

Abstract
KMT2A-rearranged infant ALL is an aggressive childhood leukemia with poor prognosis. Here, we investigated the developmental state of KMT2A-rearranged infant B-cell acute lymphoblastic leukemia (B-ALL) using bulk messenger RNA (mRNA) meta-analysis and examination of single lymphoblast transcriptomes against a developing bone marrow reference. KMT2A-rearranged infant B-ALL was uniquely dominated by an early lymphocyte precursor (ELP) state, whereas less adverse NUTM1-rearranged infant ALL demonstrated signals of later developing B cells, in line with most other childhood B-ALLs. We compared infant lymphoblasts with ELP cells and revealed that the cancer harbored hybrid myeloid–lymphoid features, including nonphysiological antigen combinations potentially targetable to achieve cancer specificity. We validated surface coexpression of exemplar combinations by flow cytometry. Through analysis of shared mutations in separate leukemias from a child with infant KMT2A-rearranged B-ALL relapsing as AML, we established that KMT2A rearrangement occurred in very early development, before hematopoietic specification, emphasizing that cell of origin cannot be inferred from the transcriptional state.
Subject terms: Cancer genomics, Leukaemia
Single-cell transcriptomic and phylogenetic analyses reveal new insights into the developmental origin and potential therapeutic targets for a particularly aggressive form of B-cell acute lymphoblastic leukemia in infants.
Main
Once a universally fatal disease, B-ALL of childhood is curable in the majority of cases. An exception is B-ALL arising in children younger than one year of age (infant B-ALL), which remains fatal in more than 50% of children1,2. Most cases (70–80%) of infant B-ALL are associated with rearrangements of the KMT2A gene (encoding a histone methyltransferase), which confers an especially poor prognosis2. Various hypotheses have been proposed to account for the aggressive nature of infant B-ALL. In particular, it has been suggested that infant lymphoblasts retain myeloid features that confer resistance to treatment strategies aimed at ALL3. Disappointingly, although protocols incorporating strategies from acute myeloid leukemia (AML) therapy marginally increased survival, additional intensification has not improved this further1. Similarly, salvage treatments that have proven successful in high-risk lymphoblastic leukemias, such as allogeneic stem cell transplantation or chimeric antigen receptor T cells targeting B-cell antigens, produce disappointing outcomes in infant B-ALL4,5. It is noteworthy that infant B-ALL not associated with KMT2A fusion, especially those with NUTM1 gene rearrangements, confer a more favorable prognosis6,7 and that KMT2A rearrangements in the setting of adult B-ALL are also considered high risk8. These observations raise the question whether the aggressive clinical behavior of KMT2A-rearranged infant B-ALL is underpinned by a distinct cellular phenotype.
Leukemias are primarily classified by their morphological appearance or immunophenotype, as assessed by flow cytometric analyses of key hematopoietic markers and cytogenetic changes. Generally, this approach is likely to capture the differentiation state of most leukemias accurately. Occasionally, it may be erroneous when cancer cells use key hematopoietic genes aberrantly, particularly in leukemias that are driven by mutations in genes that facilitate lineage plasticity, such as KMT2A. In this context, a quantitative molecular assessment of hematopoietic cell states that does not rely on any individual marker, but instead builds on entire cellular transcriptomes, would provide an unbiased readout of cell states. Such high-resolution assessments are now feasible using single-cell mRNA sequencing to directly compare cancer cells to normal cells, including to fetal and adult hematopoietic cells9–12. We set out to study the developmental phenotype of KMT2A-rearranged infant B-ALL by comparing cancer cells with normal human hematopoietic cells.
Results
Cell signal analysis of 1,665 leukemia transcriptomes
The starting point of our investigation was a meta-analysis of 1,665 bulk transcriptomes representing the entire spectrum of childhood ALL and AML across two cohorts, St. Jude Children’s Research Hospital (St Jude’s; n = 589) and TARGET (Therapeutically Applicable Research to Generate Effective Treatments; n = 1,076) (Fig. 1a and Supplementary Table 1). We determined the predominant hematopoietic cell signal within each bulk leukemia transcriptome by deconvolution. We chose a deconvolution method that uses entire transcriptomes to determine cell signals within bulk mRNA data and quantifies what proportion of the cancer bulk cannot be accounted for by normal reference cells13. As childhood leukemias, and infant ALL in particular, are generally thought to arise in utero14,15, we applied fetal hematopoietic cells as the reference in our analyses. To this end, we used recent single-cell mRNA data from ~60,000 fetal bone marrow cells, which captured the greatest breadth of fetal hematopoietic cell types to date9 (Supplementary Table 2). We adopted the annotation of normal cell types directly from the fetal bone marrow data analysis9 and supplemented the hematopoietic reference with a control fetal cell type that should not be present in human leukemia samples, Schwann cell precursors (SCPs) derived from human fetal adrenal glands16.
Fig. 1. Cell signal analysis of 1,665 leukemia transcriptomes reveals an ELP state in KMT2A-rearranged B-ALL.

a, Schematic overview of the study approach. We assessed the differentiation state of KMT2A-rearranged infant ALL by measuring signals of human fetal bone marrow cell types across the entire spectrum of childhood leukemia in data derived from two different cohorts (St Jude’s and TARGET). We then validated cell signals by single-cell mRNA sequencing for direct comparison of cancer and normal cells. b, Heatmap showing mean cell signals of human fetal bone marrow cells (y axis) in human leukemia bulk transcriptomes subdivided by genetic subtype (see labels underneath, KMT2A rearrangements shown in red text), age (gray circle, infant; black circle, noninfant) and source (S, St Jude’s; T, TARGET). Numbers next to labels refer to case load per subtype. Subtypes with only one case were excluded from analysis. baso, basophil; CMP, common myeloid progenitor; Eo, eosinophil; LMPP, lymphoid-primed multipotent progenitor; MEM progen., ; MK, megakaryocyte; mono., monocyte; MOP, monocyte progenitor; MPP, multipotent progenitor; Neut., neutrophil; NK, natural killer; Promono., promonocyte. c, Top: box and whisker plots showing proportional contribution of signals (lymphomyeloid-primed progenitor, ELP and later B-cell stages combined (i.e., pre-/pro-B, pro-B, pre-B and naive B)) to the transcriptome of leukemias (see x axis labels). Bottom: box and whisker plots summarizing the ratio of ELP to later B-cell stage signals. Center lines represent the median, box limits represent 25%/75% quartiles and whiskers represent minimum/maximum (top) and 1.5× interquartile range (bottom). n is the number of biologically independent variables, as listed below each group of plots. Risk refers to the clinical cytogenetic risk as defined in the protocol of the current European ALL trial ‘ALLTogether’ (EudraCT 2018-001795-38).
A global overview of cell signals in bulk childhood leukemia transcriptomes showed expected patterns, namely myeloid signals in myeloid leukemias, T-cell signals in T-cell ALL and imprints of the various stages of B-cell development in B-ALL (Fig. 1b and Supplementary Fig. 1). Transcriptional signatures from the control SCP population did not contribute to leukemias (negative control analysis) and matched itself (i.e., SCPs) perfectly with no unexplained signal (positive control analysis). KMT2A-rearranged infant B-ALL exhibited distinct cell signals with a marked contribution of ELPs. ELPs are oligopotent lymphoid precursors that are capable of differentiating along different lymphocyte lineages and that retain minimal myeloid differentiation capacity in vitro17,18. Defined as CD34+CD127+CD10−CD19− cells, they sit upstream of pre-/pro-B and pro-B progenitors in the B lymphopoiesis hierarchy18.
An ELP signal in KMT2A-rearranged B-ALL
To further examine the ELP signal in KMT2A-driven infant B-ALL, we examined the ratio of the ELP signal over later stages of B-cell development in each leukemia subtype (Fig. 1c). This quantification demonstrated a significant shift toward the ELP state in KMT2A-rearranged infant ALL compared to other high (cytogenetic)-risk B-ALL subtypes (P < 10−19, Student’s two-tailed t test), standard (cytogenetic)-risk subtypes (P < 10−31) and currently unstratified subtypes of B-ALL (P < 10−13) (Fig. 1c). Among KMT2A-rearranged infant B-ALL, the ELP signal was present irrespective of fusion partners of KMT2A but strongest in cases harboring the most common KMT2A rearrangement19, the KMT2A-AFF1 (MLL-AF4) gene fusion (P < 0.01 compared against other fusion partners; Mann–Whitney rank test) (Extended Data Fig. 1). The leukemias with the next highest relative ELP signals were PAX5 and MEF2D-mutated B-ALL, although the ELP signals there were accompanied by stronger signals from later B-cell stages. In contrast to KMT2A-rearranged B-ALL, differences between ELP signals and later B-cell signals were significant in PAX5- and MEF2D-mutated B-ALL (P < 0.01 and P < 0.05, respectively; Wilcoxon signed-rank test). Although MEF2D mutation results in maturation arrest at the pre-B stage, its distinct immunophenotype is recognized to overlap with both early and late B progenitor stages20. The similarity of cell signals in PAX5 and KMT2A mutant B-ALL may represent the intimate relationship of KMT2A and PAX5 in regulating B lymphopoiesis21.
Extended Data Fig. 1. ELP signal is common to KMT2A-rearranged B-ALL, independent of fusion partner.

Box and whisker plots showing contributions of cell signals – LMPP, ELP and latter B-cell stages (that is pre-pro-B, pro-B, pre-B and naive B combined) to the transcriptome of KMT2A-rearranged leukemias grouped by KMT2A fusion partner (see x axis labels). Centre lines=median, box limits=25%/75% quartiles, whiskers=min/max (top) and 1.5*interquartile range (bottom). n= biologically independent variables, as listed below each group of plots.
Studying the pattern of ELP signal across disease groups indicated that the signal was specific to KMT2A rearrangements within a B-cell context but independent of age for three reasons. First, the ELP signal was not universally associated with KMT2A rearrangements; neither myeloid nor ambiguous lineage leukemias with KMT2A rearrangements harbored appreciable ELP signals. Second, the ELP signal was not driven by young age alone, as other infant leukemias (B-ALL, ambiguous lineage leukemia and AML) exhibited no, or only minimal, ELP signal (Fig. 1c). In particular, infant B-ALL with NUTM1 rearrangement (which carries a favorable prognosis) exhibited cell signals more reminiscent of standard-risk childhood B-ALL, with a shift away from ELPs toward later B-cell stages. Third, KMT2A-rearranged B-ALL of older children did exhibit marked ELP signals akin to infant KMT2A-driven B-ALL. Overall, these findings led us to hypothesize that, relative to other B-ALL, KMT2A-rearranged B-ALL exhibits a distinct hematopoietic phenotype primarily resembling ELP cells with limited signals of B-cell development.
Direct single cancer cell to normal cell comparison
To validate and further explore this proposition, we performed single-cell RNA-sequencing (scRNA-seq) analysis (10x Genomics) of diagnostic specimens from six infants with KMT2A-rearranged infant B-ALL, including a relapse presentation (case 3) and additional day 8 specimens from responding (case 1) and nonresponding (case 2) patients. We compared these to four other leukemias: NUTM1-rearranged infant B-ALL (n = 1), KMT2A-rearranged infant AML (n = 1), megakaryoblastic neonatal AML (n = 1) and childhood ETV6-RUNX1 B-ALL (a common subtype of standard-risk childhood B-ALL; n = 1) (Supplementary Table 3). From these 12 diagnostic leukemia samples, we obtained a total of 30,242 cells, including 23,286 cancer cells that we identified based on gene expression matching patient-specific diagnostic flow cytometric profiles (Supplementary Table 4 and Extended Data Fig. 2). Using a published cell-matching method based on logistic regression12,16, we directly compared leukemia transcriptomes with mRNA profiles of human fetal bone marrow cells to determine which normal cell type the cancer cells most closely matched. We found that KMT2A-rearranged infant B lymphoblasts overwhelmingly resembled ELP cells at diagnosis and relapse and in nonresponding disease (Fig. 2a–c). By contrast, non-ELP cell signals predominated in other types of leukemia, precisely as predicted from the initial deconvolution analysis (Fig. 1b). In particular, in the aforementioned subtype of infant B-ALL with a favorable prognosis, NUTM1-rearranged infant B-ALL, single-cell analysis confirmed the shift toward pre-B-cell states and away from ELPs. To further explore the differences between KMT2A- and NUTM1-driven infant B-ALL, we performed independent differential gene expression analysis of bulk transcriptomes and single-cell data, which yielded an overlapping list of 90 differentially expressed genes (Methods). Focusing on genes used in normal fetal bone marrow, we found that in KMT2A B-ALL, genes of early B-cell development were overexpressed, whereas in NUTM1 B-ALL genes of more differentiated B cells predominated (Fig. 2d and Supplementary Table 5). These findings thus corroborate our proposition that the differentiation state of NUTM1 blasts, similar to ETV6-RUNX1 blasts, is shifted toward later stages of B-cell development.
Extended Data Fig. 2. Identification of cancer cells in leukemia scRNA-seq data using immunophenotype gene expression.

UMAP projections of leukaemia scRNA-seq data sets, coloured by Louvain cluster. Accompanying dotplots show per-cluster expression of B-ALL immunophenotype genes or AML immunophenotype genes and lineage-defining genes of monocytes (Mo.), T cells (T), NK cells (NK), B cells (B) and progenitors (Pro.). Dot colour denotes log-transformed, normalised and scaled gene expression value, while dot size indicates percentage of cells in each cluster expressing the stated gene. Immunophenotypes are provided in Supplementary Table 4.
Fig. 2. Validation of ELP signals by direct single cancer cell to normal cell comparison.

a, Heatmap comparing cell clusters from diagnostic specimens (y axis) to normal human fetal bone marrow cell types (x axis; bold labels highlight cell types shown in C). Cell clusters represent cancer (as defined by clinical diagnostic flow cytometric profiles, see Extended Data Fig. 2) and normal cells of individual patient samples (as per case ID number; see Supplementary Table 3 for an overview of patients). All are diagnostic samples at presentation, except case 3 (relapse presentation). Heat colors represent the mean probability (across the cell cluster) of a match as determined by logistic regression (red, similar; blue, different). DC, dendritic cell; GMP, granulocyte–monocyte progenitor; HSC, hematopoietic stem cell; ILC, innate lymphoid cell; Imm., immature; lin., lineage; pDC, plasmacytoid dendritic cell. b, Uniform manifold approximation and projection of B-ALL scRNA-seq data divided by genetic subtype. KMT2A-rearranged B-ALL at diagnosis and day 8 of treatment are presented separately. Within each heatmap, black dots represent cancer and gray dots noncancer. c, Per cancer cell (normal cells for day 8 remission samples of patient 1) logistic regression score against reference B-lineage cell states, with thresholds of >0.8 indicating similarity (red) and <0.2 indicating dissimilarity (blue). d, Subset of differentially expressed genes between infant KMT2A-rearranged B-ALL and NUTM1-rearranged B-ALL. x axis, gene name; y axis, fetal bone marrow cell type. Heatmap shows the average gene expression per reference cell type for genes up-regulated in NUTM1 B-ALL (left) and KMT2A B-ALL (right).
To determine the heterogeneity of B-cell states within patients, we performed logistic regression on a per-cell basis (Fig. 2c). This revealed in every case of KMT2A-rearranged ALL that the greatest proportion of blasts with a close match to a specific developing B-cell type resembled ELP cells. Similarly, very few infant KMT2A lymphoblasts were dissimilar to ELP cells. By contrast, the developmental phenotype of NUTM1 and ETV6-RUNX1 lymphoblasts was shifted toward later B-cell stages, peaking at the pro-B-cell stage in terms of the similarity and dissimilarity of individuals blasts to fetal cells. Finally, we assessed by flow cytometry a set of six KMT2A infant B-ALL samples, including four primary samples (three independent of the single-cell scRNA-seq cohort) and two xenografts derived from these patients. We demonstrated an ELP-like immunophenotype in 80–90% of cells (Extended Data Fig. 3). Together, these findings confirm that an ELP-like developmental state predominates in KMT2A infant B-ALL at diagnosis and relapse in resistance and after xenotransplantation.
Extended Data Fig. 3. Immunophenotype of KMT2A-rearranged infant B-ALL.

Left: Histograms showing surface antigen expression in a representative primary KMT2A-rearranged infant B-ALL sample relative to negative control. Right: Scatterplot showing percentage of cells in each sample expressing ELP-characteristic antigens > negative control (n = 2 xenograft, n = 4 primary KMT2A-rearranged infant B-ALL). Line= mean; 82% CD7+, 93% CD127+ and 82% FLT3+).
Phylogenetic timing of the origin of infant ALL
A key question raised by our findings is whether ELPs are the cells of origin of KMT2A-rearranged infant B-ALL or whether leukemia cells arise from another precursor and differentiate/dedifferentiate into an ELP-like state at which they arrest. A rare case of lineage switching from KMT2A-rearranged infant B-ALL to KMT2A-rearranged AML provided the opportunity to directly determine the cell of origin in phylogenetic temporal terms (Fig. 3a). We first assessed cell signals in bulk transcriptomes (in replicates) derived from a child with KMT2A-rearranged B-ALL and AML. Once again, we observed that ALL, but not AML transcriptomes, exhibited an ELP signal (Fig. 3b). To determine the phylogeny of the cancers, we performed whole-genome DNA sequencing of AML, ALL and remission bone marrow and called all classes of variants using an extensively validated mutation-calling pipeline22 (variant list in Supplementary Table 6). We determined the phylogeny of each leukemia and remission bone marrow. The remission sample and leukemias shared two mosaic (early embryonic) base substitutions, representing the first cell divisions of the zygote23,24. Thereafter, normal blood and leukemia lineages diverged. The common leukemia lineage (that is mutations shared between ALL and AML, but not the remission sample) composed only six base substitutions along with the KMT2A rearrangement (Fig. 3c,d), defining an early developmental window during which the translocation formed. Assuming a mutation rate of at least 0.9 substitutions per cell division, as recently established in human fetal hematopoietic cells25, six substitutions would place the emergence of the KMT2A rearrangement in early embryonic development, before hematopoietic cell specification. After acquisition of the KMT2A fusion, the leukemia lineages diverged and gave rise to independent cancers, each exhibiting distinct phenotypes and somatic changes (including point mutations, copy-number profiles and mutational signatures) (Fig. 3c–e). Although this single case may not be representative of infant ALL generally or lineage-switch leukemias specifically, it demonstrates that the transcriptional state of cancer cells cannot unambiguously be used to infer its cell of origin.
Fig. 3. Phylogenetic timing of the origin of infant ALL.

a, Diagnostic flow cytometry of two leukemias that arose in the same child 4 years apart: KMT2A-rearranged infant ALL (yellow, abbreviated iALL) and KMT2A-rearranged AML (red). MPO, myeloperoxidase. b, Cell signal assessment of bulk transcriptomes generated from this child (ALL in technical triplicates, AML in technical duplicates) shows that the cell signals (LMMP, ELP and B cells (i.e., the sum of all B-cell signals)) of ALL (yellow circle) and AML (red circle) follow the pattern of KMT2A-rearranged ALL (left, boxplots in background, n = 52 biologically independent samples) and KMT2A-rearranged AML (right, boxplots in background, n = 86 biologically independent samples), as defined in the St Jude’s and TARGET cohorts. Boxplot center line represents the median, whiskers represent minimum/maximum and box limits represent 25%/75% quartiles. c, Timeline with copy-number profiles of chromosomes 4 to 11 (all other chromosomes were diploid) in both leukemias showing chromosomes (x axis) and copy number (y axis), alleles (light and dark grey lines) and rearrangement breakpoints (black vertical lines and arcs), including the chromosome 4:11 translocation underpinning the KMT2A-AFF1 fusion. d, Phylogeny of blood and leukemia lineages with substitution burden defining each branch (number). e, Assessment of mutational signatures as defined by the trinucleotide context of substitutions (nomenclature as per Alexandrov et al.22) highlighting in purple the dominant contribution (as percentage of all clonal substitutions) of signature 87 to AML. This signature is thought to be due to thiopurine agents that the child had received for ALL treatment (see timeline).
Therapeutic hypotheses based on the ELP state of infant ALL
To distill the oncogenic features of the KMT2A-rearranged infant B-ALL transcriptome, we directly compared leukemia with ELP transcriptomes. We determined in independent analyses the differential gene expression between bulk KMT2A-rearranged infant B-ALL and published bulk ELP transcriptomes18 and between single lymphoblast and single ELP cell transcriptomes (Fig. 4a). The overlap of these two independent analyses (Supplementary Table 7, N = 455) provided a cross-validated gene set, hereafter referred to as the cancer core transcriptome, that differentiates KMT2A-rearranged B lymphoblasts from their closest normal cell correlate (i.e., ELPs), which we annotated in five ways. First, we queried whether the cancer core transcriptome contained known target genes of the KMT2A-AFF1 fusion26, the most common KMT2A rearrangement in B-ALL. We found 63 of 455 genes to be targets of the KMT2A-AFF1 fusion, which represents a significant enrichment (P < 10−107, as assessed in a Monte Carlo simulation; Methods and Supplementary Table 7). Second, we discerned the lineage-independent effects of KMT2A translocation by overlapping the KTM2A-rearranged B-ALL cancer core transcriptome with genes differentially expressed in KMT2A-driven AML (relative to its normal cell correlate, monocyte progenitors (MOPs); case 10, Fig. 2a). We identified an overlapping gene set of 67 genes that, according to gene ontology annotations, disrupted key regulatory processes such as cell communication, proliferation and development and promoted expression of genes maintaining a primitive state (HOXA6, BMI1 and MEIS1) (Supplementary Tables 7 and 8). Third, we asked whether the cancer core transcriptome encompassed lineage-specific genes by interrogating normal fetal bone marrow cells. We found that a subset of genes (n = 51) was lineage specific, representing either lymphoid or myeloid cell types (Fig. 4b). Fourthly, we annotated the cancer core transcriptome by gene ontology analysis. The top two disease annotations were lymphoblastic and myeloid leukemia, further suggesting that the cancer core transcriptome encoded a hybrid myeloid–lymphoid phenotype (Supplementary Table 7). Finally, we identified cell surface antigens among differentially expressed genes, as many novel treatments in childhood leukemias center on targeting blast markers through antibodies or genetically modified T cells. A total of 41 of 455 genes encoded surface markers, some of which were relatively specific to myeloid (n = 18) or lymphoid (n = 4) lineages, generating 72 potential nonphysiological marker combinations (Supplementary Table 7). Examples of nonphysiological coexpression patterns that were particularly specific to infant B-ALL are shown in Fig. 4c. Interestingly, these were centered on the lymphoid marker CD72, which a proteomic screen recently implicated as a target in infant ALL27. Coexpression of nonphysiological combinations was measured by flow cytometry, where commercial antibodies existed, confirming that dual-targeting would encompass >90% of leukemic cells (Fig. 4d). Now that surface marker therapies targeting two antigens simultaneously are already in use, nonphysiological coexpression of markers may represent an attractive therapeutic avenue in infant B-ALL.
Fig. 4. Therapeutic hypotheses based on the ELP state of infant ALL.

a, Distilling the core cancer transcriptome (i.e., differential gene expression between infant ALL and ELP cells from bulk and single-cell data) to generate a cross-validated gene list that we annotated in three ways (right). b, The core cancer transcriptome encodes a mixed myeloid–lymphoid phenotype. Shown is the log normalized expression (heat color) of genes (x axis) that have relative lineage specificity in normal fetal bone marrow cell types (y axis). Eosin, eosinophil. c, Examples of nonphysiological combinations of cell surface markers that the core cancer transcriptome encompasses. x axis, fetal bone marrow cell type or infant B-ALL lymphoblasts; y axis, marker combinations. Dots represent coexpression of the markers (average of the product of gene expression). Dot size represents the percentage of cells in the cluster that express both markers, and heat color represents the normalized coexpression level. d, Left: dotplots showing coexpression of antigen combinations in a representative primary KMT2A-rearranged infant B-ALL sample, as measured by flow cytometry on live, single CD34+CD19+ blasts. Adjunct histograms show fluorescence-minus-one (FMO) negative controls (gray) and antigen expression in the representative sample (red) compared with n = 2 xenograft samples and n = 3 further primary infant B-ALL samples (orange). APC, allophycocyanin; PE, phycoerythrin. Right: scatterplot demonstrating the percentage of cells in each sample with expression of antigen pair higher than the fluorescence-minus-one control (line represents median).
Discussion
In clinical diagnostic and therapeutic terms, KMT2A-rearranged infant B-ALL is considered to be a B-precursor leukemia. Based on independent data and analytical techniques, we arrived at the conclusion that infant lymphoblasts most closely resemble human fetal ELPs. This ELP-like transcriptional phenotype distinguishes KMT2A-rearranged infant B-ALL from other childhood B-ALLs.
A key question that our findings raise is whether the ELP-like state accounts for the poor prognosis of KMT2A-rearranged infant B-ALL. Three observations lend credence to this proposition. First, in both bulk mRNA and single-cell analyses, NUTM1-rearranged infant B-ALL, recently identified to carry a favorable prognosis6, exhibited cell signals away from the ELP state and more reminiscent of standard-risk B-ALL. Second, we observed an ELP-like state in older children with B-ALL KMT2A rearrangements, in whom KMT2A fusions are considered a high-risk cytogenetic change that mandates treatment intensification28. Third, B-ALLs with the next highest relative ELP signals (PAX5 alterations and MEF2D mutations) are also considered high risk20,29. These observations raise the possibility that ELP features confer a high-risk clinical phenotype in B-ALL while recognizing the challenge of separating this signal from the prognostic significance of cytogenetic changes.
Considerable efforts to identify the cell of origin in leukemias have arisen from the promise that targeted clearance will result in durable remission. Focusing in on the cell of origin in KMT2A-rearranged infant B-ALL, key pieces of evidence are (1) rearrangement is prenatal event, as demonstrated by Guthrie card examinations and concordance in monozygotic twins14; (2) rearrangement in the hematopoietic compartment is observed in CD34+CD19− cells30, before VDJ recombination in most cases, resulting in low frequency of clonal immunoglobulin rearrangements31,32; and (3) rearrangement may also be seen in bone marrow mesenchymal cells, suggesting a prehematopoietic origin in some33. We directly determined the phylogenetic origin of an infant leukemia in a rare case of a child in whom infant B-ALL and childhood AML developed, both harboring KMT2A rearrangements. The number of shared mutations between these leukemias suggests that the KMT2A rearrangement arose before gastrulation and specification of hematopoiesis. With the important caveat that this case will not represent all KMT2A-rearranged B-ALL, it demonstrates that the cell of origin cannot be inferred from the transcriptional phenotype of leukemia cells. Although our results demonstrate the consistency of an ELP transcriptional state in KMT2A B-ALL cells at diagnosis, in resistant disease, at relapse and in xenografts, further studies are required to establish whether an ELP signal can be traced back to disease-initiating cells.
The benefit of accurately defining the transcriptional state of KMT2A-rearranged infant B-ALL is the ability to devise novel strategies for targeted therapy. We compared leukemic blasts with fetal bone marrow ELPs from independent data sets to yield a core cancer transcriptome, which was characterized by fusion gene targets and a mixture of lymphoid and myeloid lineage genes. We identified nonphysiological combinations of surface antigen genes and demonstrated that these combinations are coexpressed as surface proteins, potentially allowing >90% of leukemic blasts to be destroyed by dual-targeting tandem-chimeric antigen receptor T-cell or bispecific antibody therapies. Targeting combinations of antigens from different lineages simultaneously may afford exquisite specificity for cancer cells.
The quantitative molecular approach we deployed here, leveraging large archives of bulk mRNAs, emerging reference catalogs of normal human cells and direct examination of single blast transcriptomes, lends itself for reappraising the phenotype of human leukemias to derive novel biological and therapeutic hypotheses. As leukemias are primarily classified by their hematopoietic phenotype, we propose that KMT2A-rearranged infant B-ALL be considered an ELP-like leukemia.
Methods
Ethics statement
Patient blood and bone marrow samples were obtained from the Newcastle Biobank (as approved by Newcastle and North Tyneside 1 Research Ethics Committee, reference 17/NE/0361) or Great Ormond Street Hospital for Children diagnostic archives (as approved by the National Research Ethics Service Committee London Brent, reference 16/LO/0960). Informed consent was obtained from all participants. Patient-derived xenograft (PDX) samples were generated in accordance with the UK Animals (Scientific Procedures) Act 1986 under project licenses PPL60/4552 and PPL60/4222 following institutional ethical review.
Sample preparation
Peripheral blood mononuclear cells were prepared from blood, bone marrow or PDX samples by density centrifugation using Lymphoprep (Stemcell) according to manufacturer’s instructions. Samples were cryopreserved in fetal bovine serum (FBS) with 10% dimethyl sulfoxide and stored in liquid nitrogen. PDXs were generated by intrafemoral transplant (under isoflurane anesthesia) of 106 patient blood or bone marrow cells into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Charles River Laboratories and bred in-house) aged 8–10 weeks old34. PDX cells were harvested from engrafted bone marrow or spleen.
Flow cytometry
Cryopreserved ALL samples (n = 4 primary samples, n = 2 PDXs; Supplementary Table 3) were thawed in RF-10 (RPMI, Sigma-Aldrich) supplemented with 10% (v/v) heat-inactivated FBS (Gibco), 100 U ml−1 penicillin (Sigma-Aldrich), 0.1 mg ml−1 streptomycin (Sigma-Aldrich) and 2 mM l-glutamine (Sigma-Aldrich). Up to one million cells were stained with antibody cocktail, incubated for 30 min on ice, washed with flow buffer (PBS containing 5% (v/v) FBS and 2 mM EDTA), and resuspended in flow buffer with DAPI (Sigma-Aldrich) added to a final concentration of 3 μM. Antibodies for immunophenotyping (Extended Data Fig. 3) were (clone, supplier) NG2 PE (9.2.27, BD Biosciences), FLT3 PEDazzle (BV10A4H2, Biolegend), CD10 PECy7 (HI10a, Biolegend), CD2 fluorescein isothiocyanate (FITC) (S5.2, BD Biosciences), CD3 FITC (SK7, BD Biosciences), CD14 FITC (MφP9, BD Biosciences), CD16 FITC (NKP15, BD Biosciences), CD56 FITC (NCAM16.2, BD Biosciences), CD235a FITC (GA-R2, BD Biosciences), CD38 PERCPCy5.5 (HB-7, Biolegend), CD45RA BV510 (HI100, BD Biosciences), CD7 BV650 M-T701, BD Biosciences), CD127 BUV737 (HIL-7R-M21, BD Biosciences), CD90 APC (5E10, Biolegend), CD19 AF700 (HIB19, Biolegend) and CD34 APCCy7 (581, Biolegend). Antibodies for nonphysiological antigen coexpression profiling (Fig. 4) were SEMA4A PE (T9-10, BD Biosciences) or LILRB1 PE (GHI/75, Biolegend); FLT3 PE Dazzle (as above); CD10 PECy7 (as above); CD19 FITC (4G7, BD Biosciences), ICOSLG BV510 (2D3/B7-H2l, BD Biosciences), CD127 BUV737 (as above), CD72 APC (SF3, Biolegend) and CD34 APCCy7 (as above). All antibodies were used at 1:25 dilution, except for CD10 PECy7 and CD127 BV650, which were used at 1:50 dilution. For fluorescence-minus-one controls, cells and antibody cocktails were prepared identically but without the antibody of interest. FACS was performed on a BD FACSAria running DIVA v.8, and data were analyzed using FlowJo (v.10.6.2, BD Biosciences). Thresholds for negative expression were set using fluorescence-minus-one controls for Fig. 4 analysis and using negative cells in PDX samples (mouse splenocytes) for Extended Data Fig. 3 analysis.
Bulk RNA sequencing
Total RNA from the lineage-switch case (Fig. 3) was extracted from peripheral blood mononuclear cells using RNeasy Mini Kit (Qiagen, 74106) and mRNA captured using NEBNext Ultra Directional RNA Kit with NEBNext poly(A) mRNA Magnetic Isolation Module. Paired-end 150-bp sequencing was performed on HiSeq 4000 (Illumina), with transcript abundance quantified from raw reads via Salmon v.0.8.2 and alignment performed against the hg38 reference human transcriptome (GENCODE release 27).
scRNA-seq
Thawed cells were manually counted and 7,000 cells added to each channel of a Single Cell Chip before loading onto the 10x Chromium Controller (10x Genomics). Reverse transcription, cDNA amplification and sequencing libraries were generated using the Single Cell 3’v2 (P1_iALL, P2_iALL, P9_iAML), Single Cell 3’v3 (P3_iALL, P4_iALL, P10_iAML) and Single Cell 5’ v1 (P5_iALL, P6_iALL, P7_iALL_NUTM1 and P8_iALL_ETV6) Reagent kit (10x Genomics) according to the manufacturer’s instructions. Libraries were sequenced using an Illumina HiSeq 4000 with v.4 SBS chemistry. All libraries were sequenced to achieve a minimum of 50,000 reads per cell.
Alignment, quantification and quality control
Raw fastq files for scRNA-seq data for P1_iALL,P2_iALL, P9_iAML (single-cell 3’ v2 kit) were processed with the Cell Ranger v2.0.2 (ref. 35) pipeline, and the rest of the samples were processed at a later time point with the Cell Ranger v3.0.2 pipeline, which aligned the reads to the reference human genome (GRCh38 v1.2.0) and produced a matrix of gene expression per cell. Ambient mRNA contamination was removed with SoupX package v1.4.8 in R with default parameters. Demultiplexing of P1_iALL/P2_iALL, P3_iALL/P10_iAML and P5_iALL/InfALL_classSwitch was performed with souporcell package v2.0 (ref. 36) with default parameters and setting number of clusters -k to 2 and–min_ref to 4 --min_alt to 4. Souporcell inferred the cluster assignment (either 0 or 1) for each cell, and given gender information (Supplementary Table 3), we were able to demultiplex the data by checking the sex-specific gene expression in each souporcell cluster (XIST for female and RPS4Y1, ZFY and a couple of others for male). Resulting gene expression matrices were further processed in python with scanpy package v1.4.4.post1 (ref. 37), and single cells were filtered to retain cells expressing >200 genes and having mitochondrial content <20%. The code used for demultiplexing and filtering is included as a Jupyter Notebook in the Code availability section.
Dimensional reduction, clustering and annotation
After filtering for low-quality genes, single-cell data were processed in a scanpy package in python, and the total number of counts per cell were normalized to 10.000 in order to correct for library size differences; normalized data were further log-transformed. Principal-component analysis was performed on log-transformed data using default parameters (N = 50), followed by computation of neighborhood graph with default parameters (N neighbors = 15) and embedding the graph in two dimensions using uniform manifold approximation and projection. Clustering of single-cell data has been performed by Louvain community detection on neighborhood graph with default resolution set to 1. Clusters were assigned as cancer or noncancer, based on expression of B-ALL or AML immunophenotype genes (derived from expression profiles in clinical diagnostic panels and lineage-defining genes of monocytes, B cells, T cells, natural killer cells or progenitors; Extended Data Fig. 2 and Supplementary Table 4).
Logistic regression analysis
To test the probability that cancer cell transcriptomes are similar normal reference transcriptomes (single-cell fetal bone marrow dataset), we used logistic regression as described previously12,16. Briefly, a logistic regression model was trained in R using cv.glmnet function on a fetal bone marrow dataset combined with SCP single cells from the fetal adrenal reference map16, setting the elastic mixing parameter alpha to 0.99, thus ensuring strong regularization. This model was then used to predict the probabilistic score of similarity of single cells in infant leukemia dataset to cell type in the fetal bone marrow dataset.
Published bulk RNA-sequencing data
Pediatric tumor bulk RNA-sequencing data for childhood leukemia was obtained from the St. Jude Cloud and TARGET, together with associated metadata. Bulk RNA-sequencing data of human fetal bone marrow ELPs18 were extracted from the Gene Expression Omnibus with accession number GSE122982. Data were quantified and mapped with Salmon v. 0.13.1 (ref. 38) with default parameters, and transcript-level estimates were summarized with tximport package v1.14.2 in R.
Deconvolution of bulk RNA-sequencing data
The fetal BM scRNA-seq dataset was used as a reference to infer the cell type composition in bulk RNA-sequencing data using a previously published method of deconvolution called cellular signal analysis13. Briefly, this method aims to predict the contribution of the normal mRNA signal to each of the bulk transcriptomes. The advantage of using cellular signal analysis over other deconvolution methods is the reporting of the ‘unexplained signal’ when the bulk transcriptome differs from all the signals in the normal reference dataset and represented as an ‘Intercept’ term. The model fit is based on tensorflow framework v1.14.0 and was run specifying gene weights using the geneWeights.tsv file that was supplied with the package and using default parameters for other arguments.
Differential gene expression analysis
Differential gene expression analysis was performed using DESeq2 package v1.26.0 (ref. 39) in R. For bulk RNA-sequencing data (childhood leukemia data and ELP data) a DESeq dataset was constructed from tximport object (from Salmon quant.sf files for both childhood leukemia and ELP and creating metadata table with ‘group’ column variables set to either ‘cancer’ or ‘ELP’). For the single-cell leukemia dataset, pseudobulk was created from single cells by summarizing counts for each patient. For the single-cell ELP, MOP or NUTM1 dataset, a matrix of counts was imported in Seurat and data were subsequently clustered using default parameters. Pseudobulk was created for each ELP cluster (five in total), MOP cluster (four in total) and NUTM1 cluster (eight in total) by summarizing raw counts. Standard differential expression analysis was run using the DESeq function, and the result was filtered to only include genes with adjusted P value less than 0.05 and log2 fold changes greater than 1.
Gene ontology analysis
Gene ontology analysis was performed using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit)40. The gene list was defined as the overlap of differentially expressed genes between bulk KMT2A-rearranged infant B-ALL and bulk ELP transcriptomes and between single lymphoblast and single ELP cell transcriptomes (N = 455). Overrepresentation analysis was run using the human genome as a reference gene set and setting the disease phenotype database OMIM as a functional database.
Analysis of enrichment of KMT2A-AFF1 targets
Gene targets for the KMT2A-AFF1 fusion (N = 1,052) were extracted from Kerry et al.26. Enrichment of these 1,052 gene targets within the core leukemia transcriptome (N = 455) was assessed using a Monte Carlo approach by randomly drawing 455 genes from the possible transcriptome of 33,660 genes. This step of randomly drawing the list of genes was repeated 1,000 times, and P values were estimated by Student’s t test.
DNA sequencing and variant calling (lineage-switch case)
DNA sequencing and alignment
Short-insert (500-bp) genomic libraries were constructed, and 150-bp paired-end sequencing clusters were generated on the Illumina HiSeq XTen platform using no-PCR library protocols. DNA sequences were aligned to the GRCh37d5 reference genome by the Burrows–Wheeler algorithm (BWA-MEM v0.7.16a)41.
Variant calling
All classes variants were called using the extensively validated pipeline of the Wellcome Sanger Institute, built on the following algorithms: CaVEMan v1.13.14 (ref. 42) for base substitutions, PINDEL v2.2.4 for insertions/deletions43, ASCAT v4.0.1 (ref. 44) and Battenberg v3.2.2 (ref. 45) for copy-number changes and BRASS v6.0.5 for structural variants46.
Phylogenetic analyses from substitutions
We applied a previously developed framework47–49. In brief, beyond the standard postprocessing flags used in CaVEMan, we filtered out substitutions affected by mapping artefacts by setting the median alignment score of reads supporting a mutation ≥140 and requiring that fewer than half of the reads were clipped (CLPM = 0, CLPM, median number of soft clipped bases in variant supporting reads). Across all samples from PD38257, we recounted substitutions that were called in either blood or tumor from the patient using a cutoff for read mapping quality (28) and base quality (25). Germline variants were removed using one-sided exact binomial test on the number of variant reads and depth present (in diploid samples) to test whether the observed counts were consistent with a true variant allele frequency of 0.5 (or 0.95 for XY chromosomes). Resulting P values were corrected for multiple testing using the Benjamini-Hochberg method and a cutoff was set at q < 10-−5. Variants were also filtered out if they were called in a region of consistently low or high depth in diploid regions. Variants were kept if their corresponding site had a mean depth of between 20 and 60 for autosomes and a mean depth of between 10 and 30 for the X and Y chromosome. Using a beta-binomial model of site-specific error rates as previously described47–49, we distinguished true presence of somatic variants from support due to noise. All shared substitutions were further visually inspected in the genome browser Jbrowse50. The final list substitutions included in our analyses can be found in Supplementary Table 6.
Classification of single-nucleotide variants
To distinguish subclonal from clonal mutations in the tumor samples, we used a binomial mixture model to deconvolve the mutation counts into separate components. For each component, the optimal binomial probability and mixing proportion was estimated using an expectation-maximization algorithm. The optimal number of components was determined by the Bayesian information criterion. If the binomial probability of a component approximated the expected variant allele frequency (0.5 for diploid regions) adjusted for tumor purity, then the mutations assigned to that cluster were classified as clonal. If the estimated binomial probability for a component was lower, it was classified as subclonal.
Mutational signature analysis
Mutation signatures were fitted to the trinucleotide counts of single-nucleotide variants in the main clone and subclone of ALL (PD38257a) and AML (PD38257c) using the SigFit algorithm51 and the COSMIC reference database of mutational signatures (https://cancer.sanger.ac.uk/signatures/sbs/, v3.2), as used previously52. Initially, all reference signatures were fitted to the mutation counts. Only signatures that contributed at least 2% were retained during the subsequent fitting. Where mutation counts were low (<100), erroneous C>T signatures, such as those from ultraviolet light exposure (SBS7a) or mismatch repair deficiency (SBS6), were attributed to the samples. Because of their biological implausibility, these signatures were removed from the final set of fitted signatures.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-022-01720-7.
Supplementary information
Supplementary Figure 1 and legends for supplementary tables.
Supplementary Tables 1–8.
Acknowledgements
We thank I. Roberts and A. Roy, both from the University of Oxford, and A. Maartens (science writer at the Wellcome Sanger Institute) for critical review of the manuscript. We thank A. Filby and his team at the Flow Cytometry Core Facility, Newcastle University, for their advice and support. Samples were contributed by the SIHMDS at Great Ormond Street Hospital and by the Newcastle Biobank http://www.ncl.ac.uk/biobanks/. We are indebted to patients and their families for participating in our research. This study was funded by Wellcome Trust grants WT206194 (Wellcome Sanger Institute), WT110104/Z/15/Z (S. Behjati) and WT107931/Z/15/Z (M.H.); the Lister Institute of Preventative Medicine (M.H.); the Newcastle National Institute for Health Research-Biomedical Research Centre (M.H. and L.J.); the National Institute for Health Research Academic Clinical Lectureship (L.J.); Medical Research Council clinician scientist fellowship MR/S021590/1 (S. Bomken); CRUK program grant C27943/A12788 (O.H.); the Kay Kendall Leukaemia Fund (KKL1142) (O.H.); Kika program grant 329 (O.H.); Children with Cancer UK grant 14-169,17-249; and the National Institute for Health Research-Great Ormond Street Hospital Biomedical Research Centre (O.W.).
Extended data
Extended Data Fig. 4. Annotation of genes shared by KMT2A-rearranged myeloid and lymphoid leukemias.
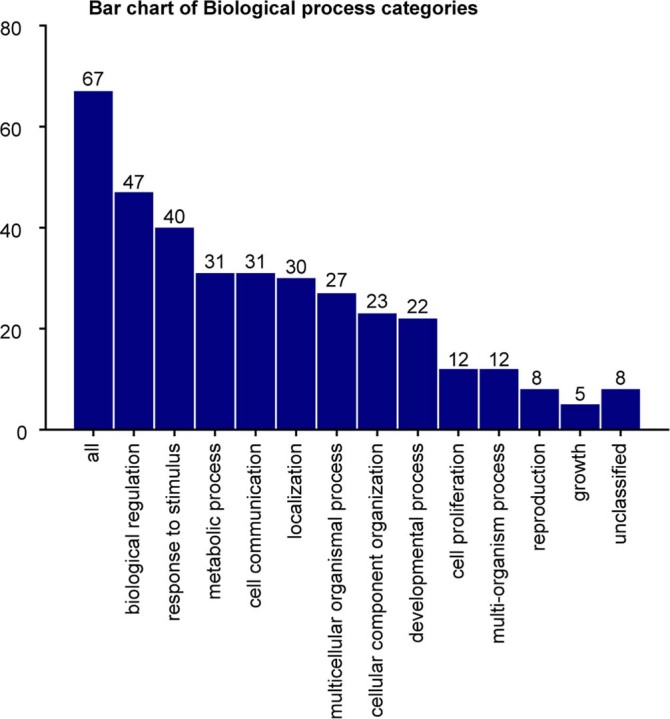
Barplot showing biological categories of genes shared between the KMT2A-rearranged B-ALL and KMT2A-rearranged AML core cancer transcriptomes.
Author contributions
Conceptualization, S. Behjati and J.B.; methodology, S. Behjati, E.K., M.D.Y., S.W. and THHC; investigation, E.K., L.J., T.D.T., T.H.H.C., J.E., T.P., E.P., K.S., S.I. and A.P.; visualization, S. Behjati, E.K. and L.J.; Funding acquisition: S. Behjati. Supervision: S. Behjati, JB, MH, S. Bomken. Writing – original draft: S. Behjati, EK, LJ. Writing – review & editing: S. Bomken, MH, GC, ST, OW, OH.
Peer review
Peer review information
Nature Medicine thanks Luca Vago, Benjamin Izar and Cheryl Willman for their contribution to the peer review of this work. Anna Maria Ranzoni was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Data availability
Single-cell RNA sequences have been deposited in the European Nucleotide Archive (accession number ERP125305) and the European Genome-phenome Archive (accession number EGAD00001007854) (Figs. 2 and 4). DNA sequences of the lineage-switch case (PD38257a, PD38257b, PD38257c) have been deposited in the European Genome-phenome Archive under study ID EGAD00001007853 and RNA sequences in the NCBI Sequence Read Archive under project IDs PRJNA547947 and PRJNA547815 (Fig. 3).
We used scRNA-seq data from developing bone marrow9, which are accessible through EMBL-European Bioinformatics Institute ArrayExpress and European Nucleotide Archive with accession codes E-MTAB-9389 and ERP125305. Scanpy h5ad objects with transformed counts are also available at https://fbm.cellatlas.io/. Bulk RNA-sequencing data on ELPs18 is available at GEO (GSE122982). TARGET leukemia RNA-sequencing data are available at dbGaP (phs000463, phs000464 and phs000465). St. Jude’s leukemia RNA-sequencing data were accessed via the St. Jude Cloud (https://stjudecloud.github.io/docs/citing-stjude-cloud/).
Code availability
Jupyter Notebook (v6.4.0) for processing single-cell data, including Cell Ranger-filtered count data and steps to reproduce Figs. 1 and 2, is available at https://github.com/kheleon/leukemia-paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Eleonora Khabirova, Laura Jardine.
These authors jointly supervised this work: Simon Bomken, Jack Bartram, Muzlifah Haniffa, Sam Behjati.
Contributor Information
Simon Bomken, Email: s.n.bomken@newcastle.ac.uk.
Jack Bartram, Email: Jack.Bartram@gosh.nhs.uk.
Muzlifah Haniffa, Email: m.a.haniffa@newcastle.ac.uk.
Sam Behjati, Email: sb31@sanger.ac.uk.
Extended data
is available for this paper at 10.1038/s41591-022-01720-7.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-022-01720-7.
References
- 1.Pieters R, et al. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the Interfant-06 protocol: results from an international phase III randomized study. J. Clin. Oncol. 2019;37:2246–2256. doi: 10.1200/JCO.19.00261. [DOI] [PubMed] [Google Scholar]
- 2.Pieters R, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–250. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 3.Ramakers-van Woerden NL, et al. In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia. 2004;18:521–529. doi: 10.1038/sj.leu.2403253. [DOI] [PubMed] [Google Scholar]
- 4.Gardner R, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sison EAR, Brown P. Does hematopoietic stem cell transplantation benefit infants with acute leukemia? Hematology. 2013;2013:601–604. doi: 10.1182/asheducation-2013.1.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boer JM, et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia. 2021;35:2978–2982. doi: 10.1038/s41375-021-01333-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pincez T, et al. Cryptic recurrent ACIN1-NUTM1 fusions in non-KMT2A-rearranged infant acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2020;59:125–130. doi: 10.1002/gcc.22808. [DOI] [PubMed] [Google Scholar]
- 8.Bassan R, et al. Updated risk-oriented strategy for acute lymphoblastic leukemia in adult patients 18-65 years: NILG ALL 10/07. Blood Cancer J. 2020;10:119. doi: 10.1038/s41408-020-00383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jardine L, et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature. 2021;598:327–331. doi: 10.1038/s41586-021-03929-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pellin D, et al. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 2019;10:2395. doi: 10.1038/s41467-019-10291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popescu D-M, et al. Decoding human fetal liver haematopoiesis. Nature. 2019;574:365–371. doi: 10.1038/s41586-019-1652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young MD, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science. 2018;361:594–599. doi: 10.1126/science.aat1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young MD, et al. Single cell derived mRNA signals across human kidney tumors. Nat. Commun. 2021;12:3896. doi: 10.1038/s41467-021-23949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gale KB, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl Acad. Sci. USA. 1997;94:13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ford AM, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363:358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- 16.Kildisiute G, et al. Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci. Adv. 2021;7:eabd3311. doi: 10.1126/sciadv.abd3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alhaj Hussen K, et al. Molecular and functional characterization of lymphoid progenitor subsets reveals a bipartite architecture of human lymphopoiesis. Immunity. 2017;47:680–696.e8. doi: 10.1016/j.immuni.2017.09.009. [DOI] [PubMed] [Google Scholar]
- 18.O’Byrne S, et al. Discovery of a CD10-negative B-progenitor in human fetal life identifies unique ontogeny-related developmental programs. Blood. 2019;134:1059–1071. doi: 10.1182/blood.2019001289. [DOI] [PubMed] [Google Scholar]
- 19.Meyer C, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. doi: 10.1038/leu.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu Z, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016;7:13331. doi: 10.1038/ncomms13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bullerwell CE, et al. EBF1 drives hallmark B cell gene expression by enabling the interaction of PAX5 with the MLL H3K4 methyltransferase complex. Sci. Rep. 2021;11:1537. doi: 10.1038/s41598-021-81000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandrov LB, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behjati S, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S, et al. Clonal dynamics in early human embryogenesis inferred from somatic mutation. Nature. 2021;597:393–397. doi: 10.1038/s41586-021-03786-8. [DOI] [PubMed] [Google Scholar]
- 25.Spencer Chapman M, et al. Lineage tracing of human development through somatic mutations. Nature. 2021;595:85–90. doi: 10.1038/s41586-021-03548-6. [DOI] [PubMed] [Google Scholar]
- 26.Kerry J, et al. MLL-AF4 spreading identifies binding sites that are distinct from super-enhancers and that govern sensitivity to DOT1L inhibition in leukemia. Cell Rep. 2017;18:482–495. doi: 10.1016/j.celrep.2016.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nix MA, et al. Surface proteomics reveals CD72 as a target for in vitro–evolved nanobody-based CAR-T cells in KMT2A/MLL1-rearranged B-ALL. Cancer Discov. 2021;11:2032–2049. doi: 10.1158/2159-8290.CD-20-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goulden, N. et al. United Kingdom National Randomised Trial For Children and Young Adults with Acute Lymphoblastic Leukaemia and Lymphoma 2011. https://www.northerncanceralliance.nhs.uk/wp-content/uploads/2019/01/UKALL2011-Protocol-v3.0-01-Oct-2013.pdf (University of Birmingham, 2013).
- 29.Gu Z, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019;51:296–307. doi: 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotfilder M, et al. Leukemic stem cells in childhood high-risk ALL/t(9;22) and t(4;11) are present in primitive lymphoid-restricted CD34+CD19- cells. Cancer Res. 2005;65:1442–1449. doi: 10.1158/0008-5472.CAN-04-1356. [DOI] [PubMed] [Google Scholar]
- 31.Agraz-Doblas A, et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica. 2019;104:1176–1188. doi: 10.3324/haematol.2018.206375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peham M, et al. Low frequency of clonotypic Ig and T-cell receptor gene rearrangements in t(4;11) infant acute lymphoblastic leukaemia and its implication for the detection of minimal residual disease. Br. J. Haematol. 2002;117:315–321. doi: 10.1046/j.1365-2141.2002.03428.x. [DOI] [PubMed] [Google Scholar]
- 33.Menendez P, et al. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J. Exp. Med. 2009;206:3131–3141. doi: 10.1084/jem.20091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bomken S, et al. Lentiviral marking of patient-derived acute lymphoblastic leukaemic cells allows in vivo tracking of disease progression. Leukemia. 2013;27:718–721. doi: 10.1038/leu.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng GXY, et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heaton H, et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods. 2020;17:615–620. doi: 10.1038/s41592-020-0820-1. [DOI] [PubMed] [Google Scholar]
- 37.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones D, et al. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Curr. Protoc. Bioinformatics. 2016;56:15.10.1–15.10.18. doi: 10.1002/cpbi.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye K, et al. Split-read indel and structural variant calling using PINDEL. Methods Mol. Biol. 2018;1833:95–105. doi: 10.1007/978-1-4939-8666-8_7. [DOI] [PubMed] [Google Scholar]
- 44.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nik-Zainal S, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coorens THH, et al. Lineage-independent tumors in bilateral neuroblastoma. N. Engl. J. Med. 2020;383:1860–1865. doi: 10.1056/NEJMoa2000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coorens THH, et al. Inherent mosaicism and extensive mutation of human placentas. Nature. 2021;592:80–85. doi: 10.1038/s41586-021-03345-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coorens THH, et al. Embryonal precursors of Wilms tumor. Science. 2019;366:1247–1251. doi: 10.1126/science.aax1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buels R, et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 2016;17:66. doi: 10.1186/s13059-016-0924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gori, K. & Baez-Ortega, A. sigfit: flexible Bayesian inference of mutational signatures. Preprint at bioRxiv10.1101/372896 (2020).
- 52.Coorens THH, et al. Clonal hematopoiesis and therapy-related myeloid neoplasms following neuroblastoma treatment. Blood. 2021;137:2992–2997. doi: 10.1182/blood.2020010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 and legends for supplementary tables.
Supplementary Tables 1–8.
Data Availability Statement
Single-cell RNA sequences have been deposited in the European Nucleotide Archive (accession number ERP125305) and the European Genome-phenome Archive (accession number EGAD00001007854) (Figs. 2 and 4). DNA sequences of the lineage-switch case (PD38257a, PD38257b, PD38257c) have been deposited in the European Genome-phenome Archive under study ID EGAD00001007853 and RNA sequences in the NCBI Sequence Read Archive under project IDs PRJNA547947 and PRJNA547815 (Fig. 3).
We used scRNA-seq data from developing bone marrow9, which are accessible through EMBL-European Bioinformatics Institute ArrayExpress and European Nucleotide Archive with accession codes E-MTAB-9389 and ERP125305. Scanpy h5ad objects with transformed counts are also available at https://fbm.cellatlas.io/. Bulk RNA-sequencing data on ELPs18 is available at GEO (GSE122982). TARGET leukemia RNA-sequencing data are available at dbGaP (phs000463, phs000464 and phs000465). St. Jude’s leukemia RNA-sequencing data were accessed via the St. Jude Cloud (https://stjudecloud.github.io/docs/citing-stjude-cloud/).
Jupyter Notebook (v6.4.0) for processing single-cell data, including Cell Ranger-filtered count data and steps to reproduce Figs. 1 and 2, is available at https://github.com/kheleon/leukemia-paper.