

Abstract
PEGylation is an attractive approach to modifying oligonucleotides intended for therapeutic purposes. The PEG conjugation reduces protein interaction with the oligonucleotide, and helps to overcome its intrinsic biopharmaceutical shortcomings, such as poor enzymatic stability, rapid body clearance, and unwanted immunostimulation. While it is known that the molecular weight and architecture of the PEG play an important role in its effectiveness, the manner in which the PEG component interferes with the hybridization of the oligonucleotide remains poorly understood. In this study, we systematically compare the hybridization thermodynamics and protein accessibility of several DNA conjugates involving linear, Y-shaped, and brush-type PEG. It is found that the PEGylated DNA experiences two opposing effects: local excluded volume effect and chemical interaction, the strengths of which are architecture-dependent. Notably, the brush architecture is able to offer significantly greater protein shielding capacity than its linear or Y-shaped counterparts, while maintain nearly identical free energy for DNA hybridization compared with free DNA.
Keywords: oligonucleotides, polymers, PEGylation, bioconjugate, hybridization thermodynamics
Graphical Abstract
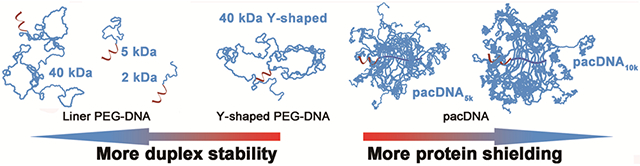
We synthesized a series of PEG-DNA conjugates and made quantitative comparisons of their hybridization thermodynamics and protein accessibility. It is found that the DNA experiences two effects from the PEG component: an excluded volume effect and a chemical interaction effect, which work in opposite directions on duplex stability and are polymer architecture-dependent. The brush architecture is found to be significantly more capable in protein shielding than linear PEG.
PEGylation is the process to covalently attach one or multiple chains of polyethylene glycol (PEG) to a target molecule.[1] Upon conjugation with PEG, the pharmacokinetics of the target molecule can oftentimes be greatly enhanced, owing to the “stealth” properties of the polymer.[2] For oligonucleotide therapeutics, which involve antisense DNA,[3] RNA interference,[4] aptamers,[5] etc., PEGylation is particularly important because oligonucleotides exhibit a range of specific or non-specific, non-hybridization interactions with cell surface receptors or serum components, which lead to unwanted hepatic uptake, poor stability, and many side effects including flu-like symptoms and coagulopathy.[6] Various types of PEG-oligonucleotide conjugates have been synthesized and studied, with some reaching clinical trials and regulatory approval.[4b,7] Pegaptanib (brand name Macugen®), a 40 kDa Y-shaped PEG-modified 28-base aptamer, has been approved by FDA and on the market since 2004.
While PEGylation in general creates a shielding effect for the conjugated oligonucleotide, protecting it from unwanted interactions, its effectiveness is tightly associated with the molecular weight and architecture of the PEG.[8] In addition, extensive PEGylation may also alter the intrinsic thermal stability of attached dsDNA.[8b] How does the PEG component affect the thermodynamics of DNA hybridization in a conjugate? What molecular parameter of the PEG is important to achieve best protein shielding while maintaining DNA hybridization? These are important questions that have not been thoroughly investigated. Recently, we have designed a novel form of PEG-DNA conjugate, termed pacDNA (Polymer-assisted Compaction of DNA), which consists of 1-5 strands of oligonucleotides tethered to the backbone of a brush polymer having dense PEG side chains.[9] The pacDNA is of a highly branched architecture, which generates much greater steric congestion compared with linear PEG-DNA conjugates and can better shield the DNA from enzymatic degradation, while allowing seemingly unhindered access by complementary DNA strands.[10] It is of significant interest to investigate whether the extensive PEGylation by the brush architecture negatively impacts the stability of the embedded dsDNA.
In order to study how different PEGylation strategies impact DNA hybridization, we synthesized a library of PEG-DNA conjugates, using linear PEG of different molecular weight (Mn=2, 5, 40 kDa, PDI<1.05), Y-shaped PEG (Mn=40 kDa, each arm 20 kDa, PDI<1.05), and brushes with 5 kDa and 10 kDa PEG side chains (Mn=197.2 and 329.1 kDa, respectively; PDI<1.15) ( Scheme 1 ). A short, 10-mer DNA strand (5’-CCC AGC CCT C-Fluo-3’) is used as a model system to study hybridization. This choice is based on the fact that association and dissociation processes for short sequences can be regarded as a two-state, all or none, fully reversible transition, which provides a more accurate model to study thermodynamic and kinetic processes.[11] In addition, a short duplex tends not to involve structural change with or without PEG attached.[12]
Scheme 1.

Structure of linear, Y-shaped, and brush-type PEG-DNA conjugates.
Different chemistries are chosen to synthesize the PEG-DNA conjugates. For linear and Y-shaped PEG, conjugates are achieved by reacting N-hydroxysuccinimide (NHS)-terminated PEG with amine-modified DNA in 0.1 M bicarbonate/carbonate buffer (pH~9) at 1 °C overnight. The resulting conjugates are then purified by electroelution from 1% agarose gel. For brush polymers, amidation reaction proved to be very low yielding, and copper-free click chemistry is instead selected. The brush polymers are synthesized via sequential ring-opening metathesis polymerization (ROMP) of norbornenyl bromide (N-Br) and norbornenyl PEG (N-PEG), followed by bromide substitution by sodium azide (Table 1, Figure S1).[13] Subsequently, 5’-dibenzocyclooctyl (DBCO)-modified oligonucleotide is reacted with the diblock brush in Nanopure™ water at 40 °C overnight, to yield the pacDNAs. Aqueous gel permeation chromatography (GPC) is used to isolate pacDNA from unreacted free DNA. The number of DNA strands per brush is determined to be ~1 for both pacDNAs by peak integration (~60% yield, Figure S2). GPC analyses and agarose gel electrophoresis indicate the successful synthesis of the PEG-DNA conjugates (Figure 1).
Table 1.
GPC Analysis for the Brush Polymer Used.
| Polymer | Composition | Mn(kDa) | Mw(kDa) | PDI |
|---|---|---|---|---|
| pacDNA5k | p(N-N3)5-b-p(N-PEG5k)35 | 178.8 | 197.2 | 1.10 |
| pacDNA10k | p(N-N3)5-b-p(N-PEG10k)28 | 285.5 | 329.1 | 1.15 |
Figure 1.

Aqueous GPC chromatograms and agarose gel (0.5%) electrophoresis of free DNA and six PEG-DNA conjugates.
To investigate the hybridization thermodynamics of PEG-DNA conjugates, we prepared duplexes of the conjugates with a dabcyl-modified complementary DNA. Hybridization brings the molecular quencher (dabcyl) to close proximity with the fluorescein at the 3’ of the nucleic acid, thus quenching its fluorescence. Upon denaturing by heating, the dabcyl-modified strand separates, and the increase of fluorescent signals allows for a melting curve to be recorded. In a typical study, PEG-DNA conjugates and free complementary DNA are mixed in 1:1 molar ratio in phosphate buffered saline (PBS, pH=7.4) at six different total DNA concentrations (CT, 31 nM to 235 nM Table S1), followed by annealing at 90 °C for 20 min and cooling to room temperature during a period of 10 h. The melting transitions (Tm) of the duplexes are determined by analyzing the first-order derivative of the melting curves (Figure 2A). With a series of Tm-CT relationships (Figure 2B), a van’t Hoff plot can be generated according to the following equation:
where Tm is the melting temperature, R is the gas constant, and CT is the total concentration of DNA. ΔH° and ΔS° are enthalpy and entropy of hybridization, respectively.[14] The hybridization thermodynamic parameters for DNA and PEG-DNA conjugates are listed in Table 2.
Figure 2.

Typical melting curves of free DNA and PEG-DNA conjugates. (B) Linear fit of concentration-dependent melting data.
Table 2.
Thermodynamics of Hybridization for Free DNA and PEG-DNA Conjugates
| ΔH° (kJ/mol) | ΔS° (J/mol·K) | ΔG° at 298k (kJ/mol) | Keq at 298K (M−1) | PEG density (g/L)[a] | |
|---|---|---|---|---|---|
| Free DNA | −264.9 ± 1.5 | −677 ± 4 | −63.0 ± 0.5 | 1.1×1011 ± 8.5×108 | 0 |
| 2 kDa PEG-DNA | −282.0 ± 4.5 | −731 ± 1 | −64.3 ± 1.4 | 1.9×1011 ± 4.2×109 | n.a. |
| 5 kDa PEG-DNA | −290.0 ± 3.2 | −754 ± 8 | −65.2 ± 1.0 | 2.6×1011 ± 4.1×109 | 118 ± 20 |
| pacDNA5k | −309.8 ± 3.8 | −822 ± 1 | −64.8 ± 1.1 | 2.2×1011 ± 3.9×109 | 159 ± 2 |
| pacDNA10k | −315.1 ± 2.6 | −839 ± 7 | −64.9 ± 0.8 | 2.3×1011 ± 2.7×109 | 143 ± 11 |
| 40 kDa YPEG-DNA | −339.0 ± 4.0 | −904 ± 1 | −69.5 ± 1.2 | 1.5×1012 ± 2.5×1010 | 116 ± 2 |
| 40 kDa PEG-DNA | −340.9 ± 4.1 | −909 ± 1 | −69.9 ± 1.2 | 1.8×1012 ± 3.0×1010 | 128 ± 7 |
Densities of PEG are estimated from the Mw and number-average hydrodynamic volume of the polymer, as determined by dynamic light scattering. Due to instrument limitations, data is not available for the 2 kDa PEG-DNA conjugate.
Analyzing the melting data, we find that, for linear and Y-shaped PEG-DNA conjugates, the binding constant (Keq) increases with increasing molecular weight of the PEG, with the 40 kDa PEG conjugate (linear and Y-shaped) showing the highest Keq (~14-16x relative to free duplex). This observation can be explained by the volume exclusion effect. It has been previously discovered that high molecular weight PEG (typically >5 kDa) can decrease the enthalpy of duplex melting and stabilize double-stranded nucleic acids as a cosolute with DNA (as opposed to being conjugated).[15] The excluded volume of the PEG increases the effective concentration of the oligonucleotide, leading to more favorable binding. However, for the conjugated systems, the increased binding is observed at PEG concentrations far below that for cosolute systems (~0.005 vs >50 g/L), indicating that there is a local, unimolecular excluded volume effect. This effect can be understood by imagining the melting of a dsDNA-PEG conjugate: at Tm, two ssDNA emerge from a duplex, and the increased volume of the DNA reduces the space that the PEG occupies, which makes the melting less favorable compared with free dsDNA. Since the PEG and DNA are conjugated, within the gyration radius of the conjugate, the weight percentage of the PEG does not change, which explains why the local volume exclusion effect is independent of concentration in dilute systems.
Comparing the pacDNA binding constants with that of linear and Y-shape conjugates, we found that the pacDNAs show only slightly increased binding constants (~2x relative to free duplex), despite the fact that both pacDNAs have higher molecular weight (197.2 and 329.1 kDa) than the highest linear PEG tested (40 kDa). This observation contradicts what one would predict based on the local excluded volume effect (vide supra), because the brush polymers are more sterically congested than linear PEG owing to the high-density side chains, which should yield a greater extent of molecular crowding and thus higher Keq. The same prediction would be made using the local PEG density argument as well (Table 2). Therefore, a counteracting interaction must be at play. We attribute this unfavorable interaction to the chemical interaction between the DNA and the PEG.[16] The brush structure provides the embedded DNA with a high local PEG density, which diminishes the stability of Watson-Crick base pairing by displacing water molecules hydrating the DNA. Therefore, while both the local excluded volume effect and chemical interaction exist for all of the PEG-DNA conjugates, their relative strength is dependent on the polymer architecture. For linear for slightly branched PEG (Y-shaped), the excluded volume effect is dominant, which favors hybridization. Highly branched PEG (brush) results in increases in both effects, with a faster increase in the destabilizing chemical interaction.
In order to validate our findings, we compared conjugates to mixtures of free DNA and PEG in a cosolute system. Previous studies using high molecular weight PEG (>1 kDa) have shown that they increase thermal stability of DNA duplexes at low concentrations, and the effect is reversed as the PEG concentration is increased.[14a] It is of interest to see if the cosolute result can be used to predict the thermal stability of conjugates. We measured the Tm of dsDNA with and without PEG (2-40 kDa linear, 50-200 g/L) at a dsDNA concentration of 235 nM (Figure 3), and the results are consistent with prior work.[17] In order to estimate the PEG density of the conjugates, dynamic light scattering (DLS) is used to measure the number-average hydrodynamic radii (Figure S3). Knowing the molecular weight of the PEG component of the conjugates, and assuming a spherical structure, the local PEG density can be estimated (Table 2). It is found that the Tm values for linear and Y-shaped PEG-DNA conjugates are similar to the corresponding cosolute solution matching their estimated local PEG density. However, both pacDNAs showed significantly lower Tm than that of the corresponding cosolutes. We attribute this result to the unique structure of pacDNA, which consists of densely-packed, high molecular weight side chains. Because of the congested environment, the side chains are forced to adopt a more extended conformation, which gives them more surface area to interact with the DNA than a coiled chain, resulting in particularly pronounced chemical interaction and thus a lower Tm. Nonetheless, all PEG-DNA conjugates remain hybridizable, and show nearly identical hybridization kinetics (Figure S4), consistent with our previous findings.[9a]
Figure 3.

Melting temperature of free dsDNA (235 nM) in PBS as a function of cosolute (PEG) concentration (dashed line: no PEG present). Stars show the estimated local concentration (density) of PEG-DNA conjugates, and their melting temperatures.
While the brush architecture does not enhance DNA duplex stability more than linear PEG, the strong local volume exclusion effect may prove to be useful for the development of oligonucleotide biopharmaceuticals, where prevention of protein access is of greater importance. In order to test how different PEG architectures can shield the conjugated DNA from proteins, the endonuclease, DNase I, is selected as a model system to act upon dsDNA modified with a quencher-fluorophore pair (Figure 4A). Upon dsDNA cleavage, the fluorophore is released, leading to an increase of fluorescence, the rate of which is indicative of the enzymatic degradation kinetics. When DNase I (0.1 unit/mL) is introduced to pre-hybridized dsDNA or PEG-dsDNA conjugates (150 nM dsDNA), it is found that the protective capability of either linear or Y-shaped PEG is limited, with half-lives (t1/2) being no greater than 7.8±0.2 min, barely above that of free dsDNA (5.9±0.2 min). On the other hand, the pacDNAs show significant enzyme inhibition, with the pacDNA5k t1/2 being 78±2 min and pacDNA10k t1/2 being 180±3 min. These results indicate that high density brush environment creates a significant kinetic barrier against large species such as proteins, while the barrier for small species (e.g. single stranded oligonucleotide) is trivial.
Figure 4.

(A) Scheme of fluorescence assay for determining DNA nuclease degradation kinetics. (B) Nuclease degradation kinetics for free DNA vs PEG-DNA conjugates. (C) Nuclease degradation kinetics for free DNA with PEG (160 g/L) as a cosolute.
It is important not to confuse the local volume exclusion with the bulk volume exclusion. Within the radius of gyration of the polymer, the volume exclusion is the same as bulk, where any volume-reducing reaction is thermodynamically favored (“squeezed together”). If a species is not stabilized by entering the brush sphere, it will instead be sterically repelled (“squeezed out”), which is not an option for bulk volume exclusion. Therefore, the local excluded volume effect works in the opposite fashion to the bulk volume exclusion for larger species. To demonstrate, we compared with rate of DNase I cleavage of dsDNA in concentrated PEG solution (160 g/L, similar to pacDNA’s local PEG density) in DNase I buffer. It is found that the cosolute PEG accelerates the enzymatic cleavage of dsDNA by DNase I, which is in stark contrast with pacDNA, which retards cleavage.
In conclusion, we have systematically characterized and compared the hybridization thermodynamics and protein accessibility of free DNA and several PEG-DNA conjugates. Our results suggest two opposing effects are at play: local excluded volume effect, which enhances hybridization, and chemical interaction, which destabilizes hybridization. The strength of the two effects is architecture-dependent. Although the brush architecture is influenced more by chemical interaction than linear conjugates, the overall free energy for pacDNA hybridization is nearly the same to that of free DNA. In addition, the pacDNAs exhibit superior shielding against proteins to linear or branched PEG-DNA conjugates, which is a critically important aspect in designing oligonucleotide therapeutics. Collectively, our data suggest that the brush architecture is more appropriate for PEGylating oligonucleotides for biopharmaceutical applications.
Supplementary Material
Acknowledgements
Financial support from Northeastern University start-up, NEU-DFCI seed grant, and NSF CAREER award 1453255 is gratefully acknowledged.
References
- [1].a) Harris JM, Chess RB, Nat. Rev. Drug Discov 2003, 2, 214–221. [DOI] [PubMed] [Google Scholar]; b) Li W, Zhan P, De Clercq E, Lou H, Liu X, Prog. Polym. Sci 2013, 38, 421–444. [Google Scholar]; c) Lee H, Polymers 2014, 6, 776–798. [Google Scholar]
- [2].a) Veronese FM, Pasut G, Drug Discov. Today 2005, 10, 1451–1458. [DOI] [PubMed] [Google Scholar]; b) Wang J, Mao W, Lock LL, Tang J, Sui M, Sun W, Cui H, Xu D, Shen Y, ACS Nano 2015, 9, 7195–7206. [DOI] [PubMed] [Google Scholar]
- [3].a) Wang S, Lee RJ, Cauchon G, Gorenstein DG, Low PS, Proc. Natl. Acad. Sci 1995, 92, 3318–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tan X, Lu X, Jia F, Liu X, Sun Y, Logan JK, Zhang K, J. Am. Chem. Soc 2016, 138, 10834–10837. [DOI] [PubMed] [Google Scholar]; c) Young KL, Scott AW, Hao L, Mirkin SE, Liu G, Mirkin CA, Nano Lett. 2012, 12, 3867–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhu G, Zheng J, Song E, Donovan M, Zhang K, Liu C, Tan W, Proc. Natl. Acad. Sci 2013, 110, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang K, Fang H, Shen G, Taylor J-SA, Wooley KL, Proc. Am. Thorac. Soc 2009, 6, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Hanessian S, Schroeder BR, Giacometti RD, Merner BL, Østergaard M, Swayze EE, Seth PP, Angew. Chem. Int. Ed 2012, 51, 11242–11245. [DOI] [PubMed] [Google Scholar]; b) Aartsma-Rus A, Mol. Ther 2016, 24, 193–194. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA, J. Am. Chem. Soc 2009, 131, 2072–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zheng D, Giljohann DA, Chen DL, Massich MD, Wang X-Q, Iordanov H, Mirkin CA, Paller AS, Proc. Natl. Acad. Sci 2012, 109, 11975–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Rush AM, Nelles DA, Blum AP, Barnhill SA, Tatro ET, Yeo GW, Gianneschi NC, J. Am. Chem. Soc 2014, 136, 7615–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Keefe AD, Pai S, Ellington A, Nat. Rev. Drug Discov 2010, 9, 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu G, Niu G, Chen X, Bioconjug. Chem 2015, 26, 2186–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fang X, Tan W, Acc. Chem. Res 2010, 43, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Ogris M, Brunner S, Schüller S, Kircheis R, Wagner E, Gene Ther. 1999, 6, 595–605. [DOI] [PubMed] [Google Scholar]; b) Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ, Nature 1992, 355, 564–566. [DOI] [PubMed] [Google Scholar]; c) Verma IM, Somia N, Nature 1997, 389, 239–242. [DOI] [PubMed] [Google Scholar]; d) Medzhitov R, Nat. Rev. Immunol 2001, 1, 135–145. [DOI] [PubMed] [Google Scholar]
- [7].Ng EWM, Shima DT, Calias P, Cunningham ET, Guyer DR, Adamis AP, Nat. Rev. Drug Discov 2006, 5, 123–132. [DOI] [PubMed] [Google Scholar]
- [8].a) Ikeda Y, Nagasaki Y, J. Appl. Polym. Sci 2014, 131, 40293. [Google Scholar]; b) Bonora GM, Ivanova E, Zarytova V, Burcovich B, Veronese FM, Bioconjug. Chem 1997, 8, 793–797. [DOI] [PubMed] [Google Scholar]
- [9].a) Lu X, Tran T, Jia F, Tan X, Davis S, Krishnan S, Amiji MM, Zhang K, J. Am. Chem. Soc 2015, 137, 12466–12469. [DOI] [PubMed] [Google Scholar]; b) Lu X, Watts E, Jia F, Tan X, Zhang K, J. Am. Chem. Soc 2014, 136, 10214–10217. [DOI] [PubMed] [Google Scholar]
- [10].Lu X, Jia F, Tan X, Wang D, Cao X, Zheng J, Zhang K, J. Am. Chem. Soc 2016, 138, 9097–9100. [DOI] [PubMed] [Google Scholar]
- [11].a) Turner DH, Sugimoto N, Freier SM, Annu. Rev. Biophys. Biophys. Chem 1988, 17, 167–192. [DOI] [PubMed] [Google Scholar]; b) Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M, Biochemistry (Mosc.) 1995, 34, 11211–11216. [DOI] [PubMed] [Google Scholar]
- [12].Nakano S, Karimata H, Ohmichi T, Kawakami J, Sugimoto N, J. Am. Chem. Soc 2004, 126, 14330–14331. [DOI] [PubMed] [Google Scholar]
- [13].a) Barner-Kowollik C, Du Prez FE, Espeel P, Hawker CJ, Junkers T, Schlaad H, Van Camp W, Angew. Chem. Int. Ed 2011, 50, 60–62. [DOI] [PubMed] [Google Scholar]; b) Liu J, Burts AO, Li Y, Zhukhovitskiy AV, Ottaviani MF, Turro NJ, Johnson JA, J. Am. Chem. Soc 2012, 134, 16337–16344. [DOI] [PubMed] [Google Scholar]; c) Jia F, Lu X, Tan X, Zhang K, Chem. Commun 2015, 51, 7843–7846. [DOI] [PubMed] [Google Scholar]; d) Tan X, Li BB, Lu X, Jia F, Santori C, Menon P, Li H, Zhang B, Zhao JJ, Zhang K, J. Am. Chem. Soc 2015, 137, 6112–6115. [DOI] [PubMed] [Google Scholar]
- [14].a) Lytton-Jean AKR, Mirkin CA, J. Am. Chem. Soc 2005, 127, 12754–12755. [DOI] [PubMed] [Google Scholar]; b) Breslauer KJ, Methods Enzymol, 1995, 269, 221–242. [DOI] [PubMed] [Google Scholar]; c) Marky LA, Breslauer KJ, Biopolymers 1987, 26, 1601–1620. [DOI] [PubMed] [Google Scholar]
- [15].a) Miyoshi D, Sugimoto N, Biochimie 2008, 90, 1040–1051. [DOI] [PubMed] [Google Scholar]; b) Nakano S, Miyoshi D, Sugimoto N, Chem. Rev 2014, 114, 2733–2758. [DOI] [PubMed] [Google Scholar]
- [16].Knowles DB, LaCroix AS, Deines NF, Shkel I, Record MT, Proc. Natl. Acad. Sci 2011, 108, 12699–12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakano S, Karimata H, Ohmichi T, Kawakami J, Sugimoto N, J. Am. Chem. Soc 2004, 126, 14330–14331 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.