

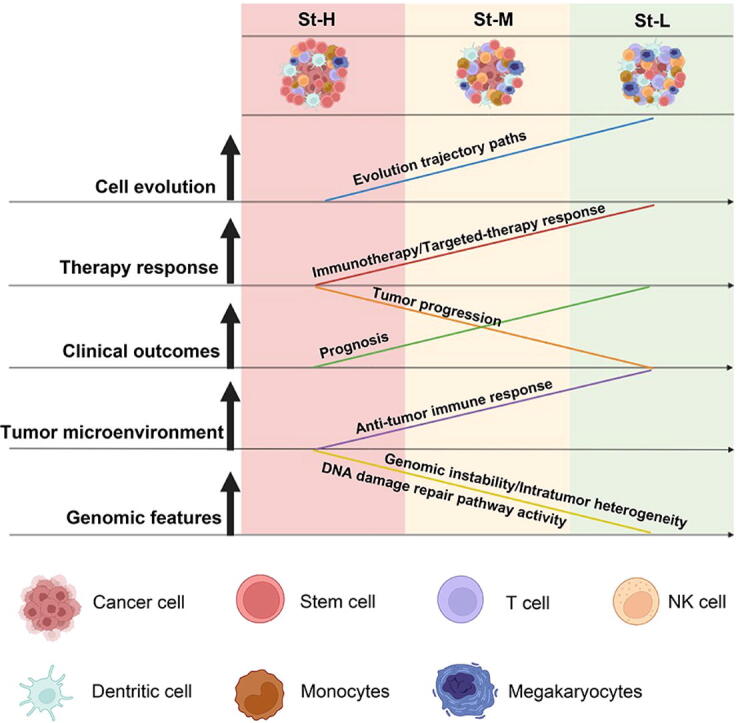
Graphical abstract
Keywords: Lung adenocarcinoma, Tumor stemness, Subtyping, Transcriptome, Clustering analysis, Immunotherapy and targeted therapy
Abbreviations: TCGA, The Cancer Genome Atlas; HLA, human leukocyte antigen; EMT, epithelial-mesenchymal transition; TMB, tumor mutation burden; SCNAs, somatic copy number alterations; ssGSEA, single-sample gene-set enrichment analysis; FDR, false discovery rate; OS, overall survival; DFS, disease free survival; HRD, homologous recombination deficiency; GO, gene ontology; RF, Random Forest; K–W, Kruskal–Wallis
Highlights
-
•
This study explores tumor stemness based on both bulk tumor and single cell transcriptome datasets.
-
•
High-stemness tumors are less responsive to immunotherapy and targeted therapy compared with low-stemness tumors.
-
•
Many high-stemness cells are at the beginning of the cell evolution trajectory, while most low-stemness cells are in the terminal or later phase.
-
•
The correlations of tumor stemness with intratumor heterogeneity and tumor immunity were in the opposite direction between bulk tumors and single cells.
Abstract
Tumor stemness is associated with tumor progression and therapy resistance. The recent advances in sequencing, genomics, and computational technologies have facilitated investigation into the tumor stemness cell-like characteristics. We identified subtypes of lung adenocarcinoma (LUAD) in bulk tumors or single cells based on the enrichment scores of 12 stemness signatures by clustering analysis of their transcriptomic profiles. Three stemness subtypes of LUAD were identified: St-H, St-M, and St-L, having high, medium, and low stemness signatures, respectively, consistently in six different datasets. Among the three subtypes, St-H was the most enriched in epithelial-mesenchymal transition, invasion, and metastasis signaling, genomically instable, irresponsive to immunotherapies and targeted therapies, and hence had the worst prognosis. We observed that intratumor heterogeneity was significantly higher in high-stemness than in low-stemness bulk tumors, but significantly lower in high-stemness than in low-stemness single cancer cells. Moreover, tumor immunity was stronger in high-stemness than in low-stemness cancer cells, but weaker in high-stemness than in low-stemness bulk tumors. These differences between bulk tumors and single cancer cells could be attributed to the non-tumor cells in bulk tumors that confounded the results of correlation analysis. Furthermore, pseudotime analysis showed that many St-H cells were at the beginning of the cell evolution trajectory, compared to most St-L cells in the terminal or later phase, suggesting that many low-stemness cells are originated from high-stemness cells. The stemness-based classification of LUAD may provide novel insights into the tumor biology as well as precise clinical management of this disease.
1. Background
Cancer stemness refers to stem cell-like characteristics in a fraction of cancer cells that confer properties of cancer progression and treatment resistance [1]. Like human embryonic stem cells (hESCs), cancer cells possess the ability to self-renew and proliferate indefinitely [2]. Acquisition of stem-cell-like characteristics in a fraction of cancer cells promotes continuous cell proliferation and dedifferentiation. Furthermore, eliminating non-stem but not stem cancer cells confers cancer resistance and relapse. The mixture of stem cell-like and non-stem cell-like cancer cells endows a tumor with intratumor heterogeneity (ITH). In addition, resistance to immune elimination has been shown to be a property of stem-cell-like cancer cells [3]. Recently, with the advances in sequencing, genomics, and computational technologies, mounting large-scale multi-omics data for cancer have emerged, such as the Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov). These multi-omics data provide valuable resource for exploring cancer features, e.g., tumor stemness [1], [4], ITH [5], [6], and tumor immunity [7]. In fact, multi-omics data analyses have shown that tumor stemness is associated with dedifferentiated oncogenic phenotype, metastasis, ITH, and immunosuppression across various cancers [1], [4]. These previous studies notwithstanding, the multi-omics data-based investigation into the association between tumor stemness and the response to cancer therapies, such as immunotherapies and targeted therapies, remains insufficient. Furthermore, the exploration of tumor stemness at the single-cell level remains lacking, although a large amount of multi-omics data for single cancer cells, particularly the transcriptomics data, have been publicly available [8], [9], [10], [11], [12], [13].
In this study, we identified subtypes of lung adenocarcinoma (LUAD) bulk tumors or single cells based on stemness scores by clustering analysis of their transcriptomic profiles. In five different datasets, we consistently identified three stemness subtypes of LUAD bulk tumors with high, medium, and low stemness signatures, respectively. Likewise, we identified three stemness subtypes of LUAD single cells in a single-cell RNA sequencing (scRNA-seq) dataset for LUAD. We further characterized the molecular and clinical features of these subtypes, as well as their association with the response to immunotherapies and targeted therapies. Our identification of the stemness subtypes of LUAD may provide novel insights into the biology of this cancer and potential clinical application for the management of this disease.
2. Methods
2.1. Datasets
We downloaded five gene expression profiling datasets for LUAD, including TCGA-LUAD, GSE31210, GSE72094, GSE37745, and OncoSG. The TCGA-LUAD data were downloaded from the genomic data commons (GDC) data portal (https://portal.gdc.cancer.gov/), the OncoSG data were from cBioportal (https://www.cbioportal.org/), and the other data were downloaded from the NCBI gene expression omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). From GDC, we also obtained the data of somatic copy number alterations (SCNAs) (“SNP6” files). We downloaded data of gene expression profiles in 1,018 pan-cancer cell lines and drug sensitivities (IC50 values) of these cell lines to 265 compounds from the Genomics of Drug Sensitivity in Cancer (GDSC) project (https://www.cancerrxgene.org/downloads). Besides, we downloaded a single-cell RNA sequencing (scRNA-seq) dataset (LUAD-scRNA) for LUAD from a recent publication [10]. In addition, we obtained gene expression profiling and clinical data for five cancer cohorts treated with immune checkpoint inhibitors (ICIs) from their associated publications, including the Jung cohort (non-small-cell lung cancer (NSCLC)) [14], Braun-Miao cohort (clear cell renal cell carcinoma (ccRCC)) [15], [16], Kim cohort (bladder carcinoma (BLCA)) [17], Ulloa-Montoya cohort (melanoma) [18], and Liu cohort (melanoma) [19]. A description of these datasets is shown in Supplementary Table S1.
2.2. Collection of stemness signatures
We collected 12 stem cell (or stemness) signatures (Homo sapiens) from the StemChecker webserver (http://stemchecker.sysbiolab.eu). These stemness signatures were derived by multiple approaches, including gene expression profiles, computationally derived, literature curation, transcription factor target genes, and RNAi screening [20]. The 12 stemness signatures and their marker genes are presented in Supplementary Table S2.
2.3. Single-sample gene-set enrichment analysis
We evaluated the enrichment score of a gene set representing a stemness signature, biological process, pathway, or phenotypic feature in a bulk tumor or single cancer cell by the single-sample gene-set enrichment analysis (ssGSEA) [21]. The ssGSEA calculates a gene set’s enrichment score in a sample based on their expression profiles. We performed the ssGSEA with the “GSVA” R package. We obtained the gene sets from their associated publications and presented them in Supplementary Table S2.
2.4. Combination of different gene expression profiling datasets
We merged two gene expression profiling datasets for ccRCC (Braun cohort [15] and Miao cohort [16]) using the merge() function in the “base” R package. We adjusted for batch effects and normalized combined data using the normalizeBetweenArrays() function in the “limma” R package.
2.5. Clustering analysis
We used hierarchical clustering to identify LUAD subtypes based on the enrichment scores of 12 stemness gene sets. We performed clustering analysis using the “hclust” R package.
2.6. Survival analysis
Kaplan–Meier curves were used to compare the survival time, and the log-rank tests were utilized to evaluate the significance of survival time differences. We implemented survival analyses using the survfit() function in the “survival” R package.
2.7. Evaluation of tumor mutation burden (TMB), SCNA, and ITH
TMB was defined as the total number of somatic mutations in the tumor. We used GISTIC2 [22] to calculate arm- and focal-level SCNAs and G-scores in tumors with the input of “SNP6” files. The G-score represents the amplitude of the CNA and the frequency of its occurrence across a group of samples [22]. We used the DITHER algorithm [6] to evaluate ITH levels, which scores ITH based on the entropies of somatic mutation profiles and SCNA profiles in the tumor.
2.8. Class prediction
We utilized the Random Forest (RF) algorithm [23] to perform class prediction. In the RF model, the number of trees was set to 500, and the features were the 12 stemness gene sets. We reported accuracy and weighted F-score for prediction performance. We performed the RF algorithm by the “randomForest” R package.
2.9. Pseudotime analysis
To infer the phylogenetic relationship between high- and low-stemness bulk tumors or cancer cells, we constructed their trajectory paths by using Monocle2 [24]. We derived differentially expressed genes from each subtype by using the differentialGeneTest() function. The genes with an adjusted P-value < 0.0001 were utilized to order the tumors or cells in pseudotime analysis.
2.10. scRNA-seq data analysis
We analyzed a LUAD scRNA-seq (SMART-seq2) dataset, which invloved gene expression profiles in 3704 cancer cells and 1231 non-cancer cells from 19 LUAD patients [10]. Before subsequent analyses, we normalized gene expression values by log2 (TPM + 1). We used the single-cell consensus clustering (SC3) method to perform unsupervised clustering of cancer cells in each stemness subtype [25]. We utilized SingleR [26] to identify cell types from the scRNA-seq data. SingleR is a method for computational recognition of cell types by the reference of transcriptomic datasets of individual cell types. We used the inferCNV algorithm [27] to infer large-scale DNA copy number variations (CNVs) in cancer cells relative to normal cells. We re-standardized the CNV values of cells by converting all values in the matrix of inferCNV to 0, 1, or 2, with “0″ representing neutral, “1” loss or addition of one copy, and “2” loss or addition of two copies. The CNV score of each cell was defined as sum of the CNV value for each gene. We utilized the t-distributed stochastic neighbor embedding (t-SNE) algorithm [28] to cluster single cells. t-SNE generates a single map to display structure at different scales, particularly useful for high-dimensional data [28].
2.11. Statistical analysis
In class comparisons, we used the Mann–Whitney U test or Kruskal–Wallis (K–W) test for non-normally distributed data (Shapiro–Wilk test, P < 0.05) and Student’s t test or ANOVA test for normally distributed data. We used Z test to assess whether there is a significant difference between two proportions. We utilized the Benjamini-Hochberg method [29] to calculate the false discovery rate (FDR) for adjusting for multiple tests. We performed all statistical analyses in the R programming environment (version 4.0.2).
3. Results
3.1. Identification of stemness subtypes of LUAD in bulk tumors
Based on the ssGSEA scores of 12 stemness signatures (gene sets), we hierarchically clustered LUAD bulk tumors in five datasets (TCGA-LUAD, GSE31210, GSE72094, GSE37745, and OncoSG), respectively. Consistently in these datasets, three clusters were clearly identified, termed St-H, St-M, and St-L, which had high, medium, and low scores of stemness signatures, respectively (Fig. 1A). We further demonstrated that the stemness levels followed the pattern: St-H > St-M > St-L, by comparing the enrichment scores of a stemness signature composed of 109 genes [1] among the subtypes (one-tailed Mann–Whitney U test, P < 0.001) (Fig. 1B). Survival analyses showed that St-H and St-L likely had the best and worst 5-year OS and/or DFS prognosis, respectively (Fig. 1C). It is consistent with that high stemness is associated with poorer survival in cancer [30], [31]. Furthermore, we found that tumor purity followed the pattern: St-H > St-M > St-L (P < 0.05) (Fig. 1D). It indicates that high-stemness bulk tumors involve a higher proportion of tumor cells than low-stemness bulk tumors. St-H harbored a higher proportion of late-stage tumors than St-L (Z test, P = 0.007, 4.09 × 10−7, 0.286, 0.5 and 0.254 for TCGA-LUAD, GSE31210, GSE72094, GSE37745, and OncoSG, respectively) (Fig. 1E). St-H had a higher mutation rate of TP53 than St-L (P = 2.2 × 10−16, 1.04 × 10−8 for TCGA-LUAD and GSE72094, respectively) (Fig. 1F). In contrast, St-H had a lower mutation rate of EGFR than St-L (P = 0.088, 0.002, and 5.04 × 10−5 for TCGA-LUAD, GSE31210, and GSE72094, respectively) (Fig. 1F). Previous studies have shown that TP53 mutations are associated with a worse prognosis [32], while EGFR mutations are associated with a better prognosis in LUAD [33]. In addition, St-H harbored a higher proportion of smokers than St-L (P = 1.45 × 10−4, 0.281, and 0.093 for GSE31210, GSE72094, and OncoSG, respectively) (Fig. 1G). We further compared several biological processes associated with tumor progression among the LUAD subtypes, including cell proliferation, epithelial-mesenchymal transition (EMT), invasion, and metastasis. Consistently, these biological processes displayed the highest enrichment scores in St-H and the lowest enrichment scores in St-L, and followed the pattern: St-H > St-M > St-L (K–W test, P < 0.05) (Fig. 1H and Supplementary Fig. S1). In contrast, the differentiation signature enrichment scores followed the pattern: St-H < St -M < St -L (K–W test, P < 0.05) (Fig. 1H). Altogether, these results support that high stemness is associated with unfavorable clinical outcomes in cancer.
Fig. 1.

Identification and characterization of stemness subtypes of LUAD in bulk tumors. A. Hierarchical clustering identifying three stemness subtypes: St-H, St-M, and St-L, consistent in five different datasets, based on the enrichment scores of 12 stemness gene sets. B. Comparison of the enrichment scores of a stemness signature composed of 109 genes [1] among the stemness subtypes of LUAD. C. Kaplan–Meier curves showing that St-H and St-L likely have the best and worst 5-year overall survival (OS) and/or disease-free survival (DFS) prognosis, respectively. D. Comparisons of tumor purity among the stemness subtypes. Comparisons of proportion of late-stage tumors (E), mutation rates of TP53 and EGFR (F), and proportion of smokers (G) between St-H and St-L. (H) Comparisons of the enrichment scores of five biological processes in TCGA-LUAD. (I) Pseudotime analysis showing the trajectory paths of the stemness subtypes in TCGA-LUAD. The one-tailed Mann–Whitney U test (B), log-rank test (C), Kruskal–Wallis test (D, H), and Z test (E, F, G) P-values are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, ns not significant.
Pseudotime analysis demonstrated that most St-H tumors were in the onset or early phase of the trajectory, while most St-L tumors were in the terminal or later phase of the trajectory (Fig. 1I). It indicates that high-stemness cancer cells are likely to be the origin of low-stemness cancer cells.
3.2. The stemness subtypes of LUAD have different levels of genomic instability and ITH
We found that DNA damage repair pathways, such as mismatch repair and homologous recombination, had the highest and lowest enrichment levels in St-H and St-L, respectively (one-tailed Mann–Whitney U test, P < 0.01) (Fig. 2A). In addition, many DNA damage response genes, including MSH2, MSH6, POLE, and POLD1, showed the highest and lowest expression levels in St-H and St-L, respectively (two-tailed Student’s t test, P < 0.05) (Fig. 2B). TMB also followed the pattern: St-H > St-M > St-L (P < 0.01) (Fig. 2C). Homologous recombination deficiency (HRD) may result in tumor aneuploidy [34]. Likewise, HRD scores showed the pattern: St-H > St-M > St-L (P < 0.01) (Fig. 2C). The G-scores of copy number amplifications and deletions were likely the highest and lowest in St-H and St-L, respectively (Fig. 2D). Furthermore, the ITH levels were the highest in St-H and the lowest in St-L, namely satisfying St-H > St-M > St-L (one-tailed Mann–Whitney U test, P < 0.01) (Fig. 2E). Taken together, these results suggest that tumor stemness is positively associated with genomic instability and ITH. Interestingly, global methylation levels [14] were the lowest in St-H while the highest in St-L (P < 0.05) (Fig. 2F).
Fig. 2.

Comparisons of genomic instability and intratumor heterogeneity (ITH) levels among the stemness subtypes of LUAD. Comparisons of the enrichment scores of DNA damage repair pathways (mismatch repair and homologous recombination) (A), the expression levels of DNA damage response genes (B), tumor mutation burden (TMB) and homologous recombination deficiency (HRD) scores (C), G-scores (D), ITH levels (E), and global methylation levels (F) among the stemness subtypes of LUAD. The Kruskal–Wallis test (A), one-way ANOVA test (B), one-tailed Mann–Whitney U test (C, E, F) P-values are shown. ** P < 0.01, *** P < 0.001.
3.3. The stemness subtypes of LUAD have different tumor immune microenvironment and immunotherapy response
Human leukocyte antigen (HLA) genes encode the major histocompatibility complex (MHC), which are responsible for immune regulation [35]. We found that the expression levels of numerous HLA genes showed the pattern: St-H < St-M < St-L (two-tailed Student’s t test, FDR < 0.05) [36] (Fig. 3A and Supplementary Fig. S2). It indicated that St-L and St-H had the most and least active anti-tumor immune microenvironment, respectively. We predicted the response rates to ICIs in the stemness subtypes of LUAD by the TIDE algorithm [37]. Interestingly, St-H tended to have the lowest response rate, while St-L likely had the highest response rate among the subtypes, consistently in the five datasets (Fig. 3B). Furthermore, we compared the response rate to ICIs in five cancer cohorts receiving anti-PD-1/PD-L1/CTLA-4 immunotherapy, including the Jung cohort (NSCLC) [14], Braun-Miao cohort (ccRCC) [15], [16], Kim cohort (BLCA) [17], Ulloa-Montoya cohort (melanoma) [18], and Liu cohort (melanoma) [19] (Fig. 3C). In three cohorts (Braun-Miao, Ulloa-Montoya, and Liu cohorts), the response rate followed the pattern: St-H < St-M < St-L. In the Jung cohort, St-H had the lowest response rate of 0%, compared to 41.7% in St-M and 37.5% in St-L. In the Kim cohort, St-L had the highest response rate of 85.7%, compared to 44.4% in St-H and 43.5% in St-M (Fig. 3C). Altogether, these results support that tumor stemness inhibits anti-tumor immune response and immunotherapy response.
Fig. 3.
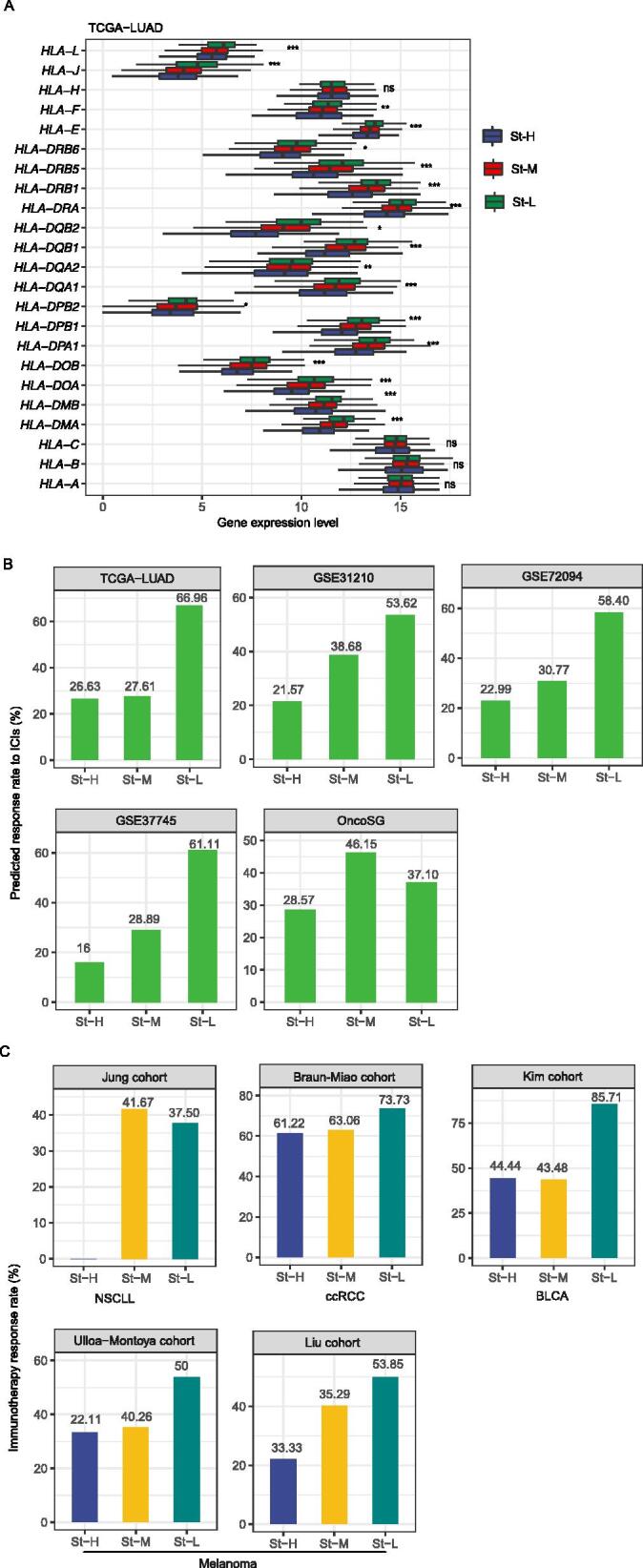
Comparisons of immune signatures and immunotherapy response among the stemness subtypes of LUAD. Comparisons of the expression levels of human leukocyte antigen (HLA) genes (A) and predicted response rates to immune checkpoint inhibitors (ICIs) (B) among the stemness subtypes of LUAD. C. Comparisons of the response rates to ICIs in five cancer cohorts treated with ICIs. The one-way ANOVA test P-values are shown in (A). NSCLC: non-small-cell lung cancer; ccRCC: clear cell renal cell carcinoma; BLCA: bladder carcinoma. * P < 0.05, ** P < 0.01, *** P < 0.001, ns not significant.
3.4. The stemness subtypes of cancer have different responses to targeted therapies
Based on the ssGSEA scores of the 12 stemness signatures, we hierarchically clustered 1,018 pan-cancer cell lines and obtained three clusters with high, medium, and low stemness scores, also termed St-H, St-M, and St-L, respectively. We compared drug sensitivities (IC50 values) of 265 anti-tumor compounds between St-H and St-L. Strikingly, we found 223 (84.2%) of the 265 compounds showing significantly higher IC50 values (lower drug sensitivities) in St-H than in St-L (one-tailed Mann–Whitney U test, FDR < 0.05) (Supplementary Table S3). In contrast, only 6 (2.3%) compounds displayed significantly lower IC50 values (higher drug sensitivities) in St-H than in St-L (FDR < 0.05) (Supplementary Table S3). These results support that tumor stemness likely confers resistance to targeted cancer therapies.
3.5. Prediction of the stemness subtypes of LUAD
We trained the model for predicting the stemness subtypes of LUAD in TCGA-LUAD and tested it in the other four datasets. The 10-fold cross-validation (CV) accuracy in TCGA-LUAD was 92.01%. The prediction accuracy in GSE31210, GSE72094, GSE37745, and OncoSG was 90.71%, 80.54%, 63.21%, and 81.07%, respectively (Fig. 4). The weighted F-score in the predictions was 92.00%, 91.20%, 87.50%, 69.50%, and 82.50% in TCGA-LUAD, GSE31210, GSE72094, GSE37745, and OncoSG, respectively. These results indicate that the subtyping of LUAD based on stemness scores is reproducible and predictable.
Fig. 4.

Prediction of the stemness subtypes of LUAD based on the enrichment scores of 12 stemness gene sets. The model for predicting the stemness subtypes of LUAD was trained in TCGA-LUAD and tested in the other four datasets by the Random Forest algorithm. The prediction accuracies and weighted F-scores are shown. In TCGA-LUAD, the 10-fold cross-validation accuracy and weighted F-score are shown.
3.6. Identification of stemness subtypes of LUAD single cells
To explore the reproducibility of the stemness-based subtyping method at the single-cell level, we analyzed a LUAD scRNA-seq dataset (LUAD-scRNA) [10]. Based on the enrichment scores of the 12 stemness signatures, we hierarchically clustered 3704 cancer cells. Likewise, these cancer cells were clearly clustered into three subgroups, also termed St-H, St-M, and St-L, which showed high, medium, and low enrichment scores of stemness signatures, respectively (Fig. 5A). Consistent with the results in bulk tumors, the stemness levels and enrichment scores of invasion and metastasis processes followed the pattern: St-H > St-M > St-L (one-tailed Mann–Whitney U test, P < 0.001) (Fig. 5B). These results support that high stemness is associated with tumor progression phenotypes in single cancer cells.
Fig. 5.

Identification of stemness subtypes of LUAD single cells in LUAD-scRNA. A. Hierarchical clustering identifying three stemness subtypes of LUAD single cells. Comparisons of the enrichment scores of three biological processes (B), expression levels of DNA damage response genes (C), and inferred CNVs by inferCNV [27] (D) among the stemness subtypes of LUAD single cells. E. Clustering analyses of LUAD single cells by SC3 [25] identifying 14, 15, and 18 cell clusters in St-H, St-M, and St-L, respectively. F. Pseudotime analysis showing the trajectory paths of the stemness subtypes of LUAD single cells. G. Gene set enrichment analysis by Monocle2 [24] identifying the gene ontology (GO) biological process (BP) terms enriched in the stemness subtypes of LUAD single cells. The one-tailed Mann–Whitney U test (B) and one-way ANOVA test (C) P-values are shown. *** P < 0.001.
The DNA damage response genes, such as MLH1, MSH2, MSH3, MSH6, PMS1, and POLD1, also showed the highest and lowest expression levels in St-H and St-L single cells, respectively (P < 0.01) (Fig. 5C). The inferred CNVs by inferCNV [27] were significantly higher in St-H than in St-L single cells and followed the pattern: St-H > St-M > St-L (one-tailed Mann–Whitney U test, P < 0.001) (Fig. 5D). These results were consistent with those obtained in bulk tumors, supporting that tumor stemness is associated with genomic instability. We further performed unsupervised clustering of cancer cells in each of the three stemness subtypes by SC3 [25]. SC3 identified 14, 15, and 18 cell clusters in St-H, St-M, and St-L, respectively (Fig. 5E). It suggests that low-stemness cancer cells are more heterogeneous than high-stemness cancer cells. We further analyzed the 3704 cancer cells by pseudotime analysis [38]. We found that most of the St-H cells were distributed in a branch and a few at another branch (Fig. 5F). In contrast, single cells in St-M and St-L were evenly distributed in three different branches. It again suggests that high-stemness cancer cells are the least heterogeneous among these subtypes. Pseudotime analysis showed that many St-H cells were at the beginning of the trajectory, while most St-L cells were at a later stage and many St-L cells at a terminal state (Fig. 5F). It indicates that many low-stemness cells are originated from high-stemness cells. In addition, we found that few St-H cells were also at a terminal state, suggesting that new cancer stem cells may arise with tumor development. GO analysis showed that low-stemness cancer cells were enriched in immune and metabolic pathways, while high-stemness cancer cells were enriched in cell cycle, DNA replication, translational regulation, and chromatin remodeling pathways (Fig. 5G).
We clustered all single cells in LUAD-scRNA by t-SNE [28]. Again, most St-H cells were in one cluster, compared to St-M and St-L cells evenly distributed in two or more clusters (Fig. 6A). Interestingly, we found that the St-H cluster was closer to the T cells cluster and dendritic cells cluster than the St-L clusters (Fig. 6A). Furthermore, we found that both immunostimulatory and immunosuppressive signatures, such as antigen processing and presentation, JAK-STAT signaling, TGF-β signaling, and PD-L1, were more enriched in St-H versus St-L cells and also followed the pattern: St-H > St-M > St-L (one-tailed Mann–Whitney U test, P < 0.001) (Fig. 6B). CXCL16 is a chemokine for recruiting T cells and was also expressed by tumor cells [39]. We found that CXCL16 expression levels were significantly higher in St-H than in St-L (P < 0.001; fold change greater than 47) and followed the pattern: St-H > St-M > St-L (one-tailed Mann–Whitney U test, P < 0.001) (Fig. 6B). Based on the clustering result in LUAD single cells, we classified a LUAD patient into one class of St-H, St-M, and St-L, which contained the most cancer cells of the patient. Finally, 7, 6, and 6 LUAD patients were classified into St-H, St-M, and St-L, respectively. We compared the enrichment of several T cell subpopulations, including activated CD8 + T cells, exhausted CD8 + T cells, activated CD4 + T cells, and resting CD4 + T cells, between St-H and St-L LUAD patients. The enrichment of an immune cell was the ssGSEA score of its gene markers. We observed that activated CD8 + T cells, exhausted CD8 + T cells, and resting CD4 + T cells were significantly enriched in St-H compared with St-L (Fig. 6C). However, the ratios of immunostimulatory over immunosuppressive signatures (activated/resting CD8 + T cells, activated/resting CD4 + T cells, and activated CD8 + T cells/resting CD4 + T cells) were significantly lower in St-H than in St-L (Fig. 6C). Altogether, these results implicate that compared with low-stemness cancer cells, high-stemness cancer cells are more immunogenetic but are more enriched in immunosuppressive versus immunostimulatory signatures.
Fig. 6.

Characterization of immune and clinical features of the stemness subtypes of LUAD single cells in LUAD-scRNA. A. Clustering all single cells in LUAD-scRNA by t-SNE [28]. Comparisons of the expression of immune signatures (B) and the ratios of immunostimulatory over immunosuppressive signatures (C) among the stemness subtypes of LUAD single cells. Comparisons of the proportions of high-stemness cells between late-stage and early-stage LUAD patients (D) and between progressive and regressive or stable LUAD patients (E). The one-tailed Mann–Whitney U test (B, D, E), two-tailed Student’s t test (B), and one-way ANOVA test P-values (B, C) are shown. *** P < 0.001.
For each LUAD patient, its cancer cells were clustered into St-H, St-M, or St-L. We calculated the proportion of St-H cells in all single cells in each LUAD patient. We found that the proportions of St-H cells were higher in late-stage than in early-stage LUAD patients (one-tailed Mann–Whitney U test, P = 0.094) (Fig. 6D). Moreover, the proportions of St-H cells were higher in the progressive LUAD patients than in the regressive or stable patients (P = 0.086) (Fig. 6E). Again, these results support that high stemness is associated with unfavorable clinical outcomes in LUAD.
4. Discussion
Based on the enrichment scores of 12 stemness gene sets, we identified three stemness subtypes (St-H, St-M, and St-L) of LUAD. We demonstrated that this classification method was stable and reproducible in five transcriptome datasets for bulk tumors and a transcriptome dataset for single cells. Notably, 11 of the 12 stemness gene sets showed significantly higher enrichment scores in cancer tissue versus normal tissue and in cancer cells versus non-cancer cells (P < 0.001). It supports that at least a fraction of cancer cells are endowed with stem cell-like characteristics [40]. Among the 19 LUAD patients in the scRNA-seq dataset, we observed the percentage of high-stemness cells ranging from 0% to 92.8%, with 5 and 2 patients whose numbers of high-stemness (St-H) cells were <10% and greater than 70%, respectively. It indicates both intratumor and intertumor heterogeneity.
An interesting finding is that high-stemness cancer cells appear to be more immunogenetic than low-stemness cancer cells in the single cell transcriptome data analysis. A potential explanation for this is that high-stemness single cancer cells are less heterogeneous than low-stemness single cancer cells, while tumor heterogeneity may dilute tumor neoantigens [41]. Nevertheless, because high stemness is associated with elevated genomic instability that plays a role in tumor immune evasion, it is justified that high-stemness single cancer cells have a stronger positive correlation with immunosuppressive than with immunostimulatory signatures. However, in the bulk tumor transcriptome data analysis, we found that high-stemness cancers were less immunogenetic and had a less active anti-tumor immune microenvironment than low-stemness cancers, an observation conflicting with that in single cancer cells. The main reason behind this could be that high-stemness bulk tumors have higher tumor purity and hence lower immune cell infiltration than low-stemness bulk tumors. Furthermore, our results showed that the association between tumor stemness and ITH was inconsistent between bulk tumors and single cancer cells in LUAD. That is, high-stemness bulk tumors had significantly higher ITH than low-stemness bulk tumors, while high-stemness single cancer cells had significantly lower ITH than low-stemness single cancer cells. Again, this difference could be attributed to the different tumor microenvironment between high- and low-stemness bulk tumors. Overall, we argue that the results for tumor stemness analysis at the single-cell level are likely more reasonable than those at the bulk-tumor level.
Abundant evidence has demonstrated that cancer cells often derive from normal or premalignant cells that gradually lose a differentiated phenotype and acquire progenitor cell-like or stem cell-like features [42]. Our analysis showed that most high-stemness cancer cells were at the beginning of the evolutionary trajectory in cancer cells, indicating that the high-stemness cancer cell population is enriched at the stage of cancer initiation. It is in agreement with previous findings [42]. In addition, our analysis showed that high tumor stemness was associated with inferior responses to immunotherapy and targeted therapy, supporting the characteristics of cancer stem cells conferring drug resistance [43], [44]. The reason why tumor stemness contributes to immunotherapy resistance could be that tumor stemness promotes the formation of immune-deprived TME [1]. Furthermore, multiple factors may contribute to the resistance of cancer stemness cells to targeted therapy and chemotherapy, such as a hyperactive DNA repair capacity, a resistance to apoptosis, overexpression of ATP-binding cassette (ABC) transporters, quiescence, EMT, and acquired genetic changes [45].
Based on immunogenomic profiles, Thorsson et al. identified six immune subtypes of TCGA pan-cancer, namely C1 (wound healing), C2 (IFN-γ dominant), C3 (inflammatory), C4 (lymphocyte depleted), C5 (immunologically quiet), and C6 (TGF-β dominant) [7]. We observed that St-H tumors mainly belonged to C2 and C1 (61.2% and 31.9%, respectively). It is consistent with that both C2 and C1 had a high proliferation rate and high adaptive immune infiltrate [7]. In contrast, St-L tumors mainly belonged to C3 (70.2%), which was characterized by low proliferation rate and low SCNAs. Again, it is consistent with the characteristics of low-stemness tumors we described.
In conclusion, LUAD can be classified into three stemness subtypes with high, medium, and low stemness signatures, respectively. High stemness is associated with tumor progressive phenotypes, unfavorable prognosis, therapy resistance, and genomic instability in LUAD (Fig. 7). A dissection of tumor stemness-associated features (such as tumor immunity and ITH) at the single-cell level could be more precise than at the bulk-tumor level.
Fig. 7.

A summary of molecular and clinical features of the LUAD stemness subtypes as well as their evolutionary relationship. The figure was created with BioRender.com.
Funding
This work was supported by the China Pharmaceutical University (grant numbers 3150120001 to XW).
Authors contributions
QL performed the research and data analyses. JL performed data analyses. XZ performed data analyses. XW conceived of the research, designed the methods, and wrote the manuscript. All authors read and approved the final manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Not applicable.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.04.004.
Contributor Information
Xiaobo Zhang, Email: zxb@cpu.edu.cn.
Xiaosheng Wang, Email: xiaosheng.wang@cpu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Comparisons of the enrichment scores of five biological processes among the LUAD stemness subtypes. The Kruskal–Wallis test P values are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, ns not significant.
Comparisons of the expression levels of human leukocyte antigen (HLA) genes among the LUAD stemness subtypes. The one-way ANOVA test P values are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, ns not significant.
References
- 1.Miranda A., et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A. 2019;116(18):9020–9029. doi: 10.1073/pnas.1818210116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X. Computational analysis of expression of human embryonic stem cell-associated signatures in tumors. BMC Res Notes. 2011;4:471. doi: 10.1186/1756-0500-4-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Codony-Servat J., Rosell R. Cancer stem cells and immunoresistance: clinical implications and solutions. Transl Lung Cancer Res. 2015;4(6):689–703. doi: 10.3978/j.issn.2218-6751.2015.12.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malta T.M., et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018;173(2):338–354 e15. doi: 10.1016/j.cell.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li M., et al. An algorithm to quantify intratumor heterogeneity based on alterations of gene expression profiles. Commun Biol. 2020;3(1):505. doi: 10.1038/s42003-020-01230-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L., Chen C., Wang X. DITHER: an algorithm for Defining IntraTumor Heterogeneity based on EntRopy. Brief Bioinform. 2021 doi: 10.1093/bib/bbab202. [DOI] [PubMed] [Google Scholar]
- 7.Thorsson V., et al. The Immune Landscape of Cancer. Immunity. 2018;48(4):812–830 e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S., et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat Cell Biol. 2021;23(1):87–98. doi: 10.1038/s41556-020-00613-6. [DOI] [PubMed] [Google Scholar]
- 9.Chung W., et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081. doi: 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maynard A., et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell. 2020;182(5):1232–1251 e22. doi: 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puram S.V., et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171(7):1611–1624 e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tirosh I., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venteicher A.S., et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355(6332) doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung H., et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun. 2019;10(1):4278. doi: 10.1038/s41467-019-12159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braun D.A., Burke K.P., Van Allen E.M. Genomic Approaches to Understanding Response and Resistance to Immunotherapy. Clin Cancer Res. 2016;22(23):5642–5650. doi: 10.1158/1078-0432.CCR-16-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miao D., et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet. 2018;50(9):1271–1281. doi: 10.1038/s41588-018-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim Y.J., et al. Gene signatures for the prediction of response to Bacillus Calmette-Guerin immunotherapy in primary pT1 bladder cancers. Clin Cancer Res. 2010;16(7):2131–2137. doi: 10.1158/1078-0432.CCR-09-3323. [DOI] [PubMed] [Google Scholar]
- 18.Ulloa-Montoya F., et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol. 2013;31(19):2388–2395. doi: 10.1200/JCO.2012.44.3762. [DOI] [PubMed] [Google Scholar]
- 19.Liu Q., et al. Identification of subtypes correlated with tumor immunity and immunotherapy in cutaneous melanoma. Comput Struct Biotechnol J. 2021;19:4472–4485. doi: 10.1016/j.csbj.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinto J.P., et al. StemChecker: a web-based tool to discover and explore stemness signatures in gene sets. Nucleic Acids Res. 2015;43(W1):W72–W77. doi: 10.1093/nar/gkv529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mermel C.H., et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breiman L. Random forests. Mach Learn. 2001;45(1):5–32. [Google Scholar]
- 24.Qiu X., et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiselev V.Y., et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14(5):483–486. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aran D., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel A.P., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Maaten L., Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 29.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B. 1995;57:289–300. [Google Scholar]
- 30.Khan A.Q., et al. F-box proteins in cancer stemness: An emerging prognostic and therapeutic target. Drug Discov Today. 2021 doi: 10.1016/j.drudis.2021.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Zeng H., et al. Stemness Related Genes Revealed by Network Analysis Associated With Tumor Immune Microenvironment and the Clinical Outcome in Lung Adenocarcinoma. Front Genet. 2020;11:549213. doi: 10.3389/fgene.2020.549213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X., Sun Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget. 2017;8(1):624–643. doi: 10.18632/oncotarget.13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W.Y., et al. The role of EGFR mutation as a prognostic factor in survival after diagnosis of brain metastasis in non-small cell lung cancer: a systematic review and meta-analysis. BMC Cancer. 2019;19(1):145. doi: 10.1186/s12885-019-5331-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knijnenburg T.A., et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018;23(1):239–254 e6. doi: 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wieczorek M., et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol. 2017;8:292. doi: 10.3389/fimmu.2017.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M., et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang P., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32(4):381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumura S., et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181(5):3099–3107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visvader J.E., Lindeman G.J. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Caswell D.R., Swanton C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017;15(1):133. doi: 10.1186/s12916-017-0900-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeulen L., et al. Cancer stem cells–old concepts, new insights. Cell Death Differ. 2008;15(6):947–958. doi: 10.1038/cdd.2008.20. [DOI] [PubMed] [Google Scholar]
- 43.Phi L.T.H., et al. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018;2018:5416923. doi: 10.1155/2018/5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y., et al. Drug resistance and Cancer stem cells. Cell Commun Signal. 2021;19(1):19. doi: 10.1186/s12964-020-00627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dean M., Fojo T., Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparisons of the enrichment scores of five biological processes among the LUAD stemness subtypes. The Kruskal–Wallis test P values are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, ns not significant.
Comparisons of the expression levels of human leukocyte antigen (HLA) genes among the LUAD stemness subtypes. The one-way ANOVA test P values are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, ns not significant.