

Abstract
Epidermal growth factor receptor inhibitors (EGFRi) are standard-of-care treatments administered to patients with non-small cell lung cancer (NSCLC) that harbor EGFR alterations. However, development of resistance post-treatment remains a major challenge. Multiple mechanisms can promote survival of EGFRi-treated NSCLC cells, including secondary mutations in EGFR and activation of bypass tracks that circumvent the requirement for EGFR signaling. Nevertheless, the mechanisms involved in bypass signaling activation are understudied and require further elucidation. In this study, we identify that loss of an epigenetic factor, lysine methyltransferase 5C (KMT5C), drives resistance of NSCLC to multiple EGFRis, including erlotinib, gefitinib, afatinib, and osimertinib. KMT5C catalyzed trimethylation of histone H4 lysine 20 (H4K20), a modification required for gene repression and maintenance of heterochromatin. Loss of KMT5C led to upregulation of an oncogenic long non-coding RNA, LINC01510, that promoted transcription of the oncogene MET, a component of a major bypass mechanism involved in EGFRi resistance. These findings underscore the loss of KMT5C as a critical event in driving EGFRi resistance by promoting a LINC01510/MET axis, providing mechanistic insights that could help improve NSCLC treatment.
Keywords: EGFR, erlotinib, resistance, epigenetic, SUV420H2, KMT5C, H4K20me3, MET, LINC01510
Introduction:
Lung cancer is the leading cause of cancer-related mortality, with an estimated 131,880 deaths predicted in 2021 in the United States (1). The majority of lung cancer patients are diagnosed with non-small cell lung cancer (NSCLC), a subtype that represents 85% of lung cancer cases. Since most lung cancer patients are diagnosed with metastatic disease, surgical resection is not curative, and thus, the most effective treatment strategies are radiotherapy, chemotherapy, and targeted therapy. Targeted therapeutics are selected based on altering genes that the cancer cells are addicted to. A few such drivers present in NSCLC include KRAS, MEK, MET, HER2, and EGFR, many of which are either mutated or amplified, resulting in constitutive pro-growth signaling (2,3).
Epidermal Growth Factor Receptor (EGFR) is a cell surface receptor required for normal cell growth and proliferation. In 10–35% of NSCLC cases EGFR is constitutively activated due to mutations, the most common of which include an amino acid substitution in exon 21 (L858R) or an in-frame deletion in exon 19. Mutant EGFR can be clinically targeted with EGFR tyrosine kinase inhibitors (EGFRi), including erlotinib and gefitinib, first generation EGFRi, afatinib, a second-generation inhibitor, or osimertinib a third-generation EGFRi that is also active against a secondary mutation in EGFR, T790M. Erlotinib binds reversibly and specifically to the ATP-binding pocket of EGFR, abrogating downstream signaling pathways. While initially beneficial, many patients develop resistance within a year, which is currently a major drawback to its use (4). The targeted gene incurs additional mutations or alternative signaling pathways are activated to evade therapy. In the case of erlotinib over 60% of tumors acquire a secondary mutation, T790M, whereas approximately 20% of tumors utilize bypass tracks. Bypass tracks allow the tumor to escape inhibition of the EGFR pathway through the use of alternative mechanisms. These include signaling through oncogenic proteins such as MET, BRAF, HER2, PIK3CA or histological transformation of cells - NSCLC transformation into small cell lung cancer or through epithelial to mesenchymal transition (4–7). In addition to an incomplete understanding of mechanisms that govern these bypass tracks there are also approximately 15–20% of NSCLC tumors that acquire erlotinib resistance by unidentified mechanisms (4).
While gain-of-function mechanisms that drive resistance have been identified, loss of tumor suppressive genes, such as PTEN, TP53, TET1, and NF1 also contributes to resistance (8–11). Indeed, many tumor suppressive proteins function as gatekeepers of the genome preventing spurious activation of oncogenes. Here, to define genes that prevent the development of resistance, a genome-wide loss of function screen was conducted using the CRISPR-Cas9 system. Our data suggest that an epigenetic factor and bona fide tumor suppressor, KMT5C can be included among the gatekeepers of the genome. KMT5C catalyzes the trimethylation of histone H4 at lysine-20 (H4K20), which is required for establishment of heterochromatin and gene repression (12–14). Loss of KMT5C has been implicated in causation of multiple cancers (15,16), but for the first time we show that KMT5C loss is a mechanism that promotes erlotinib resistance. The findings of this study determined that KMT5C mutant cells express high levels of the long non-coding RNA, LINC01510 that transcriptionally upregulates the oncogene MET, mediating resistance.
Materials and Methods:
Cell culture:
The following cell lines used in the study, were obtained from American Type Culture Collection (ATCC: HCC827, RRID:CVCL_2063; A549, RRID:CVCL_0023; CALU6, RRID:CVCL_0236; H23, RRID:CVCL_5800; H1650, RRID:CVCL_1483; H1975, RRID:CVCL_1511; H460, RRID:CVCL_0459). PC9 was obtained from Sigma Aldrich, EKVX and H322M were obtained from the NCI-DTP (EKVX, RRID:CVCL_1195; H322M, RRID:CVCL_1557), and HBEC cells were kindly provided by Dr. John Minna. All lines were tested monthly and confirmed to free of mycoplasma contamination. Cell lines generated during the study were authenticated by ATCC Cell Line Authentication. All cell lines other than HBEC cells were grown in RPMI media supplemented with 10% FBS and 1% penicillin/streptomycin. HBEC cells were cultured in Keratinocyte Serum Free Media (Life Technologies). ECas9 cells were continuously cultured in media containing 1μg/ml blasticidin, The EKVX KMT5C mutant clones A, C and E were grown in media containing 100ng/ml puromycin, inducible-KMT5C Calu6 clones were cultured in 500ng/ml puromycin containing media, and rescue clones were grown in media containing 100ng/ml puromycin and 300μg/ml G418 containing media.
Drug Preparation for in vitro studies:
Erlotinib (S7786, Selleck Chemicals), afatinib (850140-72-6, Sigma Aldrich), gefitinib (S1025, Selleck Chemicals), and osimertinib (S7297, Selleck Chemicals) were dissolved in DMSO to prepare 0.4 M stock solutions, which were aliquoted and stored at −80°C. A 200 μM working dilution of all the drugs was prepared in complete medium and were used to prepare the indicated concentrations for all in vitro experiments. A-196 (S7983, Selleck Chemicals) was dissolved in DMSO to prepare 10mg/mL stock solutions, which were aliquoted and stored at −80°C.
Knock-out CRISPR screen:
EKVX cells (4×105) were plated in 6-well plates and were transfected with 3μg of linearized lentiCas9-Blast (Addgene, 52962) using lipofectamine 2000 (11-668-019, Thermo Fisher Scientific), as per manufacturer’s instructions. Forty-eight hours later, cells were selected using 5μg/ml blasticidin. ECas9 (clone 7) cells stably expressing Cas9 plasmid were clonally selected and characterized.
Lentiviral sgRNA libraries (A and B) were generated and their titers were determined as previously described (17). The GeCKO V2 library (RRID:SCR_009001) has 6 sgRNAs targeting each protein coding gene and 4 sgRNAs targeting each microRNA. To achieve a 300-fold coverage of the libraries, seventeen 12-well plates were each seeded with 4.5×105 ECas9 cells. Nine plates were transduced with library A, and 8 plates were transduced with library B, both at a multiplicity of infection (MOI) of 0.4 in the presence of polybrene (10μg/ml). Twenty-four hours post transduction, cells were pooled and ~1.31 × 107 cells were re-plated in each of seven 15 cm plates containing complete media supplemented with 2μg/ml blasticidin. Forty-eight hours later cells were plated in six 15 cm plates in media containing 2μg/ml puromycin, to select for library-transduced cells, and 2μg/ml blasticidin. Seventy-two hours later, 2.6 × 107 cells were stored for baseline and 2.6 × 107 cells were re-plated. The following day, media was replaced with GI75 erlotinib containing media (1.23μM erlotinib) and cells were continuously exposed to GI75 erlotinib for 15 passages. Three biological replicates were performed, and genomic DNA from each baseline and erlotinib treated sample was isolated using the Genomic DNA isolation kit (K1820-01, Thermo Fisher Scientific) following the manufacturer’s protocol.
For sequencing library preparation, two sequential PCR reactions were conducted for each sample. The first PCR reaction (PCR1) specifically amplified sgRNAs from 1μg of gDNA isolated from each sample. Twenty-five such PCR reactions were conducted, pooled, and gel purified using QIAEX II Gel Extraction Kit (20021, Qiagen). Each PCR1 reaction product (10 ng) was then used for each of 20 PCR2 reactions that were pooled and gel purified. PCR2 fragment sizes and library quality were evaluated on a bioanalyzer (Agilent). Both PCR1 and PCR2 primers are listed in Supplementary Table 1 (Integrated DNA Technologies). Barcodes included in PCR2 primers were used to identify the samples after deep sequencing. All sequencing was conducted using a NovaSeq 6000 (Illumina). FastQC version 0.11.7 (RRID:SCR_014583) was used to observe sequencing data quality before and after trimming. Cutadapt version 1.13 (RRID:SCR_011841) was used to trim adapters from reads. Reads post-trimming that were shorter than 18nt were discarded. MAGeCK-VISPR v. 0.5.6 was used to perform mapping, allowing no mismatches to ensure accuracy and to reduce bias. Finally, MAGeCK was used to identify over- and under-represented sgRNAs in treated samples relative to baseline, represented as β-scores (Liu et al., 2014).
Mutant, knockdown, overexpression and rescue experiments:
For EKVX validation studies, KMT5C sgRNA were generated by annealing two oligos (see Supplementary Table 2) followed by 5’ phosphorylation (T4 Polynucleotide Kinase kit, M0201S, NEB) as described previously (LentiGuide-Puro and LentiCRISPRv2). Simultaneously, the CRISPR-Cas9 plasmid, LentiCRISPRv2 (Addgene, 52961) was digested using BsmBI (R0580, NEB), dephosporylated (Antarctic phosphatase, M0289S, NEB), and gel purified using QIAEX II Gel Extraction Kit (20021, Qiagen). The annealed oligos were ligated into the gel purified vector, transformed into Stabl3 bacteria and miniprepped, as outlined previously (LentiGuide-Puro and LentiCRISPRv2). Three micrograms of the generated pLV-sgKMT5C plasmid were linearized and forward transfected in 4×105 ECas9 (KMT5C wildtype) cells using lipofectamine 3000 (L3000015, Thermo Fisher Scientific), following the manufacturer’s protocol to generate KMT5C mutant clones A, C, and E.
For validation studies using PC9 and HCC827 cell lines, 5×104 cells were transfected with Invitrogen TrueCut Cas9 Protein v2 (A36496) along with the Invitrogen TrueGuide Synthetic gRNAs (A35534, Synthego, Supplementary Table 1) following the Lipofectamine CRISPRMAX Cas9 transfection protocol (CMAX00001, Thermo Fisher Scientific). Forty-eight hours after transfection, a limiting cell dilution was prepared and 1 cell per well was seeded in a 96-well plate, for clonal isolation and expansion.
For all siRNA-mediated knockdown experiments, 30nM of the respective siRNAs were reverse transfected into 1×104 (for dose curves and proliferation assays) or 4×105 KMT5C mutant clones using Lipofectamine RNAiMAX (13-778-150, Thermo Fisher Scientific) following the manufacturer’s protocol. siRNAs used in the study: siMET (Catalog # 4390824, Assay ID # s8700, Thermo Fisher Scientific) and siLINC01510 (Catalog #: 4392420, Assay ID # n506737 Thermo Fisher Scientific).
For generation of DOX inducible overexpression plasmid, the KMT5C sequence was amplified from an ORF expression clone for KMT5C (eGFP tagged) (EX-V0810-M98, GeneCopoeia) introducing a stop codon. The sequence was purified and ligated into the pLVX-Tetone. The oligonucleotides used to perform the sequence exchange are indicated in Supplementary Table 2. Following construction of the pLVX-Tetone-KMT5C plasmid, 3μg of the linearized plasmid was transfected into 4×105 Calu6 cells using lipofectamine 3000 to generate the KMT5C-inducible Calu6 clone.
Next, to generate the rescue lines from KMT5C mutant clone C, a puromycin resistance gene was cloned into pLVX-Tetone-KMT5C using the primers outlined in Supplementary Table 2. Following generation of the pLVX-Tetone-KMT5C-puro plasmid, 3μg of the linearized plasmid was transfected in 4×105 KMT5C mutant cells using lipofectamine 3000 for the generation of inducible-KMT5C rescue clones R1, and R2.
Finally, to test effect of MET or LINC01510 on erlotinib resistance, pT3-EF1a-c-Met (31784, Addgene, RRID:Addgene_31784) or pCMV-Hygro-LINC01510 (Twist Bioscience) were transfected using Lipofectamine 3000 in 4×105 KMT5C wildtype cells.
Genotyping of mutations:
Validation of KMT5C mutations were performed by isolating genomic DNA of each clone (K1820-01, Thermo Fisher Scientific), followed by PCR amplification in the region containing the expected KMT5C mutation using Q5 high fidelity polymerase (M0491L, NEB). PCR products were then purified using QIAquick PCR Purification Kit (28106, Qiagen) and cloned into the TOPO TA cloning vector (K457501, Thermo Fisher Scientific) and six colonies were selected and sequenced for each clone using T7 primer. Primers for amplification and sequencing are outlined in Supplementary Table 2.
Bioinformatic analysis of TCGA data:
Cancer Therapeutics Response Portal (CtRPv2) was used to validate the CRISPR-Cas9 knock-out screen (18). Gene Expression Profiling Interactive Analysis (GEPIA) database (19) (RRID:SCR_018294) was used to evaluate KMT5C, LINC01510, and MET levels in NSCLC patient samples and non-tumorigenic controls. GEPIA is a web-based tool for functional analyses of data provide from two independent resources, such as TCGA and GTEx. Spearman correlation analysis between LINC01510 and MET, or between LINC01510 or MET and KMT5C was also evaluated in LUAD tumor samples using GEPIA. Integrated Genome Viewer (IGV 2.3) was used to view bed files reported by GSE59316 using Human genome 19 (hg19) browser.
Western Blot:
Four-hundred thousand cells were grown in individual wells of a 6 well plate, and lysates were isolated at time points specified in figure legends using RIPA buffer (Sodium chloride (150 mM), Tris-HCl (pH 8.0, 50mM), N P-40 (1 %), Sodium deoxycholate (0.5 %), SDS (0.1 %), ddH2O (up to 100 mL)) containing 1X protease inhibitor cocktail (PIA32955, Thermo Fisher Scientific). Protein quantification was performed using Pierce BCA Protein Assay kit. Lysates used to generate data shown in Supplementary Figure 3B were prepared using the histone acid extraction protocol describe by Shechter et al (20). Regardless of the method of isolation, equal amounts of protein lysate were resolved through 12% or 4–20% polyacrylamide gels and transferred onto a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked using LI-COR buffer for 1 hour at room temperature, and incubated overnight in primary antibody at 4°C. The primary antibody was detected using 1:800 IR 800CW secondary antibody. Blots were scanned, and data quantified using the Odyssey LI-COR imaging system and software. Antibodies used: rabbit H4 (61299; Active Motif, RRID:AB_2650524), mouse H4K20me3 (39672; Active Motif, RRID:AB_2650526), rabbit H4K20me3 (ab9053, abcam, RRID:AB_306969), rabbit MET (D1C2) XP (8198, Cell Signaling Technology, RRID:AB_10858224), mouse β-ACTIN (3700, Cell Signaling Technology, RRID:AB_2242334)
In-Cell Western:
Ten-thousand cells were grown in individual wells of a 96-well plate. Forty-eight hours post plating, cells were fixed using cold 100% methanol for 20 minutes at 4°C. Post fixing, cells were permeabilized using 0.2% TritonX in 1X PBS at room temperature for 30 minutes. Cells were blocked using LI-COR blocking buffer for 1.5 hours followed by overnight incubation with primary antibody at 4°C. The primary antibody was detected using 1:800 IR 800CW secondary antibody (LI-COR). The IR-800 signal was quantified using the Odyssey LI-COR imaging system and software. Antibodies used: 1:400 mouse H4K20me3 (39672, Active Motif), 1:500 rabbit GAPDH (2118, Cell Signaling)
Immunofluorescence:
Two-hundred thousand cells were seeded on collagen coated coverslips that were arranged in individual wells of a 12 or 24-well plate. Forty-eight hours post plating, cells were fixed using cold 100% methanol for 20 minutes at 4°C. Post fixing, cells were permeabilized using 0.2% TritonX in 1X PBS at room temperature for fifteen minutes followed by blocking using LI-COR blocking buffer for 1 hour. For KMT5B/C inhibitor experiments, cells were fixed and permeabilized using cold 100% methanol for 10 minutes at −20°C followed by blocking using 0.2μ-filtered 1% Bovine Serum Albumin. Following blocking, cells were incubated overnight with 1:50 mouse H4K20me3 (39672, Active Motif) or 1:50 rabbit anti-H4 antibody (13919S, Cell Signaling) at 4°C. After primary antibody incubation, cells were incubated with secondary antibodies and nuclear stain for 2 hours at room temperature. 1:500 anti-mouse Alexa Fluor 647 (A-31571, Thermo Fisher Scientific) and 1:500 anti-rabbit Alexa Fluor 488 (A-11034, Thermo Fisher Scientific) was used to detect H4K20me3 and H4, respectively, and 1:1000 Hoechst dye (H3570, Thermo Fisher Scientific) was used as a nuclear stain. Coverslips were mounted on glass slides using ProLong™ Glass Antifade Mountant (P36982, Thermo Fisher Scientific). Images were acquired using Nikon A1R-MP microscope with a 40X oil objective (Nikon Inc., Melville, NY, U.S.A). The images were acquired and analyzed using the Nikon NIS-Elements imaging software (version 5.20.02) in the “.nd2” format. The acquisition settings were 1K × 1K resolution (pixels) with a scanning frame rate of 1/8 sec. All images were set to the same display lookup table (LUT) settings before exporting the files.
RNA isolation and Quantitative real time PCR (qRT-PCR):
Four-hundred thousand cells were grown in individual wells of a 6-well plate, and total RNA was isolated after 48 or 96 hours, as indicated, using the miRneasy Kit (217004, Qiagen) according to the manufacturer’s instruction. DNase I digestion (79254, Qiagen) was used in each RNA purification reaction to remove genomic DNA. RNA integrity was evaluated on a 1.5% agarose gel, and total RNA quantified using a nanodrop. For quantifying RNA from EGFR wildtype cells, cDNA was synthesized from 1μg of total RNA using MiScript Reverse Transcriptase kit (218161, Qiagen), as indicated by the manufacturer’s protocol. Q-RT-PCR was conducted using the miScript SYBR Green PCR Kit (218073, Qiagen) as indicated by the manufacturer’s protocol, to quantify target gene mRNA expression. The following primers were obtained: GAPDH (loading control) (QT00079247, Qiagen), LINC01510 (LPH09040A, Qiagen), and MET (QT00023408, Qiagen). Primers for KMT5C quantification are indicated in Supplementary Table 2.
The KMT5C transcript from EGFR mutant cell lines was quantified using Taqman assays. Briefly, cDNA was synthesized from 900ng of total RNA using SuperScript IV VILO Master Mix (11756050, Thermo Fisher Scientific). Q-RT-PCR was conducted using Taqman Fast Advanced Master Mix (4444963, Thermo Fisher Scientific). The following primers were used: KMT5C:(Hs00261961_m1, Thermo Fisher Scientific) and GAPDH (endogenous control) (Hs99999905_m1, Thermo Fisher Scientific).
ChIP-qPCR:
Briefly, a total of 2×107 cells were fixed using 1% of filter-sterilized formadehyde for 10 minutes at room temperature. The formaldehyde was quenched with 2.5M Glycine (55μL per ml of media) for 5 min. Cells were washed with cold PBS and scraped into fresh cold PBS. Cells were pelleted by centrifuging at 1500 rpm for 10 minutes at 4°C. The cell pellet was resuspended in 10 mL of freshly prepared cold cell lysis buffer (5mM PIPES, 85mM KCl, 0.5% NP40), kept on ice for 10 minutes followed by centrifuging at 1000 rm for 10 minutes at 4°C. The lysed cells were resuspended in 1 mL of nuclei lysis buffer (50mM Tris-HCl (pH 8.0),10mM EDTA, 1% SDS) containing 0.1% protease inhibitor cocktail (PIA32955, Thermo Fisher Scientific) and were transferred into 2mL eppendorf tubes, on ice. Cross-linked chromatin from the isolated nuclei was sonicated using a probe sonicator (60% duty cycle) for 10 seconds with a 1 minute rest, for 15 cycles to fragment DNA (100–500 bps). Fragmented DNA was immunoprecipitated with antibodies against mouse H4K20me3 (39672, Active Motif), or negative control mouse IgG (5415, Cell Signaling Technology) at 4°C overnight with gentle rotation. The immunoprecipitated DNA was purified using the DNA isolation kit (K1820–01; Thermo Fisher Scientific) following manufacturer’s protocol. DNA was used as a template for qRT-PCR as described above. All primer sequences used for qRT-PCR are listed in Supplementary Table 2. ChIP data are presented as fold enrichment of DNA immunoprecipitated with H4K20me3 relative to values obtained for DNA immunoprecipitated with IgG control.
Erlotinib dose response assays:
The protocol followed to evaluate erlotinib dose response was as per the NCI-60 Cell Five-Dose Screen (NCI-60 DTP). Briefly, Sulforhodamine B colorimetric assay (SRB assay)(21) was performed by exposing cells to varying concentrations of erlotinib or the highest equivalent volume of DMSO (negative control) containing media for 72 hours. To normalize data, percent of cells was calculated based on first correcting for the number of cells at the start of the assay (time zero = tz), followed by normalization of cell number to respective corrected DMSO values.
Proliferation assays:
Ten thousand cells were seeded in replicates of 6 in a 96-well plate, which was placed in a live-imaging system, Incucyte s3 2018A (ESSEN BioScience). Plates were incubated in the system for the specified times. Four images per well were obtained every 2 hours using the 10X objective. Confluence was evaluated using Incucyte s3 2018A software. To normalize data, percent of cells was calculated based on first correcting for the number of cells at the start of the assay (time zero = tz), followed by normalization of cell number to respective corrected DMSO values. Data is represented relative to controls, as described in figure legends.
Clonogenic assay:
Five thousand HCC827 cells were seeded in 6 well plates. The next day, media containing 0.1 or 0.01μM of erlotinib or the highest equivalent percentage of DMSO was added. Media containing erlotinib was changed every two days, and the plate was fixed 8 days after seeding using the DIFF-Quick Stain Kit following the manufacturer’s protocol (NC1796273, Polyscience).
Statistical analysis:
All data were analyzed using GraphPad Prism version 9 software (GraphPad Software, RIDD:SCR_002798) and are presented as mean values ± standard deviation (SD). Pearson’s correlation was utilized to evaluate linear correlation between KMT5C and/or H4K20me3 and GI50 erlotinib values. Student’s t-test or one-way ANOVA were performed, as specified in the figure legends. P-value of < 0.05 was considered significant.
Results:
Identifying mediators of erlotinib resistance
To identify mutant genes that confer resistance to erlotinib sensitive cells, a genome-wide CRISPR-Cas9 screen was performed. The screen was conducted in EKVX cells, a cell line determined to be erlotinib sensitive by the Developmental Therapeutics Program, maintained by the National Cancer Institute (NCI-60, DTP). EKVX cells were engineered to express the Cas9 protein and resulting clones were validated for erlotinib sensitivity, which was similar to parental EKVX cells (Supplementary Figure 1). Cas9-expressing EKVX clone 7 was taken forward to conduct the screen, which is hereafter referred to as ECas9. ECas9 cells were infected with the GeCKO V2 sgRNA lentiviral library (Figure 1A) (22). To obtain full coverage of the library, transduction was performed at 300-fold coverage and was conducted in triplicates to mitigate false positives. One third of the transduced cells were used to determine the library representation prior to selection in erlotinib (baseline). The remaining cells were grown for 15 passages in the presence of 1.23 uM erlotinib, a concentration that inhibits growth of 75% of ECas9 cells (GI75). Integrated sgRNAs were identified from the resulting population, and from the baseline cells, by PCR amplification and subsequent high-throughput sequencing. Combined analysis of the three replicates using the MAGeCK-VISPR algorithm identified significantly enriched sgRNAs in cells that were cultured in erlotinib (Supplementary Table 3, Figure 1B) (23). Following the analysis, multiple genes that were previously reported to be 1) downregulated during acquired resistance to chemotherapy treatment (24), 2) highly expressed in erlotinib sensitive cells (25), and 3) bona fide tumor suppressors (15,26–30), were identified among the top hits, validating the sensitivity of the screen and appropriateness of the chosen cell line.
Figure 1: A genome-wide CRISPR-Cas9 screen identifies mediators of erlotinib resistance.
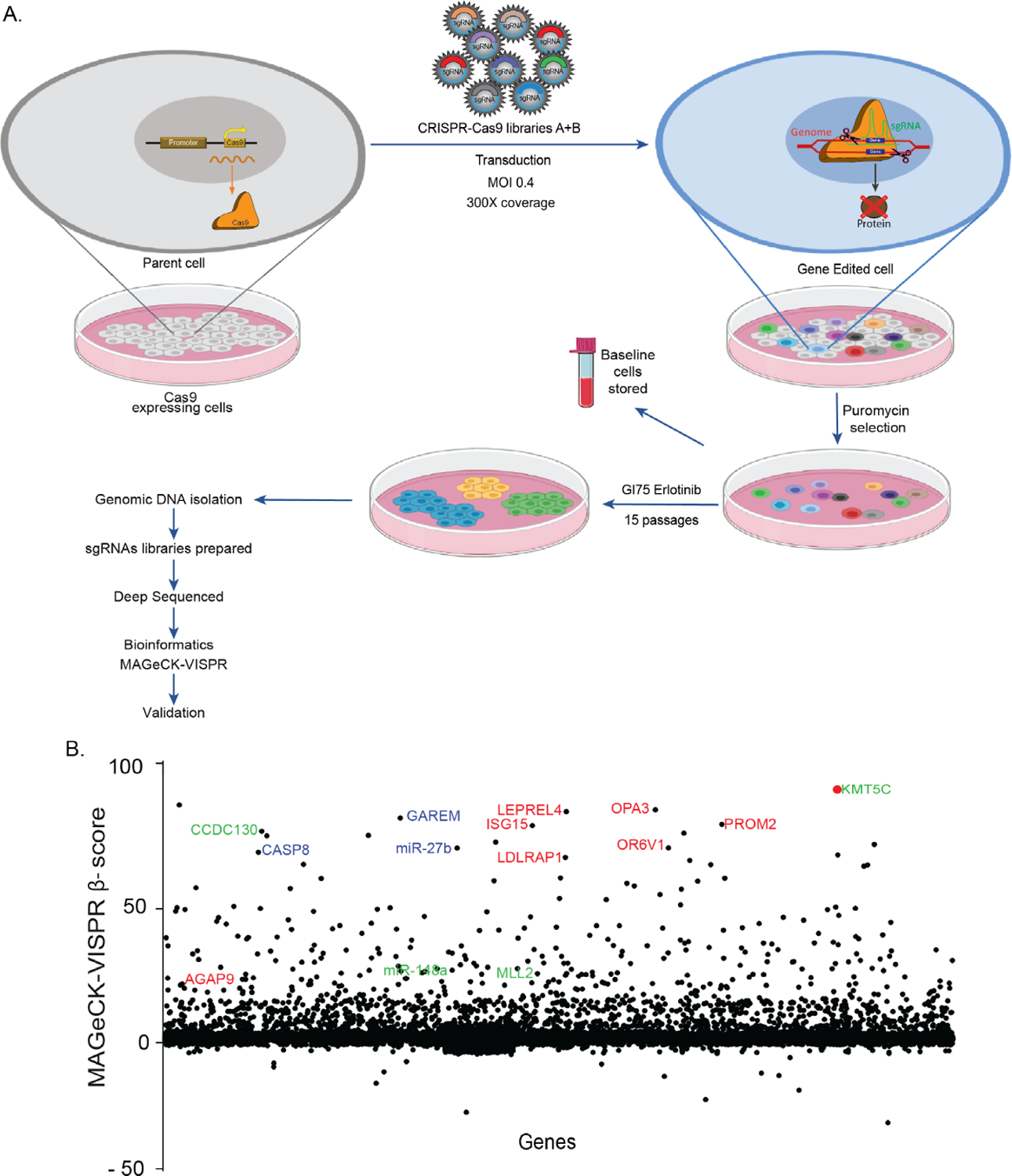
A) Outline of the screen. B) Fold enrichment (β-score) analysis of sgRNAs. Blue: genes previously been reported to be downregulated in cells post chemotherapeutic treatment; red: genes reported to be high in erlotinib sensitive cells; green genes reported as tumor suppressors.
Low expression of KMT5C is associated with erlotinib resistance, and predicts poor prognosis in NSCLC
The top hit from the screen, KMT5C is a histone methyltransferase also referred to as SUV420H2. KMT5C specifically trimethylates histone H4 lysine-20 (H4K20), which is associated with transcriptional repression and is important for establishing constitutive heterochromatic regions (12,13). Multiple studies have reported on the role of KMT5C as a tumor suppressor, and both KMT5C and H4K20 trimethylation (H4K20me3) are severely downregulated in multiple cancers (15,16,30–32). To determine if KMT5C is also a mediator of erlotinib response, various validation assays were performed. Firstly, using a panel of NSCLC cell lines, those included in the DTP and additional EGFR mutant lines, a negative correlation between KMT5C transcript and erlotinib response was determined (Figure 2A–D, Pearson r = −0.81, Supplementary Figure 2). Due to the lack of a sensitive KMT5C antibody for immunoblotting, the downstream effector of KMT5C, H4K20me3 was evaluated as a proxy for KMT5C activity (Supplementary Figure 3A–B). H4K20me3 levels positively correlate with KMT5C transcript levels (Pearson r = 0.24, Supplementary Figure 3C). Additionally, similar to the negative correlation between KMT5C transcript and erlotinib response in the NSCLC panel, H4K20me3 was also negatively correlated with erlotinib response (Pearson r = −0.47, Supplementary Figure 3D). These strong correlations suggest a possible role for KMT5C and H4K20me3 levels in mediating the response of NSCLC cells to erlotinib.
Figure 2: Reduced KMT5C transcript correlates with erlotinib resistance in NSCLC cells, and poor prognosis in NSCLC patients.
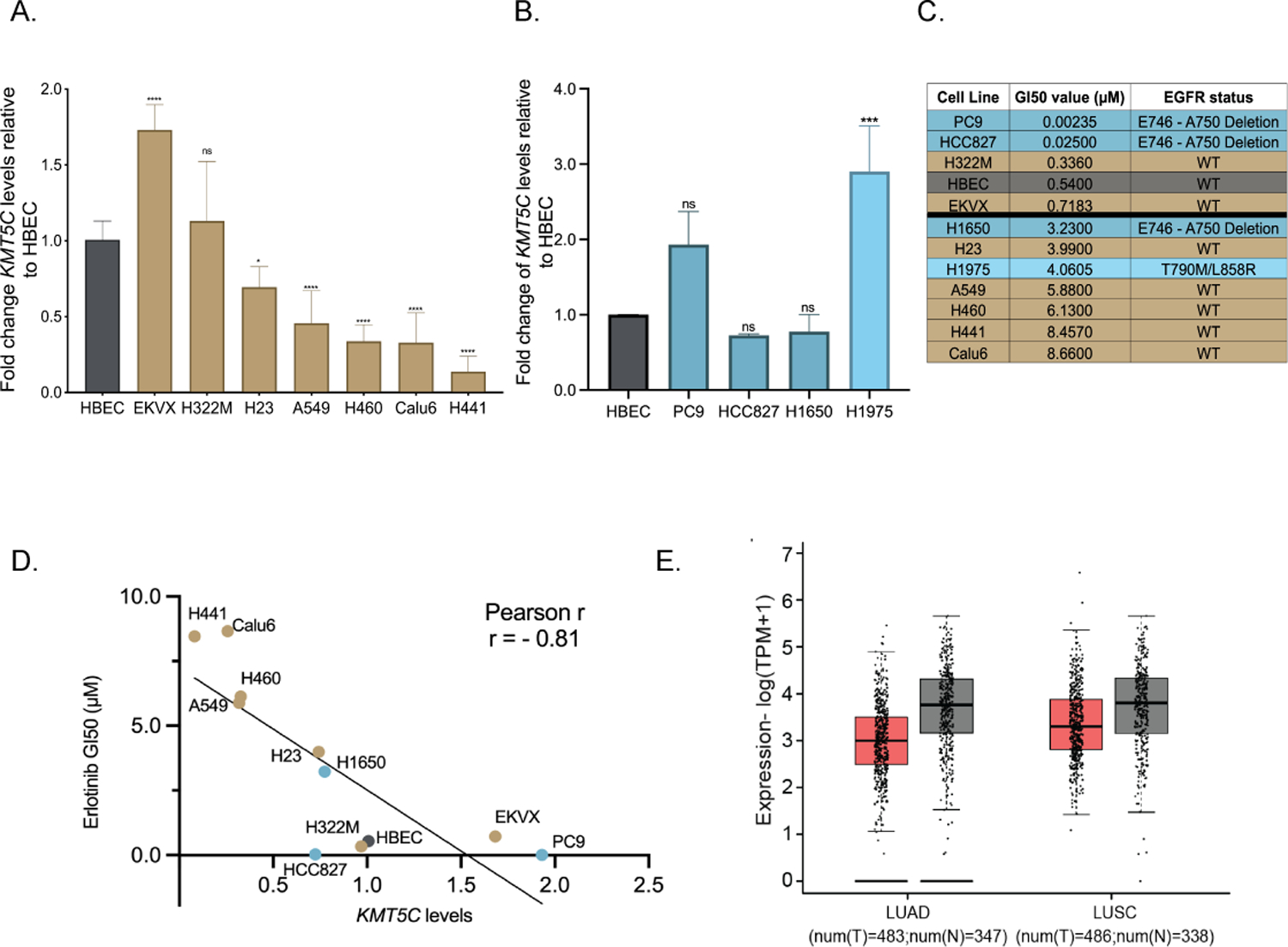
Expression of KMT5C in NSCLC cells A) represented in the DTP or B) with mutation(s) in EGFR, relative to a non-tumorigenic lung epithelial cell line (Human Bronchial Epithelial Cells, HBEC), evaluated by qRT-PCR. Data are normalized to GAPDH and relative to HBEC. One-way ANOVA followed by Dunnett’s Multiple Comparison test was used to evaluate statistical significance. Color of bars represents EGFR mutation status: gold, EGFR wt; dark teal, EGFR primary mutation; light teal, EGFR secondary mutation. C) Erlotinib dose response evaluated by exposing cell lines to varying concentrations of erlotinib or the highest equivalent volume of DMSO containing media for 72 hours followed by SRB assay. GI50 concentrations of erlotinib were calculated from respective dose curve. D) Correlation analysis between KMT5C transcript from A/B and GI50 erlotinib concentrations from C. E) GEPIA analysis for KMT5C transcript levels in normal (grey bars) and tumor samples (pink bars) from LUAD and LUSC data obtained from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) databases. TPM= Transcripts per million, T= Tumor, N=Normal.
Next, we investigated KMT5C transcript levels in NSCLC patient samples using publicly available data provided by The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) projects. Patient samples were compared to non-cancerous control tissues using Gene Expression Profiling Interactive Analysis (GEPIA, Figure 2E) (19). KMT5C transcript levels were generally lower in both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) samples relative to normal samples suggesting that KMT5C may function as a bona fide tumor suppressor.
Loss of KMT5C confers resistance to EGFR inhibitors
To further validate the findings from the CRISPR-Cas9 screen, KMT5C mutant lines, clones A, C and E were generated and validated (Supplementary Figure 4A). KMT5C transcript levels were reduced in all clones (Figure 3A) resulting in downregulation of H4K20me3 (Figure 3B, 3C and Supplementary Figure 4B). Erlotinib sensitivity of the mutant clones was 5.4 – 11.7 fold higher than wildtype cells (Figure 3D). Increased proliferation of the mutant clones in the presence of erlotinib corroborated the results (Figure 3E). We also evaluated the response of KMT5C mutant clones to other EGFRi including afatinib, gefitinib, and osimertinib. All clones were resistant to all EGFRi tested (Supplementary Figures 4C–4H). Conversely, mutant clones were unaffected in the presence of cisplatin (data not shown) suggesting that loss of KMT5C is not a global mediator of resistance, but may be specific to EGFRi or perhaps other targeted agents.
Figure 3: Loss of KMT5C confers resistance to erlotinib.
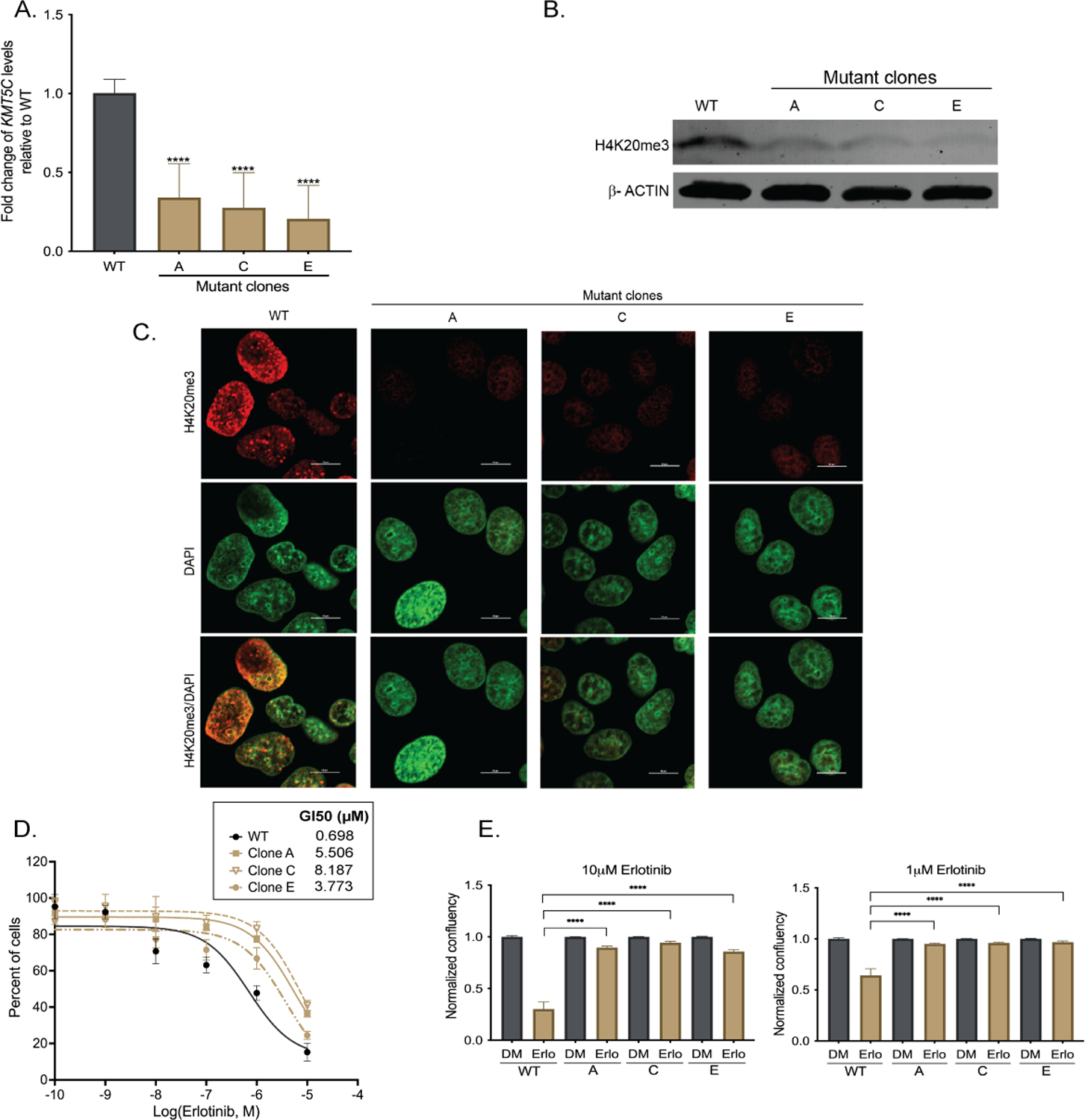
A) Expression of KMT5C transcript in EKVX mutant clones A, C, and E. Data are normalized to GAPDH and are represented relative to ECas9 (KMT5C wildtype, WT) cells. One-way ANOVA was used to evaluate statistical significance. B) Representative western blot of H4K20me3 in EKVX WT cells and KMT5C mutant clones A, C, and E. β-ACTIN serves as a loading control. C) Representative immunofluorescent image of H4K20me3 in WT cells and clones A, C, and E. Scale bar, 10μm. D) Erlotinib dose response following exposure to the indicated concentrations of erlotinib or the highest equivalent volume of DMSO for 72 hours. Following normalization, the GI50 concentration of erlotinib was calculated from the respective dose curve. E) Live cell imaging of WT or mutant clones (represented as A, C, and E) was conducted to quantify proliferating cells in the presence of erlotinib (Erlo) or vehicle control (DMSO, DM) for 72 hours. Data relative to respective normalized DMSO control treatments is represented. One-way ANOVA followed by Dunnett’s Multiple Comparison test was used to evaluate significance.
The primary screen was conducted using the EGFR wildtype cell line EKVX. Because treatment of EGFR wildtype tumors with erlotinib is no longer approved, it was imperative to determine if mutant KMT5C could also drive resistance in EGFR mutant cells. Four EGFR mutant cell lines were identified, all of which had increased EGFR signaling (Supplementary Figure 5). In two of the most sensitive cell lines, PC9 and HCC827, the SET domain of KMT5C was mutated (33) (Figure 2C, 4A–B) resulting in reduced H4K20me3 (Figure 4C). Both mutant cell lines, along with the respective parental lines, were cultured in the presence of increasing doses of erlotinib, revealing resistance of the KMT5C mutants (Figure 4D–F). HCC827 was further validated using a colony formation assay. Erlotinib treatment reduced colony formation of wildtype cells, as expected. However, colony formation from KMT5C mutant cells was similar to untreated cells, highlighting the strong effect that loss of KMT5C has in driving resistance (Figure 4D). Similar to EKVX, both PC9 and HCC827 cell lines also became resistant to osimertinib when KMT5C was mutated (Figure 4G–H).
Figure 4: Loss of KMT5C confers resistance to erlotinib and osimertinib in EGFR mutant cell lines.
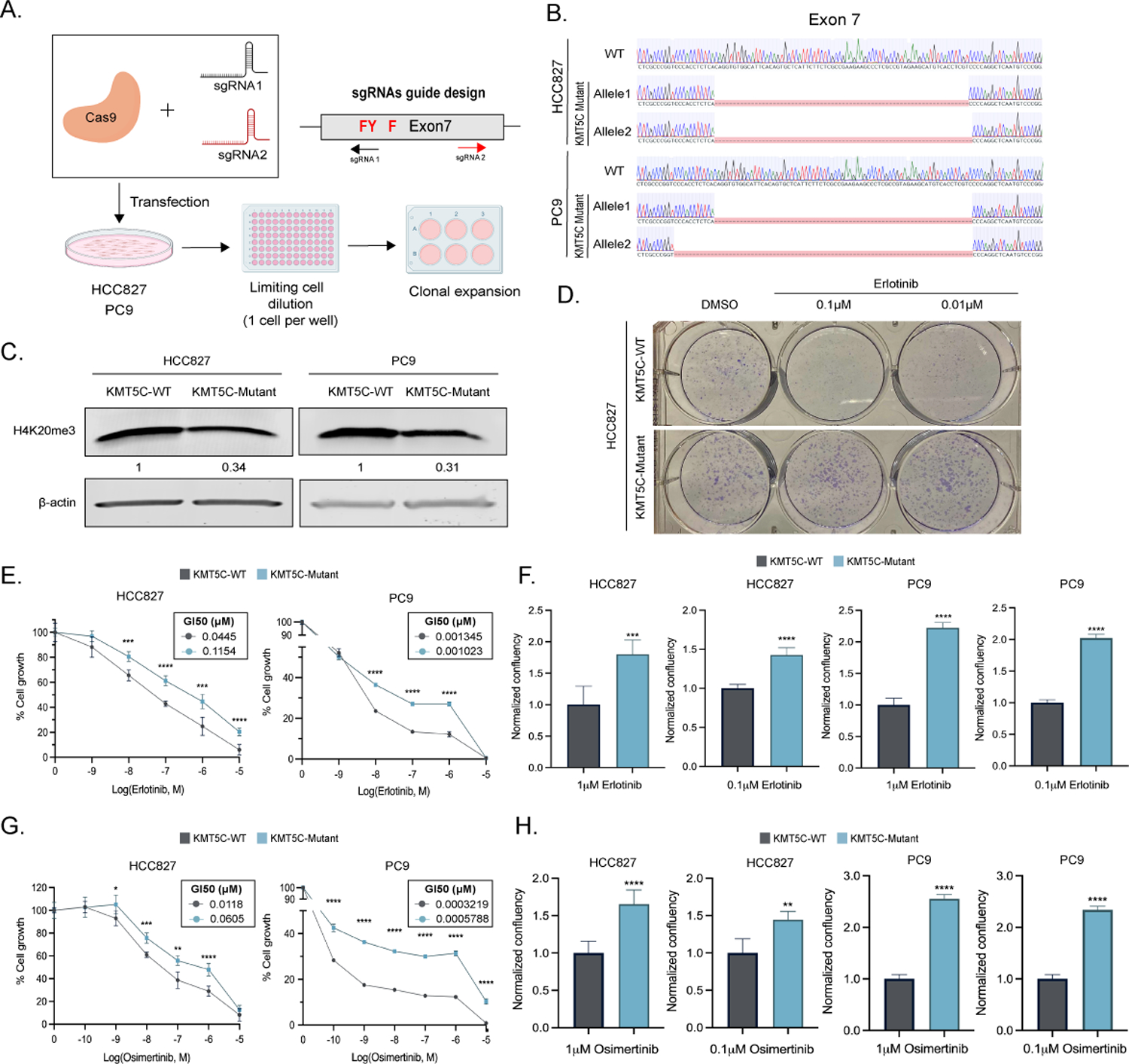
A) CRISPR Cas9 strategy to generate KMT5C SET domain mutants. SET domain active site residues are in red. B) Alignment of exon 7 sequence in WT and mutant clones using benchling (Sequence Alignment Tool, 2021) retrieved from https://benchling.com. B) Representative western blot of H4K20me3 from WT and mutant HCC827 and PC9 clones. β-actin serves as a loading control. D) Clonogenic assay in HCC827 KMT5C mutant and WT cells in the presence of 0.1 or 0.01μM erlotinib containing media for 8 days. Erlotinib E) or Osimertinib G) dose response curves following exposing the indicated cells to varying concentrations of erlotinib containing media for 72 hours. Cell confluency of KMT5C mutant cells was compared to KMT5C WT cells in the presence of 1 or 0.1μM F) erlotinib or H) osimertinib for 72h. Data relative to respective normalized DMSO control treatments is represented. Welch’s t-test was used to evaluate statistical significance.
To complement the genetic studies, HCC827 cells were exposed to A-196, a chemical inhibitor of KMT5B and KMT5C (Bromberg et al. 2017). Treatment with A-196 resulted in a dose- and time-dependent reduction in H4K20me3 (Figure 5A–C and Supplementary Figure 6) that caused resistance to both erlotinib and osimertinib (Figure 5D). Collectively, data provided following either genetic and chemical inhibition of KMT5C, suggest that KMT5C loss provides a clear advantage to both EGFR wildtype and mutant cells exposed to EGFRi.
Figure 5: Chemical inhibition of KMT5B/C increases erlotinib and osimertinib resistance in HCC827 cells line.
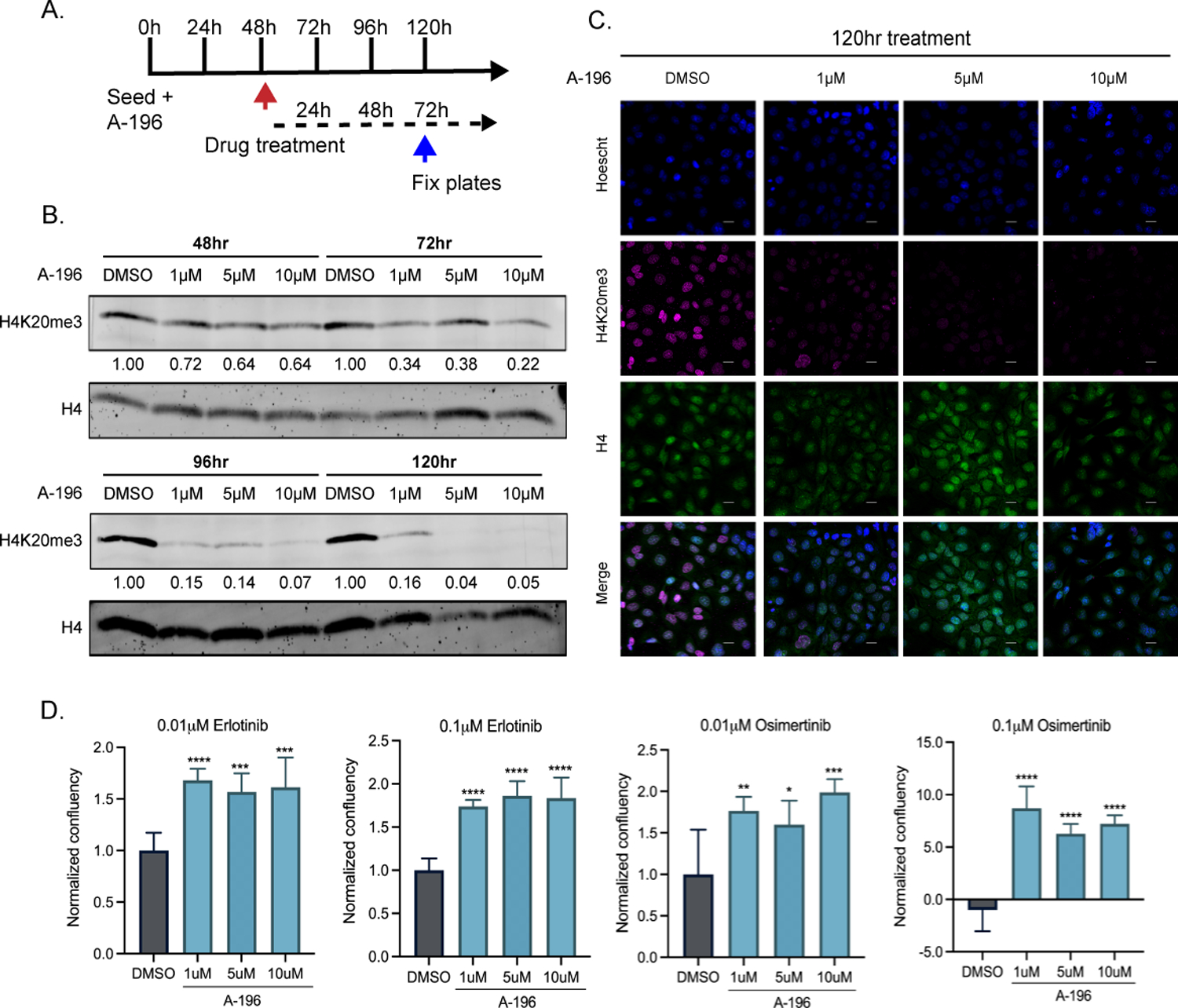
A) Experimental timeline, HCC827 cells were treated with the KMT5B/C inhibitor (A-196), 48 hours later erlotinib or osimertinib was added and cells were fixed 72h later for analysis. B) Western Blot of H4K20me3 in HCC827 cells at different time points, after treatment with A-196. H4 was used as a loading control. C) Immunofluorescence of H4K20me3 and H4 in HCC827 cells after treatment with A-196 for 120h. D) Confluency of HCC827 cells treated with A-196 in the presence of erlotinib/osimertinib for 72h. Welch’s t-test was used to evaluate statistical significance.
Ectopic expression of KMT5C partially sensitizes EGFRi resistant cells
Since loss of KMT5C led to erlotinib resistance, we evaluated if the converse holds true by overexpressing KMT5C. A doxycycline (DOX) inducible KMT5C plasmid was stably expressed in Calu6 cells, which have low levels of KMT5C (Figure 2A) and are resistant to erlotinib (Figure 2C). Culturing two clonally-derived lines in the presence of DOX resulted in a 4 to 8-fold increase of KMT5C (Supplementary Figure 7A). H4K20me3 was also significantly increased following DOX induction in both clones, but not in Calu6 parental cells (Supplementary Figure 7B). Exposure of clones to increasing concentrations of erlotinib resulted in ~2-fold increase in GI50 values for clones cultured in DOX (Supplementary Figure 7C). Live-cell proliferation analysis validated these findings (Supplementary Figure 7D). With respect to gefitinib, afatinib and osimertinib, KMT5C overexpressing clones were sensitized (Supplementary Figure 7E–F), most notably at higher concentrations.
KMT5C negatively regulates the oncogenic long non-coding RNA, LINC01510, and the oncogene, MET
Because KMT5C functions as a tumor suppressor, and is associated with repression of oncogenes (30,34), GEPIA analysis was used to determine if any of the common bypass tracks involved in erlotinib resistance were negatively correlated with KMT5C transcript levels. A significant negative correlation was identified between MET and KMT5C in LUAD (Spearman r = −0.44, p-value = 1.0e−37 Supplementary Figure 8A). MET amplification is one of the more common bypass mechanisms that cells use to overcome inhibition of EGFR signaling by erlotinib (4,35). As expected, MET transcript was higher in LUAD relative to normal tissues (Supplementary Figure 8B). To determine if the negative correlation between MET and KMT5C held true in the NSCLC cell lines, KMT5C mutant cells were evaluated for MET. Indeed, following loss of KMT5C, MET protein and transcript were increased (Figure 6Ai, Bi). Conversely, induction of KMT5C in dox-inducible clones resulted in reductions in both MET RNA and protein (Figure 6Aii, Bii).
Figure 6: KMT5C represses LINC01510 and MET via H4K20me3.
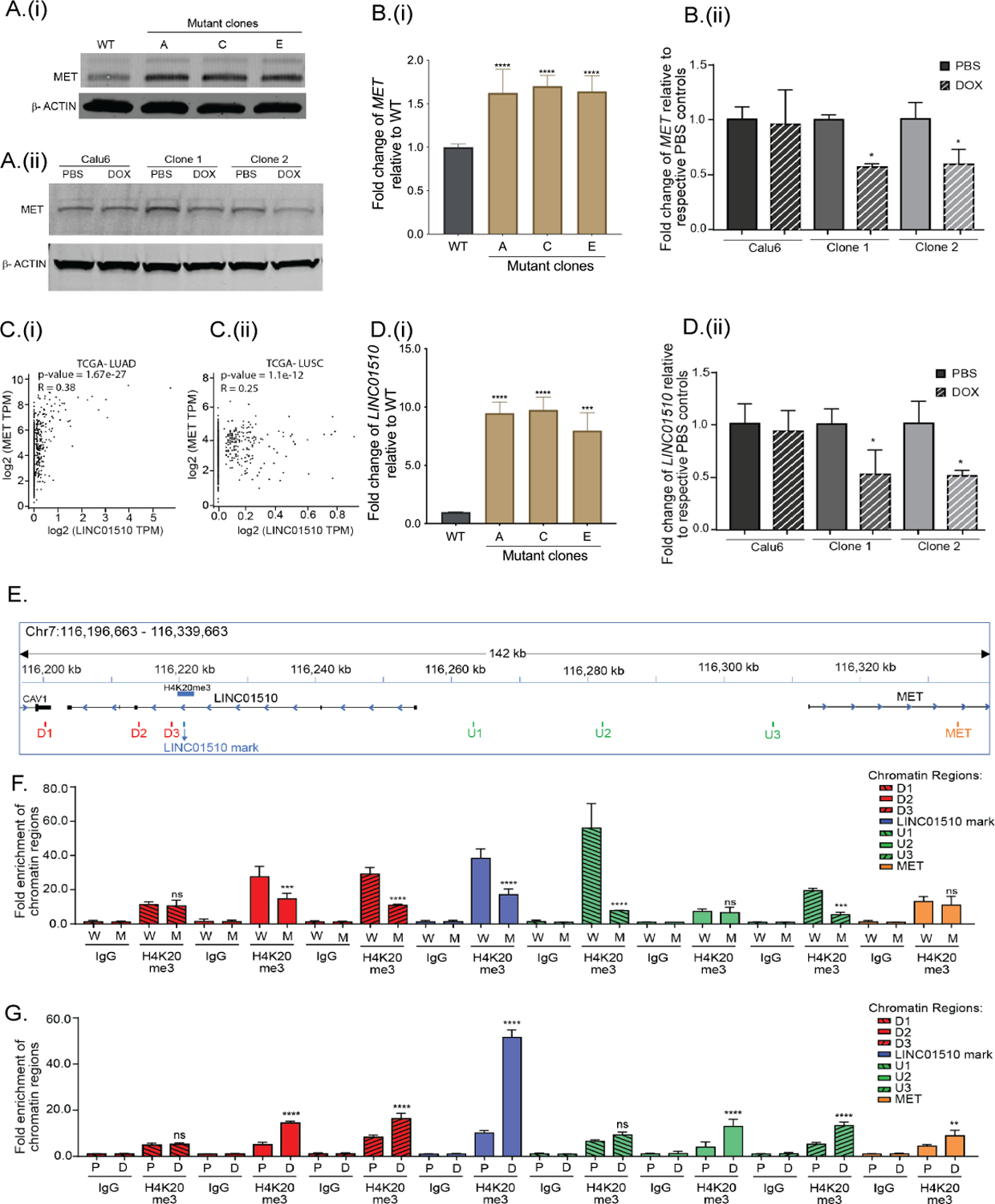
A) Representative western blot of MET in (i) EKVX KMT5C WT cells and mutant clones, and (ii) Calu6 cells and clones stably expressing a DOX-inducible KMT5C vector. B) qRT-PCR data for MET in (i) WT cells and KMT5C mutant clones, or (ii) Calu6 cells and clones stably expressing a DOX-inducible KMT5C vector. C) Correlation analysis between LINC01510 and MET transcripts obtained from (i) LUAD and (ii) LUSC datasets, evaluated using GEPIA. D) Expression of LINC01510 in (i) KMT5C mutant lines, or in (ii) KMT5C inducible clones. E) Diagram of the genomic region representing the predicted H4K20me3 modification on the LINC01510 gene body, upstream of MET, as identified from GSE59316. ChIP-qPCR primers designed on and around the H4K20me3 mark are indicated as LINC01510 mark, regions downstream (D1, D2, D3) and upstream (U1, U2, U3) of the H4K20me3 mark, and on MET. ChIP was performed on chromatin isolated from F) WT (W) or KMT5C mutant clone C (M), G) DOX inducible KMT5C cells following growth in DOX (D, induced) or PBS (P, uninduced). qPCR using the immunoprecipitated chromatin was conducted using primers depicted in E. Data is represented as fold enrichment of the chromatin region pulled-down by H4K20me3 primary antibody relative to IgG. Statistical significance is represented for fold enrichment of chromatin regions in KMT5C mutant clone C relative to WT, or DOX relative to PBS. For panels showing statistical significance, One-way ANOVA followed by Dunnett’s Multiple Comparison test was used. TPM= Transcripts per million.
MET can be induced through genomic amplification and transcriptional upregulation (35–37). And although multiple mechanisms can regulate MET transcription, recently a long non-coding RNA (lncRNA) that functions as an enhancer of MET transcription was identified (38). A short variant of the lncRNA, LINC01510, referred to as COMET (Correlated-to-MET) also positively regulates MET transcription (39). Similar to MET, high LINC01510 correlates with poor prognosis in various cancers, including NSCLC (38,40,41). And, based on the positive correlation between the LINC01510 and MET transcripts in colorectal cancer (38), we evaluated their correlation in NSCLC. A positive correlation in both LUAD (Spearman r = 0.38, p-value = 1.6e−27) and LUSC (Spearman r = 0.25, p-value = 1.1e−12) was evident (Figure 6C). Based on the reported and evaluated positive correlation between MET and LINC01510, and the negative correlation between KMT5C and MET, we hypothesized that KMT5C transcript levels would also negatively correlate with LINC01510. The correlation analysis between KMT5C and LINC01510 suggests a significant, modest negative correlation in LUAD tissues (Spearman r = −0.19, p-value = 1.8e−7, Supplementary Figure 8C). Further evaluation of LINC01510 in NSCLC via GEPIA analysis indicated that LINC01510 was higher in a subset of tumors relative to normal tissues (Supplementary Figure 8D). In condordance, in KMT5C mutant clones LINC01510 was significantly upregulated between 8 and 10-fold (Figure 6Di). Conversely, in the KMT5C inducible clones, LINC01510 was significantly lower when cells were cultured in the presence of DOX (Figure 6Dii).
KMT5C mediates its repressive effects via the H4K20me3 modification (30), hence we hypothesized that MET and/or LINC01510, are likely negatively regulated by KMT5C via H4K20me3 mediated repression. To this end, we analyzed the reported ChIP-seq profile of H4K20me3 obtained from a human lung fibroblast cell line, IMR90 (GSE59316) (34). The H4K20me3 modification in this dataset was not present within or near the MET locus but instead was localized in the gene body of LINC01510 (Figure 6E). To identify the region of the chromosome associated with the H4K20me3 modification in our erlotinib sensitive cells, chromatin immunoprecipitation followed by q-RT-PCR (ChIP-qPCR) was conducted. Sensitivity of the assay was first established using the FOXA1 locus, a target previously reported to be regulated by KMT5C (42). As expected, H4K20me3 pulldown of the FOXA1 region was dependent on the presence of KMT5C (Supplementary Figure 9).
Following the results obtained from ChIP-qPCR for FOXA1, ChIP-qPCR analysis at the LINC01510 and MET loci was conducted using primers overlapping the predicted H4K20me3 site and primers up and downstream of the predicted site (Figure 6E, Supplementary Table 2). Similar to the FOXA1 locus, pulldown varied depending on the status of KMT5C. The most abundant reduction in pulldown in the KMT5C mutant occurred just upstream of the LINC01510 locus with no obvious difference at the MET locus (Figure 6F). In concordance, induction of KMT5C followed by ChIP-qPCR resulted in enrichment of the H4K20me3 mark in regions surrounding the lncRNA, with only a marginal increase at the MET locus (Figure 6G). These results further support the hypothesis that KMT5C regulates LINC01510 expression via the H4K20me3 modification present within its gene body.
Loss of LINC01510 or MET partially re-sensitizes KMT5C mutant cells to erlotinib, conversely overexpression promotes erlotinib resistance in KMT5C wildtype cells
From Figure 6, it can be inferred that KMT5C negatively regulates both LINC01510 and MET transcript levels, and MET protein levels. Therefore, we evaluated if KMT5C negatively regulates MET indirectly through repression of LINC01510. LINC01510 or MET were knocked down in a KMT5C mutant clone, which expresses high levels of LINC01510 and MET (Figures 7Ai, 6Bi, 6Di). It was confirmed that siRNAs targeting either MET or LINC01510 downregulate MET at both the protein and transcript level (Figure 7A, B). To determine if loss of KMT5C partially mediates erlotinib resistance via upregulation of LINC01510 and MET transcripts, LINC01510 or MET were downregulated and erlotinib dose response and proliferation analyses were conducted. Both results validate that erlotinib resistant KMT5C mutant cells can be partially re-sensitized to erlotinib post knockdown of either LINC01510 or MET (Figure 7C, D).
Figure 7: Modulation of LINC01510 or MET is partially responsible for the erlotinib response.
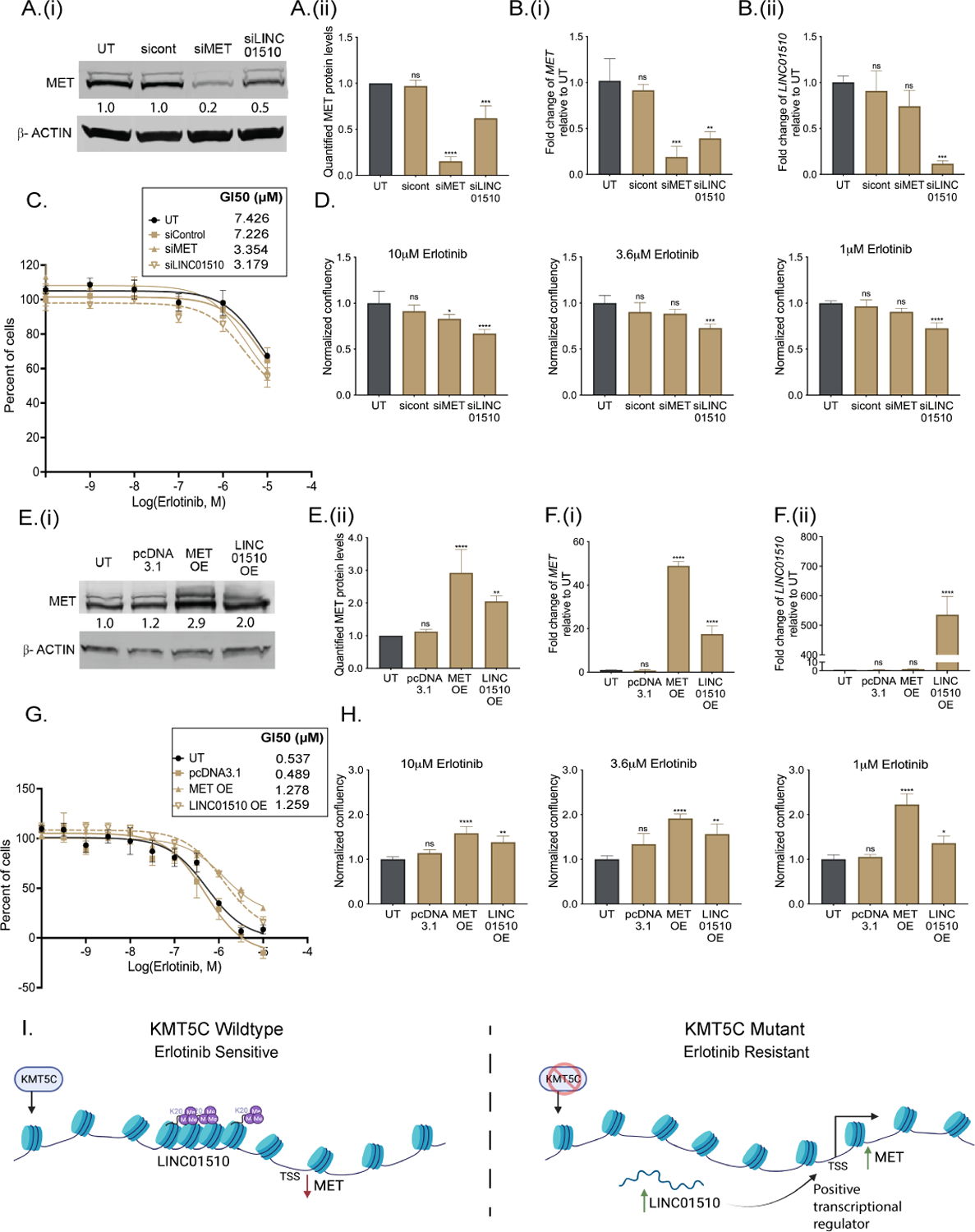
A) (i) Representative western blot of MET in KMT5C mutant cells that were either untransfected (UT) or reverse transfected with siRNA control (sicont), siRNA to MET (siMET), or siRNA to LINC01510 (siLINC01510) for 96 hours. β-ACTIN serves as a loading control. Densitometry values normalized to β-ACTIN, and relative to UT are indicated. ii) Quantification of protein levels from three biological replicates as done in A(i). B) Expression of (i) MET and (ii) LINC01510 in KMT5C mutant cells that were either UT or reverse transfected with sicont, siMET or siLINC01510 for 96 hours. Data are normalized to GAPDH and are graphed relative to data from UT cells. C) Erlotinib dose response of KMT5C mutant cells following transfection with the indicted siRNAs. Twenty-four hours post transfection, cells were exposed to varying concentrations of erlotinib or DMSO for 72 hours. Post-normalization, the GI50 concentration of erlotinib was calculated. D) Proliferation of KMT5C mutant cells following transfection with the indicted siRNAs. Twenty-four hours post transfection, cells were exposed to erlotinib for 72 hours. Normalized data is represented relative to UT. One-way ANOVA followed by Dunnett’s Multiple Comparison test was used to evaluate significance. E) (i) Representative western blot of MET in KMT5C WT cells that were untransfected (UT), or transfected with pcDNA3.1 control plasmid or plasmids to overexpress to MET (MET OE) or LINC01510 (LINC01510 OE) for 96 hours. β-ACTIN was used as a loading control. Densitometry values for the representative blots are shown below. (ii) Quantification of MET from three biological replicates as in E(i). F) Expression of (i) MET and (ii) LINC01510 in KMT5C wildtype (WT) cells that were either UT or transfected with the indicated vectors. Data are normalized to GAPDH. G) Erlotinib dose response via SRB assay was evaluated in WT cells that were either UT or that were transfected with the indicated vectors, as described in C. H) Proliferation of WT cells transfected as in G, was evaluated as described in D. I) Model depicting loss of KMT5C in NSCLC results in development of erlotinib resistance via LINC01510-mediated upregulation of MET.
Data presented in Figure 7A, B suggests that knockdown of LINC01510 reduces MET at the transcript level, therefore, we further evaluated if overexpression of LINC01510 in KMT5C wildtype cells can positively regulate MET. Following transfection of a LINC01510 or MET overexpressing plasmid, a significant increase in MET was observed (Figure 7E, F). Additionally, as hypothesized, LINC01510 or MET overexpression also led to acquired resistance in KMT5C wildtype cells (Figure 7G, H).
Overall, the findings of this study suggests that wildtype KMT5C in NSCLC cells negatively regulates LINC01510 via H4K20me3 (Figure 7I). In cells with high KMT5C, repression of LINC01510 inhibits full expression of MET. However, upon loss of KMT5C, LINC01510 becomes de-repressed due to reductions in H4K20me3, resulting in increased expression of LINC01510. Simultaneously, LINC01510 positively regulates the transcription of MET. Therefore, increased levels of LINC01510 and MET function as mediators of erlotinib resistance in KMT5C mutant cells.
Discussion:
Changes to the epigenome influence all aspects of cancer, including chemoresistance (43). However, only a few epigenetic factors have been determined to have a role in resistance (44). The aim of this study was to identify unknown mechanisms by which acquired erlotinib resistance manifests in NSCLC in an unbiased way, and loss of KMT5C was the top hit. KMT5C is a histone methyltransferase responsible for maintaining constitutive heterochromatic regions of the genome and for repressing specific genes, via the repressive mark H4K20me3.
Catalysis of H4K20me3 is a sequential process. SUV39H2 (KMT1B), another histone methyltransferase first catalyzes H3K9me3, that recruits HP1 which physically associates with KMT5C to mediate H4K20me3 (12,45). While other components of this pathway contribute to resistance, including SUV39H/1 (46,47), here, for the first time we describe a role for KMT5C in mediating drug resistance. Apart from SUV39H1/2, it is possible that other upstream regulators of KMT5C such as HP1 may have an unidentified role in mediating resistance to drugs. Indeed, the first identified demethylase for H4K20me3, mineral dust-induced gene (Mdig) was determined to be overexpressed in breast and lung cancer cells antagonizing the effects of the H4K20me3 modification which led to induction of oncogenes (48).
It has been long appreciated that genomic instability generates tumor heterogeneity and in the presence of a drug gives rise to resistant cells (43,49), also a reported mechanism of EGFRi resistance (50,51). In the current study, complete loss of KMT5C function may have led to spontaneous genetic aberrations leading to rapid establishment of resistant population of cells in the presence of EGFRi. Indeed, previous reports determined that loss of KMT5B/C impairs the DDR mechanism, inadvertently leading to accumulation of damaged DNA and increased tumorigenicity (12,13,52–56). Therefore, it is possible that in the KMT5C mutant cells, the chromatin may have suffered massive loss of H4K20me3, which disrupted the heterochromatic shield protecting the DNA from damage. On the contrary, in Calu6 cells, which still have modest amounts of H4K20me3 (Supplementary Figure 3) the regions of the chromatin lacking H4K20me3 could be localized at oncogenes leading to their upregulation, while the constitutive heterochromatic regions remained marked and compact, preventing genomic instability. Indeed, increased H4K20me3 in Calu6 cells due to DOX-induction of KMT5C resulted in reductions in MET and promoted sensitivity to EGFRi suggesting that even modest changes in H4K20me3, or other unidentified mechanisms of KMT5C can alter the response of cells to EGFRi. Additional studies addressing the dynamics of KMT5C and H4K20me3 and their role in maintaining genomic stability will need to be conducted to support these observations.
While this study defines a role for MET and LINC01510 upregulation that is mediated by loss of KMT5C in EGFRi resistance, there are likely to be several other oncogenes regulated by KMT5C that contribute to this phenotype. Using the NCI Cell Miner Database (57), multiple genes involved in NSCLC or in EGFRi resistance were found to negatively correlate with KMT5C. Some of the top genes include Annexin A5 (negative correlation, nc = −0.616), Vimentin (nc = −0.636), CD44 (nc = −0.637), AKT3 (nc = −0.612), PRKD1 (nc = −0.632) a member of the PKC family, NOTCH (nc = −0.565), JUN (nc = −.0.359) and ERK (nc = −0.343) all with p-values <0.01. The negative correlation between MET and KMT5C was −0.337. Similar to MET, many of these genes are predicted to contain a H4K20me3 modification as determined using H4K20me3 ChIP from IMR90 (GSE59316) (34). It is possible that aberrant KMT5C may alter a cohort of genes that could ultimately synergize to promote resistance, similar to the effects observed following aberrant microRNA expression (58–60). Whether or not these additional candidates are also KMT5C targets and what their contribution is to resistance remains an active area of investigation.
In conclusion, the results of this study describe that loss of KMT5C confers EGFRi resistance in NSCLC cells via a novel mechanism. Loss of KMT5C abrogates the H4K20me3 modification at an oncogenic long non-coding RNA, LINC01510, resulting in enhanced transcription of LINC01510. LINC01510 in turn functions as a positive transcriptional regulator of the oncogene MET consequently resulting in MET upregulation, a predominant mechanism of acquired resistance to erlotinib.
Supplementary Material
Statement of Significance:
Dysregulation of the epigenetic modifier KMT5C can drive MET-mediated EGFRi resistance, implicating KMT5C loss as a putative biomarker of resistance and H4K20 methylation as a potential target in EGFRi-resistant lung cancer.
Acknowledgments:
The authors acknowledge support from the Purdue Center for Cancer Research, NIH grant P30CA023168. We also thank Dr. Wen-Hung Wang for his generous help during KMT5C inducible plasmid generation, Dr. Phillip SanMiguel for his guidance during deep-sequencing of the CRISPR-Cas9 knock-out screen samples, and Dr. Andy Schaber for his input during immunofluorescence experiments.
Financial Support:
This study was funded in part by the National Institutes of Health (R01CA205420) to ALK and a grant from the NIH to the Purdue Center for cancer Research (P30CA023168). A.S.P. was supported by a Purdue Research Foundation (PRF) Research Grant award by the Department of Biological Sciences, Purdue University, a SIRG grant administered through the Purdue Center for Cancer Research, Purdue University, a Cancer Prevention Internship Program Graduate Research Assistantship funded by Purdue University, and a Bilsland Dissertation Fellowship awarded by the Department of Biological Sciences, Purdue University. A.A. was supported by a Ross Fellowship administered through Purdue University. C.L. was supported through a Bioinformatics Fellowship through the Purdue Center for Cancer Research.
Footnotes
The authors declare no potential conflicts of interest.
Declaration of Interests:
The authors declare no competing interests
References:
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. Ca Cancer J Clin. 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, et al. Highly parallel identification of essential genes in cancer cells. Proceedings of the National Academy of Sciences. 2008;105:20380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onitsuka T, Uramoto H, Ono K, Takenoyama M, Hanagiri T, Oyama T, et al. Comprehensive Molecular Analyses of Lung Adenocarcinoma with Regard to the Epidermal Growth Factor Receptor, K-ras, MET, and Hepatocyte Growth Factor Status. J Thorac Oncol. 2010;5:591–6. [DOI] [PubMed] [Google Scholar]
- 4.Liao B-C, Griesing S, Yang JC-H. Second-line treatment of EGFR T790M-negative non-small cell lung cancer patients. Ther Adv Med Oncol. 2019;11:1758835919890286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science Translational Medicine. 2011;3:75ra26–75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nature communications. 2015;6:6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc National Acad Sci. 2008;105:2070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, et al. Reduced NF1 Expression Confers Resistance to EGFR Inhibition in Lung Cancer. Cancer Discov. 2014;4:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forloni M, Gupta R, Nagarajan A, Sun L-S, Dong Y, Pirazzoli V, et al. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Reports. 2016;16:457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang S, Benavente S, Armstrong EA, Li C, Wheeler DL, Harari PM. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2011;71:7071–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, et al. PTEN Loss Contributes to Erlotinib Resistance in EGFR-Mutant Lung Cancer by Activation of Akt and EGFR. Cancer Res. 2009;69:3256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahn M, Dambacher S, Dulev S, Kuznetsova AY, Eck S, Wörz S, et al. Suv4–20h2 mediates chromatin compaction and is important for cohesin recruitment to heterochromatin. Genes & Development. 2013;27:859–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callén E, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes & Development. 2008;22:2048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weirich S, Kudithipudi S, Jeltsch A. Specificity of the SUV4–20H1 and SUV4–20H2 protein lysine methyltransferases and methylation of novel substrates. J Mol Biol. 2016;428:2344–58. [DOI] [PubMed] [Google Scholar]
- 15.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. 2005;37:391–400. [DOI] [PubMed] [Google Scholar]
- 16.Pogribny IP, Ross SA, Tryndyak VP, Pogribna M, Poirier LA, Karpinets TV. Histone H3 lysine 9 and H4 lysine 20 trimethylation and the expression of Suv4–20h2 and Suv-39h1 histone methyltransferases in hepatocarcinogenesis induced by methyl deficiency in rats. Carcinogenesis. 2006;27:1180–6. [DOI] [PubMed] [Google Scholar]
- 17.Golden RJ, Chen B, Li T, Braun J, Manjunath H, Chen X, et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature. 2017;542:197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–57. [DOI] [PubMed] [Google Scholar]
- 21.Orellana EA, Kasinski AL. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. BIO-PROTOCOL. 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR–Cas9. Nat Rev Genet. 2015;16:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.CHEN S, WANG Q, ZHOU X-M, ZHU J-P, LI T, HUANG M. MicroRNA-27b reverses docetaxel resistance of non-small cell lung carcinoma cells via targeting epithelial growth factor receptor. Mol Med Rep. 2016;14:949–54. [DOI] [PubMed] [Google Scholar]
- 25.Orzáez M, Guevara T, Sancho M, Pérez-Payá E. Intrinsic caspase-8 activation mediates sensitization of erlotinib-resistant tumor cells to erlotinib/cell-cycle inhibitors combination treatment. Cell Death Dis. 2012;3:e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aprelikova O, Palla J, Hibler B, Yu X, Greer YE, Yi M, et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene. 2013;32:3246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Zaman MS, Deng G, Majid S, Saini S, Liu J, et al. MicroRNAs 221/222 and Genistein-Mediated Regulation of ARHI Tumor Suppressor Gene in Prostate Cancer. Cancer Prev Res. 2011;4:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kantidakis T, Saponaro M, Mitter R, Horswell S, Kranz A, Boeing S, et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Gene Dev. 2016;30:408–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryu S-W, Yoon J, Yim N, Choi K, Choi C. Downregulation of OPA3 Is Responsible for Transforming Growth Factor-β-Induced Mitochondrial Elongation and F-Actin Rearrangement in Retinal Pigment Epithelial ARPE-19 Cells. Plos One. 2013;8:e63495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shinchi Y, Hieda M, Nishioka Y, Matsumoto A, Yokoyama Y, Kimura H, et al. SUV420H2 suppresses breast cancer cell invasion through down regulation of the SH2 domain-containing focal adhesion protein tensin-3. Exp Cell Res. 2015;334:90–9. [DOI] [PubMed] [Google Scholar]
- 31.Broeck AVD, Brambilla E, Moro-Sibilot D, Lantuejoul S, Brambilla C, Eymin B, et al. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:7237–45. [DOI] [PubMed] [Google Scholar]
- 32.Chekhun VF, Lukyanova NY, Kovalchuk O, Tryndyak VP, Pogribny IP. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethylated targets. Mol Cancer Ther. 2007;6:1089–98. [DOI] [PubMed] [Google Scholar]
- 33.Wu H, Siarheyeva A, Zeng H, Lam R, Dong A, Wu X-H, et al. Crystal structures of the human histone H4K20 methyltransferases SUV420H1 and SUV420H2. FEBS letters. 2013;587:3859–68. [DOI] [PubMed] [Google Scholar]
- 34.Nelson DM, Jaber-Hijazi F, Cole JJ, Robertson NA, Pawlikowski JS, Norris KT, et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome biology. 2016;17:158–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jakobsen KR, Demuth C, Madsen AT, Hussmann D, Vad-Nielsen J, Nielsen AL, et al. MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis. 2017;6:e307–e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–61. [DOI] [PubMed] [Google Scholar]
- 37.Seol DW, Chen Q, Zarnegar R. Transcriptional activation of the hepatocyte growth factor receptor (c-met) gene by its ligand (hepatocyte growth factor) is mediated through AP-1. Oncogene. 2000;19:1132–7. [DOI] [PubMed] [Google Scholar]
- 38.Cen C, Li J, Liu J, Yang M, Zhang T, Zuo Y, et al. Long noncoding RNA LINC01510 promotes the growth of colorectal cancer cells by modulating MET expression. Cancer cell international. 2018;18:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esposito R, Esposito D, Pallante P, Fusco A, Ciccodicola A, Costa V. Oncogenic properties of the antisense lncRNA COMET in BRAF- and RET-driven papillary thyroid carcinomas. Cancer Res. 2019;79:canres.2520.2018. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Wei L. Increased expression of LINC01510 predicts poor prognosis and promotes malignant progression in human non-small cell lung cancer. Biomed Pharmacother. 2019;109:519–29. [DOI] [PubMed] [Google Scholar]
- 41.Li Q, Wang X, Jin J. SOX2-induced upregulation of lncRNA LINC01510 promotes papillary thyroid carcinoma progression by modulating miR-335/SHH and activating Hedgehog pathway. Biochem Bioph Res Co. 2019;520:277–83. [DOI] [PubMed] [Google Scholar]
- 42.Viotti M, Wilson C, McCleland M, Koeppen H, Haley B, Jhunjhunwala S, et al. SUV420H2 is an epigenetic regulator of epithelial/mesenchymal states in pancreatic cancer. The Journal of cell biology. 2018;217:763–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilting RH, Dannenberg J-H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Update. 2012;15:21–38. [DOI] [PubMed] [Google Scholar]
- 45.Bosch-Presegué L, Raurell-Vila H, Thackray JK, González J, Casal C, Kane-Goldsmith N, et al. Mammalian HP1 Isoforms Have Specific Roles in Heterochromatin Structure and Organization. Cell reports. 2017;21:2048–57. [DOI] [PubMed] [Google Scholar]
- 46.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. [DOI] [PubMed] [Google Scholar]
- 47.Souza PP, Völkel P, Trinel D, Vandamme J, Rosnoblet C, Héliot L, et al. The histone methyltransferase SUV420H2 and Heterochromatin Proteins HP1 interact but show different dynamic behaviours. BMC cell biology. 2009;10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q, Thakur C, Fu Y, Bi Z, Wadgaonkar P, Xu L, et al. Mdig promotes oncogenic gene expression through antagonizing repressive histone methylation markers. Theranostics. 2020;10:602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nahar R, Zhai W, Zhang T, Takano A, Khng AJ, Lee YY, et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat Commun. 2018;9:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serizawa M, Takahashi T, Yamamoto N, Koh Y. Genomic aberrations associated with erlotinib resistance in non-small cell lung cancer cells. Anticancer Res. 2013;33:5223–33. [PubMed] [Google Scholar]
- 52.Bromberg KD, Mitchell TRH, Upadhyay AK, Jakob CG, Jhala MA, Comess KM, et al. The SUV4–20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat Chem Biol. 2017;13:317–24. [DOI] [PubMed] [Google Scholar]
- 53.Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, et al. H2AX Haploinsufficiency Modifies Genomic Stability and Tumor Susceptibility. Cell. 2003;114:371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jørgensen S, Schotta G, Sørensen CS. Histone H4 Lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kovaříková AS, Legartová S, Krejčí J, Bartová E. H3K9me3 and H4K20me3 represent the epigenetic landscape for 53BP1 binding to DNA lesions. Aging Albany Ny. 2018;10:2585–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanders SL, Portoso M, Mata J, Bähler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–14. [DOI] [PubMed] [Google Scholar]
- 57.Shankavaram UT, Varma S, Kane D, Sunshine M, Chary KK, Reinhold WC, et al. CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. Bmc Genomics. 2009;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orellana EA, Kasinski AL. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers. 2015;7:1388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams BD, Kasinski AL, Slack FJ. Aberrant Regulation and Function of MicroRNAs in Cancer. Current Biology. 2014;24:R762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kasinski AL, Slack FJ. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nature Reviews Cancer. 2011;11:849–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.