky H., Hong J. C., Busuttil R. W., Kupiec-Weglinski J. W. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nature Reviews Gastroenterology & Hepatology . 2013;10(2):79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou J., Chen J., Wei Q., Saeb-Parsy K., Xu X. The role of ischemia/reperfusion injury in early hepatic allograft dysfunction. Liver Transplantation . 2020;26(8):1034–1048. doi: 10.1002/lt.25779. [DOI] [PubMed] [Google Scholar]
- 5.Laubach V. E., Sharma A. K. Mechanisms of lung ischemia-reperfusion injury. Current Opinion in Organ Transplantation . 2016;21(3):246–252. doi: 10.1097/MOT.0000000000000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nastos C., Kalimeris K., Papoutsidakis N., et al. Global consequences of liver ischemia/reperfusion injury. Oxidative Medicine and Cellular Longevity . 2014;2014 doi: 10.1155/2014/906965.906965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soares R. O. S., Losada D. M., Jordani M. C., Évora P., Castro E. S. O. Ischemia/reperfusion injury revisited: an overview of the latest pharmacological strategies. International Journal of Molecular Sciences . 2019;20(20, article 5034) doi: 10.3390/ijms20205034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun M. S., Jin H., Sun X., et al. Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxidative Medicine And Cellular Longevity . 2018;2018 doi: 10.1155/2018/3804979.3804979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z., Yao L., Yang J., Wang Z., Du G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (review) Molecular Medicine Reports . 2018;18(4):3547–3554. doi: 10.3892/mmr.2018.9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ke Q., Costa M. Hypoxia-inducible factor-1 (HIF-1) Molecular Pharmacology . 2006;70(5):1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 11.Movafagh S., Crook S., Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. Journal of Cellular Biochemistry . 2015;116(5):696–703. doi: 10.1002/jcb.25074. [DOI] [PubMed] [Google Scholar]
- 12.Chen R., Lai U. H., Zhu L., Singh A., Ahmed M., Forsyth N. R. Reactive oxygen species formation in the brain at different oxygen levels: the role of hypoxia inducible factors. Frontiers in Cell and Developmental Biology . 2018;6:p. 132. doi: 10.3389/fcell.2018.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasai S., Shimizu S., Tatara Y., Mimura J., Itoh K. Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules . 2020;10(2):p. 320. doi: 10.3390/biom10020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim Y. W., Byzova T. V. Oxidative stress in angiogenesis and vascular disease. Blood . 2014;123(5):625–631. doi: 10.1182/blood-2013-09-512749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niture S. K., Khatri R., Jaiswal A. K. Regulation of Nrf2-an update. Free Radical Biology and Medicine . 2014;66:36–44. doi: 10.1016/j.freeradbiomed.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma G., Al-Shabrawey M., Johnson J. A., et al. Protection against myocardial ischemia/reperfusion injury by short-term diabetes: enhancement of VEGF formation, capillary density, and activation of cell survival signaling. Naunyn-Schmiedeberg's Archives of Pharmacology . 2006;373(6):415–427. doi: 10.1007/s00210-006-0102-1. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J., Wang X., Vikash V., et al. ROS and ROS-mediated cellular signaling. Oxidative medicine and cellular longevity . 2016;2016:18. doi: 10.1155/2016/4350965.4350965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalogeris T., Baines C. P., Krenz M., Korthuis R. J. Cell biology of ischemia/reperfusion injury. International Review of Cell and Molecular Biology . 2012;298:229–317. doi: 10.1016/b978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreadou I., Schulz R., Papapetropoulos A., et al. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. Journal of Cellular and Molecular Medicine . 2020;24(12):6510–6522. doi: 10.1111/jcmm.15279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodríguez-Lara S. Q., Cardona-Muñoz E. G., Ramírez-Lizardo E. J., et al. Alternative interventions to prevent oxidative damage following ischemia/reperfusion. Oxidative medicine and cellular longevity. . 2016;2016, article 7190943:16. doi: 10.1155/2016/7190943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braun M. M., Stevens W. A., Barstow C. H. Stable coronary artery disease: treatment. American Family Physician . 2018;97(6):376–384. [PubMed] [Google Scholar]
- 22.Wu M. Y., Yiang G. T., Liao W. T., et al. Current mechanistic concepts in ischemia and reperfusion injury. Cellular Physiology and Biochemistry . 2018;46(4):1650–1667. doi: 10.1159/000489241. [DOI] [PubMed] [Google Scholar]
- 23.Khoshnam S. E., Winlow W., Farzaneh M., Farbood Y., Moghaddam H. F. Pathogenic mechanisms following ischemic stroke. Neurological Sciences . 2017;38(7):1167–1186. doi: 10.1007/s10072-017-2938-1. [DOI] [PubMed] [Google Scholar]
- 24.Creager M. A., Kaufman J. A., Conte M. S. Acute limb ischemia. The New England Journal of Medicine . 2012;366(23):2198–2206. doi: 10.1056/NEJMcp1006054. [DOI] [PubMed] [Google Scholar]
- 25.Jiménez-Castro M. B., Cornide-Petronio M. E., Gracia-Sancho J., Peralta C. Inflammasome-mediated inflammation in liver ischemia-reperfusion injury. Cells . 2019;8(10) doi: 10.3390/cells8101131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caccioppo A., Franchin L., Grosso A., Angelini F., D'Ascenzo F., Brizzi M. F. Ischemia reperfusion injury: mechanisms of damage/protection and novel strategies for cardiac recovery/regeneration. International Journal of Molecular Sciences . 2019;20(20) doi: 10.3390/ijms20205024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng L., Han R., Tao L., et al. Effects of remote ischemic preconditioning on prognosis in patients with lung injury: a meta-analysis. Journal of Clinical Anesthesia . 2020;63, article 109795 doi: 10.1016/j.jclinane.2020.109795. [DOI] [PubMed] [Google Scholar]
- 28.Chistiakov D. A., Shkurat T. P., Melnichenko A. A., Grechko A. V., Orekhov A. N. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Annals of Medicine . 2018;50(2):121–127. doi: 10.1080/07853890.2017.1417631. [DOI] [PubMed] [Google Scholar]
- 29.Frank A., Bonney M., Bonney S., Weitzel L., Koeppen M., Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Seminars in Cardiothoracic and Vascular Anesthesia . 2012;16(3):123–132. doi: 10.1177/1089253211436350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesnefsky E. J., Hoppel C. L. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Archives of Biochemistry and Biophysics . 2003;420(2):287–297. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 31.Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radical Biology and Medicine . 2018;117:76–89. doi: 10.1016/j.freeradbiomed.2018.01.024. [DOI] [PubMed] [Google Scholar]
- 32.Kierans S. J., Taylor C. T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. The Journal of Physiology . 2021;599(1):23–37. doi: 10.1113/JP280572. [DOI] [PubMed] [Google Scholar]
- 33.Halestrap A. P. The mitochondrial permeability transition: its molecular mechanism and role in reperfusion injury. Biochemical Society Symposia . 1999;66:181–203. doi: 10.1042/bss0660181. [DOI] [PubMed] [Google Scholar]
- 34.Davidson S. M., Adameová A., Barile L., et al. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. Journal of Cellular and Molecular Medicine . 2020;24(7):3795–3806. doi: 10.1111/jcmm.15127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penna C., Perrelli M. G., Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxidants & Redox Signaling . 2013;18(5):556–599. doi: 10.1089/ars.2011.4459. [DOI] [PubMed] [Google Scholar]
- 36.Morciano G., Bonora M., Campo G., et al. Mechanistic role of mPTP in ischemia-reperfusion injury. In: Santulli G., editor. Mitochondrial Dynamics in Cardiovascular Medicine . Vol. 982. Cham: Springer; 2017. pp. 169–189. (Advances in Experimental Medicine and Biology). [DOI] [PubMed] [Google Scholar]
- 37.Quesnelle K. M., Bystrom P. V., Toledo-Pereyra L. H. Molecular responses to ischemia and reperfusion in the liver. Archives of Toxicology . 2015;89(5):651–657. doi: 10.1007/s00204-014-1437-x. [DOI] [PubMed] [Google Scholar]
- 38.Kalogeris T., Baines C. P., Krenz M., Korthuis R. J. Ischemia/reperfusion. Comprehensive Physiology . 2016;7(1):113–170. doi: 10.1002/cphy.c160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bompotis G. C., Deftereos S., Angelidis C., et al. Altered calcium handling in reperfusion injury. Medicinal Chemistry . 2016;12(2):114–130. doi: 10.2174/1573406411666150928112420. [DOI] [PubMed] [Google Scholar]
- 40.Murphy E., Steenbergen C. Ion transport and energetics during cell death and protection. Physiology . 2008;23:115–123. doi: 10.1152/physiol.00044.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang J. C., Lien C. F., Lee W. S., et al. Intermittent hypoxia prevents myocardial mitochondrial Ca2+ overload and cell death during ischemia/reperfusion: the role of reactive oxygen species. Cells . 2019;8(6) doi: 10.3390/cells8060564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andreyev A. Y., Kushnareva Y. E., Murphy A. N., Starkov A. A. Mitochondrial ROS metabolism: 10 years later. Biochemistry (Moscow) . 2015;80(5):517–531. doi: 10.1134/S0006297915050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreyev A. Y., Kushnareva Y. E., Starkov A. A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) . 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 44.Dan Dunn J., Alvarez L. A., Zhang X., Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biology . 2015;6:472–485. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown G. C., Borutaite V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion . 2012;12(1):1–4. doi: 10.1016/j.mito.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Forrester S. J., Kikuchi D. S., Hernandes M. S., Xu Q., Griendling K. K. Reactive oxygen species in metabolic and inflammatory signaling. Circulation Research . 2018;122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starkov A. A., Fiskum G., Chinopoulos C., et al. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. Journal of Neuroscience . 2004;24(36):7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinlan C. L., Goncalves R. L., Hey-Mogensen M., Yadava N., Bunik V. I., Brand M. D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. The Journal of Biological Chemistry . 2014;289(12):8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams L., Franco M. C., Estevez A. G. Reactive nitrogen species in cellular signaling. Experimental Biology and Medicine . 2015;240(6):711–717. doi: 10.1177/1535370215581314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schöneich C. Reactive oxygen species and biological aging: a mechanistic approach. Experimental Gerontology . 1999;34(1):19–34. doi: 10.1016/S0531-5565(98)00066-7. [DOI] [PubMed] [Google Scholar]
- 51.Dixon S. J., Lemberg K. M., Lamprecht M. R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan H. F., Tuo Q. Z., Yin Q. Z., Lei P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zoological Research . 2020;41(3):220–230. doi: 10.24272/j.issn.2095-8137.2020.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stockwell B. R., Friedmann Angeli J. P., Bayir H., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell . 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaschler M. M., Stockwell B. R. Lipid peroxidation in cell death. Biochemical and Biophysical Research Communications . 2017;482(3):419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang W. S., Stockwell B. R. Ferroptosis: death by lipid peroxidation. Trends in Cell Biology . 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muzáková V., Kandár R., Vojtísek P., et al. Antioxidant vitamin levels and glutathione peroxidase activity during ischemia/reperfusion in myocardial infarction. Physiological Research . 2001;50(4):389–396. [PubMed] [Google Scholar]
- 57.Latunde-Dada G. O. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochimica et Biophysica Acta (BBA) - General Subjects . 2017;1861(8):1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 58.Li J., Cao F., Yin H. L., et al. Ferroptosis: past, present and future. Cell death & Disease . 2020;11(2):p. 88. doi: 10.1038/s41419-020-2298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sánchez-Duarte E., Cortés-Rojo C., Sánchez-Briones L. A., et al. Nicorandil affects mitochondrial respiratory chain function by increasing complex III activity and ROS production in skeletal muscle mitochondria. The Journal of Membrane Biology . 2020;253(4):309–318. doi: 10.1007/s00232-020-00129-y. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt H. H., Stocker R., Vollbracht C., et al. Antioxidants in translational medicine. Antioxidants & Redox Signaling . 2015;23(14):1130–1143. doi: 10.1089/ars.2015.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Senoner T., Dichtl W. Oxidative stress in cardiovascular diseases: still a therapeutic target? Nutrients . 2019;11(9) doi: 10.3390/nu11092090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salvemini D., Cuzzocrea S. Superoxide, superoxide dismutase and ischemic injury. Current Opinion in Investigational Drugs . 2002;3(6):886–895. [PubMed] [Google Scholar]
- 63.Woo S. H., Kim J. C., Eslenur N., Trinh T. N., Do L. N. H. Modulations of cardiac functions and pathogenesis by reactive oxygen species and natural antioxidants. Antioxidants . 2021;10(5) doi: 10.3390/antiox10050760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lillo-Moya J., Rojas-Solé C., Muñoz-Salamanca D., Panieri E., Saso L., Rodrigo R. Targeting ferroptosis against ischemia/reperfusion cardiac injury. Antioxidants . 2021;10(5):p. 667. doi: 10.3390/antiox10050667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones S. P., Hoffmeyer M. R., Sharp B. R., Ho Y. S., Lefer D. J. Role of intracellular antioxidant enzymes after in vivo myocardial ischemia and reperfusion. American Journal of Physiology Heart and Circulatory Physiology . 2003;284(1):H277–H282. doi: 10.1152/ajpheart.00236.2002. [DOI] [PubMed] [Google Scholar]
- 66.Kumari Naga K., Panigrahi M., Prakash B. P. Changes in endogenous antioxidant enzymes during cerebral ischemia and reperfusion. Neurological Research . 2007;29(8):877–883. doi: 10.1179/016164107x181842. [DOI] [PubMed] [Google Scholar]
- 67.Rodrigo R., Fernández-Gajardo R., Gutiérrez R., et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS & Neurological Disorders Drug Targets . 2013;12(5):698–714. doi: 10.2174/1871527311312050015. [DOI] [PubMed] [Google Scholar]
- 68.Xu G., Zhao X., Fu J., Wang X. Resveratrol increase myocardial Nrf2 expression in type 2 diabetic rats and alleviate myocardial ischemia/reperfusion injury (MIRI) Annals of Palliative Medicine . 2019;8(5):565–575. doi: 10.21037/apm.2019.11.25. [DOI] [PubMed] [Google Scholar]
- 69.Rao X. G., Ma L. M., Duan M. K., Wang T., Chen B. L. Modulation of lung oxidant/antioxidant status by ischemic post-conditioning during ischemia/ reperfusion injury: experiment with rats. Zhonghua Yi Xue Za Zhi . 2008;88(22):1566–1568. [PubMed] [Google Scholar]
- 70.de Rougemont O., Dutkowski P., Clavien P.-A. Biological modulation of liver ischemia–reperfusion injury. Current Opinion in Organ Transplantation . 2010;15(2):183–189. doi: 10.1097/MOT.0b013e3283373ced. [DOI] [PubMed] [Google Scholar]
- 71.Halestrap A. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochemical Society Transactions . 2006;34(2):232–237. doi: 10.1042/BST0340232. [DOI] [PubMed] [Google Scholar]
- 72.He N., Jia J. J., Li J. H., et al. Remote ischemic perconditioning prevents liver transplantation-induced ischemia/reperfusion injury in rats: role of ROS/RNS and eNOS. World Journal of Gastroenterology . 2017;23(5):830–841. doi: 10.3748/wjg.v23.i5.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.El-Hattab A. W., Zarante A. M., Almannai M., Scaglia F. Therapies for mitochondrial diseases and current clinical trials. Molecular Genetics and Metabolism . 2017;122(3):1–9. doi: 10.1016/j.ymgme.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lesnefsky E. J., Chen Q., Tandler B., Hoppel C. L. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annual Review of Pharmacology and Toxicology . 2017;57:535–565. doi: 10.1146/annurev-pharmtox-010715-103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acín-Pérez R., Hernansanz-Agustín P., Enríquez J. A. Chapter 7 - Analyzing electron transport chain supercomplexes. Methods in Cell Biology . 2020;155:181–197. doi: 10.1016/bs.mcb.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 76.Guo R., Gu J., Zong S., Wu M., Yang M. Structure and mechanism of mitochondrial electron transport chain. Biomedical Journal . 2018;41(1):9–20. doi: 10.1016/j.bj.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmad M., Wolberg A., Kahwaji C. I. Biochemistry, electron transport chain. StatPearls . Treasure Island (FL): StatPearls Publishing LLC; 2021. [PubMed] [Google Scholar]
- 78.Dröse S., Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. In: Kadenbach B., editor. Mitochondrial Oxidative Phosphorylation . Vol. 748. New York, NY: Springer; 2012. pp. 145–169. (Advances in experimental medicine and biology). [DOI] [PubMed] [Google Scholar]
- 79.Bezawork-Geleta A., Rohlena J., Dong L., Pacak K., Neuzil J. Mitochondrial complex II: at the crossroads. Trends in Biochemical Sciences . 2017;42(4):312–325. doi: 10.1016/j.tibs.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hadrava Vanova K., Kraus M., Neuzil J., Rohlena J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox report: Communications in Free Radical Research . 2020;25(1):26–32. doi: 10.1080/13510002.2020.1752002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu B., Zhu X., Chen C. L., et al. Opening of the mitoKATP channel and decoupling of mitochondrial complex II and III contribute to the suppression of myocardial reperfusion hyperoxygenation. Molecular and Cellular Biochemistry . 2010;337(1-2):25–38. doi: 10.1007/s11010-009-0283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernandez-Vizarra E., Zeviani M. Mitochondrial complex III Rieske Fe-S protein processing and assembly. Cell Cycle . 2018;17(6):681–687. doi: 10.1016/j.bbabio.2021.148414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lobo-Jarne T., Pérez-Pérez R., Fontanesi F., et al. Multiple pathways coordinate assembly of human mitochondrial complex IV and stabilization of respiratory supercomplexes. The EMBO Journal . 2020;39(14, article e103912) doi: 10.15252/embj.2019103912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang X., Lu G. P., Cai X. D., Lu Z. J., Kissoon N. Alterations of complex IV in the tissues of a septic mouse model. Mitochondrion . 2019;49:89–96. doi: 10.1016/j.mito.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 85.Neupane P., Bhuju S., Thapa N., Bhattarai H. K. ATP synthase: structure, function and inhibition. Biomolecular Concepts . 2019;10(1):1–10. doi: 10.1515/bmc-2019-0001. [DOI] [PubMed] [Google Scholar]
- 86.Missailidis D., Annesley S. J., Allan C. Y., et al. An isolated complex V inefficiency and dysregulated mitochondrial function in immortalized lymphocytes from ME/CFS patients. International Journal of Molecular Sciences . 2020;21(3) doi: 10.3390/ijms21031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Q., Thompson J., Hu Y., Dean J., Lesnefsky E. J. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. American Journal of Physiology Cell Physiology . 2019;317(5):C910–C921. doi: 10.1152/ajpcell.00190.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kist M., Vucic D. Cell death pathways: intricate connections and disease implications. The EMBO Journal . 2021;40(5, article e106700) doi: 10.15252/embj.2020106700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ying Y., Padanilam B. J. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cellular and Molecular Life Sciences . 2016;73(11-12):2309–2324. doi: 10.1007/s00018-016-2202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schinzel A. C., Takeuchi O., Huang Z., et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proceedings of the National Academy of Sciences of the United States of America . 2005;102(34):12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Briston T., Selwood D. L., Szabadkai G., Duchen M. R. Mitochondrial permeability transition: a molecular lesion with multiple drug targets. Trends in Pharmacological Sciences . 2019;40(1):50–70. doi: 10.1016/j.tips.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 92.Colareda G. A., Ragone M. I., Bonazzola P., Consolini A. E. The mKATP channels and protein-kinase C are involved in the cardioprotective effects of genistein on estrogen-deficient rat hearts exposed to ischemia/reperfusion: energetic study. Journal of Cardiovascular Pharmacology . 2020;75(5):460–474. doi: 10.1097/FJC.0000000000000816. [DOI] [PubMed] [Google Scholar]
- 93.Xu T., Ding W., Ao X., et al. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biology . 2019;20:414–426. doi: 10.1016/j.redox.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ong S. B., Samangouei P., Kalkhoran S. B., Hausenloy D. J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. Journal of Molecular and Cellular Cardiology . 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 95.Sánchez-Martínez C., Torres-González L., Alarcón-Galván G., et al. Anti-inflammatory and antioxidant activity of essential amino acid α-ketoacid analogues against renal ischemia-reperfusion damage in Wistar rats. Biomédica . 2020;40(2):336–348. doi: 10.7705/biomedica.4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Candas D., Li J. J. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxidants & Redox Signaling . 2014;20(10):1599–1617. doi: 10.1089/ars.2013.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang H. F., Wang Z. Q., Ding Y., et al. Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neuroscience & Therapeutics . 2018;24(1):29–38. doi: 10.1111/cns.12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aghaei M., Motallebnezhad M., Ghorghanlu S., et al. Targeting autophagy in cardiac ischemia/reperfusion injury: a novel therapeutic strategy. Journal of Cellular Physiology . 2019;234(10):16768–16778. doi: 10.1002/jcp.28345. [DOI] [PubMed] [Google Scholar]
- 99.Samakova A., Gazova A., Sabova N., Valaskova S., Jurikova M., Kyselovic J. The PI3k/Akt pathway is associated with angiogenesis, oxidative stress and survival of mesenchymal stem cells in pathophysiologic condition in ischemia. Physiological Research . 2019;68(Supplement 2):S131–S1s8. doi: 10.33549/physiolres.934345. [DOI] [PubMed] [Google Scholar]
- 100.Shi B., Ma M., Zheng Y., Pan Y., Lin X. mTOR and Beclin1: two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. Journal of Cellular Physiology . 2019;234(8):12562–12568. doi: 10.1002/jcp.28125. [DOI] [PubMed] [Google Scholar]
- 101.Hollander J. M., Thapa D., Shepherd D. L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. American Journal of Physiology Heart and Circulatory Physiology . 2014;307(1):H1–14. doi: 10.1152/ajpheart.00747.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kurian G. A., Rajagopal R., Vedantham S., Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxidative medicine and cellular longevity. . 2016;2016, article 1656450:14. doi: 10.1155/2016/1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rout A., Tantry U. S., Novakovic M., Sukhi A., Gurbel P. A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert opinion on Pharmacotherapy . 2020;21(15):1851–1865. doi: 10.1080/14656566.2020.1787987. [DOI] [PubMed] [Google Scholar]
- 104.Santulli G., Xie W., Reiken S. R., Marks A. R. Mitochondrial calcium overload is a key determinant in heart failure. Proceedings of the National Academy of Sciences of the United States of America . 2015;112(36):11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shintani-Ishida K., Inui M., Yoshida K. Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. Journal of molecular and cellular cardiology . 2012;53(2):233–239. doi: 10.1016/j.yjmcc.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 106.Lascano E., Negroni J., Vila Petroff M., Mattiazzi A. Impact of RyR2 potentiation on myocardial function. American Journal of Physiology Heart and Circulatory Physiology . 2017;312(6):H1105–H11h9. doi: 10.1152/ajpheart.00855.2016. [DOI] [PubMed] [Google Scholar]
- 107.Wu S., Lu Q., Ding Y., et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation . 2019;139(16):1913–1936. doi: 10.1161/CIRCULATIONAHA.118.033552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Konishi T., Lentsch A. B. Hepatic ischemia/reperfusion: mechanisms of tissue injury, repair, and regeneration. Gene Expression . 2017;17(4):277–287. doi: 10.3727/105221617X15042750874156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rodríguez-Reynoso S., Leal-Cortés C., Portilla-de Buen E., López-De la Torre S. P. Ischemic preconditioning preserves liver energy charge and function on hepatic ischemia/reperfusion injury in rats. Archives of Medical Research . 2018;49(6):373–380. doi: 10.1016/j.arcmed.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 110.Linares I., Farrokhi K., Echeverri J., et al. PPAR-gamma activation is associated with reduced liver ischemia-reperfusion injury and altered tissue-resident macrophages polarization in a mouse model. PloS One . 2018;13(4, article e0195212) doi: 10.1371/journal.pone.0195212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsikas D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Analytical Biochemistry . 2017;524:13–30. doi: 10.1016/j.ab.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 112.Sahna E., Parlakpinar H., Ozturk F., Cigremis Y., Acet A. The protective effects of physiological and pharmacological concentrations of melatonin on renal ischemia-reperfusion injury in rats. Urological Research . 2003;31(3):188–193. doi: 10.1007/s00240-003-0314-5. [DOI] [PubMed] [Google Scholar]
- 113.Hesketh E. E., Czopek A., Clay M., et al. Renal ischaemia reperfusion injury: a mouse model of injury and regeneration. Journal of visualized experiments . 2014;88:p. 51816. doi: 10.3791/51816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shiva N., Sharma N., Kulkarni Y. A., Mulay S. R., Gaikwad A. B. Renal ischemia/reperfusion injury: an insight on in vitro and in vivo models. Life Sciences . 2020;256, article 117860 doi: 10.1016/j.lfs.2020.117860. [DOI] [PubMed] [Google Scholar]
- 115.Cruthirds D. L., Novak L., Akhi K. M., Sanders P. W., Thompson J. A., MacMillan-Crow L. A. Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Archives of Biochemistry and Biophysics . 2003;412(1):27–33. doi: 10.1016/S0003-9861(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 116.Peerapanyasut W., Thamprasert K., Wongmekiat O. Ubiquinol supplementation protects against renal ischemia and reperfusion injury in rats. Free Radical Research . 2014;48(2):180–189. doi: 10.3109/10715762.2013.858148. [DOI] [PubMed] [Google Scholar]
- 117.Hu B., Tang J., Zhang Y., et al. Glycogen synthase kinase-3β inhibitor attenuates renal damage through regulating antioxidant and anti-inflammation in rat kidney transplant with cold ischemia reperfusion. Transplantation Proceedings . 2019;51(6):2066–2070. doi: 10.1016/j.transproceed.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 118.Palutina O. A., Sharapov M. G., Temnov A. A., Novoselov V. I. Nephroprotective effect exogenous antioxidant enzymes during ischemia/reperfusion-induced damage of renal tissue. Bulletin of Experimental Biology and Medicine. . 2016;160(3):322–326. doi: 10.1007/s10517-016-3161-4. [DOI] [PubMed] [Google Scholar]
- 119.Rodríguez-Reynoso S., Leal C., Portilla-de Buen E., Castillo J. C., Ramos-Solano F. Melatonin ameliorates renal ischemia/reperfusion injury. The Journal of Surgical Research . 2004;116(2):242–247. doi: 10.1016/j.jss.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 120.Li Y., Zhong D., Lei L., Jia Y., Zhou H., Yang B. Propofol prevents renal ischemia-reperfusion injury via inhibiting the oxidative stress pathways. Cellular Physiology and Biochemistry . 2015;37(1):14–26. doi: 10.1159/000430329. [DOI] [PubMed] [Google Scholar]
- 121.Diao C., Chen Z., Qiu T., et al. Inhibition of PRMT5 attenuates oxidative stress-induced pyroptosis via activation of the Nrf2/HO-1 signal pathway in a mouse model of renal ischemia-reperfusion injury. Oxidative medicine and cellular longevity. . 2019;2019, article 2345658:18. doi: 10.1155/2019/2345658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi S., Lei S., Tang C., Wang K., Xia Z. Melatonin attenuates acute kidney ischemia/reperfusion injury in diabetic rats by activation of the SIRT1/Nrf2/HO-1 signaling pathway. Bioscience Reports . 2019;39(1) doi: 10.1042/BSR20181614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rahman N. A., Mori K., Mizukami M., Suzuki T., Takahashi N., Ohyama C. Role of peroxynitrite and recombinant human manganese superoxide dismutase in reducing ischemia-reperfusion renal tissue injury. Transplantation proceedings. . 2009;41(9):3603–3610. doi: 10.1016/j.transproceed.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 124.Jiang G. P., Liao Y. J., Huang L. L., Zeng X. J., Liao X. H. Effects and molecular mechanism of pachymic acid on ferroptosis in renal ischemia reperfusion injury. Molecular Medicine Reports . 2021;23(1) doi: 10.3892/mmr.2020.11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Su L., Jiang X., Yang C., et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. Journal of Biological Chemistry . 2019;294(50):19395–19404. doi: 10.1074/jbc.RA119.010949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bersuker K., Hendricks J. M., Li Z., et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature . 2019;575(7784):688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang J., Bi J., Ren Y., et al. Involvement of GPX4 in irisin’s protection against ischemia reperfusion-induced acute kidney injury. Journal of Cellular Physiology . 2021;236(2):931–945. doi: 10.1002/jcp.29903. [DOI] [PubMed] [Google Scholar]
- 128.Jones A. R., Overly C. C., Sunkin S. M. The Allen Brain Atlas: 5 years and beyond. Nature reviews Neuroscience . 2009;10(11):821–828. doi: 10.1038/nrn2722. [DOI] [PubMed] [Google Scholar]
- 129.Kraytsberg Y., Nekhaeva E., Bodyak N. B., Khrapko K. Mutation and intracellular clonal expansion of mitochondrial genomes: two synergistic components of the aging process? Mechanisms of ageing and Development . 2003;124(1):49–53. doi: 10.1016/S0047-6374(02)00169-0. [DOI] [PubMed] [Google Scholar]
- 130.Trifunovic A., Wredenberg A., Falkenberg M., et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature . 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 131.Allen C. L., Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. International Journal of Stroke . 2009;4(6):461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 132.Shen L., Gan Q., Yang Y., et al. Mitophagy in cerebral ischemia and ischemia/reperfusion injury. Frontiers in Aging Neuroscience . 2021;13:p. 284. doi: 10.3389/fnagi.2021.687246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li L., Tan J., Miao Y., Lei P., Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cellular and Molecular Neurobiology . 2015;35(5):615–621. doi: 10.1007/s10571-015-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Saso L., Firuzi O. Pharmacological applications of antioxidants: lights and shadows. Current Drug Targets . 2014;15(13):1177–1199. doi: 10.2174/1389450115666141024113925. [DOI] [PubMed] [Google Scholar]
- 135.Kubera M., Obuchowicz E., Goehler L., Brzeszcz J., Maes M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Progress in Neuro-Psychopharmacology & Biological Psychiatry . 2011;35(3):744–759. doi: 10.1016/j.pnpbp.2010.08.026. [DOI] [PubMed] [Google Scholar]
- 136.Salim S. Oxidative stress and the central nervous system. The Journal of pharmacology and experimental therapeutics . 2017;360(1):201–205. doi: 10.1124/jpet.116.237503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Grimm A., Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. Journal of Neurochemistry . 2017;143(4):418–431. doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rabinstein A. A. Update on treatment of acute ischemic stroke. Continuum . 2020;26(2):268–286. doi: 10.1212/CON.0000000000000840. [DOI] [PubMed] [Google Scholar]
- 139.Khan M. S., Aouad R. The effects of insomnia and sleep loss on cardiovascular disease. Sleep Medicine Clinics . 2017;12(2):167–177. doi: 10.1016/j.jsmc.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 140.Sanderson T. H., Reynolds C. A., Kumar R., Przyklenk K., Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Molecular Neurobiology . 2013;47(1):9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Papa S., Martino P. L., Capitanio G., et al. The oxidative phosphorylation system in mammalian mitochondria. In: Scatena R., Bottoni P., Giardina B., editors. Advances in Mitochondrial Medicine . Vol. 942. Dordrecht: Springer; 2012. pp. 3–37. (Advances in Experimental Medicine and Biology). [DOI] [PubMed] [Google Scholar]
- 142.Ruiz-Pesini E., López-Gallardo E., Dahmani Y., et al. Diseases of the human mitochondrial oxidative phosphorylation system. Revista de neurologia . 2006;43(7):416–424. [PubMed] [Google Scholar]
- 143.Gellerich F. N., Gizatullina Z., Gainutdinov T., et al. The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB life . 2013;65(3):180–190. doi: 10.1002/iub.1131. [DOI] [PubMed] [Google Scholar]
- 144.Monteiro J., Assis-de-Lemos G., de Souza-Ferreira E., et al. Energization by multiple substrates and calcium challenge reveal dysfunctions in brain mitochondria in a model related to acute psychosis. Journal of Bioenergetics and Biomembranes . 2020;52(1):1–15. doi: 10.1007/s10863-019-09816-5. [DOI] [PubMed] [Google Scholar]
- 145.Cogliati S., Lorenzi I., Rigoni G., Caicci F., Soriano M. E. Regulation of mitochondrial electron transport chain assembly. Journal of Molecular Biology . 2018;430(24):4849–4873. doi: 10.1016/j.jmb.2018.09.016. [DOI] [PubMed] [Google Scholar]
- 146.Krajewski S., Krajewska M., Ellerby L. M., et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proceedings of the National Academy of Sciences of the United States of America . 1999;96(10):5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nakka V. P., Gusain A., Mehta S. L., Raghubir R. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Molecular Neurobiology . 2008;37(1):7–38. doi: 10.1007/s12035-007-8013-9. [DOI] [PubMed] [Google Scholar]
- 148.Massey K. A., Nicolaou A. Lipidomics of polyunsaturated-fatty-acid-derived oxygenated metabolites. Biochemical Society Transactions . 2011;39(5):1240–1246. doi: 10.1042/BST0391240. [DOI] [PubMed] [Google Scholar]
- 149.Ayala A., Muñoz M. F., Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative medicine and cellular longevity . 2014;2014:31. doi: 10.1155/2014/360438.360438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reis A., Spickett C. M. Chemistry of phospholipid oxidation. Biochimica et Biophysica Acta . 2012;1818(10):2374–2387. doi: 10.1016/j.bbamem.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 151.Niki E. Antioxidants: basic principles, emerging concepts, and problems. Biomedical Journal . 2014;37(3):106–111. doi: 10.4103/2319-4170.128727. [DOI] [PubMed] [Google Scholar]
- 152.Abrigo J., Simon F., Cabrera D., Vilos C., Cabello-Verrugio C. Mitochondrial dysfunction in skeletal muscle pathologies. Current Protein & Peptide Science . 2019;20(6):536–546. doi: 10.2174/1389203720666190402100902. [DOI] [PubMed] [Google Scholar]
- 153.Wan J., Ren H., Wang J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke and Vascular Neurology . 2019;4(2):93–95. doi: 10.1136/svn-2018-000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Irrinki K. M., Mallilankaraman K., Thapa R. J., et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Molecular and Cellular Biology . 2011;31(18):3745–3758. doi: 10.1128/MCB.05303-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Friedman J. R., Nunnari J. Mitochondrial form and function. Nature . 2014;505(7483):335–343. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sas K., Szabó E., Vécsei L. Mitochondria, oxidative stress and the kynurenine system, with a focus on ageing and neuroprotection. Molecules . 2018;23(1):p. 191. doi: 10.3390/molecules23010191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Darlington L., Mackay G., Forrest C., Stoy N., George C., Stone T. Altered kynurenine metabolism correlates with infarct volume in stroke. European Journal of Neuroscience . 2007;26(8):2211–2221. doi: 10.1111/j.1460-9568.2007.05838.x. [DOI] [PubMed] [Google Scholar]
- 158.Lugo-Huitrón R., Blanco-Ayala T., Ugalde-Muñiz P., et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicology and Teratology . 2011;33(5):538–547. doi: 10.1016/j.ntt.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 159.Angelova P. R., Esteras N., Abramov A. Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: finding ways for prevention. Medicinal Research Reviews . 2021;41(2):770–784. doi: 10.1002/med.21712. [DOI] [PubMed] [Google Scholar]
- 160.Chavda V., Chaurasia B., Garg K., et al. Molecular mechanisms of oxidative stress in stroke and cancer. Brain Disorders . 2022;5, article 100029 doi: 10.1016/j.dscb.2021.100029. [DOI] [Google Scholar]
- 161.Siegel C. S., McCullough L. D. NAD+ and nicotinamide: sex differences in cerebral ischemia. Neuroscience . 2013;237:223–231. doi: 10.1016/j.neuroscience.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Braidy N., Smani T., Naziroglu M. Editorial: Involvements of TRP channels, oxidative stress and apoptosis in neurodegenerative diseases. Frontiers in Physiology . 2021;12, article 649230 doi: 10.3389/fphys.2021.649230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dölle C., Rack J. G., Ziegler M. NAD and ADP-ribose metabolism in mitochondria. The FEBS Journal . 2013;280(15):3530–3541. doi: 10.1111/febs.12304. [DOI] [PubMed] [Google Scholar]
- 164.Fan J., Lv H., Li J., et al. Roles of Nrf2/HO-1 and HIF-1α/VEGF in lung tissue injury and repair following cerebral ischemia/reperfusion injury. Journal of Cellular Physiology . 2019;234(6):7695–7707. doi: 10.1002/jcp.27767. [DOI] [PubMed] [Google Scholar]
- 165.Dong H., Qiang Z., Chai D., et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging . 2020;12(13):12943–12959. doi: 10.18632/aging.103378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhao H., Eguchi S., Alam A., Ma D. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. American Journal of Physiology Lung Cellular and Molecular Physiology . 2017;312(2):L155–Ll62. doi: 10.1152/ajplung.00449.2016. [DOI] [PubMed] [Google Scholar]
- 167.Jiang H., Huang Y., Xu H., Hu R., Li Q. F. Inhibition of hypoxia inducible factor-1α ameliorates lung injury induced by trauma and hemorrhagic shock in rats. Acta Pharmacologica Sinica . 2012;33(5):635–643. doi: 10.1038/aps.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shim W. S., Li W., Zhang L., et al. Angiopoietin-1 promotes functional neovascularization that relieves ischemia by improving regional reperfusion in a swine chronic myocardial ischemia model. Journal of Biomedical Science . 2006;13(4):579–591. doi: 10.1007/s11373-006-9082-x. [DOI] [PubMed] [Google Scholar]
- 169.Lin F., Pan L. H., Ruan L., et al. Differential expression of HIF-1α, AQP-1, and VEGF under acute hypoxic conditions in the non-ventilated lung of a one-lung ventilation rat model. Life Sciences . 2015;124:50–55. doi: 10.1016/j.lfs.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 170.Tan Y. B., Mulekar S., Gorodnya O., et al. Pharmacologic protection of mitochondrial DNA integrity may afford a new strategy for suppressing lung ischemia-reperfusion injury. Annals of the American Thoracic Society . 2017;14(Supplement 3):S210–S215. doi: 10.1513/annalsats.201706-438mg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Reader B. F., Dumond C., Lee Y., Mokadam N. A., Black S. M., Whitson B. A. Pegylated-catalase is protective in lung ischemic injury and oxidative stress. The Annals of Thoracic Surgery . 2021;111(3):1019–1027. doi: 10.1016/j.athoracsur.2020.05.131. [DOI] [PubMed] [Google Scholar]
- 172.Wolf P. S., Merry H. E., Farivar A. S., McCourtie A. S., Mulligan M. S. Stress-activated protein kinase inhibition to ameliorate lung ischemia reperfusion injury. The Journal of Thoracic and Cardiovascular Surgery . 2008;135(3):656–665. doi: 10.1016/j.jtcvs.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 173.Wang J., Tan J., Liu Y., Song L., Li D., Cui X. Amelioration of lung ischemia-reperfusion injury by JNK and p38 small interfering RNAs in rat pulmonary microvascular endothelial cells in an ischemia-reperfusion injury lung transplantation model. Molecular Medicine Reports . 2018;17(1):1228–1234. doi: 10.3892/mmr.2017.7985. [DOI] [PubMed] [Google Scholar]
- 174.Beckers P. A. J., Gielis J. F., Van Schil P. E., Adriaensen D. Lung ischemia reperfusion injury: the therapeutic role of dipeptidyl peptidase 4 inhibition. Annals of Translational Medicine . 2017;5(6):p. 129. doi: 10.21037/atm.2017.01.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gillani S., Cao J., Suzuki T., Hak D. J. The effect of ischemia reperfusion injury on skeletal muscle. Injury . 2012;43(6):670–675. doi: 10.1016/j.injury.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 176.Wang W. Z., Fang X. H., Stephenson L. L., Khiabani K. T., Zamboni W. A. Ischemia/reperfusion-induced necrosis and apoptosis in the cells isolated from rat skeletal muscle. Journal of Orthopaedic Research . 2008;26(3):351–356. doi: 10.1002/jor.20493. [DOI] [PubMed] [Google Scholar]
- 177.Consolini A. E., Ragone M. I., Bonazzola P., Colareda G. A. Mitochondrial bioenergetics during ischemia and reperfusion. In: Santulli G., editor. Mitochondrial Dynamics in Cardiovascular Medicine . Vol. 982. Cham: Springer; 2017. pp. 141–167. (Advances in Experimental Medicine and Biology). [DOI] [PubMed] [Google Scholar]
- 178.Michaelson L. P., Iler C., Ward C. W. ROS and RNS signaling in skeletal muscle: critical signals and therapeutic targets. Annual review of nursing research. . 2013;31:367–387. doi: 10.1891/0739-6686.31.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zuo L., Pannell B. K. Redox characterization of functioning skeletal muscle. Frontiers in Physiology . 2015;6:p. 338. doi: 10.3389/fphys.2015.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Thomas J. A., Mallis R. J. Aging and oxidation of reactive protein sulfhydryls. Experimental Gerontology . 2001;36(9):1519–1526. doi: 10.1016/S0531-5565(01)00137-1. [DOI] [PubMed] [Google Scholar]
- 181.Dubois-Deruy E., Peugnet V., Turkieh A., Pinet F. Oxidative stress in cardiovascular diseases. Antioxidants . 2020;9(9) doi: 10.3390/antiox9090864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li L., Mühlfeld C., Niemann B., et al. Mitochondrial biogenesis and PGC-1α deacetylation by chronic treadmill exercise: differential response in cardiac and skeletal muscle. Basic Research in Cardiology . 2011;106(6):1221–1234. doi: 10.1007/s00395-011-0213-9. [DOI] [PubMed] [Google Scholar]
- 183.Liu Y., Hernández-Ochoa E. O., Randall W. R., Schneider M. F. NOX2-dependent ROS is required for HDAC5 nuclear efflux and contributes to HDAC4 nuclear efflux during intense repetitive activity of fast skeletal muscle fibers. American Journal of Physiology Cell Physiology . 2012;303(3):C334–C347. doi: 10.1152/ajpcell.00152.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ndisang J. F. Synergistic interaction between heme oxygenase (HO) and nuclear-factor E2- related factor-2 (Nrf2) against oxidative stress in cardiovascular related diseases. Current Pharmaceutical Design . 2017;23(10):1465–1470. doi: 10.2174/1381612823666170113153818. [DOI] [PubMed] [Google Scholar]
- 185.Yuan P. G., Xue B. B., Lin B., et al. Nrf2/ARE pathway mediates the reducing effect of dexmedeto-midine on ischemia/reperfusion injury in skeletal muscle. Zhongguo ying yong sheng li xue za zhi = Zhongguo yingyong shenglixue zazhi = Chinese journal of applied physiology . 2016;32(3):250–254. doi: 10.13459/j.cnki.cjap.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 186.McDermott M. M., Ferrucci L., Gonzalez-Freire M., et al. Skeletal muscle pathology in peripheral artery disease: a brief review. Arteriosclerosis, thrombosis, and vascular biology. . 2020;40(11):2577–2585. doi: 10.1161/ATVBAHA.120.313831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Koekkoek W. A., van Zanten A. R. Antioxidant vitamins and trace elements in critical illness. Nutrition in Clinical Practice . 2016;31(4):457–474. doi: 10.1177/0884533616653832. [DOI] [PubMed] [Google Scholar]
- 188.Reiter R. J., Mayo J. C., Tan D. X., Sainz R. M., Alatorre-Jimenez M., Qin L. Melatonin as an antioxidant: under promises but over delivers. Journal of Pineal Research . 2016;61(3):253–278. doi: 10.1111/jpi.12360. [DOI] [PubMed] [Google Scholar]
- 189.Dennis J. M., Witting P. K. Protective role for antioxidants in acute kidney disease. Nutrients . 2017;9(7) doi: 10.3390/nu9070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lu M. C., Ji J. A., Jiang Z. Y., You Q. D. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: an update. Medicinal Research Reviews . 2016;36(5):924–963. doi: 10.1002/med.21396. [DOI] [PubMed] [Google Scholar]
- 191.Zhang Y., Murugesan P., Huang K., Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nature Reviews Cardiology . 2020;17(3):170–194. doi: 10.1038/s41569-019-0260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ndrepepa G. Myeloperoxidase - a bridge linking inflammation and oxidative stress with cardiovascular disease. Clinica Chimica Acta . 2019;493:36–51. doi: 10.1016/j.cca.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 193.Lee J., Bae E. H., Ma S. K., Kim S. W. Altered nitric oxide system in cardiovascular and renal diseases. Chonnam Medical Journal . 2016;52(2):81–90. doi: 10.4068/cmj.2016.52.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Granger D. N., Kvietys P. R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biology . 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lushchak V. I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chemico-Biological Interactions . 2014;224:164–175. doi: 10.1016/j.cbi.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 196.Schapira A. H. Complex I: inhibitors, inhibition and neurodegeneration. Experimental Neurology . 2010;224(2):331–335. doi: 10.1016/j.expneurol.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 197.Jang S., Lewis T. S., Powers C., et al. Elucidating mitochondrial electron transport chain supercomplexes in the heart during ischemia-reperfusion. Antioxidants & Redox Signaling . 2017;27(1):57–69. doi: 10.1089/ars.2016.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lee H. L., Chen C. L., Yeh S. T., Zweier J. L., Chen Y. R. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. American Journal of Physiology Heart and Circulatory Physiology . 2012;302(7):H1410–H1422. doi: 10.1152/ajpheart.00731.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lenaz G., Baracca A., Barbero G., et al. Mitochondrial respiratory chain super-complex I-III in physiology and pathology. Biochimica et Biophysica Acta (BBA) - Bioenergetics . 2010;1797(6-7):633–640. doi: 10.1016/j.bbabio.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 200.Tompkins A. J., Burwell L. S., Digerness S. B., Zaragoza C., Holman W. L., Brookes P. S. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease . 2006;1762(2):223–231. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 201.Madungwe N. B., Zilberstein N. F., Feng Y., Bopassa J. C. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. American Journal of Cardiovascular Disease . 2016;6(3):93–108. [PMC free article] [PubMed] [Google Scholar]
- 202.Chang W. T., Li J., Vanden Hoek M. S., et al. Baicalein preconditioning protects cardiomyocytes from ischemia-reperfusion injury via mitochondrial oxidant signaling. The American Journal of Chinese Medicine . 2013;41(2):315–331. doi: 10.1142/s0192415x13500237. [DOI] [PubMed] [Google Scholar]
- 203.Cheng H., Liu H. F., Yang L., et al. N-(3,5-Dichloro-4-(2,4,6-trichlorophenoxy)phenyl)benzenesulfonamide: a new dual-target inhibitor of mitochondrial complex II and complex III via structural simplification. Bioorganic & Medicinal Chemistry . 2020;28(5, article 115299) doi: 10.1016/j.bmc.2019.115299. [DOI] [PubMed] [Google Scholar]
- 204.Bordt E. A., Clerc P., Roelofs B. A., et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Developmental Cell . 2017;40(6):583–594.e6. doi: 10.3390/antiox11030450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Detaille D., Pasdois P., Sémont A., Dos Santos P., Diolez P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS One . 2019;14(5, article e0216385) doi: 10.1371/journal.pone.0216385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Skemiene K., Rekuviene E., Jekabsone A., Cizas P., Morkuniene R., Borutaite V. Comparison of effects of metformin, phenformin, and inhibitors of mitochondrial complex I on mitochondrial permeability transition and ischemic brain injury. Biomolecules . 2020;10(10, article 1400) doi: 10.3390/biom10101400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen Q., Hoppel C. L., Lesnefsky E. J. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. The Journal of Pharmacology and Experimental Therapeutics . 2006;316(1):200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 208.Ichikawa H., Takagi T., Uchiyama K., et al. Rotenone, a mitochondrial electron transport inhibitor, ameliorates ischemia-reperfusion-induced intestinal mucosal damage in rats. Redox Report . 2004;9(6):313–316. doi: 10.1179/135100004225006795. [DOI] [PubMed] [Google Scholar]
- 209.Matsuzaki S., Humphries K. M. Selective inhibition of deactivated mitochondrial complex I by biguanides. Biochemistry . 2015;54(11):2011–2021. doi: 10.1021/bi501473h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nadtochiy S. M., Burwell L. S., Ingraham C. A., et al. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. Journal of Molecular and Cellular Cardiology . 2009;46(6):960–968. doi: 10.1016/j.yjmcc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Martín-Rodríguez S., de Pablos-Velasco P., Calbet J. A. L. Mitochondrial complex I inhibition by metformin: drug-exercise interactions. Trends in Endocrinology and Metabolism . 2020;31(4):269–271. doi: 10.1016/j.tem.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 212.Mohsin A. A., Chen Q., Quan N., et al. Mitochondrial complex I inhibition by metformin limits reperfusion injury. The Journal of pharmacology and experimental therapeutics . 2019;369(2):282–290. doi: 10.1124/jpet.118.254300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Brown J. M., Quinton M. S., Yamamoto B. K. Methamphetamine-induced inhibition of mitochondrial complex II: roles of glutamate and peroxynitrite. Journal of Neurochemistry . 2005;95(2):429–436. doi: 10.1111/j.1471-4159.2005.03379.x. [DOI] [PubMed] [Google Scholar]
- 214.Fink B. D., Bai F., Yu L., et al. Oxaloacetic acid mediates ADP-dependent inhibition of mitochondrial complex II-driven respiration. The Journal of Biological Chemistry . 2018;293(51):19932–19941. doi: 10.1074/jbc.RA118.005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Lai B., Zhang L., Dong L. Y., Zhu Y. H., Sun F. Y., Zheng P. Impact of inhibition of Qo site of mitochondrial complex III with myxothiazol on persistent sodium currents via superoxide and protein kinase C in rat hippocampal CA1 cells. Neurobiology of Disease . 2006;21(1):206–216. doi: 10.1016/j.nbd.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 216.Baudry N., Laemmel E., Vicaut E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: involvement of xanthine oxidase and mitochondria. American Journal of Physiology Heart and Circulatory Physiology . 2008;294(2):H821–H828. doi: 10.1152/ajpheart.00378.2007. [DOI] [PubMed] [Google Scholar]
- 217.Tanaka-Esposito C., Chen Q., Lesnefsky E. J. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Translational Research . 2012;160(3):207–216. doi: 10.1016/j.trsl.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hough M. A., Silkstone G., Worrall J. A., Wilson M. T. NO binding to the proapoptotic cytochrome c-cardiolipin complex. Vitamins and Hormones . 2014;96:193–209. doi: 10.1016/b978-0-12-800254-4.00008-8. [DOI] [PubMed] [Google Scholar]
- 219.Nakagawa T., Shimizu S., Watanabe T., et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature . 2005;434(7033):652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 220.Karlsson L. O., Zhou A. X., Larsson E., et al. Cyclosporine does not reduce myocardial infarct size in a porcine ischemia-reperfusion model. Journal of Cardiovascular Pharmacology and Therapeutics . 2010;15(2):182–189. doi: 10.1177/1074248410362074. [DOI] [PubMed] [Google Scholar]
- 221.Lie R. H., Stoettrup N., Sloth E., Hasenkam J. M., Kroyer R., Nielsen T. T. Post-conditioning with cyclosporine A fails to reduce the infarct size in an in vivo porcine model. Acta anaesthesiologica Scandinavica . 2010;54(7):804–813. doi: 10.1111/j.1399-6576.2010.02241.x. [DOI] [PubMed] [Google Scholar]
- 222.Lindblom R. S. J., Higgins G. C., Nguyen T. V., et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clinical Science . 2020;134(2):239–259. doi: 10.1042/cs20190787. [DOI] [PubMed] [Google Scholar]
- 223.Ahmadi F., Hajihashemi S., Rahbari A., Ghanbari F. Effects of nitroglycerine on renal ischemia-reperfusion injury in adult male rats. Drug Research . 2019;69(11):612–620. doi: 10.1055/a-0958-1987. [DOI] [PubMed] [Google Scholar]
- 224.Roy S., Šileikytė J., Neuenswander B., et al. N-Phenylbenzamides as potent inhibitors of the mitochondrial permeability transition pore. ChemMedChem . 2016;11(3):283–288. doi: 10.1002/cmdc.201500545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fancelli D., Abate A., Amici R., et al. Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia-reperfusion injury protective effect in vivo. Journal of Medicinal Chemistry . 2014;57(12):5333–5347. doi: 10.1021/jm500547c. [DOI] [PubMed] [Google Scholar]
- 226.Li X., Zhang J., Zhang X., Dong M. Puerarin suppresses MPP+/MPTP-induced oxidative stress through an Nrf2-dependent mechanism. Food and Chemical Toxicology . 2020;144, article 111644 doi: 10.1016/j.fct.2020.111644. [DOI] [PubMed] [Google Scholar]
- 227.Li X., Zhang J., Rong H., Zhang X., Dong M. Ferulic acid ameliorates MPP+/MPTP-induced oxidative stress via ERK1/2-dependent Nrf2 activation: translational implications for Parkinson disease treatment. Molecular Neurobiology . 2020;57(7):2981–2995. doi: 10.1007/s12035-020-01934-1. [DOI] [PubMed] [Google Scholar]
- 228.Dhingra R., Guberman M., Rabinovich-Nikitin I., et al. Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovascular Research . 2020;116(6):1161–1174. doi: 10.1093/cvr/cvz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Janani C., Ranjitha Kumari B. D. PPAR gamma gene – a review. Diabetes & Metabolic Syndrome: Clinical Research & Reviews . 2015;9(1):46–50. doi: 10.1016/j.dsx.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 230.Shao X., Wang M., Wei X., et al. Peroxisome proliferator-activated receptor-γ: master regulator of adipogenesis and obesity. Current Stem Cell Research & Therapy . 2016;11(3):282–289. doi: 10.2174/1574888X10666150528144905. [DOI] [PubMed] [Google Scholar]
- 231.DiRenzo J., Söderstrom M., Kurokawa R., et al. Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Molecular and cellular biology . 1997;17(4):2166–2176. doi: 10.1128/MCB.17.4.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yang W., Chen J., Meng Y., Chen Z., Yang J. Novel targets for treating ischemia-reperfusion injury in the liver. International Journal of Molecular Sciences . 2018;19(5) doi: 10.3390/ijms19051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Efrati S., Berman S., Ilgiyeav E., Averbukh Z., Weissgarten J. PPAR-γ activation inhibits angiotensin II synthesis, apoptosis, and proliferation of mesangial cells from spontaneously hypertensive rats. Nephron Experimental Nephrology . 2007;106(4):e107–e112. doi: 10.1159/000104834. [DOI] [PubMed] [Google Scholar]
- 234.Yu Y., Xue B. J., Wei S. G., et al. Activation of central PPAR-γ attenuates angiotensin II-induced hypertension. Hypertension . 2015;66(2):403–411. doi: 10.1161/hypertensionaha.115.05726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sasaki S. Development of novel functional molecules targeting DNA and RNA. Chemical and Pharmaceutical Bulletin . 2019;67(6):505–518. doi: 10.1248/cpb.c19-00169. [DOI] [PubMed] [Google Scholar]
- 236.Shekhar S., Cunningham M. W., Pabbidi M. R., Wang S., Booz G. W., Fan F. Targeting vascular inflammation in ischemic stroke: recent developments on novel immunomodulatory approaches. European Journal of Pharmacology . 2018;833:531–544. doi: 10.1016/j.ejphar.2018.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ray P. D., Huang B. W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular Signalling . 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang J., Ren D., Fedorova J., He Z., Li J. SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the aging heart. Antioxidants . 2020;9(9) doi: 10.3390/antiox9090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Serocki M., Bartoszewska S., Janaszak-Jasiecka A., Ochocka R. J., Collawn J. F., Bartoszewski R. miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis . 2018;21(2):183–202. doi: 10.1007/s10456-018-9600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bandara K. V., Michael M. Z., Gleadle J. M. MicroRNA biogenesis in hypoxia. MicroRNA . 2017;6(2):80–96. doi: 10.2174/2211536606666170313114821. [DOI] [PubMed] [Google Scholar]
- 241.Zaccagnini G., Maimone B., Fuschi P., et al. Hypoxia-induced miR-210 is necessary for vascular regeneration upon acute limb ischemia. International Journal of Molecular Sciences . 2019;21(1) doi: 10.3390/ijms21010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Karshovska E., Wei Y., Subramanian P., et al. HIF-1α (hypoxia-inducible factor-1α) promotes macrophage necroptosis by regulating miR-210 and miR-383. Arteriosclerosis, Thrombosis, and Vascular Biology . 2020;40(3):583–596. doi: 10.1161/ATVBAHA.119.313290. [DOI] [PubMed] [Google Scholar]
- 243.Cullinan S. B., Diehl J. A. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. The International Journal of Biochemistry & Cell Biology . 2006;38(3):317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 244.Cheng F. Y., Lee Y. H., Hsu Y. H., et al. Promising therapeutic effect of thapsigargin nanoparticles on chronic kidney disease through the activation of Nrf2 and FoxO1. Aging . 2019;11(21):9875–9892. doi: 10.18632/aging.102437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kobayashi M., Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxidants & Redox Signaling . 2005;7(3-4):385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 246.Nguyen T., Nioi P., Pickett C. B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of Biological Chemistry . 2009;284(20):13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mitchell S., Vargas J., Hoffmann A. Signaling via the NFκB system. WIREs Systems Biology and Medicine . 2016;8(3):227–241. doi: 10.1002/wsbm.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cordani M., Butera G., Pacchiana R., et al. Mutant p53-associated molecular mechanisms of ROS regulation in cancer cells. Biomolecules . 2020;10(3) doi: 10.3390/biom10030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Berg D., Youdim M. B., Riederer P. Redox imbalance. Cell and Tissue Research . 2004;318(1):201–213. doi: 10.1007/s00441-004-0976-5. [DOI] [PubMed] [Google Scholar]
- 250.Matsui R., Watanabe Y., Murdoch C. E. Redox regulation of ischemic limb neovascularization - what we have learned from animal studies. Redox Biology . 2017;12:1011–1019. doi: 10.1016/j.redox.2017.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.De Windt L. J., Thum T. State-of-the-art on non-coding RNA bioinformatics, diagnostics and therapeutics in cardiovascular diseases. Journal of Molecular and Cellular Cardiology . 2015;89:1–2. doi: 10.1016/j.yjmcc.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 252.Kura B., Szeiffova Bacova B., Kalocayova B., Sykora M., Slezak J. Oxidative stress-responsive microRNAs in heart injury. International Journal of Molecular Sciences . 2020;21(1):p. 358. doi: 10.3390/ijms21010358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lu T. X., Rothenberg M. E. MicroRNA. The Journal of Allergy and Clinical Immunology . 2018;141(4):1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Carlomosti F., D'Agostino M., Beji S., et al. Oxidative stress-induced miR-200c disrupts the regulatory loop among SIRT1, FOXO1, and eNOS. Antioxidants & Redox Signaling . 2017;27(6):328–344. doi: 10.1089/ars.2016.6643. [DOI] [PubMed] [Google Scholar]
- 255.Gong C., Zhou X., Lai S., Wang L., Liu J. Long noncoding RNA/circular RNA-miRNA-mRNA axes in ischemia-reperfusion injury. BioMed research international. . 2020;2020, article 8838524:33. doi: 10.1155/2020/8838524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hinkel R., Penzkofer D., Zühlke S., et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation . 2013;128(10):1066–1075. doi: 10.1161/CIRCULATIONAHA.113.001904. [DOI] [PubMed] [Google Scholar]
- 257.Shi Y., Han Y., Niu L., Li J., Chen Y. MiR-499 inhibited hypoxia/reoxygenation induced cardiomyocytes injury by targeting SOX6. Biotechnology Letters . 2019;41(6-7):837–847. doi: 10.1007/s10529-019-02685-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Xiao X., Lu Z., Lin V., et al. MicroRNA miR-24-3p reduces apoptosis and regulates Keap1-Nrf2 pathway in mouse cardiomyocytes responding to ischemia/reperfusion injury. Oxidative Medicine and Cellular Longevity . 2018;2018:9. doi: 10.1155/2018/7042105.7042105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wei Z., Qiao S., Zhao J., et al. miRNA-181a over-expression in mesenchymal stem cell-derived exosomes influenced inflammatory response after myocardial ischemia-reperfusion injury. Life sciences . 2019;232, article 116632 doi: 10.1016/j.lfs.2019.116632. [DOI] [PubMed] [Google Scholar]
- 260.Zheng D., He D., Lu X., Sun C., Luo Q., Wu Z. The miR-148a alleviates hepatic ischemia/reperfusion injury in mice via targeting CaMKIIα. Xi bao yu fen zi mian yi xue za zhi = Chinese journal of cellular and molecular immunology . 2016;32(9):1202–1206. [PubMed] [Google Scholar]
- 261.Zheng D., Li Z., Wei X., et al. Role of miR-148a in mitigating hepatic ischemia-reperfusion injury by repressing the TLR4 signaling pathway via targeting CaMKIIα in vivo and in vitro. Cellular Physiology and Biochemistry . 2018;49(5):2060–2072. doi: 10.1159/000493716. [DOI] [PubMed] [Google Scholar]
- 262.Huang Z. Q., Xu W., Wu J. L., Lu X., Chen X. M. MicroRNA-374a protects against myocardial ischemia-reperfusion injury in mice by targeting the MAPK6 pathway. Life Sciences . 2019;232, article 116619 doi: 10.1016/j.lfs.2019.116619. [DOI] [PubMed] [Google Scholar]
- 263.Boon R. A., Jaé N., Holdt L., Dimmeler S. Long noncoding RNAs: from clinical genetics to therapeutic targets? Journal of the American College of Cardiology . 2016;67(10):1214–1226. doi: 10.1016/j.jacc.2015.12.051. [DOI] [PubMed] [Google Scholar]
- 264.Valadkhan S., Valencia-Hipólito A. lncRNAs in stress response. Current Topics in Microbiology and Immunology . 2016;394:203–236. doi: 10.1007/82_2015_489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Huang L., Guo B., Liu S., Miao C., Li Y. Inhibition of the LncRNA Gpr19 attenuates ischemia-reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR-324-5p/Mtfr1 axis. IUBMB Life . 2020;72(3):373–383. doi: 10.1002/iub.2187. [DOI] [PubMed] [Google Scholar]
- 266.Wang S. M., Liu G. Q., Xian H. B., Si J. L., Qi S. X., Yu Y. P. LncRNA NEAT1 alleviates sepsis-induced myocardial injury by regulating the TLR2/NF-κB signaling pathway. European Review for Medical and Pharmacological Sciences . 2019;23(11):4898–4907. doi: 10.26355/eurrev_201906_18078. [DOI] [PubMed] [Google Scholar]
- 267.Ming N., Na H. S. T., He J. L., Meng Q. T., Xia Z. Y. Propofol alleviates oxidative stress via upregulating lncRNA-TUG1/Brg1 pathway in hypoxia/reoxygenation hepatic cells. Journal of Biochemistry . 2019;166(5):415–421. doi: 10.1093/jb/mvz054. [DOI] [PubMed] [Google Scholar]
- 268.Zeng J., Zhu L., Liu J., et al. Metformin protects against oxidative stress injury induced by ischemia/reperfusion via regulation of the lncRNA-H19/miR-148a-3p/Rock2 axis. Oxidative medicine and cellular longevity. . 2019;2019, article 8768327:18. doi: 10.1155/2019/8768327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang Y., Zhang Y. lncRNA ZFAS1 improves neuronal injury and inhibits inflammation, oxidative stress, and apoptosis by sponging miR-582 and upregulating NOS3 expression in cerebral ischemia/reperfusion injury. Inflammation . 2020;43(4):1337–1350. doi: 10.1007/s10753-020-01212-1. [DOI] [PubMed] [Google Scholar]
- 270.Shaw L., Bowers L., Demers L., et al. Critical issues in cyclosporine monitoring: report of the Task Force on Cyclosporine Monitoring. Clinical Chemistry . 1987;33(7):1269–1288. doi: 10.1093/clinchem/33.7.1269. [DOI] [PubMed] [Google Scholar]
- 271.Ibanez B., Macaya C., Sánchez-Brunete V., et al. Effect of early metoprolol on infarct size in ST-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: the Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial. Circulation . 2013;128(14):1495–1503. doi: 10.1161/CIRCULATIONAHA.113.003653. [DOI] [PubMed] [Google Scholar]
- 272.Van de Werf F., Janssens L., Brzostek T., et al. Short-term effects of early intravenous treatment with a beta-adrenergic blocking agent or a specific bradycardiac agent in patients with acute myocardial infarction receiving thrombolytic therapy. Journal of the American College of Cardiology . 1993;22(2):407–416. doi: 10.1016/0735-1097(93)90044-2. [DOI] [PubMed] [Google Scholar]
- 273.Parang P., Singh B., Arora R. Metabolic modulators for chronic cardiac ischemia. Journal of Cardiovascular Pharmacology and Therapeutics . 2005;10(4):217–223. doi: 10.1177/107424840501000402. [DOI] [PubMed] [Google Scholar]
- 274.van der Horst I. C., Zijlstra F., van’t hof A. W., et al. Glucose-insulin-potassium infusion inpatients treated with primary angioplasty for acute myocardial infarction: the glucose-insulin-potassium study: a randomized trial. Journal of the American College of Cardiology . 2003;42(5):784–791. doi: 10.1016/S0735-1097(03)00830-1. [DOI] [PubMed] [Google Scholar]
- 275.Bedjaoui A., Allal K., Lounes M. S., et al. Intracoronary or intravenous abciximab after aspiration thrombectomy in patients with STEMI undergoing primary percutaneous coronary intervention. Cardiovascular Journal of Africa . 2019;30(1):45–51. doi: 10.5830/CVJA-2018-063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.O'Neill S., Gallagher K., Hughes J., Wigmore S. J., Ross J. A., Harrison E. M. Challenges in early clinical drug development for ischemia-reperfusion injury in kidney transplantation. Expert Opinion on Drug Discovery . 2015;10(7):753–762. doi: 10.1517/17460441.2015.1044967. [DOI] [PubMed] [Google Scholar]
- 277.Janssens S. P., Bogaert J., Zalewski J., et al. Nitric oxide for inhalation in ST-elevation myocardial infarction (NOMI): a multicentre, double-blind, randomized controlled trial. European Heart Journal . 2018;39(29):2717–2725. doi: 10.1093/eurheartj/ehy232. [DOI] [PubMed] [Google Scholar]
- 278.Berwanger O., Lopes R. D., Moia D. D. F., et al. Ticagrelor versus clopidogrel in patients with STEMI treated with fibrinolysis: TREAT trial. Journal of the American College of Cardiology . 2019;73(22):2819–2828. doi: 10.1016/j.jacc.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 279.Neimark M. I. Ischemia-reperfusion syndrome. Khirurgiya. Zhurnal im. N.I. Pirogova . 2021;9:71–76. doi: 10.17116/hirurgia202109171. [DOI] [PubMed] [Google Scholar]
- 280.Li S., Hafeez A., Noorulla F., et al. Preconditioning in neuroprotection: from hypoxia to ischemia. Progress in Neurobiology . 2017;157:79–91. doi: 10.1016/j.pneurobio.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bugger H., Pfeil K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease . 1866;2020(7, article 165768) doi: 10.1016/j.bbadis.2020.165768. [DOI] [PubMed] [Google Scholar]
- 282.Sharma A., Mukewar S., Chari S. T., Wong Kee Song L. M. Clinical features and outcomes of gastric ischemia. Digestive Diseases and Sciences . 2017;62(12):3550–3556. doi: 10.1007/s10620-017-4807-4. [DOI] [PubMed] [Google Scholar]
- 283.Oldenburg W. A., Lau L. L., Rodenberg T. J., Edmonds H. J., Burger C. D. Acute mesenteric ischemia: a clinical review. Archives of Internal Medicine . 2004;164(10):1054–1062. doi: 10.1001/archinte.164.10.1054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We provide editable versions of our figures and editing platform publishing license.