

Abstract
Type I toxin-antitoxin (TA) systems typically consist of a protein toxin that imbeds in the inner membrane where it can oligomerize and form pores that change membrane permeability, and an RNA antitoxin that interacts directly with toxin mRNA to inhibit its translation. In Escherichia coli, symE/symR is annotated as a type I TA system with a non-canonical toxin. SymE was initially suggested to be an endoribonuclease, but has predicted structural similarity to DNA binding proteins. To better understand SymE function, we used RNA-seq to examine cells ectopically producing it. Although SymE drives major changes in gene expression, we do not find strong evidence of endoribonucleolytic activity. Instead, our biochemical and cell biological studies indicate that SymE binds DNA. We demonstrate that the toxicity of symE overexpression likely stems from its ability to drive severe nucleoid condensation, which disrupts DNA and RNA synthesis and leads to DNA damage, similar to the effects of overproducing the nucleoid-associated protein H-NS. Collectively, our results suggest that SymE represents a new class of nucleoid-associated proteins that is widely distributed in bacteria.
Graphical Abstract
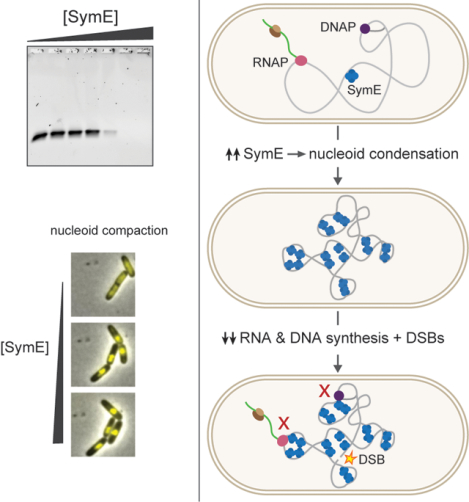
The conserved protein SymE from Escherichia coli was originally thought to be a toxin of a type I toxin-antitoxin system that cleaves RNA. However, our biochemical and cell biological studies indicate that SymE instead binds DNA and can, when overexpressed, drive severe nucleoid condensation. This condensation, similar to that observed with H-NS, disrupts bulk DNA and RNA synthesis, and produces double-strand breaks. We propose that SymE represents a new class of nucleoid-associated protein.
Introduction
Toxin-antitoxin (TA) systems are typically composed of a relatively stable toxic protein and a more labile antitoxin. These systems are widespread in bacterial genomes, with a single species often harboring multiple systems. TA systems have been suggested to participate in a wide variety of physiological processes, though the functions of most TA systems remain unknown or poorly understood. The first characterized TA systems were plasmid borne and found to help maintain plasmids within a population of cells (1–3). Chromosomally encoded TA systems have been suggested to participate in various stress responses and persistence, but a growing body of work suggests that many TA systems may protect their hosts from phage infection (4–7).
Seven different types of TA system have been identified, with the type defined by the nature of neutralization by the antitoxin (8). Type I antitoxins are small RNAs encoded antisense to or nearby their cognate toxin. These RNA antitoxins interact directly with toxin mRNA to occlude the ribosome binding site, promote degradation of the toxin/antitoxin RNA complex, or both (9). Almost all type I toxins reported to date are small (< 100 amino acids), single-domain proteins that are thought to oligomerize within the cell membrane, causing membrane depolarization and permeabilization (10). Though discovered soon after type II TA systems, type I systems are not nearly as well studied.
The E. coli MG1655 genes symE and symR are currently annotated as a type I TA system (Figure 1A). Expressing symE at high levels blocks cell growth, while symR expression can prevent SymE translation (11). symR encodes a small RNA that is transcribed largely within and antisense to the 5’ end of symE. The symR RNA is transcribed at ~10-fold higher levels than symE and likely occludes ribosome binding when bound to symE RNA. The expression of symE is also controlled at the transcriptional level by the LexA repressor, making it part of the SOS regulon (11–13). Though induced in the SOS response, there is limited evidence of a role for SymE in recovery from DNA damage. ΔsymE mutants show no growth difference in most DNA damaging conditions except for a modest defect when treated with styrene oxide (11, 14).
Figure 1. E. coli symE genetic architecture and SymE protein structural homology.
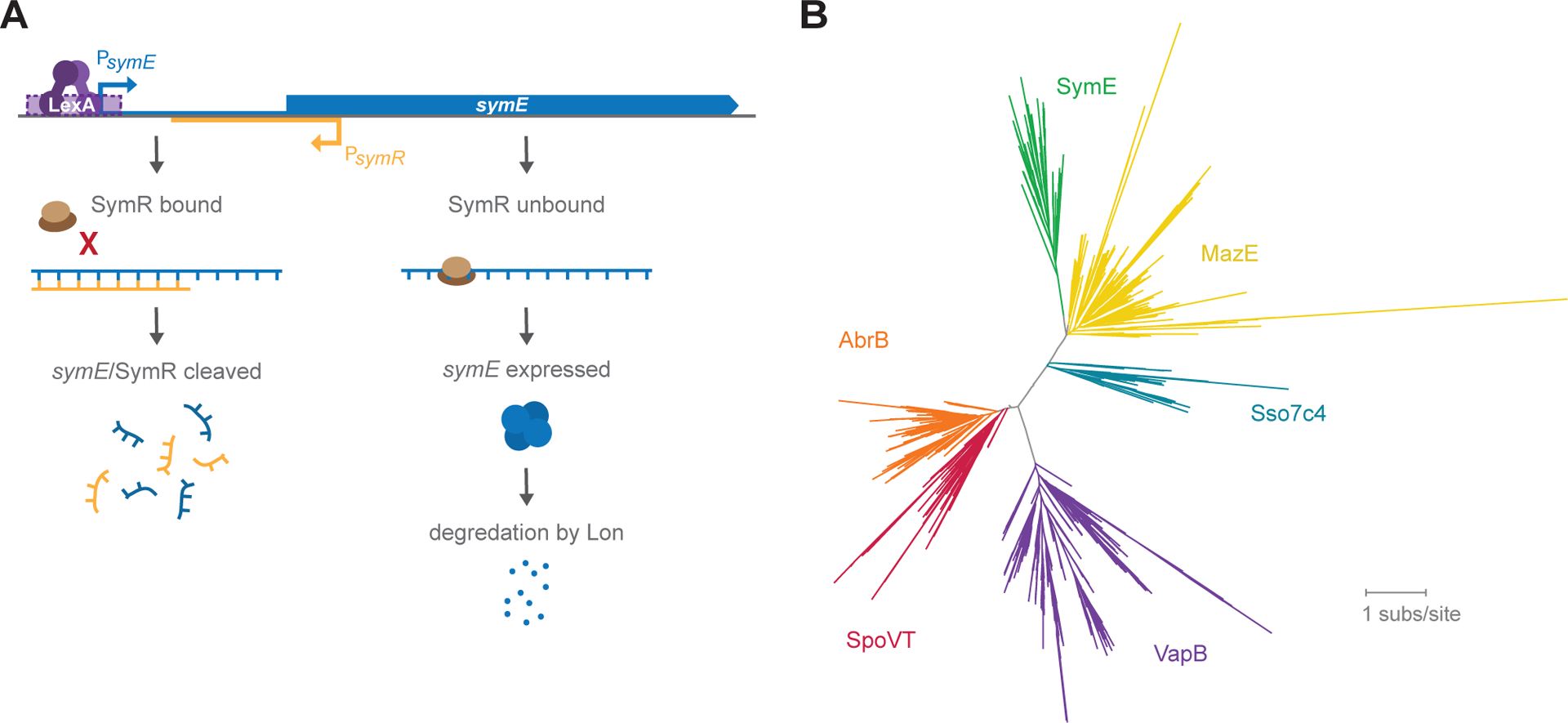
(A) symR is encoded antisense to symE and overlapping the ribosome binding site of symE. In the presence of SymR, ribosome binding to symE mRNA is occluded, and the symE/SymR complex is degraded. When there is more symE mRNA than SymR present, SymE protein is produced, but also degraded by Lon.
(B) Protein homology tree based on primary sequence alignments to SymE, indicating that SymE is likely to be most structurally similar to MazE, Sso7c4, VapB, SpoVT and AbrB.
In cells overexpressing symE, viability drops ~10-fold within 90 minutes, with a concomitant drop in global translation (11). Cells overproducing SymE also show decreases in the levels of several transcripts, including recA and ompA, with possible RNA degradation products observed in the case of ompA. These observations suggested that SymE may be an endoribonuclease. However, SymE is a 113 amino-acid protein with structural similarity to AbrB and MazE family proteins that feature a DNA-binding, swapped hairpin fold (15, 16). Most AbrB-like proteins control the expression of a specific set of genes, although the recently described Sso7c4 from the archaeon Sulfolobus solfataricus binds DNA non-sequence specifically and may function primarily to control DNA compaction (17). Among the AbrB family of proteins, SymE is most similar to the DNA-binding domain of MazE, the antitoxin of the type II TA system mazEF (Figure 1B). Our phylogenetic analyses indicated that SymE homologs are found in all bacterial phyla except actinobacteria and cyanobacteria, but only in 357 of ~9,000 representative species. As in E. coli MG1655, symE homologs are often encoded near restriction-modification systems, especially mcrBC systems. Nearly half of the species with symE have a paralogous copy, with up to 41 copies in a single species.
To further examine the molecular function of SymE, we used a previously developed RNA-seq method to test how SymE impacts E. coli transcripts. Although the levels of many RNAs change after inducing symE, we find no evidence of endoribonuclease activity. Instead, ChIP-seq and electrophoretic mobility shift assays using purified SymE indicate that SymE binds DNA in vivo and in vitro. Further, using fluorescent microscopy, we find that high levels of SymE drive nucleoid compaction. Such compaction was not seen when overproducing other known toxins, but is similar to that seen after overexpressing hns, which encodes a nucleoid-associated protein (18). Similarly, we find that overexpressing either symE or hns leads to double strand breaks in DNA. Our results indicate that SymE binds to and compacts the nucleoid in a way that disrupts both DNA replication and transcription, as well as producing double strand breaks in DNA. Taken together, our findings support the notion that SymE is not a canonical type I TA toxin and instead represents a novel DNA binding protein.
Results
SymE homologs are widespread and frequently co-located with restriction modification systems
We first sought to reexamine the conservation and phylogenetic distribution of symE homologs in bacteria (11, 16, 19). Using E. coli MG1655 SymE as a query, we searched the Progenomes representative bacterial genomes database (20) and found homologs in all bacterial phyla except for Actinobacteria and Cyanobacteria (Figure 2A) (see Methods). Note that our search was based solely on symE, and that we did not search for antisense symR homologs. Although symE is widespread in bacteria, we found homologs in only 357 of the approximately 9,000 queried species. An alignment of SymE sequences from representative species revealed a conserved 34 amino acid core that is located within the E. coli SymE AbrB domain, predicted here using PSIPRED (Figure 2B) (21). The most conserved positions featured small, hydrophobic residues (G, I, L, V) or aromatic residues (F, W). In many Proteobacteria and some Firmicutes, symE was encoded in genomes immediately adjacent to or near restriction-modification (RM) systems, especially mcrBC-like systems (Figure 2A). This co-location with RM systems might indicate that SymE plays a role in defense against foreign DNA, as phage defense systems often cluster together (22), but we were unable to identify a coliphage that symE protects against.
Figure 2. Bioinformatic analysis of SymE.
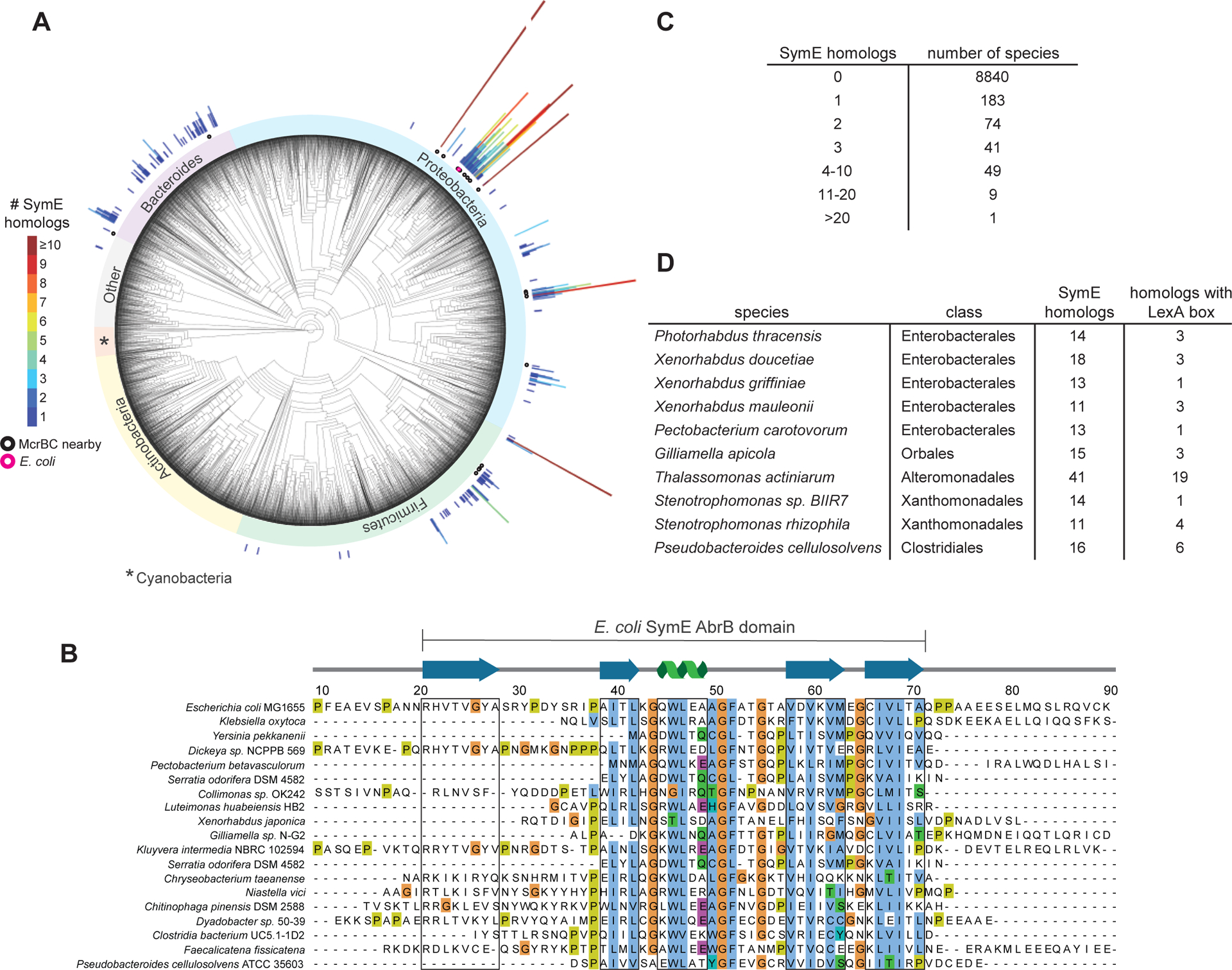
(A) Phylogenetic tree showing the distribution of symE throughout bacteria. Each radial line appears above a species that encodes a SymE homolog, with the length and color of the line denoting how many copies of symE occur in that species. Homologs encoded near an mcrBC locus are shown with a black circle under the line, and E. coli is denoted with a pink circle. The ‘Other’ group contains uncharacterized outgrouping bacteria.
(B) Sequence alignment of representative SymE homologs from Proteobacteria, Firmicutes, and Bacteroides. SymE is most conserved in the area of the protein that is predicted to have an AbrB-like fold. Sequences that correspond to β-sheets or α-helices are boxed in grey.
(C) Tabulated summary of how many species harbor the indicated number of paralogs.
(D) Summary of the species identified with more than 10 paralogs of SymE.
Strikingly, we found that symE is often highly duplicated within certain species (Figure 2A). The degree of duplication was variable; about half of the species harbor only a single symE with the other half containing 2 to 41 copies (Figure 2C). Ten species encode > 11 different SymE homologs (Figure 2D). Paralogs of symE were especially prevalent in the Enterobacterales. When we analyzed the promoters of the symE paralogs in a given species, we found that not all contained LexA binding sites based on consensus sequences from different bacterial phylum and classes (23).
Inducing SymE does not produce a signature of endoribonuclease activity in vivo
Given the wide distribution of symE homologs, we sought to better understand its function. SymE was initially suggested to be an endoribonuclease (11), but its activity has not been well characterized. To examine whether SymE drives RNA cleavage in vivo and to map its potential specificity as a nuclease, we used a previously developed, paired-end RNA-seq method for mapping cleavage events transcriptome-wide following SymE induction (24). A schematic of this technique is shown in Figure 3A using data from the well characterized RNase MazF. In this method we calculate a ‘cleavage ratio’, which is the ratio of the read counts for each nucleotide within a transcript in cells producing a potential endoribonuclease compared to cells harboring an empty vector. Large dips or “valleys” in the plot of cleavage ratio across a transcript indicate sites that were likely cleaved. In E. coli, 3’-to-5’ exonuclease activity can drive additional loss in fragment density and low cleavage ratios in regions immediately 5’ of a cleavage site. To identify cleavage events and reduce noise from 3’-to-5’ exonuclease activity, we calculated the slope of the cleavage ratio across a transcript in sliding 30 nucleotide windows. Local maxima in this slope indicate putative cleavage sites.
Figure 3. Expressing symE leads to widespread changes in RNA expression but without evidence of RNA cleavage.
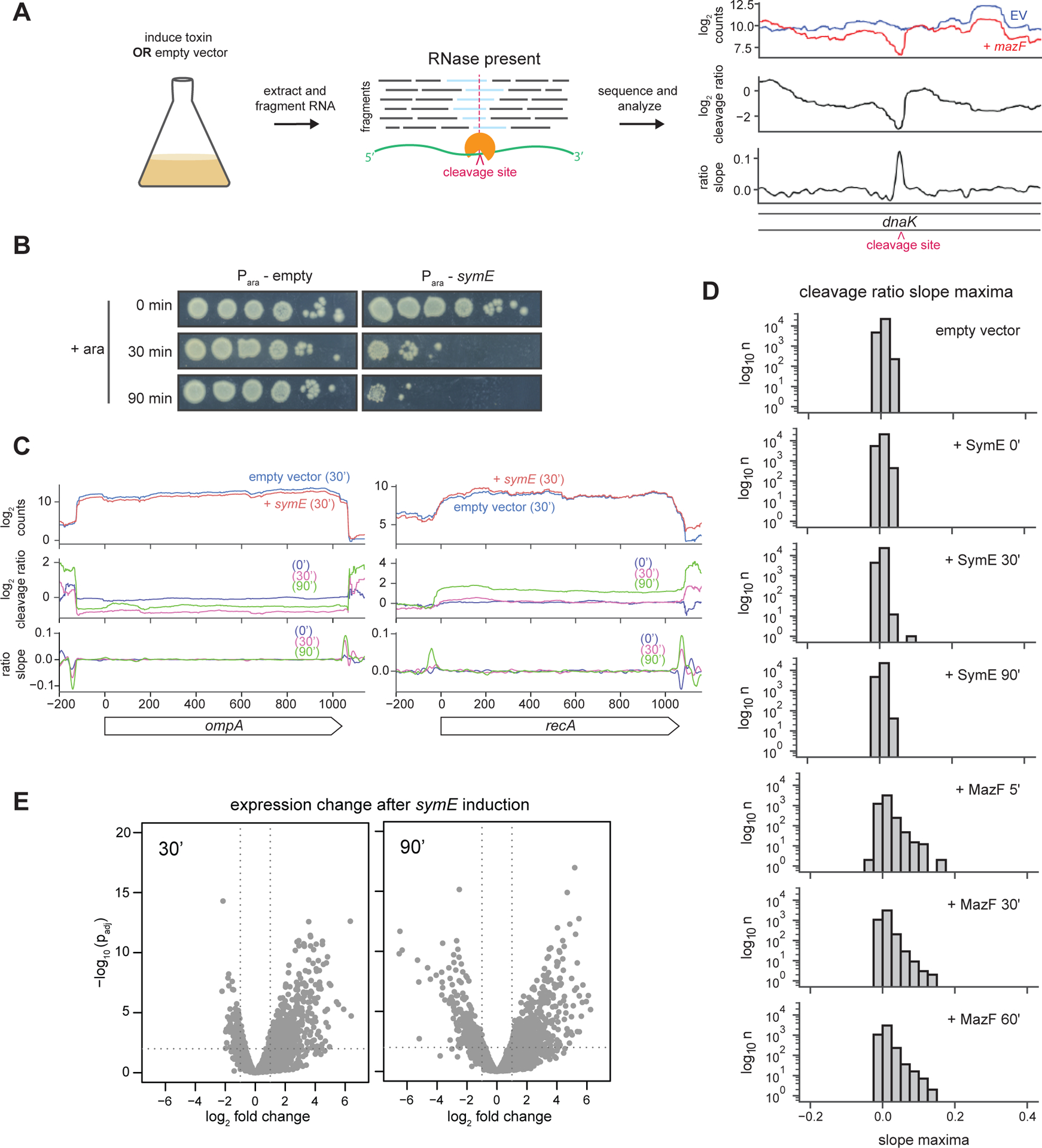
(A) Overview of the RNA-seq experiment performed, but with an example of cleavage analysis for the known RNase MazF.
(B) Plating viability of cells either harboring an empty vector or a plasmid encoding symE and grown in arabinose for the times indicated.
(C) Gene level cleavage analysis for two transcripts (ompA and recA) previously examined by Northern blotting(11) after induction of SymE.
(D) Transcriptome wide analysis of cleavage ratio maxima for cells harboring an empty vector or expressing symE or mazF for the time indicated.
(E) Expression changes after symE induction for the indicated times. Volcano plots indicate the log2 fold change in expression of each gene and their significance (-log p). symE is omitted from this plot as it was exogenously overexpressed.
To determine whether SymE cleaves mRNA in vivo, we used a strain consisting of symE on a plasmid under the control of an arabinose inducible promoter in E. coli MG1655 also harboring the PCP18-araE’ mutation that enables relatively homogeneous expression from the Para promoter (11, 25). Prior to induction, cells harboring the plasmid with symE had viability comparable to an empty vector control strain (Figure 3B). However, after adding arabinose, cells producing SymE exhibited a substantial loss in viability, consistent with prior work on symE (11). We took samples 0, 30, and 90 minutes after induction and used our paired-end RNA-seq method to look for potential RNA cleavage.
We first inspected ompA and recA, which were previously examined by Northern blotting in cells overexpressing symE (11). We found no evidence of endoribonuclease activity, with the cleavage ratio profiles remaining flat, and cleavage ratio slopes close to 0 throughout each transcript (Figure 3C). To evaluate cleavage events globally, we assessed the maximum cleavage ratio slope value in each transcript and then plotted these values in a histogram (Figure 3D). The histograms of slope maxima after 30 minutes and 90 minutes of symE expression clustered tightly around 0, similar to that seen when comparing two empty vector samples to one another. In contrast, when we performed the same analysis on similar, previously generated data for MazF, there was a clear skewing of the histograms to the right after 5, 30, and 60 minutes of mazF expression, indicating that many transcripts had been cleaved (Figure 3D) (24). These results indicated that SymE is not detectably cleaving RNA.
Although we did not detect cleavage within transcripts, we did detect changes in the expression levels of many genes. (11). Analysis of the entire transcriptome revealed significant increases and decreases in hundreds of transcripts after 30 and 90 minutes of symE induction (Figure 3E). However, there was no obvious or detectable enrichment of specific classes or functionally related sets of genes (Table S1) (26). Many of the down-regulated genes at both time points examined were related to metabolism. Up-regulated genes have even less in common and were mostly uncharacterized or putative in nature, though many are encoded on prophages and may reflect a general stress response following SymE induction (27). There was also an induction of the SOS regulon, as discussed below.
SymE binds DNA in vivo and in vitro
Given its predicted structural similarity and homology to AbrB family proteins (Figure 1B), we hypothesized that SymE binds DNA (15, 16). To test this possibility in vivo, we performed chromosome immunoprecipitation sequencing (ChIP-seq) following expression of C-terminally SPA-tagged SymE or, as a control, untagged SymE in E. coli MG1655 cells growing in LB. The SPA tag has previously been shown to not significantly affect SymE toxicity (11). The tagged and untagged SymE were each induced and ChIP-seq performed on samples taken 30- and 60 minutes post-induction. Figure 4A summarizes the enrichment of tagged SymE relative to the untagged control across the E. coli chromosome. SymE-SPA showed high occupancy relative to the untagged control sample at a broad range of genomic positions at both 30- and 60-minute time points. Peaks in the ChIP data were called using the MACS algorithm, and additional fold enrichment and maximum background thresholds were applied using MochiView (28, 29) (see Methods). A total of 61 peaks exceeded the thresholds set in both the 30- and 60-minute time points. These peaks were highly variable in their width and where within genes they occurred. We sorted them into five general categories based on the genomic region spanned: (I) from an intergenic region into the 5’ end of a coding region, (II) from the 3’ end of a coding region into an intergenic region, (III) from within one coding region into an adjacent coding region, (IV) 90% of one or more coding regions and, (V) not overlapping with any coding region (Figure 4B–C). Category I and IV were the most common, comprising > 68% of the total. On average, peaks were 466 ± 343 bp wide. We also searched for sequence motifs both within the entire set of 61 peaks, and within the subsets of peaks, but no obvious common motif nor a significant AT skew emerged (30). To assess the reproducibility of our ChIP-seq analyses, we performed two additional biological replicates of the 30-minute time point. A majority of the 61 peaks initially found were again identified by MACS; 52 of the 61 peaks were called in at least three of the four total experiments, and 47 of the 61 peaks were called in all four experiments (Table S2). The representative peaks shown in Figure 4C are shown again in Figure S1A, with all four replicates displayed.
Figure 4. SymE-SPA ChIP-seq shows widely distributed and varied binding across the E. coli genome.
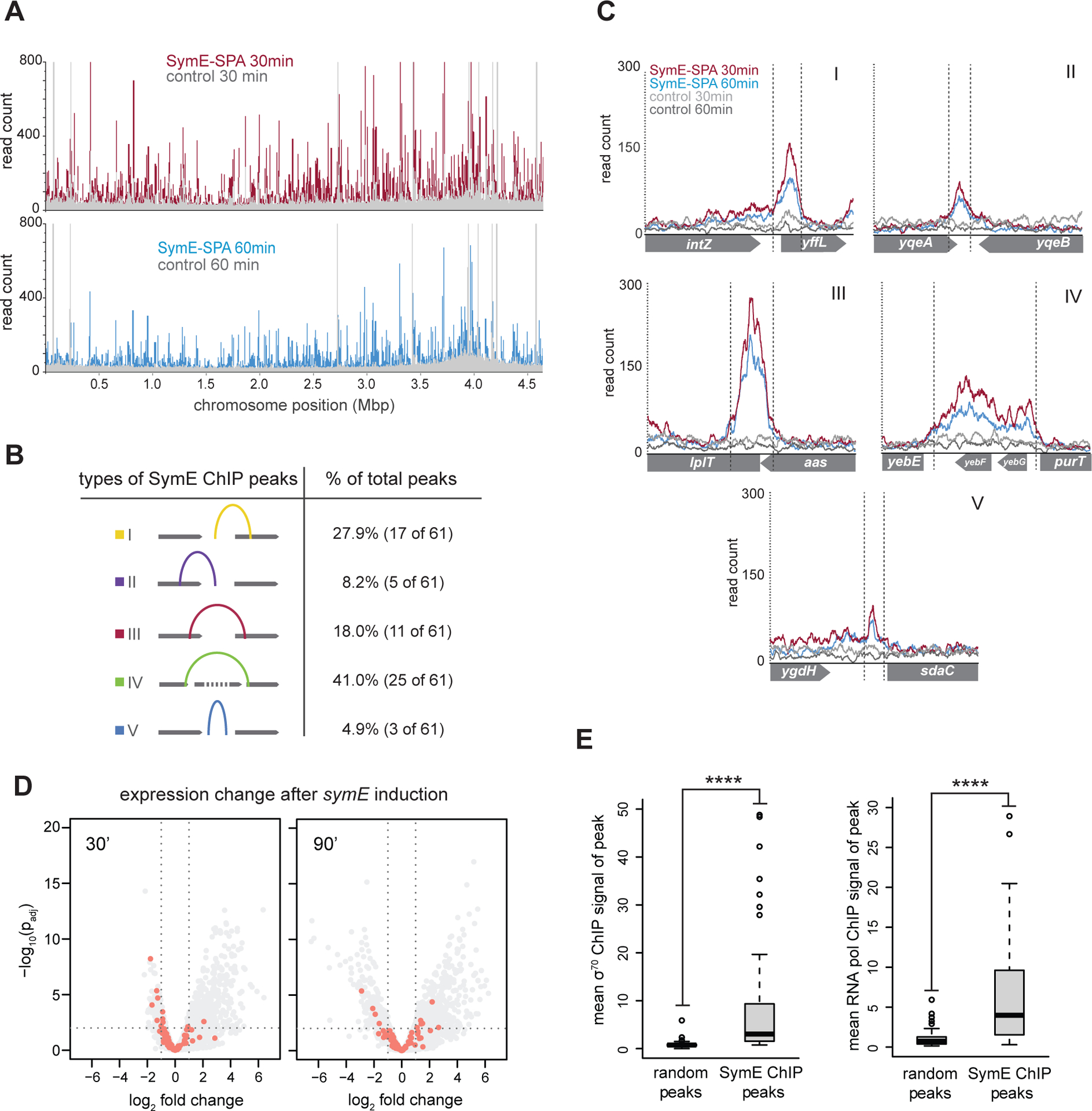
(A) SymE-SPA binding across the E. coli genome. (Top) The red trace shows ChIP-seq read counts after 30 minutes of symE-SPA expression; the grey trace shows the read counts after an untagged symE control was expressed. (Bottom) The blue trace shows ChIP-seq read counts after 60 minutes of symE-SPA expression, the grey trace shows read counts after untagged symE control expression.
(B) Summary of the different categories of SymE-SPA ChIP peaks.
(C) Examples of peaks from each category in (B).
(D) Same volcano plots as in Figure 3E, but with ChIP peak-associated genes shown in pink.
(E) Analysis of the overlap between SymE-SPA ChIP peak locations and signal from σ70 or RNA polymerase ChIP. When compared to a random set of locations, SymE-SPA ChIP locations had significantly (indicated with asterisks) higher mean signal from both σ70 and RNA polymerase ChIP.
To determine whether the 61 SymE-SPA peaks correlated with the genes whose expression changed the most in our RNA-seq analysis of symE overexpression, we replotted our RNA-seq data, highlighting genes with overlapping or upstream ChIP-seq peaks (Figure 4D). The genes associated with ChIP-seq peaks were slightly more likely to be downregulated than other genes after 30 minutes of SymE induction (Figure S1B). However, there were many regions of the genome bound by SymE whose expression did not change, suggesting that SymE is not a specific transcription factor. We therefore considered whether SymE localizes to regions of the genome that are simply more accessible, such as genes that are highly transcribed (31). To test this idea, we compared our SymE-SPA ChIP data to available σ70 and RNA polymerase ChIP datasets and found that the regions harboring SymE ChIP-seq peaks had significantly higher σ70 and RNA polymerase occupancy than 61 randomly chosen locations that were size matched to the SymE peaks (Figure 4E) (32, 33). This trend held for σ70 even if we relaxed our peak calling thresholds (Figure S1C). These findings indicate that SymE may have some preference for binding to actively transcribed regions of the genome. It is also important to consider that this ChIP experiment, as with most of our experiments and the prior study, was done using the ectopic expression of symE such that SymE is likely present at high concentrations. This high abundance likely means that high and low affinity binding sites for SymE cannot be distinguished.
To test whether SymE directly binds DNA, we used electrophoretic mobility shift assays (EMSAs). For these experiments, we purified His6-TEV-SymE and then cleaved off the His6 tag to produce untagged SymE. Gel filtration analysis of this purified SymE revealed a single dominant peak at ~57 kDa. Given the molecular weight of a SymE monomer (~12 kDa), this result suggests that SymE oligomerizes, possibly forming a tetramer, which would be consistent with other AbrB-family proteins that form various oligomeric complexes (Figure S2) (15). We first performed an EMSA for SymE binding to a 444 bp fragment of DNA corresponding to the ChIP peak identified within the lplT and aas coding regions (Figure 5A). The lplT-aas band decreased in intensity at ~0.5 μM SymE and then was completely lost at 1.0 μM SymE. Notably, the lplT-aas probe did not form discrete, lower mobility bands with increasing concentrations of SymE. Instead, no DNA was seen, suggesting that the DNA-SymE complexes were no longer entering the gel. To rule out possible DNase contamination in our SymE preparation or that SymE itself is a DNase, we repeated the experiment, but added SDS to each reaction immediately before loading the gel. In this case, all of the DNA entered the gel, indicating that it had not been degraded.
Figure 5. SymE binds DNA in vitro and is associated with the nucleoid in vivo.
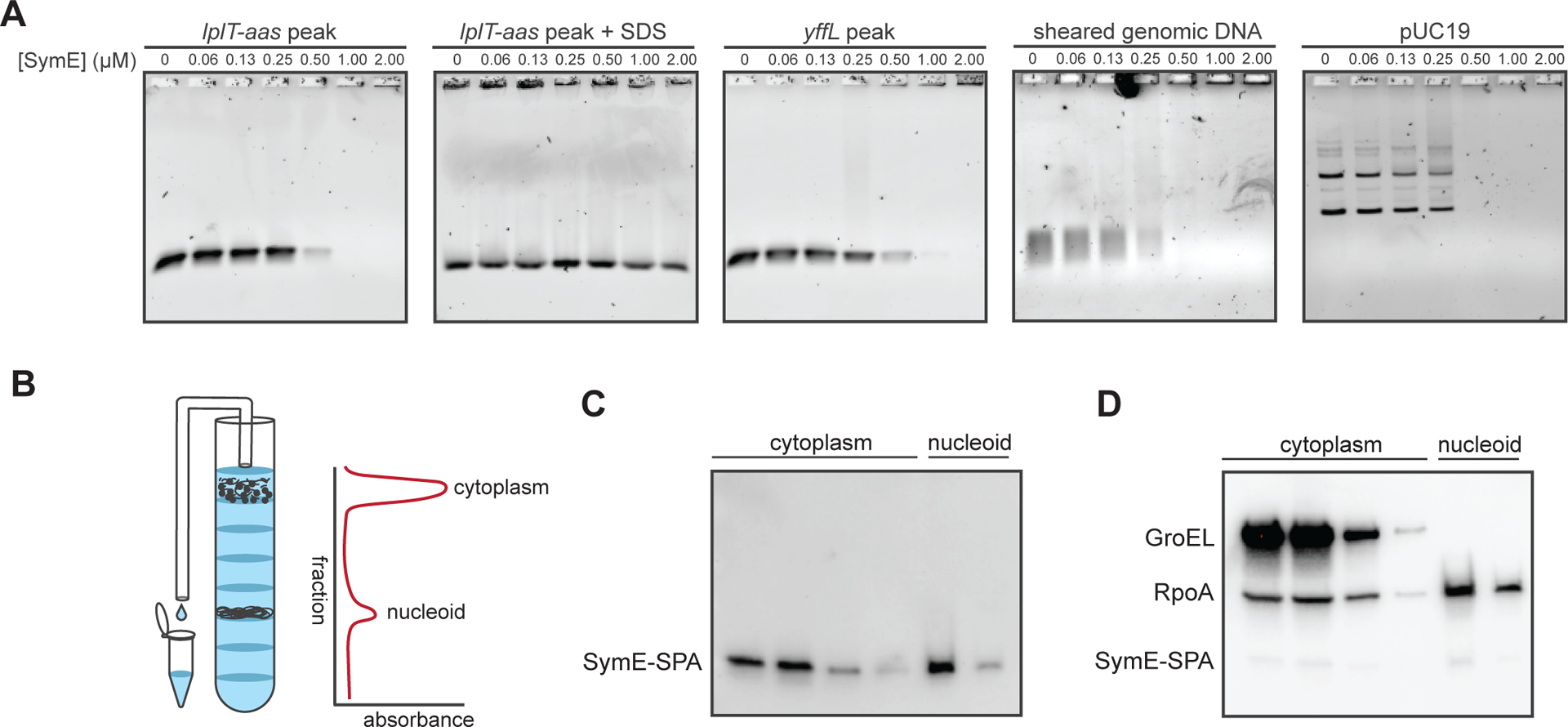
(A) Electrophoretic mobility shift assays of SymE binding to 4 ng of probe corresponding to the lplT-aas ChIP peak, the yffL peak, randomly sheared E. coli genomic DNA, or purified pUC19 plasmid.
(B) Schematic of how sucrose gradient fractions were collected. Fractions containing cytoplasmic or nucleoid-associated proteins were identified by their position in the gradient and by their increased absorbances at 260 and 280 nm.
(C) Western blot of collected fractions probed with an α-FLAG antibody (which binds to the FLAG portion of the SPA tag) showing that SymE-SPA localizes to the nucleoid and cytoplasm.
(D) Same blot as in (C), but re-probed for GroEL, a chaperone, and RpoA, the alpha subunit of RNA polymerase. The bands from SymE-SPA are still visible under the RpoA bands, though the signal is weaker as RpoA and GroEL are far more abundant.
An EMSA with another probe, this one corresponding to the ChIP-seq peak found within yffL, was almost identical to that of lplT-aas (Figure 5A). We also tested SymE binding to sheared ~200 bp fragments of E. coli genomic DNA. We found that SymE bound the sheared DNA with similar apparent affinity as the DNA corresponding to ChIP peaks. This result suggests that SymE does not strongly prefer to bind DNA in vitro corresponding to the ChIP peaks we tested and also that SymE has no preference for methylated genomic DNA over unmethylated PCR product. Finally, we performed an EMSA on closed circular DNA (the plasmid pUC19) and again found that the probe first showed diminished intensity at ~0.5 μM SymE and then was fully retained within the wells at higher concentrations.
Next, we assessed whether SymE is localized to the nucleoid in vivo. To this end, we used a sucrose gradient to separate the nucleoid from the cytoplasm in extracts taken from cells producing a SPA-tagged copy of SymE at its native locus. This strain also has mutations in the symR promoter which prevent expression of symR antisense RNA and increase SymE levels to aid in detection (11). Cells were treated with the DNA damaging agent mitomycin C (MMC), which is known to induce symE expression (11). After 90 minutes of treatment with MMC, we loaded gently lysed cells onto a sucrose gradient and centrifuged. Fractions corresponding to the cytoplasm or nucleoid were identified and isolated based on their position in the gradient and absorbance at 260 and 280 nm (Figure 5B). A Western blot for the FLAG portion of the SPA tag indicated that SymE-SPA is found in both the cytoplasm and the nucleoid (Figure 5C). We re-probed the blot with antibodies against GroEL, a chaperone, and RpoA, the alpha subunit of RNA polymerase (Figure 5D). GroEL was localized only to the cytoplasm, as expected, whereas RpoA was found in both the cytoplasmic and nucleoid fractions, similar to SymE-SPA. Thus, SymE-SPA localizes to the nucleoid in a manner comparable to RpoA, which is known to associate with the nucleoid.
We conclude that SymE directly binds DNA, consistent with its predicted structural similarity to AbrB-family proteins. The shifting of bound DNA to a form that could not enter the gel in our EMSAs likely reflects the oligomerization of SymE. Whether the bound complex represents a single tetramer of SymE bound to DNA or involves higher-order assemblies is not clear. The binding of SymE was generally non-specific in vitro, consistent with its apparent lack of sequence specificity in our ChIP-seq analyses and the notion that DNA accessibility may be a major determinant of SymE binding in vivo.
symE expression leads to nucleoid condensation
To further probe SymE function and elucidate the mechanistic basis of its toxicity when overexpressed, we performed time-lapse microscopy and examined changes in nucleoid structure and cell morphology (Figure 6A). To visualize nucleoid structure in living cells, we used an E. coli MG1655 strain producing GFP-tagged HupA, a subunit of the histone-like, nucleoid-associated protein HU, from its native chromosomal location (34). We introduced into this strain a plasmid for expressing symE from an arabinose-inducible promoter or an empty vector. To quantify condensation, we calculated the percentage of the total area of cells taken up by their nucleoid, as measured by HU-GFP staining, at each time point (Figure 6B). Cells with an empty vector grew and divided normally with nucleoids occupying a large percentage of the cell, as expected. In contrast, for cells expressing symE, the nucleoids were condensed within 30 minutes of induction and remained condensed throughout the 180-minute time course.
Figure 6. symE overexpression causes nucleoid condensation.
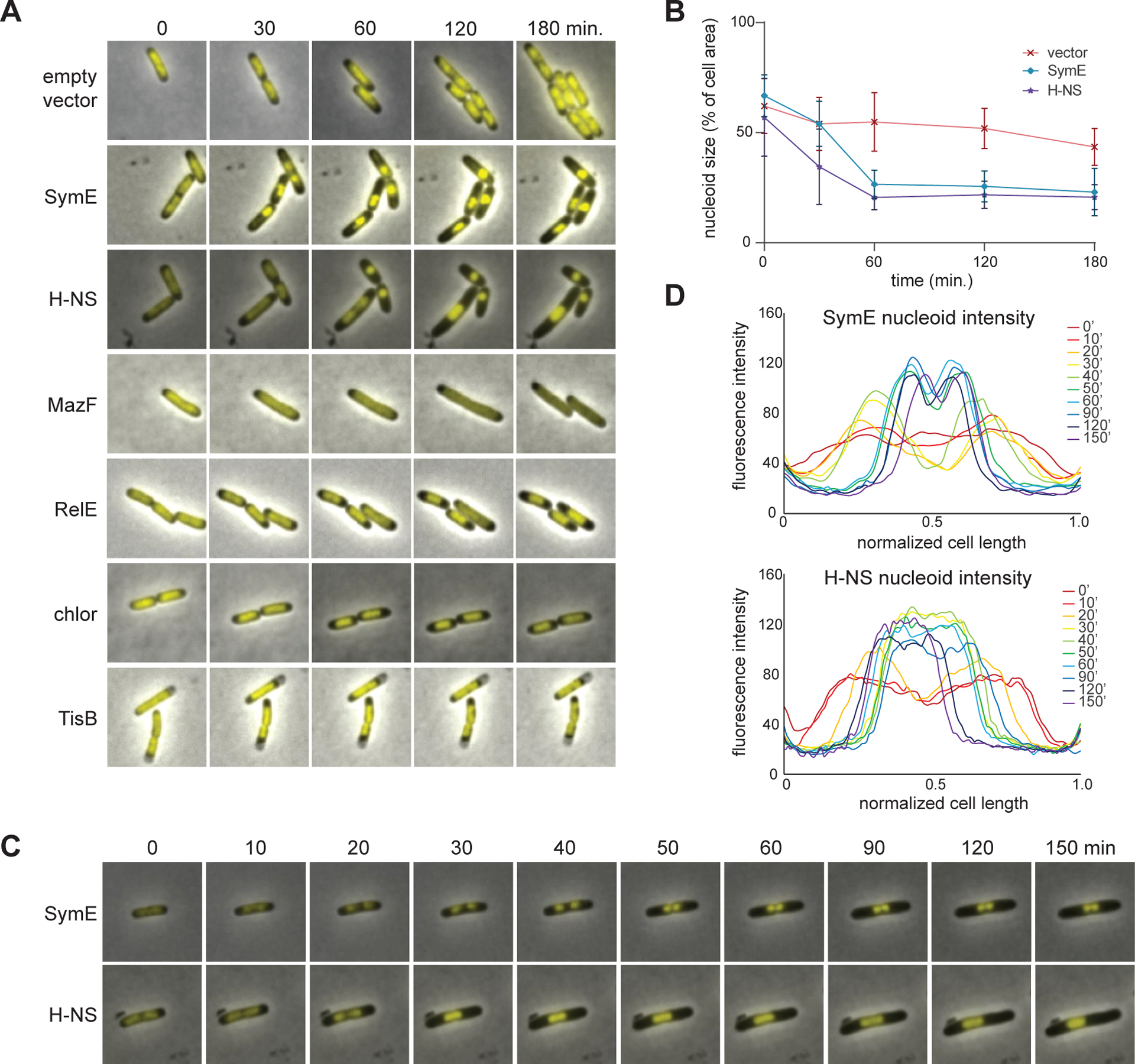
(A) Cells expressing each of the genes indicated or those treated with chloramphenicol were examined by time-lapse microscopy with HU-GFP used to stain the nucleoid.
(B) Quantification of the degree of nucleoid compaction over time in cells harboring an empty vector, or expressing symE or hns. Nucleoid size is presented as the percent of total cell area occupied by the nucleoid. n ≥ 45 cells for each data point. Quantification of all treatments is shown in Figure S3D.
(C) Additional time-lapse imaging of cells producing SymE or H-NS to illustrate how distinct nucleoids aggregate.
(D) Line graph quantifying the fluorescence intensity across the longitudinal axes of the cells in panel (C).
The nucleoid condensation in cells expressing symE did not always happen in a synchronous manner within a given cell. While the nucleoids in some cells condensed all at once, in many cells certain regions condensed first, seemingly at random. It is possible that two nucleoids existed prior to SymE accumulation and then condensed separately from one another. These condensed domains or nucleoids could then aggregate with each other (Figure 6C–D). Additional examples of this behavior are illustrated in Figure S3A. As the nucleoids were condensing after overproducing SymE, some cells finished a round of cell division, but no subsequent divisions were seen. Once condensed, the nucleoids were never seen to decondense.
Nucleoid condensation like that seen with SymE has also been reported for cells overproducing H-NS, a nucleoid-associated protein in E. coli (18). High levels of H-NS are lethal, and cause nucleoids to condense into highly compact spheres, halting bulk transcription and translation. To directly compare SymE to H-NS, we overexpressed hns from a plasmid in the strain producing HU-GFP. Note that symE was expressed from a plasmid with a higher copy number and stronger RBS (pBAD24) than the vector which hns was on (pBAD30) (35). The growth of cells in both strains stopped at comparable time scales as seen by microscopy indicating a similar amount of toxicity between the two strains. The nucleoid compaction caused by H-NS looked remarkably similar to that seen when overproducing SymE (Figure 6A–B, Figure S3B) except that H-NS appeared to compact nucleoids in a more uniform, coordinated fashion than SymE did; and two regions that initially condensed separately, could fuse into one uniform mass (Figure 6C–D). When expressed from the lower copy vector pBAD30, SymE did eventually condense the nucleoid, but it took longer to halt cell growth (Figure S3C).
We also compared cells overexpressing symE or hns to those overexpressing toxins from TA systems and to cells treated with chloramphenicol, which has been reported to trigger nucleoid condensation (Figure 6A, Figure S3D) (36). Inducing MazF, a ribosome-independent endoribonuclease toxin of the MazEF system, caused nucleoids to become relaxed and continue to fill most of the cell, even as cell division was disrupted. RelE, a ribosome-dependent endoribonuclease of the RelBE system, caused a modest condensation of the nucleoid, though not to the degree seen with SymE or H-NS. Additionally, the RelE nucleoids retained the capacity to fully decondense (Figure S3E). The nucleoids from cells treated with chloramphenicol behaved like those expressing relE. Finally, nucleoids in cells expressing the SOS-induced type I toxin tisB retained a relatively constant size and remained static within the cytoplasm, likely due to the rapid onset of cell death in these cells. Overall, none of these toxins, nor chloramphenicol, caused a nucleoid compaction phenotype comparable to H-NS or SymE. We conclude that, as with H-NS, SymE is directly compacting the nucleoid.
Double strand breaks form in the DNA of cells expressing symE or hns, and symE inhibits bulk DNA and RNA synthesis
DNA-protein roadblocks can cause stalled replication forks and DNA damage associated with that stalling, including double strand breaks (37). We hypothesized that if excessive amounts of SymE binding to DNA was sufficient to drive nucleoid compaction, it may also disrupt DNA replication and produce a signal of DNA damage. In support of this hypothesis, we found that SymE induction triggered the expression of many DNA damage inducible SOS response genes in our RNA-seq data (Figure 7A) (38). This up-regulation was statistically significant when comparing the SOS genes to the same number of randomly selected genes at both 30- and 90-minutes post symE induction (Figure S4).
Figure 7. Overproducing SymE causes double-strand breaks, and inhibits DNA and RNA synthesis.
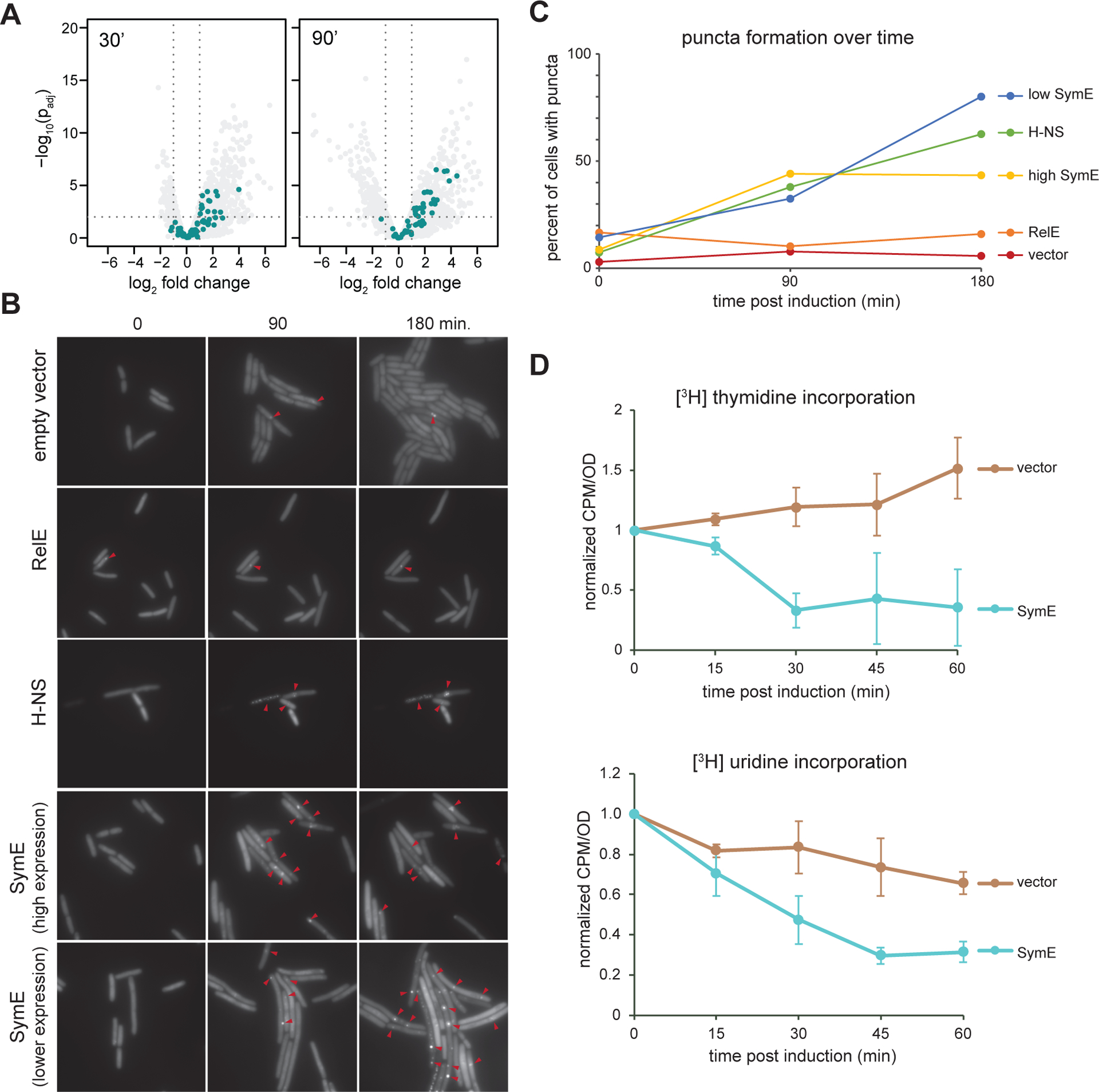
(A) Same volcano plots as in Figure 3E, but with SOS-induced genes shown in teal.
(B) Time-lapse microscopy of cells producing Gam-GFP, which forms foci at double-strand breaks, and expressing high levels of symE, lower levels of symE, relE, or hns.
(C) Graph of percentage of cells with at least one Gam-GFP focus for each condition over time. n ≥ 85 cells for each data point.
(D) [3H]-thymidine (top) and [3H]-uridine (bottom) incorporation assays, comparing the counts per minute (corresponding to incorporated [3H]) per OD600 of cells at various time points post induction of E. coli cells harboring an empty vector versus a symE expression vector (the “high symE” vector from (B) and (C)). n ≥ 3 for all conditions; error bars indicate standard deviation.
To assess whether SymE induction leads to double strand break (DSB) formation, we performed time-lapse fluorescent microscopy on strains producing GFP-tagged Gam. Gam is a protein derived from phage Mu that binds to the ends of double stranded DNA, and GFP-tagged Gam forms visible puncta in cells upon the creation of DSBs (39). We examined Gam-GFP puncta formation in cells expressing either high or low levels of symE, hns, or relE, and compared each to a strain with an empty vector (Figure 7B). In cells harboring the empty vector, we saw very few puncta as cells grew and divided over the course of 180 minutes, with the percentage of cells having at least one focus remaining relatively low throughout (Figure 7C). The occasional breaks seen are likely stochastic and expected during normal cell growth (39). In cells producing high levels of SymE, many more Gam-GFP puncta were seen, suggestive of increased DNA damage. At lower levels of symE expression, there were initially fewer Gam-GFP puncta than with higher SymE levels. However, by 180 minutes, there were many more Gam-GFP puncta than seen with higher symE expression. The lower expression of symE may lead to less initial nucleoid condensation and allow for more DNA replication to occur; as SymE gradually accumulated in these cells, it likely produced the extensive DNA damage suggested by the abundant Gam-GFP foci. In contrast, higher levels of SymE may cause a more rapid cessation of DNA replication, limiting the number of replication forks and subsequent Gam-GFP foci that can accumulate. As with SymE, when the nucleoid was condensed by overexpressing hns, most cells had at least one Gam-GFP focus by 180-minutes of expression (Figure 7B–C). In contrast, most cells expressing relE did not have Gam-GFP foci, with the percentage of cells having at least one focus only slightly higher than the empty vector control (Figure 7C). As RelE causes nucleoids to modestly condense via a mechanism other than direct DNA binding, this result suggests that condensation of the nucleoid alone does not necessarily cause DSBs to form and that the more significant nucleoid condensation, like that driven by SymE, is necessary to produce DSBs.
Our findings indicate that high levels of SymE drive nucleoid condensation, which likely blocks both DNA replication and bulk transcription. To directly test this hypothesis, we performed [3H]-thymidine and [3H]-uridine incorporation assays in which we monitored uptake of radiolabeled thymidine or uridine over time after symE induction (Figure 7D). Indeed, we found that the ectopic overexpression of symE caused a rapid and significant decrease in the incorporation of both thymidine and uridine over time when compared to an empty vector control, indicating that SymE inhibits DNA replication and transcription, likely by compacting the nucleoid.
Discussion
SymE is a nucleoid-associated protein in E. coli
SymE was originally hypothesized to be an endoribonuclease as the abundance of several transcripts decreases upon SymE induction. Using RNA-seq and [3H]-uridine incorporation assays, we also found that transcript levels decreased significantly after inducing SymE, but found no evidence of cleavage throughout the transcriptome. Instead, our results collectively indicated that SymE binds DNA, consistent with its predicted structural similarity to AbrB family proteins. Our ChIP-seq and EMSAs both indicated that SymE can bind DNA without apparent sequence specificity across the genome, similar to the AbrB-fold protein Sso7c4 from Sulfolobus solfataricus (17). However, SymE did form discrete peaks within the genome. What drives SymE to these particular locations of the genome is not yet clear. SymE binding was often associated with highly expressed genes and recent work has suggested that such regions of the genome may get localized to the periphery of the nucleoid (31), possibly making them more available to and readily bound by SymE. Alternatively, or in addition, there may be sequence features or structural elements of the genome that we could not identify that promote SymE binding.
We found that SymE can oligomerize and cause DNA to be retained in very high molecular weight complexes in EMSAs. The jump from DNA being mostly unbound at ~0.25 μM SymE to mostly bound at ~0.5 μM SymE may indicate some degree of cooperativity in SymE binding. The oligomerization of SymE may give it multivalency that could promote crosslinking of DNA fragments, thereby inhibiting large DNA-protein complexes from entering the agarose gel matrix. Based on the large and highly variable sizes of our ChIP-peaks, it is also possible that SymE oligomers can spread across DNA, or interact with other key binding partners in the cell. The precise nature and stoichiometry of the high molecular weight SymE-DNA complexes seen in vitro is not yet clear, but they seem likely to underpin the nucleoid compaction capabilities of SymE. A similar aggregation of protein and DNA likely occurs when symE is expressed in vivo. Indeed, when overproduced, SymE triggered nucleoid condensation within 30 minutes, with relatively small, tight nucleoids persisting over the duration of our time-lapse studies. In some cases, the entire nucleoid condensed at once, but in others it occurred non-uniformly with some regions of the nucleoid condensing first and then aggregating later with other condensed regions. Overproducing H-NS, a well-studied nucleoid-associated protein, caused a similar degree of condensation as SymE on a comparable time scale at levels of expression that enable both proteins to stop cell growth. However, unlike SymE, H-NS almost always caused the nucleoid to condense into a symmetric sphere.
High levels of SymE are toxic, inhibit DNA and RNA synthesis, and induce double-strand breaks
In addition to driving nucleoid condensation, overproducing SymE also led to substantial changes in gene expression. However, there were no obvious patterns or functionally related sets of genes that changed, and there was little correlation between the genes whose expression changed and those to which SymE bound in our ChIP-seq. We also saw that bulk RNA synthesis was inhibited upon SymE induction. Thus, we favor the notion that many of the gene expression changes we observed stem from SymE forming protein “roadblocks” on the DNA or disrupting chromosome accessibility that then non-specifically disrupts transcription and translation, consistent with prior reports showing decreases in transcription and bulk translation following SymE induction (11, 37, 40).
SymE-dependent obstructions on DNA and the prolonged condensation of the nucleoid likely also cause DNA damage by blocking replication forks. In E. coli, DNA replication can initiate from oriC multiple times per cell cycle. If two independent forks moving in the same direction collide after encountering SymE directly or due to the condensation of the nucleoid, it may produce double-strand breaks. We found that cells overproducing SymE accumulated multiple Gam-GFP foci, and had increased expression of SOS induced genes, consistent with this model. SymE is unlikely to be directly cleaving DNA as we saw no DNase activity in vitro during our EMSAs and it harbors homology to DNA-binding proteins, not nucleases. Although E. coli cells can tolerate some double-strand breaks, using homologous recombination to drive repair, we speculate that the excessive number of DSBs caused by SymE overproduction contributes to the observed loss in viability, in addition to its effects on transcription and DNA replication.
Functions of SymE
The functions and a physiological role for SymE remain unclear. Although often classified as a type I TA system, the evidence to support this assertion is limited. While symE/symR bears some similarity to type I TA systems given that symE is toxic when overexpressed and is regulated by an antisense RNA, cells engineered to not express symR are viable, even after SOS induction (11). For canonical type I TA systems, eliminating the antitoxin RNA typically leads to activation of the toxin and cell death (2, 41, 42). The viability of the symR mutant could reflect a requirement for additional signals that promote SymE accumulation. As noted, symE is under SOS control and prior work showed that the Lon protease may drive SymE degradation (11). However, ΔsymRΔlon cells treated with the DNA damaging agent MMC, which express and accumulate SymE, grew comparably to ΔsymERΔlon cells in the same conditions (11). Additionally, unlike canonical type I TA systems, the antitoxin symR is more stable than the toxin SymE (11).
Regardless of its categorization as a TA system or not, a physiological role for SymE remains unclear. All of the experiments performed here were done in cells overexpressing symE and conditions in which SymE accumulates to such levels are not known. Under some conditions SymE is likely to have a critical function, as homologs are widely prevalent in bacteria and found in most bacterial phyla. Prior work showed that symE is under LexA control, and it was shown early on to be DNA damage-inducible (receiving the designation of dinL) and part of the SOS regulon (12, 13). One recent study found that a ΔsymER strain has a modest growth defect after treatment with the alkylating agent styrene oxide (14), but symE is not required to survive other DNA damaging agents such as mitomycin C (11).
One potential hint of SymE’s function comes from our phylogenetic studies in which we found that symE often colocalizes with restriction-modification systems in bacterial genomes, specifically the mcrBC system. As systems that protect against foreign DNA often colocalize with one another in so-called defense islands (22, 43), it is possible that SymE protects against phage or plasmids. SymE could also function in restriction alleviation, a phenomenon in which unmodified regions of the genome, e.g. newly synthesized DNA following DNA repair, are protected against a restriction enzyme by compacting the genome (44). Such a function has been suggested previously for the nucleoid-associated protein StpA, a paralog of H-NS (45). Whatever the case, our work strongly supports the notion that SymE’s function will involve the DNA-binding and possibly the nucleoid compaction capabilities documented here.
Methods
Strains, plasmids, and oligos
All strains, plasmids and oligos used are listed in Tables S3, S4, and S5, respectively.
Plasmid construction
pMKT26 was made by first removing the maltose binding protein coding sequence from plasmid ML333 via round-the-horn PCR, leaving in the 6x-His tag and TEV cleavage site 5’ of the protein coding region to generate pMKT25. pMKT25 was then PCR linearized and the symE coding sequence was PCR amplified out of the E. coli MG1655 genome and inserted directly 3’ of the TEV cleavage site via Gibson assembly. To make pMKT57, pBAD30 was PCR linearized and the symE coding sequence was PCR amplified out of the E. coli MG1655 genome and inserted 3’ of the arabinose promoter. pMKT59 and pMKT91 were made in a similar fashion, with the tisB and hns coding sequences being PCR amplified from the E. coli MG1655 genome and inserted into the pBAD30 backbone via Gibson assemble, respectively.
Growth conditions
For cloning, strain maintenance, and ChIP-seq, E. coli was grown in LB medium at 37 °C. Cells for RNA-seq and microscopy were grown in M9 minimal media (6.4 g/L Na2HPO4-7H2O, 1.5 g/L KH2PO4, 0.25 g/L NaCl, 0.5 g/L NH4Cl medium supplemented with 0.05% casamino acids, 0.4% glycerol, 0.1 mM CaCl2, 1 mM MgSO4, and 1 μg/mL thiamine) at 37 °C. Protein expression was performed in 2x YT (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl). Unless otherwise specified the following concentrations of antibiotics were used for plasmid maintenance and strain selection (liquid; plate): carbenicillin (50 μg/mL; 100 μg/mL), chloramphenicol (20 μg/mL; 30 μg/mL), or kanamycin (30 μg/mL; 50 μg/mL)
Bioinformatics
To generate a phylogeny of the AbrB family, distant SymE homologs were identified using HHPred (46). Hits with probability > 80 were used as seeds for individual HMMER searches (e value cutoff = 0.01). Hits were pooled and highly similar sequences were eliminated using CD-HIT (0.6 cutoff, word length = 4). The resulting 1,219 sequences were aligned using MUSCLE (2 iterations) (47). Best model (WAG) was selected using ModelTest-NG and the tree was built using Fasttree (48, 49). The final tree was pruned to remove poorly supported branches.
To determine the distribution of SymE across bacteria, a bacterial species tree was generated based on a concatenated alignment of 27 ribosomal protein genes. HMMER was used to identify and align orthologs of these genes from the ProGenomes Database. The concatenated alignment was manually trimmed to remove positions represented in < 50% of sequences and positions with < 25% conservation, and a tree was generated using FastTree, and rooted on Cyanobacteria (49). SymE homologs in these species were identified and aligned using JackHMMER (5 iterations, query threshold 10). LexA boxes were identified within intergenic regions upstream of each SymE (23). McrB homologs were identified using JackHMMER, and classified as near SymE if they were within 10 genes of SymE in the gene annotation.
RNA-sequencing
Sample preparation, RNA extraction, library preparation and sequencing were performed in triplicate.
Sample preparation
E. coli MG1655 PCP18-araE cells harboring either pBAD24 or pBAD24::symE were grown overnight in M9 media containing carbenicillin and then back diluted into fresh M9 carb to OD600 0.02. Cultures were then grown to OD600 0.5 and 0-minute time point samples were collected. At every time point, OD600 was measured and samples were taken to monitor cell survival post induction and to extract RNA. Cells were then induced with arabinose at 0.02%. Further samples were taken after 30- and 90- minutes of induction. To measure cell survival, 200 μL of culture at each time point was taken and then a log10 dilution series from 100 to 10−5 was made. 5 μL of each dilution was then spotted on LB carbenicillin agar plates. For RNA samples, at each time point 1350 μL of culture was added to a tube that contained 150 μL of stop solution (95% ethanol, 5% acid-buffered phenol). Tubes were inverted to mix and then spun down for 30 seconds at 13,000 rpm in a tabletop centrifuge. Supernatant was removed and pellets were flash frozen in liquid nitrogen and then stored at −80 °C until ready for use.
RNA extraction
To extract RNA, 400 μL of Trizol reagent (Invitrogen) pre heated to 65 °C was added to pellets, vortexed and then placed on a thermomixer (Eppendorf) at 65 °C for 10 minutes at 2000 rpm. Samples were then frozen at −80 °C for at least 10 minutes. Samples were then thawed at RT and spun for 5 minutes at 13,000 rpm in a tabletop centrifuge. Supernatant was removed and added to 400 μL of ethanol. Samples were then applied to a Zymo DirectZol spin column and centrifuge for 30 seconds at 10,000 rpm in a tabletop centrifuge. Columns were washed 2x with 400 μL of PreWash buffer and 1x with 700 μL of RNA Wash Buffer. After discarding flow through, samples were centrifuged for an additional 2 minutes to remove residual wash buffer. 90 μL of DEPC treated water was added to the column matrix and the column was incubated at RT for 2 minutes, then centrifuged and collected. 10 μL of 10x Turbo DNase buffer (Invitrogen) was added the sample and then 2 μL of Turbo DNase I was added and samples were incubated at 37 °C for 20 minutes. Samples were then brought to 200 μL with DEPC water, then 200 μL of acid-phenol:chloroform IAA, pH 4.5 (Invitrogen) was added and samples were vortexed for 1 minute. They were then centrifuged at 13,000 rpm in a tabletop centrifuge for 10 minutes. The top (aqueous) layer was extracted and precipitated with 20 μL of 3 M NaOAc, 2 μL glycogen and 600 μL ice cold ethanol. Samples were then incubated at −20 °C for at least 1 hour. Samples were then centrifuged at 13,000 rpm in a table top centrifuge at 4 °C for 30 minutes. Pellets were washed with 500 μL 70% ice cold ethanol and centrifuged for an additional 5 minutes. This wash step was repeated. Pellets were then air dried for 5 minutes and resuspended in 35 μL of DEPC water. Concentration and integrity of the RNA samples were assessed with a Nanodrop and by running out the samples on 6% TBE-Urea acrylamide gels.
Library preparation
RNA-seq libraries were prepared as described in Culviner et al (24). rRNA was removed either using the Ribo-Zero rRNA Removal Kit for Bacteria (Illumina) or with a method recently described in Culviner et al (50). For samples prepared with Ribo-Zero, 225 μL of provided magnetic beads were washed with 225 μL of DEPC water twice and resuspended in 65 μL of provided resuspension solution and 1 μL of RiboGaurd RNase inhibitor was added. Then, 2.5–5 μg total RNA was mixed with 4 μL provided reaction buffer, 10 μL rRNA Removal Solution and brought to 40 μL total volume with DEPC water. This reaction was incubated for 10 minutes at 68 °C followed by 5 minutes at RT. These reactions were mixed with the previously prepared magnetic beads by pipetting followed by vortexing, and allowed to incubate for 5 minutes at RT. Bead mixtures were then vortexed briefly and incubated at 50 °C for 5 minutes. Reactions were placed on a magnet and the now rRNA depleted supernatant was transferred to a new tube. Samples were brought to 180 μL total volume with DEPC water and precipitated with 18 μL of 3 M NaOAc, 2 μL glycogen and 600 μL ice cold ethanol. Samples were then incubated at −20 °C for at least 1 hour. Samples were then centrifuged at 13,000 rpm in a tabletop centrifuge at 4 °C for 30 minutes. Pellets were washed with 500 μL 70% ice cold ethanol and centrifuged for an additional 5 minutes. This wash step was repeated. Pellets were then air dried for 5 minutes and resuspended in 10 μL of DEPC water.
To fragment RNA, 1 μL of 10x RNA fragmentation buffer (Ambion) was added to 9 μL of the resuspended RNA. Samples were mixed briefly by vortexing and spun down then incubated at 70 °C for 8 minutes. Samples were then placed on ice and 1 μL of RNA fragmentation stop solution (Ambion) was added. Reactions were then brought to 20 μL volume with DEPC water and precipitated with 2 μL of 3 M NaOAc, 2 μL glycogen and 600 μL ice cold ethanol. Samples were incubated at −20 °C for at least 1 hour then centrifuged at 13,000 rpm in a tabletop centrifuge at 4 °C for 30 minutes. Pellets were washed with 200 μL of 70% ethanol and centrifuged for an additional 5 minutes, then allowed to dry and were resuspended in 6 μL of DEPC water.
First strand synthesis was performed using 1 μL random primers (Invitrogen) added to 6 μL of fragmented RNA. Reactions were heated to 65 °C for 5 minutes then placed on ice for 1 minute. 4 μL 5x first strand synthesis buffer (Invitrogen), 2 μL of 100 mM DTT, 10 mM dNTPs, 4 μL DEPC water and 1 μL of Superase-In was added to each reaction and allowed to sit at RT for 2 minutes. 1 μL of Superscript III (Invitrogen) was added to reactions which were then heated to 25 °C for 10 minutes, 50 °C for 1 hour, 70 °C for 15 minutes and then held at 4 °C. Reactions were then brought to 200 μL with DEPC water. 200 μL of phenol-chloroform isoamyl alcohol pH 8 was added and reactions were vortexed for 1 minute then centrifuged for 10 minutes at 13,000 rpm in a tabletop centrifuge at 4 °C. ~200 μL of the top (aqueous) layer was extracted and then precipitated with 2 μL glycogen, 18.5 μL NaOAc, 600 μL ice cold ethanol, and incubated at −20 °C for 1 hour. Samples were then spun at 4 °C for 30 minutes at 13,000 rpm in a tabletop centrifuge. Pellets were washed 2x with 500 μL of ice-cold ethanol, then air dried and resuspended in 104 μL of DEPC water. Second strand synthesis was done by adding 30 μL 2x second strand synthesis buffer (Invitrogen), 4 μL 10 mM dNTPs (with dTTP replaced by dUTP), 4 μL 5x first strand buffer and 100 mM DTT to the 104 μL first strand reaction. Samples were then placed on ice for 5 minutes. After incubation on ice, 1 μL of RNase H (NEB), 1 μL of E. coli DNA Ligase (NEB), and 4 μL of E. coli DNA Pol I (NEB) was added to each reaction which were then mixed and incubated at 16 °C for 2.5 hours.
To clean up the second strand reactions, 100 μL of Ampure XP beads per reaction were placed in a magnetic rack and supernatant was replaced with 450 μL of PEG 8,000 20%/2.5 M NaCl. The PEG/bead mix was then added to each second strand synthesis reaction and samples were mixed by pipetting and vortexing briefly and then allowed to incubate at RT for 5 minutes. Samples were then placed on a magnetic rack and allowed to clear. Supernatant was removed and beads were washed 2x with 500 μL of 80% ethanol. All ethanol was carefully removed and beads were allowed to dry completely. Beads were then resuspended in 50 μL of elution buffer (10 mM Tris-Cl, pH 8.5).
End repair was done by adding 10 μL T4 DNA ligase buffer (NEB), 4 μL 10 mM dNTPs, 5 μL T4 DNA polymerase (NEB), 1 μL Klenow DNA polymerase (NEB), 5 μL T4 PNK (NEB) and 25 μL DEPC water to the 50 μL resuspended beads. Samples were then incubated at 25 °C for 30 minutes. After incubation, 300 μL PEG 8,000 20%/2.5 M NaCl was added to each reaction which were then mixed and allowed to incubate at RT for 5 minutes. Samples were placed on a magnetic rack and allowed to clear. Supernatant was removed and beads were washed 2x with 500 μL 80% ethanol. All ethanol was carefully removed and beads were allowed to completely dry. Beads were then resuspended in 32 μL elution buffer.
3’ Adenylation was done by adding 5 μL NEB buffer 2, 1 μL 10 mM dATP, 3 μL Klenow 3’ to 5’ exo- (NEB) and 9 μL DEPC water to the 32 μL resuspended beads from the previous step. Reactions were then incubated at 37 °C for 30 minutes. 150 μL of PEG 8,000 20%/2.5 M NaCl was added to each reaction which were then incubated at RT for 5 minutes. Samples were then placed on a magnetic rack and allowed to clear. Supernatant was removed and beads were washed 2x with 80% ethanol. All ethanol was carefully removed and beads were allowed to completely dry. Tubes were then removed from the magnetic rack and beads were resuspended in 20 μL of elution buffer and allowed to incubate at RT for 5 minutes. Tubes were then placed back on the magnetic rack and allowed to clear. 10 μL of supernatant was removed and stored in case of downstream failure. The beads were then resuspended in the remaining supernatant and the bead/library mixture was used in the next steps.
Next, paired end sequencing adaptors were ligated onto the library. To do that, adaptors were annealed to each other by mixing 50 μL of each 200 μM adaptor stock and then heating them to 90 °C in a PCR machine and cooling by 2 °C/minute for 30 minutes. Reactions were then placed on ice and diluted with 100 μL DEPC water to make 50 μM stocks. Working stocks of adaptors were made by diluting stocks to 5 μM. 1 μL of 5 μM adaptor mix and 10 μL of Blunt/TA ligase master mix (NEB) was added to the bead/library mixture from the previous step and reactions were incubated at 25 °C for 20 minutes. Then 60 μL of PEG 8,000 20%/2.5 M NaCl was added to reactions and they were further incubated at RT for 5 minutes. After incubation, sample tubes were placed in magnetic racks and the solution was allowed to clear. Supernatant was removed and beads were washed 2x with 500 μL of 80% ethanol. All ethanol was then carefully removed and beads were allowed to completely dry. Tubes were removed from magnetic racks and beads were resuspended in 19 μL of 10 mM Tris pH 8.0, 0.1 mM EDTA. Samples were incubated at RT for 5 minutes then returned to the magnetic rack and allowed to clear. Supernatant containing the library was carefully removed and stored for later, and beads were discarded. The second strands of the libraries containing dUTP were removed by taking 19 μL of library dsDNA and adding 1 μL of USER enzyme (NEB). Reactions were then incubated at 37 °C for 15 minutes then at 95 °C for 5 minutes.
Libraries were amplified by mixing 10 μL of library template, 2 μL of global primers, 2 μL of barcoded primers, 11 μL DEPC treated water, and 25 μL 2x KAPA master mix and running them through the following PCR procedure: 98 °C for 45 seconds, 98 °C for 15 seconds, 60 °C for 30 seconds, 72 °C for 30 seconds and 72 °C for 1 minute, with steps 2–4 repeating for 9–12 cycles. Cycle number was determined based on trial amplifications run using 10 μL reactions.
To select for the desired size of 200 to 350 bp, two sets of Ampure beads were first prepared per sample. The first 100 μL of beads were placed on a magnetic rack, supernatant was removed and beads were resuspended in 120 μL of 20% PEG 8,000/2.5 M NaCl. The second 100 μL of beads was placed on a magnetic rack, supernatant was removed and beads were resuspended in 40 μL 20% PEG 8,000/2.5 M NaCl. 50 μL of amplified library was brought to 200 μL volume with 150 μL water. The now 200 μL of library reaction was added to the beads resuspended in 120 μL of PEG solution which was then vortexed and incubated at RT for 5 minutes. Tubes were then placed on a magnetic rack and the solution was allowed to clear. The library will now be located in the supernatant. The supernatant was removed and added to the beads prepared in 40 μL of 20% PEG 8,000/2.5 M NaCl, samples were vortexed and incubated at RT for 5 minutes. Tubes were then placed in a magnetic rack and the solution was allowed to clear. Supernatant was removed and beads were washed 2x with 500 μL of 80% ethanol. All ethanol was carefully removed and beads were allowed to completely dry. Beads were then resuspended in 11 μL of elution buffer and allowed to sit for 5 minutes at RT. Tubes were then returned to magnetic racks and allowed to clear. Supernatant, now containing the appropriately sized library, was removed and submitted for paired-end sequencing performed on an Illumina NextSeq500 at the MIT BioMicroCenter.
Cleavage and gene expression analysis
RNA-seq read mapping and calculation of cleavage ratios of symE:empty vector and mazF:empty vector were done as described previously (24). Briefly, paired end reads were mapped to the E. coli MG1655 genome using Bowtie2 (51). For each fragment defined by a read pair a single count was added to all bases crossed. Samples were normalized by sequencing depth in coding regions as in (24), and the mean of the log-transformed replicates were plotted or used to calculate cleavage ratio slopes. For empty vector:empty vector cleavage ratios, we calculated the cleavage ratio directly on two empty vector samples. Cleavage ratio slopes were calculated by a least-squares-fit of the log-transformed cleavage ratio across 30 bases; slopes were plotted at the 5’-end of this region. Transcriptome-wide cleavage ratio slope maxima were also summarized by histograms. Here, we identified all local cleavage ratio slope maxima after applying a Gaussian smooth to the data (sigma = 5). To remove noise associated with low expression, we required that the 30-nucleotide region used to calculated the slope had a minimum expression value in the empty vector ≥64 depth normalized read counts and was entirely within an annotated coding region.
Differential expression analysis of empty vector versus symE induced samples was performed by analyzing mapped reads using DESeq2 and visualized in R (52). To determine if SOS genes were significantly more expressed after SymE induction, we took the log2 fold change of expression of SOS response associated genes at 30- and 90- minutes post SymE induction and compared them to an equal number of randomly chosen genes with a t-test. symE itself was not included in the list of SOS induced genes as it was being artificially overexpressed.
ChIP-seq
E. coli MG1655 cells harboring either pBAD24::symE or pBAD24::symE-SPA were grown overnight in LB containing carbenicillin at 37 °C and then back diluted into fresh LB carbenicillin to OD600 0.02. Cultures were then grown at 37 °C to OD600 0.25 at which point they were induced with 0.2% arabinose. After 30- and 60- minutes of induction, 20 mL samples of each culture were taken. 200 μL 1 M sodium phosphate pH 7.6 and 500 μL 37% formaldehyde was added to each sample. Samples were incubated at RT for 20 minutes and then quenched with 840 μL 2.5 M glycine and incubated at RT for 5 minutes. Samples were then moved to ice and incubated for 15 minutes. Cells were then centrifuged at 8,000 × g for 5 minutes at 4 °C. Supernatant was discarded and pellets were washed three times in 20 mL ice cold 1x PBS pH 7.4. Pellets were then resuspended in 500 μL ice cold TES buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 100 mM NaCl). Samples were then snap frozen and stored at −80 °C until ready for use.
Protein A Dynabeads (Thermo) were pre-blocked by aliquoting 100 μL per sample into tubes. Tubes were placed on a magnet, solution was allowed to clear and supernatant was removed. Tubes were removed from the magnet and beads were resuspended in 500 μL ChIP 0.01% SDS buffer (16.7 mM Tris-HCl pH 8.1, 167 mM NaCl, 1.1% Triton X-100, 1.2 mM EDTA, 0.01% SDS). Tubes were returned to the magnet and allowed to clear. Supernatant was removed and beads were resuspended in 1 mL ChIP 0.01% SDS buffer. 2 μL 50 mg/mL BSA was added to each sample. Beads were incubated rotating overnight at 4 °C.
Previously snap-frozen samples were thawed at RT. 1 μL 35,000 U/μL Ready-Lyse (Lucigen) was added to each sample. Samples were incubated at RT for 15 minutes. 500 μL of ChIP buffer (16.7 mM Tris-HCl pH 8.1, 167 mM NaCl, 1.1% Triton X-100, 1.2 mM EDTA) containing Sigma Protease Inhibitor Tables (1 tablet per 30 mL ChIP buffer) was added to lysate samples which were then incubated at 37 °C for 10 minutes. After incubation, samples were sonicated on ice in 6× 10 second bursts at 15% output with 10 second pauses between bursts on a Branson sonicator. Lysate was then spun at 14,000 rpm in a tabletop centrifuge for 5 minutes at 4 °C. Samples were then diluted in ChIP buffer + 0.01% SDS such that each sample was 1 mL containing 500 μg protein. 500 μL of the 1 mL of pre-blocked Protein A Dynabeads were added to each sample which were then rotated at 4 °C for 1 hour. After incubation, tubes were placed on a magnet and allowed to clear. 90 μL of supernatant was removed to determine total input chromatin. The remaining supernatant was removed from beads and moved to a new tube. 1 μL Anti-FLAG antibody (Sigma) was added to each tube. Tubes were then incubated overnight at 4 °C with rotation.
The second 500 μL of pre-blocked Protein A Dynabeads were moved into fresh tubes for each reaction. Tubes were then put on a magnet and supernatant was removed. Beads were then resuspended in the immune complex that had been incubated at 4 °C the previous night. Samples were then incubated on a rotator at 4 °C for 2 hours. Tubes were placed on a magnet and supernatant was removed. Beads were washed for 15 minutes at 4 °C consecutively with 1 mL the following buffers: 1x Low Salt wash buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl), 1x High salt wash buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl), 1x LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1mM EDTA, 10mM Tris-HCl pH 8.1), 2x TE buffer (10 mM Tris-HCl pH 8.1 and 1 mM EDTA). Complexes were eluted from the beads by adding 250 μL of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3). Beads were incubated in elution buffer rotating at 30 °C for 15 minutes. Tubes were placed on a magnet and cleared eluate (250 μL) was placed in a new tube. Another 250 μL of elution buffer was added to the beads and reactions were incubated at 30 °C for 15 minutes. The second 250 μL eluate was combined with the first for a total of 500 μL of eluate. 30 μL of 5 M NaCl and 2 μL of 0.5 mg/mL RNase A was added to the collective eluate. Samples were then incubated at 65 °C overnight to reverse crosslinking.
40 μL of 0.5 M EDTA pH 8.0, 20 μL 1M Tris-HCl pH 6.5, and 5 μL 20 mg/mL Proteinase-K (NEB) was added to each sample which were then incubated at 45 °C for 2 hours. After incubation 575 μL of phenol:chloroform:isoamyl alcohol was added to each sample which were then vortexed for 1 minute. Samples were then centrifuged for 10 minutes at 13,000 rpm in a tabletop centrifuge at 4 °C. The top (aqueous) layer was extracted and moved to a new tube. 575 μL phenol:chloroform:isoamyl alcohol was added to the extracted layer and samples were vortexed for 1 minute. Samples were centrifuged again and the top (aqueous) layer was extracted and placed in a new tube. 60 μL 3 M NaOAc, 5 μL glycogen and 600 μL ice cold isopropanol was added to each sample and they were incubated at least 1 hour at −20 °C. Samples were then centrifuged for 30 minutes at 13,000 rpm in a tabletop centrifuge at 4 °C. Pellets were washed with 500 μL ice cold 75% ethanol and then allowed to dry completely. Dry pellets were then resuspended in 50 μL of elution buffer (10 mM Tris-HCl pH 8.5, Qiagen)
To begin making sequencing libraries, 100 μL of Ampure XP beads per reaction were placed in a magnetic rack and supernatant was replaced with 150 μL of PEG 8,000 20%/2.5 M NaCl. The PEG/bead mix was then added to each 50 μL elution from the previous step and samples were mixed by pipetting and vortexing briefly and then allowed to incubate at RT for 10 minutes. Samples were then placed on a magnetic rack and allowed to clear. Supernatant was removed and beads were washed 2x with 500 μL of 80% ethanol. All ethanol was carefully removed and beads were allowed to dry completely. Beads were then resuspended in 50 μL of elution buffer (10 mM Tris-Cl, pH 8.5).
Libraries were either then made following the same protocol as was done for our RNA-seq starting with end repair, omitting the USER digest step, or were done by the MIT BioMicroCenter. Completed libraries were submitted for single-end sequencing performed on an Illumina MiSeq at the MIT BioMicroCenter.
ChIP-seq data analysis
Sequence reads were mapped to E. coli K-12 MG1655 genome (U00096.2) using the bowtie 2 software suite (51). Mapped reads were then analyzed using the MACS algorithm on default settings to call peaks and obtain fold enrichment values (28). Wiggle files of mapped reads were visualized in Mochiview (29). Peaks were further pruned using Mochiview by removing any peaks that were below 2x fold enrichment over background or whose background reached higher than 50 reads over the span of the peak. Finally, only peaks that met thresholds at both initial 30- and 60-minute time points were kept, resulting in 61 total peaks which were used as our ChIP-seq peaks for the rest of the text unless otherwise mentioned. Data from two additional 30-minute ChIP-seq replicates was analyzed using MACS on default settings and called peak locations were compared to those of the 61 significant peaks of the previously analyzed paired 30- and 60- minute data to assess reproducibility of the ChIP-seq.
ChIP peak associated genes were chosen based on the following criteria: any genes whose coding sequence was overlapped by one of the 61 ChIP peaks or any gene whose 5’ intergenic region was overlapped by one of the 61 ChIP peaks. To see if ChIP associated genes were more affected in our RNA-seq data, we took the log2 fold change of expression of ChIP associated genes at 30- and 90- minutes post SymE induction and compared them to an equal number of randomly chosen genes with a t-test.
To determine if the locations where SymE bound were also enriched for σ70 or RNA polymerase binding, we compared our ChIP peaks to the ChIP signal from two previously published data sets (32, 33). Unlike our data and the σ70 data, the RNA polymerase ChIP was mapped to E. coli K-12 MG1655 genome U00096.3. To accurately compare the RNA-polymerase ChIP data to our peaks, we took the sequences of the top 61 called peaks and obtained their locations in the U00096.3 genome. The RNA-polymerase wiggle files were provided log2 transformed, which we reverted back to non-log values. We then did the comparison by taking the mean ChIP signal from σ70 or RNA polymerase ChIP at each of the genome locations for the 61 SymE-SPA ChIP peaks using Mochiview. Next, we found the mean signals from 61 randomly generated genome locations, called “random peaks”, size matched to the SymE-SPA ChIP locations. We then performed a t-test to see if the σ70 or RNA polymerase ChIP signal from the random peaks locations was significantly different from the ChIP associated peak locations. This analysis was also done using the σ70 ChIP data for all of the peaks originally called by MACS and an equal number of randomly selected locations the same size as those original peak locations.
[3H]-thymidine and-[3H] uridine incorporation assays
E. coli MG1655 PCP18-araE cells harboring either pBAD24 or pBAD24::symE were grown to OD600 ~0.3 in M9 carb at 37 °C. Cells were back diluted to OD600 0.1 in M9 carb at 37 °C and induced with 0.02% arabinose. At 0, 15, 30, 45 and 60 minutes after induction, 250 μL of the bacterial culture was transferred to a microcentrifuge tube containing methyl- [3H] thymidine (Perkin-Elmer) (40 μCi/mL) or uridine, 5–6[3H] (Perkin Elmer) (4 μCi/mL). Tubes were incubated at 37 °C for 2 min for incorporation. Reactions were quenched by addition of nonradioactive thymidine or uridine (1.5 mM) and incubated at 37 °C for an additional 2 min. Samples were added to ice cold trichloroacetic acid (TCA) (10% w/v) and incubated 30 min on ice to allow for precipitation. The resulting sample was vacuum filtered onto a glass microfiber filter (Whatman,1820–024) that had been pre-wetted with 5% w/v TCA. Filters were washed with 35x volume of 5% w/v TCA, then with 5x volume of 100% ethanol. Air dried filters were placed in tubes with scintillation fluid and measured in a scintillation counter (Perkin Elmer). Three to four independent biological replicates were performed.
SymE expression
T7 Express (NEB) cells were transformed with pMKT26 which has a 6xHis-TEV-SymE construct under the control of a T7 promoter driven by IPTG. Colonies were scraped from transformation plates and resuspended in 6 mL of 2x YT media with carbenicillin. That 6 mL was then further diluted into 6 L of 2xYT media with carbenicillin. Cells were grown at 37 °C to OD600 0.5 at which point they were induced with 1 mM IPTG and moved to 16 °C to grow overnight. Cells were then spun down in 1 L bottles at 5,000 × g for 10 minutes, washed with lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole adjusted to pH 8.0) and pellets were snap frozen in falcon tubes and stored at −80 °C until ready to purify.
SymE purification
Buffers were stored and purification steps were performed at 4 °C. Prior to purification, 200 mL of lysis buffer was supplemented with 1 mL of 200x PMSF (180 mg PMSF in 5 mL ethanol) and 2 crushed SIGMAFAST protease inhibitor cocktail tablets (Sigma). Two pellets derived from 1 L of liquid culture each were thawed and resuspended in 20 mL of supplemented lysis buffer. Cells were then lysed in a microfluidizer at 18,000 PSI for 2–3 cycles. Lysate was centrifuged at 25,000 × g for 1 hour at 4 °C. Supernatant was then passed over a 1 mL column of Ni-NTA Agarose resin (Qiagen) which had been equilibrated in lysis buffer. The column was washed with 20 column volumes of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole adjusted to pH 8.0). Finally, protein was eluted with 5 mL of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 7 mM βME, adjusted to pH 8.0). Eluted protein was loaded onto a 5 mL HiTrap heparin column (Sigma) and eluted with a NaCl gradient (from 50 mM NaH2PO4, 100 mM NaCl, 7 mM βME pH 8.0 to 50 mM NaH2PO4, 2 M NaCl, 7 mM βME pH 8.0) using an AKTA pure FPLC. Fractions containing 6xHis-TEV-SymE were collected and combined. To remove the 6xHis tag, 1 μL of acTEV TEV protease (Invitrogen) was added per 30 μg protein to the combined fractions with 1:20 provided acTEV buffer and 1 mM DTT. The TEV protease/protein mixture was dialyzed into storage buffer (50 mM NaH2PO4, 100 mM NaCl, 7 mM βME, 10 mM imidazole, 10% glycerol, adjusted to pH 8.0) in a 3kD MWCO Slide-A-Lyzer (Thermofisher) cassette at 4 °C overnight. After dialysis, protein was removed from the dialysis cassette and run over 250 μL of Ni-NTA agarose column that had been equilibrated in storage buffer. SymE that had been cleaved and no longer had the 6x-His tag was collected in the flow through and concentrated in 3K Amicon centrifugal filters (Millipore). SymE concentration was quantified with Bradford reagent and stored at 4 °C until ready for use.
Gel filtration of SymE
To determine the oligomerization state of SymE, 500 μL of SymE purified as above was loaded onto a Superdex 75 Increase 10/300 GL column (Cytiva) on an AKTA pure FPLC and eluted with storage buffer. Elution time of SymE was compared to that of Aprotinin (6.5 kDa), Cytochrome C (12.4 kDa), Carbonic Anhydrase (29 kDa) and Albumin (66 kDa) from the Gel Filtration Markers Kit for Protein Molecular Weights 6,500–66,000 Da (Sigma).
Electrophoretic mobility shift assays
ChIP peak probes were PCR amplified using E. coli MG1655 DNA as template. Primers used to amplify ChIP peaks are listed in Table S5. pUC19 plasmid was purified from E. coli MG1655 using a ZR plasmid mini prep kit (Zymo). Sheared E. coli genomic DNA was run out on a 1.5% Agarose TAE gel (40 mM Tris, 20 mM acetic acid, 1 mM EDTA). DNA running at ~200 bp was excised and extracted from the gel using a Zymoclean Gel DNA Recovery Kit (Zymo). EMSA reactions were prepared by mixing 1 μL of 10x binding buffer (100 mM Tris pH 7.5, 10 mM EDTA, 1 M KCl, 1 mM DTT, 50% glycerol v/v, and 0.1 mg/mL BSA), 2 μL 2 ng/μL DNA probe, 6 μL nuclease free water and 1 μL of SymE at different concentrations (0 μM, 0.625 μM, 1.25 μM, 2.5 μM, 5 μM, 10 μM or 20 μM) and reactions were then incubated at 30 °C for 1 hour. Reactions were then placed on ice. 2 μL Gel Loading Dye, Purple (6X), no SDS (NEB) was added to each reaction. Reactions were loaded into a 0.8% agarose 0.5X TBE (45 mM Tris base, 45 mM boric acid, 1 mM EDTA pH 8.0) gel that had been chilled to 4 °C. Gels were run at 100V for 105 minutes at 4 °C. pUC19 EMSAs were run longer for better separation. Gels were stained by incubating them in 180 mL staining solution (18 μL SYBR Gold Nucleic Acid stain (Invitrogen) in 180 mL 0.5X TBE) for 5 minutes. Gels were then imaged on a Typhoon FLA 9500 imager (GE). To determine if DNA was being degraded during the 30 °C incubation, reactions were doubled, and after the 30 °C incubation they were split in half. One half was loaded onto the gel as before, and 2.6 μL of 10% SDS was added to the other half. SDS containing samples were then loaded onto gels as before.
Nucleoid isolation sucrose gradients
E. coli MG1655 symE-SPA ΔsymR cells were grown overnight in LB. Overnight cultures were then diluted to OD600 0.02 in LB. Cells were grown to OD600 0.3 and then treated with 1 μg/mL mitomycin C for 90 minutes. After mitomycin C treatment, cells were diluted to OD600 0.4 in 24 mL of LB. The 24 mL of diluted cells were then spun down at 8,500 rpm at 4 °C for 5 minutes. Supernatant was removed and cells were resuspended in 500 μL if ice-cold buffer A (10 mM Tris-HCl pH 8.2, 100 mM NaCl, and 20% sucrose). 100 μL of ice-cold buffer B (100 mM Tris-HCl pH 8.2, 50 mM EDTA, 0.6 mg/mL lysozyme; lysozyme added day of) was added to the sample by pipetting up and down twice, and mixtures were incubated on ice for 30 seconds. 500 μL of ice-cold buffer C (10 mM Tris-HCl pH 8.2, 10 mM EDTA, 10 mM spermidine, 1% Brij-58, and 0.4% deoxycholate; spermidine added day of) was added to the mixture by pipetting up and down twice. This was then incubated on ice for 3 minutes. The mixture was then loaded on top of 50%−15% sucrose step gradients, 1.2 mL of each step in 5% sucrose gradients increments, where each step contained the appropriate percentage of sucrose in 10 mM Tris-HCl pH 8.2 and 100 mM NaCl. Gradients were then spun for 20 minutes at 10,000 rpm in a Beckmann SW 41 Ti rotor at 4 °C. ~500 μL fractions were collected using a Biocomp piston gradient fractionator. The absorbance at 280 nm and 260 nm of each fraction was monitored using a Triax flow cell. The cytoplasmic and nucleoid fractions of the gradient were collected based on position in the gradient and 280 nm and 260 nm absorbance, and flash frozen for later use.
Western blots of sucrose gradient fractions
Frozen gradient fractions were thawed and incubated with 25 U of benzonase (Millipore sigma) for 30 minutes at room temperature. 12 μL of each fraction was then run on an AnyKD (BioRad) polyacrylamide gel and transferred to PVDF membrane using a Trans-blot turbo blotting system. Membranes were blocked with 10 mL of 5% milk in TBST for 1 hour at room temperature. Next, to probe for SymE-SPA, mouse αFLAG M2 antibody from Sigma was added directly to the blocking solution at a 1:2,000 dilution (5 μL of antibody into 10 mL of blocking solution). Blots were then incubated for 1 hour at room temperature. After incubating, blots were washed with TBST for 5 minutes. Washes were repeated a total of 3 times. After washes, blots were incubated with peroxidase conjugated goat α-mouse secondary antibody (Thermo) at a dilution of 1:20,000 in 5% milk TBST (0.5 μL antibody in 10 mL 5% milk TBST) for 1 hour at room temperature. After incubating, blots were washed with TBST for 5 minutes. Washes were repeated a total of 3 times. Blots were then imaged using SuperSignal chemiluminescent substrate (Thermo).
After imaging, blots were washed and reprobed with mouse αRpoA and mouse αGroEL antibodies to act as controls for a known nucleoid associated protein and cytoplasmic protein, respectively. To do this, blots were washed with TBST for 5 minutes. Washes were repeated a total of 3 times. Then, blots were probed simultaneously with mouse αRpoA and mouse αGroEL antibodies at 1:2,500 and 1:10,000 respectively in 10 mL of 5% milk TBST. The blot was then incubated for 1 hour at room temperature. After incubating, blots were washed with TBST for 5 minutes. Washes were repeated a total of 3 times. After washes, blots were incubated with peroxidase conjugated goat α-mouse secondary antibody (Thermo) at a dilution of 1:20,000 in 5% milk TBST (0.5 μL antibody in 10 mL 5% milk TBST) for 1 hour at room temperature. After incubating, blots were washed with TBST for 5 minutes. Washes were repeated a total of 3 times. Blots were then imaged using SuperSignal chemiluminescent substrate (Thermo).
Microscopy
All microscopy was done using an Axio Observer Microscope (Zeiss) with a Lumencor Spectra-X LED light source, Chroma filters and Hamamatsu CMOS camera using Metamorph software (Molecular Devices).
HU-GFP
E. coli MG1655 HU-GFP cells harboring either pBAD30, pBAD30::symE, pBAD30::hns, pBAD30::mazF, pBAD30::relE, pBAD30::tisB or pBAD24::symE were grown overnight in M9 media containing carbenicillin and then back diluted into fresh M9 carb to OD600 0.02. Cultures were then grown at 37 °C to OD600 0.4. A 1 μL drop of culture was then spotted onto a 35 mm glass bottom culture dish (Cellvis), and an M9 minimal media 1.5% agar pad containing carbenicillin and 0.2% arabinose was placed gently on top of the drop of culture. Time lapse microscopy was then performed; phase and FITC fluorescence images were saved every 10 minutes. Merged phase and fluorescence images were made in ImageJ (53).
Gam-GFP
E. coli MG1655 Gam-GFP cells harboring either pBAD30, pBAD30::symE, pBAD30::hns, pBAD30::relE, or pBAD24::symE were grown overnight in M9 media containing carbenicillin and then back diluted into fresh M9 carb to OD600 0.02. Cultures were then grown at 37 °C to OD600 0.15 and Gam-GFP production was induced with 10 ng/mL aTc. Cells were then further incubated at 37°C to OD600 0.4. A 1 μL drop of culture was then spotted onto a 35 mm glass bottom culture dish (Cellvis), and an M9 minimal media 1.5% agar pad containing carbenicillin and 0.2% arabinose was placed gently on top of the drop of culture. Time lapse microscopy was then performed; phase and FITC fluorescence images were saved every 10 minutes. Merged phase and fluorescence images were made in ImageJ.
Microscopy analysis
To calculate percentage of cell area that is taken up by nucleoid associated fluorescence, cells were paired from phase and fluorescence images using the MicrobeJ ImageJ plugin (54). The area taken up by the fluorescence signal associated with a cell from the phase image was divided by the area of the cell. Between ~50–150 cells were used to calculate each data point.
The nucleoid intensity over the longitudinal axis of the cells from Figure 6C shown in Figure 6D were calculated using the ImageJ Plot profile tool. The data from the ImageJ plots were normalized to cell length and re-plotted.
The percentage of cells containing at least one Gam-GFP puncta were determined by manually counting the cells in a microscopy image that contained at least one puncta vs total cells (n ≥ 85) for a given condition.
Sequencing Data
Sequencing data for RNA-seq and ChIP-seq experiments is available from the GEO repository under accession numbers GSE185892 and GSE185893, respectively.
Supplementary Material
Acknowledgements
Strains ML3577, ML3578, and ML3576 and plasmids pMKT9, pMKT10, and pMKT23 were generous gifts of Dr. Gisela Storz of The National Institute of Child Health and Human Development at the National Institutes of Health. Strains ML3582 and ML3583 were generous gifts of Dr. Anjana Badrinarayanan of The National Centre for Biological Sciences, India. Plasmids pMKT86 and pMKT87 were provided by Dr. Stephanie Jones of the Laub laboratory. We thank G. Storz for her critical feedback on the manuscript and helpful discussions. This work was supported by an NIH grant to M.T.L. (R01GM082899), who is also an Investigator of the Howard Hughes Medical Institute. The authors have no conflicts of interest to declare.
References
- 1.Gerdes K, Larsen JEL, Molin S. 1985. Stable inheritance of plasmid R1 requires two different loci. J Bacteriol 161:292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thisted T, Gerdes K. 1992. Mechanism of post-segregational killing by the hok/sok system of plasmid R1. Sok antisense RNA regulates hok gene expression indirectly through the overlapping mok gene. J Mol Biol 223:41–54. [DOI] [PubMed] [Google Scholar]
- 3.Ogura T, Hiraga S. 1983. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci USA 80:4784–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GPC. 2009. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A 106:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ronneau S, Helaine S. 2019. Clarifying the Link between Toxin–Antitoxin Modules and Bacterial Persistence. J Mol Biol 431:3462–3471. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi Y, Park J-H, Inouye M. 2011. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu Rev Genet 45:61–79. [DOI] [PubMed] [Google Scholar]
- 7.Pecota DC, Wood TK. 1996. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol 178:2044–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Yao J, Sun YC, Wood TK. 2021. Type VII Toxin/Antitoxin Classification System for Antitoxins that Enzymatically Neutralize Toxins. Trends Microbiol 29:388–393. [DOI] [PubMed] [Google Scholar]
- 9.Fozo EM, Hemm MR, Storz G. 2008. Small toxic proteins and the antisense RNAs that repress them. Microbiol Mol Biol Rev 72:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brantl S 2012. Bacterial type I toxin-antitoxin systems. RNA Biol 9:1488–90. [DOI] [PubMed] [Google Scholar]
- 11.Kawano M, Aravind L, Storz G. 2007. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 64:738–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35:1560–1572. [DOI] [PubMed] [Google Scholar]
- 13.Lewis LK, Harlow GR, Gregg-Jolly LA, Mount DW. 1994. Identification of high affinity binding sites for LexA which define new DNA damage-inducible genes in Escherichia coli. J Mol Biol 241:507–523. [DOI] [PubMed] [Google Scholar]
- 14.Muenter MM, Aiken A, Akanji JO, Baig S, Bellou S, Carlson A, Conway C, Cowell CM, Delateur NA, Hester A, Joshi C, Kramer C, Leifer BS, Nash E, Qi MH, Travers M, Wong KC, Hu M, Gou N, Giese RW, Gu AZ, Beuning PJ. 2019. The response of Escherichia coli to the alkylating agents chloroacetaldehyde and styrene oxide. Mutat Res Gen Tox En 840:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobay BG, Andreeva A, Mueller GA, Cavanagh J, Murzin AG. 2005. Revised structure of the AbrB N-terminal domain unifies a diverse superfamily of putative DNA-binding proteins. FEBS Lett 579:5669–5674. [DOI] [PubMed] [Google Scholar]
- 16.Coles M, Djuranovic S, Söding J, Frickey T, Koretke K, Truffault V, Martin J, Lupas AN. 2005. AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-Psi β barrels. Structure 13:919–928. [DOI] [PubMed] [Google Scholar]
- 17.Lin BL, Chen CY, Huang CH, Ko TP, Chiang CH, Lin KF, Chang YC, Lin PY, Tsai HHG, Wang AHJ. 2017. The arginine pairs and C-termini of the Sso7c4 from Sulfolobus solfataricus participate in binding and bending DNA. PLoS One 12:e0169627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spurio R, Dürrenberger M, Falconi M, La Teana A, Pon CL, Gualerzi CO. 1992. Lethal overproduction of the Escherichia coli nucleoid protein H-NS: ultramicroscopic and molecular autopsy. Mol Gen Genet 231:201–211. [DOI] [PubMed] [Google Scholar]
- 19.Sibley MH, Raleigh EA. 2004. Cassette-like variation of restriction enzyme genes in Escherichia coli C and relatives. Nucleic Acids Res 32:522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mende DR, Letunic I, Huerta-Cepas J, Li SS, Forslund K, Sunagawa S, Bork P. 2017. ProGenomes: A resource for consistent functional and taxonomic annotations of prokaryotic genomes. Nucleic Acids Res 45:D529–D534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchan DWA, Jones DT. 2019. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 47:W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, Sorek R. 2018. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359:eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erill I, Campoy S, Barbé J. 2007. Aeons of distress: An evolutionary perspective on the bacterial SOS response. FEMS Microbiol Rev 31:637–656. [DOI] [PubMed] [Google Scholar]
- 24.Culviner PH, Laub MT. 2018. Global Analysis of the E. coli Toxin MazF Reveals Widespread Cleavage of mRNA and the Inhibition of rRNA Maturation and Ribosome Biogenesis. Mol Cell 70:868–880.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. 2001. Homogeneous expression of the PBAD promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity araE transporter. Microbiology 147:3241–3247. [DOI] [PubMed] [Google Scholar]
- 26.Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martínez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Peralta-Gil M, Subhraveti P, Velázquez-Ramírez DA, Weaver D, Collado-Vides J, Paulsen I, Karp PD. 2017. The EcoCyc database: Reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res 45:D543–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, Wood TK. 2010. Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng J, Liu T, Qin B, Zhang Y, Liu XS. 2012. Identifying ChIP-seq enrichment using MACS. Nat Protoc 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Homann OR, Johnson AD. 2010. MochiView: Versatile software for genome browsing and DNA motif analysis. BMC Biol 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Second Int Conf Intell Syst Mol Biol 2:28–36. [PubMed] [Google Scholar]
- 31.Yang S, Kim S, Kim DK, Jeon An H, Bae Son J, Hedén Gynnå A, Ki Lee N. 2019. Transcription and translation contribute to gene locus relocation to the nucleoid periphery in E. coli. Nat Commun 10:5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myers KS, Yan H, Ong IM, Chung D, Liang K, Tran F, Keleş S, Landick R, Kiley PJ. 2013. Genome-scale Analysis of Escherichia coli FNR Reveals Complex Features of Transcription Factor Binding. PLoS Genet 9:11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freddolino PL, Goss TJ, Amemiya HM, Tavazoie S. 2020. Dynamic landscape of protein occupancy across the Escherichia coli chromosome. bioRxiv 1–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wery M, Woldringh CL, Rouviere-Yaniv J. 2001. HU-GFP and DAPI co-localize on the Escherichia coli nucleoid. Biochimie 83:193–200. [DOI] [PubMed] [Google Scholar]
- 35.Guzman L, Belin D, Carson MJ. 1995. Tight Regulation, Modulation, and High-Level Expression by Vectors Containing the Arabinose P BAD Promoter 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zusman DR, Carbonell A, Haga JY. 1973. Nucleoid condensation and cell division in Escherichia coli MX74T2 ts52 after inhibition of protein synthesis. J Bacteriol 115:1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henderson ML, Kreuzer KN. 2015. Functions that protect Escherichia coli from tightly bound DNA-protein complexes created by mutant EcoRII methyltransferase. PLoS One 10:e0128092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simmons LA, Foti JJ, Cohen SE, Walker GC. 2008. The SOS Regulatory Network. EcoSal Plus 3:1–30. [DOI] [PubMed] [Google Scholar]
- 39.Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu L-Y, Magnan D, Halliday JA, Frisch RL, Gibson JL, Nehring RB, Do HG, Hernandez M, Li L, Herman C, Hastings P, Bates D, Harris RS, Miller KM, Rosenberg SM. 2013. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife 2:e01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavco PA, Steege DA. 1990. Elongation by Escherichia coli RNA polymerase is blocked in vitro by a site-specific DNA binding protein. J Biol Chem 265:9960–9969. [PubMed] [Google Scholar]
- 41.Weel-Sneve R, Kristiansen KI, Odsbu I, Dalhus B, Booth J, Rognes T, Skarstad K, Bjoras M. 2013. Single Transmembrane Peptide DinQ Modulates Membrane-Dependent Activities. PLoS Genet 9:e1003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogel J, Argaman L, Wagner EGH, Altuvia S. 2004. The Small RNA IstR Inhibits Synthesis of an SOS-Induced Toxic Peptide. Curr Biol 14:2271–2276. [DOI] [PubMed] [Google Scholar]
- 43.Makarova KS, Wolf YI, Snir S, Koonin EV. 2011. Defense Islands in Bacterial and Archaeal Genomes and Prediction of Novel Defense Systems. J Bacteriol 193:6039–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Day RS. 1977. UV-induced alleviation of K-specific restriction of bacteriophage lambda. J Virol 21:1249–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keatch SA, Leonard PG, Ladbury JE, Dryden DTF. 2005. StpA protein from Escherichia coli condenses supercoiled DNA in preference to linear DNA and protects it from digestion by DNase I and EcoKI. Nucleic Acids Res 33:6540–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimmermann L, Stephens A, Nam S, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V. 2018. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol 430:2237–2243. [DOI] [PubMed] [Google Scholar]
- 47.Edgar RC. 2004. MUSCLE : multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darriba D, Posada D, Flouri T, Kozlov AM, Stamatakis A, Morel B. 2019. ModelTest-NG : A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models 37:291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Culviner PH, Guegler CK, Laub MT. 2020. A Simple, Cost-Effective, and Robust Method for rRNA Depletion in RNA-Sequencing Studies. MBio 11:e00010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ : 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ducret A, Quardokus EM, Brun YV. 2016. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat Microbiol 1:16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.