

Abstract
Mutational processes driving genome evolution and heterogeneity contribute to immune evasion and therapeutic resistance in viral infections and cancer. APOBEC3 enzymes promote such mutations by catalyzing the deamination of cytosines to uracils in single-stranded DNA. Chemical inhibition of APOBEC3 enzymes may yield an antimutation therapeutic approach that improves the durability of current drug therapies that are prone to resistance mutations. APOBEC3 small molecule drug discovery efforts to date have been restricted to a single high-throughput biochemical activity assay; however, the arsenal of discovery assays has significantly expanded in recent years. The assays employed to study APOBEC3 enzymes are reviewed here with an eye towards their potential for utilization in small molecule discovery efforts.
Keywords: APOBEC, chemical probes, DNA deaminase inhibitors, drug discovery, screening
1. Cellular roles of APOBEC3 enzymes
1.1. Structure and function of APOBEC3 enzymes
APOBEC3A-H (apolipoprotein B mRNA editing catalytic polypeptide-like 3A-H, A3A-H) is a seven-member subfamily of cytosine deaminase enzymes that convert cytosines to uracils in single-stranded DNA (ssDNA) and is part of a larger family of cytosine deaminases including APOBEC1, APOBEC2, APOBEC4, and Activation-Induced Deaminase (AID). The chemical mechanism of deamination (see Glossary) is inferred from work done on structurally related bacterial [1–5], yeast [6–10], and human [11] cytidine deaminase (CDA) enzymes. APOBECs utilize zinc that is tetrahedrally coordinated by a combination of conserved cysteine and histidine residues, as well as a water molecule. A conserved glutamate residue activates that water molecule to act as a nucleophile, and mediates attack on C-4 of the cytosine base, followed by conversion of the unstable tetrahedral hemiaminal intermediate to uracil and release of ammonia (Figure 1A).
Figure 1.
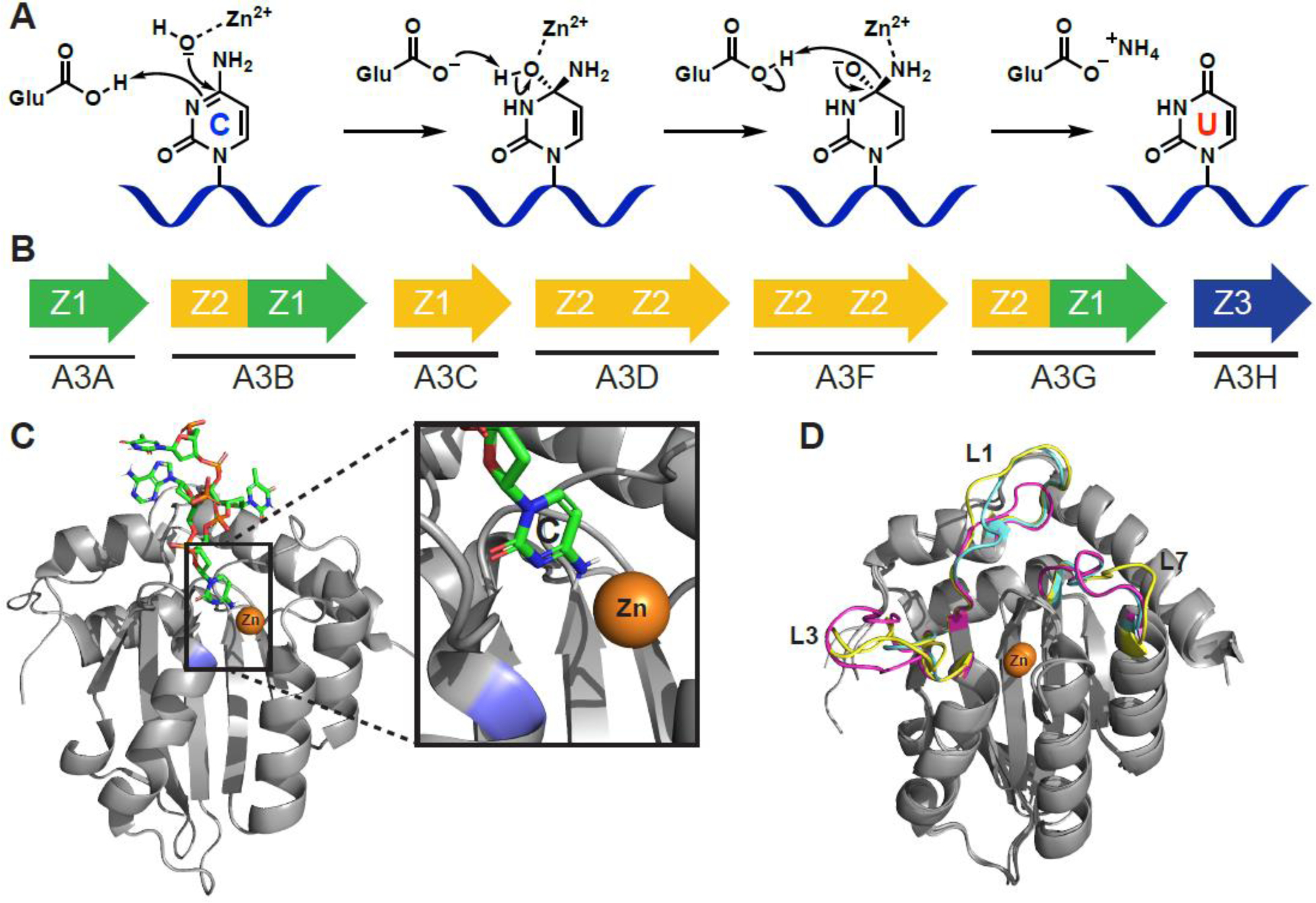
APOBEC3 structure and function. A) Proposed C-to-U hydrolytic deamination mechanism. B) The human APOBEC3 subfamily represented by arrows (single or double domain), and colors indicating phylogenetic grouping (single or double domain). C) A3B C-terminal domain co-crystal structure bound to ssDNA (PDB: 5TD5). The target cytosine is shown (inset) to be in proximity to the catalytic zinc (orange) and catalytic Glu255 (blue; mutated to Ala for crystallographic studies). D) Overlay of A3A (magenta, PDB: 4XXO), A3B C-terminal domain (cyan, PDB: 5CQI), and A3G C-terminal domain (yellow, PDB: 3IR2) showing conservationed structure between Z1 domains of different APOBEC3 enzymes of therapeutic relevance. Most of the structural variability occurs in structure is in the flexible loop regions that have significant effects on substrate binding and catalytic activity. Structure alignments performed with Pymol 2.3 align function.
APOBEC3 (A3) enzymes are categorized by phylogenetically distinguishable zinc-coordinating domains (Z1, Z2, or Z3 defined by conserved amino acid residues) and whether they are made up of one (A3A, A3C, and A3H) or two (A3B, A3D, A3G, and A3F) Z-domains (Figure 1B)[12, 13]. In the case of double-domain enzymes, only the C-terminal domain is catalytically active; the N-terminal domain is similar to the catalytic domain in structure, but not in function, and is often referred to as pseudocatalytic [14]. APOBEC3 enzymes require a minimum 5-mer oligonucleotide in order to bind [15], where the ssDNA adopts a conformation to allow the target cytosine to access the active site (Figure 1C) [16–19]. Most APOBEC3 enzymes have a 5′-TC-3′ dinucleotide sequence preference [20–26]; A3G is the exception and prefers to deaminate cytosine in a 5′-CC-3′ motif [27–29].
There is a large degree of sequence and structural conservation within and across the three Z domains, but some unique features between the protein family members may be exploited to achieve selectivity during inhibitor development. For example, multiple loop regions of A3A, A3B C-terminal domain, and A3G C-terminal domain are flexible and contain the lowest degree of sequence similarity between proteins (L1, L3, and L7 in Figure 1D). Additionally, the pseudocatalytic N-terminal domains contribute to ssDNA/RNA binding and subcellular localization [30–34], and may act in a deamination-independent manner to restrict viral infection by binding viral RNA and sterically blocking viral reverse transcriptase [35–37]. However, the deamination-dependent activity of APOBEC3 enzymes is likely the prominent mechanism of virus restriction, tumor mutagenesis, and by definition, virus and tumor evolution [38–40]. Consequently, most of the assays discussed in this review will focus on the C-terminal catalytic domain.
It should be noted that while this review focuses on the APOBEC3 subfamily of enzymes, the related enzyme AID is also a drug target with contributions to initiation and progression of lymphomas/leukemias (reviewed in [41, 42]) and shares some degree of structural similarity with APOBEC3s making it another consideration for achieving selectivity in inhibitor discovery [43].
1.2. Relevance of APOBEC3 enzymes to HIV and cancer
APOBEC3 enzymes were recognized originally as antiviral factors [44], a cluster of potential editing genes on chromosome 22 [45], and as ssDNA deaminases [46]. A3D/F/G/H package into budding virions in HIV-infected cells and after fusion to a target cell and reverse transcription of viral RNA, the transient ssDNA serves as a substrate for APOBEC3s to mutate the HIV genome to a lethal level. APOBEC3 enzymes have since been shown to restrict a variety of DNA and RNA viruses including human T-cell leukemia virus-1 (HTLV-1), hepatitis B virus (HBV), human papilloma virus (HPV), and herpesviruses (reviewed in [47–50]). In the case of HIV, the virus encodes for the virion infectivity factor (Vif) protein, which binds APOBEC3 enzymes and recruits an E3 ligase complex to polyubiquitinate and degrade APOBEC3s [51]. Other viruses, such as Epstein-Barr virus (EBV), have adapted their ribonucleotide reductase to inhibit APOBEC3 activity and prevent restriction [52]. The role of APOBEC3 enzymes in beneficially mutating the HIV genome at a sublethal level and aiding in viral evolution is debated among those that study viruses/APOBEC3 (reviewed more extensively in [53]) and studies have been published arguing both for [54–58] and against [59–61] the effect of APOBEC3s on viral evolution. The development of specific APOBEC3 inhibitors would certainly aid the field in understanding the interaction between APOBEC3 activity, HIV evolution, and drug resistance.
APOBEC3s (primarily A3A and A3B) have also been implicated in evolution and genomic heterogeneity of a variety of cancers leading to recurrence, metastasis, and therapeutic resistance (reviewed in [42, 49, 62–69]). In one study, A3B depletion enhanced the durability of tamoxifen therapy in a breast cancer murine xenograft model, whereas A3B overexpression accelerated the evolution of mutations that conferred tamoxifen resistance [40]. Whole genome sequencing and gene expression level analysis have shown that A3B is upregulated, signature APOBEC3 mutation patterns are frequently found, and that mutation clusters (kataegis) occur in many different cancer types [70–72]. Additionally, introduction of A3A to the Apcmin murine model of intestinal cancer development and Fah murine model of hepatocellular carcinoma demonstrates that A3A mutagenesis can drive tumor formation [73].
Most studies associated with manipulating APOBEC3 function for antiviral applications have focused on the development of Vif inhibitors (reviewed in [64]). Inhibition of Vif is known as a “therapy by hypermutation” strategy, because preventing Vif-mediated APOBEC3 degradation leads to increased levels of APOBEC3 enzymes in cells, thereby allowing them to act in an uncontested manner to restrict viral replication [53]. Conversely, inhibition of APOBEC3 enzymes could prove useful in what is known as “therapy by hypomutation” [53, 64]. Elimination of APOBEC3-catalyzed cytosine deamination in virus and/or tumor genomes is anticipated to prevent the accrual of drug resistance mutations that contribute to the failure of current drug therapies. We hypothesize that an APOBEC3 inhibitor used in combination with current antiretroviral or targeted cancer therapies may improve long-term clinical outcomes. Given these overarching motivations to develop APOBEC3-targeting small molecules, a robust suite of assays is required to support the discovery and development of APOBEC3 inhibitors. Accordingly, this review highlights the biochemical, biophysical, and cellular assays that have been used to study APOBEC3 structure and function along with commentary on the adaptation or potential for adaptation for small molecule drug discovery (Box 1). A limited number of these approaches have been employed to discover APOBEC3 inhibitors (Table 1).
Box 1.
High throughput screening (HTS) and fragment-based drug discovery (FBDD) approaches. HTS utilizes libraries of moderately sized molecules (300 to 500 Daltons) typically in a 384- or 1536-well assay format to assess activity against large libraries (10,000s to 1,000,000s) of small molecules. Phenotypic or biochemical activity assays are most commonly used to find hits that have moderate to high potency and usually require less structural optimizations during medicinal chemistry campaigns. FBDD uses smaller molecules (typically <300 Daltons) that are screened in microplates or alternative formats (NMR tubes, capillaries, etc.) against smaller libraries (100s to 10,000s) of molecules. As fragments tend to have weaker binding affinities and, in many cases, no inhibitory activity against a target, sensitive biophysical and/or biochemical assays are often used to detect binding events between fragments and their targets. More extensive structural optimizations during medicinal chemistry campaigns are usually required. However, FBDD offers advantages to HTS, including smaller libraries to screen, higher molecular diversity/chemical matter coverage, and more opportunities to tune the physicochemical properties of the molecule [133].
Figure I.
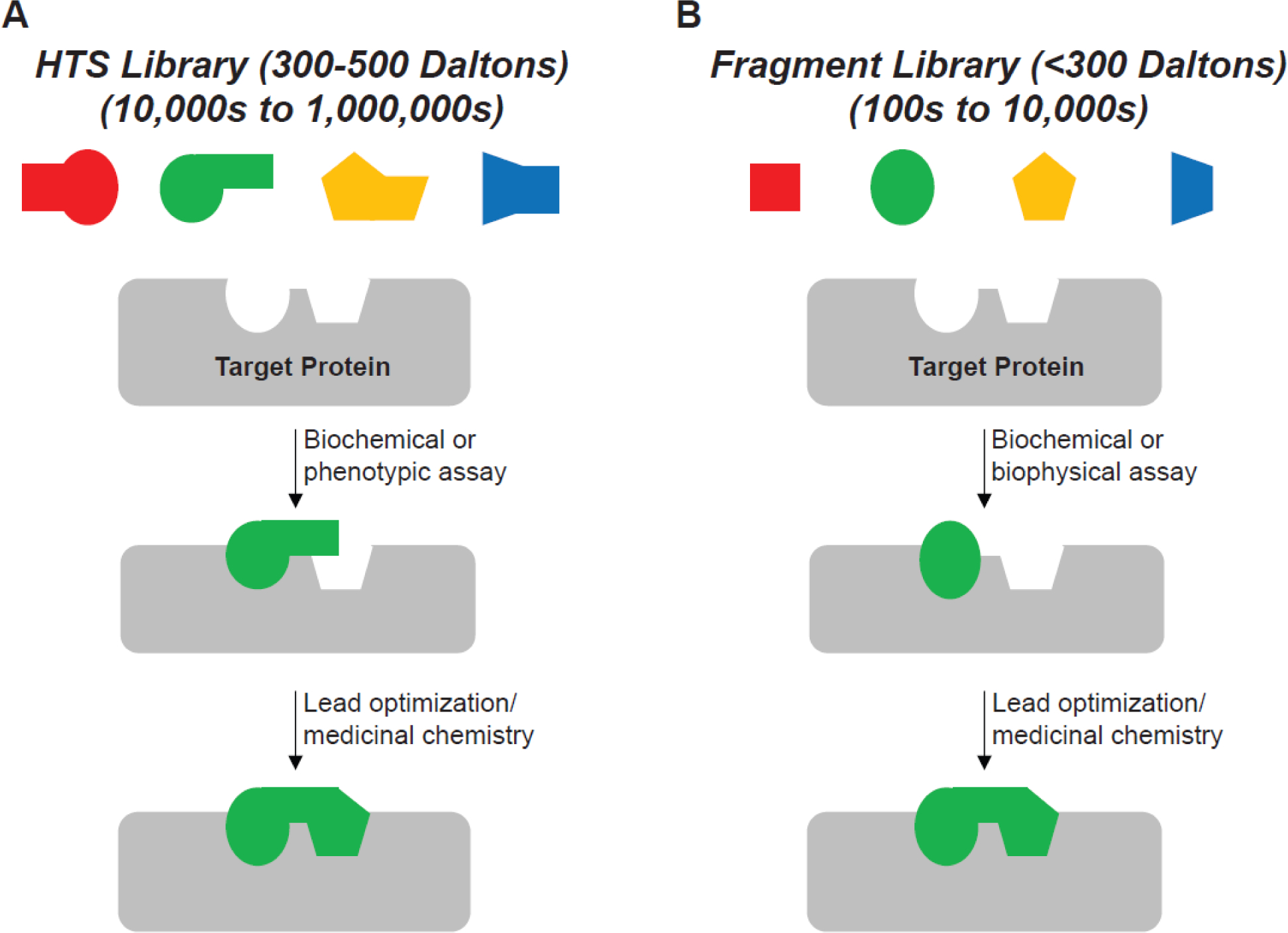
HTS and FBDD approaches. Depiction of typical library design parameters and types of assays used for A) HTS, and B) FBDD.
Table 1.
HTS efforts to discover APOBEC3 inhibitors.
| Screening Center/Campaign | Library/Size | Target APOBEC | Hit Rate | Assay Used | Z-factor | Reference |
|---|---|---|---|---|---|---|
| Institute of Medicinal Biotechnology, Chinese Academy of Medical Science (Cen) | In-house chemical library & natural product collection – 8,634 compounds | A3G | 0.02% | Cell-based, A3G-YFP degradation assay, 96-well | - | [85] |
| UMN Institute for Therapeutics Discovery & Development (Harris) | Sigma LOPAC® library – 1,280 compounds | A3G | 2.8% | FRET-based activity assay, 384-well | 0.85 | [80] |
| Sanford-Burnham Medical Research Institute (Harris) | NIH MLPCN Collection – 331,861 compounds | A3A & A3G | 0.3% (A3G) 0.4% (A3A) | FRET-based activity assay, 1,536-well | 0.85 | [81, 82] |
| Harvard Medical School – ICCB-Longwood Screening Center (Harris, Harki) | Mix of 20 commercial libraries – 168,192 compounds | A3Bctd & A3G | 0.2% (A3Bctd) 0.8% (A3G) | FRET-based activity assay, 384-well | 0.6 – 0.8 | M. E. Olson, Ph. D. Thesis, University of Minnesota, 2015 |
| Simon Fraser University/Memorial University of Newfoundland (Larijani) | ZINC “clean-leads” subset – 4.6 million compounds | AID1 | -2 | Docking, then gel-based alkaline cleavage assay | - | [84] |
Although AID was the intended target, the compounds tested also inhibited various APOBEC3 enzymes.
A hit rate was not determined.
2. In vitro biochemical and biophysical assays
2.1. Deaminase activity assays
In vitro assays for the quantitation of APOBEC3 catalytic activity in cell lysate or with purified protein are among the earliest developed and most commonly used. The first reported APOBEC3 cytosine deaminase activity assay used biotinylated ssDNA with a target cytosine and a fluorescein tag at the 3′-terminus as the substrate for incubation with A3G [27, 28, 74]. After the protein and ssDNA are incubated, the oligonucleotides are bound to magnetic streptavidin beads, washed, then incubated with uracil-DNA glycosylase (UDG) and sodium hydroxide (NaOH). If the protein is active, the target C is deaminated to U, and then UDG followed by sodium hydroxide excises the deaminated nucleobase and hydrolyzes the ssDNA strand at the abasic site, respectively. The processed ssDNA is subsequently analyzed by gel electrophoresis and imaged for fluorescence to assess turnover by the separation of the longer starting and shorter product oligonucleotides. Although this method was useful for initial studies of APOBEC3 activity, the streptavidin purification is now an unnecessary step. Multiple studies since have simplified this workflow by elimination of the biotin on the ssDNA; the APOBEC3 protein, UDG, and fluorophore-labeled ssDNA are incubated together, and the entire sample is loaded onto a polyacrylamide gel for analysis [16, 40, 75–77]. However, given that the assay readout requires gel electrophoresis, these assays lack the throughput necessary for compound screening.
An assay was subsequently developed to assess A3G activity in cell lysate in 96-well plates [78]. The assay design took advantage of Förster resonance energy transfer (FRET) by labeling the ssDNA substrate with fluorescein (FAM) and tetramethylrhodamine (TAMRA) fluorophores on the 5′ and 3′ ends, respectively. Incubation of the ssDNA with A3G-containing lysate, followed by treatment with UDG, then NaOH results in strand cleavage and alleviation of the spatial restriction of the FRET pair. This leads to an increase of FAM emission signal as a function of enzyme activity (Figure 2A). Other fluorophore/quencher pairs, including cyanine 5/Black Hole Quencher 2 (Cy5/BHQ2), have been implemented in similar assays against AID [79].
Figure 2.
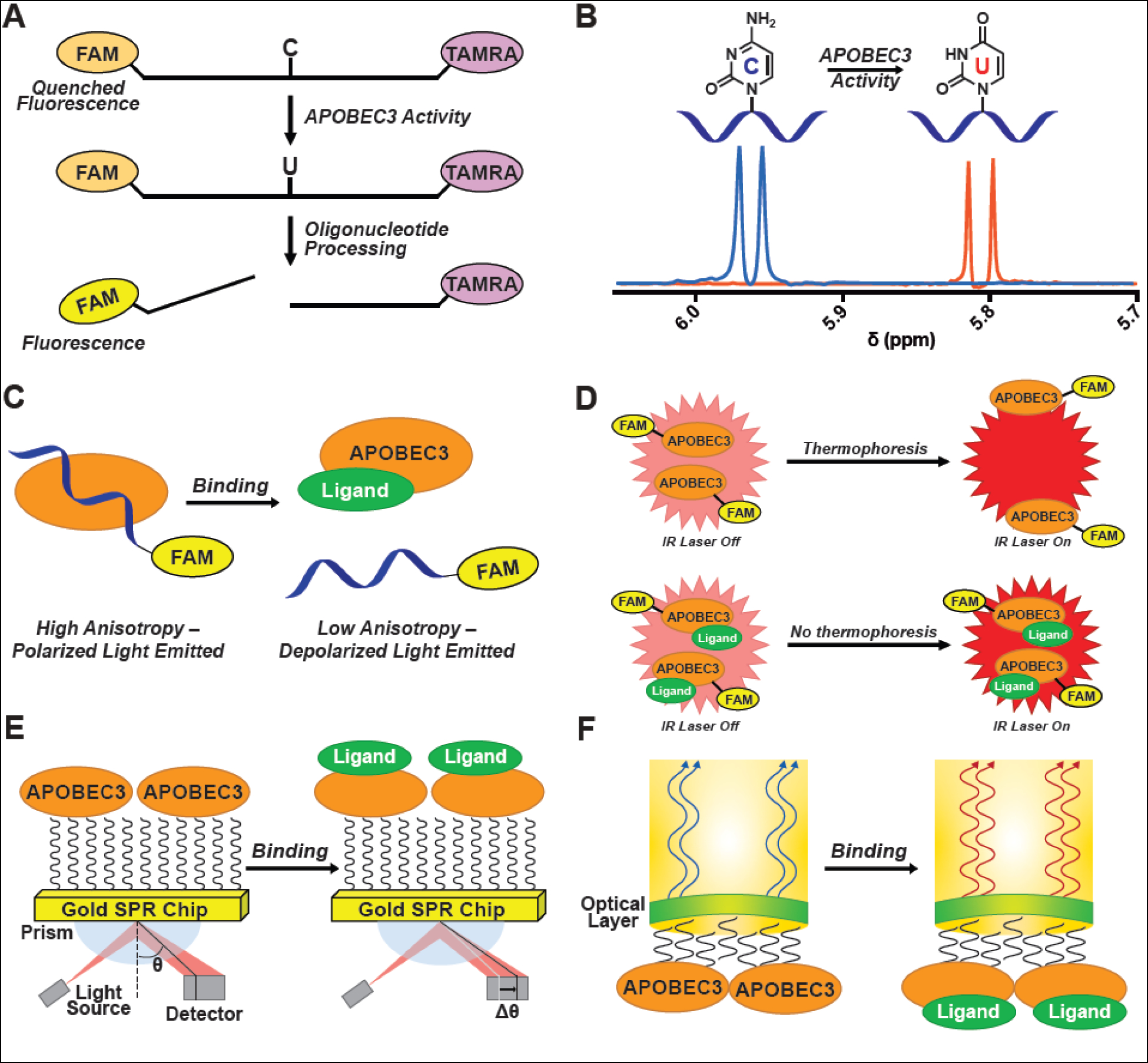
Assays used for APOBEC3 drug discovery. A) Microplate-based APOBEC3 deamination activity assay. A ssDNA containing a target cytosine is labeled on the 5′ and 3′ end, respectively, with FAM and TAMRA (FAM emission quenched by FRET with TAMRA). When incubated with active APOBEC3 enzyme, the target cytosine is converted to a uracil, and treatment with UDG, then alkaline conditions yields a cleaved oligonucleotide with an increase in measurable FAM fluorescence. B) NMR-based APOBEC3 activity assay. The C5-proton signal is monitored by 1H-NMR. Upon deamination of the substrate, the signal is shifted, and deamination is quantified. C) Fluorescence anisotropy/polarization assay (FA/FP). Fluorescence is emitted by FAM on a labeled ssDNA that is subjected to polarized light. When bound to APOBEC3, the ssDNA tumbles slowly leaving the polarized light intact (high signal). When ssDNA is displaced, the faster tumbling depolarizes the light (low signal). D) Microscale thermophoresis (MST). FAM-labeled APOBEC3 moves through a capillary in an IR-beam-generated temperature gradient (thermophoresis). Binding of a ligand changes the APOBEC3 thermophoretic property, thereby preventing APOBEC3 from moving away from the irradiated region in the capillary, thus resulting in an increase in fluorescent signal when compared to the unbound state. E) Surface plasmon resonance (SPR). APOBEC3 is immobilized to a gold chip and ligand binding to the protein results in a quantifiable change in the angle of the reflected light source (θ). F) Biolayer interferometry (BLI). A dip-and-read sensor is functionalized with APOBEC3 and the interference pattern of incident white light through the sensor is measured following binding of a ligand to the APOBEC3 enzyme.
The deaminase assay described above with recombinantly expressed and purified C-terminal myc- and hexahistidine-tagged A3A and A3G from HEK293T cells was subsequently adapted for use in 384-well microplates [80]. An impressive Z-factor of 0.85 was achieved, suggesting that the assay is robust and reproducible. Screening of a 1,280 compound library of pharmacologically active compounds (LOPAC®, Sigma) yielded several hits (Table 1) [80]. One year later, the same group reported a follow-up screen against A3A and A3G with the same assay in 1536-well format using 331,861 compounds from the NIH Molecular Libraries Probe Production Centers Network (MLPCN) collection and performed at the Sanford-Burnham Medical Research Institute (Table 1) [81, 82]. A 4-amino-1,2,4-triazole-3-thiol scaffold was identified as an A3G inhibitor with a half-maximal inhibitory concentration (IC50) value in the low micromolar range [83]. However, to date, hits from these screens have not been pursued due to issues of cellular cross-reactivity [64]. More recently, virtual screening was employed to discover inhibitors of AID using the ZINC “clean leads” subset of 4.6 million compounds [84]. Follow-up computational experiments prioritized 10 compounds that were evaluated in a deaminase activity assay against AID, APOBEC3A, APOBEC3B, and APOBEC3G. This work yielded pan-inhibitors with moderate selectivity and potency for some APOBEC3 enzymes over AID.
Although the plate-based deaminase assay has proven useful for HTS (Table 1) it has several drawbacks. Firstly, it is an endpoint activity assay that does not allow for enzyme/inhibitor kinetic analysis. Secondly, though it has a good Z-factor (> 0.5) [86], several liquid additions are required, which increases the potential for error. Finally, it requires the addition of another enzyme (UDG), necessitating a counter-screen to verify that a given hit is not inhibiting UDG. These issues may be addressed by the development of a deaminase assay that is completed in few liquid additions, does not require a second enzyme, and with real-time monitoring of a signal.
In 2009, the first use of a real-time nuclear magnetic resonance (NMR)-based assay was reported to monitor A3G enzymatic activity. A 10-mer ssDNA was incubated with the C-terminal catalytic domain of A3G, expressed in E. coli, monitoring the 1-dimensional (1D) 1H-NMR and 2-dimensional (2D) 1H-13C Heteronuclear Single Quantum Coherence (HSQC) NMR [87]. They observed changes in the resonances of H5 and the H5-C5 correlation in the 1D and 2D spectra, respectively (Figure 2B). This method has since been used to obtain enzyme kinetic parameters with ssDNA inhibitors targeting A3A/A3B [88–90]. These oligos contain the pyrimidine base analogue 2′-deoxyzebularine in place of the target cytosine, which acts as a transition-state inhibitor with low micromolar potency. While this NMR assay addresses some of the issues of the aforementioned endpoint activity assay, NMR assays in general lack throughput when compared to microplate-based assays. In order to better characterize and screen compounds, the field would best be served with an assay that combines the high-throughput capabilities of the endpoint FRET-based assay with the inhibitor kinetics information provided by the NMR activity assay. A live-read fluorescence assay that utilizes a substrate that changes fluorescence properties upon cytosine deamination could provide kinetic information without sacrificing throughput.
2.2. Fluorescence anisotropy/polarization binding assays
Fluorescence anisotropy/polarization (FA/FP) is another powerful assay for studying APOBEC3-ligand interactions. The assay measures parallel and perpendicular polarized light emitted from a fluorophore attached to a ligand (termed tracer) and anisotropy or polarization is calculated as a function of the tumbling rate of the fluorophore in solution. Differential FA/FP is observed between the bound and unbound state of the ligand-protein complex and may be used to determine binding dissociation constants (Kd). For APOBEC3s, a standard setup for this assay involves using a fluorophore-conjugated ssDNA substrate as the tracer; however, other ligands may also be used. The size difference between the fluorescent ssDNA and ssDNA-APOBEC3 complex results in a differential tumbling rate, and allows a curve to be generated upon titration of protein with a fixed fluorophore concentration (Figure 2C). The assay typically employs a catalytically inactive APOBEC3 enzyme to prevent differential binding events that result from catalytic turnover of the ssDNA substrate.
The FP assay has been developed and employed for studying RNA and/or ssDNA interactions with A3A [91, 92] A3C [93], A3F [94], A3G [77, 94, 95], and A3H [96]. It was also adapted for evaluating DNA-based inhibitors using catalytically inactive, E. coli-expressed A3A in 384-well plates with a Z-factor of 0.57 [90]. The apparent Kd values are determined for competitive ligands by running the assay in competition mode using a fluorescently labeled tracer of known Kd. It should also be noted that this assay has been deployed for studying direct A3B-ssDNA interactions [33, 34, 97, 98], though not yet for small molecule inhibitor screening. The FP assay benefits from a simple setup and run time compared to the FRET activity assays described above. Because the ssDNA tracer is competed from the active site, this assay also has the potential to provide some information about the location of ligand binding (Table 2). It should be noted that using ssDNA as a tracer may not fully elucidate the binding location of a non-competitive inhibitor, such as allosteric inhibitors, or molecules targeting an exosite.
Table 2.
Summary of practical parameters to consider for using biochemical/biophysical assays.
| Assay | Protein Consumed | Throughpust | Effective Kd Upper Limit | Information obtained |
|---|---|---|---|---|
| Enzyme activity (fluorescence) | Low | High | High μM | IC50 |
| Enzyme activity (NMR) | High | Low | High μM | K i 1 |
| Fluorescence anisotropy/polarization | Medium | High | Dependent on tracer Kd | IC50/Kd, competition2 |
| Enzyme activity (DRONE) | Low | Low | – | IC50, Ki |
| Protein-observed NMR | High | Medium | Mid mM | Kd, binding location |
| Isothermal titration calorimetry | High | Low | Low mM | Kd, ΔG, ΔH, ΔS, binding stoichiometry3 |
| Microscale thermophoresis | Low | Medium | Low mM | Kd, ΔG, ΔH, ΔS, binding stoichiometry, competition |
| Surface plasmon resonance | Low | Medium | Low mM | Kd, kon, koff, competition |
| Biolayer Interferometry | Medium | Medium | Low mM | Kd, kon, koff, competition |
Equilibrium inhibitory constant.
The assay can be used to determine if a candidate ligand can competitively displace an established fluorophore-labeled tracer ligand (e.g. fluorescent ssDNA).
The number of molecules of ligand binding to one molecule of APOBEC3 may be determined.
2.3. Liquid chromatography and mass spectrometry assays
The use of liquid chromatography-mass spectrometry (LC-MS) for HTS has traditionally not been used since this method is less high-throughput than others. However, LC-MS/MS is often used in drug development for characterization of covalent inhibitors discovered through HTS [99, 100]. A significant advancement in this area was the development of DRONE (Direct Resolution of ONE Dalton difference), which is a UHPLC-based APOBEC3 activity assay that monitors ssDNA C-to-U deamination with HPLC separation [101]. Since APOBEC3-mediated deamination results in only a single Dalton difference between substrate and product, sensitive analytical techniques are required to distinguish the two. In this assay, the authors leveraged a variety of LC parameters, including a hexafluoroisopropanol-triethylamine buffer system in the mobile phase to increase chromatographic resolution [101]. Limitations of this assay include requiring sample processing prior to analysis, which decreases adaptability for HTS, and the assay does not report real-time deamination (Table 2). Importantly though, this assay could be used to characterize APOBEC3 deamination kinetics in the presence of inhibitors at fixed time points, which is difficult with the multi-step, fluorescence-based activity assay discussed above.
2.4. Nuclear magnetic resonance binding assays
NMR binding assays have proven to be robust and reliable in drug discovery for lead generation and structure-activity relationship (SAR) studies [102–104]. Two-dimensional (2D)-NMR in particular has been found to be an asset in lead generation and optimization especially in the absence of a crystal structure [105]. While generally requiring a large quantity of NMR-active isotope enriched protein, 2D methods have the advantage of being able to elucidate the location of ligand binding (Table 2). Recently, the use of 15N-1H HSQC with A3A was reported, and revealed details of A3A-ssDNA binding termed “method of small changes” [106]. Incubation of A3A with several slightly different ssDNA substrates allowed researchers to map key A3A-ssDNA interactions and provide parameters for computational models. In addition, combinations of similar 2D-NMR methods as described above have been used to solve solution structures of A3A [107], A3B [108], and A3G [87, 109–112]. The aforementioned NMR assays have potential for aiding in APOBEC3 lead discovery and optimization given their high orthogonality to other assays reviewed here.
2.5. Thermodynamic binding assays
Thermal and calorimetry binding assays have been used for lead optimization and characterization of ligands by providing information such as the binding dissociation constant (Kd), enthalpy (ΔH), entropy (ΔS), and stoichiometry of binding. Isothermal titration calorimetry (ITC) has been employed for evaluating APOBEC3-ssDNA substrate [106] and APOBEC3-oligonucleotide inhibitor interactions [88–90]. Though it provides a large amount of information from a single series of experiments, ITC is known to be material-intensive and low throughput (Table 2).
Microscale thermophoresis (MST) provides the same information as ITC while addressing some drawbacks [113]. Taking advantage of the phenomena of thermophoresis and changes in fluorescence due to changes in temperature, MST is a method that was developed for use in understanding molecular conformation, studying biomolecular interactions, and fragment screening [114, 115]. MST typically involves mixing fluorophore-labeled protein with a ligand in a capillary and measuring fluorescence at a particular section of the capillary upon infrared irradiation. As ligand concentration increases, it causes a change in the chemical environment of the target protein and perturbs its ability to thermophoretically move through the capillary, which may be quantified through fluorescence (Figure 2D). Additionally, a method to study protein-ligand interactions was developed that relies upon the intrinsic fluorescence of protein tryptophan residues, and therefore, does not require conjugation of the protein of interest with a fluorophore [116]. A3G-ssDNA interactions were recently characterized using MST in order to evaluate and prioritize ssDNA sequences for obtaining co-structures with A3G [19]. These findings provide a strong precedent for using this method to characterize APOBEC3-ligand interactions. MST offers a number of advantages over similar techniques in that it uses very little sample and it may be performed in biological liquids such as serum or cell lysate (Table 2) [113].
2.6. Sensor-based binding assays
Biosensor-based assays such as surface plasmon resonance (SPR) and biolayer interferometry (BLI) are popular for FBDD. In SPR, a gold chip is chemically functionalized, and typically, a protein of interest is immobilized on the surface of the chip. When ligands flow over the immobilized protein and binding occurs, the change in mass results in a measured deflection of the angle of incident light (Figure 2E). This technique yields kinetic binding parameters such as kon, koff, and Kd making it useful and complementary to techniques that provide thermodynamic binding parameters (Table 2). SPR was used to follow up on a cell-based HTS campaign to discover inhibitors of the A3G-Vif interaction (Table 1) [85]. A3G or Vif was immobilized to the sensor chip and compounds were flowed over the chip. The authors found that their lead compound bound exclusively to A3G and not Vif, preventing A3G-Vif binding and leading to stabilization of A3G in cells [85].
BLI is a robust and sensitive binding assay with similar physical principles to SPR. For HTS applications, the protein of interest is immobilized onto the surface of a biosensor tip and the biosensor is dipped into a solution containing an analyte of interest. If the analyte binds to the immobilized protein, the optical thickness of the biolayer increases, which leads to a shift in the interference pattern of incident white light (Figure 2F) [117]. This technique has been used to study ssDNA-A3F and Vif-A3F interactions by immobilizing either ssDNA or Vif to a streptavidin BLI probe and immersing in buffer with varying concentrations of A3F [118]. It was also used to evaluate compounds that were designed using in silico methods to disrupt the A3G-Vif binding interaction [119]. In this application, A3G was biotinylated using NHS-ester chemistry and then immobilized to streptavidin-coated BLI sensors; compounds were then titrated to determine binding affinities. Additionally, BLI has been used to quantify binding of the BORF2-APOBEC3 interaction and aid in rationalizing the selectivity for APOBEC3B inhibition by BORF2 over other APBOEC3 enzymes [120]. These biosensor assays offer the distinct advantage of requiring little protein compared to most biophysical assays, making them powerful for screening and complementary to other (thermodynamic) binding assays (Table 2).
3. Cell-based assays for drug discovery
3.1. Virus restriction and antibiotic resistance assays
Early assays for APOBEC3 deaminase activity utilized bacterial growth on selective media and targeted genome sequencing to assess activity. E. coli transformed with various APOBEC3 expression constructs were assessed for their ability to acquire mutations that confer antibiotic resistance [46, 121]. The E. coli rpoB gene, which encodes the β-subunit of RNA polymerase, develops resistance to rifampicin upon mutation by APOBEC3. After plating a known number of cells on medium containing rifampicin, the number of surviving colonies after overnight growth are counted to determine mutation frequencies. Targeted sequencing of the rpoB gene demonstrated strong C/G-to-T/A mutation biases and preferential deamination site (hot spots) in APOBEC3-expressing cells versus controls [46]. This same general strategy has been extended to measure APOBEC3 activity against other selectable markers in E. coli and yeast [28, 122, 123]. For instance, the utilization of sacB (sucrose-resistance) instead of the rpoB helped to distinguish the local preferences of several different DNA deaminases including AID, APOBEC1, and A3G [28].
APOBEC3 activity can also be assessed as a function of retrovirus infectivity because several APOBEC3 enzymes, including A3B and A3G, can potently restrict Vif-deficient HIV-1 [124]. Co-transfection of 293T cells with a fluorescent reporter (e.g., GFP) and appropriate viral packaging plasmids (e.g., Gag-Pol, Env) results in the production of a virus that readily infects target cells and renders them fluorescent [27]. If the cells are simultaneously co-transfected with an A3B or A3G expression plasmid, these proteins also package into the virus particles and restrict the infection of target cells. The resulting decrease in fluorescence from viral restriction by APOBEC3s can be assessed by fluorescent microscopy, flow cytometry, or fluorescent plate readers. This assay can also be adapted for screening; however, a small molecule inhibitor must not only enter the cell, but also gain access to the viral capsid during assembly and maturation, which eventually becomes the site of viral reverse transcription and viral cDNA C-to-U deamination.
3.2. Real-time deamination assays for APOBEC3 activity in cells
The hunt for a real-time APOBEC3 activity assay, as discussed above, took a significant step forward with the development of cytosine base editors (CBEs) in 2016 [125]. First generation CBEs utilized rat APOBEC1 fused to Cas9 nickase (Cas9n) and uracil DNA glycosylase inhibitor (UGI) to edit efficiently while preventing undesirable outcomes such as DNA breakage and insertion/deletion formation [125]. Since then, further generations of CBEs have been developed and optimized for increased editing efficiency and reduction of off-target editing events (reviewed in [126]).
Precision CBE development for genome editing has been facilitated by the development of robust reporter systems, including the first fluorescence-based, real-time base editing reporters for APOBEC3 activity quantification in the nuclei of living cells [127]. These first-generation reporters were quickly replaced by more effective constructs, including a system termed APOBEC-mediated base editing reporter (AMBER; Figure 3A) [128]. This reporter is comprised of a constitutively expressed mCherry protein for transfection rate normalization, and an eGFP cassette that contains an L202S codon mutation that ablates eGFP fluorescence. APOBEC3 editing of the TCA codon to TTA converts the S202 back to the wildtype L202 residue and restores fluorescence. The ratio of eGFP/mCherry fluorescence is directly proportional to APOBEC3 editing activity and can be readily quantified in real-time by fluorescent microscopy or fluorescence plate readers.
Figure 3.
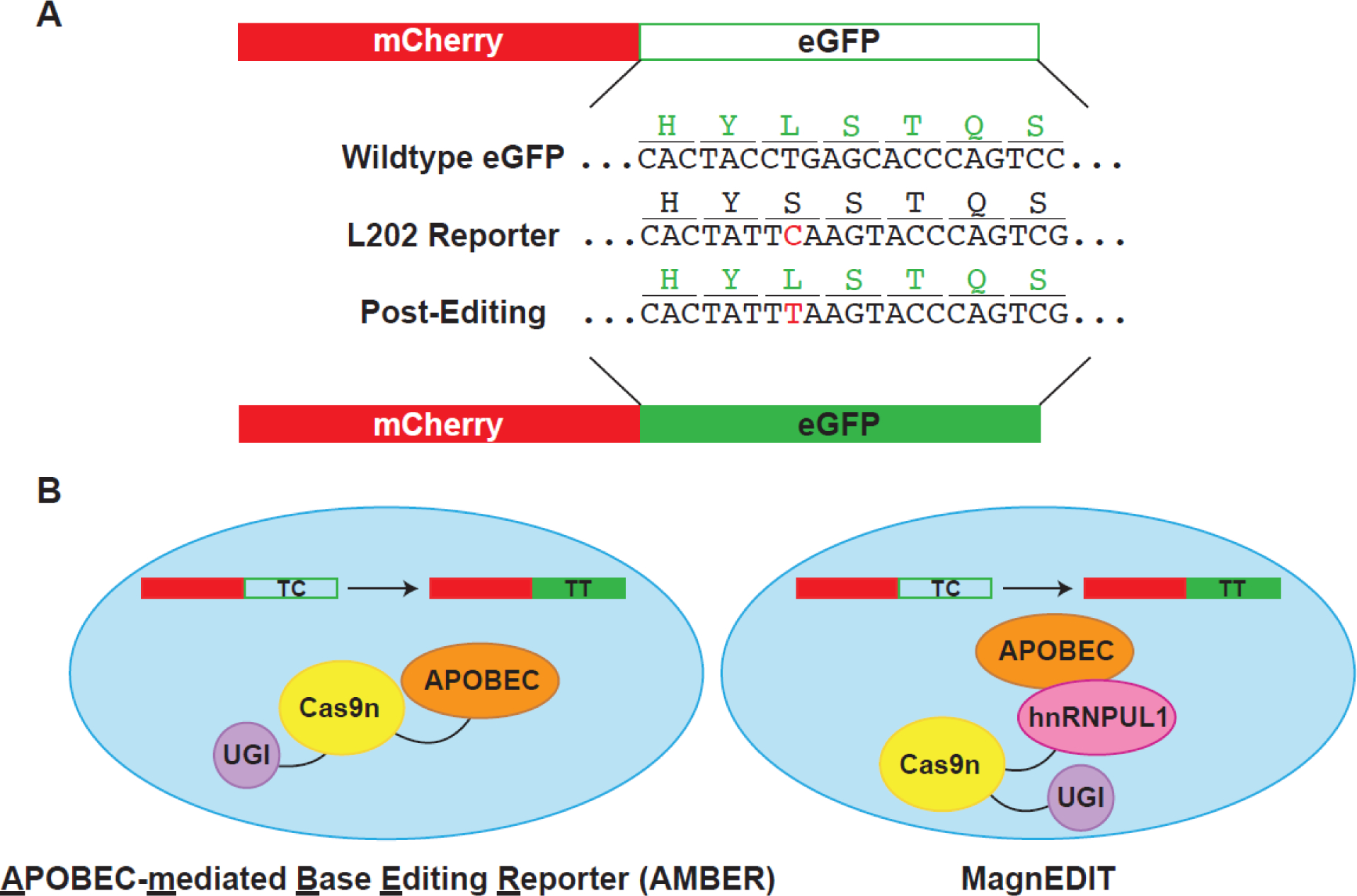
Overview of cellular assays for in vivo activity assessment. A) Plasmid editing by APOBEC-mediated Base Editing Reporter (AMBER). Reporter plasmid contains constitutively-expressed active mCherry for transfection/transduction normalization. eGFP reporter protein is inactivated by mutation of the L202 codon to encode for a serine residue, which ablates fluorescence. Silent mutations were also introduced near the editing site to reduce the opportunity for double-stranded breaks. APOBEC3 editing restores eGFP fluorescence. B) Comparison of AMBER editing system and MagnEDIT. AMBER fuses all three proteins – APOBEC3, Cas9n, and UGI – to direct editing to the eGFP reporter construct in panel A. MagnEDIT fuses Cas9n/UGI with an APOBEC-interacting protein, hnRNPUL1. gRNA directs hnRNPUL1/Cas9n/UGI to the eGFP reporter construct from panel A. hnRNPUL1 therefore acts as a “magnet” to attract the APOBEC3 protein to the reporter and restore eGFP fluorescence. Elimination of the tethering of the APOBEC3 protein to the Cas9n/UGI construct results in less off-target editing.
There are a few advantages of this system for small molecule screening: firstly, mCherry is a built-in fluorescent protein control for molecules that may be non-specific quenchers or cytotoxic; secondly, molecules screened in this assay will have to penetrate both the cell and nuclear membranes to inhibit APOBEC activity in the nucleus, helping to ensure membrane permeability of hits. However, there are also drawbacks: firstly, given that small molecules must permeate all the way to the nucleus, cell- or nucleus-impermeable compounds that might inhibit in an in vitro assay and could be developed into a cell-permeable inhibitor will be disregarded; secondly, due to the fact that APOBEC3 is covalently tethered to the Cas9n/UGI complex, an inhibitor must have a fairly strong binding affinity (low Kd) otherwise the APOBEC3 protein will still have an opportunity to edit the cassette upon molecule dissociation, and no decrease in editing will be observed; thirdly, this system is a coupled assay that requires the localization of Cas9n and activity of UGI for the fluorescent readout to occur, and if APOBEC editing is not the rate-limiting step or the screened compound inhibits another part of the system, false positives or false negatives may occur. Another version of the CBE reporter mitigates some concern over off-target editing events by creating a non-covalent editing construct [76]. In this system, the Cas9n-UGI is fused with an A3B interacting protein, hnRNPUL1, which acts as a “magnet” for A3B and attracts it transiently to the genomic site for a single base edit, which restores eGFP fluorescence (Figure 3B).
4. Concluding Remarks and Future Perspectives
Assay throughput and material consumption are critical parameters to be considered in every drug discovery campaign (Table 2). These parameters have led to challenges in the field of APOBEC3 inhibitor discovery, with issues around protein production and general protein stability making biochemical and biophysical assay development challenging. Protein expression in bacteria yields a much higher quantity as compared to mammalian cell expression, but expression of active APOBEC3 in bacteria can result in auto-mutagenesis as the APOBEC3 enzyme deaminates its own expression plasmid in the host cell [129]. The development of additional expression systems to facilitate production of active protein in E. coli would be a significant advancement for the field.
The stability and solubility of APOBEC3 proteins has significantly impacted structural studies, with the first elucidated co-crystal structures of APOBEC3s and oligonucleotide substrates only being reported recently [16, 17, 19, 130]. For A3B specifically, the protein construct used for co-crystallographic studies required significant alterations in order to improve solubility, increase binding affinity to the ssDNA substrate, and improve structural resolution [16, 75]. Given these concerns, lead compound optimization and structure-guided design of inhibitors would be accelerated by advances in achieving APOBEC3 structural resolution without extensive protein engineering and mutations. This could potentially be addressed by advances in the field of structural biology, such as the use of cryogenic electron microscopy (cryo-EM) and/or small-angle x-ray scattering (SAXS) (see Outstanding Questions). Cryo-EM structures of APOBEC3 enzymes have been challenging due to the relatively small size of APOBEC3 proteins (<50 kDa), but structures are beginning to emerge of APOBEC3 enzymes in complex with other proteins such as an A3F-Vif-CBF-β complex [131] and a wildtype A3B catalytic domain-viral ribonucleotide reductase complex [120]. A SAXS model of A3B was published recently showing the C-terminal catalytic domain of A3B in complex with a ssDNA-based inhibitor making this study the first reported solution-phase structure of an APOBEC3 enzyme with an inhibitor [132].
Outstanding Questions.
Can small molecules be developed that selectively target different APOBEC3 enzymes?
What mechanisms of APOBEC3 inhibition are possible (e.g., competitive, allosteric)?
Will cell-based APOBEC3 activity assays be predictive for the development of small molecule APOBEC3 inhibitors that ultimately prevent phenotypes caused by APOBEC3-catalyzed mutation, including tumorigenesis, metastasis, and therapeutic resistance?
Will advances in the structural biology of APOBEC3 enzymes be required for the development of small molecule inhibitors?
The development of selective probes would be useful for discerning the cellular roles of each APOBEC3 subfamily member in viral or cancer-related contexts. However, high sequence homology between APOBEC enzymes makes the development of a selective inhibitor difficult (e.g., A3A and A3B share 92% sequence identity) (see Outstanding Questions). While selectivity is a challenge in APOBEC3 drug discovery, a non-selective, pan-APOBEC3 inhibitor would still be valuable as a chemical probe; for example, a pan-APOBEC3 would be useful for understanding the role that APOBEC3 enzymatic activity plays in cancer mutagenesis.
Another challenge with the development of APOBEC3 inhibitors for anticancer therapy is that APOBEC3 enzymes are not traditional anticancer drug targets because cellular inhibition of APOBEC3 enzymatic activity has no obvious or immediate phenotype. This makes the in vivo analysis of both the biological role of APOBEC3 enzymes and the effect of APOBEC3 inhibitors difficult to assess. The utilization of APOBEC3 enzymes in CBE systems has led to the possibility of evaluating APOBEC3 inhibitors in cells. However, it remains to be seen whether APOBEC3 inhibitors that demonstrate inhibition in these assays would also show the same efficacy against endogenous APOBEC3 enzymes (see Outstanding Questions). These issues are less pronounced in assessing inhibition of APOBEC3s in viruses due to the fact that viral genomes acquire APOBEC3-catalyzed mutations at higher rates, making real-time in vivo assessments of co-treatment more feasible.
Since the recognition of APOBEC3 enzymes as antiviral and anticancer targets [64] and with the recent development of a variety of assays used to study APOBEC3 dynamics and molecular interactions, the field is poised for the discovery of novel inhibitors. Discovery campaigns can be initiated using a variety of ligand-identification approaches (HTS, virtual screening, FBDD, rational design, etc.). However, once initial hit compounds are identified, we advocate strongly for a chemistry-forward hit triage program to confirm the identified compounds are bona fide ligands for the intended APOBEC3 target enzyme. Assessment of compounds in orthogonal biochemical/biophysical assays, evaluation of compounds for APOBEC3 target engagement and inhibition in a cellular context, and ligand-APOBEC3 structural studies are all excellent methods to triage early chemical matter. An ideal discovery program also performs rigorous triaging and quality control of hit compounds, such as careful vetting of hits for assay interference moieties, aggregation, and redox potential, as well as verifying the hit compounds being evaluated are pure (we recommend > 95% by high-performance liquid chromatography, HPLC, analysis), the correct structure (determined by NMR or mass spectrometry analysis), and free of impurities that can result in false positives in assays. To address this latter concern, we routinely repurify commercially available hit compounds (by preparative HPLC) and rescreen to verify activity. In conclusion, “antimutation therapy” has the potential to become a revolutionary new approach for managing both retroviral infections and cancer and for helping to maximize clinical success.
Highlights.
APOBEC3 catalyzed cytosine-to-uracil deamination contributes to mutations in virus and cancer genomes that facilitate disease progression and the evolution of drug resistance.
Chemical inhibition of APOBEC3-catalyzed mutation is a novel therapeutic approach by preventing pro-evolutionary mutagenesis in virus and cancer genomes.
Many biophysical, biochemical, and cellular assays have been developed to study APOBEC3 structure and function.
Funding
This work was supported by the NIH (P01-CA234228 to DAH and RSH and R37-AI064046 to RSH). DAH acknowledges funding from the Masonic Cancer Center at the University of Minnesota with resources from Minnesota Masonic Charities. Salary support for KFMJ was provided by the National Science Foundation Graduate Research Fellowship Program. RSH is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished University McKnight Professor, and an Investigator of the Howard Hughes Medical Institute.
Glossary.
- Abasic site
A position in a nucleic acid that lacks a nucleobase on the sugar.
- Allosteric inhibitor
A molecule that inhibits protein function by binding outside of the active site.
- Deamination
The removal of an amino (-NH2) group from a molecule.
- Exosite
A remote binding site of a natural substrate of a protein that is outside of the active site. An exosite is distinguished from an allosteric site.
- Förster resonance energy transfer (FRET)
Energy transfer phenomenon between two light-responsive molecules.
- Heteronuclear Single Quantum Coherence (HSQC)
A 2-D NMR technique used to determine bond correlations between two different atoms that are bonded to each other.
- Kataegis
Clusters of strand-coordinated APOBEC3 signature mutations often found in cancer genomes.
- Mutagenesis
The act of causing a heritable change in the genetic information of an organism.
- Molecular Libraries Probe Production Centers Network (MLPCN) collection
A library of chemically diverse small molecules maintained in the Molecular Libraries Small Molecule Repository at the National Institutes of Health.
- Nuclear magnetic resonance (NMR)
A spectroscopic technique utilizing the magnetic properties of a molecule by perturbing it with a magnetic field.
- Nucleophile
A molecule that contributes electrons in a chemical reaction that results in formation of a covalent bond.
- Polyubiquitinate
The addition of multiple ubiquitin molecules to a protein.
- Ribonucleotide reductase
An enzyme that catalyzes the reduction of ribonucleotides to deoxyribonucleotides.
- Structure-activity relationship (SAR)
The relationship between small molecule structure and biological activity.
- Thermophoresis
The differential motion of molecules through a temperature gradient.
- Uracil-DNA glycosylase (UDG)
An enzyme that hydrolytically excises uracils from DNA.
- Vif inhibitor
A small molecule that affects the biological activity of the lentiviral protein called virion infectivity factor (Vif).
- Virus restriction
The ability of the host cell to prevent propagation of a virus.
- Z-factor
A statistical measure of assay robustness (0–1.0; >0.5 is often workable and 1.0 is perfect).
Footnotes
Declaration of interests
Reuben S. Harris and Daniel A. Harki were co-founders of ApoGen Biotechnologies, Inc., which closed operations in April 2021.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Betts L, et al. (1994) Cytidine deaminase. The 2.3 Å crystal structure of an enzyme: transition-state analog complex. J. Mol. Biol. 235, 635–656 [DOI] [PubMed] [Google Scholar]
- 2.Johansson E, et al. (2002) Crystal structure of the tetrameric cytidine deaminase from Bacillus subtilis at 2.0 Å resolution. Biochemistry 41, 2563–2570 [DOI] [PubMed] [Google Scholar]
- 3.Yao L, et al. (2005) A molecular dynamics exploration of the catalytic mechanism of yeast cytosine deaminase. J. Phys. Chem. B 109, 7500–7510 [DOI] [PubMed] [Google Scholar]
- 4.Hall RS, et al. (2011) Three-dimensional structure and catalytic mechanism of cytosine deaminase. Biochemistry 50, 5077–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manta B, et al. (2014) Reaction Mechanism of Zinc-Dependent Cytosine Deaminase from Escherichia coli: A Quantum-Chemical Study. J. Phys. Chem. B 118, 5644–5652 [DOI] [PubMed] [Google Scholar]
- 6.Ireton GC, et al. (2003) The 1.14 Å crystal structure of yeast cytosine deaminase: evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure 11, 961–972 [DOI] [PubMed] [Google Scholar]
- 7.Ko TP, et al. (2003) Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J. Biol. Chem. 278, 19111–19117 [DOI] [PubMed] [Google Scholar]
- 8.Xie K, et al. (2004) The structure of a yeast RNA-editing deaminase provides insight into the fold and function of activation-induced deaminase and APOBEC-1. Proc. Natl. Acad. Sci. U. S. A. 101, 8114–8119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sklenak S, et al. (2004) Catalytic mechanism of yeast cytosine deaminase: an ONIOM computational study. J. Am. Chem. Soc. 126, 14879–14889 [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, et al. (2017) Product release mechanism and the complete enzyme catalysis cycle in yeast cytosine deaminase (yCD): A computational study. BBA - Proteins Proteom. 1865, 1020–1029 [DOI] [PubMed] [Google Scholar]
- 11.Costanzi S, et al. (2006) Human cytidine deaminase: a three-dimensional homology model of a tetrameric metallo-enzyme inferred from the crystal structure of a distantly related dimeric homologue. J. Mol. Graph. Model. 25, 10–16 [DOI] [PubMed] [Google Scholar]
- 12.Conticello SG (2008) The AID/APOBEC family of nucleic acid mutators. Genome Biol. 9, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaRue RS, et al. (2009) Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 83, 494–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haché G, et al. (2005) The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 280, 10920–10924 [DOI] [PubMed] [Google Scholar]
- 15.Harjes S, et al. (2013) Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J. Virol. 87, 7008–7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi K, et al. (2017) Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouno T, et al. (2017) Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 8, 15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou S, et al. (2021) Structural basis of substrate specificity in human cytidine deaminase family APOBEC3s. J. Biol. Chem. 297, 100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiti A, et al. (2018) Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 9, 2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, et al. (2006) APOBEC3A Is a Potent Inhibitor of Adeno-Associated Virus and Retrotransposons. Curr. Biol. 16, 480–485 [DOI] [PubMed] [Google Scholar]
- 21.Bishop KN, et al. (2004) Cytidine Deamination of Retroviral DNA by Diverse APOBEC Proteins. Curr. Biol. 14, 1392–1396 [DOI] [PubMed] [Google Scholar]
- 22.Liddament MT, et al. (2004) APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14, 1385–1391 [DOI] [PubMed] [Google Scholar]
- 23.Yu Q, et al. (2004) APOBEC3B and APOBEC3C Are Potent Inhibitors of Simian Immunodeficiency Virus Replication*. J. Biol. Chem. 279, 53379–53386 [DOI] [PubMed] [Google Scholar]
- 24.Langlois M-A, et al. (2005) Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 33, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harari A, et al. (2009) Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 83, 295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang Y, et al. (2006) Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80, 10522–10533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris RS, et al. (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809 [DOI] [PubMed] [Google Scholar]
- 28.Beale RC, et al. (2004) Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J. Mol. Biol. 337, 585–596 [DOI] [PubMed] [Google Scholar]
- 29.Carpenter MA, et al. (2010) Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair 9, 579–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenglein MD, et al. (2008) Two Regions within the Amino-Terminal Half of APOBEC3G Cooperate To Determine Cytoplasmic Localization. J. Virol. 82, 9591–9599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pak V, et al. (2011) The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J. Virol. 85, 8538–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salamango DJ, et al. (2018) APOBEC3B Nuclear Localization Requires Two Distinct N-Terminal Domain Surfaces. J. Mol. Biol. 430, 2695–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adolph MB, et al. (2017) Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 45, 11925–11940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Y, et al. (2015) DNA cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem. J. 471, 25–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newman ENC, et al. (2005) Antiviral Function of APOBEC3G Can Be Dissociated from Cytidine Deaminase Activity. Curr. Biol. 15, 166–170 [DOI] [PubMed] [Google Scholar]
- 36.Bishop KN, et al. (2006) Antiviral Potency of APOBEC Proteins Does Not Correlate with Cytidine Deamination. J. Virol. 80, 8450–8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holmes RK, et al. (2007) APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: comparisons with APOBEC3G. J. Biol. Chem. 282, 2587–2595 [DOI] [PubMed] [Google Scholar]
- 38.Miyagi E, et al. (2007) Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. Journal of virology 81, 13346–13353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schumacher AJ, et al. (2008) The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 82, 2652–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Law EK, et al. (2016) The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2, e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitz KM and Petersen-Mahrt SK (2012) AIDing the immune system-DIAbolic in cancer. Semin. Immunol. 24, 241–245 [DOI] [PubMed] [Google Scholar]
- 42.Gao J, et al. (2018) Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like family genes activation and regulation during tumorigenesis. Cancer Sci. 109, 2375–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King Justin J., et al. (2015) Catalytic Pocket Inaccessibility of Activation-Induced Cytidine Deaminase Is a Safeguard against Excessive Mutagenic Activity. Structure 23, 615–627 [DOI] [PubMed] [Google Scholar]
- 44.Sheehy AM, et al. (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650 [DOI] [PubMed] [Google Scholar]
- 45.Jarmuz A, et al. (2002) An Anthropoid-Specific Locus of Orphan C to U RNA-Editing Enzymes on Chromosome 22. Genomics 79, 285–296 [DOI] [PubMed] [Google Scholar]
- 46.Harris RS, et al. (2002) RNA Editing Enzyme APOBEC1 and Some of Its Homologs Can Act as DNA Mutators. Mol. Cell 10, 1247–1253 [DOI] [PubMed] [Google Scholar]
- 47.Harris RS and Dudley JP (2015) APOBECs and virus restriction. Virology 479–480, 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng AZ, et al. (2021) APOBECs and Herpesviruses. Viruses 13, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green AM and Weitzman MD (2019) The spectrum of APOBEC3 activity: From anti-viral agents to anti-cancer opportunities. DNA Repair 83, 102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uriu K, et al. (2021) The Battle between Retroviruses and APOBEC3 Genes: Its Past and Present. Viruses 13, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goila-Gaur R and Strebel K (2008) HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng AZ, et al. (2019) Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol. 4, 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harris RS (2008) Enhancing immunity to HIV through APOBEC. Nat. Biotech. 26, 1089–1090 [DOI] [PubMed] [Google Scholar]
- 54.Jern P, et al. (2009) Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 5, e1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim EY, et al. (2010) Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 84, 10402–10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mulder LCF, et al. (2008) Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A. 105, 5501–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Warren CJ, et al. (2017) Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses 9, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mirabello L, et al. (2017) HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 170, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delviks-Frankenberry KA, et al. (2016) Minimal Contribution of APOBEC3-Induced G-to-A Hypermutation to HIV-1 Recombination and Genetic Variation. PLoS Pathog. 12, e1005646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohammadzadeh N, et al. (2019) Role of co-expressed APOBEC3F and APOBEC3G in inducing HIV-1 drug resistance. Heliyon 5, e01498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imahashi M, et al. (2014) Lack of association between intact/deletion polymorphisms of the APOBEC3B gene and HIV-1 risk. PLoS One 9, e92861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siriwardena SU, et al. (2016) Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases. Chem. Rev. 116, 12688–12710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knisbacher BA, et al. (2016) DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet. 32, 16–28 [DOI] [PubMed] [Google Scholar]
- 64.Olson ME, et al. (2017) APOBEC enzymes as targets for virus and cancer therapy. Cell Chem. Biol. 25, 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou J, et al. (2017) APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci. 7, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venkatesan S, et al. (2018) Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol. 29, 563–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith NJ and Fenton TR (2019) The APOBEC3 genes and their role in cancer: insights from human papillomavirus. J. Mol. Endocrinol. 62, R269–R287 [DOI] [PubMed] [Google Scholar]
- 68.Silvas TV and Schiffer CA (2019) APOBEC3s: DNA-editing human cytidine deaminases. Protein Sci. 28, 1552–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Granadillo Rodríguez M, et al. (2020) The interesting relationship between APOBEC3 deoxycytidine deaminases and cancer: a long road ahead. Open Biol. 10, 200188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nik-Zainal S, et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roberts SA, et al. (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nature Genet. 45, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burns MB, et al. (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nature Genet. 45, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Law EK, et al. (2020) APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 217, e20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petersen-Mahrt SK and Neuberger MS (2003) In Vitro Deamination of Cytosine to Uracil in Single-stranded DNA by Apolipoprotein B Editing Complex Catalytic Subunit 1 (APOBEC1)*. J. Biol. Chem. 278, 19583–19586 [DOI] [PubMed] [Google Scholar]
- 75.Shi K, et al. (2015) Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J. Biol. Chem. 290, 28120–28130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCann JL, et al. (2019) The DNA deaminase APOBEC3B interacts with the cell-cycle protein CDK4 and disrupts CDK4-mediated nuclear import of Cyclin D1. J. Biol. Chem. 294, 12099–12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chelico L, et al. (2006) APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13, 392–399 [DOI] [PubMed] [Google Scholar]
- 78.Thielen BK, et al. (2007) T cells contain an RNase-insensitive inhibitor of APOBEC3G deaminase activity. PLoS Pathog. 3, 1320–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alvarez-Gonzalez J, et al. (2021) Small Molecule Inhibitors of Activation-Induced Deaminase Decrease Class Switch Recombination in B Cells. ACS Pharmacol. Transl. Sci. 4, 1214–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li M, et al. (2012) First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 7, 506–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Genomics, B.C.f.C. (2011) Single concentration confirmation of uHTS for APOBEC3G DNA Deaminase Inhibitors via a fluorescence-based single-stranded DNA deaminase assay. https://pubchem.ncbi.nlm.nih.gov/bioassay/493152
- 82.Genomics, B.C.f.C. (2011) Single concentration confirmation of APOBEC3A DNA Deaminase Inhibitors via a UDG counterscreen. https://pubchem.ncbi.nlm.nih.gov/bioassay/504714 [Google Scholar]
- 83.Olson ME, et al. (2013) Small-molecule APOBEC3G DNA cytosine deaminase inhibitors based on a 4-amino-1,2,4-triazole-3-thiol scaffold. ChemMedChem 8, 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.King JJ, et al. (2021) Structure-Based Design of First-Generation Small Molecule Inhibitors Targeting the Catalytic Pockets of AID, APOBEC3A, and APOBEC3B. ACS Pharmacol. Transl. Sci. 4, 1390–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cen S, et al. (2010) Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J. Biol. Chem. 285, 16546–16552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Markossian S, et al. , eds (2004) Assay Guidance Manual, Eli Lilly & Company and the National Center for Advancing Translational Sciences; [PubMed] [Google Scholar]
- 87.Furukawa A, et al. (2009) Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 28, 440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kvach MV, et al. (2019) Differential Inhibition of APOBEC3 DNA-Mutator Isozymes by Fluoro- and Non-Fluoro-Substituted 2’-Deoxyzebularine Embedded in Single-Stranded DNA. Chembiochem 20, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barzak FM, et al. (2019) Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2’-deoxyzebularine. Org. Biomol. Chem. 17, 9435–9441 [DOI] [PubMed] [Google Scholar]
- 90.Kvach MV, et al. (2019) Inhibiting APOBEC3 activity with single-stranded DNA containing 2′-deoxyzebularine analogues. Biochemistry 58, 391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Love RP, et al. (2012) Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 287, 30812–30822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bohn MF, et al. (2015) The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure 23, 903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adolph MB, et al. (2017) Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res. 45, 3378–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ara A, et al. (2014) Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PLoS Pathog. 10, e1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Polevoda B, et al. (2015) RNA binding to APOBEC3G induces the disassembly of functional deaminase complexes by displacing single-stranded DNA substrates. Nucleic Acids Res. 43, 9434–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Feng Y, et al. (2015) Natural Polymorphisms and Oligomerization of Human APOBEC3H Contribute to Single-stranded DNA Scanning Ability. J. Biol. Chem. 290, 27188–27203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wan L, et al. (2017) Observation by Real-Time NMR and Interpretation of Length- and Location-Dependent Deamination Activity of APOBEC3B. ACS Chem. Biol. 12, 2704–2708 [DOI] [PubMed] [Google Scholar]
- 98.Hou S, et al. (2019) Structural Analysis of the Active Site and DNA Binding of Human Cytidine Deaminase APOBEC3B. J. Chem. Theory Comput. 15, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fell JB, et al. (2018) Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with In Vivo Activity. ACS Med. Chem. Lett. 9, 1230–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeng M, et al. (2017) Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem. Biol. 24, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 101.Sasaki T, et al. (2018) DRONE: Direct Tracking of DNA Cytidine Deamination and Other DNA Modifying Activities. Anal. Chem. 90, 11735–11740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Y and Kang C (2017) Solution NMR Spectroscopy in Target-Based Drug Discovery. Molecules 22, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sugiki T, et al. (2018) Current NMR Techniques for Structure-Based Drug Discovery. Molecules 23, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shuker SB, et al. (1996) Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 274, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 105.Erlanson DA, et al. (2019) Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol. 26, 9–15 [DOI] [PubMed] [Google Scholar]
- 106.Harjes S, et al. (2017) NMR-based method of small changes reveals how DNA mutator APOBEC3A interacts with its single-stranded DNA substrate. Nucleic Acids Res. 45, 5602–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Byeon I-JL, et al. (2013) NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 4, 1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Byeon I-JL, et al. (2016) Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 55, 2944–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen K-M, et al. (2008) Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452, 116–119 [DOI] [PubMed] [Google Scholar]
- 110.Harjes E, et al. (2009) An Extended Structure of the APOBEC3G Catalytic Domain Suggests a Unique Holoenzyme Model. J. Mol. Biol. 389, 819–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kouno T, et al. (2015) Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 22, 485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yan X, et al. (2019) Structural Investigations on the Interactions between Cytidine Deaminase Human APOBEC3G and DNA. Chem. – Asian J. 14, 2235–2241 [DOI] [PubMed] [Google Scholar]
- 113.Wienken CJ, et al. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]
- 114.Niether D and Wiegand S (2019) Thermophoresis of biological and biocompatible compounds in aqueous solution. J. Phys. - Condens. Matter 31, 503003. [DOI] [PubMed] [Google Scholar]
- 115.Asmari M, et al. (2018) Thermophoresis for characterizing biomolecular interaction. Methods 146, 107–119 [DOI] [PubMed] [Google Scholar]
- 116.Seidel SAI, et al. (2012) Label-Free Microscale Thermophoresis Discriminates Sites and Affinity of Protein–Ligand Binding. Angew. Chem. Int. Ed. 51, 10656–10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rich RL and Myszka DG (2007) Higher-throughput, label-free, real-time molecular interaction analysis. Anal. Biochem. 361, 1–6 [DOI] [PubMed] [Google Scholar]
- 118.Siu KK, et al. (2013) Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 4, 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ma L, et al. (2018) Identification of small molecule compounds targeting the interaction of HIV-1 Vif and human APOBEC3G by virtual screening and biological evaluation. Sci. Rep. 8, 8067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shaban NM, et al. (2021) CryoEM structure of the EBV ribonucleotide reductase BORF2 and mechanism of APOBEC3B inhibition. bioRxiv, 2021.2008.2030.458246. Accepted at Sci. Adv., will be in press for further citation by galley proof stage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Petersen-Mahrt SK, et al. (2002) AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418, 99–104 [DOI] [PubMed] [Google Scholar]
- 122.Schumacher AJ, et al. (2005) APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc. Natl. Acad. Sci. U. S. A. 102, 9854–9859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chan K, et al. (2012) Base Damage within Single-Strand DNA Underlies In Vivo Hypermutability Induced by a Ubiquitous Environmental Agent. PLOS Genet. 8, e1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hultquist JF, et al. (2011) Human and Rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H Demonstrate a Conserved Capacity To Restrict Vif-Deficient HIV-1. J. Virol. 85, 11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Komor AC, et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anzalone AV, et al. (2020) Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotech. 38, 824–844 [DOI] [PubMed] [Google Scholar]
- 127.St. Martin A, et al. (2018) A fluorescent reporter for quantification and enrichment of DNA editing by APOBEC–Cas9 or cleavage by Cas9 in living cells. Nucleic Acids Res. 46, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.St. Martin A, et al. (2019) A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep. 9, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Logue EC, et al. (2014) A DNA Sequence Recognition Loop on APOBEC3A Controls Substrate Specificity. PLoS One 9, e97062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shaban NM, et al. (2018) The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol. Cell 69, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hu Y, et al. (2019) Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat. Struct. Mol. Biol. 26, 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Barzak FM, et al. (2021) Small-Angle X-ray Scattering Models of APOBEC3B Catalytic Domain in a Complex with a Single-Stranded DNA Inhibitor. Viruses 13, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Erlanson DA, et al. (2016) Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15, 605–619 [DOI] [PubMed] [Google Scholar]