

Abstract
Background:
MicroRNA-150 (miR-150) plays a protective role in heart failure (HF). Long noncoding RNA (ncRNA), Myocardial Infarction-Associated Transcript (MIAT) regulates miR-150 function in vitro by direct interaction. Concurrent with miR-150 downregulation, MIAT is upregulated in failing hearts, and gain-of-function single nucleotide polymorphisms in MIAT are associated with increased risk of MI in humans. Despite the correlative relationship between MIAT and miR-150 in HF, their in vivo functional relationship has never been established, and molecular mechanisms by which these two ncRNAs regulate cardiac protection remain elusive.
Methods:
We use MIAT knockout (KO), homeobox a4 (Hoxa4) KO, MIAT transgenic (TG) and miR-150 TG mice. We also develop double transgenic (DTG) mice overexpressing MIAT and miR-150. We then employ a mouse model of MI followed by cardiac functional, structural and mechanistic studies by echocardiography, immunohistochemistry, transcriptome profiling, Western blotting and quantitative real-time RT-PCR. Moreover, we perform expression analyses in hearts from patients with HF. Lastly, we investigate cardiac fibroblast (CF) activation using primary adult human CFs and in vitro assays to define the conserved MIAT/miR-150/HOXA4 axis.
Results:
Using novel mouse models, we demonstrate that genetic overexpression of MIAT worsens cardiac remodeling, while genetic deletion of MIAT protects hearts against MI. Importantly, miR-150 overexpression attenuates the detrimental post-MI effects caused by MIAT. Genome-wide transcriptomic analysis of MIAT null mouse hearts identifies Hoxa4 as a novel downstream target of the MIAT/miR-150 axis. Hoxa4 is upregulated in CFs isolated from ischemic myocardium and subjected to hypoxia/reoxygenation. HOXA4 is also upregulated in patients with HF. Moreover, Hoxa4 deficiency in mice protects the heart from MI. Lastly, protective actions of CF miR-150 are partially attributed to the direct and functional repression of pro-fibrotic Hoxa4.
Conclusions:
Our findings delineate a pivotal functional interaction among MIAT, miR-150 and Hoxa4 as a novel regulatory mechanism pertinent to ischemic HF.
Keywords: Cardiac fibroblast gene regulation, carvedilol, homeobox A4, noncoding RNA
Classification: Basic-Cardiac Biology and Remodeling, Noncoding RNA Biology, Receptor Signaling & Cell Biology, Pharmacology
Introduction
Controlling microRNA (miR) biogenesis in the heart is an important underlying mechanism of heart failure (HF)1–5. Intriguingly, novel miR therapies are being used in clinical trials for other diseases6–9 and more recently for HF10. Our previous study revealed that miR-150–5p (hereinafter referred to as miR-150) is upregulated by the β-adrenergic receptor (βAR) antagonist (β-blocker), carvedilol (Carv) acting through the β-arrestin1-biased β1AR cardioprotective signaling11. Using a global miR-150 knockout (KO) mouse model, we also reported that β1AR/β-arrestin1-responsive miR-150 confers cardiac protection against myocardial infarction (MI)12. Interestingly, miR-150 is downregulated in patients with multiple cardiovascular diseases (CVDs) such as acute MI (AMI), atrial fibrillation, dilated cardiomyopathy and ischemic cardiomyopathy13–16, as well as in multiple mouse models of HF (MI, transverse aortic constriction [TAC] and ischemia/reperfusion [I/R] injury)12, 17, 18. MiR-150 is conserved between mice and humans, and significantly associated with HF severity and outcomes in humans19. Collectively, previous studies established the strong clinical relevance and potential diagnostic/prognostic/therapeutic application of miR-150 as a key mediator of cardiac protection. However, the upstream regulators controlling miR-150 have not been well elucidated. Also, there is a lack of mechanistic insight by which miR-150 confers cardiac protection.
Long noncoding RNAs (LncRNAs) have recently emerged as important regulators of cardiac development and disease20, 21. LncRNA therapeutics are accordingly under development by pharmaceutical companies22–25. Emerging evidence also suggests that the crosstalk among lncRNAs, miRs, and messenger RNAs (mRNAs) represents a novel regulatory mechanism underlying the pathogenesis of CVDs. Specifically, lncRNAs act as competing endogenous RNAs (ceRNAs) that sponge miRs, thereby activating target genes of miRs26. It was reported that MIAT (also known as Gomafu27), an intergenic and highly conserved lncRNA, directly interacted with miR-150 in vitro (evident by luciferase assays as well as RNA pull-down and immunoprecipitation assays) and functioned as a ceRNA for miR-150 in ocular cells28, 29. Interestingly, previous studies uncovered the association between gain-of-function single nucleotide polymorphisms (SNPs) in MIAT and an increased risk of MI30, 31. MIAT upregulation was found in patients with Chagas cardiomyopathy32 and directly associated with maladaptive cardiac remodeling in patients with type 2 diabetes33. Rodent studies also showed that MIAT was upregulated in mouse models of MI, angiotensin II (AngII)- or isoproterenol (ISO)-induced cardiac hypertrophy, and diabetic cardiomyopathy, as well as in cardiomyocytes (CMs) or cardiac fibroblasts (CFs) subjected to AngII, ISO, high glucose, or simulated ischemia/reperfusion (sI/R)34–38. Lentivirus-mediated knockdown of MIAT improved cardiac function and structure in post-MI36 and diabetic hearts34, as well as reduced I/R-induced myocardial infarct size and apoptosis35. In cultured CMs, MIAT knockdown inhibited CM apoptosis against high glucose or sI/R34, 35, and MIAT positively regulated CM hypertrophy in vitro in part by suppressing miR-150 expression37, 38. Despite these increasing data from previous correlative studies, direct evidence firmly establishing in vivo functional relationship between MIAT and miR-150 in HF is lacking, and mechanisms by which MIAT mediates cardiac pathology have not been adequately defined using appropriate mouse models.
Homeobox a4 (HOXA4) is a transcription factor and highly conserved in vertebrates. Previous studies suggested that Hoxa4 is involved in patterning embryonic mouse lungs39 and early chick hearts40. Accumulated evidences also indicated the overexpression of this development-associated gene in colorectal and ovarian cancers41, 42. Knockdown of a myelopoiesis-associated regulatory lncRNA (HOTAIRM1), which is expressed specifically in the myeloid lineage, was shown to inhibit retinoic acid-induced expression of Hoxa4 during the myeloid differentiation in vitro43. However, little is known about regulatory mechanisms of Hoxa4 by ncRNAs. Especially, whether Hoxa4 is functionally regulated by the Carv-responsive miR-150, as well as contributes to maladaptive cardiac cell function and cardiac pathology are unknown.
Using novel mouse models, an unbiased genome-wide profiling, and primary human CFs (HCFs), we demonstrate here that (i) genetic overexpression of MIAT exacerbates maladaptive post-MI remodeling, while global genetic deletion of MIAT in mice protects hearts against MI; (ii) miR-150 overexpression attenuates maladaptive post-MI remodeling caused by overexpression of MIAT; (iii) Hoxa4 is a novel downstream target of the MIAT/miR-150 axis; (iv) Hoxa4 loss protects the mouse heart against MI; (v) the expression of Hoxa4 is upregulated selectively in CFs isolated from ischemic myocardium; (vi) HOXA4 is upregulated in patients with HF with reduced ejection fraction (HFrEF), but its expression is downregulated in mouse hearts and HCFs by Carv, which is inversely associated with the expression of miR-150; and (vii) the protective action of miR-150 is mediated by the direct and functional repression of pro-fibrotic HOXA4 in HCFs. Therefore, the MIAT/miR-150/Hoxa4 axis may be considered as a novel therapeutic option for ischemic heart disease.
Methods
Availability of data and materials
The microarray data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series access number GSE185396. All other data that are not included in this publication, analytical methods and study materials will be made available to other researchers for purposes of reproducing results or replicating procedures. Other methods are provided in Supplemental Material.
MiR-150 knockout (KO), MIAT KO and transgenic (TG) mice, and generation of MIAT/miR-150 double TG (DTG) mice
Systemic miR-150 KO mice were purchased from the Jackson Laboratory (007750). MIAT/Gomafu KO mice lacking PGK-Neomycin cassette were generated by crossing the Gomafu KO mice described in Ip et al.44 with a transgenic mouse expressing Flp recombinase. CAG promoter-driven MIAT TG mice were generated by injecting pCAG-Gomafu27 into the pronuclei of fertilized mouse eggs using standard methods. For genotyping, following primers were used (MIAT_TG_Forward: TACAGCTCCTGGGCAACGTGCTGGTTA, MIAT_TG_Reverse: ACACGAACAAGCACCCATCT, MIAT_KO_Forward: GCCTCTCCACTGGCCAGCGT, and MIAT_KO_Reverse: CCAATCTTGGCTCACCAGCAACTC). A CAG promoter-driven miR-150 TG mouse line45 was kindly provided by Dr. Jennifer Richer at University of Colorado, and was then bred to MIAT TG mice to generate the novel MIAT/miR-150 DTG mouse line. Mice were maintained on a C57BL/6J background, and wild type (WT) littermates were used as controls.
Hoxa4 mutant mouse strain
Hoxa4 heterozygous mutant mice were obtained from the Mutant Mouse Resource & Research Centers (RRID: MMRRC_067050-UCD). This mutant mouse line was generated using the CRISPR/Cas9 strategy. In brief, CRISPR guide(s) and the Cas9 protein were microinjected into C57BL/6NCrl zygotes and progeny were screened for the desired mutation. Founders were mated to C57BL/6NCrl breeders, and derived F1 offspring were identified by PCR and sequencing. F1 mice were then mated again to C57BL/6NCrl breeders to generate F2 mice, which were identified by PCR and sequencing. The CRISPR-mediated genetic alteration resulted in the deletion of exon 1, the coding region of exon 2 and flanking splicing regions from the Hoxa4 gene locus. The obtained Hoxa4 heterozygous mutant mice were bred to generate Hoxa4 KO mice and WT littermates for the current study. For genotyping, the following primers were used. WT Forward: 5’- GAA AGC ACA AAC TCA CAG CCC ACA C −3’, WT Reverse: 5’- TTT GGT GTC TCG GGT TTA CTT AGG −3’, KO Forward: 5’- AGC CTG GTT GGA CTG GAG ATC G −3’, and KO Reverse: 5’- CAC ACC TAC CAT CAA GGT CTA CAC ACT −3’.
Human heart samples
Left ventricle (LV) samples of failing human hearts were collected from ischemic cardiomyopathy and non-ischemic cardiomyopathy patients, who were hospitalized with heart failure with reduced ejection fraction (HFrEF) and underwent orthotopic cardiac transplantation as previously described46. LV tissues were dissected and snap-frozen in liquid nitrogen. The frozen samples were then stored in the specimen storage facility at the Indiana Clinical and Translational Sciences Institute located at Indiana University. Non-failing LV tissues were obtained from donor hearts not suitable for transplantation, as well as were collected and stored in the same manner. Demographic characteristics of these LV tissue samples are provided in Supplemental Table I.
Ethics committee approval
The use of animals in this study was conformed to the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health in USA. Animals were euthanized by thoracotomy under 1–4% inhalant isoflurane. All animal experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee at Indiana University (approval reference #19018). Eight to sixteen-week-old C57BL/6J mice of both genders were used. One dose of buprenorphine SR Lab (0.05mg/kg; ZooPharm) was given subcutaneously immediately before the surgery. We used responses to toe/skin pinch and heart rate for optimal anesthesia and appropriate post-operative monitoring plans. All of the procedures involving human samples were conformed to the principles outlined in the Declaration of Helsinki and the Guidelines for the Health Insurance Portability and Accountability Act (HIPAA), as well as approved by the Indiana University Institutional Review Board (approval reference #08–018). All participants provided informed written consent prior to inclusion in the study.
Statistics
Data are shown as mean ± SEM (unless noted otherwise in the figure legend) from independent experiments with different biological samples per group. Normality was assessed with the Kolmogorov-Smirnov test. Statistical significance was determined by unpaired 2-tailed t-test for comparisons between 2 groups, 1-way ANOVA with Tukey multiple comparison test for multiple groups, 2-way ANOVA with Tukey multiple comparison test for comparisons between 2 groups with different treatments, and 2-way repeated-measures ANOVA with Bonferroni post hoc test for 2 groups over time. A P value <0.05 was considered statistically significant. P values are indicated as follows: *, # or §P<0.05, **, ## or §§P<0.01, and ***, ### or §§§P<0.001.
Results
MIAT deletion in mice attenuates cardiac dysfunction and remodeling post-MI
To define in vivo roles of MIAT in cardiac injury, we first conducted the permanent ligation of LAD artery in mice to induce MI. In consistent with a previous report in the heart at 1, 2 and 4 weeks post-MI36, we observe cardiac upregulation of MIAT in WT mice subjected to 4 weeks of MI (Figure 1A), concurrent with miR-150 downregulation12. We next examined the expression of MIAT in different cardiac cell types, and we find that its expression at baseline is significantly higher in cardiac inflammatory cells (CIs) and CFs than CMs and cardiac endothelial cells (CEs). Interestingly, MIAT expression is upregulated only in CMs and CFs isolated from ischemic myocardium (Figure 1B). We then examined MIAT KO mice and show that MIAT KO mice exhibit normal cardiac function at baseline (Supplemental Table II and Figure 1C–D) but respond differently to ischemic cardiac injury. Despite the normal cardiac function of MIAT KO mice at post-MI 1 week (Supplemental Table III), MI results in a significant improvement of cardiac function at 2 weeks (Supplemental Table IV) and 4 weeks (Figure 1C–D and Supplemental Table V), which are indicated by increased EF and FS as compared to WT controls.
Figure 1. Myocardial Infarction-Associated Transcript (MIAT) deletion attenuates, while MIAT overexpression exacerbates cardiac dysfunction after myocardial infarction (MI) in part by repressing miR-150.
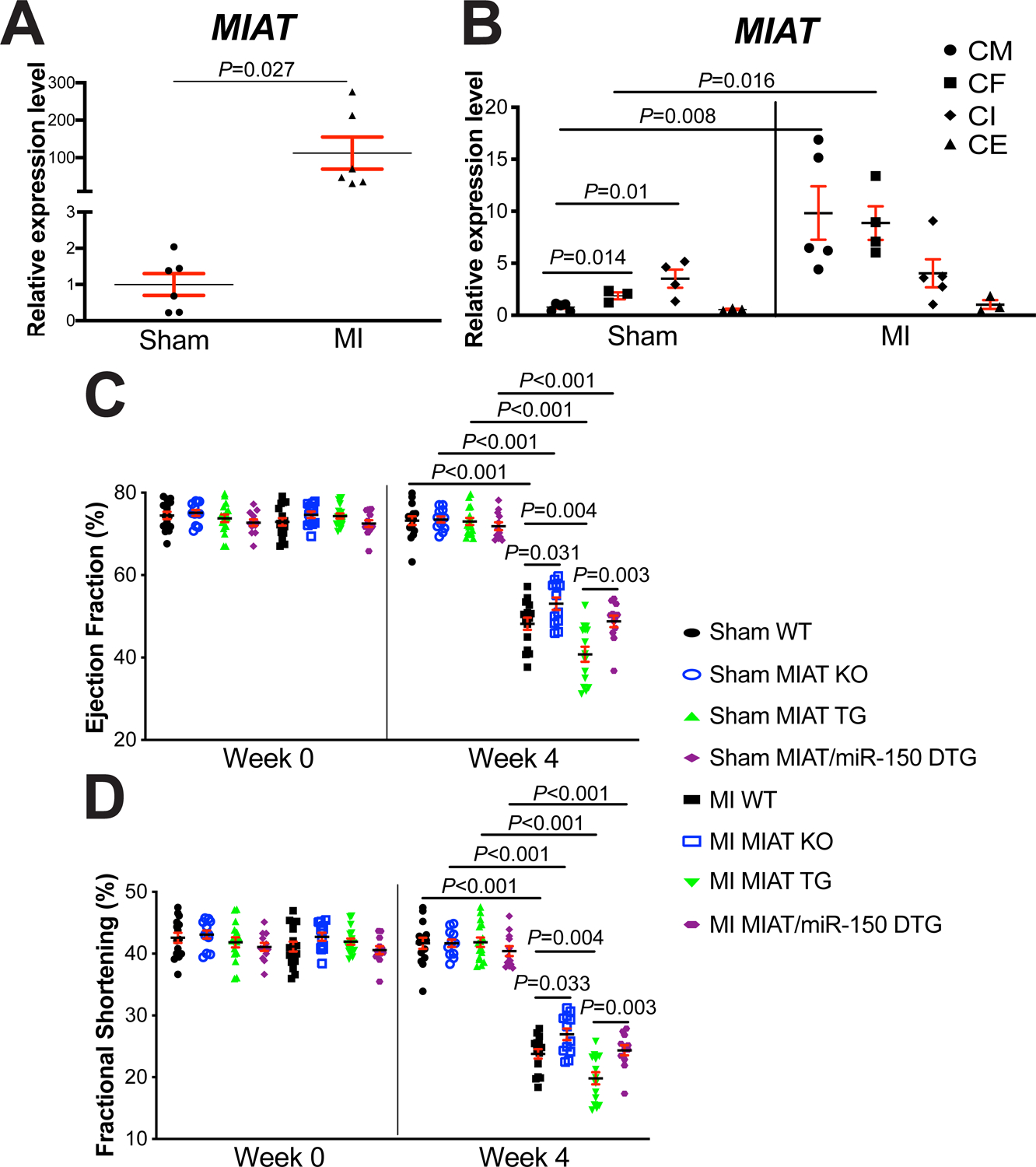
A, The expression of MIAT in left ventricles (LVs) from wild type (WT) adult mice at 4 weeks post-surgery (N=6). Data are shown as fold induction of MIAT expression normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Unpaired 2-tailed t-test. B, Real-Time Quantitative Reverse Transcription (QRT)-PCR expression analysis of MIAT in cardiomyocytes (CMs), cardiac fibroblasts (CFs), cardiac inflammatory cells (CIs) and cardiac endothelial cells (CEs) isolated from adult mouse hearts at 7 days post-MI. N=3–5 per group. MIAT expression compared to Gapdh was calculated using 2−ΔΔCt, and data are shown as fold induction of MIAT expression levels normalized to CM sham. Two-way ANOVA with Tukey multiple comparison test. C-D, Transthoracic echocardiography was performed to 8 experimental groups [sham and MI of WT, MIAT knockout (KO), MIAT transgenic (TG) and MIAT/miR-150 double TG (DTG)] at week 0 and 4 post-MI. Quantification of LV ejection fraction (C) and fractional shortening (D) is shown. N=12–17 per group. Two-way repeated-measures ANOVA with Bonferroni post hoc test.
We also find that MIAT KO MI hearts exhibit a decrease in loss of normal architecture and cellular integrity (Figure 2A), which is consistent with decreased mRNA levels of fetal Nppa (Supplemental Figure IA) after 4 weeks of MI compared to WT MI hearts. To further determine the response of MIAT KO mice to MI, we assessed the degree of fibrosis by Masson’s trichrome staining of the hearts at 4 weeks post-MI. We observe that MIAT KO hearts contain significantly smaller fibrotic regions than WT hearts post-MI (Figure 2B–C and Supplemental Figure II). MIAT KO MI hearts also exhibit decreased expression of fibrotic Ctgf (Supplemental Figure IB) compared to WT controls. Lastly, we examined evidence of apoptosis, and we find that MIAT KO hearts display decreased numbers of TUNEL-positive cells post-MI (Supplemental Figure IIIA–B) and mRNA levels of apoptotic Bak1 (Supplemental Figure IIIC) compared to WT hearts. Collectively, these results indicate for the first time that genetic deletion of MIAT in mice improves cardiac structural and functional abnormalities associated with post-MI remodeling.
Figure 2. Myocardial Infarction-Associated Transcript (MIAT) deletion decreases, while MIAT overexpression increases cardiac stress and fibrosis post-myocardial infarction (MI) in part by repressing miR-150.
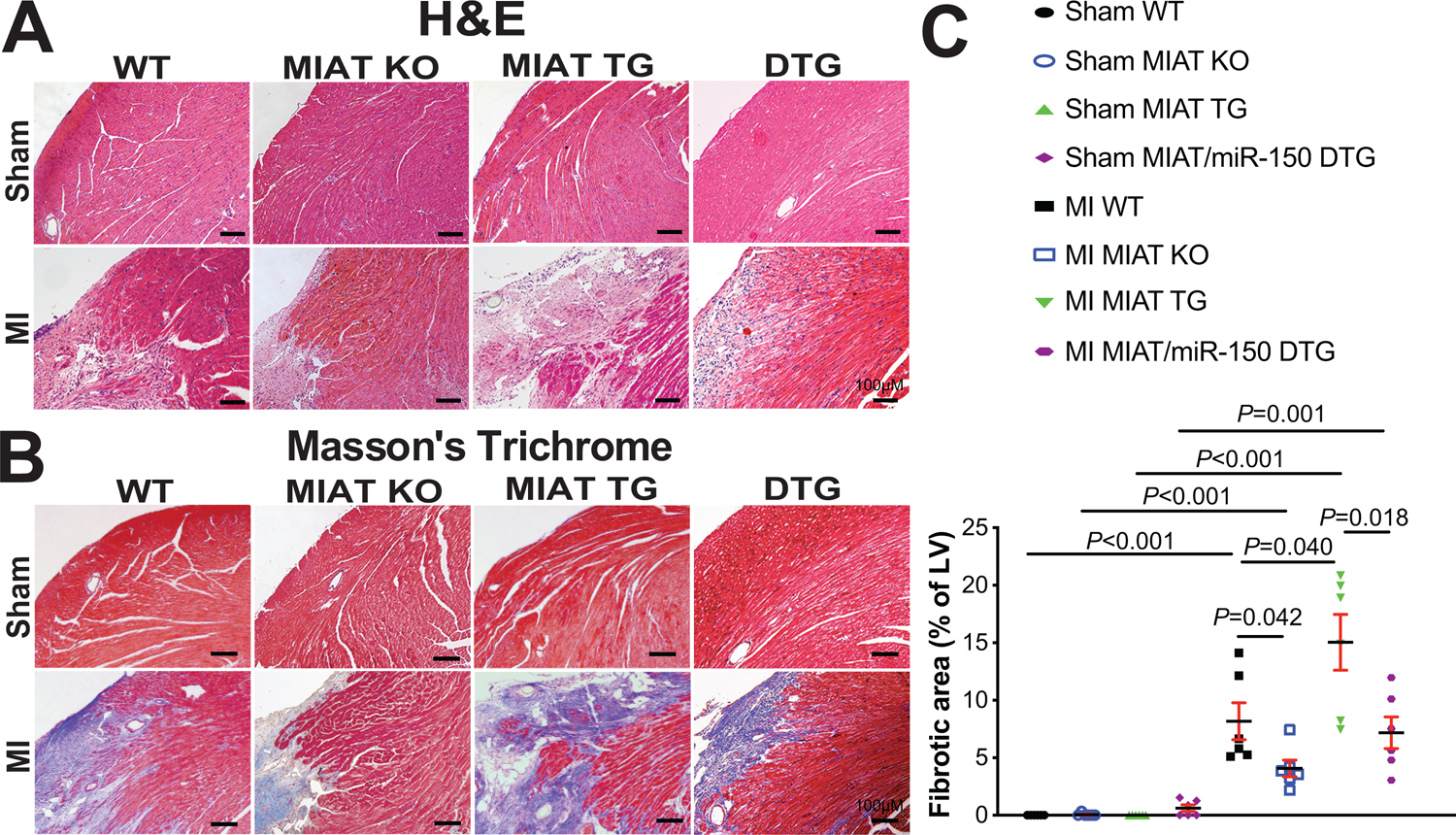
A, Representative hematoxylin and eosin (H&E) images in heart sections of peri-ischemic border area from 8 experimental groups at 4 weeks post-MI. Scale bars: 100μm. B-C, Representative Masson’s trichrome images in heart sections of peri-ischemic border area in 8 experimental groups at 4 weeks post-MI [B] and fibrosis quantification in whole left ventricles (LVs) [C]. Scale bars: 100μm. N=6 per group. Two-way ANOVA with Tukey multiple comparison test.
Genetic overexpression of MIAT exacerbates maladaptive post-MI remodeling
To test if MIAT augments ischemic cardiac injury, we next subjected MIAT TG mice to MI. We first observe that cardiac function of MIAT TG mice is indistinguishable from WT controls at baseline (Supplemental Table II and Figure 1C–D). However, MI significantly worsens cardiac function of MIAT TG mice at 1 week, which is indicated by decreased EF and FS, as well as an increase in end-diastolic volume (EDV), end-systolic volume (ESV) and systolic left ventricular interior diameter [LVID] (Supplemental Table III). MIAT TG mice also display impaired cardiac function at 2 weeks post-MI, which is shown by a significant decrease in EF, FS and systolic left ventricular anterior wall thickness (LVAW), as well as a significant increase in ESV, diastolic LVID and systolic LVID (Supplemental Table IV). MI also causes augmented cardiac dysfunction in MIAT TG mice at 4 weeks, as evidenced by a significant decrease in EF, FS and diastolic LVAW, as well as a significant increase in EDV, ESV, diastolic LVID and systolic LVID (Figure 1C–D and Supplemental Table V). In contrast, WT controls show less functional impairment at 1 week (Supplemental Table III), 2 weeks (Supplemental Table IV) and 4 weeks following MI (Figure 1C–D and Supplemental Table V). Notably, MIAT TG mice have no increase in post-MI mortality (Supplemental Table II–V: see n for animal numbers per each group at Week 0, Week 1, Week 2 and Week 4 after MI. N=2 for MIAT TG and WT are died within 1 week after MI).
We also find that MIAT TG hearts exhibit an increase in loss of normal architecture and cellular integrity post-MI (Figure 2A), as well as increased expression levels of fetal Nppb and inflammatory Tnf-α (Supplemental Figure IVA–B) after 4 weeks of MI compared to WT MI hearts. Using Masson’s trichrome staining of the hearts at 4 weeks post-MI, we observe small regions of fibrosis in WT hearts, while MIAT TG hearts have significantly larger fibrotic areas (Figure 2B–C and Supplemental Figure II). Lastly, we show that MIAT TG MI hearts contain higher numbers of TUNEL-positive cells (Supplemental Figure IIIA–B) and increased mRNA levels of apoptotic Bak1 (Supplemental Figure IIIC) compared to WT MI hearts. Taken together, our data using a novel mouse model suggest that MIAT overexpression significantly augments cardiac dysfunction, as well as stress, fibrosis, and apoptosis in the heart after MI.
MiR-150 overexpression attenuates excessive maladaptive post-MI remodeling mediated by MIAT overexpression
Although MIAT and miR-150 are associated with HF in humans19, 30, 31 and their correlative relationship in CMs in vitro is shown37, 38, in vivo functional relationship between these two ncRNAs in the heart has not been established. We first observe that cardiac miR-150 is upregulated in MIAT KO mice (Supplemental Figure VA–B), while both miR-150 loss and miR-150 overexpression do not affect the cardiac expression of MIAT (Supplemental Figure VC–D). We also confirm that miR-150 is downregulated in MIAT TG mouse hearts (Supplemental Figure VIA–B), and two known direct targets of miR-150, egr2 and p2×7r12, are upregulated in MIAT TG mouse hearts (Supplemental Figure VIC–D). These results suggest that MIAT suppresses miR-150 in mouse hearts, presumably acting through a target RNA‐directed miR decay mechanism with multiple binding sites47. This notion is supported by the facts that MIAT has three potential binding sites of miR-15037, and that MIAT directly interacts with miR-150 in vitro evident by luciferase assays as well as RNA pull-down and immunoprecipitation assays28, 48. Moreover, mouse and human MIAT genes have almost identical genomic organization, indicating evolutionary conservation of MIAT’s regulation of miR-150 and their roles in both species.
To directly investigate in vivo functional interaction between MIAT and miR-150 in the heart, we generated a novel MIAT/miR-150 DTG mouse line by breeding MIAT TG mice with miR-150 TG mice. We first show that MIAT/miR-150 DTG mouse hearts are functionally normal at baseline (Supplemental Table II and Figure 1C–D). However, a significant improvement of cardiac function at 1 week after MI is observed in MIAT/miR-150 DTG mice compared to MIAT TG mice, which is indicated by an increase in EF and FS, as well as a decrease in EDV, ESV, diastolic LVID and systolic LVID (Supplemental Table III). MIAT/miR-150 DTG mice also display enhanced cardiac function at 2 weeks post-MI, as evidenced by a significant increase in EF, FS, and systolic LVAW, as well as a significant decrease in ESV and systolic LVID (Supplemental Table IV) as compared to MIAT TG mice. MI also causes improved cardiac function in MIAT/miR-150 DTG mice at 4 weeks compared to MIAT TG mice, as shown by a significant increase in EF, FS and systolic LVAW, as well as a significant decrease in EDV, ESV, diastolic LVID and systolic LVID (Figure 1C–D and Supplemental Table V).
MIAT/miR-150 DTG hearts also display a decrease in loss of normal architecture and cellular integrity (Figure 2A), as well as decreased mRNA levels of fetal Nppb (Supplemental Figure IVA) after 4 weeks of MI compared to MIAT TG hearts. The expression of inflammatory Tnf-α is also reduced in MIAT/miR-150 DTG hearts (Supplemental Figure IVB), as compared to MIAT TG hearts. We next observe reduced fibrosis post-MI in MIAT/miR-150 DTG hearts compared to MIAT TG hearts (Figure 2B–C and Supplemental Figure II). Lastly, MIAT/miR-150 TG hearts contain lower numbers of TUNEL-positive cells post-MI (Supplemental Figure IIIA–B) and decreased mRNA levels of apoptotic Bak1 (Supplemental Figure IIIC) compared to MIAT TG hearts. Altogether, our data suggest that miR-150 overexpression ameliorates maladaptive post-MI remodeling caused by overexpression of MIAT, and that MIAT is a functionally important upstream negative regulator of miR-150 in the heart, thereby competitively sequestering miR-150 from its targets.
MIAT regulates the expression of a subset of cardiac genes post-MI involved in dilated cardiomyopathy, hypertrophic cardiomyopathy, renin-angiotensin system and extracellular matrix-receptor interaction
Although MIAT and miR-150 are known to be involved in HF19, 30, 31, the detailed mechanisms of their actions remain elusive. To investigate how cardiac MIAT exerts its function, we performed transcriptome profiling of LVs from WT and MIAT KO mice subjected to sham or MI for 4 weeks. Among 24,881 mouse genes that we profiled, 304 genes are significantly upregulated (Supplemental Figure VIIA and Supplemental Table VI; see up_Group2 vs Group1 sheet), and 283 genes are significantly downregulated (Supplemental Figure VIIA and Supplemental Table VI; see down_Group2 vs Group1 sheet) in sham MIAT KO compared to sham WT. We also find that 1,825 genes are significantly upregulated (Supplemental Figure VIIB and Supplemental Table VI; see up_Group3 vs Group1 sheet), and 1,517 genes are significantly downregulated (Supplemental Figure VIIB and Supplemental Table VI; see down_Group3 vs Group1 sheet) in MI WT compared to sham WT. In addition, 90 genes are significantly upregulated, and 78 genes are significantly downregulated (Supplemental Figure VIIC) in MI MIAT KO compared to sham MIAT KO. Lastly, we observe that 1,108 genes are significantly upregulated (Supplemental Figure VIID and Supplemental Table VI; see up_Group4 vs Group3 sheet), and 1,233 genes are significantly downregulated (Supplemental Figure VIID and Supplemental Table VI; see down_Group4 vs Group3 sheet) in MI MIAT KO compared to MI WT.
To discover the functional roles of differentially regulated genes in MIAT KO, we then classified differentially expressed genes by the signaling pathway classification system of Kyoto Encyclopedia of Genes and Genomes (KEGG). Our signaling pathway analysis in sham MIAT KO compared to sham WT demonstrates that upregulated genes are involved in extracellular matrix (ECM)-receptor interaction, NF-κB signaling pathway, cytokine-cytokine receptor interaction, and thyroid hormone signaling pathway (Supplemental Figure VIIIA). In contrast, downregulated genes are related to leukocyte trans-endothelial migration, adrenergic signaling in CMs, cAMP signaling pathway, and MAPK signaling pathway (Supplemental Figure IXA). We also find that the top canonical signaling pathways for upregulated genes in MI WT compared to sham WT include chemokine signaling pathway, viral myocarditis, and relaxin signaling pathway (Supplemental Figure VIIIB). In contrast, downregulated genes are involved in signaling pathways regulating carbon metabolism, propanoate metabolism, TCA cycle, glyoxylate and dicarboxylate metabolism, and arrhythmogenic right ventricular cardiomyopathy [ARVC] (Supplemental Figure IXB). Moreover, the expression of genes involved in ECM-receptor interaction, focal adhesion, and AGE-RAGE signaling pathway are significantly upregulated (Supplemental Figure VIIIC), while the expression of genes involved in glycolysis/gluconeogenesis, synthesis and degradation of ketone bodies, metabolism of xenobiotics by cytochrome P450, drug metabolism, and butanoate metabolism are significantly downregulated (Supplemental Figure IXC) in MI MIAT KO compared to sham MIAT KO. Lastly, we observe that genes involved in dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM) and renin-angiotensin system are significantly increased (Supplemental Figure VIIID) in MI MIAT KO compared to MI WT. In contrast, genes involved in relaxin signaling pathway, focal adhesion, renin secretion, ECM-receptor interaction and gap junction are significantly decreased (Supplemental Figure IXD). Overall, our transcriptomic data suggest that MIAT regulates the expression of a subset of genes/signaling pathways in the heart to increase maladaptive cardiac remodeling.
MIAT activates Hoxa4, Fmo2, Lrrn4, Marveld3 and Fat4 in mouse hearts
To identify novel MIAT targets that promote cardiac pathology and impair myocardial cell responses, we filtered 21 significantly dysregulated genes from our array dataset based on the correlation between genotypes and transcript signatures (Figure 3A–B, I), as well as the correlation between cardiac phenotypes shown in Figure 1–2 and transcript signatures from MI WT vs. Sham WT (Figure 3A–B, II) or MI MIAT KO vs. MI WT (Figure 3A–B, III). The rationale to focus on these 18 potentially maladaptive genes (Ces1f, Cdh19, Seh1l, Postn, Echdc1, Ctla2b, Luc7i, Fmo2, Hoxa4, Nap1l1, Sgce, Ifih1, Prpf31, Lrrn4, Marveld3, Itsn1, Ergic2, and Fat4) is that they are downregulated in both sham MIAT KO vs. Sham WT (i.e., genotype effects) and MI MIAT KO vs. MI WT (i.e., phenotypic effects of KO), but upregulated in MI WT vs. sham WT (i.e., MI effects) (Figure 3A–C). The rationale to focus on these 3 potentially beneficial genes (Chn2, Sycp1, and Cbfa2t3) is that they are upregulated in both sham MIAT KO vs. Sham WT (i.e., genotype effects) and MI MIAT KO vs. MI WT (i.e., phenotypic effects of KO), but downregulated in MI WT vs. sham WT (i.e., MI effects) (Figure 3A–C).
Figure 3. Transcriptome profiling in Myocardial Infarction-Associated Transcript (MIAT) knockout (KO) mice identifies novel target genes of cardiac MIAT.
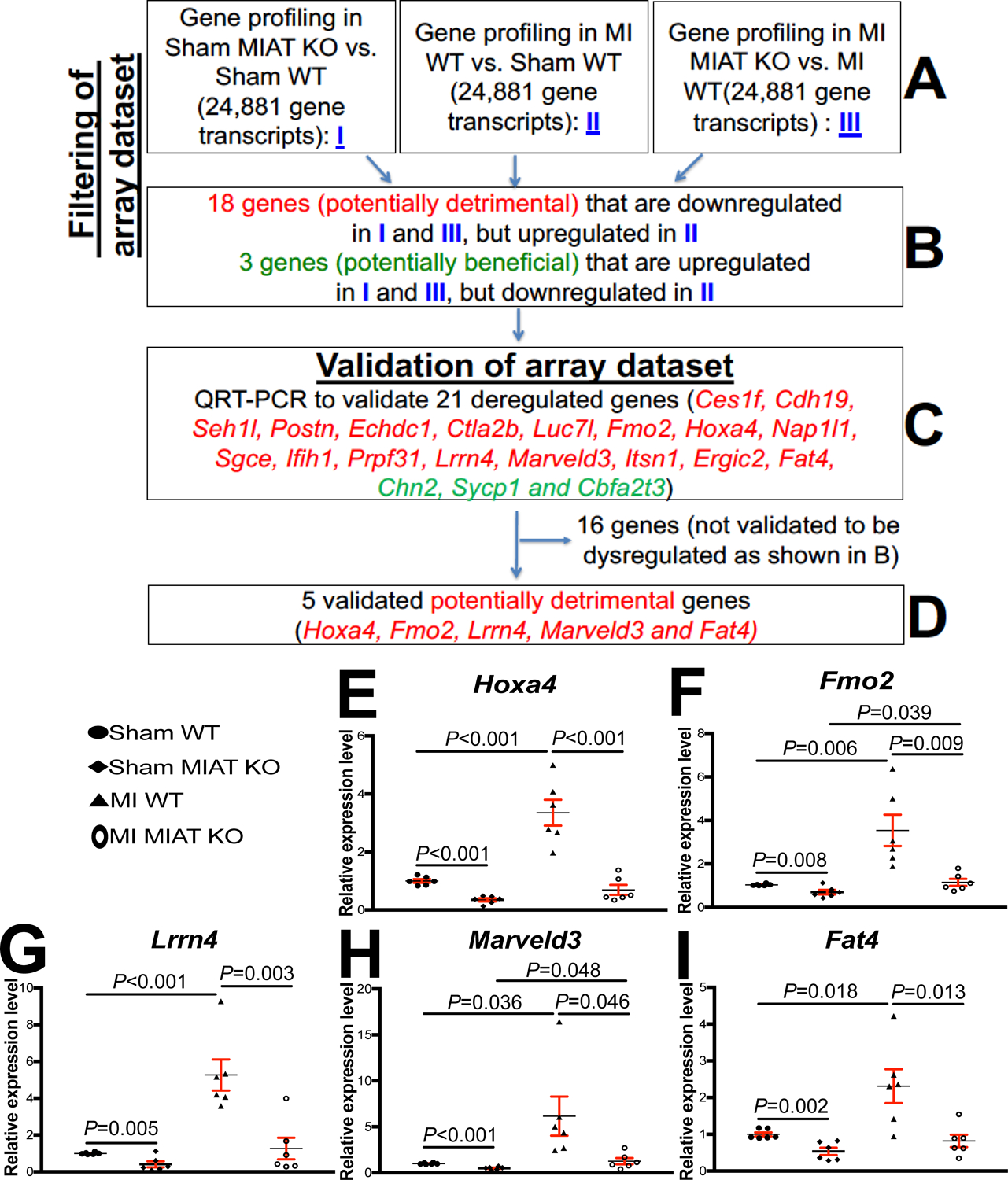
A-B, Filtering strategy of array dataset based on the correlation between genotypes and transcript signatures (I) or between cardiac phenotypes and transcript signatures (II and III). Eighteen genes, which are downregulated in both I (sham MIAT KO compared to sham wild type [WT] controls) and III (myocardial infarction [MI] MIAT KO compared to MI WT) but are upregulated in II (MI WT compared to Sham WT) at 4 weeks post-MI, were chosen for further analyses. Three genes, which are upregulated in both I (sham MIAT KO compared to sham WT controls) and III (MI MIAT KO compared to MI WT) but are downregulated in II (MI WT compared to Sham WT) at 4 weeks post-MI, were also chosen for further analyses. C-D, Validation strategy of array dataset. Twenty-one dysregulated (DE) genes were validated by Real-Time Quantitative Reverse Transcription (QRT)-PCR analyses. Note that other sixteen genes are not validated to be dysregulated as shown B. E-I, The expression of five potentially detrimental genes [homeobox a4 (Hoxa4: E), flavin-containing dimethylaniline monoxygenase 2 (Fmo2: F), leucine-rich repeat neuronal 4 (Lrrn4: G), MARVEL domain containing 3 (Marveld3: H) and FAT atypical cadherin 4 (Fat4: I)] in left ventricles (LVs) from WT and MIAT KO mice at 4 weeks post-MI. Data are shown as fold induction of gene expression normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). N=6 per group. Two-way ANOVA with Tukey multiple comparison test.
Using real-time PCR analyses to validate the expression of the 21 filtered genes, we find that homeobox a4 (Hoxa4), flavin-containing dimethylaniline monooxygenase 2 (Fmo2), leucine-rich repeat neuronal 4 (Lrrn4), MARVEL domain containing 3 (Marveld3) and FAT atypical cadherin 4 (Fat4) in LVs are upregulated after MI. However, the 5 genes are downregulated in MIAT KO compared to WT and in MI MIAT KO compared to MI WT controls (Figure 3D–I). Notably, the expression of these 5 genes is upregulated in post-MI hearts (Figure 3E–I), concurrent with miR-150 downregulation12 and MIAT upregulation in the heart (Figure 1A). Thus, our transcript validation analyses suggest that Hoxa4, Fmo2, Lrrn4, Marveld3 and Fat4 are novel targets of MIAT post-MI.
MiR-150 directly represses a novel target, Hoxa4
Given that MIAT upregulates Hoxa4, Fmo2, Lrrn4, Marveld3 and Fat4, and represses miR-150 in the heart, we next tested whether these 5 genes are downregulated by miR-150. We observe that Hoxa4 is indeed downregulated in miR-150 TG mouse hearts (Figure 4A). Our in vivo protein analysis also reveals significantly decreased levels of HOXA4 in miR-150 TG mouse hearts compared to WT controls (Supplemental Figure XA). Interestingly, cardiac Fmo2 and Fat4 are increased in miR-150 TG mice (Figure 4B–C), and Lrrn4 and Marveld3 are not dysregulated in miR-150 TG mouse hearts as described in Figure 4 legend. These results suggest that MIAT activates Fmo2, Fat4, Lrrn4 and Marveld3 in a miR-150-independent mechanism (i.e., not by downregulating miR-150).
Figure 4. Homeobox a4 (Hoxa4) is a novel target of miR-150 in the heart and HOXA4 is upregulated in patients with heart failure.
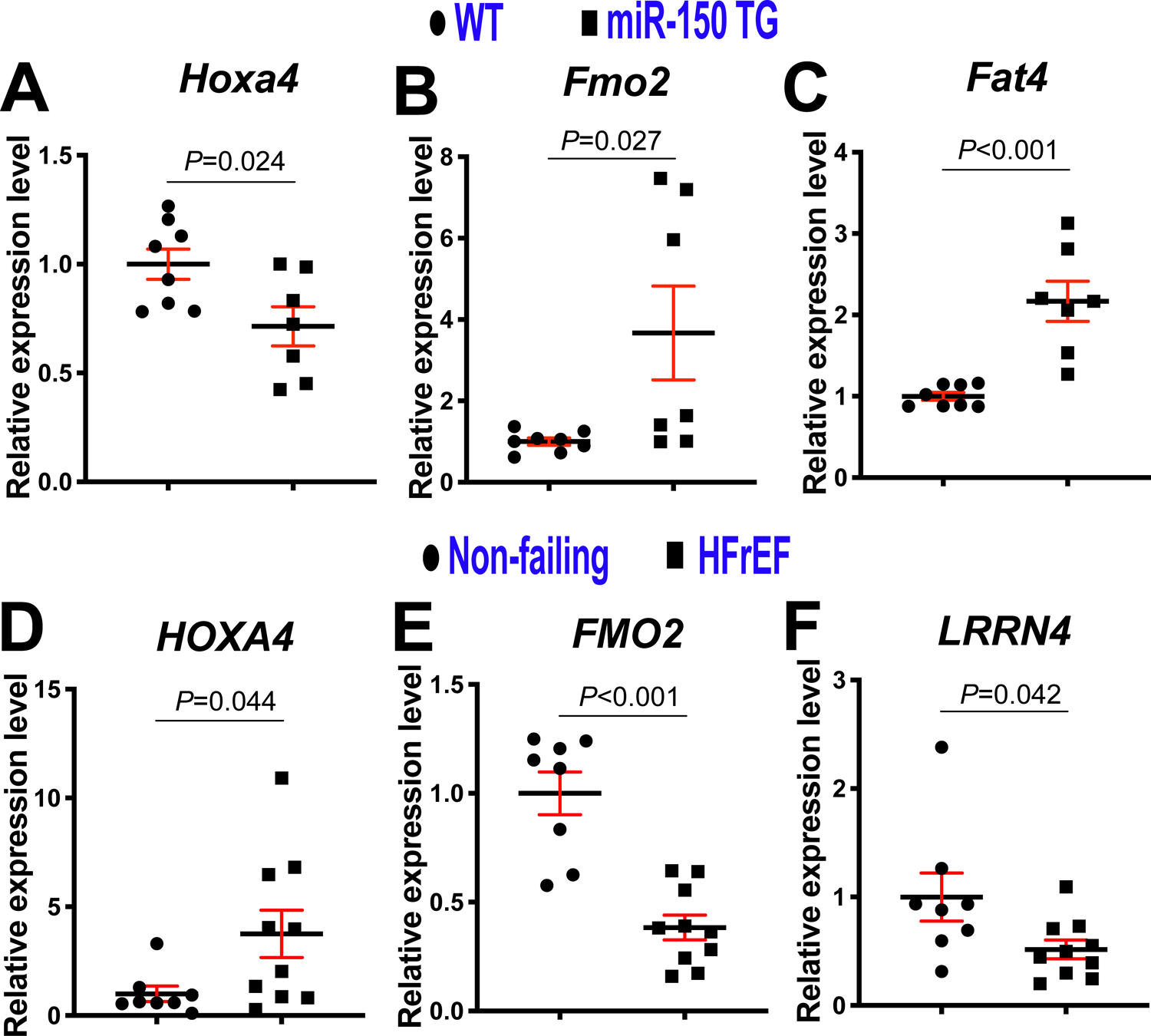
A-C, Real-Time Quantitative Reverse Transcription (QRT)-PCR expression analysis of Hoxa4 (A), flavin-containing dimethylaniline monoxygenase 2 (Fmo2: B) and FAT atypical cadherin 4 (Fat4: C) in left ventricles (LVs) from miR-150 transgenic (TG) and wild type (WT) mice. Data are shown as fold induction of gene expression normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). N=7–8. Unpaired 2-tailed t-test. Note that leucine-rich repeat neuronal 4 (Lrrn4) and MARVEL domain containing 3 (Marveld3) shown in Figure 3G–H are not dysregulated in miR-150 TG mouse LVs. D–F, QRT-PCR expression analysis of HOXA4 (D), FMO2 (E) and LRNN4 (F) in LVs from patients with heart failure with reduced ejection fraction (HFrEF) relative to non-failing heart tissues (N=8–10). Data are shown as fold induction of gene expression normalized to GAPDH. Unpaired 2-tailed t-test. Note that FAT4 shown in Figure 3I is not dysregulated in HFrEF patients and MARVELD3 in Figure 3H is undetectable in human LVs.
Next, our in vivo protein analysis reveals significantly decreased levels of HOXA4 in MIAT KO mouse hearts compared to WT controls and significantly elevated levels of HOXA4 at 4 weeks post-MI (Supplemental Figure XB–C), which are consistent with mRNA data (Figure 3E). Interestingly, Hoxa4 is also upregulated at 3 days and 7 days after MI (Supplemental Figure XI). We also find for the first time that LV HOXA4 is upregulated in patients with HFrEF (Figure 4D), concurrent with downregulation of cardiac miR-15013, 14 and upregulation of cardiac MIAT32. Our human data on cardiac upregulation of HOXA4 is in agreement with studies from colorectal and ovarian cancer patients41, 42, as well as our mRNA and protein data on mouse hearts (Figure 3E, Supplemental Figure XB–C and Supplemental Figure XI). Interestingly, LV FMO2 is decreased in patients with HFrEF (Figure 4E). We also show that LV LRRN4 is downregulated in patients with HFrEF (Figure 4F), which is consistent with a previous report on hearts from patients with DCM and mice subjected to TAC49. Notably, FAT4 is not dysregulated in HFrEF patients and MARVELD3 is undetectable in human LVs as described in Figure 4 legend. Based on these results, we focused on Hoxa4 as a novel regulatory target of miR-150.
We accordingly employed bioinformatic miR target prediction tools50–53 and identify a putative binding site for miR-150 in mouse Hoxa4 3’-untranslated region (UTR). Interestingly, mouse and human Hoxa4 genes have one miR-150 binding site, suggesting the conserved regulation of Hoxa4 by miR-150 and their roles in mice and humans. To examine whether Hoxa4 is a direct target of miR-150, we co-transfected miR-150 mimics and constitutively active luciferase (LUC) reporter constructs containing the binding site of miR-150 in mouse Hoxa4 (Figure 5A). We observe the repressed LUC activity by miR-150 for the WT Hoxa4 reporter. When we mutated seed binding sites for miR-150, miR-150 overexpression does not decrease LUC activity (Figure 5B), indicating that miR-150 suppresses Hoxa4 in a sequence-specific manner. Together with previous in vitro studies reporting the direct interaction between MIAT and miR-150 as evidenced by LUC assays28, 37, 48, our data thus suggest that Hoxa4 is a novel direct target of the MIAT/miR-150 axis.
Figure 5. Homeobox a4 (Hoxa4) is a novel direct target of miR-150, and HOXA4 is necessary for miR-150-dependent regulation of human cardiac fibroblast (HCF) proliferation.
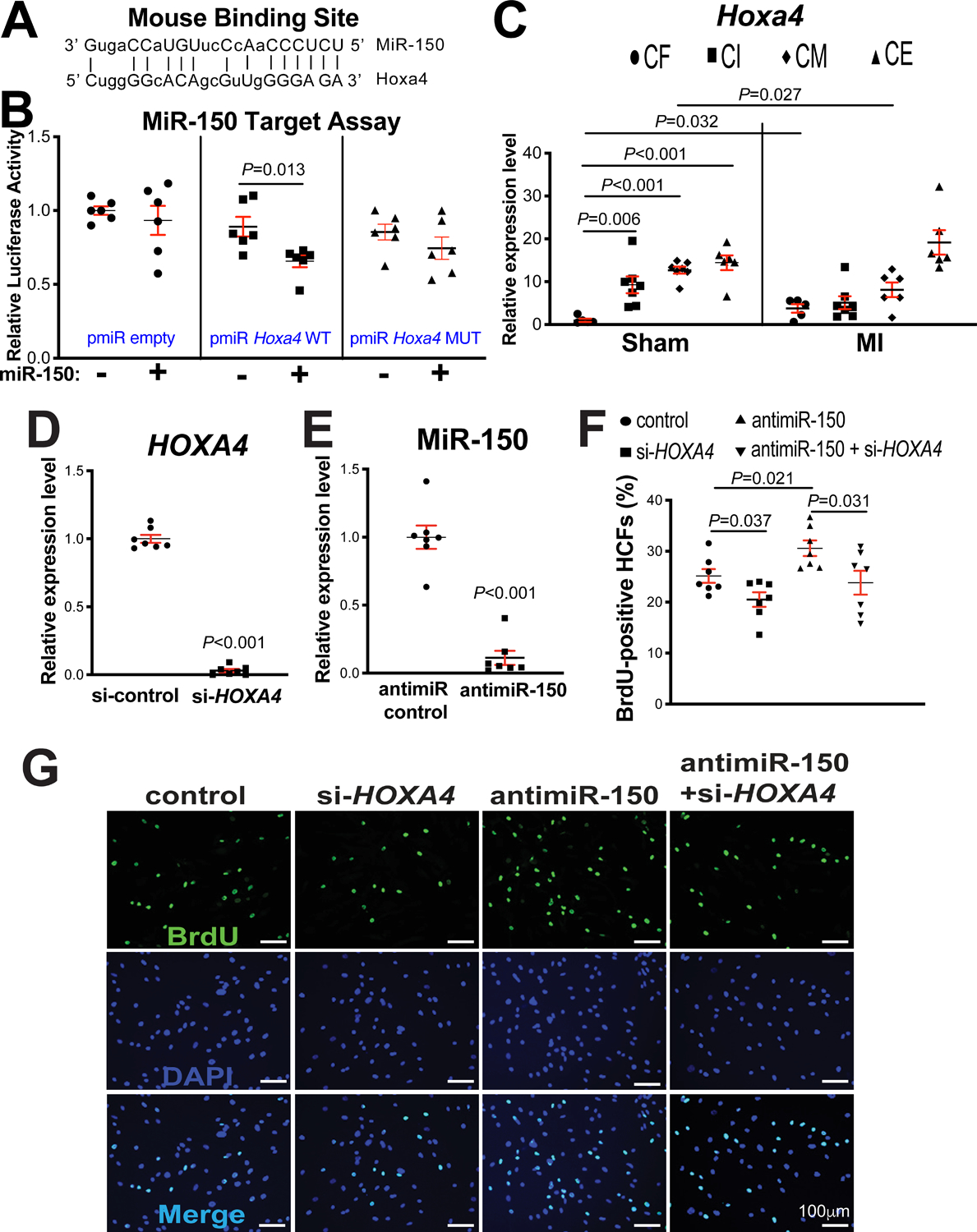
A, Mouse Hoxa4 has a miR-150 binding site in the 3’-untranslated region (3’UTR). MiR-150 seed pairing in the target region is shown as vertical lines. B, MiR-150’s ability to directly repress the activity of luciferase (LUC) reporter constructs that contain either wild type (WT) or mutated (MUT) binding site for Hoxa4. Transfection with or without miR-150 mimic in H9c2 cells is indicated. Firefly LUC activity was normalized to Renilla LUC activity and compared with empty vector measurements. N=6. Unpaired 2-tailed t-test. C, Real-Time Quantitative Reverse Transcription (QRT)-PCR expression analysis of Hoxa4 in cardiac fibroblasts (CFs), cardiac inflammatory cells (CIs), cardiomyocytes (CMs), and cardiac endothelial cells (CEs) isolated from adult mouse hearts at 7 days post-myocardial infarction (MI). N=5–7. Hoxa4 expression compared to glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was calculated using 2−ΔΔCt, and data are shown as fold induction of Hoxa4 expression levels normalized to CF sham. Two-way ANOVA with Tukey multiple comparison test. D–E, HCFs were transfected with control scramble siRNA (si-control) or HOXA4 siRNA (si-HOXA4) (D) and with antimiR control or antimiR-150 (E). QRT-PCR analyses for HOXA4 (D) or miR-150 (E) were performed to check the knockdown efficiency. Data were normalized to GAPDH (D) or U6 SNRNA (E) and expressed relative to controls. N=7 per group. Unpaired 2-tailed t-test. F-G, RNA interference with HOXA4 protects HCFs from the increased proliferation mediated by antimiR-150. HCFs were transfected as indicated, and bromodeoxyuridine (BrdU) assays were then performed. The percentage of proliferating nuclei (green) was calculated by normalizing total nuclei (blue). N=7 per group. One-way ANOVA with Tukey multiple comparison test.
HOXA4 is a transcription factor and highly conserved in vertebrates. Hoxa4 was reported to be involved in the patterning of the early chick heart40. Notably, our fractionation studies of different cardiac cell types show that Hoxa4 expression at baseline is significantly lower in CFs than CIs, CMs and CEs. Interestingly, Hoxa4 is upregulated selectively in CFs isolated from mouse hearts post-MI (Figure 5C), which is similarly associated with MIAT expression (Figure 1B). Interestingly, Hoxa4 is downregulated selectively in CMs isolated from mouse hearts post-MI (Figure 5C), which is not correlated with MIAT expression (Figure 1B). Thus, our cardiac cell fractionation results (Figure 1B and Figure 5C) and in vivo data (Figure 1A, Figure 3E and Supplemental Figure X) suggest that MIAT activates Hoxa4 in CFs, not in CMs. Given that cardiac miR-150 is upregulated by cardioprotective β-blocker Carv11, we next asked whether Carv inversely regulates the novel target of miR-150, Hoxa4 in hearts of WT mice. Indeed, Hoxa4 is downregulated in mouse hearts after Carv (Supplemental Figure XIIA–B). Together with our in vivo data revealing significantly decreased levels of Hoxa4 and HOXA4 in MIAT KO mouse hearts (Figure 3E and Supplemental Figure XB) and miR-150 TG mouse hearts (Figure 4A and Supplemental Figure XA) compared to WT controls, these results indicate that Hoxa4 is a critical downstream target of the cardiac MIAT/miR-150 dyad.
MiR-150 negatively regulates CF activation in part by functionally inhibiting pro-fibrotic Hoxa4
Due to cardiac upregulation of miR-150 by Carv11 concurrent downregulation of Hoxa4 (Supplemental Figure XIIA–B), and miR-150’s downregulation in CFs isolated from TAC mice54 concurrent upregulation of Hoxa4 selectively in CFs during MI (Figure 5C), we next focused on CFs to test whether miR-150 and Hoxa4 are inversely regulated in CFs treated with Carv, as well as CFs subjected to hypoxia/reoxygenation conditions. Indeed, HOXA4 is downregulated in HCFs subjected to hypoxia/reoxygenation conditions after Carv (Supplemental Figure XIIC), concurrent upregulation of miR-150 (Supplemental Figure XIID). We also observe that HOXA4 is increased in HCFs after hypoxia/reoxygenation (Supplemental Figure XIIC), which is consistent with our in vivo results in post-MI hearts (Figure 3E) and isolated CFs from ischemic myocardium (Figure 5C). Notably, we find that miR-150 is downregulated in HCFs after hypoxia/reoxygenation (Supplemental Figure XIID). Together with previous reports on miR-150 downregulation in hypoxia/reoxygenation and MI12, as well as I/R55, 56, our results strongly suggest that Hoxa4 is a critical functional target of miR-150 in CFs.
Since Hoxa4 expression is selectively upregulated in CFs isolated from ischemic myocardium (Figure 5C), concurrent with downregulation of miR-150 in CFs isolated from TAC mice54, and miR-150 negatively regulates CF activation in vitro54, we next investigated if the novel target of miR-150, HOXA4 regulates HCF activation. Our loss-of-function studies uncover that compared to controls, HOXA4 knockdown decreased HCF proliferation (Figure 5D–G) and migration (Figure 6A–B). Our data suggest that HOXA4 is required for HCF activation. Finally, to establish the functional relationship between miR-150 and HOXA4 in HCF activation, we applied an antimiR/siRNA-based rescue strategy for validating the functional relevance of the novel miR-150 target, HOXA4. The antimiR-150 treatment promotes HCF proliferation (Figure 5F–G) and migration (Figure 6A–B), which are attenuated by siRNA against HOXA4 (Figure 5F–G and Figure 6A–B). Taken together, these data indicate that miR-150 in HCFs confers protective effects in part through direct functional repression of pro-fibrotic HOXA4.
Figure 6. Homeobox a4 (Hoxa4) is necessary for miR-150-dependent regulation of human cardiac fibroblast (HCF) migration.
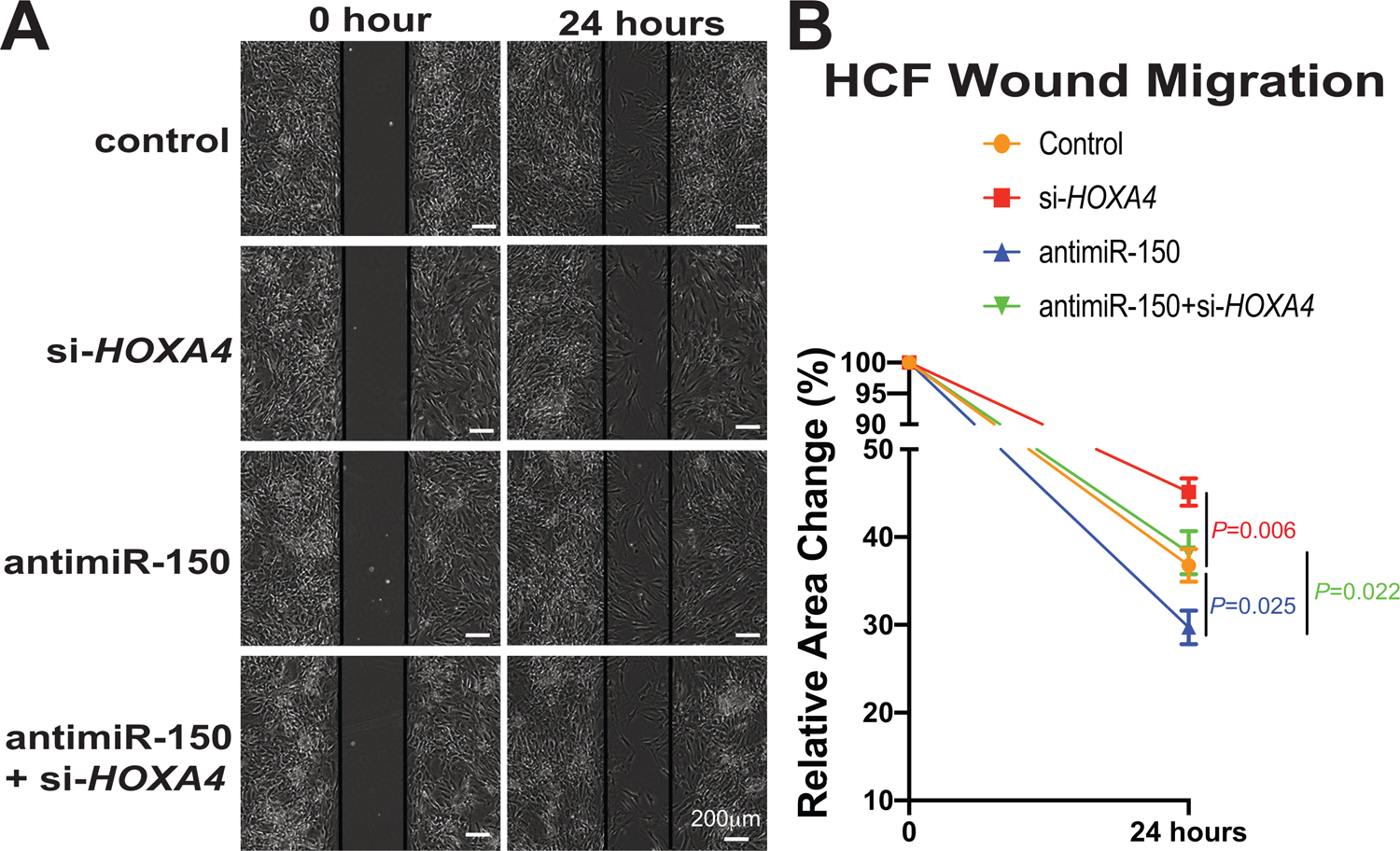
A-B, HCFs were transfected as indicated in Figure 5F–G, and scratch migration assays were then performed. RNA interference with HOXA4 protects HCFs from the increased migration mediated by antimiR-150. N=6 per group. Two-way repeated-measures ANOVA with Bonferroni post hoc test.
Loss of Hoxa4 in mice attenuates cardiac dysfunction after MI
To evaluate in vivo roles of Hoxa4 in cardiac stress, we obtained a novel Hoxa4 mutant mouse line. We first confirm that Hoxa4 KO mouse hearts have undetectable HOXA4 levels (Figure 7A). We next show that Hoxa4 KO mice have normal cardiac function at baseline (Supplemental Table VII and Figure 7B–F). However, Hoxa4 deficiency subjected to MI results in a significant improvement of cardiac function at 1 week, which is indicated by an increase in EF, FS, cardiac output [CO] and stroke volume (SV), as well as a decrease in ESV and systolic LVID (Supplemental Table VIII and Figure 7B–F). Hoxa4 KO mice also display enhanced cardiac function at 2 weeks post-MI, as evidenced by a significant increase in EF, FS and SV, as well as a significant decrease in ESV and systolic LVID (Supplemental Table IX and Figure 7B–F) as compared to WT controls. Taken together, these results demonstrate for the first time that deficiency of Hoxa4 in mice improves cardiac function post-MI.
Figure 7. Homeobox a4 (Hoxa4) deficiency protects the mouse heart against myocardial infarction (MI).
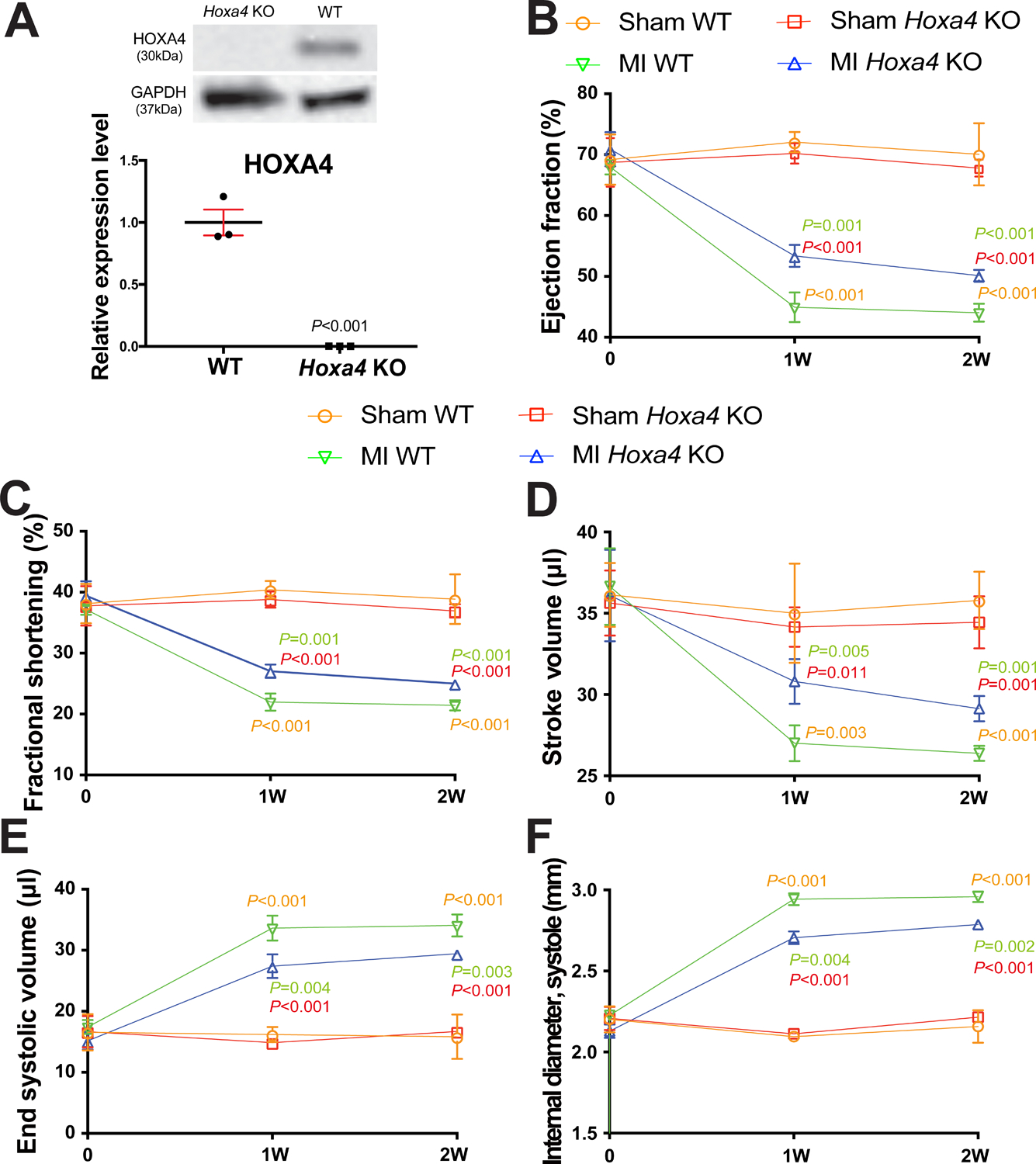
A, HOXA4 protein levels were measured in left ventricle (LV) lysates from Hoxa4 knockout (KO) mice compared to wild type (WT) controls. Data are shown as mean ± SEM from N=3 per group. Unpaired 2-tailed t-test. B-F, Transthoracic echocardiography was performed at 0, 1 week and 2 weeks post-MI. Quantification of ejection fraction (EF: B), fractional shortening (FS: C), stroke volume (SV: D), end systolic volume (ESV: E) and internal diameter, systole (LVIDs: F) is shown. Data are shown as mean ± SD from N=4 per group. Two-way repeated-measures ANOVA with Bonferroni post hoc test.
Discussion
Here, we identify the functional interaction among MIAT, miR-150 and Hoxa4 as a novel regulatory mechanism pertinent to MI. Mice overexpressing MIAT are sensitized to MI, as indicated by increased cardiac fibrosis, apoptosis and impairment of ventricular function, while mice deficient for MIAT are protected. Using a novel DTG mouse model, we demonstrate that miR-150 overexpression attenuates excessive maladaptive post-MI remodeling mediated by MIAT. Mechanistically, we discover that miR-150 directly inhibits pro-fibrotic Hoxa4 and MIAT blocks the inhibitory effect of miR-150 on Hoxa4 via its ceRNA action. Thus, the decreased expression of Hoxa4 in MIAT KO mice causes a lower degree of maladaptive post-MI remodeling, while the increased expression of HOXA4 in HCFs lacking miR-150 causes a higher degree of sustained CF activation.
The β1AR is predominantly expressed in cardiac tissues, and β-arrestin-mediated β1AR signaling confers cardioprotective effects57. We previously reported that Carv-mediated β1AR/β-arrestin1 signaling upregulates miR-15011. Together with the results presented in the current study, we postulate that miR-150 may be an important downstream mechanism by which β1AR-mediated β-arrestin signaling pathways confer cardiac protection, and that β-arrestin1-mediated β1AR regulatory mechanisms of miR-150 activation elicit beneficial remodeling in failing hearts by repressing CF activation through inhibition of pro-fibrotic genes such as Hoxa4. Interestingly, we reported that systemic miR-150 deletion leads to maladaptive post-MI remodeling by increasing cardiac apoptosis without affecting post-MI neovascularization12, whereas another group showed that systemic overexpression of miR-150 mediated by AgomiR injection protects the mouse heart against AMI by suppressing monocyte migration17. Moreover, cardiac-specific overexpression of miR-150 attenuated TAC-induced cardiac hypertrophy and dysfunction18, while systemic miR-150 loss caused a higher degree of cardiac fibrosis after TAC and miR-150 was downregulated in CFs isolated from TAC mice54. MiR-150 was regulated by MIAT by directly interacting with miR-150 and functioning as its ceRNA in vitro28, 29. Gain-of-function SNPs in MIAT were associated with increased susceptibility to MI in humans30, 31. Moreover, MIAT expression was increased in mouse hearts after MI, and lentivirus-mediated knockdown of MIAT improved cardiac function and structure in post-MI hearts36 and diabetic hearts34, as well as reduced I/R-induced myocardial infarct size and apoptosis35. In cultured CFs, MIAT was also upregulated after AngII treatment, and MIAT knockdown abrogated fibrogenesis by inhibiting collagen production and CF proliferation36. Although these previous studies showed the importance of miR-150 and MIAT in HF, our overall knowledge of their actions remains in its infancy in part due to (i) a lack of mechanistic insight by which these two ncRNAs regulate cardiac protection; and (ii) the absence of definitive and rigorous studies using appropriate mouse models to establish their in vivo functional relationship in HF. Here, we make novel discoveries to establish the functional MIAT/miR-150 axis in cardiac pathology and to identify its novel direct and functional target, Hoxa4 in CFs.
By using transcriptomic profiling approaches, we identify that MIAT regulates the expression of a relatively small number of cardiac genes post-MI. These genes are enriched in previously known and unknown pathways for MIAT, including DCM, HCM, renin-angiotensin system and ECM-receptor interaction (Supplemental Figure VII–IX and Supplemental Table VI). Although additional studies will be needed to validate the relationship between MIAT’s maladaptive actions in hearts and the individual genes/pathways identified in our study, our current filtering and validation analyses show that Hoxa4 is downregulated in the LV tissues of sham MIAT KO when compared with sham WT controls, and in the LVs of MI MIAT KO when compared with MI WT controls. In contrast, Hoxa4 is upregulated in the LV tissues of MI WT when compared with sham WT controls (Figure 3A–E). Our further QRT-PCR analyses demonstrate that cardiac HOXA4 is upregulated in patients with HFrEF (Figure 4D), and cardiac Hoxa4 is downregulated in miR-150 TG mice (Figure 4A). This leads us to discover Hoxa4 as a novel target of the MIAT/miR-150 axis.
HOXA4 is a highly conserved transcription factor. Hoxa4 was shown to be involved in the patterning of embryonic mouse lungs39 and early chick hearts40. Previous studies also indicated the overexpression of this fatal gene in colorectal and ovarian cancers41, 42. Our novel data likewise show that Hoxa4 expression and HOXA4 protein levels are increased in post-MI hearts (Figure 3E, Supplemental Figure XC and Supplemental Figure XI), concurrent with upregulation of MIAT (Figure 1A) and downregulation of miR-15012. Interestingly, our cardiac cell fractionation data also show that Hoxa4 is selectively upregulated in CFs isolated from mouse hearts post-MI (Figure 5C), suggesting an important role of Hoxa4 in CFs. In agreement with this notion, we show for the first time that HOXA4 knockdown suppresses HCF proliferation and migration (Figure 5D–G and Figure 6). In the current study, we also show for the first time that LV HOXA4 is upregulated in patients with HF (Figure 4D), which is consistent with reports in patients with renal clear cell carcinoma58, high-grade serous ovarian carcinoma59, and acute myeloid leukemia60. Interestingly, HOXA4 expression was inversely linked with survival of cancer patients59, 61. Here, we also show that Hoxa4 is downregulated in hearts and HCFs by a cardioprotective β-blocker, Carv (Supplemental Figure XIIA–C), concurrent with upregulation of miR-150 in hearts11 and HCFs (Supplemental Figure XIID). Notably, we demonstrate for the first time that Hoxa4 loss in mice protects hearts against MI (Figure 7). Thus, the finding of selective upregulation of Hoxa4 in CFs during MI supports that Hoxa4 inhibition could be therapeutically beneficial for cardiac fibrosis. Notably, retinoic acid-induced expression of Hoxa4 was shown to be regulated by a myelopoiesis-associated regulatory lncRNA, HOTAIRM1 in vitro43. Except for this report, little is known about the regulation of Hoxa4 by ncRNAs. Given our data that pro-fibrotic Hoxa4 is a novel direct and functional target for the MIAT/miR-150 dyad in hearts and CFs (Figure 3–7), MI patients with increased levels of MIAT or decreased levels of miR-150 could be considered for future targeted treatment options based on Hoxa4.
Limitations
Although we demonstrate that systemic overexpression of miR-150 in mice attenuates maladaptive post-MI remodeling caused by MIAT overexpression and that miR-150 is an important negative regulator of HCF activation in vitro by directly and functionally repressing pro-fibrotic HOXA4, it is possible that MIAT or miR-150 expression in other myocardial cells also plays a prominent role. Future studies using conditional cell-specific mouse models are thus warranted to fully understand the possible contribution of miR-150 expression in other cell types to miR-150’s protective actions on maladaptive cardiac remodeling observed in MIAT TG mice. Moreover, RNA-FISH and protein-RNA double labeling studies would be needed to determine the subcellular localization of the MIAT/miR-150 axis and their colocalization in the various cells of myocardium post-MI. To fully define the detailed underlying mechanisms of extra-CF actions mediated by MIAT or miR-150 and to get a much more complete understanding of the sequence of events, additional histopathological and immunohistochemical assessments (e.g., inflammation and apoptosis), as well as gene expression studies at earlier time points post-MI are also needed. Moreover, MIAT/miR-150 DTG mice overexpress the transgenes in all cell types, which may introduce confounders. In future studies, we could use loss-of-function approaches (MIAT/miR-150 DKO mice) to confirm their in vivo functional relationship in the heart. Notably, the MIAT/miR-150 axis may also have other targets mediating distinct functions. MIAT indeed downregulates miR-22–3p, miR-24, and miR-93 in the heart34, 36, 62, which could be explored as miR-150-independent mechanisms in our future studies with MIAT TG mice and myocardial cells. We will also investigate additional novel functional targets (i.e., Hoxa4-independent mechanisms) by cross-referencing the gene signature from MIAT KO mice (Figure 3 and Supplemental Table VI) with prediction analyses of miR-150 binding sites in our future mechanistic studies. Importantly, downstream targets of Hoxa4 to regulate CF activation remain elusive. Whether Hoxa4 is functionally regulated by the MIAT/miR-150 axis in other myocardial cell types and hearts also remains to be determined and beyond the scope of the current study. Lastly, additional in vivo injury models (e.g., I/R), as well as detailed studies on other roles of the two ncRNAs in CFs and non-CF cell types are required before pursuing this ncRNA axis as a therapeutic modality.
Conclusions
Our results using novel loss- and gain-of-function mouse models suggest that MIAT exacerbates maladaptive post-MI remodeling, miR-150 attenuates excessive maladaptive post-MI remodeling mediated by MIAT, and miR-150 plays a vital protective role in part by blunting CF activation through its direct functional repression of pro-fibrotic Hoxa4. Although MIAT and miR-150 are associated with HF in humans19, 30, 31 and their correlative relationship in CMs in vitro is shown37, 38, our studies using novel mouse models and unbiased transcriptome analyses directly establish in vivo functional relationship between these two ncRNAs in MI and define the underlying mechanism by which miR-150 affects CF activation. Given that upregulation of MIAT32 or downregulation of miR-15014, 55, 56, 63 also underlies other forms of cardiac disease, the deleterious action of MIAT and the protective action of miR-150 in hearts are likely applicable to diverse stress settings. Therefore, lessening MIAT levels via MIAT knockdown, and boosting miR-150 levels via miR-150 overexpression, in part to attenuate CF activation, could be an attractive adjunctive strategy to provide therapeutic benefits.
Supplementary Material
WHAT IS NEW?
Genetic deletion of MIAT in mice protects hearts against MI, while genetic overexpression of MIAT exacerbates maladaptive post-MI remodeling.
MiR-150 overexpression ameliorates maladaptive post-MI remodeling mediated by MIAT.
We identify HOXA4 as a novel target of the MIAT/miR-150 axis.
HOXA4 expression is upregulated in left ventricles from HF patients but is downregulated in human CFs (HCFs) and mouse hearts by carvedilol, which is inversely associated with miR-150 expression.
Hoxa4 loss in mice protects hearts against MI.
We identify a mechanism for the protective action of miR-150 to be mediated by the direct and functional repression of pro-fibrotic HOXA4 in HCFs.
WHAT ARE THE CLINICAL IMPLICATIONS?
The β-arrestin-biased β-blocker, carvedilol has been shown to increase the expression of miR-150 and to decrease the expression of MIAT or Hoxa4 in the heart, respectively.
Given our findings, we may anticipate that the β-arrestin-biased β-blockers and future treatments targeting the novel MIAT/miR-150/HOXA4 axis may have higher benefits in patients with HF and acute MI that are associated with enhanced MIAT or repressed miR-150, which can be identified by circulating levels of increased MIAT or reduced miR-150.
Sources of Funding
This work was supported by the National Institutes of Health [R01HL124251 and R01HL146481 to I.K.]; and the American Heart Association [18POST34030054 to T.A., 20POST34990024 to B.M., and 18TPA34170104 to I.K.].
Non-Standard Abbreviations and Acronyms
- βARs
β-adrenergic receptors
- β-blockers
β-adrenergic receptor antagonists
- AMI
acute myocardial infarction
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- BW
body weight
- Carv
carvedilol
- CE
cardiac endothelial cell
- ceRNAs
competing endogenous RNAs
- CF
cardiac fibroblast
- CI
cardiac inflammatory cell
- CM
cardiomyocyte
- CO
cardiac output
- CVDs
cardiovascular diseases
- DCM
dilated cardiomyopathy
- DTG
double transgenic
- ECM
extracellular matrix
- EDV
end-diastolic volume
- EF
ejection fraction
- ESV
end-systolic volume
- FS
fractional shortening
- HCM
hypertrophic cardiomyopathy
- HF
heart failure
- HFrEF
heart failure with reduced ejection fraction
- HOXA4
homeobox a4
- HR
heart rate
- HW
heart weight
- I/R
ischemia/reperfusion
- KO
knockout
- LAD
left anterior descending
- LUC
luciferase
- LV
left ventricle
- LVAW
left ventricular anterior wall thickness
- LVID
left ventricular interior diameter
- LVPW
left ventricular posterior wall thickness
- LVW
left ventricular weight
- MI
myocardial infarction
- MiRNAs or MiRs
microRNAs
- sI/R
simulated ischemia/reperfusion
- SV
stroke volume
- TAC
transverse aortic constriction
- TG
transgenic
- WT
wild type
Footnotes
Supplemental Materials:
Supplemental Tables I–V and VII–IX
Supplemental Table VI (datasets for Differentially Expressed Genes in Excel file)
References
- 1.Lei Z, Sluijter JP and van Mil A. MicroRNA Therapeutics for Cardiac Regeneration. Mini Rev Med Chem. 2015;15:441–451. [DOI] [PubMed] [Google Scholar]
- 2.Catalucci D, Gallo P and Condorelli G. MicroRNAs in cardiovascular biology and heart disease. Circ Cardiovasc Genet. 2009;2:402–408. [DOI] [PubMed] [Google Scholar]
- 3.van Rooij E The art of microRNA research. Circ Res. 2011;108:219–234. [DOI] [PubMed] [Google Scholar]
- 4.Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA and Olson EN. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J Clin Invest. 2012;122:1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arunachalam G, Upadhyay R, Ding H and Triggle CR. MicroRNA Signature and Cardiovascular Dysfunction. J Cardiovasc Pharmacol. 2015;65:419–429. [DOI] [PubMed] [Google Scholar]
- 6.Rupaimoole R and Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty C, Sharma AR, Sharma G, Doss CGP and Lee SS. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol Ther Nucleic Acids. 2017;8:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, Smith S, Bader AG, Kim S and Hong DS. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017;35:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA and Hodges MR. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. [DOI] [PubMed] [Google Scholar]
- 10.Taubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, Rode L, Weigt H, Genschel C, Lorch U, Theek C, Levin AA, Bauersachs J, Solomon SD and Thum T. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2020;42:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, Traynham CJ, Pironti G, Mao L, Su H, Johnson JA, Koch WJ and Rockman HA. beta-arrestin1-biased beta1-adrenergic receptor signaling regulates microRNA processing. Circ Res. 2014;114:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Y, Wang Y, Park KM, Hu Q, Teoh JP, Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, Tang Y, Ramesh G and Kim IM. MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc Res. 2015;106:387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreth S, Ledderose C, Schutz S, Beiras A, Heyn J, Weis F and Beiras-Fernandez A. MicroRNA-150 inhibits expression of adiponectin receptor 2 and is a potential therapeutic target in patients with chronic heart failure. J Heart Lung Transplant. 2014;33:252–260. [DOI] [PubMed] [Google Scholar]
- 14.Devaux Y, Vausort M, McCann GP, Zangrando J, Kelly D, Razvi N, Zhang L, Ng LL, Wagner DR and Squire IB. MicroRNA-150: a novel marker of left ventricular remodeling after acute myocardial infarction. Circ Cardiovasc Genet. 2013;6:290–298. [DOI] [PubMed] [Google Scholar]
- 15.Rhodes CJ, Wharton J, Boon RA, Roexe T, Tsang H, Wojciak-Stothard B, Chakrabarti A, Howard LS, Gibbs JS, Lawrie A, Condliffe R, Elliot CA, Kiely DG, Huson L, Ghofrani HA, Tiede H, Schermuly R, Zeiher AM, Dimmeler S and Wilkins MR. Reduced microRNA-150 is associated with poor survival in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187:294–302. [DOI] [PubMed] [Google Scholar]
- 16.Goren Y, Meiri E, Hogan C, Mitchell H, Lebanony D, Salman N, Schliamser JE and Amir O. Relation of reduced expression of MiR-150 in platelets to atrial fibrillation in patients with chronic systolic heart failure. Am J Cardiol. 2014;113:976–981. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Ye P, Wang S, Wu J, Sun Y, Zhang A, Ren L, Cheng C, Huang X, Wang K, Deng P, Wu C, Yue Z and Xia J. MicroRNA-150 Protects the Heart From Injury by Inhibiting Monocyte Accumulation in a Mouse Model of Acute Myocardial Infarction. Circ Cardiovasc Genet. 2015;8:11–20. [DOI] [PubMed] [Google Scholar]
- 18.Liu W, Liu Y, Zhang Y, Zhu X, Zhang R, Guan L, Tang Q, Jiang H, Huang C and Huang H. MicroRNA-150 protects against pressure overload-induced cardiac hypertrophy. J Cell Biochem. 2015;116:2166–2176. [DOI] [PubMed] [Google Scholar]
- 19.Scrutinio D, Conserva F, Passantino A, Iacoviello M, Lagioia R and Gesualdo L. Circulating microRNA-150–5p as a novel biomarker for advanced heart failure: A genome-wide prospective study. J Heart Lung Transplant. 2017;36:616–624. [DOI] [PubMed] [Google Scholar]
- 20.Archer K, Broskova Z, Bayoumi AS, Teoh JP, Davila A, Tang YL, Su HB and Kim IM. Long Non-Coding RNAs as Master Regulators in Cardiovascular Diseases. Int J Mol Sci. 2015;16:23651–23667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thum T and Condorelli G. Long Noncoding RNAs and MicroRNAs in Cardiovascular Pathophysiology. Circ Res. 2015;116:751–762. [DOI] [PubMed] [Google Scholar]
- 22.Hsiao J, Yuan TY, Tsai MS, Lu CY, Lin YC, Lee ML, Lin SW, Chang FC, Liu Pimentel H, Olive C, Coito C, Shen G, Young M, Thorne T, Lawrence M, Magistri M, Faghihi MA, Khorkova O and Wahlestedt C. Upregulation of Haploinsufficient Gene Expression in the Brain by Targeting a Long Non-coding RNA Improves Seizure Phenotype in a Model of Dravet Syndrome. EBioMedicine. 2016;9:257–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goyal A, Myacheva K, Gross M, Klingenberg M, Duran Arque B and Diederichs S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017;45:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL and Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo CJ, Maier VK, Davey R, Brennan J, Li G, Brothers J 2nd, Schwartz B, Gordo S, Kasper A, Okamoto TR, Johansson HE, Mandefro B, Sareen D, Bialek P, Chau BN, Bhat B, Bullough D and Barsoum J. Gene activation of SMN by selective disruption of lncRNA-mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proc Natl Acad Sci U S A. 2017;114:E1509–E1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinn JL and Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sone M, Hayashi T, Tarui H, Agata K, Takeichi M and Nakagawa S. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J Cell Sci. 2007;120:2498–2506. [DOI] [PubMed] [Google Scholar]
- 28.Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q and Jiang Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–1156. [DOI] [PubMed] [Google Scholar]
- 29.Shen Y, Dong LF, Zhou RM, Yao J, Song YC, Yang H, Jiang Q and Yan BA. Role of long non-coding RNA MIAT in proliferation, apoptosis and migration of lens epithelial cells: a clinical and in vitro study. J Cell Mol Med. 2016;20:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohnishi Y, Tanaka T, Yamada R, Suematsu K, Minami M, Fujii K, Hoki N, Kodama K, Nagata S, Hayashi T, Kinoshita N, Sato H, Sato H, Kuzuya T, Takeda H, Hori M and Nakamura Y. Identification of 187 single nucleotide polymorphisms (SNPs) among 41 candidate genes for ischemic heart disease in the Japanese population. Hum Genet. 2000;106:288–292. [DOI] [PubMed] [Google Scholar]
- 31.Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, Saito S, Nakamura Y and Tanaka T. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006;51:1087–1099. [DOI] [PubMed] [Google Scholar]
- 32.Frade AF, Laugier L, Ferreira LR, Baron MA, Benvenuti LA, Teixeira PC, Navarro IC, Cabantous S, Ferreira FM, da Silva Candido D, Gaiotto FA, Bacal F, Pomerantzeff P, Santos RH, Kalil J, Cunha-Neto E and Chevillard C. Myocardial Infarction-Associated Transcript, a Long Noncoding RNA, Is Overexpressed During Dilated Cardiomyopathy Due to Chronic Chagas Disease. J Infect Dis. 2016;214:161–165. [DOI] [PubMed] [Google Scholar]
- 33.de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, Smit JW, Lamb HJ, Llorente-Cortes V and Thum T. Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci Rep. 2016;6:37354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Zhang W, Jin M, Chen J, Xu W and Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22–3p in diabetic cardiomyopathy. Cell Death Dis. 2017;8:e2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Zhang D, Yu L and Dong H. Targeting MIAT reduces apoptosis of cardiomyocytes after ischemia/reperfusion injury. Bioengineered. 2019;10:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S, Yang T, Zhan L, Yuan Y, Chu W, Pan Z, Wang Z, Yang B and Lu Y. MIAT Is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci Rep. 2017;7:42657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu XH, Yuan YX, Rao SL and Wang P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur Rev Med Pharmacol Sci. 2016;20:3653–3660. [PubMed] [Google Scholar]
- 38.Li Z, Liu Y, Guo X, Sun G, Ma Q, Dai Y, Zhu G and Sun Y. Long noncoding RNA myocardial infarctionassociated transcript is associated with the microRNA1505p/P300 pathway in cardiac hypertrophy. Int J Mol Med. 2018;42:1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Packer AI, Mailutha KG, Ambrozewicz LA and Wolgemuth DJ. Regulation of the Hoxa4 and Hoxa5 genes in the embryonic mouse lung by retinoic acid and TGFbeta1: implications for lung development and patterning. Dev Dyn. 2000;217:62–74. [DOI] [PubMed] [Google Scholar]
- 40.Searcy RD and Yutzey KE. Analysis of Hox gene expression during early avian heart development. Dev Dyn. 1998;213:82–91. [DOI] [PubMed] [Google Scholar]
- 41.Bhatlekar S, Addya S, Salunek M, Orr CR, Surrey S, McKenzie S, Fields JZ and Boman BM. Identification of a developmental gene expression signature, including HOX genes, for the normal human colonic crypt stem cell niche: overexpression of the signature parallels stem cell overpopulation during colon tumorigenesis. Stem Cells Dev. 2014;23:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamashita T, Tazawa S, Yawei Z, Katayama H, Kato Y, Nishiwaki K, Yokohama Y and Ishikawa M. Suppression of invasive characteristics by antisense introduction of overexpressed HOX genes in ovarian cancer cells. Int J Oncol. 2006;28:931–938. [PubMed] [Google Scholar]
- 43.Zhang X, Lian Z, Padden C, Gerstein MB, Rozowsky J, Snyder M, Gingeras TR, Kapranov P, Weissman SM and Newburger PE. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood. 2009;113:2526–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ip JY, Sone M, Nashiki C, Pan Q, Kitaichi K, Yanaka K, Abe T, Takao K, Miyakawa T, Blencowe BJ and Nakagawa S. Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci Rep. 2016;6:27204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP and Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. [DOI] [PubMed] [Google Scholar]
- 46.Yu CC, Corr C, Shen C, Shelton R, Yadava M, Rhea IB, Straka S, Fishbein MC, Chen Z, Lin SF, Lopshire JC and Chen PS. Small conductance calcium-activated potassium current is important in transmural repolarization of failing human ventricles. Circ Arrhythm Electrophysiol. 2015;8:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ulitsky I Interactions between short and long noncoding RNAs. FEBS Lett. 2018;592:2874–2883. [DOI] [PubMed] [Google Scholar]
- 48.Shen Y, Dong LF, Zhou RM, Yao J, Song YC, Yang H, Jiang Q and Yan B. Role of long non-coding RNA MIAT in proliferation, apoptosis and migration of lens epithelial cells: a clinical and in vitro study. J Cell Mol Med. 2016;20:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li R, Fang J, Huo B, Su YS, Wang J, Liu LG, Hu M, Cheng C, Zheng P, Zhu XH, Jiang DS and Wei X. Leucine-rich repeat neuronal protein 4 (LRRN4) potentially functions in dilated cardiomyopathy. Int J Clin Exp Pathol. 2017;10:9925–9933. [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP and Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. [DOI] [PubMed] [Google Scholar]
- 52.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M and Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. [DOI] [PubMed] [Google Scholar]
- 53.Dweep H, Sticht C, Pandey P and Gretz N. miRWalk--database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44:839–847. [DOI] [PubMed] [Google Scholar]
- 54.Deng P, Chen L, Liu Z, Ye P, Wang S, Wu J, Yao Y, Sun Y, Huang X, Ren L, Zhang A, Wang K, Wu C, Yue Z, Xu X and Chen M. MicroRNA-150 Inhibits the Activation of Cardiac Fibroblasts by Regulating c-Myb. Cell Physiol Biochem. 2016;38:2103–2122. [DOI] [PubMed] [Google Scholar]
- 55.Topkara VK and Mann DL. Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc Drugs Ther. 2011;25:171–182. [DOI] [PubMed] [Google Scholar]
- 56.Duan Y, Zhou B, Su H, Liu Y and Du C. MiR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp Cell Res. 2013;319:173–84. [DOI] [PubMed] [Google Scholar]
- 57.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ and Rockman HA. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007;117:2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui Y, Yan M, Zhang C, Xue J, Zhang Q, Ma S, Guan F and Cao W. Comprehensive analysis of the HOXA gene family identifies HOXA13 as a novel oncogenic gene in kidney renal clear cell carcinoma. J Cancer Res Clin Oncol. 2020;146:1993–2006. [DOI] [PubMed] [Google Scholar]
- 59.Miller KR, Patel JN, Zhang Q, Norris EJ, Symanowski J, Michener C, Sehouli J, Braicu I, Destephanis DD, Sutker AP, Jones W, Livasy CA, Biscotti C, Ganapathi RN, Tait DL and Ganapathi MK. HOXA4/HOXB3 gene expression signature as a biomarker of recurrence in patients with high-grade serous ovarian cancer following primary cytoreductive surgery and first-line adjuvant chemotherapy. Gynecol Oncol. 2018;149:155–162. [DOI] [PubMed] [Google Scholar]
- 60.Larramendy ML, Niini T, Elonen E, Nagy B, Ollila J, Vihinen M and Knuutila S. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica. 2002;87:569–577. [PubMed] [Google Scholar]
- 61.Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, Zeng C, Baron A, Hunger SP, Varella-Garcia M, Gemmill R, Brizard F, Brizard A and Roche J. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16:186–195. [DOI] [PubMed] [Google Scholar]
- 62.Li Y, Wang J, Sun L and Zhu S. LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol. 2018;818:508–517. [DOI] [PubMed] [Google Scholar]
- 63.Liu Z, Zhou C, Liu Y, Wang S, Ye P, Miao X and Xia J. The expression levels of plasma micoRNAs in atrial fibrillation patients. PLoS ONE. 2012;7:e44906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD and Rockman HA. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci U S A. 2008;105:14555–14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bayoumi AS, Teoh JP, Aonuma T, Yuan Z, Ruan X, Tang Y, Su H, Weintraub NL and Kim IM. MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc Res. 2017;113:1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y, Su H, Weintraub NL and Kim IM. A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol. 2017;114:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramakrishna S, Kim IM, Petrovic V, Malin D, Wang IC, Kalin TV, Meliton L, Zhao YY, Ackerson T, Qin Y, Malik AB, Costa RH and Kalinichenko VV. Myocardium defects and ventricular hypoplasia in mice homozygous null for the Forkhead Box M1 transcription factor. Dev Dyn. 2007;236:1000–1013. [DOI] [PubMed] [Google Scholar]
- 68.Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, Major ML, Gusarova GA, Yoder HM, Costa RH and Kalinichenko VV. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–2161. [DOI] [PubMed] [Google Scholar]
- 69.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J and Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. [DOI] [PubMed] [Google Scholar]
- 70.Teoh JP, Park KM, Broskova Z, Jimenez FR, Bayoumi AS, Archer K, Su H, Johnson J, Weintraub NL, Tang Y and Kim IM. Identification of gene signatures regulated by carvedilol in mouse heart. Physiol Genomics. 2015;47:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu H, Han C, Lu D and Wu T. MiR-17–92 cluster promotes cholangiocarcinoma growth: evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am J Pathol. 2014;184:2828–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cossette SM, Gastonguay AJ, Bao X, Lerch-Gaggl A, Zhong L, Harmann LM, Koceja C, Miao RQ, Vakeel P, Chun C, Li K, Foeckler J, Bordas M, Weiler H, Strande J, Palecek SP and Ramchandran R. Sucrose non-fermenting related kinase enzyme is essential for cardiac metabolism. Biol Open. 2014;4:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liao JM, Zeng SX, Zhou X and Lu H. Global effect of inauhzin on human p53-responsive transcriptome. PLoS ONE. 2012;7:e52172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stueckle TA, Lu Y, Davis ME, Wang L, Jiang BH, Holaskova I, Schafer R, Barnett JB and Rojanasakul Y. Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol Appl Pharmacol. 2012;261:204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, Collins PJ, Chu TM, Bao W, Fang H, Kawasaki ES, Hager J, Tikhonova IR, Walker SJ, Zhang L, Hurban P, de Longueville F, Fuscoe JC, Tong W, Shi L and Wolfinger RD. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat Biotechnol. 2006;24:1140–1150. [DOI] [PubMed] [Google Scholar]
- 76.Park KM, Teoh JP, Wang Y, Broskova Z, Bayoumi AS, Tang Y, Su H, Weintraub NL and Kim IM. Carvedilol-responsive microRNAs, miR-199a-3p and −214 protect cardiomyocytes from simulated ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2016;311:H371–H383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH and Kalinichenko VV. The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem. 2005;280:22278–22286. [DOI] [PubMed] [Google Scholar]
- 78.Kim IM, Wolf MJ and Rockman HA. Gene deletion screen for cardiomyopathy in adult Drosophila identifies a new notch ligand. Circ Res. 2010;106:1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Teoh JP, Bayoumi AS, Aonuma T, Xu Y, Johnson JA, Su H, Weintraub NL, Tang Y and Kim IM. beta-arrestin-biased agonism of beta-adrenergic receptor regulates Dicer-mediated microRNA maturation to promote cardioprotective signaling. J Mol Cell Cardiol. 2018;118:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series access number GSE185396. All other data that are not included in this publication, analytical methods and study materials will be made available to other researchers for purposes of reproducing results or replicating procedures. Other methods are provided in Supplemental Material.