

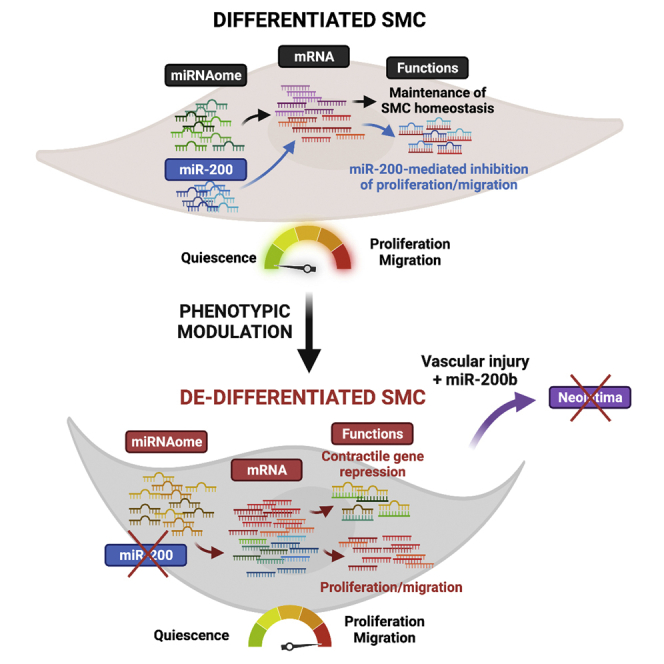
Summary
miRNAs are versatile regulators of smooth muscle cell (SMC) fate and behavior in vascular development and disease. Targeted loss-of-function studies have established the relevance of specific miRNAs in controlling SMC differentiation or mediating phenotypic modulation. Our goal was to characterize SMC miRNAome and its contribution to transcriptome changes during phenotypic modulation. Small RNA sequencing revealed that dedifferentiation led to the differential expression of over 50 miRNAs in cultured SMC. miRNA/mRNA comparison predicted that over a third of SMC transcript expression was regulated by differentially expressed miRNAs. Our screen identified the miR-200 cluster as highly downregulated during dedifferentiation. miR-200 maintains SMC quiescence and represses proliferation, migration, and neointima formation, in part by targeting Quaking, a central SMC phenotypic switching mediator. Our study unraveled the substantial contribution of miRNAs in regulating the SMC transcriptome and identified the miR-200 cluster as a pro-quiescence mechanism and a potential inhibitor of vascular restenosis.
Subject areas: Pathophysiology, Molecular biology, Complex system biology
Graphical abstract
Highlights
-
•
Small RNAseq uncovers global changes in miRNA during SMC phenotypic modulation
-
•
miRNA/mRNA co-analysis reveals that a third of transcripts are regulated by miRNA
-
•
Our screen identifies the miR-200 cluster repression during SMC dedifferentiation
-
•
miR-200 inhibits SMC proliferation, migration, and neointima formation in vivo
Pathophysiology; Molecular biology; Complex system biology
Introduction
Control of SMC gene expression plays a critical role in the maintenance of SMC basal functions of contraction, maintenance of vasculature integrity, and regulation of systemic blood pressure (Owens et al., 2004). SMC transcriptome undergoes profound and diverse modifications during phenotypic modulation linked with atherosclerosis or other vascular diseases (Alencar et al., 2020; Dobnikar et al., 2018). These transcriptional changes correlate with the acquisition of properties such as proliferation, clonal expansion, migration, and lipid uptake (Espinosa-Diez et al., 2021). Multiple transcriptional and posttranscriptional mechanisms have been identified as drivers of activation or repression of subsets of genes responsible for context-dependent SMC functions (Bauer and Martin, 2017). Transcriptional and epigenetic regulatory mechanisms have been implicated in regulating the SMC differentiation state. Among them, the formation of the serum response factor (SRF)-myocardin complex, the enrichment of histones modifications, including H3K4me2, and the recruitment of the methylcytosine dioxygenase ten-eleven translocation 2, TET2, on the SMC contractile gene repertoire are essential for the maintenance of SMC differentiation, lineage identity, and contractile function (Liu et al., 2013, 2021; McDonald et al., 2006; Wang et al., 2004). Conversely, disruption of these epigenetic and transcriptional complexes by factors such as KLF4, TCF21, histone deacetylases, and DNA methyltransferases leads to repression of the SMC contractile genes and the activation of the previously repressed genes associated with proliferation, migration, inflammation, and other processes involved in both SMC adaptive and maladaptive response (Nagao et al., 2020; Salmon et al., 2012; Shankman et al., 2015; Zhuang et al., 2017). In addition to these transcription and epigenetic mechanisms, noncoding RNAs such as miRNAs, long noncoding RNAs, and circular RNAs are directly involved in the posttranscriptional regulation of transcript abundance in SMC (Vacante et al., 2021; Wang and Atanasov, 2019; Zeng et al., 2021).
miRNAs are a group of abundantly expressed small noncoding RNAs that degrade mRNA or repress protein translation by complexing with the RNA-induced silencing complex (RISC). The subset of transcripts targeted by a given miRNA (“targetome”) is distinctly based on complementary binding between mRNA 3’ UTR and miRNA seed sequence, although it can be partially shared among several miRNAs displaying homology in their seed sequence (O'Brien et al., 2018). miRNA are categorized into families and clusters based on (1) evolutionary homology and sequence conservation (Ding et al., 2011; Nawrocki et al., 2015); (2) genomic location, genomic clustering, or polycistronic expression (Olena and Patton, 2010); (3) similarity of secondary structure (Kaczkowski et al., 2009; Will et al., 2007); and (4) similarity in function and targetome (Chen and Rajewsky, 2007; Lewis et al., 2003). Based on sequence homology, it has been estimated that miRNAs regulate around 30% of expressed transcripts in human (Lewis et al., 2005). Functionally, clustered miRNAs can enhance targeting efficacy by the synergistic regulation of shared targets and pathways (Gam et al., 2018). The impact of miRNAs on the transcriptome fluctuates based on the abundance of both miRNAs and their targets, which can be a variable subset of transcripts depending on the cell type and environmental cues (Nam et al., 2014; Sood et al., 2006). A large body of research has delineated the role of an expanding set of miRNAs controlling SMC function in various pathological conditions, including atherosclerosis, ischemic disease, and diabetes. For example, proper differentiation of the SMC lineage during embryonic development is dependent on the expression of the miR-143/145 cluster (Cordes et al., 2009). Several other miRNAs have been identified as regulators of SMC functions, including migration (miR-29b (Sun et al., 2017)), proliferation (miR-146a (Sun et al., 2011)), and calcification (miR-125 (Goettsch et al., 2011)).
miRNA studies are often based on single miRNA loss- or gain-of-function approaches that are critical for examining the functional relevance of miRNAs and evaluating their potential as therapeutic targets. However, this may be suboptimal for capturing the synergistic or antagonistic effects of the context-dependent ensemble of miRNA expressed in the cell (miRNAome). Thus, there is a need for unbiased and exhaustive characterization of miRNA expression and abundance in SMC to identify functionally relevant miRNAs and the relative contribution of the miRNAome in regulating transcript expression during VSMC phenotypic modulation. Although the majority of SMC miRNA screening published to date have been done with microarray technology, the development of high-throughput small RNA sequencing and single-cell microRNA sequencing should provide more sensitive, reproducible, and comparable datasets (Giraldez et al., 2018; Wang et al., 2019; Witwer and Halushka, 2016). Co-profiling of miRNA and mRNA expression provides decisive information for assessing the miRNAome functional contribution to transcript expression, as well as synergy, antagonism, or redundancy between expressed miRNAs, which is highly relevant for the successful development of miRNA-based therapeutic strategies. Although RIP sequencing or CLIP sequencing allows for the rigorous assessment of miRNA/mRNA interactions, miRNA/mRNA co-profiling provides comprehensive transcriptomic information and serves as a highly effective prediction tool, which helps guide prediction-assisted miRNA functional discovery (Khorshid et al., 2013; Tan et al., 2009). In that regard, a recent study of miRNA/mRNA co-profiling established the transcriptomic signature of major vascular cell types subjected to proatherogenic cues and allowed the identification of miR100-5p as a functionally relevant microRNA in atherosclerosis (Moreau et al., 2021).
In the present study, we characterized modifications of miRNA and mRNA profiles and assessed the overall contribution of miRNAs in regulating transcript expression and downstream functions during SMC dedifferentiation. We compared two models of SMC dedifferentiation: reversible dedifferentiation induced by platelet-derived growth factor-BB (PDGF-BB) and a profound loss of lineage identity caused by the erasure of the H3K4me2 signature on the myocardin-regulated genes (Liu et al., 2021).
By performing integrative miRNA/mRNA co-profiling, we were able to (1) establish the miRNAome in SMC and its fluctuation driven by PDGF-BB-driven dedifferentiation or H3K4me2-editing-driven loss of lineage identity or both; (2) predict the contribution of differentially expressed miRNAs (DEmiRs) in regulating the SMC transcriptome; (3) screen differentially expressed and functionally relevant miRNA; (4) identify the downregulation of miR-200a, miR-200b, and miR-429 as a shared process in various models of SMC dedifferentiation; and (5) finally, establish the pro-quiescence, anti-proliferation, and anti-migration properties of the miR-200 cluster in SMC in vitro and in vivo during injury-induced vascular remodeling.
Results
SMC differentiation status influences miRNA expression profile
We performed small RNA sequencing (small RNAseq) for the unbiased characterization of the miRNAome during SMC phenotypic modulation (Figure 1A). We also evaluated the contribution of DEmiRs in regulating transcript abundance by comparing and co-analyzing small RNAseq data and a previously generated RNAseq dataset (GEO: GSE186972, GSE179220) (Liu et al., 2021). Although different RNA extracts were used for sequencing, similar cell lines and conditions were used to generate both datasets. miRNA and mRNA profiles were analyzed in contractile SMC and two models of SMC phenotypic modulation: the well-established PDGF-BB-induced reversible dedifferentiation and an epigenome-editing-mediated loss of lineage identity consisting in the demethylation of the histone posttranslational modification of H3K4me2 on the SMC gene repertoire (Holycross et al., 1992; Liu et al., 2021; McDonald et al., 2006) (Figure S1A). These two models are characterized by a marked reduction in SMC contractile gene (Acta2, Myh11, Cnn1, Tagln) expression, whereas H3K4me2 editing is also associated with loss of lineage identity as exemplified by the further decrease in expression in SMC differentiation master regulators (Mef2c, Notch3, Rbpms) (Figures S1B and S1C). Small RNAseq performed in these three groups showed a similar proportion of small noncoding RNA expression, including miRNAs, piRNAs, rRNAs, and tRNAs (Figure 1B). Interestingly, a decrease in modified miRNAs (isomiRs) was observed in dedifferentiated SMC, which could influence miRNA functions (Cloonan et al., 2011) (Figure 1C). This decrease in isomiRs level was associated with the reduction in expression of several miRNA processing and editing enzymes (Figures 1D, S2A, and S2B). Next, we analyzed the differential expression of miRNAs in contractile, PDGF-BB-treated, and H3K4me2-edited SMCs (Figures 1E–1G). SMC phenotypic modulation led to significant changes in miRNA expression profile. Principal component analysis revealed distinct miRNA signatures in the three groups analyzed (Figure 1E). More precisely, a total of 50 and 148 differentially expressed miRNAs (DEmiRs) were identified in PDGF-BB-treated and H3K4me2-edited SMC as compared with differentiated SMC, respectively (Figure 1F). When comparing DEmiRs in both dedifferentiation models, we observed a partial overlap in downregulated and upregulated DEmiRs (Figure 1G); 86.7% (13/15) of downregulated and 48.6% (17/35) of upregulated DEmiRs in PDGF-BB-treated SMC were similarly regulated in H3K4me2-edited SMC. This percentage of similarly regulated miRNAs was lower in the H3K4me2-edited SMC due to the larger number of DEmiRs; this suggests that loss of lineage identity induces a more profound alteration of miRNA expression as compared with reversible phenotypic modulation induced by PDGF-BB.
Figure 1.

Small RNA/mRNA co-profiling predicts miRNA contribution in regulating SMC transcriptional states during phenotypic modulation
(A) Schematics representing the experimental pipeline for small RNAseq and RNAseq co-analysis.
(B) Percentage of small RNA subtypes normalized to total small RNA amount in control, PDGF-BB-treated, and H3K4me2-edited SMC. Data are expressed as mean ± SEM. Groups were compared by one-way ANOVA. p < 0.05 is considered statistically significant.
(C) Percentage of edited miRNA (isomiR) in control, PDGF-BB-treated, and H3K4me2-edited SMC. Data are expressed as mean ± SEM. Groups were compared by one-way ANOVA. p < 0.05 is considered statistically significant.
(D) Heatmap representing the differential expression of miRNA processing and editing enzymes. Data are expressed as log2 fold change compared with control SMC.
(E) Principal component analysis comparing control, PDGF-BB-treated, and H3K4me2-edited SMC small RNA expression profiles.
(F) Heatmap of differentially expressed miRNA in control, PDGF-BB-treated, and H3K4me2-edited SMC. Minimum cutoff value of fold-change > 1.5 and p value < 0.05.
(G) Venn diagram representing the overlap of DEmiR in PDGF-BB-treated and H3K4me2-edited SMC (compared with control SMC).
(H) Venn diagram representing the percentage of DEG targeted by DEmiR targets. Data were filtered using miRNA database and co-profiling considering differential expression directions.
(I) Gene Ontology analysis performed on DEG targeted by DEmiRs revealed enrichment in pathways associated with cell division (upregulated genes, red) and SMC differentiation (downregulated genes, blue).
mRNA/miRNA co-profiling predicts a major contribution of miRNA in regulating SMC transcriptome
To further evaluate the relative contribution of miRNAs in regulating differentially expressed genes (DEG) in SMC and define their context-dependent targetome, we compared RNAseq and small RNAseq datasets. As previously described (Liu et al., 2021), PDGF-BB treatment and H3K4me2 editing induced profound alterations in transcript expression with remarkably similar segregation compared with miRNAs (Figures S3A–S3C). Using publicly available miRNA target databases (TargetScan), we assess the association between DEmiRs and DEG. We found that 40 and 66% of genes that are predicted targets of DEmiRs display opposite changes in expression in PDGF-BB-treated and H3K4me2-edited SMC, respectively (Figures 1H and S3D). These results suggest that miRNAs are central regulators of transcript expression. After PDGF-BB treatment, 24.8% of upregulated transcripts were established targets of downregulated miRNAs, and 50.1% of downregulated transcripts were defined targets of upregulated miRNAs. This broad mRNA/miRNA antipodal coregulation in SMC coincided with results obtained in other cellular models (Lewis et al., 2005). Notably, gene ontology pathway and network analysis showed that DEG targets of DEmiRs are involved in processes and functions relevant to SMC homeostasis (Figures 1I, S3E, and S4). For example, downregulated genes targeted by upregulated miRNAs in SMC treated in PDGF-BB are involved in processes relevant to SMC differentiation, including regulation of vascular development and angiogenesis. In contrast, upregulated genes targeted by downregulated miRNAs in response to PDGF-BB are involved in cell division and migration. These data suggest that the miRNAome in SMC is highly context dependent and sensitive to SMC differentiation states. Moreover, our studies predict that the subset of miRNAs expressed in differentiated SMC controls pathways associated with quiescence, whereas the miRNAs upregulated in response to PDGF-BB exposure repress the SMC differentiation program.
The top-20 miRNA signature expressed in differentiated SMC regulates SMC homeostasis and cell-specific properties
The cell miRNAome depends on lineages, cell types, and phenotypic state (McCall et al., 2017; Nam et al., 2014; Sood et al., 2006; Sun et al., 2018). It also maintains central cellular biological processes by buffering aberrant transcript expression in response to environmental perturbations (Ebert and Sharp, 2012). We sought to determine the signature of the most abundantly expressed miRNAs in contractile SMC and evaluate their potential role in maintaining SMC homeostasis. Among 474 detected miRNAs in contractile SMC, the top 20 expressed miRNAs represent over 80% of total miRNAs (Figures 2A and 2B). By analyzing the targetome of these top 20 miRNAs, we predicted that they positively regulate SMC differentiation and quiescence by repressing genes involved in proliferation, migration, biosynthesis, and other developmental programs (Figures 2C, S5A, and S5B). Pairwise gene ontology analysis assessed the similarity of top-20 miRNA targets and demonstrated that these miRNAs could be grouped in several clusters with redundant functions (Figure 2D).
Figure 2.

The most abundant miRNAs in differentiated SMC are associated with the maintenance of SMC homeostasis and quiescence
(A and B) Expression of the most abundant top 20 miRNAs in differentiated SMC (control SMC). Data are expressed as mean ± SEM. (B) Chart representing the relative percentage of the most abundantly expressed miRNAs in differentiated SMC compared with total miRNA.
(C) Gene Ontology analysis of predicted targets of the top 20 expressed miRNA in differentiated SMC showed enrichment in cell proliferation (red bar graph), migration (orange bar graph), and biosynthesis (blue bar graph).
(D) Pairwise Gene Ontology term similarities for top expressed miRNAs using GOSemSim, for the enriched GOBP terms. p < 0.001 (Fisher exact test) and clustered with parameters as Euclidean distance.
(E) Volcano plot showing miRNA differential expression in PDGF-BB-treated group versus control group. Data are presented as Log2 fold change and −Log10 p-value.
(F) Prediction of upregulated (green) and downregulated (red) miRNAs based on RNAseq data and differentially expressed coding transcripts in PDGF-BB treated SMC (versus control).
(G) Venn diagram representing the overlap of DEmiRs identified in PDGF-BB-treated SMC (versus control) by small RNAseq quantification and RNAseq-based prediction.
(H) Validation of miR-145-5p and miR-200b-3p expression in PDGF-BB-treated, H3K4me2-editing-induced dedifferentiated SMC and control SMC by qPCR. Data are expressed as mean ± SEM. Groups were compared by one-way ANOVA. Threshold for statistical significance: p < 0.05.
As shown in Figure 1, PDGF-BB- and H3K4me2-editing-induced SMC dedifferentiation were associated with the differential expression of a large number of miRNAs (Figures 2E and S6A). To prioritize functional investigation of these miRNAs, we compared small RNAseq data and prediction of miRNA expression based on transcripts expression (RNAseq). We predicted DEmiR relevance based on target expression (Figures 2F and S6B). Interestingly, a partial overlap of DEmiRs identified by small RNAseq and coding-transcript-based prediction was observed (Figures 2G and S6C). These DEmiRs, at the intersection of quantification and prediction, are likely associated with the regulation of SMC phenotypic and functional states during dedifferentiation. Among these DEmiRs, we identified the miR-200 family (miR-200a, miR-200b, and miR-429) as markedly downregulated in our models of SMC dedifferentiation (Figures 2H and S7A). Besides miR-200, we confirmed the downregulation of miRNAs that have previously been implicated in SMC differentiation, including miR-145 and miR-124 (Choe et al., 2017; Cordes et al., 2009). Together, our analysis defined the SMC miRNA signature and identified the miR-200 cluster as a highly repressed miRNA family. Yet, the function of miR-200 in the context of SMC dedifferentiation has not been characterized.
The miR-200 cluster expression is tightly associated with SMC differentiation status
We further characterized the miR-200 family expression in differentiated and dedifferentiated SMC from rat, mouse, and human aorta. The miR-200 family is comprised of five highly conserved miRNA members, namely miR-200a, miR-200b, miR-200c, miR-429, and miR-141, which are grouped into two genomic clusters: miR429, miR200a, and miR200b form the genomic cluster I, whereas the genomic cluster 2 is composed of miR141 and miR200c (Figure 3A). Functionally, these miRNAs target two different sequences and are divided into functional groups I (miR-200b, miR-200c, and miR-429) and II (miR-141 and miR-200a). Of note, the sequence of miR-200a, miR-200b, and miR-429 among rat, mouse, and human is highly conserved (Figure S7B). We observed that both PDGF-BB treatment and H3K4me2 editing induced the marked repression of miR-200 genomic cluster I (miR-200a-3p, miR-200b-3p, and miR-429) (Figures 3B, 3C, and S7C). In contrast, the expression of genomic cluster II miRNAs, miR-141-3p and miR-200c-3p, was unchanged in response to PDGF-BB or increased in H3K4me2-edited SMC (Figures 3B, 3C, and S7C). Using publicly available data from the FANTOM miRNA atlas (de Rie et al., 2017), we found that miR-200a, miR-200b, and miR-429 are highly expressed in vascular smooth muscle cells compared with other major cell types (Figure 3D). The downregulation of miR-200 genomic cluster I was also observed in human and mouse SMC subjected to dedifferentiation stimuli (Figure 3E). We confirmed the expression of miR-200b in differentiated SMC in mouse aortas by in situ hybridization (ISH). ISH revealed the high expression of miR-200b in SMC within the medial layer (Figure 3F). Next, we assessed the expression of miR-200a, miR-200b, and miR-429 in rat aortic SMC using several models of dedifferentiation, including PDGF-BB, serum supplementation, and knockdown of myocardin or TET2, two central master regulators of SMC differentiation (Figures 3G and 3H). We found a robust and consistent downregulation of miR-200 genomic cluster I in these models, providing further evidence that the expression of miR-200 is tightly regulated by SMC differentiation status.
Figure 3.

Expression of mir200 genomic cluster I miRNAs is strongly repressed during SMC dedifferentiation
(A) Schematic of the mir200 family in rat. miR-200 family’s miRNAs are grouped in to two distinct genomic clusters based on different genomic locations and two functional clusters based on seed sequence similarities.
(B) Heatmap of miR-200 family expression in control, PDGF-BB-treated, and H3K4me2 SMC. Data presented as counts per million extracted from the small RNAseq dataset.
(C) Differential expression of miR200-cluster-associated miRNAs in rat PDGF-BB-treated SMC compared with control SMC. Data are expressed as mean ± SEM. Groups were compared by Student’s t test. ∗p < 0.05 was considered the threshold for significance.
(D) miR-200a, miR-200b, and miR-429 expression ranking among 250 cell types and subtypes in human. Dataset source: FANTOM miRNA atlas.
(E) Human and mouse SMC expression of miR-200a, miR-200b, and miR-429 subjected to serum- (human SMC; top graph) or PDGF-BB-induced (mouse SMC; bottom graph) dedifferentiation. Data are expressed as mean ± SEM. Groups were compared by multiple Student’s t tests. Threshold for statistical significance: p < 0.05.
(F) In situ hybridization staining using miR-200b or scramble control LNA probes in mouse carotid arteries. Merge with DAPI nuclear counterstaining. Scale bar: 50 μm.
(G) Expression of miR-200a, miR-200b, and miR-429 in rat SMC treated with PDGF-BB or 10% serum as compared with vehicle and 0% serum, respectively. Data are expressed as mean ± SEM. Groups were compared by multiple Student’s t tests. Threshold for statistical significance: p < 0.05.
(H) Expression of miR-200a, miR-200b, miR-429, myocardin, and TET2 in rat SMC transfected with siMyocd or siTET2 and compared with control siRNA-transfected SMC. Data are expressed as mean ± SEM. Groups were compared by multiple Student’s t tests. Threshold for statistical significance: p < 0.05.
The miR-200 cluster is a potent repressor of SMC proliferation, migration, and neointima formation after vascular injury
Based on mRNA/miRNA co-profiling and pathway analysis, we found that miR-200 predictively targets a cohort of genes involved in cell proliferation and migration, which were upregulated by PDGF-BB treatment (Figures 4A and S8A). To validate these findings, we analyzed SMC proliferation and migration capacities after miR-200b knockdown or overexpression (Figure 4B). First, we found that overexpression of miR-200b abolished the increase of cells in the G2/M phase induced by PDGF-BB (Figure 4C). This marked repression of cell proliferation was confirmed by quantifying Ki67 expression (Figure 4D). Although a large proportion of SMC treated with PDGF-BB expressed Ki67, the addition of miR-200b mimic markedly decreased active cell division. Conversely, inhibition of miR-200b was sufficient to increase Ki67 expression and cell division in SMC (Figure 4D). Of note, overexpression of miR-200b did not cause a decrease in SMC survival, apoptosis, or senescence (Figures S8B and S8C). Similarly, PDGF-BB-induced migration was abolished by miR-200b overexpression, whereas miR-200b knockdown increased SMC migration capacities in the absence of PDGF-BB (Figures 4E, S8D, and S8E). Interestingly, using inhibitors targeting specifically miR-200a, miR-200b, or miR-429, we found that inhibition of SMC migration was predominantly mediated by miR-200b and miR-429, which are part of the same functional cluster I, whereas inhibition of miR-200a had no significant effect on migration (Figure S8E). The combination of miR-200b and miR-429 inhibitors showed a similar impact on migration compared with a single inhibitor treatment, providing evidence of the redundancy in miR-200b and miR-429 function. In contrast, triple inhibition of miR-200a/200b/429 significantly reduces SMC migration compared with miR-200b and miR-429 inhibitors. Because miR-200a and miR-200b/429 belong to two different functional clusters and regulate different transcripts and pathways (Figures 3A and S8A), the triple inhibition may lead to the expression of transcripts with antagonistic effects on migration.
Figure 4.

miR-200b is a potent repressor of SMC proliferation and migration in vitro
(A) Chord plot of miR-200b targeted genes whose expression are upregulated in PDGF-BB-treated SMC and associated Gene Ontology pathways.
(B) Expression of miR-200b after treatment of rat SMC with control or miR-200b inhibitors (left graph) or mimics (right graph). Data are expressed as mean ± SEM. Groups were compared by Student’s t test. Threshold for statistical significance: p < 0.05.
(C) Cell-cycle analysis in rat SMC treated with PDGF-BB and miR-200b mimic by propidium iodide (PI) staining combined with flow cytometry. Data are expressed as mean ± SEM. Groups were compared by two-way ANOVA. ∗∗∗p < 0.001: comparison percentage in G1. ### p < 0.001: comparison percentage in G2/M.
(D) Cell proliferation assessed by Ki67 staining. Merged with DAPI nuclear counterstaining. Scale bar: 100 μm. The Ki67 staining mean intensity was normalized to DAPI. Data are expressed as mean ± SEM. Groups were compared by Student’s t test (miR-200b inhibitor) or one-way ANOVA (miR-200b mimic). Threshold for statistical significance: p < 0.05.
(E) Scratch wound assay performed on rat SMC treated with PDGF-BB or vehicle, in the absence or presence of miR-200b inhibitor or mimic and compared with their respective controls. Scratch area was measured at baseline, 8 h, and 24 h. Scale bar: 200 μm. Data are expressed as % scratch closure and mean ± SEM. Groups were compared by Student’s t test (miR-200b inhibitor) or one-way ANOVA (miR-200b mimic). Threshold for statistical significance: p < 0.05. ∗ <0.05, ∗∗ <0.01, ∗∗∗ <0.001.
Given its potent pro-quiescence and anti-migration functions in vitro and its marked decrease in expression during SMC phenotypic modulation, we investigated the impact of miR-200b modulation in vivo. First, we found that unilateral ligation of the carotid artery induced a decrease in miR-200b expression both in the media and in the neointima (Figure 5A). Next, we tested the impact of miR-200b overexpression on neointima formation. Using SMC fate mapping Myh11-CreERT2 YFP+/+ mice, we locally delivered lentivirus encoding miR-200b and the mCherry reporter protein concomitantly to carotid artery ligation (Figure 5B). mCherry staining showed that a large percentage of cells transduced by the control or miR-200b encoding lentivirus (Figure S9). Overexpression of miR-200b markedly reduced neointima formation without impacting the medial area (Figures 5C and 5D). Together, these data showed that miR-200b exerts pro-quiescent, anti-proliferative, and anti-migratory properties, and overexpression of miR-200b successfully attenuates neointima formation during injury-induced vascular remodeling in vivo.
Figure 5.

miR-200b overexpression prevents neointima formation after carotid artery ligation
(A) In situ hybridization staining using miR-200b LNA probes in uninjured and injured mouse carotid arteries 21 days post ligation. Merge with DAPI nuclear counterstaining. Scale bar: 50 μm.
(B) Schematics of the experimental design consisting in simultaneous unilateral ligation of the right carotid artery and delivery of lentiviral particles encoding for miR-200b-mCherry or control-mCherry in Myh11-CreERT2 R26R-EYFP mice.
(C) Immunofluorescent staining of ligated right carotid arteries stained with DAPI, YFP, and ACTA2 antibodies. Scale bar: 100 μm.
(D) Morphometric analysis of ligated carotid artery lesion and medial area in mice receiving miR-200b lentivirus (n = 6) or control lentivirus (n = 5). Data are expressed as mean ± SEM. Groups were compared by Student’s t test. Threshold for statistical significance: p < 0.05.
The miR-200 cluster targets Quaking, a central driver of SMC phenotypic modulation
We sought to define more precisely the miR-200b targetome in SMC and identify the mechanisms by which miR-200b prevents SMC proliferation and migration. First, we validated that the expression of multiple miR-200b predicted targets implicated in cell division and migration was directly impacted by miR-200b expression (Figure 6A). These targets were upregulated by PDGF-BB stimulation, but this effect was abolished by the co-treatment with miR-200b mimic. By comparing their respective targetome networks, we identified miR-200a and miR-200b/429, selective and shared targets whose expression was increased in dedifferentiated SMC (Figures 6B and S10A). The RNA binding protein Quaking (QKI) appeared as a common and central target of the mir200 genomic cluster I, targeted by both miR-200a and miR-200b/429 due to the presence of multiple conserved binding sites on QKI transcript (Figures S10B–S10D). QKI has been previously identified as a central regulator of SMC phenotype and an SMC dedifferentiation inducer by targeting, at least in part, myocardin transcript splicing (Veer et al., 2013). miR-200b overexpression potently abrogated the PDGF-BB-induced increase in QKI expression (Figures 6C and 6D). QKI 3’UTR luciferase assay confirmed that miR-200b directly targets QKI transcript (Figure 6E). miR-200b overexpression prevented the increase in QKI expression observed in SMC treated with siTET2 and siMyocd (Figure 6F). Next, we evaluated the contribution of QKI in regulating SMC proliferation. QKI knockdown was associated with a marked decrease in proliferation assessed by Ki67 staining (Figures 6G and S10E). To further determine the contribution of QKI in regulating SMC proliferation among miR-200b targets, we expressed a Quaking construct devoid of miR-200b sites and resistant to miR-200b targeting (QKIRES) (Figure S10F). Expression of QKIRES increased SMC proliferation. This augmentation of SMC proliferation was partially abolished by miR-200b mimic (Figure 6H). These findings provide evidence that miR-200b prevents SMC proliferation by targeting several central mechanisms, including Quaking. Our data also suggest that other mechanisms promoting SMC proliferation are targeted by miR-200b, including possible transcripts regulated by Quaking (Figure S10G).
Figure 6.

miR-200b inhibits SMC proliferation by direct targeting of Quaking
(A) Expression of miR-200b target genes associated with proliferation and migration in PDGF-BB-treated SMC in the absence or presence of miR-200b mimic.
(B) Network analysis of miR-200a and miR-200b targets upregulated in PDGF-BB-treated SMC. Quaking (QKI) is a shared target of miR-200a and miR-200b.
(C) Validation of the expression of Qki mRNA in rat PDGF-BB-treated SMC in the absence or presence of miR-200b mimic. Data are expressed as mean ± SEM. Groups were compared by one-way ANOVA. Threshold for statistical significance: p < 0.05.
(D) QKI protein expression in rat SMC treated with miR-200b mimic. Data are expressed as mean ± SEM. Groups were compared by Student’s t test. Threshold for statistical significance: p < 0.05.
(E) Luciferase activity assay using QKI-3′UTR or scramble UTR luciferase constructs transfected in rat SMC in the absence or presence of miR-200b mimic. Data are expressed as mean ± SEM. Groups were compared by multiple Student’s t test. Threshold for statistical significance: p < 0.05.
(F) Qki mRNA expression in rat SMC treated with siTET2 or siMyocd combined with miR-200b mimic treatment and compared with combination of negative control mimic and scramble siRNA. Data are expressed as mean ± SEM. Groups were compared by one-way ANOVA. Threshold for statistical significance: p < 0.05.
(G) Ki67 staining in rat SMC treated with siQKI or siCtrl. Scale bar: 100μm. Data are expressed as mean ± SEM. Groups were compared by Student’s t test. Threshold for statistical significance: p < 0.05.
(H) Ki67 staining in SMC overexpression QKIRES and transfected with miR-200b mimic as compared with control. Scale bar: 100μm. Data are expressed as mean ± SEM. Groups were compared by one-way ANOVA. Threshold for statistical significance: p < 0.05.
Discussion
miRNAs play a central regulatory role in maintaining and modulating cell behavior and function (Ebert and Sharp, 2012). This essential function of miRNAs is well illustrated by the lethality of Drosha deletion, specifically in SMC, demonstrating that miRNA biogenesis is required for vascular development (Fan et al., 2013). As detailed previously, specific miRNA gene deletion or antigomiR loss-of-function studies have delineated the active role of miRNAs regulating SMC differentiation state during development and vascular diseases, including miR143/145 and miR128 (Cordes et al., 2009; Farina et al., 2020; Goettsch et al., 2011; Sun et al., 2011, 2017). In this study, we unbiasedly characterized the miRNAome in differentiated and dedifferentiated SMC and predicted that the signature of abundantly expressed miRNA representing 80% of expressed miRNAs serves for maintaining SMC homeostasis and quiescence. Interestingly, the top miRNA identified in our study in rat aortic SMC partially overlaps with the most abundantly expressed miRNAs in human aortic SMC reported in a recent screen (Moreau et al., 2021). Of interest, the authors of this study found that the top miRNAs were partially similar across SMC, endothelial cells, and CD14+ macrophages. Together, these observations may indicate that a common core of abundantly expressed miRNAs across lineages and conditions may be required for basic functions or cell survival, rather than being lineage-specific and state-specific signatures. Yet, it is unclear whether a subset of miRNAs ubiquitously highly expressed regulates similar or distinct downstream cellular functions in different cell types. Comparing the miRNAome among different cell types and cell populations found in diverse physiological and pathological environments might help answer these remaining questions: (1) what is the function of abundantly expressed miRNA in different lineages? (2) Beyond commonly expressed miRNAs, can we identify reliable lineage-specific or differentiation-state-specific miRNA signatures? And (3) what is the functional relevance of such signature? The recent and ongoing development of single-cell miRNA sequencing and single-cell multiplexing platforms will help gain insight regarding alteration of miRNA expression during phenotypic modulation in vivo (Wang et al., 2019).
With the advances in genome sequencing and analysis capabilities, mRNA-miRNA co-profiling has become a feasible and effective approach for the prediction of mRNA/miRNA networks under certain physiological and pathological conditions (Liao et al., 2019; Wang et al., 2019). Although CLIPseq or RIPseq remains an accurate and evidential method for direct targeting identification (Clark et al., 2014; Wen et al., 2011), mRNA-miRNA co-profiling provides informative data to evaluate transcript abundance and differential expression, as well as regulatory networks based on mRNA-miRNA targeting with the help of bioinformatics tools and public databases. By co-profiling transcript and miRNA expression, we analyzed the overall mRNA-miRNA modulation during SMC dedifferentiation and found that 37.8% of DEG are direct targets of DEmiR after PDGF-BB treatment, a similar contribution as previously described in human cells (Lewis et al., 2005). This approach also led to the identification of the miR200 genomic cluster I as a central mechanism controlling SMC quiescence, based on not only the expression of miR-200 but also the directionality and pathway analysis of its putative targets.
The miR-200 family includes five miRNA members, miR-200a, miR-200b, miR-200c, miR-429, and miR-141, grouped in genomic and functional clusters based on their chromosomal location and seed sequence (Magenta et al., 2017). We provide evidence that the dedifferentiation-induced repression of miR200 is restricted to miRNAs included in the genomic cluster I (miR-200a, miR-200b, and miR-429). These miRNAs were highly sensitive to various dedifferentiation stimuli, including PDGF-BB and serum supplementation, but also TET2 and myocardin knockdown. This coordinate downregulation of miR200a, miR200b, and miR429 could be due to transcriptional and epigenetic regulation on a common promoter or enhancer regions controlling these genes distant of a few hundred base pairs. Future studies will be needed to determine the transcriptional and possible posttranscriptional mechanisms regulating miR-200 expression and function. The miR-200 cluster acts as a potent repressor of SMC proliferation and migration by targeting, at least in part, QKI, which has been previously identified as a critical regulator of SMC phenotypic switching (Veer et al., 2013). QKI is an RNA binding protein regulating splicing of pre-mRNA. In SMC, expression of QKI during phenotypic switching induces myocardin pre-mRNA alternative splicing and reduces the expression of pro-contractile and pro-quiescent myocardin transcript variants. Our study suggests that miR-200a, miR-200b, and miR-429 serve to maintain a low level of QKI. Importantly, our understanding of the transcripts targeted by QKI in SMC during dedifferentiation is limited. Whether some of these targets regulate proliferation and migration and are subject to miR-200b-mediated repression must be experimentally explored.
The miR-200 family is expressed by many cell types and implicated in various pathological processes, such as cancer and cardiovascular diseases. In cancer, miR-200 has been implicated as an essential regulator in controlling endothelial-to-mesenchymal transition by targeting ZEB1 and ZEB2 (Humphries and Yang, 2015; Park et al., 2008). In cardiovascular disease, downregulation of miR-200c and concomitant upregulation of ZEB1/2 have been associated with aneurysm development in patients with bicuspid valves (Maleki et al., 2019). Interestingly, an increase of miR-200b in SMC isolated from diabetic (db/db) mice has been reported (Reddy et al., 2012). In that context, the increase of miR-200b promotes a proinflammatory phenotype by suppressing ZEB-1 and promoting the expression of the proinflammatory genes MCP-1 and COX2. Compared with our study, these results indicate that miR-200b expression in SMC is highly sensitive to different stimuli and can be either downregulated (PDGF-BB treatment) or upregulated (diabetes). The subsets of genes regulated by miR-200b in diabetic/proinflammatory and healthy SMC are likely different, as the SMC transcription profiles in these conditions markedly diverge (Alexander et al., 2012). Together, these studies support the involvement of the miR-200 family in vascular homeostasis and disease and the versatility in miR-200 function, which depends on its “targetome” expressed in SMC under various conditions.
The versatility of the miR-200 family function in SMC is also remarkable in light of our in vivo data. miR-200b overexpression in SMC lineage-tracing mice provided strong evidence that miR-200b restrains neointima formation. The overexpression of miR-200b in the carotid artery, including SMC comprised within the medial layer, prevented SMC participation in vascular remodeling and their population of the neointima, likely due to impairment of proliferation and migration abilities, as no SMC (YFP+) were observed within the neointimal space. These observations highlight the anti-proliferative potential of miR-200b-based therapeutic therapies for treating diseases and conditions such as restenosis or atherosclerosis. In that latter context, further investigations are needed to evaluate the effects of forced miR-200b expression on both atherosclerotic plaque formation and established plaque regression/remodeling. Of note, miR-200a, miR-200b, and miR-429 levels of expression fluctuate in different SMC subtypes, including aortic, carotid, and coronary SMC. Whether these differences impact the role of miR-200b in these SMC subpopulations should be rigorously investigated.
In conclusion, our study established the mRNA-miRNA co-profiling during SMC dedifferentiation, predicted the overall contribution of miRNA in modulating mRNA transcriptome, identified miR-200 as a functional miRNA cluster in SMC differentiation, and demonstrated the underlying mechanism controlling SMC quiescence. Notably, miR-200b overexpression significantly prevented SMC investment into injury-induced neointimal formation, which provides a potential therapeutic basis for alleviating neointimal proliferative diseases.
Limitations of the study
A limitation of our study is that the lentivirus-mediated overexpression of miR-200b is not restricted to medial SMC but also occurs in endothelial and adventitial cells. As miR-200b also inhibits endothelial-to-mesenchymal transition, the reduction of neointima lesion size may result from a lack of participation of both SMC and endothelial cells (Sinha et al., 2015; Vanchin et al., 2021). Furthermore, QKI appears as a central and common target of miR-200b in SMC and endothelial cells, miR-200b. miR200b also inhibits QKI-mediated endothelial cell proliferation in endothelial cells (Azam et al., 2019). Although future studies should focus on deciphering the cell-specific role of miR-200b in vascular remodeling, miR-200b function in both endothelial cells and SMC positions this microRNA as a potential therapeutic strategy to prevent neointima formation in the event of arterial occlusions, such as stent restenosis or atherosclerosis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-QKI antibody | Invitrogen | Cat# PA5-53930, RRID:AB_2646184 |
| Anti-GAPDH antibody | Abcam | Cat# ab9485, RRID:AB_307275 / Cat# ab8245, RRID:AB_2107448 |
| Anti-GFP antibody | Abcam | Cat# ab6673, RRID:AB_305643 |
| Anti-mCherry antibody | Abcam | Cat# ab167453, RRID:AB_2571870 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Molecular Probes | Cat# A-21447, RRID:AB_141844 |
| Secondary donkey anti-rabbit 555 antibody | Invitrogen | Cat# A31572, RRID:AB_162543 |
| Anti-ki67 antibody | Abcam | Cat# ab15580, RRID:AB_443209 |
| Anti-DIG-POD antibody | Roche | Cat# 11207733910, RRID:AB_514500 |
| Mouse IgG - Isotype Control | Abcam | Cat# ab37355, RRID:AB_2665484 |
| Rabbit IgG - Isotype Control | Abcam | Cat# ab171870, RRID:AB_2687657 |
| Donkey Anti-Rat IgG H&L (Alexa Fluor 555) | Abcam | Cat# ab150154, RRID:AB_2813834 |
| IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody | LI-COR | Cat# 926-32211, RRID:AB_621843 |
| IRDye 680RD Donkey anti-Mouse IgG Secondary Antibody | LI-COR | Cat# 926-68072, RRID:AB_10953628 |
| Monoclonal Anti-Actin, a-Smooth Muscle - FITC antibody produced in mouse | Sigma-Aldrich | Cat# F3777, RRID:AB_476977 |
| Bacterial and virus strains | ||
| Lenti-mmu-miR-200b-3p | Genecopoeia | Cat# MmiR-SN0300-LVE004 |
| Lenti-miR-Ctrl | Genecopoeia | Cat# CmiR-SN0001-LVE004 |
| Lenti-Myocd-LSD1 | Liu et al., 2021 | N/A |
| Lenti-Myocd-LSD1NF | Liu et al., 2021 | N/A |
| Biological samples | ||
| Human coronary artery smooth muscle cells | Lonza | Cat# CC-2583 |
| Chemicals, peptides, and recombinant proteins | ||
| Corning Fetal Bovine Serum, 500 mL, Premium, United States Origin | Corning | Cat# 35-015-CV |
| L-Glutamine (200 mM) | Gibco | Cat# 25030081 |
| L-Ascorbic acid | Sigma-Aldrich | Cat# A4403; CAS: 50-81-7 |
| apo-Transferrin human | Sigma-Aldrich | Cat# T5391; CAS: 11096-37-0 |
| Sodium selenite | Sigma-Aldrich | Cat# S5261; CAS: 10102-18-8 |
| PDGF-BB human | Sigma-Aldrich | Cat# SRP3138 |
| Rapamycin from Streptomyces hygroscopicus | Sigma-Aldrich | Cat# R0395; CAS: 53123-88-9 |
| Lipofectamine 3000 Transfection Reagent | Invitrogen | Cat# L3000015 |
| Lipofectamine RNAiMax Reagent | ThermoFisher | Cat# 13778075 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat# A25742 |
| mirVana miRNA mimic miR-145-5p | ThermoFisher | Cat# MC11480 |
| mirVana miRNA mimic negative control | ThermoFisher | Cat# 4464058 |
| mirVana miRNA mimic miR-200b-3p | ThermoFisher | Cat# MC11073 |
| mirVana miRNA inhibitor miR-200a-3p | ThermoFisher | Cat# MH10991 |
| mirVana miRNA inhibitor miR-200b-3p | ThermoFisher | Cat# MH11073 |
| mirVana miRNA inhibitor miR-429 | ThermoFisher | Cat# MH10759 |
| mirVana miRNA inhibitor negative control | ThermoFisher | Cat# 4464079 |
| Silencer Select siTet2 siRNA | ThermoFisher | Cat# s160756 |
| Silencer Select siMyocardin siRNA | ThermoFisher | Cat# s140957 |
| Silencer Select siQKI siRNA | ThermoFisher | Cat# s177598 |
| Silencer Select control siRNA | ThermoFisher | Cat# 4390843 |
| Duo-luciferase miRNA 3' UTR target clone control | Genecopoeia | Cat# CmiT000001-MT06 |
| Invitrogen Alexa Fluor 647 Phalloidin | Invitrogen | Cat# A22287 |
| Duolink In Situ mounting medium with DAPI | Sigma-Aldrich | Cat# DUO82040 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648; CAS: 10540-29-1 |
| Pluronic F-127 | Sigma-Aldrich | Cat# P2443; CAS: 9003-11-6 |
| 16% Paraformaldehyde Aqueous Solution, EM Grade, Ampoule 10 ML | Electron Microscopy Sciences | Cat# 15710 |
| Crystal violet solution | Sigma-Aldrich | Cat # V5265; CAS: 548-62-9 |
| Antigen Unmasking Solution, Citrate-Based | Vector Laboratories | Cat# H-3300 |
| DMEM:F12 cell culture medium | Gibco | Cat# 11320-033 |
| Penicillin-streptomycin antibiotics | Gibco | Cat# 15140122 |
| rhEGF | Biolegend | Cat# 713008 |
| rhFGF | Biolegend | Cat# 713034 |
| M199 medium | Gibco | Car# 11150-067 |
| QIAzol | Qiagen | Cat# 79306 |
| TaqMan™ Universal Master Mix II, no UNG | Applied Biosystems | Cat# 4440043 |
| NuPAGE 4-12% Bis-Tris Gels | Invitrogen | Cat# NP0321PK2 |
| 0.45 μm Nitrocellulose Membranes | Bio-Rad | Cat# 1620115 |
| Prolong Gold Antifade Reagent | Fisher | Cat# P36930 |
| SSC solution | Sigma | Cat# S6639-1L |
| TSA-plus Cyanine 3 | Akoya Bioscience | Cat# NEL744001KT |
| Critical commercial assays | ||
| Qubit RNA BR Assay Kit | Invitrogen | Cat# Q10210 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| TaqMan MicroRNA Reverse Transcription Kit | Applied Biosystems | Cat# 4366596 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# 7570 |
| Luciferase assay kit | Promega | Cat# E1500 |
| Propidium Iodide (PI) Flow Cytometry Kit | Abcam | Cat# ab139418 |
| Luc-Pair Duo-Luciferase Assay Kits 2.0 | Genecopoeia | Cat# LF001 |
| miRCURY LNA miRNA ISH Optimization Kits | Geneglobe | Cat# 339459 |
| Deposited data | ||
| Bulk RNAseq | Liu et al., 2021 | NCBI’s Gene Expression Omnibus GSE179220 |
| Small RNAseq | This paper | NCBI’s Gene Expression Omnibus GSE186972 |
| Experimental models: Cell lines | ||
| Human aortic smooth muscle cells (hASMCs) | Lonza | Cat# CC-2571 |
| Experimental models: Organisms/strains | ||
| Myh11-CreERT2+/-R26R-EYFP+/+ male mice | This paper | N/A |
| Myh11-CreERT2-/-R26R-EYFP+/+ female mice | This paper | N/A |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| pLX317-QKI expression plasmid | Addgene | Cat# pLX317-QKI |
| Duo-luciferase miRNA 3' UTR target clone for QKI | Genecopoeia | Cat# MmiT073700-MT06 |
| Duo-luciferase miRNA 3' UTR target clone control | Genecopoeia | Cat# CmiT000001-MT06 |
| Software and algorithms | ||
| Prism 9 | Graph Pad | https://www.graphpad.com/ |
| Adobe Illustrator 2021 | Adobe | https://www.adobe.com/products/illustrator.html |
| Office 365 | Microsoft | https://www.microsoft.com/en-us/microsoft-365 |
| Adobe Photoshop 2021 | Adobe | https://www.adobe.com/products/photoshop.html |
| R version 4.0.4 | http://www.r-project.org/ | RRID:SCR_001905 |
| QIAGEN CLC Genomics Workbench | Qiagen | https://www.qiagen.com/us/products/discovery-and-translational-research/next-generation-sequencing/informatics- and-data/analysis-and-visualization/clc-genomics-workbench/ |
| BaseSpace Correlation Engine | Illumina | https://www.illumina.com/products/by-type/informatics-products/basespace-correlation-engine.html |
| Targetscan | http://targetscan.org | RRID:SCR_010845 |
| Ocular Advanced Scientific Camera Control software | Digital Opitcs Limited | https://www.photometrics.com/products/ocular |
| Image Pro Premier | Media Cybernetics | https://www.mediacy.com/support/imagepropremier |
| Circlize | Github | RRID:SCR_002141 |
| GOSemSim | Github | https://github.com/YuLab-SMU/GOSemSim |
| miRNA atlas | FAMTOM 5.0 | https://fantom.gsc.riken.jp/5/ |
| Ingenuity Pathway Analysis | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Cytoscape | National Institute of General Medical Sciences of NIH | https://cytoscape.org |
| ImageJ | NIH Image | https://imagej.nih.gov/ij/index.html |
| Facsdiva 8 | BD Bioscience | https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software |
| Other | ||
| LEICA Dmi8 Inverted Fluorescent Microscope | LEICA | https://www.leica-microsystems.com/products/light-microscopes/p/leica-dmi8-id/ |
| Synergy-HTX multi-mode reader | BioTek | https://www.biotek.com/products/detection-multi-mode-microplatereaders/synergy-htx-multi-mode-reader/ |
| 6.5 mm Transwell with 8.0 mm Pore | Corning | Cat# 3422 |
| CFX Connect Realtime System | Bio-Rad | Cat# 1855201 |
| Odyssey CLx Imaging System | LI-COR | https://www.licor.com/bio/odyssey-dlx/ |
| BD LSR II Flow Cytometer | BD Bioscience | Cat# BF-LSRII |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Delphine Gomez (gomezd@pitt.edu).
Materials availability
All unique materials generated in this study are available from the lead contact upon completion of materials transfer agreement with the University of Pittsburgh.
Data and code availability
-
•
The small RNAseq data generated in this study has been deposited on the NCBI GEO Datasets platform (accession number: GSE179220) and are publicly available as of the date of publication. The RNAseq data originate from a previous publication (NCBI GEO; accession number: GSE186972). Please note that the small RNAseq and RNAseq were not performed on the same total RNA extraction but from independent experiments, though cell line, passage, culture conditions, and treatments were identical.
-
•
The paper does not report original codes or reagents. All codes or reagents used in this study are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact.
Experimental model and subject details
All the mice used in this study and related mouse protocols were reviewed and approved by University of Pittsburgh Institutional Animal Care and Use Committee. SMC lineage tracing mice were generated by crossing Myh11-CreERT2 and R26R-EYFP mice as previously described (Gomez et al., 2013). All the mice used in this study were offsprings of Myh11-CreERT2+/− R26R-EYFP+/+ males and Myh11-CreERT2−/− R26R-EYFP+/+ females. All mice were genotyped by PCR. Only male mice were used experimentally in due to the location of the Myh11-CreERT2 transgene on the Y chromosome. Male mice were injected daily at 6 weeks of age with tamoxifen dissolved in peanut oil at 10 mg/mL, for a continuous period of 10 days. One week after the last tamoxifen injection, ligation of the right carotid artery ligation was performed. Complete ligation was performed to the right common carotid artery using 7-0 silk suture. At the time of the ligation, pluronic gel containing lentivirus encoding miR-200b-3p or negative control mimic (107 pfu/carotid artery; Genecopoeia) was delivered to the same injured carotid. 3 weeks following surgery, the mice were euthanized by CO2 asphyxiation and full perfusion of PBS and 4% PFA was done using gravity perfusion system through left ventricle. The left and right common carotid arteries were excised and fixed in 4% PFA at 4°C overnight. Tissues were processed and embedded in paraffin and serial sections (thickness: 10 μm) of embedded carotid were collected starting from the suture.
miR-200b lentivirus
Lentivirus encoding miR-200b or control mimic were purchased from Genecopoeia. The lentivirus backbone pEZX-MR13 includes the EF1a promoter and the mCherry reporter sequence. The miRNA sequences cloned into the lentivirus backbone were the following:
-
1.
CCATCATTACCAGGCAGTATTATAGTGAAGCCACAGATGTATAATACTGCCTGGTAATGATGA: miR-200b, MmiR-SN0300-LVE004
-
2.
CAACGTGACACGTTCGGAGAATAGTGAAGCCACAGATGTATTCTCCGAACGTGTCACGTTT: miRNA scramble control, CmiR-SN0001-LVE004
Cell culture
Rat and mouse aortic smooth muscle cells were cultured in growth medium (DMEM:F12, Gibco, 11320-033) supplemented with fetal bovine serum (10%, Corning, 35-015-CV), L-glutamine (1.6 mM, Gibco, 25030081), and penicillin-streptomycin (100 U/mL, Gibco, 15140122) in a humidified sterile incubator at 37°C with 5% CO2. Rat aortic smooth muscle cells constitutively expressing Myocd-LSD1 were generated by retroviral transduction as previously described (Liu et al., 2021). Prior to all stimulations and treatments, SMC were starved in a serum-free, insulin-free medium supplemented with L-glutamine (1.6 mM, Gibco, 25030081), L-ascorbic acid (0.2 mM, Sigma Aldrich, A4403), Apo-Transferrin (5 mg/ml, Sigma Aldrich, T5391) and Na Selenite (6.25 ng/ml, Sigma-Aldrich, S5261) for 24 to 48 h at 37°C with 5% CO2. Independent experiments were performed using SMC from constitutive passages. Human recombinant PDGF-BB (Sigma Aldrich, SRP3138) was reconstituted in 10 mM Acetic Acid at a final concentration of 30 ng/ml for treatment. SMC were treated with PDGF-BB for 24h before being harvested for analysis.
Human aortic smooth muscle cells (hASMCs, Lonza) were cultured in SmBMTM Basal Medium (CC-3181, Lonza) supplemented with SmGMTM-2 SingleQuotsTM (CC-4149, Lonza), 10% FBS and 100 U/ml each penicillin-streptomycin. Cells were starved in serum-free medium for 16 h before treatment.
Method details
RT-qPCR for mRNA and miRNA quantification
SMC were collected and total RNA was extracted by Qiazol Reagent (Qiagen, 79306) or miRNeasy Mini Kit (Qiagen, 217004) according to manufacturer’s protocol. RNA concentration was quantified by Qubit RNA Broad Range Assay kit (Invitrogen, Q10210). For mRNA quantification, cDNA was synthesized from 1μg of RNA using iScript cDNA Synthesis Kit (Bio-Rad, 1708891) and real-time quantitative PCR was performed with PowerUpTM SYBRTM Green Master Mix (Applied Biosystems, A25742) using CFX Connect Realtime System (Bio-Rad, 1855201). mRNA levels of target genes were normalized to 18s expression. For miRNA quantification, 100 ng RNA was reversely transcribed using TaqMan™ MicroRNA Reverse Transcription Kit (Applied Biosystems, 4366596) and specific RT probe included in TaqMan™ microRNA assays (U6 assay ID: #001973, miR-145 assay ID: #002278, miR-200a assay ID: #000502, miR-200b assay ID: #001800, miR-429 assay ID: #001077) according to the manufacturer’s instructions. qPCR was performed using TaqMan™ Universal Master Mix II, no UNG (Applied Biosystems, 4440043) and specific microRNA TaqMan primers included in the assays. The relative quantification of microRNA expression was normalized to snRNA U6 using the 2−ΔΔCt method and fold change was normalized to control group from the same independent experiments. Primers used for qPCR are listed in Table S1.
Small RNA sequencing and data analysis
Total small RNA was extracted, and column purified from control SMC (n = 3), Myocd-LSD1 SMC (n = 3), and SMC treated with PDGF-BB (30 ng/ml, Sigma Aldrich, SRP3138) for 24 h (n = 3) using miRNeasy Mini Kit (Qiagen, 217004) following manufacture’s protocol. Genomic DNA was removed using DNase I (Qiagen). RNA quality control, library preparation and sequencing were performed by Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh. RNA quality was checked with Agilent RNA Screen Tape Assay Tape Station System. The library of small RNA was prepared by QIAseq small RNA according to manufacturer’s protocol and sequencing was performed with illumina NextSeq High Output 75 cycle kit (1 × 75 bp) with reading depth of 30 million reads per sample. The 75 bp single end reads and gene ID were mapped to Rnor 6.0 using default setting by CLC Genomic Workbench 21 (Qiagen). All mapped reads were normalized by Trimmed Mean of M values (TMM normalization) for differential gene expression analysis in CLC Genomic Workbench. Analysis and plotting of principal component analysis, volcano plots and heatmaps were created by CLC Genomic Workbench at default setting. Global heatmap was generated with parameters as Euclidean distance measure, complete linkage cluster. For all the analysis of small RNAseq data, the criteria for data filtration and cut-off are fold-change > 1.5 and p value < 0.05. Small RNA percentage was generated from Geneglobe primary analysis and isomiR analysis was obtained using default setting in CLC Genomic Workbench (Qiagen).
RNAseq data analysis was performed on previously published data (NCBI GEO Accession number: GSE179220) using the same cell lines under identical treatment conditions as previously described (Liu et al., 2021). Analysis and plotting method of RNAseq data are same as small RNAseq analysis. The cut-off values for RNAseq are fold-change > 1.7 and p value < 0.05.
Bioinformatic analysis and plotting
All data were acquired from mRNAseq and small RNAseq with the criteria described above. Network of mRNA-miRNA pairing was created on Ingenuity Pathway Analysis (IPA) platform, using filtered mRNAseq and small RNAseq data combined with miRbase, Targescan databases. Briefly, the mRNA-miRNA targeting relationship was predicted and collected in IPA platform. Subsequently, the mRNA-miRNA network was filtered by mRNAseq and small RNAseq data taking into account 1) mRNA/miRNA abundance (max group mean >10), 2) mRNA/miRNA direction (e.g. miRNA is upregulated and mRNA is downregulated) and 3) mRNA/miRNA differentially expression significance (fold change >1.5 and p value < 0.05) 4) strength of miRNA-mRNA targeting (experimentally validated and highly predicted). Pathway enrichment analysis (Gene Ontology) and gene lists associated with single or overall mRNA-miRNA network were acquired from the Biogroups app from Correlation Engine (IIlumina). miRNA expression ranking data was acquired from FANTOM 5.0 miRNA atlas. Pie chart and bar graph for target-associated pathways and biological process were generated on PantherDB. Probability of conserved targeting data was acquired from TargetScan. Chord plots were generated by RStudio using Circlize package. Pairwise similarities for top expressing miRNAs were analyzed using GOSemSim, for the enriched GOBP terms. p < 0.001 (Fisher’s exact test) and clustered with parameters as euclidean distance. Network plots were generated by Cytoscape.
MicroRNA, siRNA and Plasmid Transfection
SMC were seeded at 1.5 × 105 cells per well in routine growth medium with 10% FBS into 6-well plates. When cells reached 70-80% confluency, the culture media was switched to transfection medium which excluded antibiotics from the routine growth media. For miRNA and siRNA transfection, miR-200b mimic (MC11073), miR-200a inhibitor (MH10991), miR-200b inhibitor (MH11073), miR-429 inhibitor (MH10759) with their corresponding negative control mimic (4464058) / inhibitor (4464079) (ThermoFisher, miRVana) and siTet2 (s160756), siMyocardin (s140957), siQKI (s177598) (ThermoFisher, Silencer Select siRNA), and siControl were transfected into cells using Lipofectamine RNAiMax Reagent (ThermoFisher, 13778075) according to manufacturer’s instructions. All mimics, inhibitors, and siRNA were used at 50 nM and 0.1 nmol were transfected into one well of 6-well plate. For plasmid transfection, Quaking expression plasmid pLX317-QKI (Addgene) and duo-luciferase plasmid carrying QKI 3’UTR (Genecopeia, #MmiT073700-MT06, GenBank: NM_001159517.1) were transfected with Lipofectamine 3000 Transfection Reagent (Invitrogen, L3000015). 2 μg of plasmid was transfected in each well of a 6-well plate. 24 hours after transfection, cells were switched to serum-free insulin-free culture medium for cell state recovery for 24 h and were then used for downstream experiments and analysis.
Western blot
Total proteins were extracted using CHAPS buffer (1% CHAPS hydrate, 150 mM NaCl, 25 mM HEPES Buffer). Extracts were separated by electrophoresis on NuPAGE 4-12% Bis-Tris Gels (Invitrogen, NP0321PK2), and transferred onto 0.45 μm Nitrocellulose Membranes (Bio-Rad, 1620115). Membranes were incubated with antibodies specific to QKI (Invitrogen, PA5-53930, RRID:AB_2646184) and GADPH (Cat# ab9485, RRID:AB_307275 / Cat# ab8245, RRID:AB_2107448). IRDye® anti-rabbit IgG secondary antibody (LI-COR, RRID: AB_621843) or anti-mouse IgG secondary antibody (LI-COR, RRID: AB_10953628) were used for immune-blotting. Membranes were imaged on the LI-COR Odyssey CLx imaging system.
Immunofluorescent staining
Tissues were embedded vertically. Serial 10μm sections were collected beginning from the level of visible suture. For immunofluorescent staining of tissue slides, sections covering 180μm from the ligation site were collected. Following deparaffinization, antigen retrieval was performed by heat method (Vector Laboratories H-3300). Staining was performed to evaluate gene expression in cells using primary antibodies for GFP (Abcam ab6673 1:250), mCherry (Abcam ab167453 1:500) or control IgG from same species. Secondary antibodies included donkey anti-goat 647 (Life Technologies A21447) and donkey anti-rabbit 555 (Invitrogen A31572). Along with the secondary antibodies, sections were also stained for ACTA2 (Sigma-Aldrich F3777 1:500) and DAPI (1:1000). Slides were mounted using Prolong Gold Antifade Reagent (Fisher P36930). For immunofluorescent staining on cultured cells, cells were fixed with 4% Paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.5% Triton X-100 (Sigma). For proliferation studies, Ki67 (Abcam ab15580 1:500) were used as primary antibody and secondary donkey anti-rabbit 555 (Invitrogen A31572) were used subsequently.
Images were acquired on a fluorescent microscope (Leica, DMi8) using the Ocular Advanced Scientific Camera Control software (Digital Optics Limited) or Nikon A1 Confocal microscope using NIS-Elements software (Nikon). Image processing was performed using ImageJ using similar and standardized parameters.
Transwell assay
SMCs were seeded (1.2 × 105 cells/transwell) with serum-free insulin-free media in 8 μm pore size polycarbonate transwell inserts (Corning, 3422). Serum-free insulin-free media supplemented with PDGF-BB (Sigma-Aldrich, SRP3138) was added to the lower compartment in a final concentration of 30 ng/ml as the chemo-attractant. After 24 h incubation, cells were fixed with 4% paraformaldehyde (Electrion Microscopy Science, 15710) and stained with 0.1% crystal violet (Sigma-Aldrich, V5265). Cells on the upper compartment were carefully removed by a cotton swab. The membrane was excised and mounted onto a glass slide with Prolong Gold Mounting Media (ThermoFisher, P10144). Images were taken by microscope (Leica DM500) with the Leica LAS EZ software and processed using ImageJ software under same parameters for each batch experiment.
Wound assay
SMCs were seeded at 1.5 × 105 cells per well in routine growth medium with 10% FBS into 6-well plates. SMCs were then transfected with miRNA mimic or inhibitor as described above. 24 h after transfection, the cells were switched to serum-free insulin-free culture medium and incubated for 24 h until the cells reached 90-95% confluency. Wound was made uniformly by a 2-mm wide comb to the center of the culture well to create a gap allowing cell migration. The culture media was then switched to serum-free insulin-free culture medium containing control vehicle or PDGF-BB to induce dedifferentiation and migration of SMC. Images were collected at 0, 6 and 24 h on a fluorescent microscope (Leica, DMi8) using the Ocular Advanced Scientific Camera Control software (Digital Optics Limited). Image processing and measurement was accomplished on ImageJ.
Cell cycle assay
SMCs were seeded in routine growth medium with 10% FBS into 6-well plate and was transfected at 70-80% confluency as described above. After transfection and PDGF-BB or vehicle treatment, cells were trypsinized into single cell suspension and collect into a cell pellet by centrifugation. Cells were fixed with cold 70% ethanol at −20C for at least 16 h before staining. DNA content was stained with Propidium Iodide (PI) Flow Cytometry Kit (Abcam, ab139418) according to manufacturer’s instructions. Analysis was performed by FACS in a BD LSR II Flow Cytometer in the Unified Flow Cytometry Core at University of Pittsburgh School of Medicine. Data were acquired and analyzed on Facsdiva 8. According to instructions and data distribution, the PI signal was clustered and converted to 2N, 2N–4N, and 4N, which represented cell population of phase G1, G2/Mitotic and S.
Luciferase assay
The dual-luciferase constructs were purchased from Genecopoeia: miR-200b 3’UTR #MmiT073700-MT06 optimized for miRNA targeting at Genbank: NM_001159517.1 3’UTR and control scramble 3’UTR # CmiT000001-MT06. Briefly, the plasmids were transfected using Lipofectamine 3000 (Invitrogen, L3000015) and incubated for 48 h at 37°C with 5% CO2. Cells were lysed and luciferase activity was assessed by Luc-Pair Duo-Luciferase Assay Kits 2.0 (Genecopoeia, LF001). Renilla luciferase activity was normalized to the corresponding firefly luciferase activity and plotted as a percentage of the control.
Fluorescence microRNA in situ hybridization
Fluorescence microRNA in situ hybridization was performed on tissue sections from healthy or injured mouse carotid arteries using miRCURY LNA miRNA ISH Optimization Kits (Geneglobe, 339459) according to manufacturer’s instructions. Briefly, the sectioning slides were deparaffinized and digested by proteinase K approach at 37°C for 15 min. LNA miR-200b (YD00619853) and LNA scrambled microRNA (YD00699004) probes were hybridized at final concentration of 1 nM, 40nM and 40nM respectively, followed by stringent washes of serial diluted SSC (Sigma, S6639-1L). After 30 min blocking, the slides were incubated with anti-DIG-POD (Roche, 11207733910) at 1:400 dilution for 60 min at room temperature according to manufacturer’s instructions. TSA-plus Cyanine 3 (Akoya Bioscience, NEL744001KT) substrate was added to the sections and incubated two times for 5 min each to amplify the fluorescence signal. NucBlue Live ReadyProbes Reagent was applied to the slides at last to facilitate cell nuclei staining. Images were acquired on a fluorescent microscope (Leica, DMi8) using the Ocular Advanced Scientific Camera Control software (Digital Optics Limited) or Nikon A1 Confocal microscope using NIS-Elements software (Nikon).
Cell Titer-Glo
Rat aortic smooth muscle cells were seeded in a 96-well-plate at a confluency of 1000 cells/well and were cultured in growth medium (DMEM:F12, Gibco) supplemented with fetal bovine serum (10%, Corning), L-glutamine (1.6 mM, Gibco), and penicillin-streptomycin (100 U/mL, Gibco). Cell viability was assessed by measuring ATP content at 24, 48 and 72 h by using the Cell Titer-Glo luminescent cell viability assay following manufacture’s recommendations (Promega, G7570). Luminesce measurements were performed in a Synergy-HTX multi-mode reader (Biotek).
Quantification and statistical analysis
All data are presented as the mean ± SEM. In vitro experiments were repeated independently at least 3 times with technical duplicates. One single data symbol represented the mean value of technical repeats for one experiment. For in vivo experiments, 5 and 6 independent mouse littermates were used and randomly assigned as control and experimental group. All statistics were performed using GraphPad Prism 8. Two-tailed unpaired Student’s t-tests with a confidence level of 95% were used to compare two groups with continuous variables, normal distribution, and equal variances (tested by Kolmogorov-Smirnov test). Two-tailed unpaired Student’s t-tests followed by a Welch’s correction with the confidence level of 95% were performed if two groups have unequal variances. Two-tailed unpaired Mann-Whitney U-tests with the confidence level of 95% were used if variables were non-normally distributed. For comparison between multiple groups with a single factor or two factors, we used one-way or two-way ANOVA, respectively. For categorial data, we used two-sided Fisher’s exact test. p ≤ 0.05 was considered as statistically significant.
Acknowledgments
We thank the Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh, the Health Sciences Library System at the University of Pittsburgh, and the Rangos Research Center Histopathology Core at Children’s Hospital of Pittsburgh. Schematics were created with Biorender. This work is supported by the National Institute of Health R01HL146465 and the American Heart Association 20IPA35310394 grants to D.G., the National Institute of Health T32 HL129964 to C.E-D., the Third Xiangya Hospital of Central South University to M.D. (#2204130712), the National Institutes of Health R01 HL124021, HL122596 and American Heart Association Established Investigator Award 18EIA33900027 to S.Y.C.
Author contributions
Conceptualization, D.G. and M.D.; Methodology, D.G., M.D., C.E-D., and A.H.; Investigation, D.G., M.D., C.E-D., M.L., I.A.A., J.W., and S.M.; Formal analysis, M.D., C.E., M.L., I.A.A., and A.H.; Writing—Original Draft, M.D. and D.G.; Writing—Review & Editing, M.D., C.E., M.L., I.A.A., S.M, J.W., A.H., S.Y.C, and D.G; Funding Acquisition, D.G. and S.Y.C; Supervision, D.G. and S.Y.C.
Declaration of interests
S.Y.C. has served as a consultant for Acceleron Pharma and United Therapeutics. S.Y.C. is a director, officer, and shareholder in Synhale Therapeutics. S.Y.C. has held grants from Actelion, Bayer, and Pfizer. S.Y.C. has filed patent applications regarding metabolism and next-generation therapeutics in pulmonary hypertension.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104169.
Supplemental information
References
- Alencar G.F., Owsiany K.M., Karnewar S., Sukhavasi K., Mocci G., Nguyen A.T., Williams C.M., Shamsuzzaman S., Mokry M., Henderson C.A., et al. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation. 2020;142:2045–2059. doi: 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander M.R., Murgai M., Moehle C.W., Owens G.K. Interleukin-1beta modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-kappaB-dependent mechanisms. Physiol. Genom. 2012;44:417–429. doi: 10.1152/physiolgenomics.00160.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam S.H., Porrello A., Harrison E.B., Leslie P.L., Liu X., Waugh T.A., Belanger A., Mangala L.S., Lopez-Berestein G., Wilson H.L., et al. Quaking orchestrates a post-transcriptional regulatory network of endothelial cell cycle progression critical to angiogenesis and metastasis. Oncogene. 2019;38:5191–5210. doi: 10.1038/s41388-019-0786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A.J., Martin K.A. Coordinating regulation of gene expression in cardiovascular disease: interactions between chromatin modifiers and transcription factors. Front. Cardiovasc. Med. 2017;4:19. doi: 10.3389/fcvm.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Choe N., Kwon D.H., Shin S., Kim Y.S., Kim Y.K., Kim J., Ahn Y., Eom G.H., Kook H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4) FEBS Lett. 2017;591:1041–1052. doi: 10.1002/1873-3468.12606. [DOI] [PubMed] [Google Scholar]
- Clark P.M., Loher P., Quann K., Brody J., Londin E.R., Rigoutsos I. Argonaute CLIP-Seq reveals miRNA targetome diversity across tissue types. Sci. Rep. 2014;4:1–11. doi: 10.1038/srep05947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N., Wani S., Xu Q., Gu J., Lea K., Heater S., Barbacioru C., Steptoe A.L., Martin H.C., Nourbakhsh E., et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011;12:1–20. doi: 10.1186/gb-2011-12-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes K.R., Sheehy N.T., White M.P., Berry E.C., Morton S.U., Muth A.N., Lee T.H., Miano J.M., Ivey K.N., Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rie D., Abugessaisa I., Alam T., Arner E., Arner P., Ashoor H., Åström G., Babina M., Bertin N., Burroughs A.M., et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017;35:872–878. doi: 10.1038/nbt.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Zhou S., Guan J. miRFam: an effective automatic miRNA classification method based on n-grams and a multiclass SVM. BMC Bioinf. 2011;12:216. doi: 10.1186/1471-2105-12-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnikar L., Taylor A.L., Chappell J., Oldach P., Harman J.L., Oerton E., Dzierzak E., Bennett M.R., Spivakov M., Jorgensen H.F. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat. Commun. 2018;9:4567. doi: 10.1038/s41467-018-06891-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert M.S., Sharp P.A. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149:515–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Diez C., Mandi V., Du M., Liu M., Gomez D. Smooth muscle cells in atherosclerosis: clones but not carbon copies. JVS Vasc. Sci. 2021;2:136–148. doi: 10.1016/j.jvssci.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P., Chen Z., Tian P., Liu W., Jiao Y., Xue Y., Bhattacharya A., Wu J., Lu M., Guo Y., et al. miRNA biogenesis enzyme Drosha is required for vascular smooth muscle cell survival. PLoS One. 2013;8:e60888. doi: 10.1371/journal.pone.0060888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina F.M., Hall I.F., Serio S., Zani S., Climent M., Salvarani N., Carullo P., Civilini E., Condorelli G., Elia L., Quintavalle M. miR-128-3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ. Res. 2020;126:e120–e135. doi: 10.1161/CIRCRESAHA.120.316489. [DOI] [PubMed] [Google Scholar]
- Gam J.J., Babb J., Weiss R. A mixed antagonistic/synergistic miRNA repression model enables accurate predictions of multi-input miRNA sensor activity. Nat. Commun. 2018;9:1–12. doi: 10.1038/s41467-018-04575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez M.D., Spengler R.M., Etheridge A., Godoy P.M., Barczak A.J., Srinivasan S., De Hoff P.L., Tanriverdi K., Courtright A., Lu S., et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018;36:746–757. doi: 10.1038/nbt.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettsch C., Rauner M., Pacyna N., Hempel U., Bornstein S.R., Hofbauer L.C. miR-125b regulates calcification of vascular smooth muscle cells. Am. J. Pathol. 2011;179:1594–1600. doi: 10.1016/j.ajpath.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D., Shankman L.S., Nguyen A.T., Owens G.K. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat. Methods. 2013;10:171–177. doi: 10.1038/Nmeth.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holycross B.J., Blank R.S., Thompson M.M., Peach M.J., Owens G.K. Platelet-derived growth factor-BB-induced suppression of smooth muscle cell differentiation. Circ. Res. 1992;71:1525–1532. doi: 10.1161/01.res.71.6.1525. [DOI] [PubMed] [Google Scholar]
- Humphries B., Yang C. The microRNA-200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget. 2015;6:6472–6498. doi: 10.18632/oncotarget.3052. https://www.oncotarget.com/article/3052/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczkowski B., Torarinsson E., Reiche K., Havgaard J.H., Stadler P.F., Gorodkin J. Structural profiles of human miRNA families from pairwise clustering. Bioinformatics. 2009;25:291–294. doi: 10.1093/bioinformatics/btn628. [DOI] [PubMed] [Google Scholar]
- Khorshid M., Hausser J., Zavolan M., van Nimwegen E. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods. 2013;10:253–255. doi: 10.1038/nmeth.2341. [DOI] [PubMed] [Google Scholar]
- Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lewis B.P., Shih I.H., Jones-Rhoades M.W., Bartel D.P., Burge C.B. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Liao J., Wang J., Liu Y., Li J., Duan L. Transcriptome sequencing of lncRNA, miRNA, mRNA and interaction network constructing in coronary heart disease. BMC Med. Genom. 2019;12:1–12. doi: 10.1186/s12920-019-0570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Espinosa-Diez C., Mahan S., Du M., Nguyen A.T., Hahn S., Chakraborty R., Straub A.C., Martin K.A., Owens G.K., Gomez D. H3K4 di-methylation governs smooth muscle lineage identity and promotes vascular homeostasis by restraining plasticity. Dev. Cell. 2021;56:2765–2782.e10. doi: 10.1016/j.devcel.2021.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Jin Y., Tang W.H., Qin L., Zhang X., Tellides G., Hwa J., Yu J., Martin K.A. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128:2047–2057. doi: 10.1161/CIRCULATIONAHA.113.002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magenta A., Ciarapica R., Capogrossi M.C. The emerging role of miR-200 family in cardiovascular diseases. Circ. Res. 2017;120:1399–1402. doi: 10.1161/CIRCRESAHA.116.310274. [DOI] [PubMed] [Google Scholar]
- Maleki S., Cottrill K.A., Poujade F.A., Bhattachariya A., Bergman O., Gadin J.R., Simon N., Lundstromer K., Franco-Cereceda A., Bjorck H.M., et al. The mir-200 family regulates key pathogenic events in ascending aortas of individuals with bicuspid aortic valves. J. Intern. Med. 2019;285:102–114. doi: 10.1111/joim.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M.N., Kim M.S., Adil M., Patil A.H., Lu Y., Mitchell C.J., Leal-Rojas P., Xu J., Kumar M., Dawson V.L., et al. Toward the human cellular microRNAome. Genome Res. 2017;27:1769–1781. doi: 10.1101/gr.222067.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald O.G., Wamhoff B.R., Hoofnagle M.H., Owens G.K. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J. Clin. Invest. 2006;116:36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau P.R., Tomas Bosch V., Bouvy-Liivrand M., Ounap K., Ord T., Pulkkinen H.H., Polonen P., Heinaniemi M., Yla-Herttuala S., Laakkonen J.P., et al. Profiling of primary and mature miRNA expression in atherosclerosis-associated cell types. Arterioscler. Thromb. Vasc. Biol. 2021;41:2149–2167. doi: 10.1161/ATVBAHA.121.315579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao M., Lyu Q., Zhao Q., Wirka R.C., Bagga J., Nguyen T., Cheng P., Kim J.B., Pjanic M., Miano J.M., Quertermous T. Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ. Res. 2020;126:517–529. doi: 10.1161/CIRCRESAHA.119.315968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J.W., Rissland O.S., Koppstein D., Abreu-Goodger C., Jan C.H., Agarwal V., Yildirim M.A., Rodriguez A., Bartel D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell. 2014;53:1031–1043. doi: 10.1016/j.molcel.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki E.P., Burge S.W., Bateman A., Daub J., Eberhardt R.Y., Eddy S.R., Floden E.W., Gardner P.P., Jones T.A., Tate J., Finn R.D. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J., Hayder H., Zayed Y., Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne) 2018;9:402. doi: 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olena A.F., Patton J.G. Genomic organization of microRNAs. J. Cell Physiol. 2010;222:540–545. doi: 10.1002/jcp.21993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Park S.M., Gaur A.B., Lengyel E., Peter M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M.A., Jin W., Villeneuve L., Wang M., Lanting L., Todorov I., Kato M., Natarajan R. Pro-inflammatory role of microrna-200 in vascular smooth muscle cells from diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2012;32:721–729. doi: 10.1161/ATVBAHA.111.241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon M., Gomez D., Greene E., Shankman L., Owens G.K. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22alpha promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ. Res. 2012;111:685–696. doi: 10.1161/CIRCRESAHA.112.269811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman L.S., Gomez D., Cherepanova O.A., Salmon M., Alencar G.F., Haskins R.M., Swiatlowska P., Newman A.A., Greene E.S., Straub A.C., et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M., Ghatak S., Roy S., Sen C.K. microRNA-200b as a switch for inducible adult angiogenesis. Antioxidants Redox Signal. 2015;22:1257–1272. doi: 10.1089/ars.2014.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood P., Krek A., Zavolan M., Macino G., Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. U S A. 2006;103:2746–2751. doi: 10.1073/pnas.0511045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Zhao M., Zhang J., Lv M., Li Y., Yang X., Liu A., Wu Z. MiR-29b downregulation induces phenotypic modulation of vascular smooth muscle cells: implication for intracranial aneurysm formation and progression to rupture. Cell Physiol. Biochem. 2017;41:510–518. doi: 10.1159/000456887. [DOI] [PubMed] [Google Scholar]
- Sun S.G., Zheng B., Han M., Fang X.M., Li H.X., Miao S.B., Su M., Han Y., Shi H.J., Wen J.K. miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011;12:56–62. doi: 10.1038/embor.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.Y., Qin S.S., Cheng Y.H., Wang C.Y., Liu X.J., Liu Y., Zhang X.L., Zhang W., Zhan J.X., Shao S., et al. MicroRNA expression profile and functional analysis reveal their roles in contact inhibition and its disruption switch of rat vascular smooth muscle cells. Acta Pharmacol. Sin. 2018;39:885–892. doi: 10.1038/aps.2018.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L.P., Seinen E., Duns G., de Jong D., Sibon O.C., Poppema S., Kroesen B.J., Kok K., van den Berg A. A high throughput experimental approach to identify miRNA targets in human cells. Nucleic Acids Res. 2009;37:e137. doi: 10.1093/nar/gkp715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacante F., Rodor J., Lalwani M.K., Mahmoud A.D., Bennett M., De Pace A.L., Miller E., Van Kuijk K., de Bruijn J., Gijbels M., et al. CARMN loss regulates smooth muscle cells and accelerates atherosclerosis in mice. Circ. Res. 2021;128:1258–1275. doi: 10.1161/CIRCRESAHA.120.318688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanchin B., Sol M., Gjaltema R.A.F., Brinker M., Kiers B., Pereira A.C., Harmsen M.C., Moonen J.A.J., Krenning G. Reciprocal regulation of endothelial-mesenchymal transition by MAPK7 and EZH2 in intimal hyperplasia and coronary artery disease. Sci. Rep. 2021;11:17764. doi: 10.1038/s41598-021-97127-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veer E.P.v.d., Bruin R.G.d., Kraaijeveld A.O., Vries M.R.d., Bot I., Pera T., Segers F.M., Trompet S., Gils J.M.v., Roeten M.K., et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 2013;113:1065–1075. doi: 10.1161/CIRCRESAHA.113.301302. [DOI] [PubMed] [Google Scholar]
- Wang D., Atanasov A.G. The microRNAs regulating vascular smooth muscle cell proliferation: a minireview. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Zheng J., Chen Z., Liu Y., Dura B., Kwak M., Xavier-Ferrucio J., Lu Y.C., Zhang M., Roden C., et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat. Commun. 2019;10:95. doi: 10.1038/s41467-018-07981-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wang D.Z., Hockemeyer D., McAnally J., Nordheim A., Olson E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428:185–189. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- Wen J., Parker B.J., Jacobsen A., Krogh A. MicroRNA transfection and AGO-bound CLIP-seq data sets reveal distinct determinants of miRNA action. RNA. 2011;17:820–834. doi: 10.1261/rna.2387911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will S., Reiche K., Hofacker I.L., Stadler P.F., Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer K.W., Halushka M.K. Toward the promise of microRNAs - enhancing reproducibility and rigor in microRNA research. RNA Biol. 2016;13:1103–1116. doi: 10.1080/15476286.2016.1236172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z., Xia L., Fan S., Zheng J., Qin J., Fan X., Liu Y., Tao J., Liu Y., Li K., et al. Circular RNA CircMAP3K5 acts as a MicroRNA-22-3p sponge to promote resolution of intimal hyperplasia via TET2-mediated smooth muscle cell differentiation. Circulation. 2021;143:354–371. doi: 10.1161/CIRCULATIONAHA.120.049715. [DOI] [PubMed] [Google Scholar]
- Zhuang J., Luan P., Li H., Wang K., Zhang P., Xu Y., Peng W. The Yin–Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2017;37:84–97. doi: 10.1161/ATVBAHA.116.307923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The small RNAseq data generated in this study has been deposited on the NCBI GEO Datasets platform (accession number: GSE179220) and are publicly available as of the date of publication. The RNAseq data originate from a previous publication (NCBI GEO; accession number: GSE186972). Please note that the small RNAseq and RNAseq were not performed on the same total RNA extraction but from independent experiments, though cell line, passage, culture conditions, and treatments were identical.
-
•
The paper does not report original codes or reagents. All codes or reagents used in this study are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact.