

Abstract
Background
Venlafaxine is a dual serotonin (5-HT) and norepinephrine reuptake inhibitor. The specific dose at which it begins to efficiently engage the norepinephrine transporter (NET) remained to be determined. Paroxetine is generally considered as a selective 5-HT reuptake inhibitor but exhibits some affinity for NET. Atomoxetine is a NET inhibitor but also has some affinity for the 5-HT reuptake transporter (SERT).
Methods
This study examined the effects of forced titration of venlafaxine from 75 to 300 mg/d, paroxetine from 20 to 50 mg/d, or atomoxetine from 25 to 80 mg/d in 32 patients with major depressive disorder. Inhibition of SERT was estimated using the depletion of whole-blood 5-HT. Inhibition of NET was assessed using the attenuation of the systolic blood pressure produced by i.v. injections of tyramine.
Results
All 3 medications significantly reduced 5-HT levels at the initiating regimens: venlafaxine and paroxetine by approximately 60% and atomoxetine by 16%. The 3 subsequent regimens of venlafaxine and paroxetine reduced 5-HT levels by over 90%, but the highest dose of atomoxetine only reached a 40% inhibition. Atomoxetine dose dependently inhibited the tyramine pressor response from the lowest dose, venlafaxine from 225 mg/d, and paroxetine left it unaltered throughout.
Conclusion
These results confirm that venlafaxine and paroxetine are potent SERT inhibitors over their usual therapeutic range but that venlafaxine starts inhibiting NET only at 225 mg/d, whereas paroxetine remains selective for SERT up to 50 mg/d. Atomoxetine dose dependently inhibits NET from a low dose but does not inhibit SERT to a clinically relevant degree.
Keywords: Antidepressant doses, blood pressure, plasma levels, serotonin levels, tyramine
Significance Statement.
Serotonin and/or norepinephrine reuptake inhibitors are the most commonly used medications for the treatment of major depressive disorder. Their regimens necessary to inhibit the reuptake of one or both neurotransmitters to a clinically relevant level have been a matter of controversy. We report here, using physiological methodologies validated with a variety of medications in humans, the regimens of venlafaxine, paroxetine, and atomoxetine necessary to inhibit serotonin and/or norepinephrine in depressed patients to obtain their optimal reuptake inhibiting potential in clinical management.
Introduction
Various approaches have been used to evaluate the extent of serotonin (5-hydroxytryptamine [5-HT]) and norepinephrine (NE) reuptake inhibition produced by medications used commonly to treat major depressive disorder (MDD). In general, there is a proportional relationship between the affinity of such drugs to bind to 5-HT and NE transporters (5-HTT, NET) and their potency to inhibit the actual reuptake processes. In the case of the 5-HT system, there is consistency between the affinity of various medications to bind to human 5-HTT in vitro, their capacity to inhibit 5-HT reuptake in various tissue preparations, the ex vivo assessment of 5-HT reuptake inhibition using the serum of patients undergoing pharmacological treatment of MDD, and the capacity of 5-HT reuptake inhibitors to displace positron emission tomography (PET) ligands from 5-HT reuptake sites in the brain of healthy participants and of patients with MDD (Owens et al., 1997; Tatsumi et al., 1997; Meyer et al., 2001; Gilmor et al., 2002). The latter brain imaging approach has documented that minimal effective doses of selective serotonin reuptake inhibitors (SSRIs) and 5-HT and NE reuptake inhibitors result in fairly high occupancy of 5-HTT (approximately 80%), but higher regimens achieve even greater occupancy (Meyer et al., 2001; Voineskos et al., 2007). Such results imply that there is a high threshold in the proportion of 5-HTT that need to be inhibited before 5-HT transmission is enhanced in the brain, thus indicating the presence of transporter reserve (Blier 2008).
In the case of the NE system, there are some inconsistencies between different approaches to assess NE reuptake. In particular, venlafaxine exhibits low in vitro affinity for NET (i.e., in the micromolar range) that stands in contrast with its relatively high in vivo potency to inhibit NE reuptake compared with the high-affinity NET inhibitor desipramine (Owens et al., 1997; Béïque et al., 1998, 1999). Indeed, NE reuptake has been shown to be dose dependently inhibited in patients undergoing desipramine or venlafaxine treatment when their serum was used in a cell line expressing the human NET (Gilmor et al., 2002; Owens et al., 2008). The latter study showed that NE reuptake was inhibited by venlafaxine ex vivo to approximately 25% with 75 mg/d, 45% with 150 mg/d, 50% with 225 mg/d, 55% with 300 mg/d, and 60% with 375 mg/d. These results, however, do not indicate what degree of inhibition is necessary to inhibit NET to obtain a physiological effect in vivo, even more so as there is a high degree of inter-individual variability in the resultant steady-state serum concentrations produced by identical dosages. In contrast to the brain imaging data for the 5-HT system, the threshold of NET occupancy to obtain a therapeutic action in MDD through this mechanism is not as clear. For instance, repeated administration of the NET inhibitor nortriptyline to MDD patients leads to constant NET occupancy of approximately 60% with regimens of 75 to 200 mg/d without evidence of a concentration-dependent relationship in that range (Takano et al., 2014). Furthermore, single doses of the NET inhibitor atomoxetine of 25, 50, and 100 mg in the same healthy participants occupied the NET to approximately 40% with no significant differences between doses for any brain region (Logan et al., 2007).
The use of the tyramine pressor response as a proxy of physiological NET activity has provided consistent results for the capacity of various regimens of reuptake inhibitors to block NET (Ghose and Turner 1975). In brief, it consists of injecting small amounts of tyramine i.v. and assessing the increase in systolic blood pressure (SBP) resulting from tyramine entering peripheral NE terminals through their NET and releasing NE in a calcium-independent manner (Hoffman and Lefkowitz 1990). Minimal effective regimens of paroxetine, sertraline, and nefazodone for MDD do not alter this pressor response, whereas desipramine, clomipramine, venlafaxine (225 but not 75 mg/d), and duloxetine (120 but not 60 and 80 mg/d) inhibit it by preventing the uptake of tyramine (Harvey et al., 2000; Turcotte et al., 2001; Vincent et al., 2004; Blier et al., 2007; Debonnel et al., 2007). In some of these studies, a surrogate marker for functional 5-HT reuptake inhibition was the depletion of whole-blood 5-HT. Since 90% of the blood 5-HT is in platelets and finds its way there through a 5-HT reuptake transporter, a lowering of whole-blood 5-HT is a good indicator of 5-HT reuptake inhibition in the brain (Demet et al., 1978; Artigas et al., 1985). Minimal effective antidepressant regimens of paroxetine, venlafaxine, duloxetine, and clomipramine lower 5-HT levels by more than 70%, whereas desipramine does so by less than 20% (Turcotte et al., 2001; Vincent et al., 2004; Blier et al., 2007; Debonnel et al., 2007).
The main objective of the present study was to determine the in vivo potency of venlafaxine and paroxetine in patients with MDD using a wide dose range to determine at which dose(s) these medications begin to exert an impact through 5-HT and NE reuptake inhibition to produce a clinical response. To this end, the degree of whole-blood 5-HT depletion and the attenuation of the tyramine pressor response were used, respectively. Atomoxetine was also included in this study as an active comparator to block NET (Bymaster et al., 2002) and for allowing a comparison of its selectivity for NET and SERT vs the PET methodology (Nogami et al., 2013; Ding et al., 2014).
METHODS
Patients
Adults (aged 18–65 years) of both genders who met the DSM-IV-TR (American Psychiatric Association, 2000) criteria for MDD using the Structured Clinical Interview for Diagnostic and Statistical Manual-IV (Spitzer and Williams, 1988) were enrolled in the study. All patients had an initial global score of at least 18 on the 17-item Hamilton Depression Rating Scale (HAMD-17; Hamilton, 1960). Exclusion criteria included pregnancy and evidence of physical illness (by clinical interview, physical examination, electrocardiogram, clinical chemistry, hematology, urinalysis, or thyroid function) contraindicating the use of venlafaxine, paroxetine, atomoxetine, or tyramine. Patients were not allowed to use any concurrent psychotropic medication during the study, except small doses of lorazepam or clonazepam (<2 mg/d) not 8 hours before the tyramine pressor test. The patients could not be intolerant, or lack a prior antidepressant response, to optimal doses of venlafaxine, paroxetine, or atomoxetine. Written informed consent was obtained as approved by the Royal Ottawa Health Care Group Research Ethics Board. The study was registered and received the International Standard Randomized Controlled Trial Number of ISRCTN87057460.
Clinical Design
This study was conducted on an outpatient basis at the Mood Disorders Research Unit of the University of Ottawa Institute of Mental Health Research Institute. After a thorough screening process and a 1-week washout period (if needed), patients were randomly assigned to 1 of 3 treatment groups in blocks of 6 patients throughout the study. The investigators involved in the tyramine test or collection of biochemical data were blind to the medications used by patients. Clinical raters and patients were not blind to treatment as the study was not powered to detect differences in clinical efficacy but only changes in physiological parameters.
At baseline and each subsequent visit, every patient underwent a clinical interview using the Montgomery-Asberg Depression Rating Scale (MADRS; Montgomery and Asberg 1979), the HAMD-17 (Hamilton 1960), and the Clinical Global Severity scale (CGI; Guy 1976).
Medication Regimens
Doses were increased weekly as tolerated. Patients could remain at the same daily dose for an additional 1 to 2 weeks but had to reach the maximal daily dose by the sixth week. Venlafaxine XR (Effexor XR) was initiated at 37.5 mg/d am for the first 3 days and increased to 75 mg/d AM for the remaining 4 days of the first week. The dose was then increased to 150 mg/d (75 mg BID), 225 mg/d (150 mg am and 75 mg pm), and 300 mg/d (150 mg BID). Paroxetine (Paxil) was initiated at dose of 20 mg/d am and then increased to 30 mg/d (20 mg am and 10 pm), 40 mg/d (20 mg BID), and then 50 mg/d, the maximal recommended dose for MDD in Canada (20 mg in am and 30 mg in pm). Atomoxetine (Strattera) was initiated at 25 mg am for the first week and then increased to 43 (25 mg in am and 18 mg in pm), 65 (40 mg am and 25 mg pm), and 80 mg/d (40 mg BID). The last dose was taken 2–3 hours before blood drawing to determine plasma levels of drugs and 5-HT concentrations and prior to initiation of the tyramine pressor test.
Plasma Drug Levels
To ascertain the levels of medications in circulation at the time of the tyramine pressor test, blood levels of these drugs were measured. Before the procedure, plasma samples were collected, and the serum was maintained frozen at −80° C until analyzed. The levels of venlafaxine and desvenlafaxine were determined by the method described in Newport et al. (2009), those of paroxetine in Ritchie et al. (2009), and those of atomoxetine by an adapted assay of Farid et al. (1985). Since venlafaxine and O-desmethyl-venlafaxine (ODV) have approximately the same affinity for SERT and NET, their concentrations were combined (Owens et al., 2008).
Assessment of 5-HT Reuptake
Although lacking the enzymatic synthesis and some 5-HT receptor subtype, platelets can take up and store 5-HT similarly to 5-HT neurons (Demet et al., 1978; Pletscher 1986). The lifespan of circulating platelets is 10 days (Harker and Finch, 1969). Following blockade of the 5-HT reuptake process, platelet 5-HT content gradually decreases as older platelets that have previously taken up 5-HT are removed from the circulation and replaced by new ones that are rendered incapable of effective 5-HT uptake by the reuptake-inhibiting drugs. Moreover, as a very large proportion (90%–95%) of blood 5-HT is sequestrated in the platelets, whole-blood 5-HT content largely reflects platelet 5-HT content and avoids biases that could be introduced by platelet isolation (Demet et al., 1978; Artigas et al., 1985; Pletscher, 1986). Whole-blood 5-HT content (in nmol/L) was thus determined, as previously described (Blier et al., 2007), at least 1 week after each dose change and compared with the baseline level. The changes were used as an index of the degree of SERT inhibition. Although there are seasonal variations in whole-blood 5-HT (Brewerton et al., 1993), it is unlikely that this could have influenced the overall results over a period of 4 to 6 weeks given the randomization process.
Assessment of NE Reuptake
The tyramine pressor test examines the activity of the NE transporter by measuring the increase in SBP produced by a small i.v. injection of tyramine (Hoffman and Lefkowitz, 1990). Tyramine penetrates peripheral NE terminals through their NE transporters and then releases NE, which transiently increases blood pressure (Hoffman and Lefkowitz, 1990). NE reuptake inhibition therefore attenuates this response (Ghose, 1984). Supine blood pressure and heart rate were automatically measured every 2 minutes using the Criticare 507 apparatus. The tyramine test (Ghose et al., 1975) consists of measuring the transient change in the SBP of patients after 3 escalating doses of i.v. tyramine infusion (3, 4, and 6 mg) separated by at least 10 minutes to allow the blood pressure to return to baseline level. The extrapolated dose of tyramine to increase SBP by 30 mm of Hg (ED30) has been used as a safe and reliable measure to estimate SBP changes produced by low doses of tyramine, for example, following monoamine oxidase inhibition (Ghose 1984). Nevertheless, in the presence of optimal NET inhibition, this approach can lead to extrapolated very high ED30 values. The procedural details of this experiment can be reviewed in previous work (Blier et al., 2007; Debonnel et al., 2007).
Genotyping of the Transporters
Since single nucleotides polymorphisms for SERT and NET have been linked to their function and possibly to clinical responses, genotyping of the most commonly studied variants for SERT (SLC6A4, rs25531; Greenberg et al., 1996; Ren et al., 2020) and NET (SLC6A2, rs2242446; Yoshida et al., 2004; Zhao et al., 2020) were carried out for these variants as previously described by our group (Phillips et al., 2015).
Statistical Analyses
At baseline and after each subsequent dose escalation, the reduction of whole blood 5-HT was analyzed using 2-way ANOVA for repeated measures. The difference in the SBP pre- and 2 minutes post-tyramine (3, 4, and 6 mg) doses was considered the primary efficacy variable. Two-way ANOVA for repeated measures for doses of tyramine and treatments were conducted on the data to assess the effects of the different drug regimens on the pressor response to 3, 4, and 6 mg tyramine. Post-hoc analyses were conducted where appropriate with Holm-Sidak method tests in all of the multiple comparisons. ED30 values of tyramine were estimated using regression analysis. All values are expressed as means ± SEM unless otherwise stated. The level of significance was set at P < .05. One-way ANOVAs were used to compare the clinical efficacy of the different doses of each medication. Statistical analyses and graphs were done using SigmaStat Version 3.5 (2006 Systat Software, Inc., San Jose, CA) and SigmaPlot Version 11.0 (2008 Systat Software, Inc., San Jose, CA).
RESULTS
Patients Demographics and Clinical Characteristics
Thirty-two patients were randomized in the trial, 30 of whom (94%) completed the study. Only 2 male patients dropped out of the study because of intolerance of atomoxetine (at a dose of 60 mg/d) and venlafaxine (at a dose of 75 mg/d). Their partial response data were not included in the final analyses.
Table 1 summarizes the demographic and baseline clinical data for the 3 groups. There were no statistically significant differences between the 3 groups in terms of age at randomization, age of first depressive episode, duration of present episode, gender, weight, or body mass index (BMI). However, the 3 groups differed in terms of baseline scores of HAMD-17 (F2,28 = 4.57, P = .019), HAMD-24 (F2,28 = 5.86, P = .007), MADRS (F2,28 = 5.89, P = .007), and CGI (F2,28 = 3.73, P = .036). With respect to HAMD-17 scores, post-hoc analysis showed that the venlafaxine group differed from the paroxetine group (Punadjusted = .007 [t = 2.92]), but the atomoxetine group did not differ from either of the 2 groups. With respect to CGI, the venlafaxine group differed from the atomoxetine group using post-hoc analysis (Punadjusted = .012 [t = 2.67]).
Table 1.
Demographics and Clinical Data at Baseline
| Demographic | Atomoxetine (n = 9) | Paroxetine (n = 11) | Venlafaxine (n = 10) | F |
|---|---|---|---|---|
| Mean (SD) | ||||
| Age at randomization (y) | 43.6 (±12.2) | 41.6 (±10.2) | 41.3 (±10.1) | F 2,28 = 0.14, P = .87 |
| Age at onset of first depressive episode (y) | 31.0 (±13.5) | 34.5 (±14.8) | 30.4 (±12.9) | F 2,28 = 0.28, P = .76 |
| Duration of present episode (mo) | 14.3 (±9.2) | 13.3 (±10.7) | 14.9 (±16.0) | F 2,28 = 0.05, P = .95 |
| Gender | Chi square: 41, P = .82 | |||
| Male | 5 | 7 | 7 | |
| Female | 4 | 4 | 3 | |
| Weight (kg) | 75.7 (±15.1) | 90.8 (±20.2) | 80.7 (±12.9) | F 2,28 = 2.36, P = .11 |
| BMIa | 25.3 (±5.0) | 30.6 (±4.9) | 28.3 (±4.2) | F 2,26 = 3.02, P = .07 |
| HAMD-17 | 24.0 (±4.1) | 24.9 (±2.0) | 21.4 (±2.1) | F 2,28 = 4.57, P = .019 |
| MADRS | 33.9 (±3.3) | 34.7 (±2.7) | 30.6 (±2.8) | F 2,28 = 5.89, P = .007 |
| CGI-Severity | 5.0 (± 0.5) | 4.8 (± 0.4) | 4.5 (±0.5) | F 2,28 = 3.73, P = .036 |
Abbreviations: CGI, Clinical Global Improvement scale; HAM-D, Hamilton Rating Scale for Depression; MADRS, Montgomery-Asberg Depression Rating Scale.
a BMI score was missing in 1 patient in the atomoxetine group and 1 in the paroxetine group.
As shown in Table 1, the gender distribution among the 3 treatment groups was relatively equal. However, the female to male ratio for this sample was approximately 1:2, which is the reverse of the usual 2:1 ratio in the general population of depressed patients. The effect of gender on baseline SBP responses to the different doses of tyramine (3, 4, and 6 mg) was analyzed. There was no significant difference between the 2 genders with respect to baseline SBP response to tyramine dose of 3 mg (F1,30 = 3.96, P = .056), 4 mg (F1,30 = 2.69, P = .11), and 6 mg (F1,30 = 0.41, P = .53).
Clinical Responses
As shown in Figure 1, there was significant difference in the MADRS mean scores considering the treatment daily doses (F4,108 = 30.11, P < .001), but not when considering the treatment group (F2,108 = 1.19, P = .32). As well, there was no statistically significant interaction between treatment group and dose level (F8,108 = 0.93, P = .50). Considering each daily dose, there was no difference among the treatment groups. Post-hoc tests (Holm-Sidak method) showed that MADRS mean scores at baseline were significantly different compared with MADRS mean values at treatment dose 1 (Punadjusted = .009, t = 2.67), dose 2 (Punadjusted < .001, t = 4.89), dose 3 (Punadjusted < .001, t = 7.73), and dose 4 (Punadjusted < .001, t = 9.73). Changes of scores on the HAMD-17 and CGI-severity mirrored those obtained with MADRS (data not shown).
Figure 1.

Evolution over time of the depressive symptoms, as assessed on the Montgomery Asberg Depression Rating Scale (MADRS). The scores of 2 randomized patients were not included as they dropped out due to side effects. Medication regimens were titrated weekly, but they could remain on the same dose level for an additional 1 or 2 weeks, but all patients reached level 4 for at least for 1 week after 6 weeks. Atomoxetine levels were 25, 43, 65, and 80 mg/d; venlafaxine levels were 75, 150, 225, and 375 mg/d; paroxetine levels were: 20, 30, 40, and 50 mg/d. *P < .05 denotes significant improvement between baseline scores and levels 2–4 in each group.
The criterion for response was set as at least 50% reduction in MADRS score at the last visit compared with baseline MADRS score. Remission criterion was set at ≤10 on MADRS at the last visit. All 3 treatment groups showed a significant response (Figure 1). Among the patients who received venlafaxine, 5 patients responded (50%) and 2 of them remitted (20%). In the paroxetine group, 5 patients responded (45%) and only 1 patient remitted (9%). Four patients responded to atomoxetine (44%) and 3 of them remitted (33%).
Plasma Levels of Active Compounds
The total plasma levels of venlafaxine plus ODV were positively correlated with the corresponding dosages (r = 0.69, [F1,38 = 33.34, P < .001]). One male patient, however, did not have detectable level of venlafaxine but did have one for ODV 1 week after being on the lowest dose (Table 2). He had the second highest BMI (32) and third highest weight (89 kg) among the venlafaxine group patients. The ODV to venlafaxine ratio means at the 4 different doses were 2.0 at 75 mg, 4.0 at 150 mg, 2.5 at 225 mg, and 2.0 at 300 mg. Two patients had relatively reversed ratios, suggesting that they were poor P-450 2D6 metabolizers.
Table 2.
Plasma Levels of Active Compounds
| Treatment group | Atomoxetine (n = 9) | Paroxetine (n = 10)a | Total venlafaxine and ODV (n = 10) | |||
|---|---|---|---|---|---|---|
| Dose level | Plasma level mean (ng/mL ± SD) | Plasma level range (ng/mL) | Plasma level mean (ng/mL ± SD) | Plasma level range (ng/mL) | Plasma level mean (ng/mL ± SD) | Plasma level range (ng/mL) |
| I | 114 (70) | 33–235 | 17 (15) | 0–52 | 101 (69) | 0–224 |
| II | 106 (50) | 0–160 | 38 (20) | 10–66 | 242 (115) | 107–497 |
| III | 198 (90) | 50–462 | 61 (22) | 33–91 | 376 (143) | 164–580 |
| IV | 184 (101) | 54–325 | 81(28) | 40–119 | 472 (243) | 159–849 |
Abbreviation: ODV, O-desmethyl-venlafaxine.
a Data for 1 patient not included due to a detection issue (see results section).
All but one of the patients in the paroxetine group had detectable paroxetine levels after receiving paroxetine (Table 2). Linear regression analysis (determined from the Pearson correlation coefficient) showed the paroxetine plasma levels were positively correlated with paroxetine doses; (r = 0.76, F1,38 = 51.54, P < .001). The 1 patient in whom paroxetine level was not detected in any of the samples obtained nevertheless affirmed that she was compliant, reported the typical SSRI side effects, and had a gradual and sustained clinical response. Furthermore, her total 5-HT level decreased but only 1 week after being on 40 mg by 68% and 88% on 50 mg, compared with baseline. As well, all of the study patients had a drop in total 5-HT starting at the lowest dose except this patient. Again, the only unusual clinical feature of this patient is that she was obese; she had the highest weight (129 kg) and the highest BMI (42) among all the study participants. It is thus clear that there can be factors that can alter detection of plasma levels of medications, as was reported by Owens et al., 2008 for paroxetine.
All 9 patients on atomoxetine had detectable levels at all 4 dose levels, except 1 patient who had undetectable level while on 43 mg/d during that specific week (Table 2). Linear regression analysis (determined from the Pearson correlation coefficient) showed the atomoxetine plasma levels were positively correlated with atomoxetine doses (r = 0.66, [F1,34 = 22.1, P < .001]).
Alterations of 5-HT Blood Levels
The percentage reduction in whole-blood 5-HT was statistically significant at all the treatment levels of all 3 treatment groups compared with baseline. A simple main effect test indicated there was a difference in the 5-HT percentage reduction in relation to the treatment group (F2,130 = 252.89; P < .001) and treatment doses (F4,130 = 238.68; P < .001). As well, there was a statistically significant interaction between the treatment group and treatment doses (F8,130 = 16.76; P < .001).
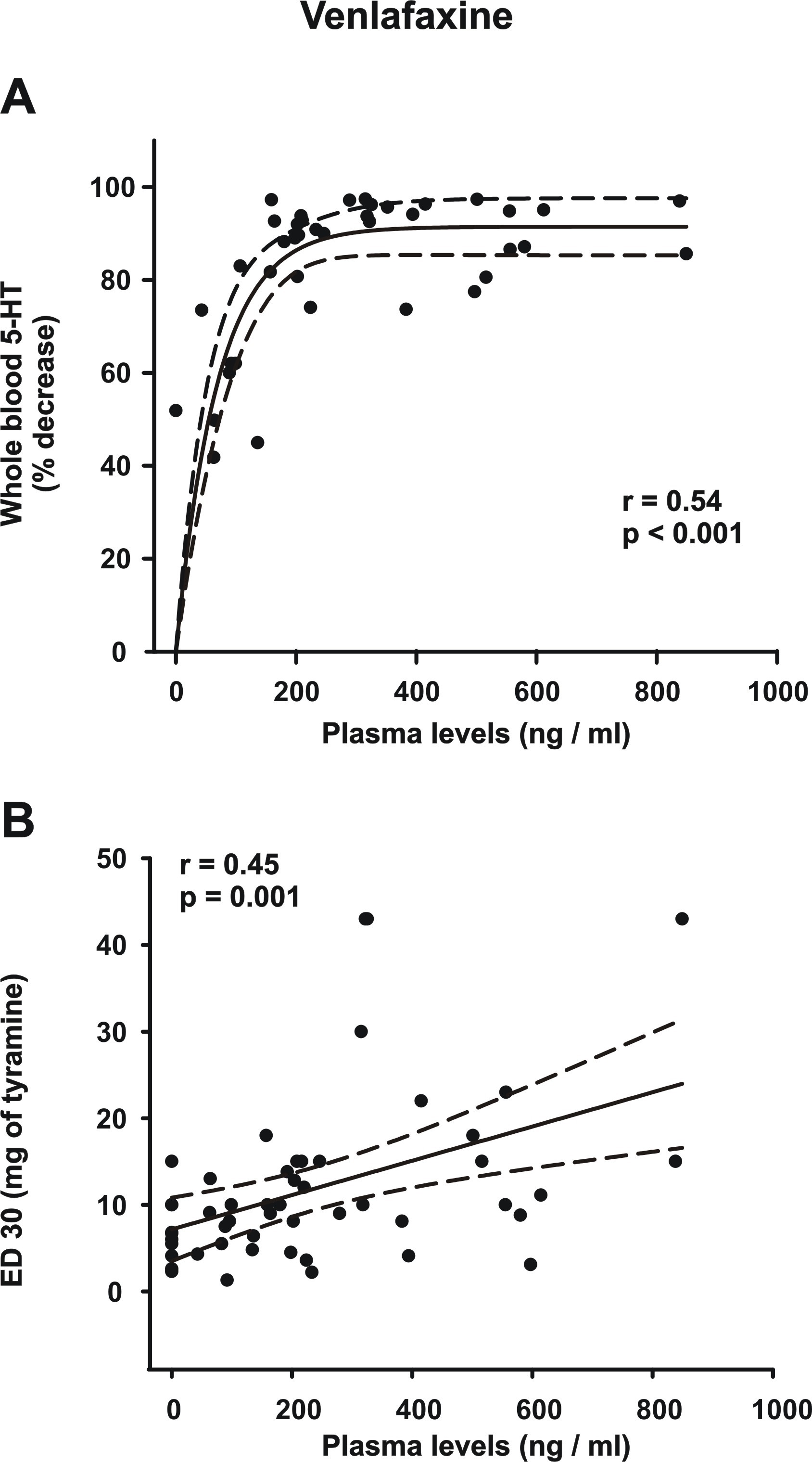
All doses of venlafaxine exerted a dose-dependent reduction in the total 5-HT counts in all patients (Figure 2). Most of the patients (8 out of 10) had >90% reduction in total 5-HT at a dose of 225 mg/d and >95% reduction at dose of 300 mg/d. There was a highly significant correlation between plasma levels of venlafaxine + ODV and the percentage of whole-blood 5-HT depletion (r = 0.54, P < .001), reaching more than 80% with the plasma levels ≥200 ng/mL (supplemental Figure 1A). These results are consistent with an estimated ex vivo Ki value of 85 ng/mL of venlafaxine + ODV for [3H]5-HT uptake inhibition (Owens et al., 2008).
Figure 2.

Percentage decrease of whole blood serotonin (5-HT) over a 6-week period. Medication regimens were titrated weekly, but they could remain on the same dose level for an additional 1 or 2 weeks, but all patients reached level 4 for at least for 1 week after 6 weeks. The baseline levels of 5-HT in nmol/L were 788 ± 107, 897 ± 253, and 719 ± 129 in the atomoxetine, paroxetine, and venlafaxine groups, respectively. Atomoxetine levels were: 25, 43, 65, and 80 mg/d; venlafaxine levels were 75, 150, 225, and 375 mg/d; paroxetine levels were: 20, 30, 40, and 50 mg/d. *P < .05 indicates statistical differences within each treatment group when compared with their respective baseline. There were no statistically significant differences between the venlafaxine and paroxetine groups, but atomoxetine produced significantly lesser decreases of 5-HT levels than the other 2 medications.
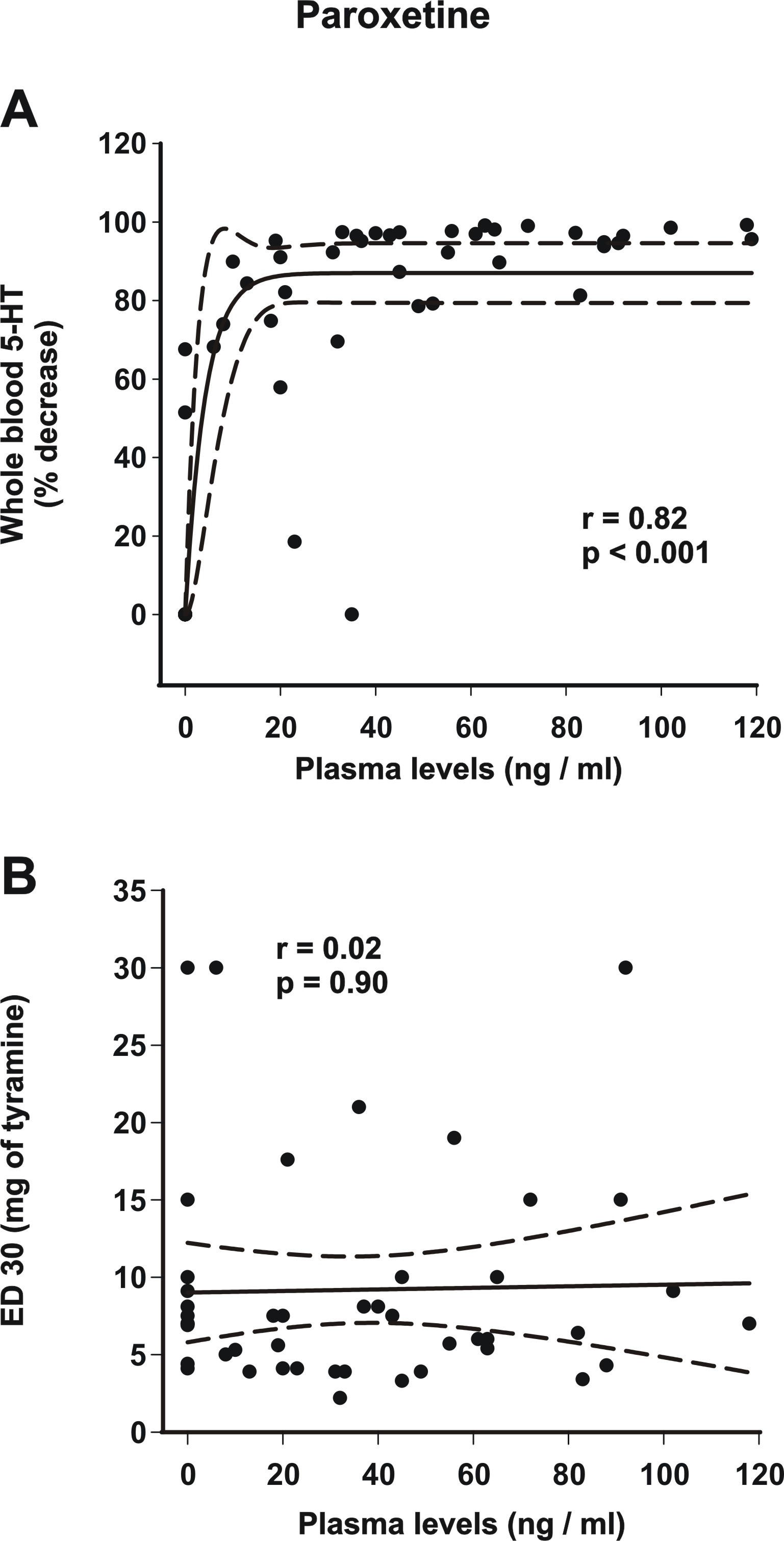
All the doses of paroxetine exerted a dose-dependent reduction in the total 5-HT levels in all patients (Figure 2). Nearly all plasma levels >20 ng/mL led to whole-blood 5-HT depletion ≥80% (supplemental Figure 2B). These results are consistent with an estimated ex vivo Ki value of 9 ng/mL of paroxetine for [3H]5-HT uptake inhibition (Owens et al., 2008). However, there was only 1 patient who had elevated total 5-HT counts at doses of 20 mg and 30 mg by 35% and 13%, respectively, but then her total 5-HT levels reduced at doses of 40 and 50 mg by 68% and 88%, respectively. She is the only paroxetine-treated patient who had undetectable plasma levels at all visits. As mentioned above, she had the highest weight and BMI compared with all the study patients. Interestingly, another patient with a body weight of 129 kg and BMI of 35 had the highest percentages of 5-HT reductions by 91%, 97%, 98%, and 99%, respectively, compared with all other patients treated by paroxetine. Having had higher plasma levels (20, 43, 65, and 102 ng/mL, respectively) compared with the group means (Table 2) might explain these data of robust and potent inhibition of 5-HT reuptake in this patient.
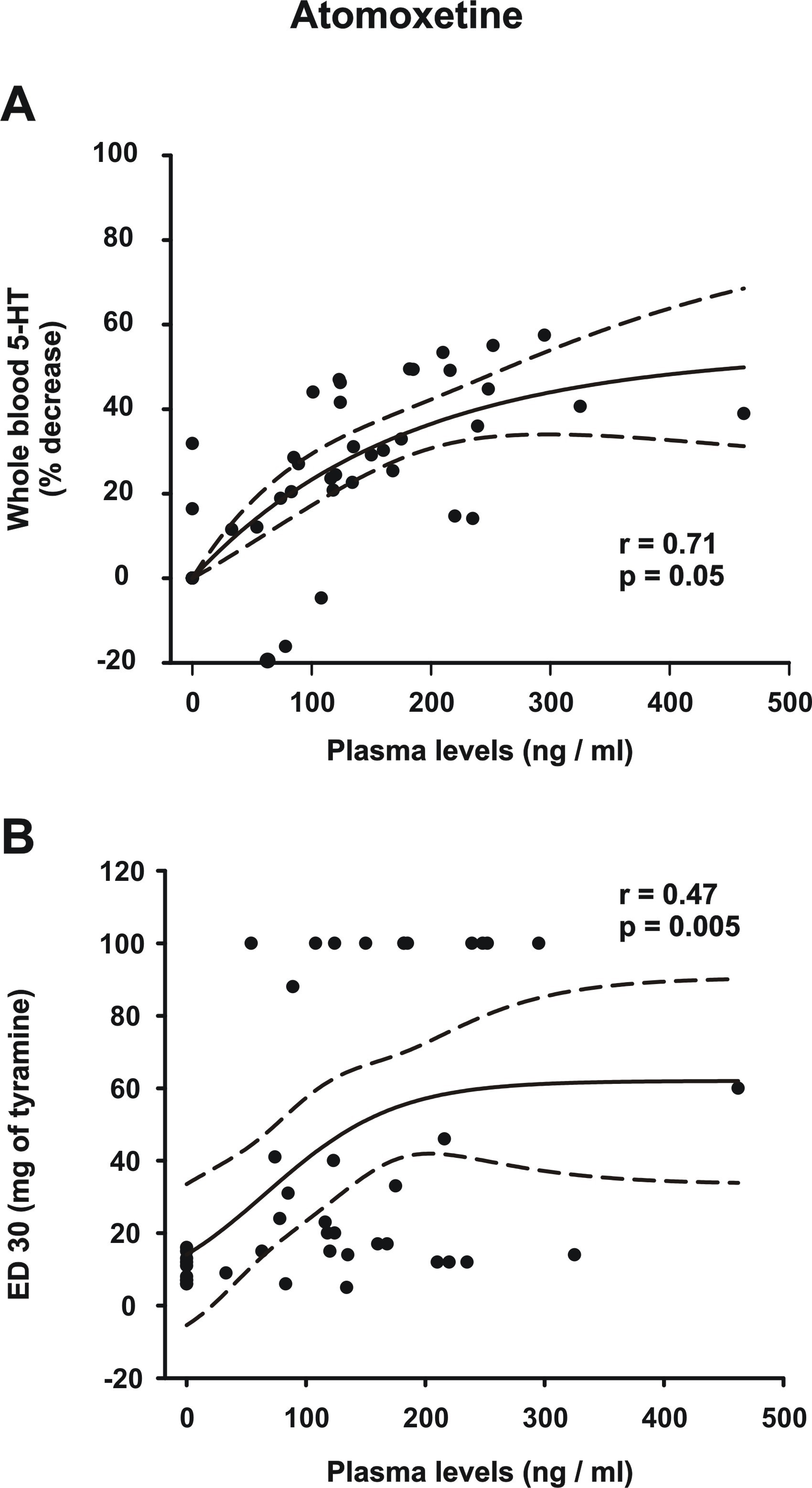
All the atomoxetine dose regimens exerted a dose-dependent reduction in the total 5-HT levels, albeit much smaller than in the venlafaxine and paroxetine groups and not surpassing the 50% mark in most cases, irrespective of the plasma levels (Figure 2; supplemental Figure 3A). There was only 1 patient who had an elevation of 5-HT levels at daily doses of 25 mg (19%), 43 mg (16%), and 65 mg (4%) compared with baseline, but at a dose of 80 mg/d her total 5-HT level was reduced by 12% compared with baseline. This patient had the lowest plasma levels of atomoxetine compared with all other patients.
Assessment of Changes in Tyramine Responses
In the venlafaxine group, there was a simple main effect test indicating that there was a difference in the SBP increase in relation to the dose of tyramine (F2,72 = 17.3; P < .001) as well as to the venlafaxine regimen (F4,72 = 10.6; P < .001). There was also a statistically significant interaction between venlafaxine regimen and tyramine dose (F8,72 = 3.5; P = .002). Post-hoc analysis (Holm-Sidak method) indicated a significant difference in the means of BP increase (P < .05) only when comparing venlafaxine regimens of 225 mg/d and 300 mg/d with the baseline. Moreover, regimens of 225 mg/d and 300 mg/d showed significant differences when each was compared with the regimens of 75 mg/d and 150 mg/d (P < .05; Figure 3). As previously reported with the 75- and 375-mg/d regimens (Debonnel et al., 2007), there was a significant correlation between the ED30 values and the plasma levels of venlafaxine + ODV (supplemental Figure 1B).
Figure 3.

Alterations of systolic blood pressure within 2 minutes of the i.v. injection of 3, 4, and 6 mg of tyramine in a bolus manner. All 10 patients were tested after at least 1 week after achieving the indicated regimens of venlafaxine, 2 to 3 hours after taking the last dose. *P < .05 indicates that only the 225- and 300-mg/d regimens differed significantly from baseline and the 75- and 150-mg/d regimens.
In the paroxetine group, there was a simple main effect test indicating that there was a difference in the SBP increase in relation to the dose of tyramine (F2,80 = 75.9; P < .001), but not to the paroxetine dose (F4,80 = 1.3; P = 0.28). Post-hoc analysis (Holm-Sidak method) indicated an absence of any significant difference in the means of BP increase when comparing all paroxetine regimens with the baseline and comparing the different regimens of paroxetine (P > .05; Figure 4). In accordance with these results, there was no correlation (r = 0.02, P = .9) between the ED30 values and the plasma levels of paroxetine (supplemental Figure 2B). It is unlikely that the patient with a 95 ng/mL plasma level of paroxetine had significant NE reuptake blockade since the ex vivo Ki value of paroxetine to inhibit [3H]NE uptake is 227 ng/mL (Owens et al., 2008); it thus represents an outlying value due to an experimental condition.
Figure 4.

Alterations of systolic blood pressure within 2 minutes of the i.v. injection of 3, 4, and 6 mg of tyramine in a bolus manner. All 11 patients were tested after at least 1 week after achieving the indicated regimens of paroxetine, 2 to 3 hours after taking the last dose. There were no statistical differences between any of the groups.
In the atomoxetine group, there was a simple main effect test indicating that there was a difference in the SBP increase in relation to the dose of tyramine (F2,64 = 9.6; P = .002) and the atomoxetine dose (F4,64 = 15.9; P < .001). As well, there was a statistically significant interaction between the dose of tyramine and atomoxetine dose (F8,64 = 2.6; P = .018). Post-hoc analysis (Holm-Sidak method) indicated significant differences in the means of SBP increase (P < .05) when comparing all atomoxetine regimens with the baseline. Moreover, the 25-mg/d regimen showed a significant difference when compared with the regimens of 65 mg/d and 80 mg/d. However, there was no significant difference between the pressor response between the doses of 43, 65, and 80 mg/d (P > .05; Figure 5). In general, patients had progressive inhibition of the pressor response, but several patients had a complete loss of response to tyramine, thus making it impossible to calculate ED30 values, and a dose of 100 mg was assigned to such outcomes for graphical purposes (supplemental Figure 3B).
Figure 5.

Alterations of systolic blood pressure within 2 minutes of the i.v. injection of 3, 4, and 6 mg of tyramine in a bolus manner. All 9 patients were tested after at least 1 week after achieving the indicated regimens of atomoxetine, 2 to 3 hours after taking the last dose. *P < .05 indicates that all groups statistically differed from the baseline assessment.
Genotypes for SERT and NET
There was 1 patient in each of the 3 groups for whom genotyping was not available. For the SERT alleles, no LA was found in the 27 subjects. The distribution of the genotypes was as follows: L(Long)L (LGLG) 5 patients, S(Short) 4 patients, and SS 18 patients. That distribution is quite different from the usual 1:2:1 ratio commonly observed in large samples (Kato and Serretti 2010). The 3 genotype groups did not statistically differ in terms of the baseline 5-HT levels. (F2,24 = 0.9; P = .4). This is consistent with prior results showing a lack of difference in platelet content with respect to the SERT genotype and with similar SERT occupancy in the brain among the 3 polymorphisms (Greenberg et al., 1999; Parsey et al., 2006). Regardless of the treatment group, the 3 genotypes did not differ in terms of the percentage reduction in the 5-HT levels at regimen level 4 compared with baseline (F2,24 = 0.54; P = .6). Because atomoxetine has less affinity to the 5-HT transporter compared with paroxetine and venlafaxine, it was deemed appropriate to compare the 3 genotypes in terms of percentage reduction in 5-HT levels at the end of the study, excluding the patients who were taking atomoxetine. Nevertheless, the different genotypes still did not statistically differ (F2,16 = 0.35; P = .7) after excluding the atomoxetine group.
With respect to the T182C alleles for the NET polymorphism, no TC genotypes were found in the 27 patients, 24 had the TT genotype, and 3 had the CC genotype. The 2 genotype groups did not statistically differ in terms of their baseline SBP in response to the 3 doses of tyramine. Because paroxetine lacks significant affinity for the NET, the 2 genotypes were compared in terms of SBP change after the highest dose of the venlafaxine and atomoxetine regimens, thus excluding the paroxetine group. There were no significant differences between the 2 genotypes (data not shown), although the sample size was too small to rule out any contribution of this polymorphism to the physiological parameters.
There were no significant correlations between rate of response or remission rates among the different genotypes of either of the 2 transporters in relation to the mechanism of action (SERT vs NET vs both; data not shown). Nevertheless, among the paroxetine group, all 6 of the non-responders had the SS genotype, while the only remitter had a LL genotype. The only other LL-genotyped patient on paroxetine had improved from 34 to 13 on MADRS at the last visit. The results are consistent with lower response rate to SSRIs in patients with SS than LL genotypes (Ren et al., 2020). Among the 3 atomoxetine group remitters, 2 of them both had the TT genotype, and no genotyping was available for the third one. In the venlafaxine group, the 2 remitters had the SS polymorphism for the SERT but the TT genotype for the NET.
DISCUSSION
The results of the present study indicate that for venlafaxine, a dose of 225 mg/d is a threshold regimen to engage reliably the NET, whereas atomoxetine exerted a dose-dependent inhibition starting at 25 mg/d up to 80 mg/d; paroxetine did not inhibit the NET up to dose of 50 mg/d. Both venlafaxine and paroxetine produced a robust 60% inhibition of the SERT starting at their minimal effective doses, reaching >90% reduction of platelet 5-HT at 300 and 50 mg/d, respectively, whereas the degree of inhibition produced by atomoxetine achieved a 40% plateau at 65 mg/d.
It is important to emphasize that this study was not designed to examine clinical effectiveness of the 3 medications but to put into evidence their physiological impact based on the number of participants used included in prior studies using the 2 methodologies. Nevertheless, all 3 medications produced a significant improvement over several weeks. This was expected for venlafaxine and paroxetine, but not atomoxetine. An initial large trial of 600 patients comparing (a)tomoxetine and desipramine with placebo failed with no difference between any of the 3 arms (Eli Lilly & Co, data on file) and was thus difficult to interpret. The only double-blind placebo-controlled study supporting some hint of therapeutic benefit of atomoxetine in MDD was in patients who had not responded to the SSRI sertraline and randomized to addition of atomoxetine or placebo. Patients with 2 short alleles for SERT had a better response to atomoxetine, presumably because of the potency of the adjunct to enhance NE transmission (Michelson et al., 2007). The potent NET inhibitor reboxetine, in contrast, has been shown to be efficacious in MDD, albeit exerting a more modest effect than other medications (Cipriani et al., 2018).
The decrease in whole blood 5-HT reached a plateau with both venlafaxine and paroxetine from the second level up. This time-delayed phenomenon using this approach to estimate SERT inhibition, however, could be due to the turnover rate of platelets. Indeed, it is likely that the SERT on the platelets is near optimally inhibited with the minimal effective doses of these 2 drugs, but the older platelets still in circulation had significant levels of 5-HT, whereas the newly produced ones lacked the capacity to store 5-HT. Indeed, when venlafaxine was administered to patients for 28 days, the same degree of whole-blood 5-HT depletion was observed with 75 mg/d as with 375 mg/d achieved in the last 3 weeks of treatment (Debonnel et al., 2007). As brain PET studies have documented, the threshold of approximately 80% occupancy of SERT, at least in the striatum, is necessary to presumably increase 5-HT transmission and yield an antidepressant action. Nevertheless, higher doses of SERT inhibitors produce a greater occupation of SERT and may produce greater increases of 5-HT transmission (Meyer et al., 2001; Voineskos et al., 2007). This principle can be applied to conceptualize why higher doses of SERT inhibitors are necessary to produce more consistent benefits in obsessive compulsive disorder (Katzman et al., 2014). Accordingly, a more recent PET imaging study showed that the occupancy of SERT by minimal effective doses of es/citalopram in depression is higher in the anterior/posterior/subgenual cingulate cortex, regions more intimately involved in depression, than in the orbitofrontal gyrus, which has been linked more closely with obsessive compulsive disorder (Baldinger et al., 2014). With this caveat in mind, the 40% decrease in whole-blood 5-HT produced by atomoxetine, or the 18% decrease observed with a 7-day regimen of desipramine (Blier et al., 2007), would presumably not be sufficient to increase 5-HT transmission through this mechanism, given the significant reserve of SERT able to clear extracellular 5-HT. Taken together, these results clearly support and confirm the principle of SERT reserve with respect to the degree of inhibition necessary to even begin to modify synaptic 5-HT.
In a prior clinical study from our research unit, regimens of 75, 225, and 375 mg/d of venlafaxine were studied using the tyramine pressor response (Debonnel et al., 2007). Using the ED30 method, only the 2 higher regimens were efficacious, thus leaving the question open whether 150 mg/d would still be enough to inhibit NET. The present results show that 150 mg/d is still primarily a SSRI dose in most individuals. This interpretation is consistent with similar remission rates obtained with a switch to either sertraline (mean: 136 mg/d) or venlafaxine (mean: 194 mg/d) after failing to respond to an average of 42 mg/d of citalopram (Rush et al., 2006). However, the superiority of venlafaxine (mean: 272 mg/d) over paroxetine (mean: 36 mg/d) was shown in a sample of 125 patients after failing prior antidepressant attempts (Poirier and Boyer, 1999). In contrast, using PET imaging of the NET, recent results indicate that doses >150 mg/d of venlafaxine did not show greater occupancy (Arakawa et al., 2019). As mentioned in the Introduction, there seem to be signal to noise problems with the PET ligand used to label NET in the brain. For instance, occupancies or non-displaceable binding potentials of that reboxetine derivative used as a PET ligand cannot be estimated in regions such as the hippocampus and the nucleus accumbens because of very high or even negative values (Sekine et al., 2010). Therefore, using PET imaging in the brain currently does not necessarily provide a reliable ratio of differential occupancy of SERT to NET given a plateau of approximately 50% obtained with the PET ligand to label NET using drugs such as nortriptyline and milnacipran (Nogami et al., 2013; Takano et al., 2014). Thus, given the correlations between plasma levels of venlafaxine + ODV and the functional measure of NET inhibition (the ED30 values) reported herein, it appears that the least cumbersome way to ensure that NET is engaged with venlafaxine is to use the daily dose of 225 mg/d. This dose yielded the same mean plasma level of 376 ng/mL as in the prior report of Owens et al. (2008).
The present study has some limitations. First, the number of subjects tested with each medication was relatively small. Nevertheless, the results of the physiological estimates of SERT and NET inhibition were fully consistent with those of a prior study with twice the sample size in each venlafaxine group (Debonnel et al., 2007). Second, both models used herein estimated reuptake inhibition in the periphery and not in the brain. However, even assessing SERT occupation in the brain using PET imaging has revealed differential degrees of occupation in various brain areas (Baldinger et al., 2014; Silberbauer et al., 2019). It will thus be interesting to examine the selectivity ratio using the whole-blood 5-HT content and the tyramine pressor test with other agents such as milnacipran and levomilnacipran, which have in vitro/in vivo selectivity ratios more oriented towards NET than SERT (Koch et al., 2003; Auclair et al., 2013).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Interest Statement
This research was initially funded by Wyeth Pharmaceuticals. Blier was supported by a Tier I Canada Research Chair and a departmental Research Chair, received grant funding and/or honoraria for lectures, expert testimony, and/or participation in advisory boards for Allergan, Bristol Myers Squibb, Eli Lilly, Janssen, Lundbeck, Otsuka, Pfizer, and Wyeth. Norris, Tremblay, James, Ritchie, and Aldosary declare no conflict of interest.
References
- American Psychiatric Association (2000) Diagnostic and statistical manual of mental disorders. 4th ed., text revision. Washington, DC: American Psychiatric Association. [Google Scholar]
- Arakawa R, Stenkrona P, Takano A, Svensson J, Andersson M, Nag S, Asami Y, Hirano Y, Halldin C, Lundberg J (2019) Venlafaxine ER blocks the norepinephrine transporter in the brain of patients with major depressive disorder: a PET study using [18F]FMeNER-D2. Int J Neuropsychopharmacol 22:278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artigas F, Sarrias MJ, Martínez E, Gelpí E (1985) Serotonin in body fluids: characterization of human plasmatic and cerebrospinal fluid pools by means of a new HPLC method. Life Sci 37:441–447. [DOI] [PubMed] [Google Scholar]
- Auclair AL, Martel JC, Assié MB, Bardin L, Heusler P, Cussac D, Marien M, Newman-Tancredi A, O’Connor JA, Depoortère R (2013) Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology 70:338–347. [DOI] [PubMed] [Google Scholar]
- Baldinger P, Kranz GS, Haeusler D, Savli M, Spies M, Philippe C, Hahn A, Höflich A, Wadsak W, Mitterhauser M, Lanzenberger R, Kasper S (2014) Regional differences in SERT occupancy after acute and prolonged SSRI intake investigated by brain PET. Neuroimage 88:252–262. [DOI] [PubMed] [Google Scholar]
- Béïque JC, de Montigny C, Blier P, Debonnel G (1998) Blockade of 5-hydroxytryptamine and noradrenaline uptake by venlafaxine: a comparative study with paroxetine and desipramine. Br J Pharmacol 125:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béïque JC, de Montigny C, Blier P, Debonnel G (1999) Venlafaxine: discrepancy between in vivo 5-HT and NE reuptake blockade and affinity for reuptake sites. Synapse 32:198–211. [DOI] [PubMed] [Google Scholar]
- Blier P (2008) Resiliency of monoaminergic systems: the 80% rule and its relevance to drug development. J Psychopharmacol 22:587–589. [DOI] [PubMed] [Google Scholar]
- Blier P, Saint-André E, Hébert C, de Montigny C, Lavoie N, Debonnel G (2007) Effects of different doses of venlafaxine on serotonin and norepinephrine reuptake in healthy volunteers. Int J Neuropsychopharmacol 10:41–50. [DOI] [PubMed] [Google Scholar]
- Brewerton TD, Flament MF, Rapoport JL, Murphy DL (1993) Seasonal effects on platelet 5-HT content in patients with OCD and controls. Arch Gen Psychiatry 50:409. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, Morin SM, Gehlert DR, Perry KW (2002) Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27:699–711. [DOI] [PubMed] [Google Scholar]
- Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, Leucht S, Ruhe HG, Turner EH, Higgins JPT, Egger M, Takeshima N, Hayasaka Y, Imai H, Shinohara K, Tajika A, Ioannidis JPA, Geddes JR (2018) Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet 391:1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debonnel G, Saint-André E, Hébert C, de Montigny C, Lavoie N, Blier P (2007) Differential physiological effects of a low dose and high doses of venlafaxine in major depression. Int J Neuropsychopharmacol 10:51–61. [DOI] [PubMed] [Google Scholar]
- Demet EM, Halaris AE, Bhatarakamol S (1978) Indoleamine compartmentation in human blood. Clin Chim Acta 89:285–292. [DOI] [PubMed] [Google Scholar]
- Ding YS, Naganawa M, Gallezot JD, Nabulsi N, Lin SF, Ropchan J, Weinzimmer D, McCarthy TJ, Carson RE, Huang Y, Laruelle M (2014) Clinical doses of atomoxetine significantly occupy both norepinephrine and serotonin transports: implications on treatment of depression and ADHD. Neuroimage 86:164–171. [DOI] [PubMed] [Google Scholar]
- Farid NA, Bergstrom RF, Ziege EA, Parli CJ, Lemberger L (1985) Single-dose and steady-state pharmacokinetics of tomoxetine in normal subjects. J Clin Pharmacol 25:296–301. [DOI] [PubMed] [Google Scholar]
- Ghose K (1984) Tyramine pressor test: implications and limitations. Methods Find Exp Clin Pharmacol 6:455–464. [PubMed] [Google Scholar]
- Ghose K, Turner P (1975) Intravenous tyramine pressor response in depression. Lancet 1:1317–1318. [DOI] [PubMed] [Google Scholar]
- Gilmor ML, Owens MJ, Nemeroff CB (2002) Inhibition of norepinephrine uptake in patients with major depression treated with paroxetine. Am J Psychiatry 159:1702–1710. [DOI] [PubMed] [Google Scholar]
- Greenberg BD, Tolliver TJ, Huang SJ, Li Q, Bengel D, Murphy DL (1999) Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet 88:83–87. [PubMed] [Google Scholar]
- Guy W (1976) Assessment manual of psychopharmacology. Revised. Rockville, MD: National Institute of Mental Health. [Google Scholar]
- Hamilton M (1960) A rating scale for depression. J Neurol Neurosurg Psychiatry 23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker LA, Finch CA (1969) Thrombokinetics in man. J Clin Invest 48:963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AT, Rudolph RL, Preskorn SH (2000) Evidence of the dual mechanisms of action of venlafaxine. Arch Gen Psychiatry 57:503–509. [DOI] [PubMed] [Google Scholar]
- Hoffman BB, Lefkowitz RJ (1990) Catecholamines and sympathomimetic drugs. In: Goodman and Gilman’s the pharmacological basis of therapeutics, 8th ed. (Gilman AG, Rall TW, Nies AS, Taylor P, eds.), pp. 187–220. New York, NY: Macmillan Publishing Co. [Google Scholar]
- Kato M, Serretti A (2010) Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 15:473–500. [DOI] [PubMed] [Google Scholar]
- Katzman MA, Bleau P, Blier P, Chokka P, Kjernisted K, Van Ameringen M (2014) Canadian clinical practice guidelines for the management of anxiety, posttraumatic stress and obsessive-compulsive disorders. BMC Psychiatry 14(Suppl 1):S1–S83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Hemrick-Luecke SK, Thompson LK, Evans DC, Threlkeld PG, Nelson DL, Perry KW, Bymaster FP (2003) Comparison of effects of dual transporter inhibitors on monoamine transporters and extracellular levels in rats. Neuropharmacology 45:935–944. [DOI] [PubMed] [Google Scholar]
- Logan J, Wang GJ, Telang F, Fowler JS, Alexoff D, Zabroski J, Jayne M, Hubbard B, King P, Carter P, Shea C, Xu Y, Muench L, Schlyer D, Learned-Coughlin S, Cosson V, Volkow ND, Ding YS (2007) Imaging the norepinephrine transporter in humans with (S,S)-[11C]O-methyl reboxetine and PET: problems and progress. Nucl Med Biol 34:667–679. [DOI] [PubMed] [Google Scholar]
- Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K, Houle S (2001) Occupancy of serotonin transporters by paroxetine and citalopram during treatment of depression: a [(11)C]DASB PET imaging study. Am J Psychiatry 158:1843–1849. [DOI] [PubMed] [Google Scholar]
- Michelson D, Adler LA, Amsterdam JD, Dunner DL, Nierenberg AA, Reimherr FW, Schatzberg AF, Kelsey DK, Williams DW. Michelson D (2007) Addition of atomoxetine for depression incompletely responsive to sertraline: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry 68:582–587. [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Asberg M (1979) A new depression scale designed to be sensitive to change. Br J Psychiatry 134:382–389. [DOI] [PubMed] [Google Scholar]
- Newport DJ, Ritchie JC, Knight BT, Glover BA, Zach EB, Stowe ZN (2009) Venlafaxine in human breast milk and nursing infant plasma: determination of exposure. J Clin Psychiatry 70:1304–1310. [DOI] [PubMed] [Google Scholar]
- Nogami T, Takano H, Arakawa R, Ichimiya T, Fujiwara H, Kimura Y, Kodaka F, Sasaki T, Takahata K, Suzuki M, Nagashima T, Mori T, Shimada H, Fukuda H, Sekine M, Tateno A, Takahashi H, Ito H, Okubo Y, Suhara T (2013) Occupancy of serotonin and norepinephrine transporter by milnacipran in patients with major depressive disorder: a positron emission tomography study with [(11)C]DASB and (S,S)-[(18)F]FMeNER-D(2). Int J Neuropsychopharmacol 16:937–943. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Morgan WN, Plott SJ, Nemeroff CB (1997) Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther 283:1305–1322. [PubMed] [Google Scholar]
- Owens MJ, Krulewicz S, Simon JS, Sheehan DV, Thase ME, Carpenter DJ, Plott SJ, Nemeroff CB (2008) Estimates of serotonin and norepinephrine transporter inhibition in depressed patients treated with paroxetine or venlafaxine. Neuropsychopharmacology 33:3201–3212. [DOI] [PubMed] [Google Scholar]
- Parsey RV, Hastings RS, Oquendo MA, Hu X, Goldman D, Huang YY, Simpson N, Arcement J, Huang Y, Ogden RT, Van Heertum RL, Arango V, Mann JJ (2006) Effect of a triallelic functional polymorphism of the serotonin-transporter-linked promoter region on expression of serotonin transporter in the human brain. Am J Psychiatry 163:48–51. [DOI] [PubMed] [Google Scholar]
- Phillips JL, Batten LA, Tremblay P, Aldosary F, Du L, Blier P (2015) Impact of monoamine-related gene polymorphisms on hippocampal volume in treatment-resistant depression. Acta Neuropsychiatr 27:353–361. [DOI] [PubMed] [Google Scholar]
- Pletscher A (1986) Blood platelets as neuronal models: use and limitations. Clin Neuropharmacol 9(Suppl 4):344–346. [PubMed] [Google Scholar]
- Poirier MF, Boyer P (1999). Venlafaxine and paroxetine in treatment-resistant depression. Double-blind, randomised comparison. Br J Psychiatry 175:12–16. [DOI] [PubMed] [Google Scholar]
- Ren F, Ma Y, Zhu X, Guo R, Wang J, He L (2020) Pharmacogenetic association of bi- and triallelic polymorphisms of SLC6A4 with antidepressant response in major depressive disorder. J Affect Disord 273:254–264. [DOI] [PubMed] [Google Scholar]
- Ritchie JC, Glover B, Ramsey C, Scott-Harrell P (2009) A routine UPLC-ms/ms assay for the newer antidepressants. Ther Drug Monit 31:646. [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, Ritz L, Biggs MM, Warden D, Luther JF, Shores-Wilson K, Niederehe G, Fava M; STAR*D Study Team (2006) Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 354:1231–1242. [DOI] [PubMed] [Google Scholar]
- Sekine M, Arakawa R, Ito H, Okumura M, Sasaki T, Takahashi H, Takano H, Okubo Y, Halldin C, Suhara T (2010) Norepinephrine transporter occupancy by antidepressant in human brain using positron emission tomography with (S,S)-[18F]FMeNER-D2. Psychopharmacology (Berl) 210:331–336. [DOI] [PubMed] [Google Scholar]
- Silberbauer LR, Gryglewski G, Berroterán-Infante N, Rischka L, Vanicek T, Pichler V, Hienert M, Kautzky A, Philippe C, Godbersen GM, Vraka C, James GM, Wadsak W, Mitterhauser M, Hacker M, Kasper S, Hahn A, Lanzenberger R (2019) Serotonin transporter binding in the human brain after pharmacological challenge measured using PET and PET/MR. Front Mol Neurosci 12:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer RL, Williams JB (1988) Revised diagnostic criteria and a new structured interview for diagnosing anxiety disorders. J Psychiatr Res 22 Suppl 1:55–85. [DOI] [PubMed] [Google Scholar]
- Takano H, Arakawa R, Nogami T, Suzuki M, Nagashima T, Fujiwara H, Kimura Y, Kodaka F, Takahata K, Shimada H, Murakami Y, Tateno A, Yamada M, Ito H, Kawamura K, Zhang MR, Takahashi H, Kato M, Okubo Y, Suhara T (2014) Norepinephrine transporter occupancy by nortriptyline in patients with depression: a positron emission tomography study with (S,S)-[18F]FMeNER-D₂. Int J Neuropsychopharmacol 17:553–560. [DOI] [PubMed] [Google Scholar]
- Tatsumi M, Groshan K, Blakely RD, Richelson E (1997) Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 340:249–258. [DOI] [PubMed] [Google Scholar]
- Turcotte JE, Debonnel G, de Montigny C, Hébert C, Blier P (2001) Assessment of the serotonin and norepinephrine reuptake blocking properties of duloxetine in healthy subjects. Neuropsychopharmacology 24:511–521. [DOI] [PubMed] [Google Scholar]
- Vincent S, Bieck PR, Garland EM, Loghin C, Bymaster FP, Black BK, Gonzales C, Potter WZ, Robertson D (2004) Clinical assessment of norepinephrine transporter blockade through biochemical and pharmacological profiles. Circulation 109:3202–3207. [DOI] [PubMed] [Google Scholar]
- Voineskos AN, Wilson AA, Boovariwala A, Sagrati S, Houle S, Rusjan P, Sokolov S, Spencer EP, Ginovart N, Meyer JH (2007) Serotonin transporter occupancy of high-dose selective serotonin reuptake inhibitors during major depressive disorder measured with [11C]DASB positron emission tomography. Psychopharmacology 193:539–545. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Takahashi H, Higuchi H, Kamata M, Ito K, Sato K, Naito S, Shimizu T, Itoh K, Inoue K, Suzuki T, Nemeroff CB (2004) Prediction of antidepressant response to milnacipran by norepinephrine transporter gene polymorphisms. Am J Psychiatry 161:1575–1580. [DOI] [PubMed] [Google Scholar]
- Zhao X, Cui W, Liu Q, Cao S, Pang J, Li H (2020) The T-182C polymorphism enhances promoter activity of the norepinephrine transporter gene, but may not be associated with antidepressant response. Genet Test Mol Biomarkers 24:812–818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.