

Abstract
Introgression is an important biological process affecting at least 10% of the extant species in the animal kingdom. Introgression significantly impacts inference of phylogenetic species relationships where a strictly binary tree model cannot adequately explain reticulate net-like species relationships. Here, we use phylogenomic approaches to understand patterns of introgression along the evolutionary history of a unique, nonmodel insect system: dragonflies and damselflies (Odonata). We demonstrate that introgression is a pervasive evolutionary force across various taxonomic levels within Odonata. In particular, we show that the morphologically “intermediate” species of Anisozygoptera (one of the three primary suborders within Odonata besides Zygoptera and Anisoptera), which retain phenotypic characteristics of the other two suborders, experienced high levels of introgression likely coming from zygopteran genomes. Additionally, we find evidence for multiple cases of deep inter-superfamilial ancestral introgression. [Gene flow; Odonata; phylogenomics; reticulate evolution.]
In recent years, numerous studies have showed that multiple parts of the Tree of Life did not evolve according to a strictly bifurcating phylogeny (Hallstrom and Janke 2010; Mallet et al. 2016). Instead, many organisms experience reticulate network-like evolution that is caused by an exchange of interspecific genetic information via various biological processes. In particular, lateral gene transfer, incomplete lineage sorting (ILS), and introgression can result in gene trees that are discordant with the species tree (Maddison 1997; Posada and Crandall 2001; Degnan and Rosenberg 2009). Lateral transfer and introgression both involve gene flow following speciation, thereby producing “reticulate” phylogenies. ILS, on the other hand, occurs when lineages fail to coalesce within their ancestral population. Since this process does not involve any postspeciation gene flow, it does not contribute to reticulate evolution, even though it often results in discordant gene trees. Phylogenetic species-gene tree incongruence observed in empirical data can provide insight into underlying biological factors that shape the evolutionary trajectories of a set of taxa. The major source of reticulate evolution for eukaryotes is introgression where it affects approximately 25% of flowering plant and 10% of animal species (Mallet 2005; Mallet et al. 2016). Introgressed alleles can be fitness-neutral, deleterious (Petr et al. 2019), or adaptive (Norris et al. 2015; Oziolor et al. 2019). For example, adaptive introgression has been shown to provide an evolutionary rescue from polluted habitats in gulf killifish (Fundulus grandis; Oziolor et al. 2019), yielded mimicry adaptations among Heliconius butterflies (Heliconius Genome Consortium 2012) and archaic introgression has facilitated adaptive evolution of altitude tolerance (Huerta-Sanchez et al. 2014), immunity and metabolism in modern humans (Gouy and Excoffier 2020). Additionally, hybridization and introgression are important and often overlooked mechanisms of invasive species establishment and spread (Perry et al. 2002).
Odonata, the insect order that contains dragonflies and damselflies, lacks a strongly supported backbone tree to clearly resolve higher-level phylogenetic relationships (Dijkstra, Kalkman et al. 2014; Carle et al. 2015). Current evidence places odonates together with Ephemeroptera (mayflies) as the living representatives of the most ancient insect lineages to have evolved wings and active flight (Thomas et al. 2013). Odonates possess unique anatomical and morphological features such as a specialized body form, specialized wing venation, a distinctive form of muscle attachment to the wing base (Busse et al. 2013) allowing for direct flight and accessory (secondary) male genitalia that support certain unique behaviors (e.g., sperm competition). They are among the most adept flyers of all animals and are exclusively carnivorous insects relying primarily on vision to capture prey (Chauhan et al. 2014; Suvorov et al. 2017). During their immature stage, they are fully aquatic and spend much of their adult life in flight. Biogeographically, odonates exhibit species ranges varying from worldwide dispersal (Troast et al. 2016) to island-endemic. Odonates also play crucial ecological roles in local freshwater communities, being a top invertebrate predator as both adults and immatures (Dijkstra, Monaghan et al. 2014). Due to this combination of characteristics, odonates are quickly becoming model organisms to study specific questions in ecology, physiology, and evolution (Cordoba-Aguilar 2008; Bybee et al. 2016). However, the extent of introgression at the genomic scale within Odonata remains largely unknown.
In various biological systems, the empirical evidence shows that hybridization can potentially lead to intermediate phenotypes (Runemark et al. 2019) observed at molecular level (e.g. semidominant expression in interspecific hybrids; Landry et al. 2005) as well as organismal morphology (e.g. Lemmon and Lemmon 2010; Rothfels et al. 2015; Káldy et al. 2020). The Anisozygoptera suborder, which contains only three extant species, retains traits shared with both dragonflies and damselflies (hence its taxonomic name), ranging from morphology and anatomical structures (Busse et al. 2015) to behavior and flight biomechanics (Ruppell and Hilfert 1993). These characteristics could suggest either a hybrid origin of this suborder or substantial introgression at loci governing key morphological and behavioral traits shortly after the suborder’s formation. The potential introgression scenario for Anisozygoptera is yet to be formally tested using genome-wide data. Two early attempts to tackle introgression/hybridization patterns within Odonata were undertaken in (Monetti et al. 2002; Sánchez-Guillén et al. 2005). The studies showed that two closely related species of damselflies, Ischnura graellsii and Ischnura elegans, can hybridize under laboratory conditions and that genital morphology of male hybrids shares features with putative hybrids from I. graellsii to I. elegans natural allopatric populations (Monetti et al. 2002). The existence of abundant hybridization and introgression in natural populations of I. graellsii and I. elegans has received further support from an analysis of microsatellite data (Sanchez-Guillen et al. 2011). Putative hybridization events have also been identified in a pair of calopterygoid damselfly species, Mnais costalis and Mnais pruinosa based on the analyses of two molecular loci (mtDNA and nucDNA; Hayashi et al. 2005), and between Calopteryx virgo and Calopteryx splendens using 16S ribosomal DNA and 40 random amplified polymorphic DNA markers (Tynkkynen et al. 2008). A more recent study identified an interspecific hybridization between two cordulegasterid dragonfly species, Cordulegaster boltonii and Cordulegaster trinacriae using two molecular markers (mtDNA and nucDNA) and geometric morphometrics (Solano et al. 2018).
Here, we present a comprehensive analysis of transcriptomic data from 83 odonate species. First, we reconstruct a robust phylogenetic backbone using up to 4341 genetic loci for the order and discuss its evolutionary history spanning from the Carboniferous period (~360 Ma) to present day. Furthermore, in light of the “intermediate” phenotypic nature of Anisozygoptera, we investigate phylogenetic signatures of introgression within Odonata. Most notably, we identify a strong signal of deep introgression in the Anisozygoptera suborder, species of which possess traits of both main suborders, Anisoptera and Zygoptera. Although the strongest signatures of introgression are found in Anisozygoptera, we find evidence that introgression was pervasive in Odonata throughout its entire evolutionary history.
Materials and Methods
Taxon Sampling and RNA-seq
In this study, we used 85 distinct species (83 ingroup and 2 outgroup taxa). Thirty-five RNA-seq libraries were obtained from NCBI (Supplementary Table S1 available on Dryad at https://doi.org/10.5061/dryad.j3tx95xdp). The remaining 58 libraries were sequenced in the Bybee Lab (some species have several RNA-seq libraries; Supplementary Table S1 available on Dryad). Total RNA was extracted for each taxon from eye tissue using NucleoSpin columns (Clontech) and reverse-transcribed into cDNA libraries using the Illumina TruSeq RNA v2 sample preparation kit that both generates and amplifies full-length cDNAs. Prepped mRNA libraries with insert size of ~200 bp were multiplexed and sequenced on an Illumina HiSeq 2000 producing paired-end reads with average length of 275 bp by the Microarray and Genomic Analysis Core Facility at the Huntsman Cancer Institute at the University of Utah, Salt Lake City, UT, USA. Quality scores, tissue type, and other information about RNA-seq libraries are summarized in Supplementary Table S1 available on Dryad and NCBI BioProject PRJNA641626.
Transcriptome Assembly and CDS Prediction
RNA-seq libraries were trimmed and de novo assembled using Trinity (Grabherr et al. 2011; Haas et al.
2013) with default parameters. Then only the longest isoform was selected from
each gene for downstream analyses using the Trinity utility script. In order to identify
potentially coding regions within the transcriptomes, we used TransDecoder with default
parameters specifying to predict only the single best ORF. Each predicted proteome was
screened for contamination using DIMOND BLASTP (Buchfink
et al. 2015) with an 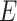 -value cutoff of
10
-value cutoff of
10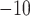 against custom protein database.
Nonarthropod hits were discarded from proteomes (amino acid, AA sequences) and
corresponding CDSs. To mitigate redundancy in proteomes and CDSs, we used CD-HIT (Fu et al. 2012) with the identity threshold of 0.99.
Such a conservative threshold was used to prevent exclusion of true paralogous sequences;
thus, reducing possible false-positive detection of 1:1 orthologs during homology
searches.
against custom protein database.
Nonarthropod hits were discarded from proteomes (amino acid, AA sequences) and
corresponding CDSs. To mitigate redundancy in proteomes and CDSs, we used CD-HIT (Fu et al. 2012) with the identity threshold of 0.99.
Such a conservative threshold was used to prevent exclusion of true paralogous sequences;
thus, reducing possible false-positive detection of 1:1 orthologs during homology
searches.
Homology Assessment
In the present study, three types of homologous loci (gene clusters), namely conserved single-copy orthologs (CO), all single-copy orthologs (AO), and paralogy-parsed orthologs (PO) identified by BUSCO v1.22 (Simao et al. 2015), OrthoMCL (Li et al. 2003), and (Yang and Smith, 2014) pipelines, respectively, were used in phylogenetic inference.
BUSCO arthropod Hidden Markov Model Profiles of 2675 single-copy orthologs were used to find significant COs matches within CDS data sets by HMMER’s hmmersearch v3 (Eddy 2011) with group-specific expected bit-score cutoffs. BUSCO classifies loci into complete [duplicated] and fragmented. Thus, only complete single-copy loci were extracted from CDS data sets and corresponding AA sequences for further phylogenetic analyses. Since loci were identified as true orthologs if they score above expected bit-score, and complete if their lengths lie within ~95% of BUSCO group mean length, many partial erroneously assembled sequences were filtered out.
OrthoMCL v2.0.9 (Li et al. 2003) was used to
compute AOs in all species using predicted AA sequences by TransDecoder. AA sequences were
used in an all-vs-all BLASTP with an 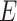 -value cutoff of
10
-value cutoff of
10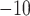 to find putative orthologs and
paralogs. The Markov Cluster algorithm (MCL) inflation point parameter was set to 2. Only
1:1 orthologs were used in further analyses. In order to exclude false-positive homology
clusters identified by OrthoMCL, we applied machine learning filtering procedure (Fujimoto et al. 2016) implemented in OGCleaner software
v1.0 (Fujimoto et al. 2017) using a metaclassifier
with logistic regression.
to find putative orthologs and
paralogs. The Markov Cluster algorithm (MCL) inflation point parameter was set to 2. Only
1:1 orthologs were used in further analyses. In order to exclude false-positive homology
clusters identified by OrthoMCL, we applied machine learning filtering procedure (Fujimoto et al. 2016) implemented in OGCleaner software
v1.0 (Fujimoto et al. 2017) using a metaclassifier
with logistic regression.
Finally, to identify additional clusters, we used Yang and Smith’s tree-based orthology
inference pipeline (Yang and Smith 2014) that was
specifically designed for nonmodel organisms using transcriptomic data. Yang and Smith’s
algorithm is capable of parsing paralogous gene families into “orthology” clusters that
can be used in phylogenetic analyses. It has been shown that paralogous sequences
encompass useful phylogenetic information (Hellmuth et al.
2015). First, the Transdecoder-predicted AA sequences were trimmed using CD-HIT
with the identity threshold of 0.995. Then, all-vs-all BLASTP with an
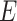 -value cutoff of 10
-value cutoff of 10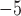 search was implemented. The raw BLASTP
output was filtered by a hit fraction of 0.4. Then, MCL clustering was performed with an
inflation point parameter of 2. Each cluster was aligned using iterative algorithm of
PASTA (Mirarab et al. 2015) and then was used to
infer a maximum-likelihood (ML) gene tree using IQ TREE v1.5.2 (Nguyen et al. 2015) with an automatic model selection. Tree tips that
were longer than relative and absolute cutoffs of 0.4 and 1, respectively, were removed.
Mono- and paraphyletic tips that belonged to the same species were masked as well. To
increase quality of homology clusters realignment, tree inference, and tip masking steps
were iterated with more stringent relative and absolute masking cutoffs of 0.2 and 0.5,
respectively. Finally, POs (AA sequences and corresponding CDSs) were extracted by rooted
ingroups (RI) procedure using Ephemera danica as an outgroup (for details
see Yang and Smith 2014).
search was implemented. The raw BLASTP
output was filtered by a hit fraction of 0.4. Then, MCL clustering was performed with an
inflation point parameter of 2. Each cluster was aligned using iterative algorithm of
PASTA (Mirarab et al. 2015) and then was used to
infer a maximum-likelihood (ML) gene tree using IQ TREE v1.5.2 (Nguyen et al. 2015) with an automatic model selection. Tree tips that
were longer than relative and absolute cutoffs of 0.4 and 1, respectively, were removed.
Mono- and paraphyletic tips that belonged to the same species were masked as well. To
increase quality of homology clusters realignment, tree inference, and tip masking steps
were iterated with more stringent relative and absolute masking cutoffs of 0.2 and 0.5,
respectively. Finally, POs (AA sequences and corresponding CDSs) were extracted by rooted
ingroups (RI) procedure using Ephemera danica as an outgroup (for details
see Yang and Smith 2014).
Cluster Alignment, Trimming, and Supermatrix Assembly
For most of the analyses, only clusters with 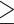 42 (~50%) species
present were retained. In total, we obtained five cluster types, namely DNA (CDS) and AA
COs, AA AOs and DNA and AA POs. Each cluster was aligned using PASTA (Mirarab et al. 2015) for the DNA and AA alignments and
PRANK v150803 (Loytynoja and Goldman 2008; Loytynoja 2014) for the codon alignments and alignments
where either 1st and 2nd or 3rd codon positions were removed. In order to reduce the
amount of randomly aligned regions, we implemented ALISCORE v2.0 (Misof and Misof 2009) trimming procedure (for PASTA alignments)
followed by masking any site with
42 (~50%) species
present were retained. In total, we obtained five cluster types, namely DNA (CDS) and AA
COs, AA AOs and DNA and AA POs. Each cluster was aligned using PASTA (Mirarab et al. 2015) for the DNA and AA alignments and
PRANK v150803 (Loytynoja and Goldman 2008; Loytynoja 2014) for the codon alignments and alignments
where either 1st and 2nd or 3rd codon positions were removed. In order to reduce the
amount of randomly aligned regions, we implemented ALISCORE v2.0 (Misof and Misof 2009) trimming procedure (for PASTA alignments)
followed by masking any site with 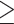 42 gap characters (for
both PASTA and PRANK alignments). Also, since fragmentary data may have a negative effect
on accuracy of gene and hence species tree inference (Wickett et al. 2014), sequence fragments with >50% gap characters were
removed from clusters that were used to estimate trees with ASTRAL v4.10.12 (Mirarab et al. 2014). For each of the cluster type, we
assembled supermatrices from trimmed gene alignments. Additionally, completely untrimmed
supermatrices were generated from DNA and AA COs with
42 gap characters (for
both PASTA and PRANK alignments). Also, since fragmentary data may have a negative effect
on accuracy of gene and hence species tree inference (Wickett et al. 2014), sequence fragments with >50% gap characters were
removed from clusters that were used to estimate trees with ASTRAL v4.10.12 (Mirarab et al. 2014). For each of the cluster type, we
assembled supermatrices from trimmed gene alignments. Additionally, completely untrimmed
supermatrices were generated from DNA and AA COs with 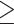 5
species present.
5
species present.
Phylogenetic Tree Reconstruction
Four different in spirit tree-building methods (ML:IQTREE, Bayesian:ExaBayes, Supertree:ASTRAL, Alignment-Free [AF]: Co-phylog) were used to infer odonate phylogenetic relationships using different input data types (untrimmed and trimmed supermatrices, codon supermatrices, codon supermatrices with 1st and 2nd or 3rd positions removed, gene trees, and assembled transcriptomes). In total, we performed 48 phylogenetic analyses and compared topologies to identify stable and conflicting relationships (Supplementary Table S2 available on Dryad).
We inferred phylogenetic ML trees from each supermatrix using IQTREE implementing two partitioning schemes: single partition and those identified by PartitionFinder v2.0 (three GTR models for DNA and a large array of protein models for AA; Lanfear et al. 2017) with relaxed hierarchical clustering option (Lanfear et al. 2014). In the first case, IQTREE was run allowing model selection and assessing nodal support with 1000 ultrafast bootstrap (UFBoot; (Minh et al. 2013)) replicates. In the second case, IQTREE was run with a given PartitionFinder partition model applying gene and site resampling to minimize false positives (Gadagkar et al. 2005) for 1000 UFBoot replicates.
For Bayesian analyses implemented in ExaBayes (Aberer et
al. 2014), we used highly trimmed (retaining sites only with occupancy of
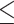 5 gap characters) and original DNA and
AA CO supermatrices assuming a single partition. We initiated four independent runs with
four Markov Chain Monte Carlo (MCMC) coupled chains sampling every 500th iteration. Due to
high computational demands of the procedure, only the GTR and JTT substitution model
priors were applied to DNA and AA CO supermatrices, respectively, with the default
topology, rate heterogeneity, and branch lengths priors. However, all supported protein
substitution models as a prior were specified for the trimmed AA CO supermatrix. For
convergence criteria, an average standard deviation of split frequencies (ASDSF; Lakner et al. 2008), a potential scale reduction factor
(PSRF; Brooks and Gelman 1998), and an effective
sample size (ESS; Lanfear et al. 2017) were
utilized. Values of 0% <ASDSF <1% and 1% <ASDSF <5% indicate excellent and
acceptable convergence, respectively; ESS >100 and PSRF ~1 represent good convergence
(see ExaBayes manual; Aberer et al. 2014).
5 gap characters) and original DNA and
AA CO supermatrices assuming a single partition. We initiated four independent runs with
four Markov Chain Monte Carlo (MCMC) coupled chains sampling every 500th iteration. Due to
high computational demands of the procedure, only the GTR and JTT substitution model
priors were applied to DNA and AA CO supermatrices, respectively, with the default
topology, rate heterogeneity, and branch lengths priors. However, all supported protein
substitution models as a prior were specified for the trimmed AA CO supermatrix. For
convergence criteria, an average standard deviation of split frequencies (ASDSF; Lakner et al. 2008), a potential scale reduction factor
(PSRF; Brooks and Gelman 1998), and an effective
sample size (ESS; Lanfear et al. 2017) were
utilized. Values of 0% <ASDSF <1% and 1% <ASDSF <5% indicate excellent and
acceptable convergence, respectively; ESS >100 and PSRF ~1 represent good convergence
(see ExaBayes manual; Aberer et al. 2014).
ASTRAL analyses were conducted using two input types: 1) gene trees obtained by IQTREE allowing model selection for fully trimmed DNA and AA clusters and 2) gene trees obtained from the alignment-tree coestimation process in PASTA. Nodal support was assessed by local posterior probabilities (Sayyari and Mirarab 2016). In addition to standard phylogenetic inferential approaches, we applied an AF species tree estimation algorithm using Co-phylog (Yi and Jin 2013). Raw Transdecoder CDS outputs were used in this analysis using k-mer size of 9 as the half context length required for Co-phylog. Bootstrap replicate trees were obtained by running Co-Phylog with the same parameter settings on each subsampled with replacement CDS Transdecoder libraries and were used to assess nodal support.
Assessment of Phylogenetic Support via Quartet Sampling
As an additional phylogenetic support, we implemented quartet sampling (QS) approach (Pease et al. 2018). Briefly, this method provides three scores for internal nodes: 1) the quartet concordance (QC) score gives an estimate of how sampled quartet topologies agree with the putative species tree; 2) quartet differential (QD) estimates frequency skewness of the discordant quartet topologies, which can be indicative of introgression if a skewed frequency is observed; and 3) quartet informativeness (QI) quantifies how informative sampled quartets are by comparing likelihood scores of alternative quartet topologies. Finally, QS provides a quartet fidelity score for terminal nodes that measures a taxon “rogueness.” We performed QS analysis with all 48 putative species phylogenies using the SuperMatrix_50BUSCO_dna_pasta_ali_trim supermatrix, specifying the IQTREE engine for quartet likelihood calculations with 100 replicates (i.e., number of quartet draws per focal branch).
Fossil Dating
A Bayesian algorithm of MCMCTree v4.9h (Yang
2007) with approximate likelihood computation was implemented to estimate
divergence times within Odonata using 20 crown node fossil constraints with corresponding
prior distributions (Supplementary Table S3 available on Dryad). First, we estimated
branch lengths by ML and then the gradient and Hessian matrix around these ML estimates in
MCMCTree using SuperMatrix_50BUSCO_dna_pasta_ali_trim supermatrix. Second, we used these
gradient and Hessian matrices to construct an approximate likelihood function by Taylor
expansion (Dos Reis and Yang 2011) and perform
fossil calibration in MCMC framework under the uncorrelated clock model. For this step, we
specified GTR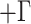 substitution model with four gamma
categories, along with birth, death, and sampling parameters of 1, 0.5, and 0.01,
respectively. To ensure convergence, the analysis was run independently five times for
6
substitution model with four gamma
categories, along with birth, death, and sampling parameters of 1, 0.5, and 0.01,
respectively. To ensure convergence, the analysis was run independently five times for
6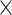 10
10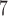 generations, logging every 1000th
generation and then removing 50% as a burn-in. Convergence (ESS >200) of the MCMC
chains was verified using Tracer v1.7.1 (Rambaut et al.
2018). Visualization of the calibrated tree was performed in R using the
MCMCtreeR package (Puttick 2019).
generations, logging every 1000th
generation and then removing 50% as a burn-in. Convergence (ESS >200) of the MCMC
chains was verified using Tracer v1.7.1 (Rambaut et al.
2018). Visualization of the calibrated tree was performed in R using the
MCMCtreeR package (Puttick 2019).
Analyses of Introgression
In order to address the scope of possible reticulate evolution across odonate phylogeny,
we used various methods of introgression detection such as HyDe/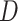 (Blischak et al. 2018),
(Blischak et al. 2018), 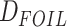 (Pease and Hahn 2015),
(Pease and Hahn 2015), 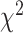 goodness-of-fit test, branch length test (BLT), QuIBL (Edelman et al. 2019), and PhyloNet
(Than et al. 2008; Wen et al. 2018). Furthermore, we used the methodological consensus of
HyDe/
goodness-of-fit test, branch length test (BLT), QuIBL (Edelman et al. 2019), and PhyloNet
(Than et al. 2008; Wen et al. 2018). Furthermore, we used the methodological consensus of
HyDe/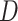 ,
, 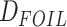 ,
,
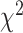 goodness-of-fit test, and BLT
approaches to provide more conservative inferences of introgression across the order (see
Discussion section). Specifically, we compared sets of unique introgressing species pairs
that were identified by each of the aforementioned methods. The significance of overlap
among the signals from these different methods was then assessed using an exact test of
multiset interactions (Wang et al. 2015).
goodness-of-fit test, and BLT
approaches to provide more conservative inferences of introgression across the order (see
Discussion section). Specifically, we compared sets of unique introgressing species pairs
that were identified by each of the aforementioned methods. The significance of overlap
among the signals from these different methods was then assessed using an exact test of
multiset interactions (Wang et al. 2015).
The HyDe framework allows detection of hybridization events which relies on
quantification of phylogenetic site patterns. HyDe estimates whether a putative hybrid
population (or taxon) H is sister to either population P1 with probability
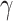 or to P2 with probability
1-
or to P2 with probability
1-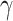 in a 4-taxon (quartet) tree
((P1,H,P2),O), where O denotes an outgroup. Then, it conducts a formal statistical test of
H
in a 4-taxon (quartet) tree
((P1,H,P2),O), where O denotes an outgroup. Then, it conducts a formal statistical test of
H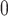 :
: 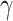
 0 vs. H
0 vs. H :
:
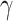 >0 using Z-test, where
>0 using Z-test, where
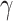
 0
(
0
( 1) is indicative of nonsignificant
introgression. We applied HyDe to the concatenated supermatrix
SuperMatrix_50BUSCO_dna_pasta_ali_trim of 1603 BUSCO genes under default parameters
specifying E. danica as an outgroup. Under this setup, HyDe evaluates all
possible taxa quartets. Since HyDe only allows indication of a single outgroup taxon (i.e.
E. danica), we excluded all quartets that contained the
Isonychia kiangsinensis outgroup from the HyDe output. Additionally, we
calculated Patterson’s
1) is indicative of nonsignificant
introgression. We applied HyDe to the concatenated supermatrix
SuperMatrix_50BUSCO_dna_pasta_ali_trim of 1603 BUSCO genes under default parameters
specifying E. danica as an outgroup. Under this setup, HyDe evaluates all
possible taxa quartets. Since HyDe only allows indication of a single outgroup taxon (i.e.
E. danica), we excluded all quartets that contained the
Isonychia kiangsinensis outgroup from the HyDe output. Additionally, we
calculated Patterson’s 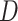 statistic (Patterson et al. 2012) for every quartet from the frequency
(
statistic (Patterson et al. 2012) for every quartet from the frequency
(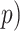 of ABBA-BABA site patterns estimated by
HyDe as
of ABBA-BABA site patterns estimated by
HyDe as 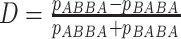 .
To test significance of
.
To test significance of 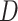 statistics, we used a
statistics, we used a
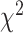 , test to assess whether the
proportions
, test to assess whether the
proportions 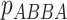 and
and 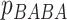 were significantly different. To minimize effect of false-positive cases (type I error) in
the output, we first applied a Bonferroni correction to the
were significantly different. To minimize effect of false-positive cases (type I error) in
the output, we first applied a Bonferroni correction to the 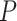 -values
derived from
-values
derived from 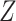 - and
- and 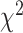 tests and then filtered the results based on a significance level of 0.05 and
10
tests and then filtered the results based on a significance level of 0.05 and
10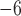 for
for 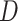 and
and
 , respectively. Additionally, we
excluded all quartets that did not match the species topology. Furthermore, we ran HyDe on
SuperMatrix_50BUSCO_dna_prank_trim excluding 3rd codon position to investigate a potential
impact of the saturation effect on introgression inference.
, respectively. Additionally, we
excluded all quartets that did not match the species topology. Furthermore, we ran HyDe on
SuperMatrix_50BUSCO_dna_prank_trim excluding 3rd codon position to investigate a potential
impact of the saturation effect on introgression inference.
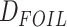 is an alternative site pattern-based approach that detects introgression
using symmetric 5-taxon (quintet) trees, i.e. (((P1,P2),(P3,P4)),O).
is an alternative site pattern-based approach that detects introgression
using symmetric 5-taxon (quintet) trees, i.e. (((P1,P2),(P3,P4)),O).
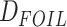 represents a collection of
statistics for quintet trees that are similar in spirit to the Patterson’s
represents a collection of
statistics for quintet trees that are similar in spirit to the Patterson’s
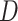 statistic; if considered simultaneously,
these statistics provide a powerful approach to identify introgression including ancestral
as well as donor and recipient taxa (i.e. introgression directionality). Moreover,
statistic; if considered simultaneously,
these statistics provide a powerful approach to identify introgression including ancestral
as well as donor and recipient taxa (i.e. introgression directionality). Moreover,
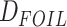 exhibits exceptionally low
false-positive rates (Pease and Hahn 2015). Since
the number of possible quintet topologies for a phylogeny of 85 taxa is >
32
exhibits exceptionally low
false-positive rates (Pease and Hahn 2015). Since
the number of possible quintet topologies for a phylogeny of 85 taxa is >
32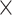 10
10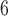 , for
analysis we extracted them only for every odonate suborder individually using custom R
scripts. Note that for Anisozygoptera, we only considered quintets that can be formed
between Anisozygoptera, Anisoptera, and Zygoptera taxonomic groups. As the number of
Anisozygoptera quintets is highly disproportional (34,619 out of all 72,971 tested Odonata
quintets), for downstream analyses, we randomly selected 4000 Anisozygoptera quintets
which approximately matches the number of quintets for an individual species. Analogously
to HyDe, we applied
, for
analysis we extracted them only for every odonate suborder individually using custom R
scripts. Note that for Anisozygoptera, we only considered quintets that can be formed
between Anisozygoptera, Anisoptera, and Zygoptera taxonomic groups. As the number of
Anisozygoptera quintets is highly disproportional (34,619 out of all 72,971 tested Odonata
quintets), for downstream analyses, we randomly selected 4000 Anisozygoptera quintets
which approximately matches the number of quintets for an individual species. Analogously
to HyDe, we applied 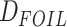 to the concatenated supermatrix
SuperMatrix_50BUSCO_dna_pasta_ali_trim of 1603 BUSCO genes under default parameters
specifying E. danica as an outgroup. Also, since
to the concatenated supermatrix
SuperMatrix_50BUSCO_dna_pasta_ali_trim of 1603 BUSCO genes under default parameters
specifying E. danica as an outgroup. Also, since
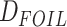 requires that every quintet has a
symmetric topology, we considered only those quintets within our phylogeny that met this
criterion (Fig. 1). Additionally,
requires that every quintet has a
symmetric topology, we considered only those quintets within our phylogeny that met this
criterion (Fig. 1). Additionally,
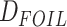 requires that the divergence time
of P3 and P4 precedes divergence of P1 and P2, i.e.
requires that the divergence time
of P3 and P4 precedes divergence of P1 and P2, i.e. 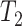 >
> 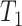 , thus we filtered out quintets that
violated this assumption using divergence times from our fossil calibrated phylogeny. In
order to correct the
, thus we filtered out quintets that
violated this assumption using divergence times from our fossil calibrated phylogeny. In
order to correct the 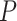 -values resulted from
-values resulted from
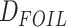 analysis for multiple testing, we
applied the Benjamini–Hochberg procedure at a false discovery rate (FDR) cutoff of
0.05.
analysis for multiple testing, we
applied the Benjamini–Hochberg procedure at a false discovery rate (FDR) cutoff of
0.05.
Figure 1.

Evolutionary history of odonata. Fossil calibrated ML phylogenetic tree of Odonata using a DNA supermatrix consisting of 1603 BUSCO genes with a total of 2,167,861 aligned sites. The blue densities at each node represent posterior distributions of ages estimated in MCMCTree using 20 fossil calibration points. The red-dashed vertical line indicates the beginning of establishment for major odonate lineages originating in and spanning the Cretaceous. The histogram (blue bars) represents temporal distribution of fossil Odonatoptera samples. The black-dashed vertical lines mark major extinction events, namely Permian-Triassic (P-Tr, ~251 Ma), Triassic-Jurassic (Tr-J, ~201.3 Ma), and Cretaceous-Paleogene (K-Pg, ~66 Ma)
As an alternative test for introgression, we performed a simple yet conservative
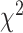 goodness-of-fit test on the gene
count values for each triplet. Specifically, we asked whether one of the two possible
discordant gene tree topologies was supported by a greater number of genes than the other
discordant topology (i.e. a significant difference between the number of discordant gene
trees showing ((P1,P3),P2) vs. ((P2,P3),P1), where the ((P1,P2),P3) topology corresponds
to the species tree). Under ILS alone, the fractions of genes supporting each discordant
topology are expected to be the same, while in the presence of introgression they may
differ. We, therefore, used a
goodness-of-fit test on the gene
count values for each triplet. Specifically, we asked whether one of the two possible
discordant gene tree topologies was supported by a greater number of genes than the other
discordant topology (i.e. a significant difference between the number of discordant gene
trees showing ((P1,P3),P2) vs. ((P2,P3),P1), where the ((P1,P2),P3) topology corresponds
to the species tree). Under ILS alone, the fractions of genes supporting each discordant
topology are expected to be the same, while in the presence of introgression they may
differ. We, therefore, used a 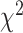 test to determine if these
fractions differed significantly, and we considered triplets where the null hypothesis was
rejected to be suggestive of introgression. Because we tested many triplets for
introgression, we corrected the
test to determine if these
fractions differed significantly, and we considered triplets where the null hypothesis was
rejected to be suggestive of introgression. Because we tested many triplets for
introgression, we corrected the 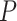 -values resulted from
these
-values resulted from
these 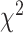 tests using the Benjamini–Hochberg
procedure and applied an FDR cutoff of 0.05. Second, we used a BLT to identify cases of
introgression (Suvorov et al. 2021). This test
examines branch lengths to estimate the age of the most recent coalescence event (measured
in substitutions per site). Introgression should result in more recent coalescences than
expected under the concordant topology with complete lineage sorting, while ILS yields
older coalescence events. Importantly, ILS alone is not expected to result in different
coalescence times between the two discordant topologies, and this forms the null
hypothesis for the BLT. For a given triplet, for each gene tree, we calculated the
distance
tests using the Benjamini–Hochberg
procedure and applied an FDR cutoff of 0.05. Second, we used a BLT to identify cases of
introgression (Suvorov et al. 2021). This test
examines branch lengths to estimate the age of the most recent coalescence event (measured
in substitutions per site). Introgression should result in more recent coalescences than
expected under the concordant topology with complete lineage sorting, while ILS yields
older coalescence events. Importantly, ILS alone is not expected to result in different
coalescence times between the two discordant topologies, and this forms the null
hypothesis for the BLT. For a given triplet, for each gene tree, we calculated the
distance 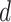 (a proxy for the divergence time between
sister taxa) by averaging the external branch lengths leading to the two sister taxa under
that gene tree topology. We calculated
(a proxy for the divergence time between
sister taxa) by averaging the external branch lengths leading to the two sister taxa under
that gene tree topology. We calculated 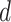 for each gene tree and
denote values of
for each gene tree and
denote values of 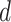 from the first discordant topology
from the first discordant topology
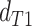 and those from the second discordant
topology
and those from the second discordant
topology 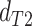 . We then compared the distributions
of
. We then compared the distributions
of 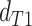 and
and 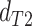 using a Wilcoxon Rank Sum Test. Under ILS alone the expectation is that
using a Wilcoxon Rank Sum Test. Under ILS alone the expectation is that
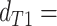
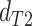 ,
while in the presence of introgression
,
while in the presence of introgression 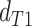 <
<
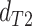 (suggesting introgression consistent
with discordant topology T
(suggesting introgression consistent
with discordant topology T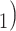 or
or 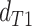 >
> 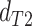 (suggesting introgression with
consistent with discordant topology T
(suggesting introgression with
consistent with discordant topology T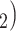 . The BLT is
conceptually similar to the D3 test (Hahn and Hibbins
2019), which transforms the values of
. The BLT is
conceptually similar to the D3 test (Hahn and Hibbins
2019), which transforms the values of 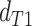 and
and
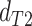 in a manner similar to the
in a manner similar to the
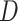 statistic for detecting introgression. As
with the
statistic for detecting introgression. As
with the 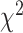 test, we performed the BLT on all
triplets within a clade and used a Benjamini–Hochberg correction with an FDR cutoff of
0.05. We note that both the
test, we performed the BLT on all
triplets within a clade and used a Benjamini–Hochberg correction with an FDR cutoff of
0.05. We note that both the 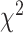 test and BLT may be conservative
in cases where there is introgression between both tested species pairs (i.e.
introgression between P1-P3 and P2-P3 for a given species topology ((P1,P2),P3)) depending
on the fraction of affected loci (affects the
test and BLT may be conservative
in cases where there is introgression between both tested species pairs (i.e.
introgression between P1-P3 and P2-P3 for a given species topology ((P1,P2),P3)) depending
on the fraction of affected loci (affects the 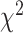 test) and timing
of introgression between each species pair (affects the BLT).
test) and timing
of introgression between each species pair (affects the BLT).
QuIBL is based on the analysis of branch length distributions across gene trees to infer
putative introgression patterns. Briefly, under coalescent theory, internal branches of
rooted gene trees for a set of 3 taxa (triplet) can be viewed as a mixture of two
distributions with the underlying parameters. Each mixture component generates branch
lengths corresponding to either ILS or introgression/speciation. Thus, estimated mixing
proportions (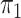 for ILS
and
for ILS
and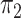 for
introgression/speciation;
for
introgression/speciation; 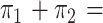 1) of those
distribution components show what fraction of the gene trees were generated through ILS or
non-ILS processes. For a given triplet, QuIBL computes frequency of gene trees that
support three alternative topologies. Then for every alternative topology, QuIBL estimates
mixing proportions along with other relevant parameters via Expectation-Maximization and
computes Bayesian Information Criterion (BIC) scores for ILS-only and introgression
models. For concordant topologies, elevated values of
1) of those
distribution components show what fraction of the gene trees were generated through ILS or
non-ILS processes. For a given triplet, QuIBL computes frequency of gene trees that
support three alternative topologies. Then for every alternative topology, QuIBL estimates
mixing proportions along with other relevant parameters via Expectation-Maximization and
computes Bayesian Information Criterion (BIC) scores for ILS-only and introgression
models. For concordant topologies, elevated values of 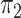 are expected whereas for discordant ones
are expected whereas for discordant ones 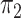 can vary
depending on the severity of ILS/intensity of introgression. In extreme cases when the
gene trees were generated exclusively under ILS,
can vary
depending on the severity of ILS/intensity of introgression. In extreme cases when the
gene trees were generated exclusively under ILS, 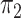 will approach zero and the expected
gene tree frequency for each alternative topology of a triplet will be approximately 1/3.
To identify significant cases of introgression here we used a stringent cutoff of
will approach zero and the expected
gene tree frequency for each alternative topology of a triplet will be approximately 1/3.
To identify significant cases of introgression here we used a stringent cutoff of
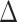 BIC <
BIC <  30
(Edelman et al. 2019). We ran QuIBL on every
triplet individually under default parameters with number of steps (numsteps parameter) is
equal to 50 and specifying one of the Ephemeroptera species (I.
kiangsinensis and E. danica) for triplet rooting. For
computational efficiency, we extracted triplets only from the odonate superfamilies in a
similar manner as we did for
30
(Edelman et al. 2019). We ran QuIBL on every
triplet individually under default parameters with number of steps (numsteps parameter) is
equal to 50 and specifying one of the Ephemeroptera species (I.
kiangsinensis and E. danica) for triplet rooting. For
computational efficiency, we extracted triplets only from the odonate superfamilies in a
similar manner as we did for 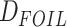 (see above). For this analysis, we
used 1603 ML gene trees estimated from CO orthology clusters. We note that most of the
phylogenomic-based introgression detection methods, including approaches used here (namely
HyDe,
(see above). For this analysis, we
used 1603 ML gene trees estimated from CO orthology clusters. We note that most of the
phylogenomic-based introgression detection methods, including approaches used here (namely
HyDe, 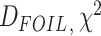 test, BLT, and
QuIBL) are not able to infer gene flow between sister lineages (Hibbins and Hahn 2021) as they rely on topological discordance at
either the gene or site level, which can only be examined for topologies with more than
two taxa.
test, BLT, and
QuIBL) are not able to infer gene flow between sister lineages (Hibbins and Hahn 2021) as they rely on topological discordance at
either the gene or site level, which can only be examined for topologies with more than
two taxa.
To identify patterns of reticulate evolution for Anisozygoptera, we estimated
phylogenetic networks from the 1603 ML gene trees estimated from CO orthology clusters
using pseudolikelihood (InferNetwork_MPL; Yu and Nakhleh
2015) and likelihood (CalGTProb) approaches implemented in PhyloNet (Than et al. 2008; Wen
et al. 2018). For scalability purposes, we subsampled our taxon set to eight
Zygoptera species, nine Anisoptera species, and Epiophlebia superstes.
For all network searches, we explicitly indicated E. superstes as a
putative hybrid (-h option). For both pseudolikelihood and likelihood analyses, we only
selected gene trees that had at least one of the outgroup species (I.
kiangsinensis and E. danica) and at least three ingroup taxa.
For pseudolikelihood analysis, we ran PhyloNet allowing a single reticulation event, with
the starting tree that corresponds to the species phylogeny (-s option), 100 iterations
(-x option), 0.9 bootstrap threshold for gene trees (-b option), and optimization of
branch lengths and inheritance probabilities on the inferred networks (-po option). To
ensure convergence, the network searches were repeated three times. For the full
likelihood estimation, we fixed the topology (equivalent to the species tree topology) and
calculated likelihood scores for possible networks with a single reticulation (generated
with a custom script) using CalGTProb. Additionally, to assess significance of networks,
we used difference of BIC scores (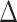 BIC) derived from
network without reticulation (i.e. tree) and a network with a reticulation (Supplementary
Table S4 available on Dryad).
BIC) derived from
network without reticulation (i.e. tree) and a network with a reticulation (Supplementary
Table S4 available on Dryad).
Dimensionality Reduction and Visualization
To uncover and visualize complex relationships between site pattern frequencies and
Patterson’s D statistic and HyDe 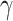 parameter, we
implemented a dimensionality reduction technique t-distributed stochastic neighbor
embedding (tSNE; van der Maaten and Hinton 2008)
under default parameters in R. Specifically, we estimated tSNE maps from counts of 15
quartet site patterns calculated by HyDe (“AAAA,” “AAAB,” “AABA,” “AABB,” “AABC,” “ABAA,”
“ABAB,” “ABAC,” “ABBA,” “BAAA,” “ABBC,” “CABC,” “BACA,” “BCAA,” and “ABCD”).
parameter, we
implemented a dimensionality reduction technique t-distributed stochastic neighbor
embedding (tSNE; van der Maaten and Hinton 2008)
under default parameters in R. Specifically, we estimated tSNE maps from counts of 15
quartet site patterns calculated by HyDe (“AAAA,” “AAAB,” “AABA,” “AABB,” “AABC,” “ABAA,”
“ABAB,” “ABAC,” “ABBA,” “BAAA,” “ABBC,” “CABC,” “BACA,” “BCAA,” and “ABCD”).
Results
Phylogenetic Inference
We compiled transcriptomic data for 83 odonate species including 49 new transcriptomes
sequenced for this study (Supplementary Table S1 available on Dryad). To assess effects of
various steps of our phylogenetic pipeline on species tree inference, we examined
different methods of sequence homology detection, multiple sequence alignment strategies,
postprocessing filtering procedures, and tree estimation methods. Specifically, three
types of homologous loci (gene clusters) were used to develop our supermatrices, namely
1603 conserved single-copy orthologs (CO), 1643 all single-copy orthologs (AO), and 4341
paralogy-parsed orthologs (PO) with 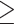 42 (~50%) species
present (for more details, see Materials and Methods section). To date, our data represent
the most comprehensive resource available for Odonata in terms of gene sampling. Each gene
cluster was aligned, trimmed, and concatenated resulting in five main supermatrices, CO
(DNA/AA), AO (AA), and PO (DNA/AA), which included 2,167,861 DNA (682,327 amino acid [AA]
sites), 882,417 AA sites, 6,202,646 DNA (1,605,370 AA) sites, respectively. Thus, the
largest alignment that we used to infer the odonate phylogeny consists of 4341 loci
concatenated into a supermatrix with > 6 million nucleotide sites. All supermatrices
are summarized in Supplementary Table S2 available on Dryad; the inferred odonate
relationships are shown in Supplementary Figure S1a available on Dryad whereas topologies
of all inferred phylogenies are plotted in Supplementary Figure S1b available on Dryad and
topologies of 1603 CO gene trees are shown in Supplementary Figure S1c available on Dryad.
Additionally, we performed nodal dating of the inferred phylogeny using 20 fossil
calibration points (Supplementary Table S3 available on Dryad).
42 (~50%) species
present (for more details, see Materials and Methods section). To date, our data represent
the most comprehensive resource available for Odonata in terms of gene sampling. Each gene
cluster was aligned, trimmed, and concatenated resulting in five main supermatrices, CO
(DNA/AA), AO (AA), and PO (DNA/AA), which included 2,167,861 DNA (682,327 amino acid [AA]
sites), 882,417 AA sites, 6,202,646 DNA (1,605,370 AA) sites, respectively. Thus, the
largest alignment that we used to infer the odonate phylogeny consists of 4341 loci
concatenated into a supermatrix with > 6 million nucleotide sites. All supermatrices
are summarized in Supplementary Table S2 available on Dryad; the inferred odonate
relationships are shown in Supplementary Figure S1a available on Dryad whereas topologies
of all inferred phylogenies are plotted in Supplementary Figure S1b available on Dryad and
topologies of 1603 CO gene trees are shown in Supplementary Figure S1c available on Dryad.
Additionally, we performed nodal dating of the inferred phylogeny using 20 fossil
calibration points (Supplementary Table S3 available on Dryad).
The inferred ML phylogenetic tree of Odonata using DNA supermatrix of 1603 BUSCO loci
(Fig. 1) was used as a primary phylogenetic
hypothesis throughout this study as it agrees with the majority of relationships inferred
by other methods (Supplementary Fig. S1a,b available on Dryad). Divergence of Zygoptera
and Epiprocta (Anisozygoptera Anisoptera) from the Most Recent Common
Ancestor (MRCA) was estimated to have occurred in the Middle Triassic ~226 Ma (95%
Credible Interval [CI]: 221.8–231.1 Ma, Fig. 1),
which is in line with recent estimates (Thomas et al.
2013; Misof et al. 2014). Comprehensive
phylogenetic coestimation of subordinal relationships within Odonata showed that the
suborders were well supported (Supplementary Fig. S1a available on Dryad), as they were
consistently recovered as monophyletic clades in all analyses. In several previous
studies, paraphyletic relationships of Zygoptera had been proposed based on wing vein
characters derived from fossil odonatoids and extant Odonata (Trueman 1996), analysis of 12S (Saux
et al. 2003), analysis of 18S, 28S, Histone 3 (H3), and morphological data (Ogden and Whiting 2003) and analysis of 16S and 28S
data (Hasegawa and Kasuya 2006). In most of these
studies, Lestidae was inferred to be sister to Anisoptera. Functional morphology
comparisons of flight systems, secondary male genitalia, and ovipositors also supported a
nonmonophyletic Zygoptera with uncertain phylogenetic placement of multiple groups (Pfau 1991). Nevertheless, the relationships inferred
from these previous data sets seem to be highly unlikely due to apparent morphological
differentiation (e.g., eye spacing, body robustness, wing shape) between the suborders and
support for monophyletic Anisoptera and Zygoptera from more recent morphological (Busse et al. 2015), molecular (Carle et al. 2008; Thomas et al.
2013; Kim et al. 2014; Suvorov et al. 2017), and combined studies using both
data types (Bybee et al. 2008). Our analyses
recover Zygoptera as monophyletic consistently (Supplementary Fig. S1a available on
Dryad).
Anisoptera) from the Most Recent Common
Ancestor (MRCA) was estimated to have occurred in the Middle Triassic ~226 Ma (95%
Credible Interval [CI]: 221.8–231.1 Ma, Fig. 1),
which is in line with recent estimates (Thomas et al.
2013; Misof et al. 2014). Comprehensive
phylogenetic coestimation of subordinal relationships within Odonata showed that the
suborders were well supported (Supplementary Fig. S1a available on Dryad), as they were
consistently recovered as monophyletic clades in all analyses. In several previous
studies, paraphyletic relationships of Zygoptera had been proposed based on wing vein
characters derived from fossil odonatoids and extant Odonata (Trueman 1996), analysis of 12S (Saux
et al. 2003), analysis of 18S, 28S, Histone 3 (H3), and morphological data (Ogden and Whiting 2003) and analysis of 16S and 28S
data (Hasegawa and Kasuya 2006). In most of these
studies, Lestidae was inferred to be sister to Anisoptera. Functional morphology
comparisons of flight systems, secondary male genitalia, and ovipositors also supported a
nonmonophyletic Zygoptera with uncertain phylogenetic placement of multiple groups (Pfau 1991). Nevertheless, the relationships inferred
from these previous data sets seem to be highly unlikely due to apparent morphological
differentiation (e.g., eye spacing, body robustness, wing shape) between the suborders and
support for monophyletic Anisoptera and Zygoptera from more recent morphological (Busse et al. 2015), molecular (Carle et al. 2008; Thomas et al.
2013; Kim et al. 2014; Suvorov et al. 2017), and combined studies using both
data types (Bybee et al. 2008). Our analyses
recover Zygoptera as monophyletic consistently (Supplementary Fig. S1a available on
Dryad).
Divergence time estimates suggest a TMRCA of Anisoptera and Anisozygoptera (we occasionally refer these two suborders as “Epiprocta”) in the Late Triassic (~204 Ma; 95% CI 201.7–207.8 Ma; Fig. 1). Epiprocta as well as Anisoptera were consistent with more recent studies and recovered as monophyletic with very high support. We also note here that our divergence time estimates of Anisoptera tend to be younger than those found by (Letsch et al. 2016). The fossil-calibration approach based on penalized likelihood that was used by (Letsch et al. 2016) has been shown to overestimate true nodal age (Britton et al. 2007) preventing direct comparison between our dates derived from the Bayesian framework MCMCTree and those estimated by (Letsch et al., 2016). Additionally, our divergence time estimate for Epiprocta is older than inferred by (Misof et al., 2014), which can be explained by the differences in calibration schemes. Specifically, for the Epiprocta crown node, we specified Liassophlebia sp. fossil using an informative skewed normal distribution prior (see Supplementary Table S3 available on Dryad).
The phylogenetic position of Gomphidae and Petaluridae, both with respect to each other
and the remaining anisopteran families, has long been difficult to resolve. Several
phylogenetic hypotheses have been proposed in the literature based on molecular and
morphological data regarding the placement of Gomphidae as sister to the remaining
Anisoptera (Blanke et al. 2013) or to Libelluloidea
(Misof et al. 2001). Petaluridae has exhibited
stochastic relationships with different members of Anisoptera, including sister to
Gomphidae (Misof et al. 2001), sister to
Libelluloidea (Carle et al. 2008), sister to
Chlorogomphidae Cordulegasteridae (Bybee et al. 2008), and sister to all other Anisoptera (Trueman 1996; Rehn
2003). The most recent analyses of the major anisopteran lineages using several
molecular markers (Carle et al. 2015) suggest
Gomphidae and Petaluridae as a monophyletic group, but without strong support. Here, the
majority of our supermatrix analyses (Supplementary Fig. S1a available on Dryad) strongly
support a sister relationship between the two families, and in our phylogeny (Fig. 1) they split from the MRCA ~161 Ma (95% CI:
156.6–165.5 Ma) in the Middle Jurassic (Fig. 1). We
further investigated the species tree topologies that were estimated by the
coalescent-based tree summary method, ASTRAL. We found that almost all these species trees
reject such a relationship with high confidence (Supplementary Fig. S1a available on
Dryad). In the presence of ILS, concatenation methods can be statistically inconsistent
(Roch and Steel 2014) leading to an erroneous
species tree topology with unreasonably high support (Kubatko and Degnan 2007). This inconsistency in the recovery of a sister group
relationship between Gomphidae and Petaluridae can be explained by elevated levels of ILS
between the families and/or possible introgression events (Maddison 1997).
Cordulegasteridae (Bybee et al. 2008), and sister to all other Anisoptera (Trueman 1996; Rehn
2003). The most recent analyses of the major anisopteran lineages using several
molecular markers (Carle et al. 2015) suggest
Gomphidae and Petaluridae as a monophyletic group, but without strong support. Here, the
majority of our supermatrix analyses (Supplementary Fig. S1a available on Dryad) strongly
support a sister relationship between the two families, and in our phylogeny (Fig. 1) they split from the MRCA ~161 Ma (95% CI:
156.6–165.5 Ma) in the Middle Jurassic (Fig. 1). We
further investigated the species tree topologies that were estimated by the
coalescent-based tree summary method, ASTRAL. We found that almost all these species trees
reject such a relationship with high confidence (Supplementary Fig. S1a available on
Dryad). In the presence of ILS, concatenation methods can be statistically inconsistent
(Roch and Steel 2014) leading to an erroneous
species tree topology with unreasonably high support (Kubatko and Degnan 2007). This inconsistency in the recovery of a sister group
relationship between Gomphidae and Petaluridae can be explained by elevated levels of ILS
between the families and/or possible introgression events (Maddison 1997).
New zygopteran lineages originated in the Early Jurassic ~189 Ma (95% CI: 182.5–197.7 Ma) with the early split of Lestoidea and the remaining Zygoptera (Fig. 1). A subsequent occurrence of two large zygopteran groups, Calopterygoidea and Coenagrionoidea, was estimated within the Cretaceous (~67 Ma; 95% CI: 61.5–71.6 Ma and 115.8 Ma; 95% CI: 112.7–121.2 Ma for Calopterygoidea and Coenagrionoidea, respectively) and culminated with the rapid radiation of the majority of extant lineages in the Paleogene in the interval between ~23 and ~66 Ma. Our calibrated divergences generally agree with estimates in (Thomas et al. 2013). However, any further comparisons are precluded by the lack of comprehensive divergence time estimation for Odonata in the literature. The backbone of the crown group Calopterygoidea that branched off from Coenagrionoidea ~129 Ma (95% CI: 121.9–134.8 Ma) in the Early Cretaceous was well supported as monophyletic in most of our inferred phylogenies (Fig. 1 and Supplementary Fig. S1a available on Dryad). Previous analyses struggled to provide convincing support for the monophyly of the superfamily (Bybee et al. 2008; Carle et al. 2008; Dijkstra, Kalkman et al. 2014), whereas only 11 out of 48 phylogenetic reconstructions rejected Calopterygoidea (Supplementary Fig. S1a available on Dryad).
We used QS (Pease et al. 2018) to provide
additional information about nodal support and investigate biological explanations for
alternative evolutionary histories that received some support. We found that for most
odonate key radiations, the majority of quartets (i.e. Frequency > 0.5) support the
proposed phylogenetic hypothesis with QC scores > 0 (Supplementary Fig. S2 available on
Dryad) across all estimated putative species trees (Supplementary Fig. S1b available on
Dryad). The few exceptions consist of the Gomphidae Petaluridae split and the
A
Petaluridae split and the
A B split, where we have Frequency < 0.5
and QC < 0, which suggests that alternative relationships are possible. QD, inspired by
B split, where we have Frequency < 0.5
and QC < 0, which suggests that alternative relationships are possible. QD, inspired by
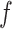 and
and 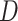 statistics for introgression, provides an indicator of how much the proportions of
discordant quartets are skewed (i.e. whether one of the two discordant relationships is
more common than the other), suggestive of introgression and/or substitution rate
heterogeneity (Pease et al. 2018). Interestingly,
we identified skewness (i.e. QD < 0.5) for almost every major radiation (Supplementary
Fig. S2 available on Dryad), which suggests that alternative relationships can be a result
of additional underlying processes (e.g. introgression) rather than ILS alone (Pease et al. 2018); however, this score may not be
highly informative if the majority of quartets agree with the focal topology (i.e.,
Frequency > 0.5 and QC > 0). For both the Gomphidae
statistics for introgression, provides an indicator of how much the proportions of
discordant quartets are skewed (i.e. whether one of the two discordant relationships is
more common than the other), suggestive of introgression and/or substitution rate
heterogeneity (Pease et al. 2018). Interestingly,
we identified skewness (i.e. QD < 0.5) for almost every major radiation (Supplementary
Fig. S2 available on Dryad), which suggests that alternative relationships can be a result
of additional underlying processes (e.g. introgression) rather than ILS alone (Pease et al. 2018); however, this score may not be
highly informative if the majority of quartets agree with the focal topology (i.e.,
Frequency > 0.5 and QC > 0). For both the Gomphidae Petaluridae and the A
Petaluridae and the A B
splits, we have Frequency > 0.5, QC < 0, and QD < 0.5, implying that alternative
phylogenetic relationships are plausibly not only due to ILS but also possible ancestral
introgression.
B
splits, we have Frequency > 0.5, QC < 0, and QD < 0.5, implying that alternative
phylogenetic relationships are plausibly not only due to ILS but also possible ancestral
introgression.
Major Trends in Evolutionary History of Odonata
Investigation of diversification rates in Odonata highlighted two major trends correlated with two mass extinction events in the Permian-Triassic (P-Tr) ~251 Ma and Cretaceous-Paleogene (K-Pg) ~66 Ma. First, it appears that P-Tr mass extinction event might have reduced the extent of biodiversity that had been present in Odonata as reflected in the fossil record (Rohde and Muller 2005) for that period (see the temporal distribution of fossil samples in Fig. 1) as was also the case for multiple insect lineages (Labandeira and Sepkoski 1993). According to the fossil record, at least two major odonatoid lineages went extinct (Protodonata and Protanisoptera; Grimaldi and Engel 2005) and likely many genera from other lineages as well (e.g., Kargalotypidae from Triadophlebimorpha; Nel et al. 2001). The establishment of major odonate lineages was observed during the Cretaceous starting ~135 Ma (Fig. 1, red line). This coincided with the radiation of angiosperm plants that, in turn, triggered the formation of herbivorous insect lineages (Misof et al. 2014). Odonates are exclusively carnivorous insects, and their diversification was likely driven by the aforementioned sequence of events. Interestingly, molecular adaptations in the odonate visual systems are coupled with their diversification during the Cretaceous as well (Suvorov et al. 2017).
Overview of Introgression Hypotheses Tested
The scope of introgression within Odonata remains largely unknown, where previous studies
looked for its patterns only within certain species relying on inference from a limited
number of genetic loci. Thus, we searched for signatures of introgression using
genome-scale data sets between lineages at several different taxonomic levels: between
different suborders, between superfamilies, and within superfamilies. We used six
different methods to test for introgression within Odonata, as exemplified for the
Anisozygoptera suborder in Figure 2. Specifically, we
searched for signatures of ancestral inter-superfamilial introgression within the
Zygoptera and Anisoptera suborders. Also, we tested the hypothesis of inter-subordinal
gene flow between Anisozygoptera and Zygoptera. Finally, we tested introgression within
superfamilies of Zygoptera and Anisoptera that included several species (Lestoidea,
Calopterygoidea, Coenagrionoidea, Aeshnoidea (Aeshnidae), Aeshnoidea
(Gomphidae Petaluridae), Cordulegastroidea, and
Libelluloidea; Fig. 1). The introgression results for
the entire Odonata order either comprise of all tests performed within the entire
phylogeny (HyDe/
Petaluridae), Cordulegastroidea, and
Libelluloidea; Fig. 1). The introgression results for
the entire Odonata order either comprise of all tests performed within the entire
phylogeny (HyDe/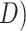 or of a union of tests performed within
Anisoptera, Zygoptera, and between Anisozygoptera-Zygoptera (
or of a union of tests performed within
Anisoptera, Zygoptera, and between Anisozygoptera-Zygoptera (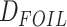 , QuiBL, and
, QuiBL, and 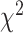 count test/BLT).
count test/BLT).
Figure 2.

Detection of introgression/hybridization trajectories by different methods in
Anisozygoptera. Three site pattern-based (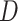 statistic, HyDe,
and
statistic, HyDe,
and 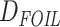 ), gene tree count/branch
length-based (
), gene tree count/branch
length-based (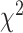 test/BLT and QuIBL), and network
ML inference (PhyloNet) methods were used to test for introgression/hybridization.
Arrows denote introgression. The figure panel represents larval and adult stages for
three Odonata suborders. Species from top to bottom: Lestes
australis, Epiophlebia superstes, and Anax
junius. Image credit: Epiophlebia superstes adult by
Christian Dutto Engelmann; L. australis, and A.
junius adults by John Abbott.
test/BLT and QuIBL), and network
ML inference (PhyloNet) methods were used to test for introgression/hybridization.
Arrows denote introgression. The figure panel represents larval and adult stages for
three Odonata suborders. Species from top to bottom: Lestes
australis, Epiophlebia superstes, and Anax
junius. Image credit: Epiophlebia superstes adult by
Christian Dutto Engelmann; L. australis, and A.
junius adults by John Abbott.
Site Pattern-Based Methods Strongly Suggest Multiple Instances of Introgression within Odonata
Initially, we tested the above hypotheses of introgression in quartet topologies
(Supplementary Fig. S3 available on Dryad) within Odonata using two site pattern-based
methods: the ABBA-BABA test (Patterson et al.
2012) and HyDe (Meng and Kubatko 2009). The
ABBA-BABA test and HyDe rely on computation of 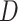 and
and
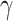 statistics, respectively, where
their significant deviation from 0 may indicate the presence of introgression between the
tested pair of taxa. Additionally, estimated
statistics, respectively, where
their significant deviation from 0 may indicate the presence of introgression between the
tested pair of taxa. Additionally, estimated 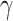 and
(1-
and
(1-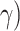 of HyDe’s hybrid speciation model
(Meng and Kubatko 2009) corresponds to the
parental fractions in a putatively hybrid genome. Note that HyDe’s hybrid speciation model
is appropriate for detecting introgression with sufficient statistical power and can
produce reasonable estimates of
of HyDe’s hybrid speciation model
(Meng and Kubatko 2009) corresponds to the
parental fractions in a putatively hybrid genome. Note that HyDe’s hybrid speciation model
is appropriate for detecting introgression with sufficient statistical power and can
produce reasonable estimates of 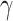 (Blischak et al. 2018; Kong and Kubatko 2021). The analysis of the ABBA-BABA test results revealed
possible gene flow events throughout the entire evolutionary history of Odonata
(Supplementary Table S5, Fig. S4a and Results available on Dryad). We also highlight the
positive relationship between the values of
(Blischak et al. 2018; Kong and Kubatko 2021). The analysis of the ABBA-BABA test results revealed
possible gene flow events throughout the entire evolutionary history of Odonata
(Supplementary Table S5, Fig. S4a and Results available on Dryad). We also highlight the
positive relationship between the values of 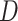 and
and
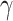 statistics (Spearman’s rank
correlation test,
statistics (Spearman’s rank
correlation test, 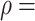 0.308,
0.308, 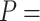 0),
demonstrating their broad concordance in identifying signatures of introgression
(Supplementary Fig. S4b available on Dryad).
0),
demonstrating their broad concordance in identifying signatures of introgression
(Supplementary Fig. S4b available on Dryad).
There are a variety of other test statistics that have been developed to detect
introgression (e.g., Green et al. 2010; Durand et al. 2011; Martin et al. 2015; Kubatko and Chifman
2019). Because, like 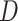 and
and
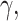 many of these statistics are
computed from different invariants, we attempted to visualize the relationships between
all 15 site patterns computed by HyDe using
many of these statistics are
computed from different invariants, we attempted to visualize the relationships between
all 15 site patterns computed by HyDe using 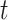 -distributed stochastic
neighbor embedding (tSNE; van der Maaten and Hinton
2008) for dimensionality reduction, along with the corresponding values of
-distributed stochastic
neighbor embedding (tSNE; van der Maaten and Hinton
2008) for dimensionality reduction, along with the corresponding values of
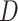 and
and 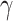 (Supplementary Fig. S4c,d available on Dryad, respectively). We found that the clustering
of quartets with significant introgression according to a two-dimensional representation
of their site patterns may suggest the presence of additional site pattern signatures of
introgression (Supplementary Fig. S4c,d available on Dryad). These results suggest that
powerful dimensionality reduction techniques could serve as a useful tool for the
exploration and visualization of complex signatures of introgression simultaneously
estimated from a large set of site patterns.
(Supplementary Fig. S4c,d available on Dryad, respectively). We found that the clustering
of quartets with significant introgression according to a two-dimensional representation
of their site patterns may suggest the presence of additional site pattern signatures of
introgression (Supplementary Fig. S4c,d available on Dryad). These results suggest that
powerful dimensionality reduction techniques could serve as a useful tool for the
exploration and visualization of complex signatures of introgression simultaneously
estimated from a large set of site patterns.
In order to assess the extent of preservation of ancestrally introgressed genetic
material within contemporary taxa, we compared inferred average values of significant
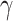 statistic from Odonata with the
averages derived from different intra- and inter-superfamilial taxonomic levels (Fig. 3a). We found significantly higher values of
statistic from Odonata with the
averages derived from different intra- and inter-superfamilial taxonomic levels (Fig. 3a). We found significantly higher values of
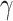 for several intra- and
inter-superfamilial comparisons including those that involve Anisozygoptera (Wilcoxon
rank-sum test [WRST], all
for several intra- and
inter-superfamilial comparisons including those that involve Anisozygoptera (Wilcoxon
rank-sum test [WRST], all 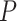 < 0.05, Fig. 3a, Supplementary Table S5 and Results available on Dryad). Additionally,
Anisozygoptera exhibits the largest average
< 0.05, Fig. 3a, Supplementary Table S5 and Results available on Dryad). Additionally,
Anisozygoptera exhibits the largest average 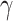 (0.27) across all
the inter-superfamilial comparisons (Fig. 3a,
Supplementary Table S5 available on Dryad). We also found an excess (Fisher exact test
[FET], all
(0.27) across all
the inter-superfamilial comparisons (Fig. 3a,
Supplementary Table S5 available on Dryad). We also found an excess (Fisher exact test
[FET], all 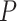 < 0.05, Supplementary Table S5
available on Dryad) of significant triplets that support introgression (Fig. 3b) based on both the ABBA-BABA and HyDe hybrid
speciation model (Supplementary Fig. S3 available on Dryad) tests for Anisozygoptera,
Aeshnoidea (Aeshnidae), Lestoidea, and Calopterygoidea (between and within). We note that
the accuracy of introgression detection for site pattern-based methods may be impaired by
saturation, which will be exacerbated on larger timescales. Thus, we additionally
performed HyDe analysis using only the 1st and 2nd codon positions and obtained highly
similar results for
< 0.05, Supplementary Table S5
available on Dryad) of significant triplets that support introgression (Fig. 3b) based on both the ABBA-BABA and HyDe hybrid
speciation model (Supplementary Fig. S3 available on Dryad) tests for Anisozygoptera,
Aeshnoidea (Aeshnidae), Lestoidea, and Calopterygoidea (between and within). We note that
the accuracy of introgression detection for site pattern-based methods may be impaired by
saturation, which will be exacerbated on larger timescales. Thus, we additionally
performed HyDe analysis using only the 1st and 2nd codon positions and obtained highly
similar results for 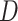 and
and 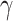 (Supplementary Fig. S5 available on Dryad) suggesting that the potential saturation effect
in 3rd codon position did not severely impact our inferences. Despite these results, the
(Supplementary Fig. S5 available on Dryad) suggesting that the potential saturation effect
in 3rd codon position did not severely impact our inferences. Despite these results, the
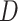 statistic distributions, if considered
individually, should be interpreted with caution: the ABBA-BABA test can produce false
positives within genomic regions of reduced interspecific divergence, and can also be
significantly affected by demographic parameters, genetic drift, and variation in
recombination rates (Martin et al. 2015).
statistic distributions, if considered
individually, should be interpreted with caution: the ABBA-BABA test can produce false
positives within genomic regions of reduced interspecific divergence, and can also be
significantly affected by demographic parameters, genetic drift, and variation in
recombination rates (Martin et al. 2015).
Figure 3.

Distributions of HyDe and quartets fractions supporting introgression across odonate
taxonomic levels. a) Distribution of significant (Bonferroni corrected
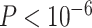 values for each quartet
estimated by HyDe. In general,
values for each quartet
estimated by HyDe. In general, 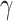 values that
are not significantly different from 0 denote no relation of a putative hybrid species
to either of the parental species P1 (1-
values that
are not significantly different from 0 denote no relation of a putative hybrid species
to either of the parental species P1 (1-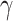 ) or P2
(
) or P2
(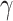 ) in a quartet. Asterisks
indicate significantly greater (red) or lower (blue) averages of various tested cases
compared to average of the entire order. b) Proportions of quartets that support
introgression based on simultaneous significance of D statistics and
) in a quartet. Asterisks
indicate significantly greater (red) or lower (blue) averages of various tested cases
compared to average of the entire order. b) Proportions of quartets that support
introgression based on simultaneous significance of D statistics and
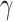 . Asterisks indicate
significantly greater (red) and smaller (blue) fraction of quartets that support
introgression compared to the entire order.
. Asterisks indicate
significantly greater (red) and smaller (blue) fraction of quartets that support
introgression compared to the entire order.
Furthermore, we tested introgression within Odonata using an alternative site
pattern-based method,  (Supplementary Fig. S6a and
Results available on Dryad), which is capable of detecting ancestral as well as intergroup
introgression and inferring its polarization (see Materials and Methods section).
Specifically, we observed a highly skewed distribution of
(Supplementary Fig. S6a and
Results available on Dryad), which is capable of detecting ancestral as well as intergroup
introgression and inferring its polarization (see Materials and Methods section).
Specifically, we observed a highly skewed distribution of  statistics for Anisozygoptera (Supplementary Fig. S6b and Results available on Dryad) that
may suggest specific directionality of introgression: all 61 (1157 out of 1158 without FDR
correction) quintets with positive evidence for ancestral introgression suggest that
Anisozygoptera is the recipient lineage whereas Zygoptera is the donor (Fig. 2, red and blue arrows). Additionally, 1030 out of
1091 (4473 out of 4738 without FDR correction) evaluated quintets with significant
intergroup introgression are indicative of one-way introgression from Zygoptera lineages
to Anisozygoptera as well, whereas the directionality of remaining 59 (254 without FDR
correction) quintets support Anisozygoptera as a donor and Zygoptera as a recipient and
only two (11 without FDR correction) show introgression between Zygoptera and
Anisoptera.
statistics for Anisozygoptera (Supplementary Fig. S6b and Results available on Dryad) that
may suggest specific directionality of introgression: all 61 (1157 out of 1158 without FDR
correction) quintets with positive evidence for ancestral introgression suggest that
Anisozygoptera is the recipient lineage whereas Zygoptera is the donor (Fig. 2, red and blue arrows). Additionally, 1030 out of
1091 (4473 out of 4738 without FDR correction) evaluated quintets with significant
intergroup introgression are indicative of one-way introgression from Zygoptera lineages
to Anisozygoptera as well, whereas the directionality of remaining 59 (254 without FDR
correction) quintets support Anisozygoptera as a donor and Zygoptera as a recipient and
only two (11 without FDR correction) show introgression between Zygoptera and
Anisoptera.
Signatures of Introgression Revealed by Phylogenetic Gene Tree-Based Discordance Methods
Besides specific site patterns, introgression will also generate certain patterns of gene
tree-species tree discordance and reduce the genetic divergence between introgressing
taxa, which is reflected in gene tree branch lengths. Thus, the footprints of
introgression can be detected using phylogenetic discordance methods. First, for a triplet
of species, under ILS alone one would expect equal proportions of gene tree topologies
supporting the two topologies disagreeing with the species tree, and any imbalance may
suggest introgression (Supplementary Fig. S7 available on Dryad). Thus, deviation from
equal frequencies of gene tree counts among discordant gene trees can be assessed using a
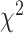 test, similar to previously
proposed methods leveraging discordant gene tree counts (Huson et al. 2005; Degnan and Rosenberg
2009). One would expect the average distance between putatively introgressing
taxa in discordant trees to be significantly smaller than the distances derived from the
concordant as well as alternative discordant triplets (see Materials and Methods section
and Supplementary Fig. S7 available on Dryad). We used both the
test, similar to previously
proposed methods leveraging discordant gene tree counts (Huson et al. 2005; Degnan and Rosenberg
2009). One would expect the average distance between putatively introgressing
taxa in discordant trees to be significantly smaller than the distances derived from the
concordant as well as alternative discordant triplets (see Materials and Methods section
and Supplementary Fig. S7 available on Dryad). We used both the 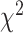 test of discordant gene tree counts and a test based on the distribution of branch lengths
for concordant and discordant gene tree triplets (BLT) to identify introgression within
Odonata testing different scenarios (Fig. 4a). With
this combination of methods, we identified a significant fraction of triplets that support
ancestral introgression for scenario 1 that involves inter-subordinal gene flow between
Anisozygoptera and Zygoptera as well as scenarios 2 through 4, which correspond to
inter-superfamilial instances of introgression within Epiprocta (Fig. 4b). Within superfamilies, we found signatures of introgression for
Calopterygoidea and Libelluloidea (Fig. 4b). For
scenario 1 (introgression between Zygoptera and Anisozygoptera), examination of the
genetic divergence distributions (Fig. 4c) for
concordant and discordant triplets showed that discordant triplets that may have resulted
from gene flow between Anisozygoptera and Zygoptera (the topology labeled “discord2”),
have markedly smaller average divergence between these two taxa, as expected in the
presence of introgression. Similarly, based on the distribution of mean divergence between
putatively introgressing taxa (Supplementary Fig. S8 available on Dryad) as well as
fraction of significant triplets (Fig. 4b) gene flow
is supported for scenarios 2, 3, and 4.
test of discordant gene tree counts and a test based on the distribution of branch lengths
for concordant and discordant gene tree triplets (BLT) to identify introgression within
Odonata testing different scenarios (Fig. 4a). With
this combination of methods, we identified a significant fraction of triplets that support
ancestral introgression for scenario 1 that involves inter-subordinal gene flow between
Anisozygoptera and Zygoptera as well as scenarios 2 through 4, which correspond to
inter-superfamilial instances of introgression within Epiprocta (Fig. 4b). Within superfamilies, we found signatures of introgression for
Calopterygoidea and Libelluloidea (Fig. 4b). For
scenario 1 (introgression between Zygoptera and Anisozygoptera), examination of the
genetic divergence distributions (Fig. 4c) for
concordant and discordant triplets showed that discordant triplets that may have resulted
from gene flow between Anisozygoptera and Zygoptera (the topology labeled “discord2”),
have markedly smaller average divergence between these two taxa, as expected in the
presence of introgression. Similarly, based on the distribution of mean divergence between
putatively introgressing taxa (Supplementary Fig. S8 available on Dryad) as well as
fraction of significant triplets (Fig. 4b) gene flow
is supported for scenarios 2, 3, and 4.
Figure 4.

Results of the 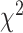 Count-BLT for Odonate Taxonomic
Levels and PhyloNet result for Anisozygoptera. a) Scenarios of deep (numbered red
arrows) and intra-superfamilial (white triangles) introgression in Odonata tested
using
Count-BLT for Odonate Taxonomic
Levels and PhyloNet result for Anisozygoptera. a) Scenarios of deep (numbered red
arrows) and intra-superfamilial (white triangles) introgression in Odonata tested
using 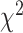 test and BLT. Red arrows mark
the location of ancestral introgression events that were tested between lineages (e.g.
for scenario 8, we tested whether contemporary species of Platystictoidea shared
introgressed genetic material with either Coenagrionoidea or Calopterygoidea). b)
Classification of triplets based on the
test and BLT. Red arrows mark
the location of ancestral introgression events that were tested between lineages (e.g.
for scenario 8, we tested whether contemporary species of Platystictoidea shared
introgressed genetic material with either Coenagrionoidea or Calopterygoidea). b)
Classification of triplets based on the 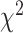 test and BLT
results. Introgression+ILS cases are those significant according to both the
test and BLT
results. Introgression+ILS cases are those significant according to both the
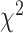 test and BLT (FDR corrected
test and BLT (FDR corrected
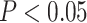 ); all the remaining cases were
those where any discordance was inferred to be due to ILS alone. c) Normalized genetic
divergence between sister taxa (shown in triplets above the panel) averaged across all
BUSCO gene trees supporting each topology involving three lineages representing
Anisoptera, Anisozygoptera, and Zygoptera. d) Phylogenetic network estimated from a
set of ML gene trees using maximum likelihood. Epiophlebia superstes, the sole
representative of Anisozygoptera in our study, was specified to be involved in a
reticulation with two other (unspecified) lineages. Blue lines indicate the
reticulation event and are labeled with PhyloNet’s estimate of
); all the remaining cases were
those where any discordance was inferred to be due to ILS alone. c) Normalized genetic
divergence between sister taxa (shown in triplets above the panel) averaged across all
BUSCO gene trees supporting each topology involving three lineages representing
Anisoptera, Anisozygoptera, and Zygoptera. d) Phylogenetic network estimated from a
set of ML gene trees using maximum likelihood. Epiophlebia superstes, the sole
representative of Anisozygoptera in our study, was specified to be involved in a
reticulation with two other (unspecified) lineages. Blue lines indicate the
reticulation event and are labeled with PhyloNet’s estimate of
 . The number above the network
indicates the log-likelihood score.
. The number above the network
indicates the log-likelihood score.
Additionally, we used a gene tree branch length-based approach, QuIBL (Edelman et al. 2019), which also detected multiple instances of introgression within the entire order (Supplementary Fig. S9 available on Dryad). We note that particularly for introgression involving Anisozygoptera we observed a larger fraction of triplets suggestive of introgression than for any other scenario tested.
Phylogenetic Network Analyses Support Anisozygoptera-Zygoptera Introgression
As an alternative approach to identify the lineage experiencing gene flow with
Anisozygoptera, we performed phylogenetic network inference in PhyloNet (Than et al. 2008; Wen
et al. 2018). For this analysis, we specified that Anisozygoptera was involved in
a reticulation event—the only such event occurring on the tree—and inferred for the other
two nodes that were most likely to be involved in the reticulation as well as the value of
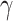 , the fraction of Anisozygoptera’s
genetic material derived from this reticulation. The full maximum likelihood (Fig. 4d) and maximum pseudolikelihood (Supplementary Fig.
S10 available on Dryad) approaches recovered topologically similar networks with
comparable values of
, the fraction of Anisozygoptera’s
genetic material derived from this reticulation. The full maximum likelihood (Fig. 4d) and maximum pseudolikelihood (Supplementary Fig.
S10 available on Dryad) approaches recovered topologically similar networks with
comparable values of 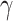 . Pseudolikelihood analysis suggests
a reticulation event between Aeshnidae (a family within Anisoptera) and Zygoptera with 38%
of genetic material coming from Zygoptera (Supplementary Fig. S10 available on Dryad),
whereas full likelihood infers reticulation between Anisoptera and Zygoptera suborders 33%
(Fig. 4d) of genetic material from Zygoptera.
Overall, we note that the full likelihood approach returned networks with higher
log-likelihood scores. This observation is most likely due to the fact that we performed
full likelihood analysis in an exhaustive manner (Supplementary Table S4 available on
Dryad) testing every possible reticulation event within a focal clade topology congruent
with the species tree (Fig. 1), whereas
pseudolikelihood analysis used the hill-climbing algorithm to search the full network
space but is not guaranteed to retrieve the most optimal solution. Moreover, differences
in objective function within likelihood and pseudolikelihood frameworks could also lead to
distinct network topologies and
. Pseudolikelihood analysis suggests
a reticulation event between Aeshnidae (a family within Anisoptera) and Zygoptera with 38%
of genetic material coming from Zygoptera (Supplementary Fig. S10 available on Dryad),
whereas full likelihood infers reticulation between Anisoptera and Zygoptera suborders 33%
(Fig. 4d) of genetic material from Zygoptera.
Overall, we note that the full likelihood approach returned networks with higher
log-likelihood scores. This observation is most likely due to the fact that we performed
full likelihood analysis in an exhaustive manner (Supplementary Table S4 available on
Dryad) testing every possible reticulation event within a focal clade topology congruent
with the species tree (Fig. 1), whereas
pseudolikelihood analysis used the hill-climbing algorithm to search the full network
space but is not guaranteed to retrieve the most optimal solution. Moreover, differences
in objective function within likelihood and pseudolikelihood frameworks could also lead to
distinct network topologies and 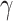 estimates.
estimates.
Discussion
Using a comprehensive multi-locus transcriptomic data set, we reconstructed a
fossil-calibrated deep evolutionary history of dragonflies and damselflies (Odonata; Fig. 1). Although our phylogenetic analyses resolve many
major radiations within the order with high confidence, we note that a strictly bifurcating
phylogeny could be positively misleading (i.e. asserting erroneous relationships with high
support) or provide a poor fit for genomic data undergoing biological processes such as ILS
and introgression, respectively. Thus, the phylogenies presented in this paper should be
interpreted with a degree of caution. Untangling patterns of phylogenetic discordance within
our data set revealed multiple gene flow events that have been impacted the course of
Odonata evolution for the past 200 myr. We examined the agreement across site- and gene
tree-based methods for detecting introgression (i.e., HyDe/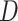 , and
, and
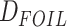 and the
and the 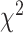 test/BLT; Fig. 5 and Supplementary Fig. S11 available
on Dryad). Overall, all methods individually were able to identify abundant introgression
within the entire Odonata order with strong agreement. We found considerable overlap across
methods in their support of introgression within Epiprocta (scenarios 2 and 3), gene flow
involving Anisozygoptera (scenario 1), and intra-superfamilial introgression within
Libelluloidea. Within Zygoptera, our methods showed strong overlap in identifying signatures
of introgression for Calopterygoidea only. Notably, our
test/BLT; Fig. 5 and Supplementary Fig. S11 available
on Dryad). Overall, all methods individually were able to identify abundant introgression
within the entire Odonata order with strong agreement. We found considerable overlap across
methods in their support of introgression within Epiprocta (scenarios 2 and 3), gene flow
involving Anisozygoptera (scenario 1), and intra-superfamilial introgression within
Libelluloidea. Within Zygoptera, our methods showed strong overlap in identifying signatures
of introgression for Calopterygoidea only. Notably, our 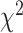 test/BLT produced very few predictions that were not in agreement with HyDe,
test/BLT produced very few predictions that were not in agreement with HyDe,
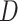 , and/or
, and/or 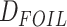 across different comparisons. Our deep-time
introgression analyses suggest that ancestral introgression events may leave genetic
footprints that are preserved in the genomes and observed in the phenotypes of contemporary
species. Below, we discuss the implications of our phylogenetic results in the context of
previous studies in Odonata, before turning to the importance and challenges associated with
the detection and interpretation of signatures of introgression within phylogenomic studies
in light of our findings.
across different comparisons. Our deep-time
introgression analyses suggest that ancestral introgression events may leave genetic
footprints that are preserved in the genomes and observed in the phenotypes of contemporary
species. Below, we discuss the implications of our phylogenetic results in the context of
previous studies in Odonata, before turning to the importance and challenges associated with
the detection and interpretation of signatures of introgression within phylogenomic studies
in light of our findings.
Figure 5.

Overlap between putatively introgressed species pairs inferred by
HyDe/ ,
,  , and
BLT/
, and
BLT/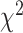 test. The tree shows tested
scenarios of deep (numbered red arrows) introgression for Anisoptera, Anisozygoptera,
and Zygoptera. The numbers within sets represent the number of unique introgressing
species pairs identified by a corresponding method. Significance of an overlap between
all methods (intersection of all sets) for each scenario was determined by the exact
multiset interactions test. Significant
test. The tree shows tested
scenarios of deep (numbered red arrows) introgression for Anisoptera, Anisozygoptera,
and Zygoptera. The numbers within sets represent the number of unique introgressing
species pairs identified by a corresponding method. Significance of an overlap between
all methods (intersection of all sets) for each scenario was determined by the exact
multiset interactions test. Significant  -values are indicated
in red. Note that due to the limitations of
-values are indicated
in red. Note that due to the limitations of 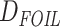 ,
introgression could not be tested for scenarios 7 and 8 using this method.
,
introgression could not be tested for scenarios 7 and 8 using this method.
Major Radiations on Odonata Phylogeny
Several competing hypotheses of evolutionary relationships within Odonata have been
proposed by multiple authors regarding various taxonomic levels (Supplementary Fig. S12a
available on Dryad; Trueman 1996; Misof et al. 2001; Saux et al. 2003; Hasegawa and Kasuya
2006; Ware et al. 2007; Bybee et al. 2008; Carle et al. 2008, 2015; Dumont et al. 2010; Blanke Greve et al. 2013; Dijkstra, Kalkman et
al. 2014; Suvorov et al. 2017). Our
phylogenetic analyses recovered Epiprocta (Anisoptera Anisozygoptera) and Zygoptera as monophyletic
with high support (Supplementary Fig. S1a available on Dryad) agreeing with other recent
studies (Bybee et al. 2008; Carle et al. 2008; Dumont et al.
2010; Suvorov et al. 2017). Our estimated
superfamilial relationships within Zygoptera support hypothesis of Dijkstra, Kalkman et
al. (2014) recovering monophyly of Lestoidea, Platystictoidea, Coenagrionoidea, and
Calopterygoidea (Supplementary Fig. S12b available on Dryad) with high support
(Supplementary Fig. S1a available on Dryad). Inferred higher-level phylogenetic
classification of anisopteran families was highly congruent with (Carle et al. 2015) and well-supported (Supplementary Fig. S1a available
on Dryad) with the exception of Gomphidae and Petaluridae radiations. The phylogenetic
position of Gomphidae and Petaluridae, both with respect to each other and the remaining
anisopteran families, has long been difficult to resolve (Supplementary Fig. S12c
available on Dryad). The most recent analyses of major anisopteran lineages, using several
molecular markers (Carle et al. 2015), suggest
Gomphidae and Petaluridae as a monophyletic group, but without strong branch support.
Anisozygoptera) and Zygoptera as monophyletic
with high support (Supplementary Fig. S1a available on Dryad) agreeing with other recent
studies (Bybee et al. 2008; Carle et al. 2008; Dumont et al.
2010; Suvorov et al. 2017). Our estimated
superfamilial relationships within Zygoptera support hypothesis of Dijkstra, Kalkman et
al. (2014) recovering monophyly of Lestoidea, Platystictoidea, Coenagrionoidea, and
Calopterygoidea (Supplementary Fig. S12b available on Dryad) with high support
(Supplementary Fig. S1a available on Dryad). Inferred higher-level phylogenetic
classification of anisopteran families was highly congruent with (Carle et al. 2015) and well-supported (Supplementary Fig. S1a available
on Dryad) with the exception of Gomphidae and Petaluridae radiations. The phylogenetic
position of Gomphidae and Petaluridae, both with respect to each other and the remaining
anisopteran families, has long been difficult to resolve (Supplementary Fig. S12c
available on Dryad). The most recent analyses of major anisopteran lineages, using several
molecular markers (Carle et al. 2015), suggest
Gomphidae and Petaluridae as a monophyletic group, but without strong branch support.
The multispecies coalescent model (MSC) provides a probabilistic framework for estimation
of species trees that accounts for genealogical discord occurring as a result of ILS.
Close scrutinization of Gomphidae and Petaluridae relationships inferred under
concatenation and ASTRAL (a supertree method that is statistically consistent under MSC)
showed that the majority of the supermatrix analyses strongly support a sister
relationship between the two families (Supplementary Fig. S1a available on Dryad);
however, almost all the ASTRAL species tree analyses reject such a relationship with high
confidence (Supplementary Fig. S1a available on Dryad). In the presence of ILS
concatenation methods can be statistically inconsistent (Roch and Steel 2014) leading to an erroneous species tree topology with
unreasonably high support (Kubatko and Degnan
2007). Thus, inconsistency in the recovery of a sister group relationship between
Gomphidae and Petaluridae between concatenation and ASTRAL could be a result of elevated
levels of ILS between the families. Furthermore, gene tree-species tree conflict can occur
in the presence of nonsister species gene flow (Leaché et
al. 2014), which violates assumptions of the MSC by skewing frequency
distributions of incongruent gene tree topologies thereby making the MSC framework
inconsistent for species tree estimation (Eckert and
Carstens 2008; Solís-Lemus et al. 2016),
although we note that studies of this phenomenon have examined gene flow at shallower
phylogenetic timescales than those investigated here. We found that the Gomphidae and
Petaluridae lineages exhibited inter-superfamilial introgression events (scenario 3, Fig. 5) involving the Cordulegastroidea
 Libelluloidea clade. Additionally, the
familial relationships within Calopterygoidea significantly varied between estimated
phylogenies using either ASTRAL or concatenation methods (Supplementary Fig. S1a available
on Dryad). For example, within clade C, monophyly of the Calopterygoidae and
Chlorocyphidae families was suggested by ASTRAL trees but rejected by the majority of ML
phylogenies estimated from the supermatrices (Fig. 1
and Supplementary Fig. S1a available on Dryad). Interestingly, for this superfamily
further analyses similarly yielded abundant instances of interfamilial introgression
(Fig. 5). Together, these results may support the
proposition that purely bifurcating species topologies inferred under concatenation/MSC
methods in the presence of postspeciation introgression on recent as well deep temporal
scales may inadequately model taxonomic relationships (Fontaine et al. 2015; McVay et al. 2017;
Zhang et al. 2021). To alleviate this problem, a
unifying multispecies coalescent network model, implemented in the program PhyloNet (Yu et al. 2011), has been developed to describe
evolutionary history via species networks where reticulation nodes correspond to instances
of introgression. Unfortunately, we were not able to infer a full species network topology
using PhyloNet because the taxon and gene sampling presented in our study would render
this analysis unfeasible even when using the more tractable pseudolikelihood
implementation (Yu and Nakhleh 2015). However, we
were able to use PhyloNet to test patterns of reticulate evolution involving
Anisozygoptera (see below).
Libelluloidea clade. Additionally, the
familial relationships within Calopterygoidea significantly varied between estimated
phylogenies using either ASTRAL or concatenation methods (Supplementary Fig. S1a available
on Dryad). For example, within clade C, monophyly of the Calopterygoidae and
Chlorocyphidae families was suggested by ASTRAL trees but rejected by the majority of ML
phylogenies estimated from the supermatrices (Fig. 1
and Supplementary Fig. S1a available on Dryad). Interestingly, for this superfamily
further analyses similarly yielded abundant instances of interfamilial introgression
(Fig. 5). Together, these results may support the
proposition that purely bifurcating species topologies inferred under concatenation/MSC
methods in the presence of postspeciation introgression on recent as well deep temporal
scales may inadequately model taxonomic relationships (Fontaine et al. 2015; McVay et al. 2017;
Zhang et al. 2021). To alleviate this problem, a
unifying multispecies coalescent network model, implemented in the program PhyloNet (Yu et al. 2011), has been developed to describe
evolutionary history via species networks where reticulation nodes correspond to instances
of introgression. Unfortunately, we were not able to infer a full species network topology
using PhyloNet because the taxon and gene sampling presented in our study would render
this analysis unfeasible even when using the more tractable pseudolikelihood
implementation (Yu and Nakhleh 2015). However, we
were able to use PhyloNet to test patterns of reticulate evolution involving
Anisozygoptera (see below).
We also note that discordance caused by ILS, if not accounted for, may lead to inaccurate inference of substitution rates (Mendes and Hahn 2016) and thus overestimation of divergence times using relaxed clock models (Ogilvie et al. 2016). Moreover, a failure to incorporate introgression effects into a probabilistic framework of a time calibration method will result in underestimated divergence times (Leaché et al. 2014). Several recent fully Bayesian approaches have been proposed to perform divergence time estimation using the MSC model alone (Heled and Drummond 2010; Ogilvie et al. 2017) or together with introgression (Jones 2019), however, they would not be scalable to our data set.
Widespread Introgression within Odonata
Introgression among Odonata has been previously thought to be uncommon (Tennessen 1982; Lowe
et al. 2008) as a result of probable reproductive isolation mechanisms such as
ethological barriers, phenotypic divergence (e.g., variable morphology of genitalia; Hosken and Stockley 2004; Barnard et al. 2017) and habitat and temporal isolation. Most of the
introgression and hybridization research in Odonata has been done within the zygopteran
Coenagrionidae family and especially between populations of genus Ishnura
focusing on mechanisms of reproductive isolation (e.g. Sanchez-Guillen et al. 2011; Sanchez-Guillen et
al. 2014). These studies established “hybridization thresholds” for these closely
related species and showed positive correlation between isolation and genetic divergence.
Furthermore, rapid karyotype evolution can also contribute to postmating isolation (Lai et al. 2005); however, recent cytogenetic studies
across Odonata phylogeny indicate very stable chromosome number with most prevalent
karyotype of 2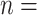 25 (Kuznetsova and Golub 2020). Additionally, in recent years, there has been a
growing body of evidence for introgression throughout evolutionary histories of various
groups. This includes more recent hybridization events observed in
Heliconius butterflies (Edelman et al.
2019), cats (Li et al. 2016), cichlid
fishes (Svardal et al. 2020) as well as deeper
ancestral introgression in primates (Vanderpool et al.
2020), Drosophila (Suvorov et
al. 2021), and vascular plants (Pease et al.
2018).
25 (Kuznetsova and Golub 2020). Additionally, in recent years, there has been a
growing body of evidence for introgression throughout evolutionary histories of various
groups. This includes more recent hybridization events observed in
Heliconius butterflies (Edelman et al.
2019), cats (Li et al. 2016), cichlid
fishes (Svardal et al. 2020) as well as deeper
ancestral introgression in primates (Vanderpool et al.
2020), Drosophila (Suvorov et
al. 2021), and vascular plants (Pease et al.
2018).
The extent of gene flow and maintenance of introgressed variation together determine the
fraction of introgressed material, or 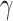 , in a focal
genome (Martin and Jiggins 2017). However, with
increased divergence time the fixation or loss of the ancestrally introduced genetic
material will primarily depend on the strength and direction of selection (Norris et al. 2015; Oziolor et al. 2019; Petr et al. 2019).
Here, our taxon sampling allowed to test ultra-deep ancestral introgression scenarios
where the MRCA of tested taxa can be traced as far back as to the Triassic period (~251 to
~201 Ma). We argue that for the cases where we infer that a large amount of introgressed
genetic material was preserved (e.g. > 25% in Anisozygoptera) the ancestral effective
migration rates (Martin and Jiggins 2017) were high
(similar conclusions can be made for Aeshnoidea (Aeshnidae), Calopterygoidea and within
Libelluloidea and Lestoidea; Fig. 3a); whereas for
the remaining taxa with lower or nonsignificant deviations of
, in a focal
genome (Martin and Jiggins 2017). However, with
increased divergence time the fixation or loss of the ancestrally introduced genetic
material will primarily depend on the strength and direction of selection (Norris et al. 2015; Oziolor et al. 2019; Petr et al. 2019).
Here, our taxon sampling allowed to test ultra-deep ancestral introgression scenarios
where the MRCA of tested taxa can be traced as far back as to the Triassic period (~251 to
~201 Ma). We argue that for the cases where we infer that a large amount of introgressed
genetic material was preserved (e.g. > 25% in Anisozygoptera) the ancestral effective
migration rates (Martin and Jiggins 2017) were high
(similar conclusions can be made for Aeshnoidea (Aeshnidae), Calopterygoidea and within
Libelluloidea and Lestoidea; Fig. 3a); whereas for
the remaining taxa with lower or nonsignificant deviations of 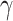 (Fig. 3a), the rates of introgression may have been
substantially lower or its signatures were purged from the genome by selection.
(Fig. 3a), the rates of introgression may have been
substantially lower or its signatures were purged from the genome by selection.
We found compelling evidence for patterns of ancestral introgression (Fig. 5) among distantly related odonate taxa. Our conservative analysis of agreement between different introgression tests shows the presence of gene flow within the entire order (Fig. 5 and Supplementary Fig. S11 available on Dryad). This observation may be explained by reduced sexual selection pressures in the early stages of odonate evolution that may have inhibited rapid genital divergence (Eberhard 2004), which is probably a primary source of reproductive isolation in Odonata (Cordero Rivera et al. 2004).
Phenotypic Consequences of Deep Time Introgression
Perhaps the most compelling evidence that we uncovered in this study was for deep
ancestral introgression between the Zygoptera and Anisozygoptera suborders, which based on
the fossil record most likely became genetically isolated after the Lower Jurassic (Carle 2012). Species of Anisozygoptera exhibit
anatomical characteristics of both Anisoptera and Zygoptera suborders. Some general
features of Anisozygoptera that relate them to Zygoptera include dorsal folding of wings
during perching in adults, characteristic anatomy of proventriculus (a part of digestive
system that is responsible for grinding of food particles), and absence of specific muscle
groups in the larval rectal gills; whereas abdominal tergite shape, rear wing geometry and
larval structures are similar to Anisoptera (Asahina et al.
1954). More recent studies also revealed that Anisozygoptera ovipositor
morphology shares similarity with Zygoptera (Matushkina
2008); muscle composition of the head resemble characteristics of both Anisoptera
and Zygoptera (Blanke et al. 2013); thoracic
musculature of Anisozygoptera larva exhibit similarity between Anisoptera and Zygoptera
(Busse et al. 2015). Thus, Anisozygoptera
represent a morphological and behavioral “intermediate” (Liu et al. 2011), which is supported by our findings where we recovered strong
evidence of introgression between Zygoptera and Anisozygoptera (Figs. 2–4). Moreover,
according to the  method the Anisozygoptera was
inferred to be a recipient taxon from a zygopteran donor, though we cannot rule out a
lesser degree of gene flow in the opposite direction as well. In fact, such a phenotypic
“intermediate” can occur as a result of homoploid hybrid speciation (Elgvin et al. 2017), which generates recombinant (mosaic) genotypes
from two parental genomes while preserving their ploidy. For example, empirical studies
have suggested that sex chromosome mosaicism in hybrid tiger swallowtail butterflies was
linked to phenotypic mosaicism (e.g. female dimorphism and duration of a life cycle; Kunte et al. 2011), and that hybrid speciation is the
cause of an intermediate plumage phenotype in sparrows (Hermansen et al. 2011). A significant fraction of genetic material from both
parental genomes is one of the main conditions required to establish hybrid speciation
(Schumer et al. 2014). Strikingly, we found that
the average HyDe probability parameter
method the Anisozygoptera was
inferred to be a recipient taxon from a zygopteran donor, though we cannot rule out a
lesser degree of gene flow in the opposite direction as well. In fact, such a phenotypic
“intermediate” can occur as a result of homoploid hybrid speciation (Elgvin et al. 2017), which generates recombinant (mosaic) genotypes
from two parental genomes while preserving their ploidy. For example, empirical studies
have suggested that sex chromosome mosaicism in hybrid tiger swallowtail butterflies was
linked to phenotypic mosaicism (e.g. female dimorphism and duration of a life cycle; Kunte et al. 2011), and that hybrid speciation is the
cause of an intermediate plumage phenotype in sparrows (Hermansen et al. 2011). A significant fraction of genetic material from both
parental genomes is one of the main conditions required to establish hybrid speciation
(Schumer et al. 2014). Strikingly, we found that
the average HyDe probability parameter 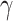 ~0.27 (Fig. 3a) inferred from multiple
quartets is very similar to PhyloNet’s inheritance probability
~0.27 (Fig. 3a) inferred from multiple
quartets is very similar to PhyloNet’s inheritance probability 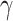 ~0.33
~0.33 This may suggest that a sizeable fraction
of the Anisozygoptera genome descends from zygopteran lineages. However, we note that
genetic mosaicism and intermediate traits can be also acquired via postspeciation gene
flow (vonHoldt et al. 2011; Bonfante et al. 2021). Thus, to fully test the Anisozygoptera hybrid
speciation hypothesis the other two conditions need to be examined: 1) the presence of
reproductive barriers between hybrid and parental lineages and 2) the establishment of
reproductive isolation was caused by hybridization events (Schumer et al. 2014). It is impossible to empirically test these
conditions for gene flow events as ancient as the one we identified for
Anisozygoptera.
This may suggest that a sizeable fraction
of the Anisozygoptera genome descends from zygopteran lineages. However, we note that
genetic mosaicism and intermediate traits can be also acquired via postspeciation gene
flow (vonHoldt et al. 2011; Bonfante et al. 2021). Thus, to fully test the Anisozygoptera hybrid
speciation hypothesis the other two conditions need to be examined: 1) the presence of
reproductive barriers between hybrid and parental lineages and 2) the establishment of
reproductive isolation was caused by hybridization events (Schumer et al. 2014). It is impossible to empirically test these
conditions for gene flow events as ancient as the one we identified for
Anisozygoptera.
Taken together, our observations strongly suggest a xenoplasious origin (Wang Y. et al. 2020) of Anisozygopteran traits (i.e. traits introduced to a recipient taxon via introgression) that are shared with Zygoptera. However, we do not reject the possibility that some trait hemiplasy may have resulted from ILS (Guerrero and Hahn 2018). Based on the gathered evidence for introgression, we suggest that ancestral lineages that gave rise to modern-day Anisozygoptera and Zygoptera experienced introgression in their past evolutionary history.
According to the hybrid swarm hypothesis (Seehausen 2004), introgression can trigger a rapid cladogenesis resulting in the establishment of ecologically different species with niche-specific adaptive phenotypes. Some of the notable examples can include adaptive radiations caused by ancestral introgression in cichlid fishes (Meier et al. 2017), and intricate interspecific introgression patterns in big cats that could be linked to rapid diversification of modern-day species in that lineage (Figueiro et al. 2017). Interestingly, the large fraction of introgressed material estimated within the Anisozygopteran lineage does not appear to have facilitated any adaptive radiation. This proposition is supported by an extremely low species diversification within the suborder (just three extant species) and also by the lack of fossil record for Anisozygoptera.
Conclusions
A rapidly growing body of empirical evidence strongly indicates that introgression is a widespread phenomenon observed not only in plants but also in the animal kingdom. Over the past decade, this biological process has received tremendous attention from the phylogenetics community, as it fundamentally changes how we view and reconstruct evolutionary histories of different organisms and may even redefine our understanding of species concepts (Wang X. et al. 2020). Introgression is an important source of novel variation that can lead to speciation, trigger rapid species diversifications, facilitate adaptation to novel environments (Nolte and Tautz 2010) as well as affect the course of species boundary establishment (Harrison and Larson 2014). From a practical perspective, introgression is one of the key factors that need to be considered during the development of biodiversity protection and conservation programs (Quilodrán et al. 2020). Furthermore, a failure to accurately detect introgression scenarios may also lead to ineffective domestication and breeding programs (Dempewolf et al. 2017; Glémin et al. 2019; Janzen et al. 2019).
Here, we investigated a unique and ancient insect lineage, Odonata, and provided the first insight into global patterns of introgression that were pervasive throughout their evolutionary history. Moreover, we note that signatures of introgression were most likely underestimated as majority of phylogenomic methods (including those used in the current study) are unable to detect gene flow between sister lineages (Hibbins and Hahn 2021). Our findings further exemplify the evidence that postspeciation gene flow can be a fairly common process occurring in various taxa, including the most diverse group of animals, that is insects. The abundance of this biological phenomenon across the Tree of Life creates patterns of reticulate evolution which make the tree model itself ill-suited for explaining historic relationships between species (Doolittle and Bapteste 2007). It is possible that ancestral sequence reconstruction methods may also be affected by introgression as they typically assume character evolution along a single species tree (Mendes and Hahn 2016). Additionally, introgression poses new challenges for phylogenetic time calibration by biasing age estimates (Leaché et al. 2014). The fraction of the introgressed variation from a donor to a recipient taxon can vary significantly (Runemark et al. 2019), including cases where the most of the ancestry originates from donor genomes (Fontaine et al. 2015). In our case, we show that in Odonata introgressed genetic material can account for a large fraction of the genome (around ~30% in Anisozygoptera) and persist for millions of years following the gene flow event(s). This pattern is indicative of a high rate of gene flow and potentially of positive selection in favor of some hybrid genotypes.
We provide a roadmap of analyses to help identify introgression at both shallow and deep phylogenetic scales that is practical, timely, and much needed. Our research highlights the importance of development of tractable models and methods that scale to modern phylogenomic data sets derived from high-throughput molecular data. Moreover, we argue there is an urgent need to develop a theoretical framework that unifies various sources of phylogenetic discordance for more accurate and comprehensive inferences in phylogenetics (Charles-Elie et al. 2020).
Acknowledgments
The authors thank Gavin Martin, Nathan Lord, and Camilla Sharkey for the generation of sequence data. They also thank the DNA Sequencing Center and Fulton Supercomputer Lab (BYU) for assistance. Additionally, they thank Aaron Comeault for his invaluable comments on the manuscript. They are grateful for thoughtful comments and suggestions from the Editors and two anonymous Reviewers that helped improve this manuscript.
Contributor Information
Anton Suvorov, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Celine Scornavacca, Institut des Sciences de l’Evolution Universiteì de Montpellier, CNRS, IRD, EPHE CC 064, Place Eugène Bataillon, 34095 Montpellier Cedex 05, France.
M Stanley Fujimoto, Department of Computer Science, Brigham Young University, Provo, UT 84602, USA.
Paul Bodily, Department of Computer Science, Idaho State University, Pocatello, ID 83209, USA.
Mark Clement, Department of Computer Science, Brigham Young University, Provo, UT 84602, USA.
Keith A Crandall, Computational Biology Institute, Department of Biostatistics and Bioinformatics, Milken Institute School of Public Health, George Washington University, Washington, DC 20052, USA.
Michael F Whiting, Department of Biology, Brigham Young University, Provo, UT 84602, USA; M.L. Bean Museum, Brigham Young University, Provo, UT 84602, USA.
Daniel R Schrider, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Seth M Bybee, Department of Biology, Brigham Young University, Provo, UT 84602, USA; M.L. Bean Museum, Brigham Young University, Provo, UT 84602, USA.
Data Availability
All raw RNA-seq read libraries generated for this study have been uploaded to the Short Read Archive (SRA) (SRR12086708-SRR12086765). All assembled transcriptomes, alignments, inferred gene, and species trees, fossil calibrated phylogenies (including paleontological record assessed from PaleoDB used to plot the fossil distribution in Fig. 1), PhyloNet results, QS results, and introgression results are available on figshare (https://doi.org/10.6084/m9.figshare.12518660). Custom scripts that were used to create various input files as well as the analysis pipeline are available on GitHub https://github.com/antonysuv/odo_introgression.
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.j3tx95xdp
Funding
This work was supported by the National Science Foundation (NSF) grant DEB-1265714 to S.M.B. and the National Institutes of Health (NIH) grants R00HG008696 and R35GM138286 to D.R.S. C.S. was funded by grant ANR-19-CE45-0012 from French Agence Nationale de la Recherche.
Author Contributions
A.S., C.S., D.R.S., and S.M.B. conceived the research. A.S., C.S., M.S.F., and P.B. analyzed the data. M.C., K.A.C., M.F.W., and D.R.S. supervised the project. A.S., C.S., D.R.S., and S.M.B. wrote the manuscript.
References
- Aberer A.J., Kobert K., Stamatakis A. 2014. ExaBayes: massively parallel bayesian tree inference for the whole-genome era. Mol. Biol. Evol. 31:2553–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina S., Asahina S., Nihon Gakujutsu S. 1954. A morphological study of a relic dragonfly Epiophlebia superstes selys: (Odonata, Anisozygoptera). Jpn. Soc. Promot. Sci. [Google Scholar]
- Barnard A.A., Fincke O.M., McPeek M.A., Masly J.P. 2017. Mechanical and tactile incompatibilities cause reproductive isolation between two young damselfly species. Evolution 71:2410–2427. [DOI] [PubMed] [Google Scholar]
- Blanke A., Beckmann F., Misof B. 2013. The head anatomy of Epiophlebia superstes (Odonata: Epiophlebiidae). Org. Divers. Evol. 13:55–66. [Google Scholar]
- Blanke A., Greve C., Mokso R., Beckmann F., Misof B. 2013. An updated phylogeny of Anisoptera including formal convergence analysis of morphological characters. Syst. Entomol. 38:474–490. [Google Scholar]
- Blischak P.D., Chifman J., Wolfe A.D., Kubatko L.S. 2018. HyDe: a python package for genome-scale hybridization detection. Syst. Biol. 67:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfante B., Faux P., Navarro N., Mendoza-Revilla J., Dubied M., Montillot C., Wentworth E., Poloni L., Varón-González C., Jones P., Xiong Z., Fuentes-Guajardo M., Palmal S., Chacón-Duque J.C., Hurtado M., Villegas V., Granja V., Jaramillo C., Arias W., Barquera R., Everardo-Martínez P., Sánchez-Quinto M., Gómez-Valdés J., Villamil-Ramírez H., Cerqueira C.C.S. de Hünemeier T., Ramallo V., Liu F., Weinberg S.M., Shaffer J.R., Stergiakouli E., Howe L.J., Hysi P.G., Spector T.D., Gonzalez-José R., Schüler-Faccini L., Bortolini M.-C., Acuña-Alonzo V., Canizales-Quinteros S., Gallo C., Poletti G., Bedoya G., Rothhammer F., Thauvin-Robinet C., Faivre L., Costedoat C., Balding D., Cox T., Kayser M., Duplomb L., Yalcin B., Cotney J., Adhikari K., Ruiz-Linares A. 2021. A GWAS in Latin Americans identifies novel face shape loci, implicating VPS13B and a Denisovan introgressed region in facial variation. Sci. Adv. 7:eabc6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton T., Anderson C.L., Jacquet D., Lundqvist S., Bremer K. 2007. Estimating divergence times in large phylogenetic trees. Syst. Biol. 56:741–752. [DOI] [PubMed] [Google Scholar]
- Brooks S.P., Gelman A. 1998. General methods for monitoring convergence of iterative simulations. J. Comput. Graphical Stat. 7:434–455. [Google Scholar]
- Buchfink B., Xie C., Huson D.H. 2015. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12:59–60. [DOI] [PubMed] [Google Scholar]
- Busse S., Genet C., Hornschemeyer T. 2013. Homologization of the flight musculature of zygoptera (insecta: odonata) and neoptera (insecta). PLoS One 8:e55787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse S., Helmker B., Hornschemeyer T. 2015. The thorax morphology of Epiophlebia (Insecta: Odonata) nymphs—including remarks on ontogenesis and evolution. Sci. Rep. 5:12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bybee S., Córdoba-Aguilar A., Duryea M.C., Futahashi R., Hansson B., Lorenzo-Carballa M.O., Schilder R., Stoks R., Suvorov A., Svensson E.I.. et al. 2016. Odonata (dragonflies and damselflies) as a bridge between ecology and evolutionary genomics. Front. Zool. 13:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bybee S.M., Ogden T.H., Branham M.A., Whiting M.F. 2008. Molecules, morphology and fossils: a comprehensive approach to odonate phylogeny and the evolution of the odonate wing. Cladistics 24:477–514. [DOI] [PubMed] [Google Scholar]
- Carle F. 2012. A new Epiophlebia (Odonata: Epiophlebioidea) from China with a review of epiophlebian taxonomy, life history, and biogeography. Arthropod Syst. Phylog. 70:75–83. [Google Scholar]
- Carle F.L., Kjer K.M., May M.L. 2008. Evolution of Odonata, with special reference to Coenagrionoidea (Zygoptera). Arthropod Syst. Phylog. 66:37–44. [Google Scholar]
- Carle F.L., Kjer K.M., May M.L. 2015. A molecular phylogeny and classification of Anisoptera (Odonata). Arthropod Syst. Phylog. 73:281–301. [Google Scholar]
- Charles-Elie R., Vincent B., Jean-Christophe G., Fabio P., Scornavacca C. 2020. On the inference of complex phylogenetic networks by Markov Chain Monte-Carlo. bioRxiv 2020.2010.2007.329425. [Google Scholar]
- Chauhan P., Hansson B., Kraaijeveld K., de Knijff P., Svensson E.I., Wellenreuther M. 2014. De novo transcriptome of Ischnura elegans provides insights into sensory biology, colour and vision genes. BMC Genomics 15:808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero Rivera A., Andres J.A., Cordoba-Aguilar A., Utzeri C. 2004. Postmating sexual selection: allopatric evolution of sperm competition mechanisms and genital morphology in calopterygid damselflies (Insecta: Odonata). Evolution 58:349–359. [DOI] [PubMed] [Google Scholar]
- Cordoba-Aguilar A. 2008. Dragonflies and damselflies: model organisms for ecological and evolutionary research. Oxford; New York: Oxford University Press. [Google Scholar]
- Degnan J.H., Rosenberg N.A. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Dempewolf H., Baute G., Anderson J., Kilian B., Smith C., Guarino L. 2017. Past and future use of wild relatives in crop breeding. Crop Sci. 57:1070–1082. [Google Scholar]
- Dijkstra K.D., Kalkman V.J., Dow R.A., Stokvis F.R., Van Tol J. 2014. Redefining the damselfly families: a comprehensive molecular phylogeny of Zygoptera (Odonata). Syst. Entomol. 39:68–96. [Google Scholar]
- Dijkstra K.D., Monaghan M.T., Pauls S.U. 2014. Freshwater biodiversity and aquatic insect diversification. Ann. Rev. Entomol. 59:143–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Bapteste E. 2007. Pattern pluralism and the Tree of Life hypothesis. Proc. Natl. Acad. Sci. USA 104:2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Reis M., Yang Z. 2011. Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol. 28:2161–2172. [DOI] [PubMed] [Google Scholar]
- Dumont H.J., Vierstraete A., Vanfleteren J.R. 2010. A molecular phylogeny of the Odonata (Insecta). Syst. Entomol. 35:6–18. [Google Scholar]
- Durand E.Y., Patterson N., Reich D., Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard W.G. 2004. Rapid divergent evolution of sexual morphology: comparative tests of antagonistic coevolution and traditional female choice. Evolution 58:1947–1970. [DOI] [PubMed] [Google Scholar]
- Eckert A.J., Carstens B.C. 2008. Does gene flow destroy phylogenetic signal? The performance of three methods for estimating species phylogenies in the presence of gene flow. Mol. Phylogenet. Evol. 49:832–842. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. 2011. Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman N.B., Frandsen P.B., Miyagi M., Clavijo B., Davey J., Dikow R.B., Garcia-Accinelli G., Van Belleghem S.M., Patterson N., Neafsey D.E.. et al. 2019. Genomic architecture and introgression shape a butterfly radiation. Science 366:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgvin T.O., Trier C.N., Tørresen O.K., Hagen I.J., Lien S., Nederbragt A.J., Ravinet M., Jensen H., Sætre G.-P. 2017. The genomic mosaicism of hybrid speciation. Sci. Adv. 3:e1602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiro H.V., Li G., Trindade F.J., Assis J., Pais F., Fernandes G., Santos S.H.D., Hughes G.M., Komissarov A., Antunes A.. et al. 2017. Genome-wide signatures of complex introgression and adaptive evolution in the big cats. Sci. Adv. 3:e1700299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine M.C., Pease J.B., Steele A., Waterhouse R.M., Neafsey D.E., Sharakhov I.V., Jiang X., Hall A.B., Catteruccia F., Kakani E.. et al. 2015. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347:1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S., Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M.S., Suvorov A., Jensen N.O., Clement M.J., Bybee S.M. 2016. Detecting false positive sequence homology: a machine learning approach. BMC Bioinformatics 17:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M.S., Suvorov A., Jensen N.O., Clement M.J., Snell Q., Bybee S.M. 2017. The OGCleaner: filtering false-positive homology clusters. Bioinformatics 33:125–127. [DOI] [PubMed] [Google Scholar]
- Gadagkar S.R., Rosenberg M.S., Kumar S. 2005. Inferring species phylogenies from multiple genes: concatenated sequence tree versus consensus gene tree. J. Exp. Zool. B Mol. Dev. Evol. 304:64–74. [DOI] [PubMed] [Google Scholar]
- Glémin S., Scornavacca C., Dainat J., Burgarella C., Viader V., Ardisson M., Sarah G., Santoni S., David J., Ranwez V. 2019. Pervasive hybridizations in the history of wheat relatives. Sci. Adv. 5:eaav9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy A., Excoffier L. 2020. Polygenic patterns of adaptive introgression in modern humans are mainly shaped by response to pathogens. Mol. Biol. Evol. 37:1420–1433. [DOI] [PubMed] [Google Scholar]
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q.. et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.. et al. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi D.A., Engel M.S. 2005. Evolution of the insects. Cambridge (UK); New York (NY): Cambridge University Press. [Google Scholar]
- Guerrero R.F., Hahn M.W. 2018. Quantifying the risk of hemiplasy in phylogenetic inference. Proc. Natl. Acad. Sci. USA. 115:12787–12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M.. et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc., 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M.W., Hibbins M.S. 2019. A three-sample test for introgression. Mol. Biol. Evol. 36:2878–2882. [DOI] [PubMed] [Google Scholar]
- Hallstrom B.M., Janke A. 2010. Mammalian evolution may not be strictly bifurcating. Mol. Biol. Evol. 27:2804–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R.G., Larson E.L. 2014. Hybridization, introgression, and the nature of species boundaries. J. Heredity 105:795–809. [DOI] [PubMed] [Google Scholar]
- Hasegawa E., Kasuya E. 2006. Phylogenetic analysis of the insect order Odonata using 28S and 16S rDNA sequences: a comparison between data sets with different evolutionary rates. Entomol. Sci. 9:55–66. [Google Scholar]
- Hayashi F., Dobata S., Futahashi R. 2005. Disturbed population genetics: suspected introgressive hybridization between two Mnais damselfly species (Odonata). Zool. Sci. 22:869–881. [DOI] [PubMed] [Google Scholar]
- Heled J., Drummond A.J. 2010. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmuth M., Wieseke N., Lechner M., Lenhof H.P., Middendorf M., Stadler P.F. 2015. Phylogenomics with paralogs. Proc. Natl. Acad. Sci. USA 112:2058–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermansen J.S., Sæther S.A., Elgvin TO, Borge T, Hjelle E, Sætre G-P. 2011. Hybrid speciation in sparrows I: phenotypic intermediacy, genetic admixture and barriers to gene flow. Mol. Ecol. 20:3812–3822. [DOI] [PubMed] [Google Scholar]
- Hibbins M., Hahn M., 2021. Phylogenomic approaches to detecting and characterizing introgression. EcoEvoRxiv doi: 10.32942/osf.io/uahd8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosken D.J., Stockley P. 2004. Sexual selection and genital evolution. Trends Ecol. Evol. 19:87–93. [DOI] [PubMed] [Google Scholar]
- Huerta-Sanchez E., Jin X., Asan, Bianba Z., Peter B.M., Vinckenbosch N., Liang Y., Yi X., He M., Somel M.. et al. 2014. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512:194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H., Klöpper T., Lockhart P.J., Steel M.A. 2005. Reconstruction of reticulate networks from gene trees. In: Miyano S., Mesirov J., Kasif S., Istrail S., Pevzner P.A., Waterman M., editors. Research in computational molecular biology. Berlin (Heidelberg): Springer Berlin Heidelberg. p. 233–249. [Google Scholar]
- Janzen G.M., Wang L., Hufford M.B. 2019. The extent of adaptive wild introgression in crops. New Phytol. 221:1279–1288. [DOI] [PubMed] [Google Scholar]
- Jones G.R. 2019. Divergence estimation in the presence of incomplete lineage sorting and migration. Syst. Biol. 68:19–31. [DOI] [PubMed] [Google Scholar]
- Káldy J., Mozsár A., Fazekas G., Farkas M., Fazekas D.L., Fazekas G.L., Goda K., Gyöngy Z., Kovács B., Semmens K., Bercsényi M., Molnár M., Patakiné Várkonyi E. 2020. Hybridization of Russian Sturgeon (Acipenser gueldenstaedtii, Brandt and Ratzeberg, 1833) and American Paddlefish (Polyodon spathula, Walbaum 1792) and evaluation of their progeny. Genes (Basel) 11:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Kim M.J.,
Jung K.S.,
Park N.S.,
Wan X.,
Kim K.-G.,
Jun J.,
Yoon T.J.,
Bae Y.J.,
Lee S.M.,
Kim
I. 2014.
Molecular phylogeny of the higher taxa of Odonata (Insecta) inferred from
COI, 16S rRNA, 28S rRNA, and EF1-
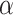 sequences. Entomol. Res.
44:65–79. [Google Scholar]
sequences. Entomol. Res.
44:65–79. [Google Scholar] - Kong S., Kubatko L.S. 2021. Comparative performance of popular methods for hybrid detection using genomic data. Syst. Biol. 70:891–907. [DOI] [PubMed] [Google Scholar]
- Kubatko L.S., Chifman J. 2019. An invariants-based method for efficient identification of hybrid species from large-scale genomic data. BMC Evol. Biol. 19:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatko L.S., Degnan J.H. 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56:17–24. [DOI] [PubMed] [Google Scholar]
- Kunte K., Shea C., Aardema M.L., Scriber J.M., Juenger T.E., Gilbert L.E., Kronforst M.R. 2011. Sex chromosome mosaicism and hybrid speciation among tiger swallowtail butterflies. PLoS Genet. 7:e1002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova V.G., Golub N.V. 2020. A checklist of chromosome numbers and a review of karyotype variation in Odonata of the world. Comp. Cytogenet. 14:501–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira C.C., Sepkoski J.J. Jr. 1993. Insect diversity in the fossil record. Science 261:310–315. [DOI] [PubMed] [Google Scholar]
- Lai Z., Nakazato T., Salmaso M., Burke J.M., Tang S., Knapp S.J., Rieseberg L.H. 2005. Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics 171:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakner C., van der Mark P., Huelsenbeck J.P., Larget B., Ronquist F. 2008. Efficiency of Markov chain Monte Carlo tree proposals in Bayesian phylogenetics. Syst. Biol. 57:86–103. [DOI] [PubMed] [Google Scholar]
- Landry C.R., Wittkopp P.J., Taubes C.H., Ranz J.M., Clark A.G., Hartl D.L. 2005. Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila. Genetics 171:1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R., Calcott B., Kainer D., Mayer C., Stamatakis A. 2014. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol. Biol. 14:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R., Frandsen P.B., Wright A.M., Senfeld T., Calcott B. 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34:772–773. [DOI] [PubMed] [Google Scholar]
- Lanfear R., Hua X., Warren D.L. 2017. Estimating the effective sample size of tree topologies from bayesian phylogenetic analyses. Genome Biol. Evol. 8:2319–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaché A.D., Harris R.B., Rannala B., Yang Z. 2014. The influence of gene flow on species tree estimation: a simulation study. Syst. Biol. 63:17–30. [DOI] [PubMed] [Google Scholar]
- Lemmon E.M., Lemmon A.R. 2010. Reinforcement in chorus frogs: lifetime fitness estimates including intrinsic natural selection and sexual selection against hybrids. Evolution 64:1748–1761. [DOI] [PubMed] [Google Scholar]
- Letsch H., Gottsberger B., Ware J.L. 2016. Not going with the flow: a comprehensive time-calibrated phylogeny of dragonflies (Anisoptera: Odonata: Insecta) provides evidence for the role of lentic habitats on diversification. Mol. Ecol. 25:1340–1353. [DOI] [PubMed] [Google Scholar]
- Li G., Davis B.W., Eizirik E., Murphy W.J. 2016. Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res. 26:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Stoeckert C.J. Jr., Roos D.S. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Yu L., Arnold M.L., Wu C.H., Wu S.F., Lu X., Zhang Y.P. 2011. Reticulate evolution: frequent introgressive hybridization among Chinese hares (genus lepus) revealed by analyses of multiple mitochondrial and nuclear DNA loci. BMC Evol. Biol. 11:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe C., Harvey I., Thompson D., Watts P. 2008. Strong genetic divergence indicates that congeneric damselflies Coenagrion puella and C. pulchellum (Odonata: Zygoptera: Coenagrionidae) do not hybridise. Hydrobiologia 605:55–63. [Google Scholar]
- Loytynoja A. 2014. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079:155–170. [DOI] [PubMed] [Google Scholar]
- Loytynoja A., Goldman N. 2008. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320:1632–1635. [DOI] [PubMed] [Google Scholar]
- Maddison W.P. 1997. Gene trees in species trees. Syst. Biol. 46:523–536. [Google Scholar]
- Mallet J. 2005. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20:229–237. [DOI] [PubMed] [Google Scholar]
- Mallet J., Besansky N., Hahn M.W. 2016. How reticulated are species? Bioessays 38:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.H., Davey J.W., Jiggins C.D. 2015. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol. 32:244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.H., Jiggins C.D. 2017. Interpreting the genomic landscape of introgression. Curr. Opin. Genet. Dev. 47:69–74. [DOI] [PubMed] [Google Scholar]
- Matushkina N.A. 2008. The ovipositor of the relic dragonfly Epiophlebia superstes: a morphological re-examination (Odonata: Epiophlebiidae). Int. J. Odonatol. 11:71–80. [Google Scholar]
- McVay J.D., Hipp A.L., Manos P.S. 2017. A genetic legacy of introgression confounds phylogeny and biogeography in oaks. Proc. R. Soc. B Biol. Sci. 284:20170300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J.I., Marques D.A., Mwaiko S., Wagner C.E., Excoffier L., Seehausen O. 2017. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 8:14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes F.K., Hahn M.W. 2016. Gene tree discordance causes apparent substitution rate variation. Syst. Biol. 65:711–721. [DOI] [PubMed] [Google Scholar]
- Meng C., Kubatko L.S. 2009. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theor. Popul. Biol. 75:35–45. [DOI] [PubMed] [Google Scholar]
- Minh B.Q., Nguyen M.A., von Haeseler A. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30:1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Nguyen N., Guo S., Wang L.S., Kim J., Warnow T. 2015. PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J. Comput. Biol. 22:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Reaz R., Bayzid M.S., Zimmermann T., Swenson M.S., Warnow T. 2014. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30:i541–i548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B., Liu S., Meusemann K., Peters R.S., Donath A., Mayer C., Frandsen P.B., Ware J., Flouri T., Beutel R.G.. et al. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science 346:763–767. [DOI] [PubMed] [Google Scholar]
- Misof B., Misof K. 2009. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 58:21–34. [DOI] [PubMed] [Google Scholar]
- Misof B., Rickert A.M., Buckley T.R., Fleck G., Sauer K.P. 2001. Phylogenetic signal and its decay in mitochondrial SSU and LSU rRNA gene fragments of Anisoptera. Mol. Biol. Evol. 18: 27–37. [DOI] [PubMed] [Google Scholar]
- Monetti L., Sánchez-Guillén R.A., Rivera A.C. 2002. Hybridization between Ischnura graellsii (Vander Linder) and I. elegans (Rambur) (Odonata: Coenagrionidae): are they different species? Biol. J. Linn. Soc. 76:225–235. [Google Scholar]
- Nel A., Bechly G., Martinez-Delclos X., Fleck G. 2001. A new family of Anisoptera from the Upper Jurassic of Karatau in Kazakhstan (Insecta: Odonata: Juragomphidae n. fam.). Stuttgart. Beitr. Naturkd. B (Geol. Palaeontol.), 314:1–9. [Google Scholar]
- Nguyen L.T., Schmidt H.A., von Haeseler A., Minh B.Q. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte A.W., Tautz D. 2010. Understanding the onset of hybrid speciation. Trends Genet. 26:54–58. [DOI] [PubMed] [Google Scholar]
- Norris L.C., Main B.J., Lee Y., Collier T.C., Fofana A., Cornel A.J., Lanzaro G.C. 2015. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc. Natl. Acad. Sci. USA 112:815–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden T.H., Whiting M.F. 2003. The problem with “the Paleoptera Problem:” sense and sensitivity. Cladistics 19:432–442. [DOI] [PubMed] [Google Scholar]
- Ogilvie H.A., Bouckaert R.R., Drummond A.J. 2017. StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol. Biol. Evol. 34:2101–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie H.A., Heled J., Xie D., Drummond A.J. 2016. Computational performance and statistical accuracy of *BEAST and comparisons with other methods. Syst. Biol. 65:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oziolor E.M., Reid N.M., Yair S., Lee K.M., Guberman VerPloeg S., Bruns P.C., Shaw J.R., Whitehead A., Matson C.W. 2019. Adaptive introgression enables evolutionary rescue from extreme environmental pollution. Science 364:455–457. [DOI] [PubMed] [Google Scholar]
- Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D. 2012. Ancient admixture in human history. Genetics 192:1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J.B., Brown J.W., Walker J.F., Hinchliff C.E., Smith S.A. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105:385–403. [DOI] [PubMed] [Google Scholar]
- Pease J.B., Hahn M.W. 2015. Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64:651–662. [DOI] [PubMed] [Google Scholar]
- Perry W.L., Lodge D.M., Feder J.L. 2002. Importance of hybridization between indigenous and nonindigenous freshwater species: an overlooked threat to North American biodiversity. Syst. Biol. 51:255–275. [DOI] [PubMed] [Google Scholar]
- Petr M., Paabo S., Kelso J., Vernot B. 2019. Limits of long-term selection against Neandertal introgression. Proc. Natl. Acad. Sci. USA 116:1639–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau H.K. 1991. Contributions of functional morphology to the phylogenetic systematics of Odonata. Adv. Odonatol. 5:109–141. [Google Scholar]
- Posada D., Crandall K.A. 2001. Intraspecific gene genealogies: trees grafting into networks. Trends Ecol. Evol. 16:37–45. [DOI] [PubMed] [Google Scholar]
- Puttick M.N. 2019. MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 35:5321–5322. [DOI] [PubMed] [Google Scholar]
- Quilodrán C.S., Montoya-Burgos J.I., Currat M. 2020. Harmonizing hybridization dissonance in conservation. Commun. Biol. 3:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. 2018. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67:901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehn A.C. 2003. Phylogenetic analysis of higher-level relationships of Odonata. Syst. Entomol. 28:181–240. [Google Scholar]
- Roch S., Steel M. 2014. Likelihood-based tree reconstruction on a concatenation of aligned sequence data sets can be statistically inconsistent. Theor. Popul. Biol. 100C:56–62. [DOI] [PubMed] [Google Scholar]
- Rohde R.A., Muller R.A. 2005. Cycles in fossil diversity. Nature 434:208–210. [DOI] [PubMed] [Google Scholar]
- Rothfels C.J., Johnson A.K., Hovenkamp P.H., Swofford D.L., Roskam H.C., Fraser-Jenkins C.R., Windham M.D., Pryer K.M. 2015. Natural hybridization between genera that diverged from each other approximately 60 million years ago. Am. Nat. 185:433–442. [DOI] [PubMed] [Google Scholar]
- Runemark A., Vallejo-Marin M., Meier J.I. 2019. Eukaryote hybrid genomes. PLoS Genet. 15:e1008404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppell G., Hilfert D. 1993. The flight of the relict dragonfly Epiophlebia superstes in comparison with that of the modern Odonata. Odonatologica 22:295–309. [Google Scholar]
- Sanchez-Guillen R.A., Cordoba-Aguilar A., Cordero-Rivera A., Wellenreuther M. 2014. Genetic divergence predicts reproductive isolation in damselflies. J. Evol. Biol. 27:76–87. [DOI] [PubMed] [Google Scholar]
- Sánchez-Guillén R.A., Van Gossum H., Cordero Rivera A. 2005. Hybridization and the inheritance of female colour polymorphism in two ischnurid damselflies (Odonata: Coenagrionidae). Biol. J. Linn. Soc. 85:471–481. [Google Scholar]
- Sanchez-Guillen R.A., Wellenreuther M., Cordero-Rivera A., Hansson B. 2011. Introgression and rapid species turnover in sympatric damselflies. BMC Evol. Biol. 11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saux C., Simon C., Spicer G.S. 2003. Phylogeny of the dragonfly and damselfly order odonata as inferred by mitochondrial 12S ribosomal RNA sequences. Ann. Entomol. Soc. Am. 96:693–699. [Google Scholar]
- Sayyari E., Mirarab S. 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M., Rosenthal G.G., Andolfatto P. 2014. How common is homoploid hybrid speciation? Evolution 68:1553–1560. [DOI] [PubMed] [Google Scholar]
- Seehausen O. 2004. Hybridization and adaptive radiation. Trends Ecol. Evol. 19:198–207. [DOI] [PubMed] [Google Scholar]
- Simao F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Solano E., Hardersen S., Audisio P., Amorosi V., Senczuk G., Antonini G. 2018. Asymmetric hybridization in Cordulegaster (Odonata: Cordulegastridae): secondary postglacial contact and the possible role of mechanical constraints. Ecol. Evol. 8:9657–9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís-Lemus C., Yang M., Ané C. 2016. Inconsistency of species tree methods under gene flow. Syst. Biol. 65:843–851. [DOI] [PubMed] [Google Scholar]
- Suvorov A., Jensen N.O., Sharkey C.R., Fujimoto M.S., Bodily P., Wightman H.M., Ogden T.H., Clement M.J., Bybee S.M. 2017. Opsins have evolved under the permanent heterozygote model: insights from phylotranscriptomics of Odonata. Mol. Ecol. 26:1306–1322. [DOI] [PubMed] [Google Scholar]
- Suvorov A., Kim B.Y., Wang J., Armstrong E.E., Peede D., D’Agostino E.R.R., Price D.K., Wadell P., Lang M., Courtier-Orgogozo V.. et al. 2021. Widespread introgression across a phylogeny of 155 “Drosophila” genomes. bioRxiv: 2020.2012.2014.422758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svardal H., Quah F.X., Malinsky M., Ngatunga B.P., Miska E.A., Salzburger W., Genner M.J., Turner G.F., Durbin R. 2020. Ancestral hybridization facilitated species diversification in the Lake Malawi Cichlid fish adaptive radiation. Mol. Biol. Evol. 37:1100–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen K. 1982. Review of reproductive isolating barriers in Odonata. Adv. Odonatol. 1:251–265. [Google Scholar]
- Than C., Ruths D., Nakhleh L. 2008. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics 9:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J.A., Trueman J.W., Rambaut A., Welch J.J. 2013. Relaxed phylogenetics and the palaeoptera problem: resolving deep ancestral splits in the insect phylogeny. Syst. Biol. 62:285–297. [DOI] [PubMed] [Google Scholar]
- Troast D., Suhling F., Jinguji H., Sahlen G., Ware J. 2016. A global population genetic study of Pantala flavescens. PLoS One, 11:e0148949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueman J.W.H. 1996. A preliminary cladistic analysis of odonate wing venation. Odonatologica 25:59–72. [Google Scholar]
- Tynkkynen K., Grapputo A., Kotiaho J.S., Rantala M.J., Väänänen S., Suhonen J. 2008. Hybridization in Calopteryx damselflies: the role of males. Anim. Behav. 75:1431–1439. [Google Scholar]
- van der Maaten L., Hinton G. 2008. Visualizing data using t-SNE. J. Mach. Learn. Res. 9:2579–2605. [Google Scholar]
- Vanderpool D., Minh B.Q., Lanfear R., Hughes D., Murali S., Harris R.A., Raveendran M., Muzny D.M., Hibbins M.S., Williamson R.J.. et al. 2020. Primate phylogenomics uncovers multiple rapid radiations and ancient interspecific introgression. PLoS Biol. 18:e3000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonHoldt B.M., Pollinger J.P., Earl D.A., Knowles J.C., Boyko A.R., Parker H., Geffen E., Pilot M., Jedrzejewski W., Jedrzejewska B.. et al. 2011. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21:1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Zhao Y., Zhang B. 2015. Efficient test and visualization of multi-set intersections. Sci. Rep. 5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., He Z., Shi S., Wu C.-I. 2020. Genes and speciation: is it time to abandon the biological species concept? Natl. Sci. Rev. 7:1387–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Cao Z., Ogilvie H.A., Nakhleh L. 2020. Phylogenomic assessment of the role of hybridization and introgression in trait evolution. bioRxiv: 2020.2009.2016.300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware J., May M., Kjer K. 2007. Phylogeny of the higher Libelluloidea (Anisoptera: Odonata): an exploration of the most speciose superfamily of dragonflies. Mol. Phylogenet. Evol. 45:289–310. [DOI] [PubMed] [Google Scholar]
- Wen D., Yu Y., Zhu J., Nakhleh L. 2018. Inferring phylogenetic networks using PhyloNet. Syst. Biol. 67:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett N.J., Mirarab S., Nguyen N., Warnow T., Carpenter E., Matasci N., Ayyampalayam S., Barker M.S., Burleigh J.G., Gitzendanner M.A.,et al. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 111:E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Smith S.A. 2014. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 31:3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yi H., Jin L. 2013. Co-phylog: an assembly-free phylogenomic approach for closely related organisms. Nucleic Acids Res. 41:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Nakhleh L. 2015. A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 16:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Than C., Degnan J.H., Nakhleh L. 2011. Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting. Syst. Biol. 60:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Rheindt F.E., She H., Cheng Y., Song G., Jia C., Qu Y., Alström P., Lei F. 2021. Most genomic loci misrepresent the phylogeny of an avian radiation because of ancient gene flow. Syst. Biol. 70:961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw RNA-seq read libraries generated for this study have been uploaded to the Short Read Archive (SRA) (SRR12086708-SRR12086765). All assembled transcriptomes, alignments, inferred gene, and species trees, fossil calibrated phylogenies (including paleontological record assessed from PaleoDB used to plot the fossil distribution in Fig. 1), PhyloNet results, QS results, and introgression results are available on figshare (https://doi.org/10.6084/m9.figshare.12518660). Custom scripts that were used to create various input files as well as the analysis pipeline are available on GitHub https://github.com/antonysuv/odo_introgression.