

Abstract
Hypoglycemia almost never develops in healthy individuals, because multiple hypoglycemia sensing systems, located in the periphery and in the central nervous system, trigger a coordinated counterregulatory hormonal response to restore normoglycemia. This involves not only the secretion of glucagon, but also of epinephrine, norepinephrine, cortisol and growth hormone. Increased hepatic glucose production is also stimulated by direct autonomous nervous connections to the liver that stimulate glycogenolysis and gluconeogenesis. This counterregulatory response, however, becomes deregulated in a significant fraction of diabetes patients that receive insulin therapy. This leads to the risk of developing hypoglycemic episodes, of increasing severity, which negatively impact the quality of life of the patients. How hypoglycemia is detected by the central nervous system is being actively investigated. Recent studies using novel molecular biological, optogenetic and chemogenetic techniques allow the characterization of glucose‐sensing neurons, the mechanisms of hypoglycemia detection, the neuronal circuits in which they are integrated and the physiological responses they control. This review discusses recent studies aimed at identifying central hypoglycemia sensing neuronal circuits, how neurons are activated by hypoglycemia and how they restore normoglycemia.
Keywords: Glucagon, Hypoglycemia, Hypothalamus
This article reviews recent data on the identification of hypoglycemia‐sensing neurons in the central nervous system. It discusses how these neurons are connected together to control autonomous nervous activity. It describes key hormonal and nervous responses that prevent hypoglycemia development.
INTRODUCTION
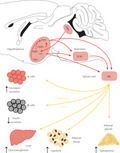
Diabetes mellitus is a hyperglycemic condition that leads to the development of micro‐ and macrovascular dysfunctions, which cause multiple secondary complications that negatively impact health and lifespan. Insulin treatment is the only option for type 1 diabetes patients and is required for an important fraction of type 2 diabetes patients. One major drawback of insulin therapy is the risk of hypoglycemia, with antecedent hypoglycemic episodes increasing the incidence and severity of subsequent episodes 1 . In healthy individuals, hypoglycemia rarely occurs, as a counterregulatory response is immediately activated to restore euglycemia when blood glucose levels fall below ~5 mmol/L. This response involves the secretion, under the control of the autonomic nervous system and the hypothalamic–pituitary–adrenal axis, of glucagon, epinephrine, norepinephrine, cortisol and growth hormone. These hormones stimulate hepatic glucose production through the induction of glycogenolysis and neoglucogenesis; epinephrine also activates glucagon secretion and suppresses insulin release, and cortisol and growth hormone induce insulin resistance in muscle and fat; these actions ensure that sufficient glucose is channeled to the brain to sustain its metabolic energy requirements. In insulin‐treated diabetes patients, this counterregulatory response progressively wanes because of the failure of as yet poorly characterized hypoglycemia sensing systems. Those are located not only in peripheral sites, such as the hepatoportal vein area or the carotid bodies, but also, in large part, in the central nervous system, where they control the activity of the autonomous nervous system and of the hypothalamic–pituitary–adrenal axis 1,2 . Another aspect of the response to hypoglycemia is to trigger a feeding response aimed at replenishing the body glucose stores 3 , 4 . Whereas the hormonal response is required for a fast prevention of hypoglycemia, the feeding response, which leads to glycogen deposition in the liver, is required for the long‐term availability of glucose.
Because of the very significant impact of hypoglycemia on the quality of life of patients, there is an important need to identify the sites of hypoglycemia sensing in the brain, the cells involved, how they control the glucose homeostatic and feeding responses, and how they become deregulated in diabetic patients.
MECHANISMS OF GLUCOSE SENSING BY THE BRAIN
All neurons can utilize glucose as a source of metabolic energy. However, a subgroup of neurons has the specific ability to regulate their firing activity in response to physiological changes in extracellular glucose concentrations. These glucose‐responsive neurons can be activated by a rise (glucose excited, GE neurons) or by fall (glucose inhibited, GI neurons) in extracellular glucose concentrations 3 , 4 . Such GE and GI neurons are classically identified in electrophysiological recording experiments as being responsive to variations in glucose concentrations between 0.1–0.5 mmol/L glucose and 2.5 mmol/L glucose ,5 . These conditions are considered to reflect the brain parenchymal glucose concentrations, which are ~30% of the blood glucose levels 6 . There is evidence that the glucose signaling pathways that lead to these neurons firing are diverse 7 . GE neurons have been suggested to respond to increase in glucose by a metabolism‐dependent signaling pathway that requires glucose uptake, glucose metabolism and a K+ ATP channel‐dependent depolarization of the plasma membrane. Although this pathway resembles that of the pancreatic β‐cells, the presence of glucose transporter 2 (Glut2) can be replaced by another glucose transporter isoform (Glut1 or Glut3), and glucokinase is not required for their glucose responsiveness 8 . Glucose sensing by GE neurons has also been reported to depend on the presence and activity of the Na+/glucose symporters sodium–glucose cotransporter 1 or sodium–glucose cotransporter 3 ,9 , or of the sweet taste receptor T1R2/T1R3 10 . GI neurons are activated by hypoglycemia by a mechanism that recruits adenosine monophosphate‐dependent protein kinase (AMPK). Subsequent depolarization of the plasma membrane and induction of firing activity has been found to rely on the activity of Na+/K+ATPase 11 , 12 , two pore‐domain K+ channels 13 , anoctamine 4 14 or the cystic fibrosis transmembrane regulator 15 . In addition, to this complexity in neuronal glucose sensing, there is evidence that astrocytes might participate in the normal response to hypoglycemia 16 . Tanycytes, ependymoglial cells lining the bottom of the third ventricle and that are in direct contact with hypothalamic neurons and blood vessels of the median eminence, also respond to variations in extracellular glucose concentrations, and participate in the control of feeding and glucose homeostasis 17 .
ANATOMICAL DISTRIBUTION OF GLUCOSE‐SENSING NEURONS
The hypothalamus
Glucose‐sensing neurons are present in several hypothalamic nuclei, including the arcuate (ARH), dorsomedial (DMH), paraventricular (PVN), ventromedial, lateral (LH) and supraoptic nuclei 2 , 18 . Connections between hypothalamic neurons and the parasympathetic nervous system are through the dorsal vagal complex (DVC), which consists in the nucleus of the tractus solitarius (NTS), the area postrema and the dorsal motor nucleus of the vagus (DMNX), which is formed by the cell bodies of the vagal nerve neurons. Neurons from the ARH, PVN, DMH and LH send direct projections to the DVC, thus forming an anatomical connection between nuclei containing glucose‐sensing neurons and the vagal nerve. In contrast, neurons from the PVN and LH can activate the sympathetic nerve by sending projections to the intermediolateral cell column (IML) of the spinal cord, either directly or through a relay in the ventrolateral medulla (VLM). Glucose‐sensing cells of the ARH, DMH and ventromedial are indirectly connected to the sympathetic nerve through their projections to the PVN and the LH (see Stanley et al.18 and Figures 1 and 2).
Figure 1.
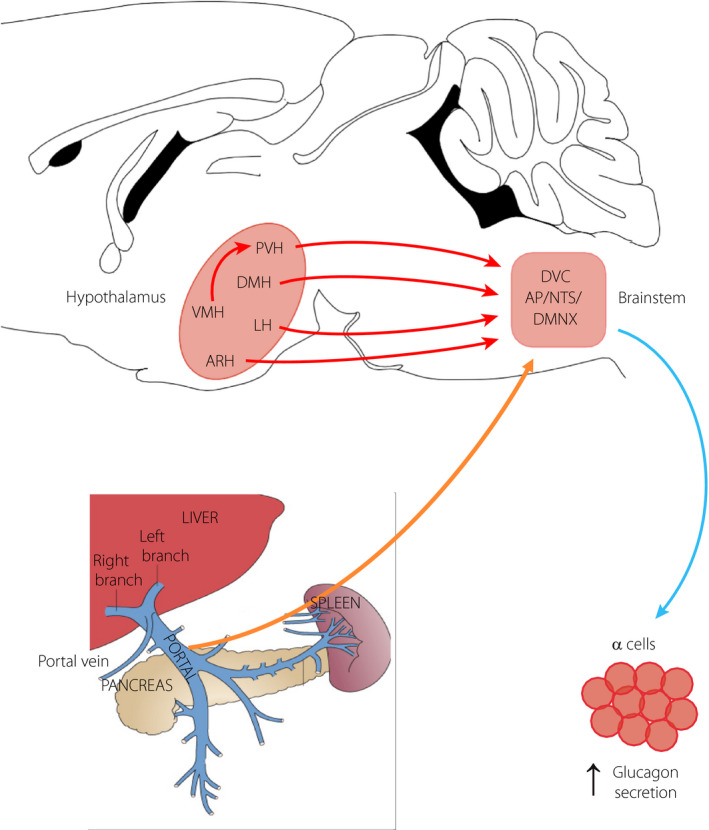
Pathways for hypoglycemia activation of the vagal nerve. Glucose‐sensing neurons that transmit a hypoglycemia signal are located in the portal vein area, the brainstem and the hypothalamus. In the portal vein area, hypoglycemia‐sensitive nerve terminals send projections to the DVC and this signal is further conveyed to the hypothalamus. At the level of the brainstem, hypoglycemia activates neurons in all three nuclei of the dorsal vagal complex (DVC), and these can directly or indirectly through hypothalamic nuclei control the activity of the dorsal motor nucleus of the vagus (DMNX) and, thus, the vagal nerve. Hypoglycemia can also activate neurons in the indicated nuclei of the hypothalamus, which send direct or indirect projections to the DVC for the control of vagal activity and glucagon secretion. AP, area postrema; ARH, arcuate nucleus; DMH, dorsomedial nucleus; LH, lateral nucleus; NTS, nucleus of the tractus solitarius; PVN, paraventricular nucleus; VMN, ventromedial nucleus.
Figure 2.
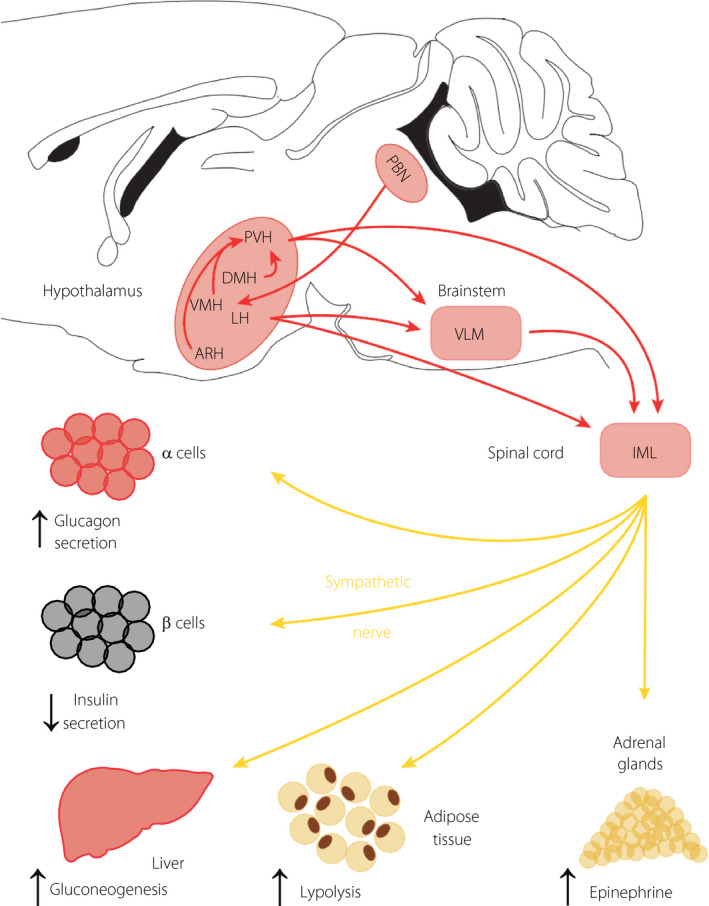
Pathways for hypoglycemia activation of the sympathetic nerve. Glucose‐sensing neurons that transmit hypoglycemia signals to the sympathetic nerve are located in the brainstem and the hypothalamus. In the brainstem, epinephrine and norepinephrine neurons of the ventrolateral medulla (VLM) can be activated by hypoglycemia or neuroglucopenia. They activate the sympathetic glucagon response indirectly through projections to the hypothalamus or directly through connections to the intermediolateral cell column (IML). Cholecystokinin neurons of the parabrachial nucleus (PBN) send projections to the ventromedial nucleus (VMN) to control glucagon secretion, as discussed in the text. The indicated hypothalamic nuclei also contain neurons that can detect hypoglycemia and activate the sympathetic nerve either through intrahypothalamic connections to the paraventricular nucleus (PVN), which send direct projections to the IML or indirect projections through the VLM. The lateral nucleus (LH) also sends projection to the VLM and IML to control sympathetic activity. The activated sympathetic nerve stimulates epinephrine secretion from the adrenals, which increases glucagon secretion and hepatic glucose production; it activates lipolysis in adipose tissue to provide free fatty acids required to fuel hepatic neoglucogenesis; it inhibits insulin secretion and directly stimulates glucagon secretion; it also directly stimulates hepatic glucose production.
The brainstem
In the brainstem, glucose‐sensing neurons are found in the DVC, the VLM and the parabrachial nucleus (PBN). In the NTS – one of the nuclei of the DVC – glucose‐responsive neurons are directly sensitive to variations in blood glucose concentrations 19 , as this structure is not protected by a blood–brain barrier. In the VLM, epinephrine and norepinephrine neurons are glucose‐sensitive and participate to the control of the counterregulatory response to hypoglycemia or 2‐deoxy‐D‐glucose (2DG)‐induced neuroglucopenia 20 . Their role depends on projections to the hypothalamus, in particular the PVN, and to the IML to control the secretion of epinephrine by adrenal glands. They also send projections to the supraoptic nuclei to control vasopressin neurons. The PBN has connections to both the DVC and hypothalamus, in particular the ventromedial nucleus (VMN), and contains neurons that are activated by hypoglycemia to control the glucagon secretion 21 (see Figures 1 and 2).
The thalamus
The paraventricular part of the thalamus (PVT) integrates interoceptive signals to the control of feeding behavior 22 . Anatomically, the PVT receives inputs from several hypothalamic nuclei involved in the regulation of glucose homeostasis and feeding, and sends dense projections to the nucleus accumbens 23 , which controls motivated feeding behavior. In humans, the PVT has been found to be activated by hypoglycemia 24 and, in a model of HAAF, synaptic activity in the PVT was found to be markedly increased, suggesting that this brain region contributes to the overall response to hypoglycemia 25 . In mice, hypoglycemia‐sensing neurons, characterized by the expression of Glut2, have been identified in the PVT and found to play a role in the motivated feeding response to hypoglycemia: when activated by hypoglycemia or by optogenetics, these neurons, through their projections to the nucleus accumbens, increase sucrose‐seeking behavior 26 . GE neurons have also been identified in the PVT that are characterized by the expression of glucokinase 27 . When activated by optogenetics, they suppress glucose‐seeking behavior; in contrast, when inhibited by chemogenetics, they increase feeding. Thus, two populations of glucose‐responsive neurons in the PVT show opposite responses to extracellular glucose levels with Glut2 neurons playing an important role to activate a feeding response to hypoglycemia.
GLUCOSE SENSING NEURONS IN GLUCAGON SECRETION
Glucose sensing cells of the DMNX/NTS
The parasympathetic or vagal nerve is activated by small decreases in glycemia and triggers a rapid secretion of glucagon 28 . The vagal nerve originates from the DMNX, a nucleus of the DVC. Insulin‐induced hypoglycemia or 2DG‐induced neuroglucopenia rapidly and strongly induce c‐fos expression in the three structures of the DVC, the DMNX, NTS and area postrema 16 , and electrophysiological measurements with extracellular electrodes show the acute glucose responsiveness of neurons of this region 29 . In search for markers identifying specific glucose responsive neurons in the DVC, Lamy et al., 13 carried out patch clamp experiments on Glut2‐expressing neurons of the NTS identified by the genetically‐driven expression of a green fluorescent protein. Previous studies had indeed shown that inactivation of Glut2 in the nervous system leads to a prediabetic phenotype characterized by lower β‐cell mass, suppressed first phase of insulin secretion, glucose intolerance associated with decreased insulin secretion and increased plasma glucagon levels 30 . Glut2 neurons of the NTS were found to be activated by hypoglycemia, showing progressively higher plasma membrane depolarization as extracellular glucose concentrations decreased from 5 to 0.5 mmol/L. This response was dependent on a decrease in glucose metabolism, leading to activation of AMPK and the control of a leak K+ channel. Glut2 neurons of the NTS are GABAergic and send projections to the DMNX. Their optogenetic activation in living mice that express channelrhodopsin‐2, a light‐activated cation channel ,31 , in Glut2 neurons of the NTS led to stimulation of vagal nerve activity and a strong induction of glucagon secretion 13 .
Glucose‐sensing cells of the VMN
The ventromedial nucleus of the hypothalamus was recognized early on as an important site for the regulation of feeding and glucose homeostasis (for review, see Shimazu et al.32). For instance, electrical stimulation of the VMN increases blood glucose levels and reduces hepatic glycogen content 33 . It is also involved in the control of glucagon secretion, as shown by the fact that injection of 2DG directly in the VMN induces glucagon secretion, and that glucagon secretion triggered by systemic insulin‐induced hypoglycemia can be prevented by VMN injections of glucose 34 , 35 . These early observations have been refined using current genetic technologies. Most VMN neurons express the transcription factor Sf1, and Sf1‐Cre mice have been widely used as a genetic tool to target VMN neurons for specific gene expression or inactivation. Glucokinase is also expressed by most VMN neurons, and these largely, but not completely, overlap with the Sf1‐expressing neuron population. Gck‐Cre mice have, thus, also been used for the Cre‐dependent controlled expression of various genes in the VMN. For instance, it is possible to express an activating or inhibiting channelrhodopsin in VMN neurons, and test the effect of light delivered through an optic fiber placed in the VMN of living mice. Using this approach with Sf1‐Cre mice, Meek et al. 36 showed that activation of Sf1 neurons of the VMN increases glucagon secretion and blood glucose levels, whereas inhibiting them decreases the glycemic recovery after insulin‐induced hypoglycemia. A similar observation was made using Gck‐Cre mice to activate Gck neurons by a magnetic field‐controlled system 37 .
VMN neurons are mostly glutamatergic. Thus, another way of inhibiting them is to genetically inactivate the expression of the vesicular glutamate transporter, vGlut2. Mice with genetic inactivation of vGlut2 in Sf1 neurons show lower glycemic levels in the fasted state than control mice and defective counterregulatory response to insulin‐induced hypoglycemia or 2DG‐induced neuroglucopenia, with lower glucagon secretion 38 . Thus, the VMN is an important nucleus involved in the counterregulatory response to hypoglycemia through the control of glucagon secretion and hepatic glucose production.
Interesting questions that are currently being investigated are to define the circuit in which VMN neurons are integrated to control glucagon secretion and endogenous glucose production, and to determine whether their intrinsic glucose sensing properties are required for the physiological response to hypoglycemia. VMN neurons send projections to numerous brain regions 39 , and those neurons involved in counterregulatory response are integrated in a circuit that includes afferent neurons able to sense glucose variations and efferent neurons that transmit the hypoglycemia signal to the endocrine pancreas and/or the liver. In one study, it has been shown that cholecystokinin neurons of the PBN are activated by hypoglycemia. These neurons send projections to the VMN, and this connection between the brainstem and the hypothalamus is required for the normal glucagon response to hypoglycemia 21 . It has been further shown that the VMN neurons receiving inputs from the cholecystokinin neurons control glucagon secretion through projections to the bed nucleus of the stria terminalis. Thus, a primary hypoglycemic signal generated in the PBN can induce glucagon secretion through a relay to the VMN. Hypoglycemia can, however, also be detected by peripheral sensors, such as those located in the hepatoportal vein region, and which send information about local glycemic levels to the brainstem and hypothalamus to control the glucagon response 40 , 41 . That VMN neurons are part of such a global neuronal circuit is attested by the experiments mentioned above, where the glucagon response to peripheral hypoglycemia can be suppressed by glucose infusion of the VMN or by the genetic inactivation of the vGlut2 gene in Sf‐1 neurons 35 , 38 .
To determine the physiological importance of hypoglycemia sensing by VMN neurons in counterregulation, Quenneville et al., 42 searched for a possibility to selectively inactivate GI neuron function. They reported that inactivation in Sf1 neurons of both the α1 and α2 subunits of AMPK, a metabolic sensor activated by hypoglycemia, suppressed GI neuron activity while leaving GE neuron activity intact. Mice with knockout of both AMPK α1 and α2 subunits were then used to address two questions. The first was to assess whether specific genes could be identified that are dysregulated by AMPK inactivation and that could account for the loss of hypoglycemia detection. The second was to determine whether the loss of GI neurons from the VMN would negatively impact the normal counterregulatory response to hypoglycemia. To address the first question, a transcribing ribosome affinity purification technique 43 was used, which allows to specifically isolate messenger ribonucleic acid (mRNA) from Sf1 neurons. This technique is based on the targeted expression in Sf1 neurons of a gene encoding the L10 ribosomal protein fused to a green fluorescent protein. This fusion protein assembles within functional ribosomes, allowing their immunopurification with anti‐GFP antibodies. Analysis of the ribosome‐associated mRNAs and their differential expression between Sf1 neurons expressing or not AMPK led to the identification of thioredoxin 2 as one of the most downregulated mRNAs in the neurons of knockout mice. Thioredoxin 2 is transcriptionally regulated by AMPK; it encodes a mitochondrial reactive oxygen species scavenger protein, and its overexpression in the VMN of AMPK knockout mice restores GI neuron activation by hypoglycemia. This, therefore, suggests that reactive oxygen species, which are induced by hypoglycemia, might prevent neuronal firing if a specific protection mechanism is not in place. In rats, overexpression in the VMN of thioredoxin 1, a cytoplasmic form of thioredoxin, has also been shown to protect against defects in hypoglycemia‐induced glucagon secretion in type 1 diabetes 44 .
To investigate the impact of suppressing GI neuron activity in the VMN, α1 and α2 AMPK knockout mice were exposed to insulin‐induced hypoglycemia or 2DG‐induced neuroglucopenia. No difference in the stimulation of glucagon secretion could be observed between control and knockout mice. Thus, the glucose‐sensing property of VMN GI neurons is not required for the normal counterregulatory response to hypoglycemia. This can be explained by the fact that hypoglycemia is detected at multiple sites in the multisynaptic neuronal circuit in which VMN neurons are integrated and that peripheral glucose sensing is sufficient to generate a counterregulatory response. VMN neurons are nevertheless required to transmit this signal, as suppressing glutamatergic synaptic transmission by vGlut2 gene inactivation prevents the glucagon response 38 . It can, thus, be hypothesized that hypoglycemia sensing by VMN GI neurons constitutes a fail‐safe system recruited in case of defective peripheral hypoglycemia sensing.
Although AMPK could be shown to be required for GI signaling, the mechanism of hyperglycemia detection by GE neurons is still not fully characterized. Genetic studies have shown that the adenosine triphosphate‐sensitive potassium channel is required 45 , suggesting that GE signaling shares similarities with glucose‐induced insulin secretion in pancreatic β‐cells. In these cells, glucokinase plays an essential role in controlling the glucose dose–response of insulin secretion. In neurons, glucokinase has been postulated to play a similar role, and to be required for the glucose responsiveness of both GE and GI neurons 46 . However, the formal testing of this hypothesis by genetic inactivation of glucokinase specifically in Sf1 neurons showed that this enzyme is fully dispensable for the GE or GI response to, respectively, high or low extracellular glucose levels 8 . Nevertheless, VMN glucokinase knockout mice still show defects in hypoglycemia‐induced glucagon secretion, increased fat mass and absence of glucose‐regulated autonomous nervous system activity. However, these phenotypes were observed only in female, and not in male mice. Thus, Gck is not required for glucose‐sensing by GE or GI neurons. Why inactivation of Gck in Sf1 neurons impacts, in a sex‐dependent manner, glucose homeostasis, fat mass and bodyweight still needs to be explained.
Fgf15 neurons of the DMH control glucagon secretion and gluconeogenesis
To identify novel hypothalamic regulators of glucagon secretion Picard et al., carried out a genetic screen in BXD recombinant inbred mice 47 (Figure 3). BXD mouse lines are derived from the cross between C57Bl/6 and DBA/2 mice 48 , and each line contains a specific and characterized combination of the parental strain genomes. The BXD lines are, thus, easily amenable to genetic screens for the identification of quantitative trait loci controlling any particular phenotype. A set of 35 mouse lines were assessed for glucagon quantitative trait loci in response to 2DG‐induced neuroglucopenia. This led to the identification of a quantitative trait loci on the distal part of chromosome 7. This region contains 128 genes, 65 of which are expressed in the hypothalamus, as determined by hypothalamus RNASeq analysis. Correlation analysis then led to the identification of the Fgf15 mRNA as the most anti‐correlated with the glucagon trait. In situ hybridization analysis showed that Fgf15 was expressed in a subset of neurons of the DMH and in the perifornical region. Intracerebroventricular injection of FGF19 (the stable human ortholog of Fgf15) reduced 2DG‐induced glucagon secretion. In contrast, silencing Fgf15 expression in the DMH by recombinant lentivirus delivery of a short hairpin RNA increased glucagon secretion. Furthermore, i.c.v. injection of FGF19 reduced 2DG‐induced c‐fos expression in the DVC and vagal nerve firing.
Figure 3.
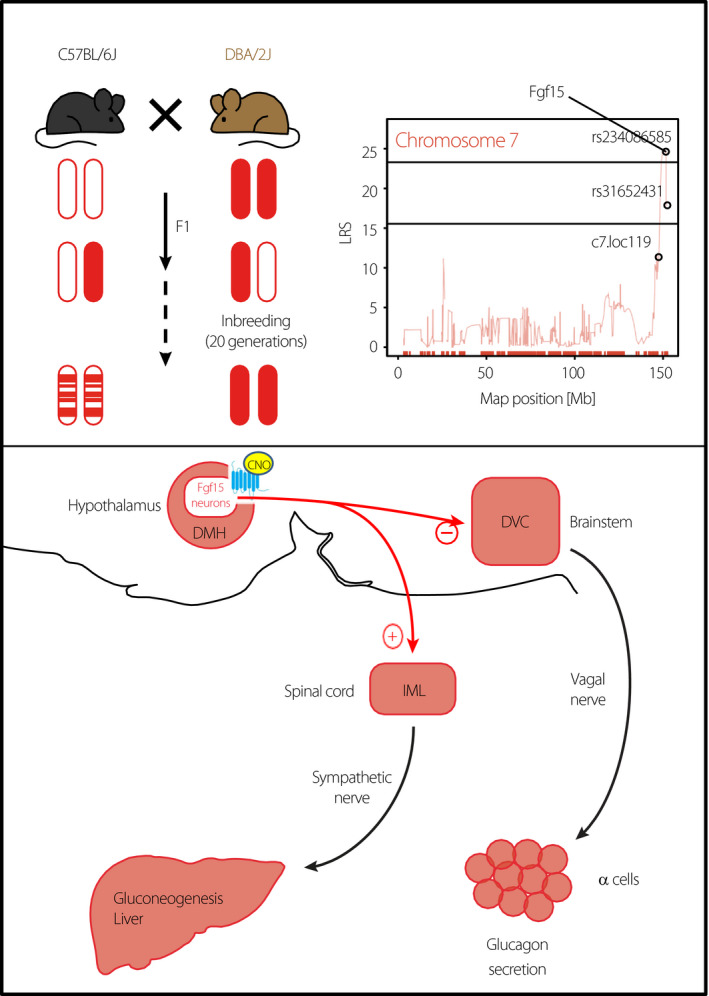
Identification of Fgf15 neurons that control glucagon secretion and hepatic glucose production. Recombinant inbred BXD mouse lines derived from the initial cross of C57Bl/6 and DBA/2J mice have been used to search for genomic intervals (QTL) that control neuroglucopenia‐induced glucagon secretion (top left). This led to the identification of Fgf15 on the distal part of chromosome 7 (top right). Fgf15 was found to be expressed in a subpopulation of dorsomedial (DMH) neurons (lower part). Expression of the hM3Dq receptor (blue) specifically in these neurons allowed their specific activation by i.p. injection of clozapine‐N‐oxide. This suppressed hypoglycemia‐induced glucagon secretion by blocking the activation of dorsal vagal complex (DVC) neurons. In contrast, activation of the Fgf15 neurons led to increased sympathetic nerve firing, leading to the induction of neoglucogenic genes in the liver and increased hepatic glucose production.
To better understand the role of the DMH Fgf15 neurons, mice were generated that express the Cre recombinase from the Fgf15 locus. These mice allowed for the use of a chemogenetic approach 31 to specifically stimulate Fgf15 neurons of the DMH. This is based on the stereotactic injections of a recombinant adeno‐associated virus driving the Cre‐dependent expression of hM3Dq 49 , a modified muscarinic receptor that can be activated by clozapine‐N‐oxide injected intraperitoneally. Chemogenetic activation of the Fgf15 neurons suppressed hypoglycemia‐induced vagal activity and glucagon secretion, in line with the results of the genetic screen. However, surprisingly, their activation also led to a strong activation of the sympathetic nerve, leading to stimulation of the cyclic AMP pathway in the liver, with increased phosphorylation of the transcription factor Crebp, induction of Pepck, the rate‐limiting enzyme in neoglucogenesis, and increased hepatic glucose production (Figure 3); this raised glycemia by ~1 mmol/l.
Patch clamp analysis of the Fgf15 neurons showed that most are glucose non‐responders, approximately 25% are GE and <10% are GI 50 . It is, thus, unlikely that activation of the Fgf15 neurons to generate the responses described above is through their glucose‐sensing properties. These neurons are also part of a neuronal network and receive inputs from the PVN, the ARH, the VMN and the LH, which all have glucose‐responsive neurons that might control the activity of the Fgf15 neurons. These neurons send projections to the medial preoptic area, the locus coeruleus and the ARH, and might therefore reach the DVC and IML indirectly to control vagal and sympathetic activity, respectively.
Therefore, the Fgf15 neurons of the DMH increase hepatic glucose production while suppressing glucagon secretion. This direct effect on the liver might be important to trigger a fast response that avoids the effects of glucagon on other physiological functions, such as suppression of feeding.
CONCLUSION
Present evidence describes the existence of a highly distributed hypoglycemia‐monitoring system, with glucose‐sensing cells located in peripheral and central locations, mainly in the hepatoportal vein area, the brainstem and hypothalamus, with others present in the ventral thalamic regions. Importantly, these cells are connected together and whereas the PVT cells appear to mainly control motivated feeding behavior, the other sensing cells are all contributing to the counterregulatory response to hypoglycemia. The mechanisms recruited to normalize glycemia include activation of the vagal nerve to stimulate glucagon secretion, of sympathetic nerves that can also stimulate glucagon secretion, but also the hypothalamic–pituitary–adrenal axis that triggers the release of epinephrine by adrenal glands; or directly stimulate hepatic glucose production by activating the cyclic AMP‐dependent expression of neoglucogenic genes. Genetic screens using recombinant inbred mice have proven to be a powerful approach to identify novel regulatory mechanisms. In addition to the mentioned 2DG‐induced neuroglucopenia screen described above, Picard et al. also carried out a screen in BXD mice for insulin‐induced hypoglycemia‐stimulated glucagon secretion. This led to the identification of several additional QTL and genes acting as regulators of glucagon secretion. Thus, much more work is required to fully characterize the mechanisms of central hypoglycemia detection and their deregulation in insulin‐treated diabetes patients to be in a position to prevent hypoglycemia‐associated autonomic failure.
DISCLOSURE
The author declares no conflict of interest.
Approval of the research protocol: N/A.
Informed consent: N/A.
Registry and the registration no. of the study/trial: N/A.
Animal studies: All animal experiments in the author's laboratory have received authorization from the Service Vétérinaire du Canton de Vaud, Lausanne.
ACKNOWLEDGMENTS
The work in the author laboratory was supported by grants from a European Research Council Advanced Grant (INTEGRATE, No. 694798), a Swiss National Science Foundation grant (310030‐182496) and has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 777460 (HypoRESOLVE). The JU receives support from the European Union’s Horizon 2020 research and innovation program, and EFPIA and T1D Exchange, JDRF, International Diabetes Federation (IDF), The Leona M. and Harry B. Helmsley Charitable Trust. The author is grateful to Dr G Labouèbe for the preparation of the figures.
J Diabetes Investig. 2022; 13: 599–607
REFERENCES
- 1. Cryer PE. Mechanisms of hypoglycemia‐associated autonomic failure in diabetes. N Engl J Med 2013; 369: 362–372. [DOI] [PubMed] [Google Scholar]
- 2. Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology 2007; 22: 241–251. [DOI] [PubMed] [Google Scholar]
- 3. Anand BK, Chhina GS, Sharma KN, et al. Activity of single neurons in the hypothalamic feeding centers: effect of glucose. Am J Physiol 1964; 207: 1146–1154. [DOI] [PubMed] [Google Scholar]
- 4. Oomura Y, Yoshimatsu H. Neural network of glucose monitoring system. J Auton Nerv Syst 1984; 10: 359–372. [DOI] [PubMed] [Google Scholar]
- 5. Routh VH. Glucose sensing neurons in the ventromedial hypothalamus. Sensors 2010; 10: 9002–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silver IA, Erecinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo‐, hypo‐, and hyperglycemic animals. J Neurosci 1994; 14: 5068–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thorens B. Sensing of glucose in the brain. Handb Exp Pharmacol 2012; 209: 277–294. [DOI] [PubMed] [Google Scholar]
- 8. Steinbusch LKM, Picard A, Bonnet MS, et al. Sex‐specific control of fat mass and counterregulation by hypothalamic glucokinase. Diabetes 2016; 65: 2920–2931. [DOI] [PubMed] [Google Scholar]
- 9. O’Malley D, Reimann F, Simpson AK, et al. Sodium‐coupled glucose cotransporters contribute to hypothalamic glucose sensing. Diabetes 2006; 55: 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lemon CH, Margolskee RF. Contribution of the T1r3 taste receptor to the response properties of central gustatory neurons. J Neurophysiol 2009; 101: 2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurita H, Xu KY, Maejima Y, et al. Arcuate Na+, K+‐ATPase senses systemic energy states and regulates feeding behavior through glucose‐inhibited neurons. Am J Physiol Endocrinol Metab 2015; 309: E320–E333. [DOI] [PubMed] [Google Scholar]
- 12. Silver IA, Erecinska M. Glucose‐induced intracellular ion changes in sugar‐sensitive hypothalamic neurons. J Neurophysiol 1998; 79: 1733–1745. [DOI] [PubMed] [Google Scholar]
- 13. Lamy C, Sanno H, Labouèbe G, et al. Hypoglycemia‐activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 2014; 19: 527–538. [DOI] [PubMed] [Google Scholar]
- 14. He Y, Xu P, Wang C, et al. Estrogen receptor‐α expressing neurons in the ventrolateral VMH regulate glucose balance. Nat Commun 2020; 11: 2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirschberg PR, Sarkar P, Teegala SB, et al. Ventromedial hypothalamus glucose‐inhibited neurones: a role in glucose and energy homeostasis? J Neuroendocrinol 2020; 32: e12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marty N, Dallaporta M, Foretz M, et al. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte‐dependent glucose sensors. J Clin Investig 2005; 115: 3545–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolborea M, Langlet F. What is the physiological role of hypothalamic tanycytes in metabolism? Am J Physiol Regul Integr Comp Physiol 2021; 320: R994–R1003. [DOI] [PubMed] [Google Scholar]
- 18. Stanley S, Moheet A, Seaquist ER. Central mechanisms of glucose sensing and counterregulation in defense of hypoglycemia. Endocr Rev 2019; 40: 768–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dallaporta M, Himmi T, Perrin J, et al. A solitary tract nucleus sensitivity to moderate changes in glucose level. NeuroReport 1999; 10: 1–4. [DOI] [PubMed] [Google Scholar]
- 20. Ritter S, Li A‐J, Wang Q, et al. Minireview: the value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology 2011; 152: 4019–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garfield A, Shah B, Madara J, et al. A parabrachial‐hypothalamic cholecystokinin neurocircuit controls counterregulatory responses to hypoglycemia. Cell Metab 2014; 20: 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Millan EZ, Ong Z, McNally GP. Paraventricular thalamus: gateway to feeding, appetitive motivation, and drug addiction. Prog Brain Res 2017; 235: 113–137. [DOI] [PubMed] [Google Scholar]
- 23. Kirouac GJ. Placing the paraventricular nucleus of the thalamus within the brain circuits that control behavior. Neurosci Biobehav Rev 2015; 56: 315–329. [DOI] [PubMed] [Google Scholar]
- 24. Arbeláez AM, Rutlin JR, Hershey T, et al. Thalamic activation during slightly subphysiological glycemia in humans. Diabetes Care 2012; 35: 2570–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arbelaez AM, Powers WJ, Videen TO, et al. Attenuation of counterregulatory responses to recurrent hypoglycemia by active thalamic inhibition: a mechanism for hypoglycemia‐associated autonomic failure. Diabetes 2008; 57: 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Labouèbe G, Boutrel B, Tarussio D, et al. Glucose‐responsive neurons of the paraventricular thalamus control sucrose‐seeking behavior. Nat Neurosci 2016; 19: 999–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kessler S, Labouèbe G, Croizier S, et al. Glucokinase neurons of the paraventricular nucleus of the thalamus sense glucose and decrease food consumption. iScience 2021; 24: 103122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taborsky GJ Jr, Mundinger TO. Minireview: the role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 2012; 153: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 2006; 570: 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tarussio D, Metref S, Seyer P, et al. Nervous glucose sensing regulates postnatal beta cell proliferation and glucose homeostasis. J Clin Investig 2014; 124: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinbusch L, Labouebe G, Thorens B. Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol Metab 2015; 26: 455–466. [DOI] [PubMed] [Google Scholar]
- 32. Shimazu T, Minokoshi Y. Systemic glucoregulation by glucose‐sensing neurons in the ventromedial hypothalamic nucleus (VMH). J Endocr Soc 2017; 1: 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimazu T, Fukuda A, Ban T. Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature 1966; 210: 1178–1179. [DOI] [PubMed] [Google Scholar]
- 34. Borg WP, Sherwin RS, During MJ, et al. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 1995; 44: 180–184. [DOI] [PubMed] [Google Scholar]
- 35. Borg MA, Sherwin RS, Borg WP, et al. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Investig 1997; 99: 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meek TH, Nelson JT, Matsen ME, et al. Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci USA 2016; 113: E2073–E2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stanley SA, Kelly L, Latcha KN, et al. Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 2016; 531: 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tong Q, Ye ChianPing, McCrimmon RJ, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 2007; 5: 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris‐leucoagglutinin study in the rat. J Comp Neurol 1994; 348: 41–79. [DOI] [PubMed] [Google Scholar]
- 40. Bohland MaryAnn, Matveyenko AV, Saberi M, et al. Activation of hindbrain neurons is mediated by portal‐mesenteric vein glucosensors during slow‐onset hypoglycemia. Diabetes 2014; 63: 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ionut V, Castro AVB, Woolcott OO, et al. Essentiality of portal vein receptors in hypoglycemic counterregulation: direct proof via denervation in male canines. Endocrinology 2014; 155: 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quenneville S, Labouèbe G, Basco D, et al. Hypoglycemia‐sensing neurons of the ventromedial hypothalamus require AMPK‐induced Txn2 expression but are dispensable for physiological counterregulation. Diabetes 2020; 69: 2253–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heiman M, Schaefer A, Gong S, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 2008; 135: 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou C, Routh VH. Thioredoxin‐1 overexpression in the ventromedial nucleus of the hypothalamus preserves the counterregulatory response to hypoglycemia during type 1 diabetes in male rats. Diabetes 2018; 67: 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miki T, Liss B, Minami K, et al. ATP‐sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci 2001; 4: 507–512. [DOI] [PubMed] [Google Scholar]
- 46. Dunn‐Meynell AA, Routh VH, Kang L, et al. Glucokinase is the likely mediator of glucosensing in both glucose‐excited and glucose‐inhibited central neurons. Diabetes 2002; 51: 2056–2065. [DOI] [PubMed] [Google Scholar]
- 47. Picard A, Soyer J, Berney X, et al. A Genetic screen identifies hypothalamic Fgf15 as a regulator of glucagon secretion. Cell Rep 2016; 17: 1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peirce JL, Lu L, Gu J, et al. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 2004; 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 2015; 55: 399–417. [DOI] [PubMed] [Google Scholar]
- 50. Picard A, Metref S, Tarussio D, et al. Fgf15 neurons of the dorsomedial hypothalamus control glucagon secretion and hepatic gluconeogenesis. Diabetes 2021; 70: 1443–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]