

Abstract
We report four neoplasms of the kidney (2 cases) and uterus (2 cases) harboring rearrangements or amplifications of the GLI1 gene, which due to their unusual clinical presentation, morphology and immunoprofile mimicked other neoplasms, causing significant diagnostic challenge. The neoplasms occurred in four female patients ages 33–88 years. Histologically they all demonstrated nodular growth, solid architecture, bland epithelioid to ovoid spindle cells with pale cytoplasm set in a variably myxoid or hyalinized stroma. One uterine tumor also demonstrated a focal round cell pattern, while another demonstrated focal pleomorphism. Unlike most previously reported neoplasms with these genetic abnormalities, the neoplasms in the current series were negative for S100 protein and minimally reactive for actin. All labeled for CD10 and cyclin D1, while two labeled for estrogen receptor and BCOR and one labeled for desmin, raising consideration of endometrial stromal sarcoma, myxoid leiomyosarcoma, metastatic breast carcinoma, and glomus tumor. One renal neoplasm demonstrated a GLI1-FOXO4 gene fusion and the other harbored a GLI1 gene rearrangement (unknown partner). The two uterine neoplasms exhibited GLI1 gene amplifications. GLI1-altered neoplasms (particularly those with GLI1-amplification) show variable morphology and lack a consistent immunophenotype, and thus may trigger diagnostic challenges which can be resolved by molecular testing.
Keywords: GLI1, Translocation, Amplification
INTRODUCTION
The glioma-associated oncogene homolog 1 (GLI1) gene on chromosome 12q13 encodes for a zinc-finger transcription factor which is a downstream effector of the Sonic hedgehog (SHH) signaling pathway1,2. The SHH pathway plays a key role in embryo development and regulates stem cell differentiation, and it was shown to be aberrantly dysregulated in many human cancers. Inactivating mutations in the negative SHH signaling regulator patched-1 membrane receptor (PTCH1) gene or gain of function mutations in the positive regulator smoothened (SMOH) gene activate GLI1 signaling in many tumor types3.
Over the past 15–20 years, structural alterations affecting the GLI1 gene resulting in oncogenic activation and GLI1 overexpression have been identified in several neoplasms. In mesenchymal neoplasia, GLI1-related fusions were first described in a so-called ‘pericytoma with t(7;12) translocation’, a neoplasm which shows predilection for tongue and stomach4–7. Despite its consistent actin reactivity and benign clinical behavior, its morphologic appearance did not fit within the spectrum of other pericytic neoplasms8. Subsequently, an identical MALAT1-GLI1 fusion was identified in two distinctive, well-characterized gastric neoplasms that typically affect young patients, plexiform fibromyxoma (a benign bland spindle cell neoplasm with fibromyxoid stroma and prominent capillary vasculature)9,10 and gastroblastoma (a malignant biphasic stromal-epithelial neoplasm featuring primitive spindle cell fascicles and cords or tubules of epithelial cells)11. Another evolving subset of neoplasms with GLI1 fusions with various gene partners was recently described under the provisional term ‘malignant epithelioid neoplasms with GLI1 gene rearrangements’8,12. These neoplasms are characterized by a distinctive nested growth of epithelioid cells with pale eosinophilic cytoplasm, associated with an arborizing thin-walled capillary network, frequent immunoreactivity for S100 protein and potential for metastatic spread. Other morphologically similar and likely pathogenetically related neoplasms have demonstrated GLI1 gene amplifications, though they are characterized by a wider histologic spectrum and less consistent immunoprofile13.
Most of the previously reported neoplasms with structural GLI1 gene alterations have arisen in soft tissue, stomach (the site of origin of plexiform fibromyxoma and gastroblastoma), or tongue. Only a handful of cases have been reported in other visceral sites, including single case reports of GLI1 rearranged tumors in the genitourinary tract (kidney)14 or gynecological tract (ovary)15,16. We report herein a small series of neoplasms with GLI1 gene rearrangements and amplifications arising in the genitourinary and gynecologic tracts which resulted in diagnostic challenges.
MATERIALS AND METHODS
IRB Approval
This study was approved by the institutional review boards at our respective institutions.
Cases
The four cases reported herein were retrieved from the consultation files of two authors (P. A. and C.R.A.) and the routine files of one hospital (MGH). The cases were identified during review of unclassified primary renal and gynecologic tract neoplasms. Case 1 was the index case originally diagnosed descriptively 15 years ago. Cases 2–4 were subsequently identified during this review over the past 2 years. Case 1 was investigated by targeted RNA-sequencing, which revealed a GLI1-FOXO4 gene fusion candidate. This result was validated with florescence in situ hybridization (FISH) using custom bacterial artificial chromosomes (BAC) probes, showing break-apart signals for both GLI1 and FOXO4 genes. Case 2 demonstrated a GLI1 gene rearrangement by break-apart FISH, with no adequate tissue available for RNA-sequencing, however, FISH studies excluded MALAT1, ACTB, PTCH1, or FOXO4 as fusion partners. Case 3 was analyzed by MSK-IMPACT targeted DNA sequencing, which revealed amplification of GLI1 gene locus that was further confirmed by FISH. Case 4 was studied by FISH alone.
Immunohistochemistry
Given the difficulty in classifying these lesions on morphologic grounds, a broad panel of immunohistochemical stains was applied to each case. Immunohistochemistry for epithelial markers [cytokeratins AE1/3 and Cam5.2, epithelial membrane antigen (EMA)], melanocytic markers (S100 protein, HMB45, melan A), muscle markers (desmin, smooth muscle actin, caldesmon), vascular markers (CD34), and endometrial stromal sarcoma markers (cyclin D1, estrogen receptor (ER), CD10, and BCOR) were performed as previously described17.
RNA and DNA sequencing
Case #1 was subjected to RNA sequencing. RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA). Fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-sequencing libraries were prepared using 20 to 100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA)18. Each sample was subjected to targeted RNA sequencing on an Illumina MiSeq (∼3 million reads per sample). All reads were independently aligned with STAR (version 2.3) and BowTie2 against the human reference genome (hg19) for Manta-Fusion and TopHat-Fusion analysis, respectively.
Case #3 was evaluated using MSK-IMPACT (Integrated Mutational Profiling of Actionable Cancer Targets), a targeted ultra-deep next generation sequencing platform designed to capture all exons and selected introns of 468 cancer-associated genes including oncogenes, tumor suppressor genes, and members of targetable pathways19.
Fluorescence in Situ Hybridization (FISH)
FISH on interphase nuclei from paraffin-embedded 4-micron sections was performed by applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking GLI1 and potential partner genes that were identified as potential fusion partners in the RNA-seq experiment. BAC clones were chosen according to UCSC genome browser (http://genome.ucsc.edu), see Supplementary Table 1 and as previously published8. The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org) and Life Technologies Corporation (Carlsbad, CA). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described20. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were omitted from the score. Samples were considered positive for amplification when one of the following signal patterns of amplification was noted: HSR-homogeneous-stained-region, DM-double minutes, ring chromosomes, and MDA-multiple dot-like amplicons in various sizes as previously described13.
RESULTS
Clinical History and Gross Appearance:
Case 1 was a 49 year-old female with a history of breast cancer (subtype unknown) who was noted on follow-up to have solid 4 cm left renal mass. The patient underwent radical nephrectomy which revealed a well-delineated 4 cm fleshy renal mass with a 2 cm cystic component associated with calcification at its border. The lesion appeared centered in the renal medulla but did not invade the renal sinus. The patient has no evidence of recurrent disease at 7 years follow-up.
Case 2 was a 33 year-old female who presented with renal colic. CT scan revealed a small tumor at the uretero-pelvic junction associated with mild hydronephrosis. After biopsy revealed evidence of a neoplasm, the patient underwent left nephrectomy. This demonstrated a 2.7 cm polypoid myxoid neoplasm confined to the renal pelvis. At 25 months follow-up, there was evidence of recurrent disease.
Case 3 was a 49 year-old female with a history of anemia who was found to have a large uterine mass. The patient underwent hysterectomy, which revealed a 7 cm fleshy lobulated uterine tumor grossly resembling leiomyoma secondarily involving the left ovary and fallopian tube and bilateral parametria, extending to all margins. Two years later, the patient developed multifocal pelvic recurrence, and five months later the patient developed brain (right parietal lobe) metastases. The patient died of disease three years after initial diagnosis.
Case 4 was an 88 year-old female who presented with pelvic pain and altered bowel habits, and was found to have a uterine mass. She underwent total abdominal hysterectomy with bilateral salpingo-oophorectomy, revealing a 12 cm gelatinous tumor with lobulated cut surfaces, focal hemorrhage and necrosis that was deeply invasive of the myometrium, with an additional 2.5cm nodule involving the rectal adventitia. The patient is alive at 2 months follow-up, having received post-operative adjuvant radiation therapy.
Histopathology
All four neoplasms demonstrated nodular growth and were composed of bland ovoid-spindle cells with short cell processes, arranged in solid sheets with accentuation around a prominent capillary vasculature. This was the predominant pattern in the renal neoplasms (cases 1 and 2). Moreover, in these two neoplasms, prominent fibrinoid material often separated the neoplastic cells from the capillary vessels. This fibrinoid material merged with more myxoid stroma or hyalinized stromal collagen, as highlighted on Masson trichrome stains. In both neoplasms, the hyalinized material formed confluent areas of paucicellular sclerosis, associated with calcification. Mitotic activity was negligible (<1/10 high power fields) (Figures 1 and 2).
Figure 1. Pathologic findings of the renal neoplasm (case 1).
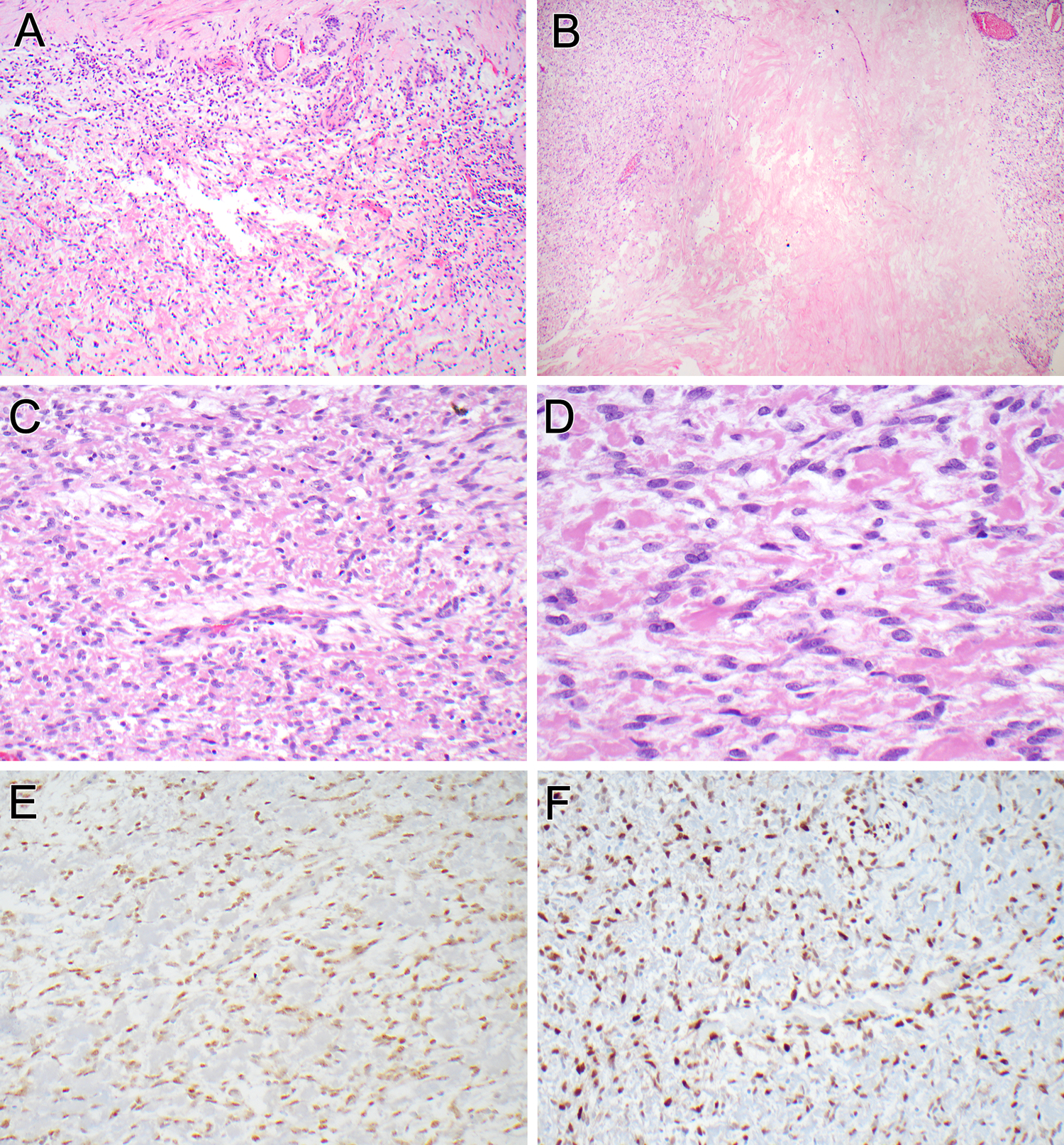
The tumor was surrounded by a variably thick fibrous capsule (top). Entrapped native renal tubules were seen embedded within the capsule and intermingled with neoplastic cells at the periphery (A). Areas of confluent, dense hyalinization were evident within the neoplasm (B). The neoplastic cells showed an ovoid to short spindle cell appearance, arranged in a perivascular distribution around intra-tumoral arterioles (C). The tumor cells were bland, with thin, short cytoplasmic extensions and ovoid nuclei with fine chromatin. The stroma separating the neoplastic cells was either myxoid or hyalinized, resembling fibrin (D). The neoplastic cells labeled diffusely for estrogen receptor (E) and cyclin D1 (E).
Figure 2. Microscopic and immunophenotypic findings of the renal pelvic neoplasm (case 2).
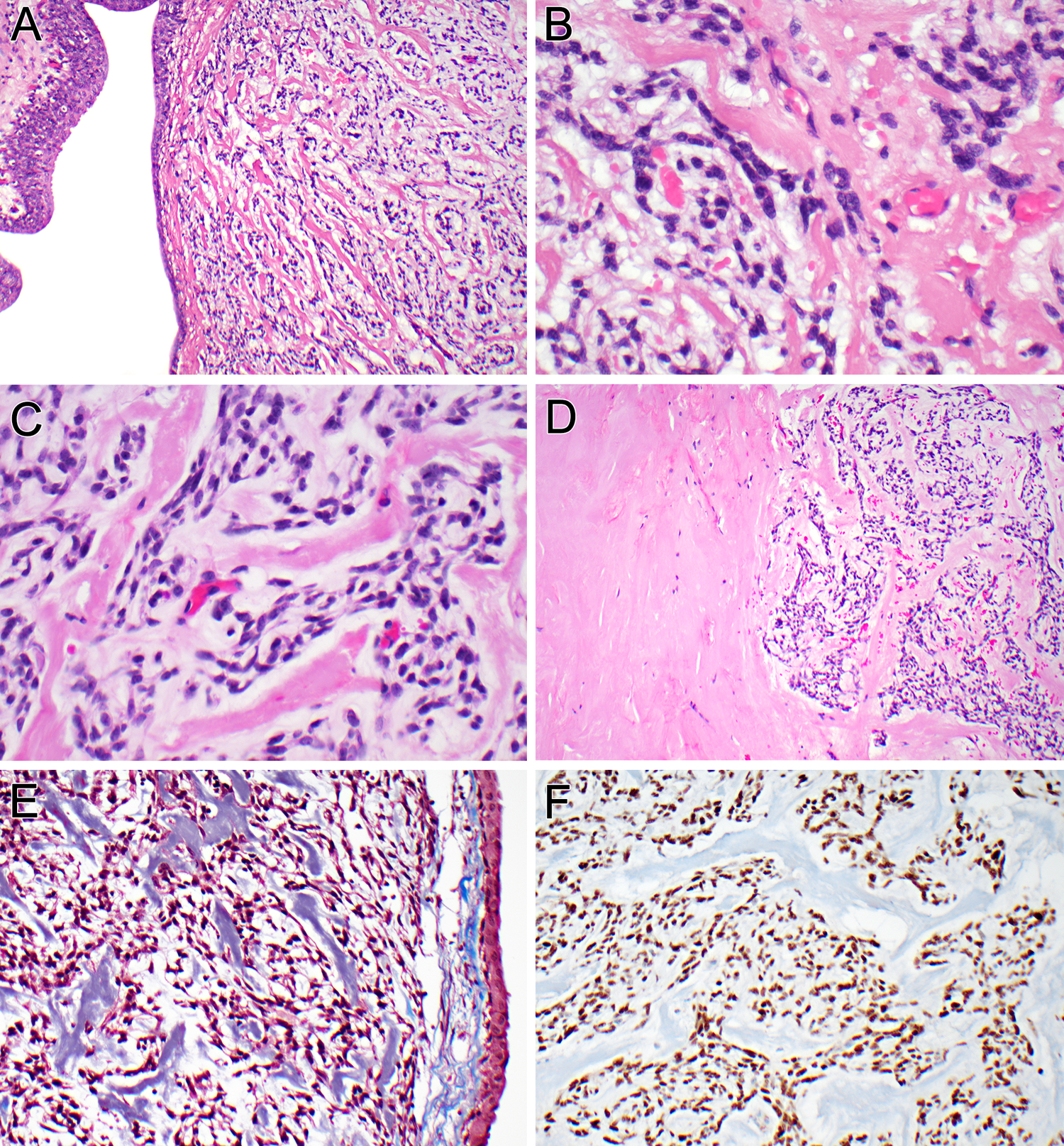
The neoplasm undermines the urothelium of the renal pelvis (A). The bland neoplastic cells are separated from intra-tumoral capillaries by hyaline and myxoid stroma, imparting a pericytic growth pattern (B). The neoplastic cells have delicate ovoid nuclei and ill-defined cytoplasmic extensions, within a myxoid and hyalinized stroma (C). The hyalinized stroma between individual neoplastic cells (right) merges with large sclerotic, hypocellular zones within the neoplasm (left) (D). The hyalinized material is basophilic on Masson trichrome stain, indicating collagen (E). The neoplastic cells label diffusely for cyclin D1 (F).
In addition to a focal ovoid-spindle cell component, one uterine neoplasm (case 3) revealed a second predominant component of round to epithelioid cells arranged in a distinctive nested architecture with nuclei showing fine chromatin. Mitotic activity was higher in the ovoid-spindle cell component (16 per 10 high power fields) than the round cell component (1 per 10 high power fields) (Figure 3). This neoplasm also demonstrated coagulative necrosis and prominent vascular invasion, which was present at both parametrial margins. Of note, while both ovoid-spindle and round cell components were present in the primary uterine tumor and pelvic recurrence, the brain metastasis demonstrated only the nested round/ epithelioid cell component.
Figure 3. Pathologic features of uterine neoplasm (case 3).
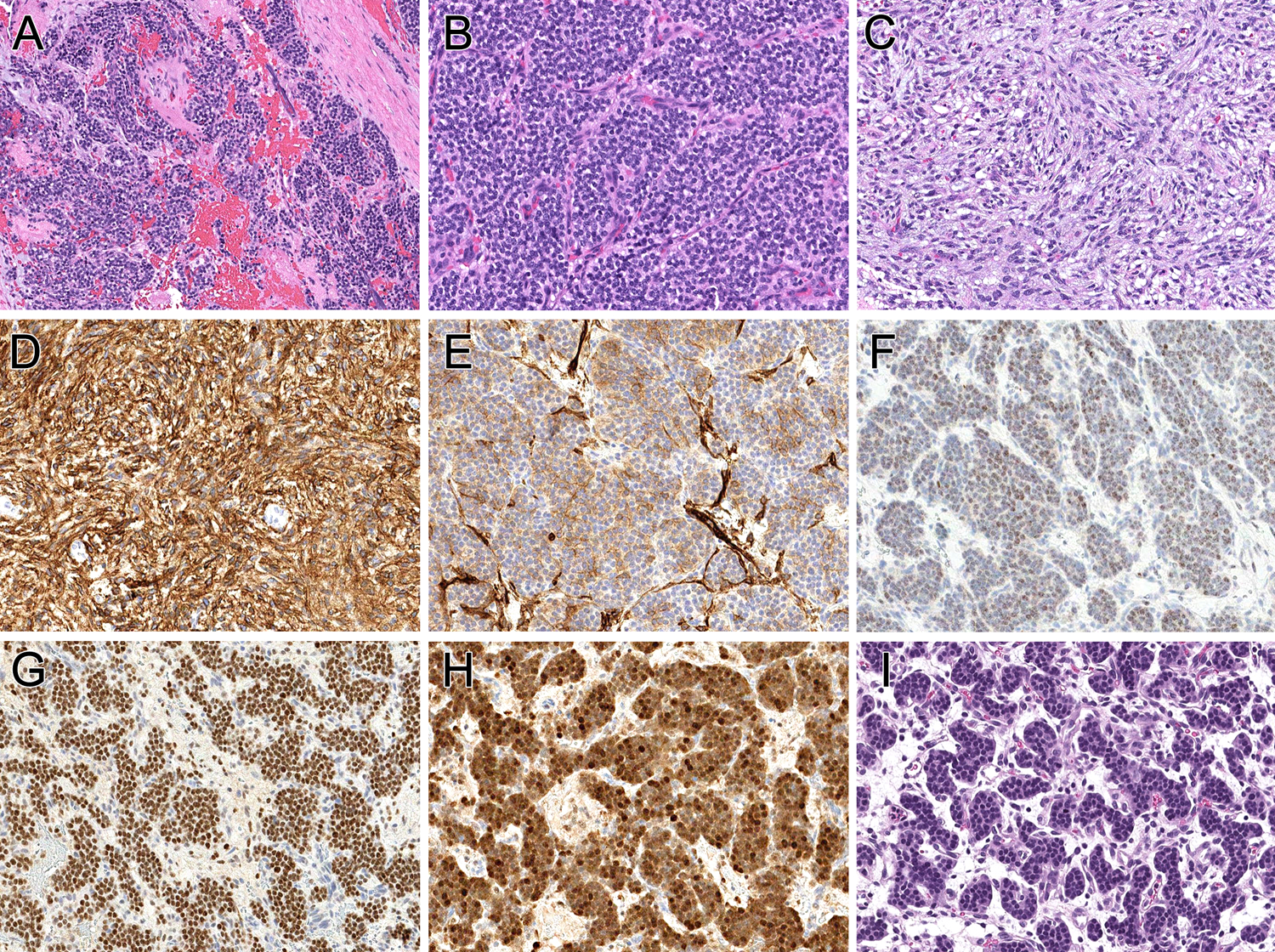
The neoplastic cells permeate the myometrium (A), having either a small round cell phenotype with prominent capillary vasculature (B) and a bland spindle cell appearance (C). The spindle cell component is diffusely positive for CD10 (D), while the small round cell component is focally positive (E). The tumor shows patchy nuclear labeling for estrogen receptor (F), strong nuclear labeling for BCOR (G) and cyclin D1 (H). The brain metastasis (I) is purely nested epithelioid.
In addition to the bland ovoid-spindle cell component within a myxoid stroma, the second uterine neoplasm (case 4) exhibited both a bland non-myxoid spindle cell component with fibrillary eosinophilic cytoplasm, as well as cellular areas with focal nuclear pleomorphism,increased mitotic activity (15 mitoses/10 high power fields) and focal coagulative necrosis (Figure 4)
Figure 4. Pathologic features of uterine neoplasm (case 4).
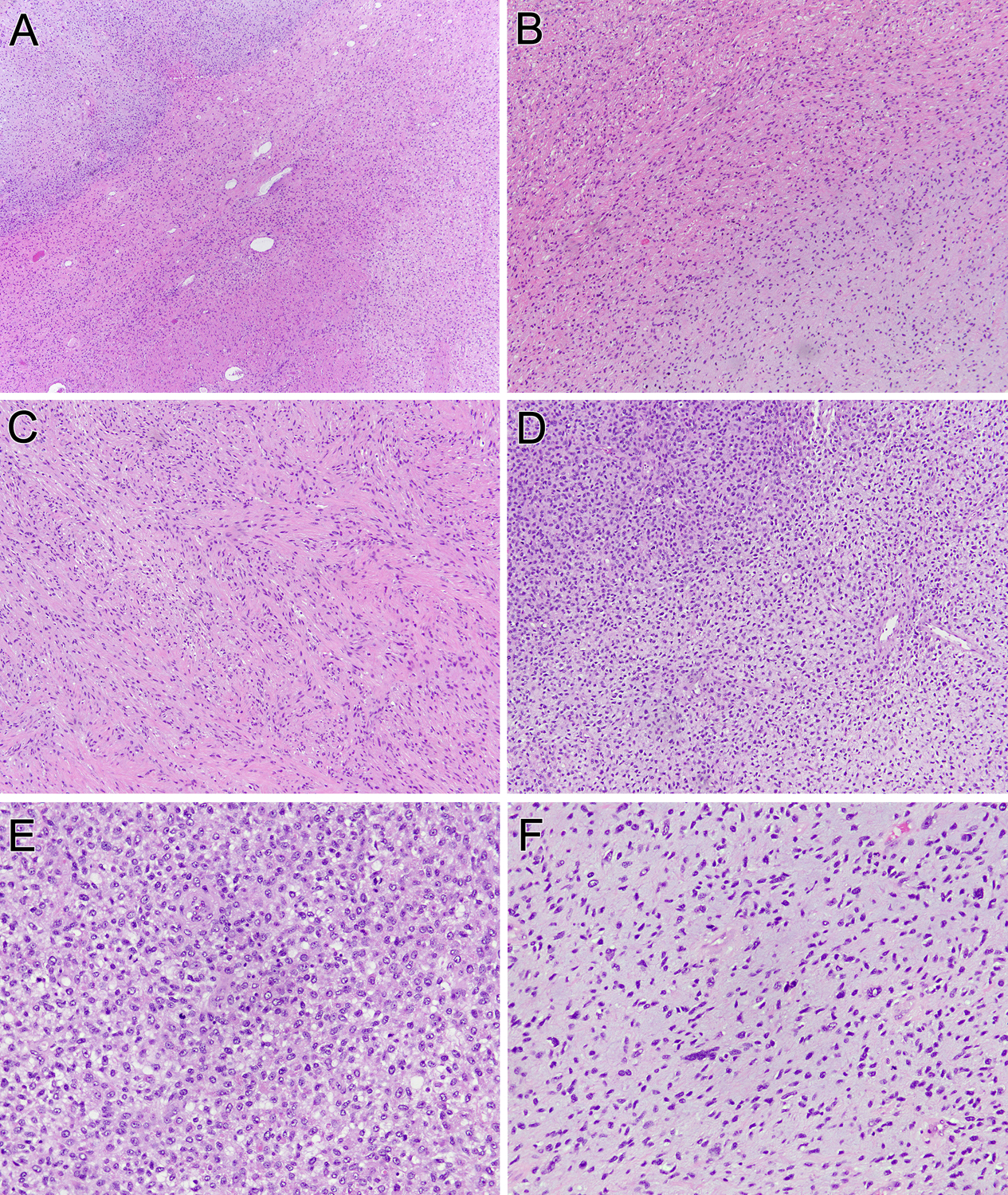
This uterine neoplasm contains three components: a bland myxoid area typical of GLI1-altered neoplasms (lower right), a bland spindle cell area (middle), and a more pleomorphic myxoid area (upper left) (A). At intermediate power, one can see the transition from the bland epithelioid myxoid area (lower right) and the bland spindled area (upper left) (B). The latter component shows spindle cells with eosinophilic cytoplasm, which along with patchy desmin immunoreactivity raised the possibility of a myxoid smooth muscle neoplasm (C). The bland myxoid areas transition into more cellular areas (upper left) (D). These more overtly malignant areas demonstrate high cellularity and mitotic activity (E), as well as greater nuclear atypia (F).
The immunohistochemical profiles of these neoplasms were not specific, but raised various differential diagnoses at these visceral sites of origin (Table 1). All four neoplasms demonstrated diffuse strong labeling for cyclin D1 (nuclear) and CD10 (membranous), while the 2 neoplasms tested were also positive for Bcl2 (cases 1 and 2). All four neoplasms were negative for S100 protein and all three tested were negative for epithelial markers cytokeratin AE1/3, cytokeratin Cam 5.2 and EMA, along with HMB45. CD34 was negative in two tested neoplasms (cases 1 and 2). Two neoplasms (cases 1 and 3) were immunoreactive for estrogen receptor (one patchy) while the other two were negative. One neoplasm (case 1) demonstrated focal weak labeling for actin but was negative for desmin, while another (case 4) demonstrated patchy staining for desmin, but caldesmon and actin were negative.
Table 1:
Clinicopathologic Features of GLI1-Altered Neoplasms in This Study
| Case | Age/Sex | Location | IHC Positive | IHC Negative | Genetics | Clinical |
|---|---|---|---|---|---|---|
| 1 | 49/F | Kidney | Cyclin D1, CD10, ER, Bcl2, SMA (weak) | S100, AE1/3, Cam5.2, CK903, EMA, PR, GCDFP, p63, HMB45, CD34, ALK | GLI1-FOXO4 fusion | History of breast cancer; NED 7 years |
| 2 | 33/F | Kidney (renal pelvis) |
Cyclin D1, CD10, Bcl2, Vimentin | S100, SMA, ER, BCOR, AE1/3, EMA, calponin, desmin, synaptophysin, p40, SOX10, CD34, HMB45 | GLI1 fusion (partner not known) | NED 25 months |
| 3 | 49/F | Uterus | Cyclin D1, CD10, ER (patchy), SMA | S100, PR, AE1/3, Cam5.2, EMA, HMB45, Melan A, CD117, pan-NTRK, ALK, calretinin | GLI1 amplification | Pelvic recurrence, brain metastasis, DOD 3 years |
| 4 | 88/F | Uterus | Cyclin D1, CD10, BCOR, MDM2, desmin (focal) | S100, SMA, caldesmon, ER, PR, ALK, ROS | GLI1 amplification | Recent case |
IHC= immunohistochemistry; DOD=dead of disease; NED=no evidence of disease
The working diagnoses were as follows. In case 1, a definitive diagnosis was not reached, but the possibilities of a smooth muscle neoplasm, inflammatory myofibroblastic tumor (despite the negative stain for ALK), and even sarcomatoid metastatic breast cancer were listed as diagnostic possibilities. Case 2 was noted to demonstrate focal labeling for collagen type 4 and actin, and was favored to represent a glomus tumor. As case 3 originated in the uterus, labeled diffusely and strongly for BCOR, CD10 and cyclin D1 and labeled focally for estrogen receptor, it was initially diagnosed as a high grade endometrial stromal sarcoma. Case 4 was initially considered a myxoid leiomyosarcoma given its eosinophilic spindled areas, patchy labeling for desmin and diffuse nuclear labeling for PLAG1 until molecular studies were performed.
Molecular Findings
Targeted RNA sequencing identified an in-frame fusion transcript composed of the first 10 exons of GLI1 gene fused to the last two exons of FOXO4 (GLI1 exon 10-FOX4 exon 2) (Figure 5, case 1). Thus, the predicted fusion oncoprotein retains all GLI1 DNA binding domains, while the GLI1 transactivation-domain is absent21. In contrast, FOXO4 retains a truncated portion of the forkhead box domain and an intact transactivation domain (TAD). These results were further confirmed by FISH break-apart assays, showing gene rearrangements for GLI1 and FOXO4 genes (Figure 6).
Figure 5. Diagrammatic representation of the GLI1-FOXO4 fusion.
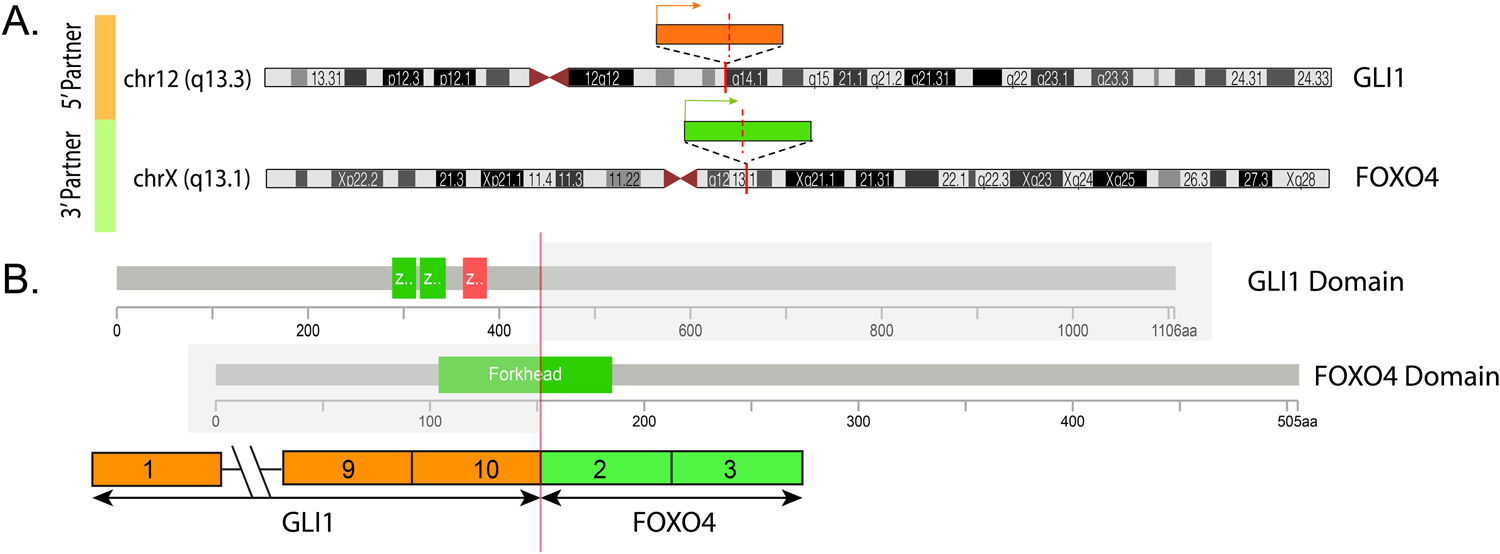
A. Schematic view of GLI1 gene locus (orange box) on 12q13.3 and FOXO4 gene (green box) on Xq13.1. The directions of transcription of each gene is shown by an arrow line. B. The resulting fusion transcript is composed of GLI1 exon 10 fused to exon 2 of FOXO4. The protein domains of the participating genes are also displayed. From the FOXO4 partner, only a truncated portion of the Forkhead box domain is retained in the fusion, while the transactivation domain is preserved. In contrast, GLI1 retains the DNA binding domains, but not the transactivation domain in the projected fusion oncoprotein21.
Figure 6. FISH analysis validating the targeted RNA and DNA sequencing studies.
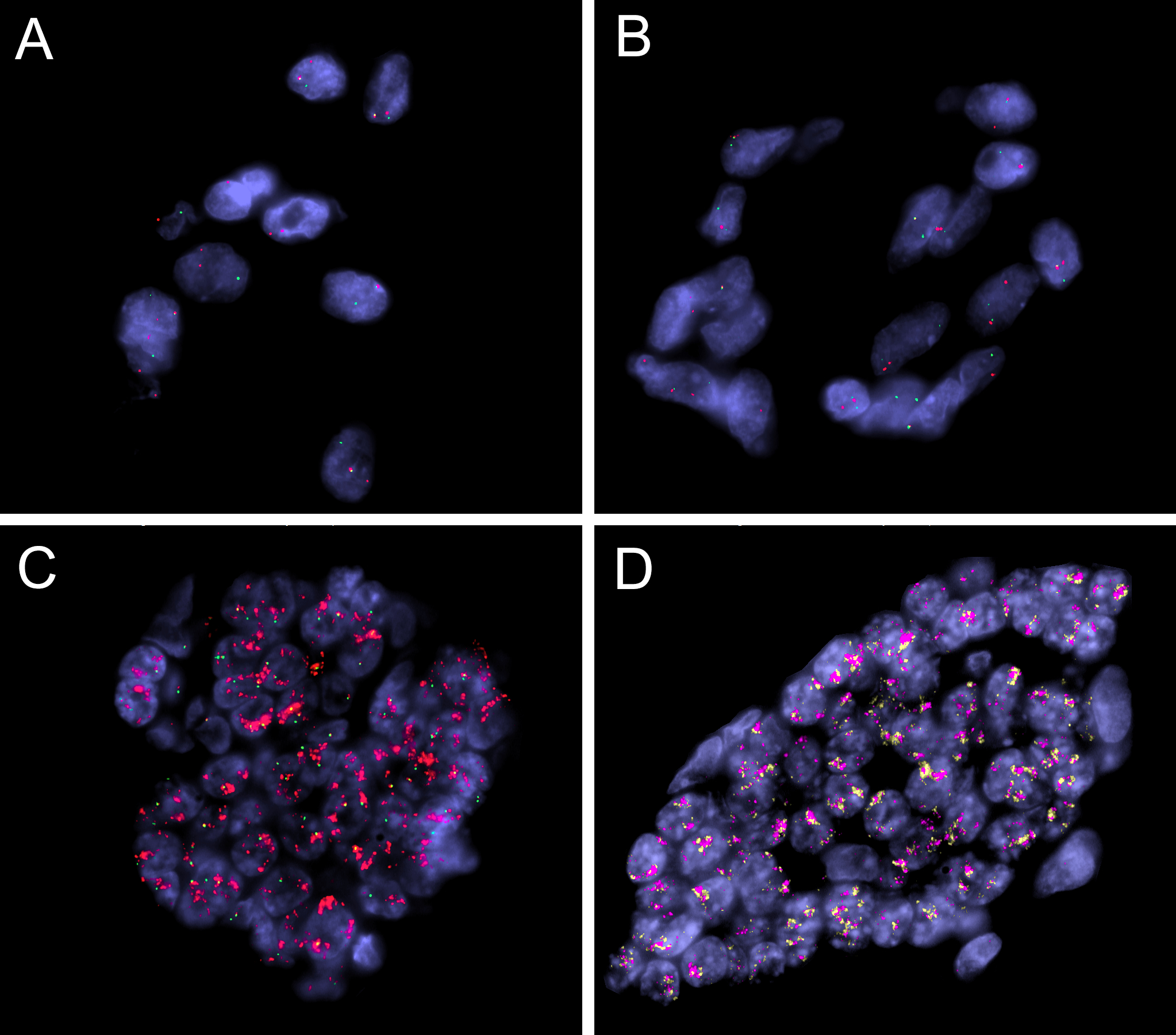
FISH analyses using break-apart assays showing GLI1 (A) and FOXO4 (B) gene rearrangements (case 1, green, centromeric; red, telomeric). FISH in case 3 confirmed the GLI1 gene amplification (C)(red, telomeric, DDIT3; green, centromeric), as well as co-amplifications of the nearby genes on 12q13–15 genes (D)(red, CDK4; orange, MDM2).
In case 2, FISH assay also revealed the presence of GLI1 gene rearrangements, while no abnormalities were detected in ACTB, PTCH1, MALAT1 and FOXO4 genes. No material was available for further RNA sequencing.
Case 3 was subjected to MSK IMPACT DNA targeted sequencing and found to have GLI1 gene amplifications. This result was further validated by FISH which showed co-amplifications of GLI1, MDM2 and CDK4 genes (Figure 6). No evidence of BCOR internal tandem duplication (ITD) or BCOR, YWHAE, or BCORL1 rearrangement was identified in case 3. Case 4 was similarly found to have co-amplifications of the GLI1, MDM2 and CDK4 genes by FISH, while lacking alterations in PLAG1, BCOR, NCOA1/2 and PGR genes.
DISCUSSION
In 2004, an ACTB-GLI1 gene fusion was identified in a distinctive neoplasm designated as ‘pericytoma with t(7;12) translocation’4,5. These neoplasms demonstrated bland uniform ovoid cells with a distinctive perivascular growth surrounding arborizing thin walled vessels. While they showed immunoreactivity for actin and ultrastructural features suggesting pericytic lineage, their overall features were distinct from the more common pericytic tumors, such as glomus and myopericytoma, which are characterized by NOTCH gene fusions or PDGFRB mutations, respectively22,23. In 2016–17, an identical MALAT1-GLI1 fusion was identified in two distinctive gastric neoplasms, including the benign plexiform fibromyxoma and the malignant biphasic stromal-epithelial neoplasm, designated as gastroblastoma9–11. In 2018, GLI1 gene fusions involving either MALAT1, ACTB, or PTCH1 partners were reported in a novel subset of soft tissue neoplasms associated with a distinctive nested architecture and predominantly epithelioid cells8. These tumors showed some morphologic overlap with the previously described pericytomas with the ACTB-GLI1 fusions, but differed in that they were frequently S100 protein positive, demonstrated a wider spectrum of histologic features and a subset followed a malignant clinical course with distant metastases. In 2019, a related group of malignant soft tissue tumors showing similar morphology to the GLI1-fusion positive neoplasms but instead harboring GLI1 gene amplifications were described, often associated with co-amplifications of nearby genes, including MDM2, CDK4, HMGA2, STAT6, resulting in mRNA and protein overexpression13.
The immunophenotype of these neoplasms harboring GLI1 gene fusions or GLI1 amplifications has been variable; while occasional cases have labeled for actin, more frequently these neoplasms have labeled for S100 protein. The main entities included in the differential diagnosis are myoepithelial neoplasms (suggested by the overlapping morphology and frequent S100/actin immunoreactivity), and pericytic neoplasms (suggested by the frequent perivascular distribution of growth and occasional actin immunoreactivity). As GLI1-amplified tumors may show immunoreactivity for MDM2 and STAT6, additional potential pitfalls include confusion with dedifferentiated liposarcoma and solitary fibrous tumor, respectively13.
Excluding the stomach (site of origin of plexiform fibromyxoma and gastroblastoma), the majority of previously reported mesenchymal neoplasms with structural GLI1 alterations have arisen in the soft tissue or head and neck, particularly the tongue4,12. Only rare examples have been reported in the genitourinary and gynecological tracts. Of note, Pettus et al.14 recently reported a primary myxoid and epithelioid mesenchymal tumor of the kidney with a GLI1-FOXO4 gene fusion, similar to our case #1. In the reported case of Pettus et al., the fusion transcript includes exon 1–10 of GLI1, and exon 2–3 of FOXO4, resulting in a predicted fusion protein that contains the N terminal amino acids 1–436 of GLI1 protein and amino acids 152–505 of the FOXO4 protein. The reported exonic composition of the fusion transcript is identical with our case 1 (Figure 5). Of note, the translocation in both cases result in a fusion involving the 5’ domains of GLI1, while all the other GLI1-related fusions reported to date, involving ACTB, MALAT1 and PTCH1, encompass GLI1 as the 3’ partner8. This phenomenon might impact on the somewhat distinct morphology and immunophenotype of these cases compared to our previous series8. Although the illustrated morphology of the case by Pettus et al.14 is very similar to that found in our primary renal neoplasm (case 1), their tumor was focally positive for melan A, while our case was negative. The two previously reported neoplasms involving the gynecological tract have involved the ovary. Koh et al.15 reported a neoplasm with ACTB-GLI1 gene fusion in an 11 year-old female. The neoplasm was 16.5 cm, solid and cystic, and diagnosed as a ‘pericytoma’ despite the immunoreactivity for S100 protein and absence of immunoreactivity for actin. Kerr et al. 16 reported a tumor with similar morphology in a 41 year-old female. The neoplasm measured 7 cm, was solid and cystic, and was described as a pericytoma with focal labeling for both actin and S100 protein.
Our series broadens the differential diagnosis of neoplasms with GLI1 genomic alterations, based upon clinical presentation in the genitourinary or gynecologic tracts and variant morphologies and immunoprofiles. First, none of our cases labeled for S100 protein, and actin was only focally positive in two of the four neoplasms, highlighting the variable immunophenotype of these neoplasms. Second, two of our four cases demonstrated nuclear labeling for estrogen receptor, which in case 1 raised the differential diagnosis of a metastatic metaplastic mammary carcinoma involving the kidney or a smooth muscle neoplasm, the latter of which frequently expresses estrogen receptor when originating in the peritoneum/retroperitoneum of females24. While the absence of cytokeratin immunoreactivity in this case argued against metaplastic carcinoma, it should be noted that not all mammary metaplastic carcinomas express cytokeratin in their spindle components so cytokeratin negativity does not exclude that diagnosis25,26. Third, the immunoreactivity for BCOR27, CD10 and cyclin D1, along with the focal immunoreactivity for estrogen receptor, lead to a diagnosis of a high grade endometrial stromal sarcoma in case 3. Along these lines, one recent study28 identified two cases of GLI1-rearranged neoplasms in a large RNA sequencing study of putative endometrial stromal sarcomas, though the morphology or immunohistochemical profiles of these cases was not illustrated. We do not believe that our GLI1-amplified uterine neoplasms are high grade endometrial stromal sarcomas based on morphology (greater pleomorphism, absence of characteristic sinusoidal vasculature of YWHAE-NUT 2A/B fusion high-grade endometrial stromal sarcoma or collagen plaques of BCOR-altered high-grade endometrial stromal sarcoma), immunohistochemical profile (CD10 is typically negative in high-grade endometrial stromal sarcomas associated with YWHAE-NUT 2A/B fusion, unlike our cases), and genetics (absence of gene fusions otherwise associated with endometrial stromal sarcoma in our cases). It should be noted that while neither BCOR nor estrogen receptor immunoreactivity are consistent findings in GLI1 associated neoplasms, immunoreactivity for CD10 and cyclin D1 were seen in all four cases in our series. Cyclin D1 immunoreactivity is somewhat expected, as cyclin D1 is known to be upregulated by GLI1 overexpression3. Fourth, the extensive perivascular hyalinization of case 2 raised the differential diagnosis of glomus tumor. In both cases 1 and 2, stromal hyalinization was focally extensive, resulting in hyalinized hypocellular zones within the neoplasm, a finding has not previously been reported. Fifth, while most GLI1-rearranged neoplasms are epithelioid and some show spindled cell features, the predominant/exclusive ovoid to short spindle phenotype of neoplasms 1 and 2 is also distinctly unusual and has not been well described. Finally, the eosinophilic spindle cell component of case 4, along with pleomorphism and patchy immunoreactivity for desmin, have not been described in GLI1-altered neoplasms and raised the differential diagnosis of myxoid leiomyosarcoma. The absence of immunoreactivity for caldesmon and actin argued against this possibility. Moreover, PLAG1 immunoreactivity is not specific for myxoid leiomyosarcoma29. Overall, the wider morphologic spectrum seen in our two GLI1-amplified tumors compared to the two GLI1-rearranged lesions is analogous to what has been observed in GLI1-altered soft tissue tumors13.
In summary, neoplasms with GLI1 gene rearrangement or amplification have a broad morphologic and immunohistochemical spectrum. The immunohistochemical profile is not specific, and reliance upon positive immunoreactivity for relatively nonspecific markers (such as cyclin D1, CD10, BCOR and estrogen receptor) can be misleading. The diagnosis of GLI1 associated neoplasia should be suspected in a neoplasm with nodular growth, myxoid stroma and bland epithelioid to spindled cells which does not fit an existing established diagnostic entity. Variant morphology may be seen, particularly in those neoplasms with GLI1 amplification. Molecular analysis of such cases can lead to the correct diagnosis. The correct classification may have clinical relevance, as GLI1 inhibitors are now approved by the US Food and Drug Administration and are currently the subject of clinical trials in a variety of different malignant neoplasms30.
Supplementary Material
Acknowledgement:
We thank Norman Barker MA, MS, for expert photographic assistance.
Disclosures: Supported in part by: P50 CA217694 (CRA), P50 CA 140146–01 (CRA), P30 CA 008748 (CRA); Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA), Dahan Translocation Carcinoma Fund and Joey’s Wings (PA)
REFERENCES
- 1.Timmis AJ, Riobo-Del Galdo NA. Another twist to the GLI code. Biochem J. 2020;477:4343–4347. [DOI] [PubMed] [Google Scholar]
- 2.Chetty R Gene of the month: GLI-1. J Clin Pathol. 2020;73:228–230. [DOI] [PubMed] [Google Scholar]
- 3.Katoh M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clin Sci (Lond). 2019;133:953–970. [DOI] [PubMed] [Google Scholar]
- 4.Dahlen A, Fletcher CD, Mertens F, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12). Am J Pathol. 2004;164:1645–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahlen A, Mertens F, Mandahl N, et al. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12). Biochem Biophys Res Commun. 2004;325:1318–1323. [DOI] [PubMed] [Google Scholar]
- 6.Bridge JA, Sanders K, Huang D, et al. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum Pathol. 2012;43:1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro E, Cortes-Santiago N, Ferguson LM, et al. Translocation t(7;12) as the sole chromosomal abnormality resulting in ACTB-GLI1 fusion in pediatric gastric pericytoma. Hum Pathol. 2016;53:137–141. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR, Agaram NP, Sung YS, et al. A Distinct Malignant Epithelioid Neoplasm With GLI1 Gene Rearrangements, Frequent S100 Protein Expression, and Metastatic Potential: Expanding the Spectrum of Pathologic Entities With ACTB/MALAT1/PTCH1-GLI1 Fusions. Am J Surg Pathol. 2018;42:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miettinen M, Makhlouf HR, Sobin LH, et al. Plexiform fibromyxoma: a distinctive benign gastric antral neoplasm not to be confused with a myxoid GIST. Am J Surg Pathol. 2009;33:1624–1632. [DOI] [PubMed] [Google Scholar]
- 10.Spans L, Fletcher CD, Antonescu CR, et al. Recurrent MALAT1-GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J Pathol. 2016;239:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham RP, Nair AA, Davila JI, et al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol. 2017;30:1443–1452. [DOI] [PubMed] [Google Scholar]
- 12.Xu B, Chang K, Folpe AL, et al. Head and Neck Mesenchymal Neoplasms With GLI1 Gene Alterations: A Pathologic Entity With Distinct Histologic Features and Potential for Distant Metastasis. Am J Surg Pathol. 2020;44:729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agaram NP, Zhang L, Sung YS, et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod Pathol. 2019;32:1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pettus JR, Kerr DA, Stan RV, et al. Primary myxoid and epithelioid mesenchymal tumor of the kidney with a novel GLI1-FOXO4 fusion. Genes Chromosomes Cancer. 2021;60:116–122. [DOI] [PubMed] [Google Scholar]
- 15.Koh NWC, Seow WY, Lee YT, et al. Pericytoma With t(7;12): The First Ovarian Case Reported and a Review of the Literature. Int J Gynecol Pathol. 2019;38:479–484. [DOI] [PubMed] [Google Scholar]
- 16.Kerr DA, Pinto A, Subhawong TK, et al. Pericytoma With t(7;12) and ACTB-GLI1 Fusion: Reevaluation of an Unusual Entity and its Relationship to the Spectrum of GLI1 Fusion-related Neoplasms. Am J Surg Pathol. 2019;43:1682–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith NE, Illei PB, Allaf M et al. t(6;11) renal cell carcinoma (RCC): expanded immunohistochemical profile emphasizing novel RCC markers and report of 10 new genetically confirmed cases. Am J Surg Pathol. 2014;38: :604–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Argani P, Reuter VE, Kapur P, et al. Novel MEIS1-NCOA2 Gene Fusions Define a Distinct Primitive Spindle Cell Sarcoma of the Kidney. Am J Surg Pathol. 2018;42:1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niewiadomski P, Niedziolka SM, Markiewicz L, et al. Gli Proteins: Regulation in Development and Cancer. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosquera JM, Sboner A, Zhang L, et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer. 2013;52:1075–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung YP, Fletcher CDM. Myopericytomatosis: Clinicopathologic Analysis of 11 Cases With Molecular Identification of Recurrent PDGFRB Alterations in Myopericytomatosis and Myopericytoma. Am J Surg Pathol. 2017;41:1034–1044. [DOI] [PubMed] [Google Scholar]
- 24.Miettinen M. Smooth muscle tumors of soft tissue and non-uterine viscera: biology and prognosis. Mod Pathol. 2014;27 Suppl 1:S17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rakha EA, Quinn CM, Foschini MP, et al. Metaplastic carcinomas of the breast without evidence of epithelial differentiation: a diagnostic approach for management. Histopathology. 2021;78:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter MR, Hornick JL, Lester S, et al. Spindle cell (sarcomatoid) carcinoma of the breast: a clinicopathologic and immunohistochemical analysis of 29 cases. Am J Surg Pathol. 2006;30:300–309. [DOI] [PubMed] [Google Scholar]
- 27.Kao YC, Sung YS, Zhang L, et al. BCOR Overexpression Is a Highly Sensitive Marker in Round Cell Sarcomas With BCOR Genetic Abnormalities. Am J Surg Pathol. 2016;40:1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brahmi M, Franceschi T, Treilleux I, Pissaloux D, Ray-Coquard I, Dufresne A, Vanacker H, Carbonnaux M, Meeus P, Sunyach MP, Bouhamama A, Karanian M, Meurgey A, Blay JY, Tirode F. Molecular Classification of Endometrial Stromal Sarcomas Using RNA Sequencing Defines Nosological and Prognostic Subgroups with Different Natural History. Cancers (Basel). 2020;12:2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arias-Stella JA 3rd, Benayed R, Oliva E, et al. Novel PLAG1 Gene Rearrangement Distinguishes a Subset of Uterine Myxoid Leiomyosarcoma From Other Uterine Myxoid Mesenchymal Tumors Am J Surg Pathol. 2019. 43:382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dusek CO, Hadden MK. Targeting the GLI family of transcription factors for the development of anti-cancer drugs. Expert Opin Drug Discov. 2021;16:289–302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.