

Summary
Misfolding and aggregation of disease-specific proteins, resulting in the formation of filamentous cellular inclusions, is a hallmark of neurodegenerative disease with characteristic filament structures, or conformers, defining each proteinopathy. Here we show that a previously unsolved amyloid fibril comprised of a 135 amino acid C-terminal fragment of TMEM106B is a common finding in distinct human neurodegenerative diseases, including cases characterized by abnormal aggregation of TDP-43, tau, or α-synuclein protein. A combination of cryo-electron microscopy and mass spectrometry was used to solve the structures of TMEM106B fibrils at a resolution of 2.7 Å from postmortem human brain tissue afflicted with frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP, N=8), progressive supranuclear palsy (PSP, N=2), or dementia with Lewy bodies (DLB, N=1). The commonality of abundant amyloid fibrils composed of TMEM106B, a lysosomal/endosomal protein, to a broad range of debilitating human disorders indicates a shared fibrillization pathway that may initiate or accelerate neurodegeneration.
Graphical Abstract

In Brief:
Cryo-EM and mass spectrometry-based proteomics of insoluble amyloid fibrils derived from postmortem human brains afflicted with diverse neurodegenerative diseases reveals widespread fibrillization of an endolysosomal membrane protein, TMEM106B, pointing towards a potentially pathogenic commonality between distinct proteinopathies.
Introduction
Neurodegenerative proteinopathies are characterized by the deposition of filamentous protein aggregates in neurons and/or glia (Jucker and Walker, 2013). The transactive response DNA-binding protein-43 (TDP-43), the microtubule-associated phosphoprotein tau, and α-synuclein protein can each misfold and accrue intracellularly into tangled filamentous inclusions manifesting in neurodegenerative diseases known collectively as TDP-43 proteinopathies, tauopathies, and synucleinopathies, respectively (Lee et al., 2001). The structures of TDP-43 and tau filaments in frontotemporal lobar degeneration (FTLD) (Arakhamia et al., 2020; Arseni et al., 2022; Shi et al., 2021; Zhang et al., 2020) and α-synuclein fibrils in multiple system atrophy (MSA) (Schweighauser et al., 2020) have recently been reported. What has emerged from these studies is that each disease has a homotypic molecular fold, or conformer, characteristic of the underlying neuropathology (Arakhamia et al., 2020; Falcon et al., 2018; Falcon et al., 2019; Fitzpatrick et al., 2017; Zhang et al., 2020). The evidence for the “one conformer per disease” paradigm is growing, with the presence of unknown buried cofactors (Arakhamia et al., 2020; Falcon et al., 2019; Zhang et al., 2020) and diverse patterns of posttranslational modification (Arakhamia et al., 2020; Kametani et al., 2020) mediating the structural diversity of fibrillar polymorphs.
Here, using a combination of cryo-electron microscopy (cryo-EM) and mass spectrometry (MS), we show that a previously unsolved amyloid fibril found in cases representing a variety of neurodegenerative conditions is comprised of a 135 amino acid C-terminal fragment of TMEM106B; a known risk gene for FTLD-TDP and aging (Feng et al., 2021; Van Deerlin et al., 2010). The fibrillization of TMEM106B into identical fibrillar structures in a wide range of sporadic or genetic TDP-43 proteinopathies (FTLD-TDP), a tauopathy (PSP), and a synucleinopathy (DLB) points towards a commonality between these diverse neurodegenerative diseases. This suggests that the formation of amyloid fibrils composed of TMEM106B, a lysosomal/endosomal protein, may contribute to pathogenicity via a loss or gain of function.
Results
Structures of TMEM106B amyloid fibrils in diverse neurodegenerative diseases
TMEM106B is a 274 amino acid type II transmembrane protein (Figure 1A) that under physiological conditions spans the lysosomal and endosomal membranes of neurons, glia, endothelial cells and pericytes (Figure 1B) (Busch et al., 2013; Lang et al., 2012). Current knowledge points to a physiological role for TMEM106B in the proper sorting and transport of endosomes and lysosomes in these various cell types (Feng et al., 2020; Lang et al., 2012; Lüningschrör et al., 2020; Rademakers et al., 2021; Schwenk et al., 2014; Stroobants et al., 2021; Zhou et al., 2020). TMEM106B consists of a C-terminal domain that projects in the lysosomal lumen, a single-pass transmembrane domain, and an N-terminal domain that extends into the cytosol (Figure 1B) (Lang et al., 2012).
Figure 1. Overview of native TMEM106B.

(A), Schematic view of TMEM106B with its N-terminal domain (NTD), transmembrane domain (TM), and C-terminal domain (CTD). (B), A predicted structure of TMEM106B from ROBETTA (Kim et al., 2004) colored with the same scheme as (A), indicating sites at which the C-terminal domain may get cleaved. (C) Aggregation propensity mapped onto a predicted structure of TMEM106B(120–254) from AlphaFold (Jumper et al., 2021) (positive values in blue indicate soluble regions and negative values in red correspond to aggregation-prone regions).
The N-terminus, TMEM106B(1–96) (Figures 1A and 1B), is involved in lysosomal and endosomal trafficking via interactions with MAP6 and can interact with itself as well as with TMEM106C to form homo- and hetero-multimers at the surface of the lysosome (Schwenk et al., 2014; Stagi et al., 2014). The transmembrane domain, TMEM106B(97–117), is a single-pass α-helix (Figures 1A and 1B). Although there is in vitro evidence that the lumenal C-terminus, TMEM106B(118–274), may undergo proteolytic cleavage to release a C-terminal fragment (Figure 1B) (Brady et al., 2014), this has not been confirmed in vivo under physiological or disease conditions. Importantly, the 135 amino acid C-terminal fragment that we identified in fibrils from FTLD-TDP, PSP and DLB cases, TMEM106B(120–254), is predicted to have a high β strand content and to be prone to aggregation (Figure 1C) (Jumper et al., 2021). TMEM106B has many known posttranslational modifications, with a heavily glycosylated C-terminus (Lang et al., 2012). Here, we show that a truncated C-terminal fragment, TMEM106B(120–254), aggregates into amyloid fibrils in diverse genetic and sporadic proteinopathies and present experimentally determined atomic models of the fibrils extracted from eleven postmortem human brains.
Protein filaments were isolated from insoluble protein fractions derived from human postmortem brain tissue representing a number of neurodegenerative disorders. Remarkably, cryo-EM 3D reconstructions of fibrils extracted from five FTLD-TDP type A cases (four with different GRN mutations and one sporadic case), two FTLD-TDP type B cases, one FTLD-TDP type C case, two cases of PSP (a 4R tauopathy) and one case of DLB (Table S1, Figures S1 and S2), found two common fibril subtypes (Figures 2 and S3). In each of these cases, there were wide, two-fold symmetric, twisted ribbons with a helical pitch of ~2,100 Å, a maximum width of ~260 Å, and a minimum width of ~120 Å, which we designate “doublet” fibrils (Figures 2 and S3A). There were also narrow, twisted rods displaying a regular helical pitch of ~2,100 Å, and an approximately uniform width of ~125 Å, which we refer to as “singlet” fibrils (Figures 2 and S3B). A structural variation of the singlet fibril was found in one case (Figure 2, Case 7/Type B case 2), with a shorter helical pitch of ~1,250 Å (Figure S3C). The ratio of singlet to doublet fibril populations were found to vary between 1:2 and 2:1 for the majority of cases. All density maps show clear inter-β sheet separation perpendicular to the long axis of the fibrils, with separation of β strands discernible at resolutions better than 4.5 Å (Figure 2) (Fitzpatrick et al., 2017). The backbone trace of all cross-sections is clear (Falcon et al., 2018) and shows a generic fibrillar fold common to TDP-43 proteinopathies (FTLD-TDP), a tauopathy (PSP), and a synucleinopathy (DLB) (Figure 2).
Figure 2. Cryo-EM reconstructions of TMEM106B(120–254) fibrils.

Cross-sections (10 z-slice average) of the TMEM106B(120–254) singlet and doublet fibril unsharpened density maps from eight cases of FTLD-TDP, two cases of PSP, and one case of DLB. See also Figures S1–S3 and S6 and Tables S2 and S3.
Density maps of the unknown singlet and doublet fibril subtypes were determined to resolutions of 3.0 Å and 2.7 Å, respectively (Figures 2–5 and S4, Tables S2 and S3). To identify this unknown fibril, proteomic analysis of fractionated tissue from FTLD-TDP case 1 (Supplementary File) was used to identify 600 potential protein candidates. The well resolved sidechain density of the doublet fibril map was mapped to this reduced proteome using cryoID (Ho et al., 2020) and findMySequence (Chojnowski et al., 2022) software which unambiguously identified an amino acid sequence corresponding to residues 120–254 of TMEM106B (See STAR Methods). Fibrils formed by TMEM106B(120–154) exhibit ultrastructural polymorphism with a common protofilament (Figure 4A) existing as either a singlet fibril (Figure 5A and 5C) or in a two-fold symmetrical juxtaposition as a doublet fibril (Figure 3A). The inter-protofilament interface of the doublet fibrils is mediated by an apparently non-proteinaceous anionic cofactor that binds to the sidechains of residues K178 and R180 of each protofilament with near-stoichiometric occupancy (Figure 3B).
Figure 5. Cryo-EM structure of highly twisted TMEM106B singlet fibril and comparison of singlet fibril molecular polymorphs.
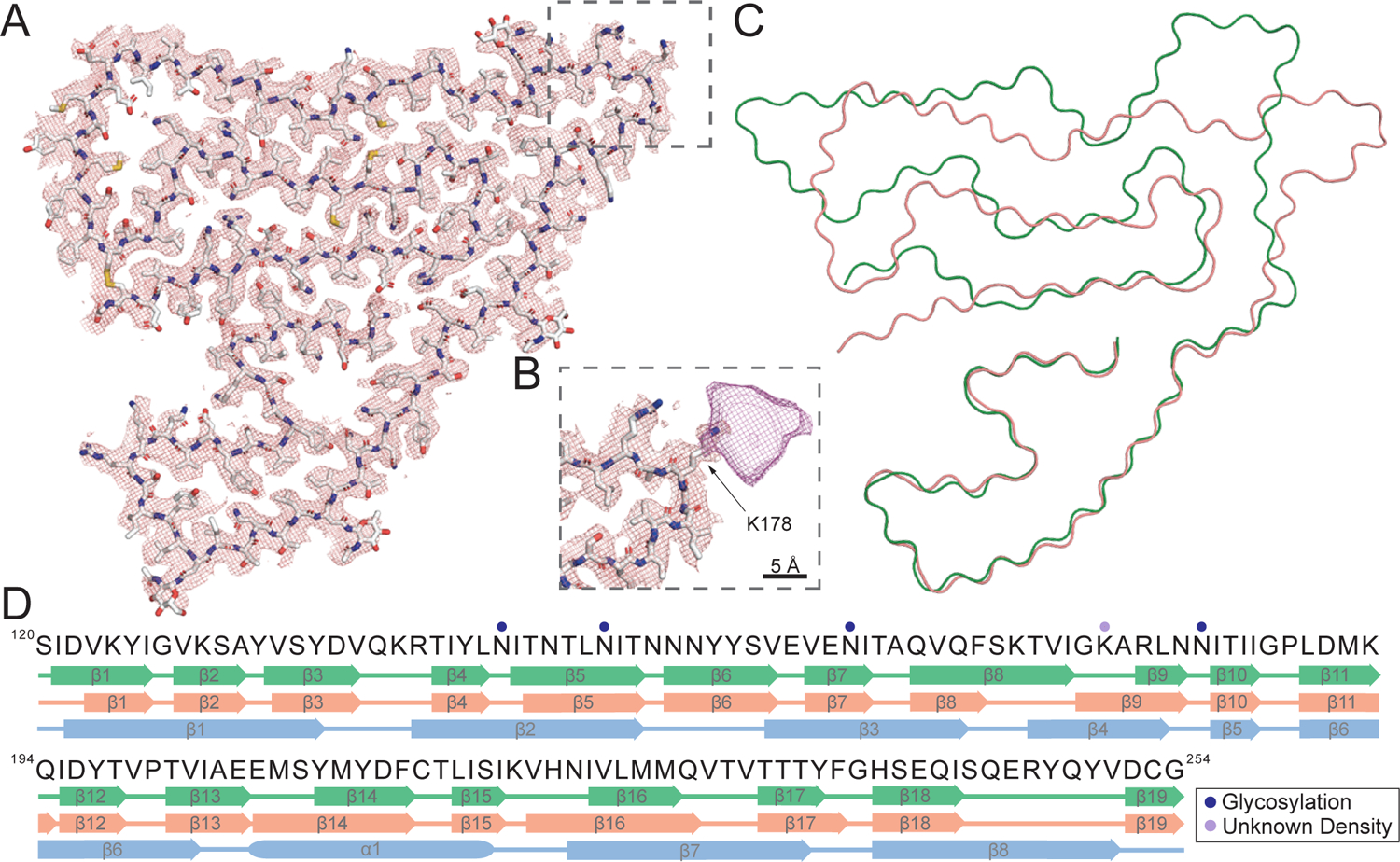
(A) Cryo-EM mesh density (mesh) and atomic model (sticks) of the highly twisted TMEM106B singlet fibril. (B) Magnified view of an unknown density bound to K178 in the highly twisted TMEM106B singlet fibril. (C) Overlay of the atomic models (Cα chain shown) of the low-twist (green) and high-twist (pink) singlet fibrils. (D) Comparison of secondary structure motifs formed by the low-twist (green), high-twist (pink) singlet fibrils, and the native protein predicted by AlphaFold (blue) with experimentally determined post-translational modifications of the highly twisted TMEM106B singlet fibril. See also Figure S5 and Table S2.
Figure 4. Cryo-EM structure of a TMEM106B protofilament highlighting the key structural features.
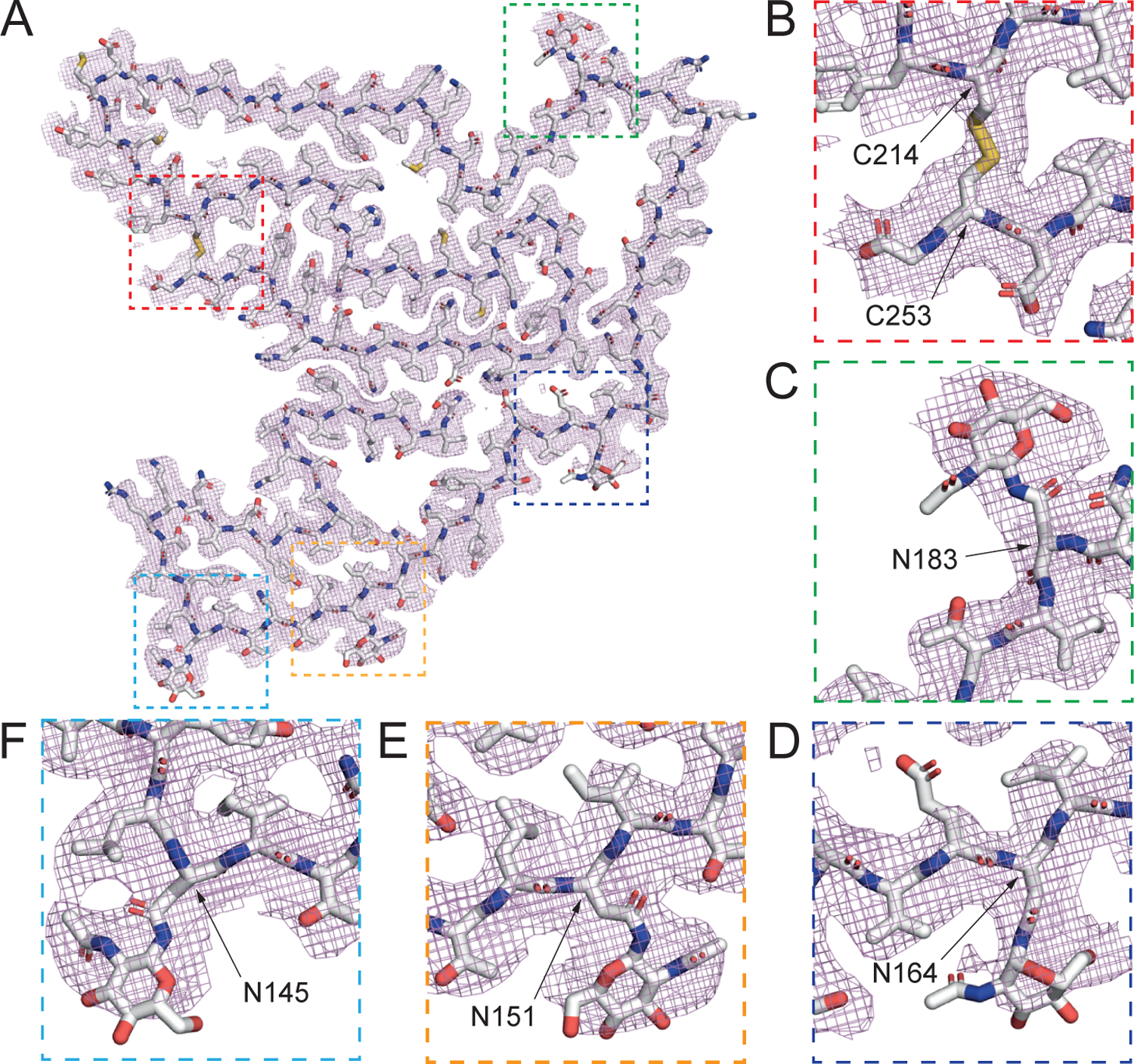
(A) Cryo-EM density (mesh) and atomic model (sticks) of a TMEM106B protofilament. (B) A disulfide bond between C214 and C253. (C) Polymorphic site T185S and glycosylated asparagine at N183. (D) Glycosylated N164, (E) N151, and (F) N145. See also Figures S4 and S5 and Table S2.
Figure 3. Cryo-EM structure of a TMEM106B doublet fibril.
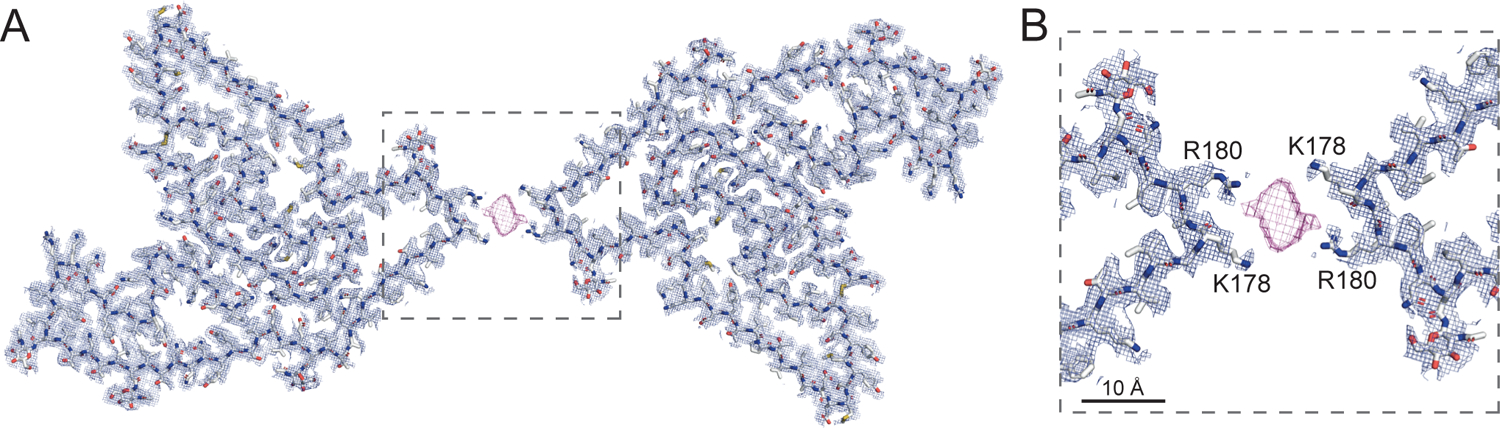
Cryo-EM mesh density (mesh) and atomic model (sticks) of (A) a TMEM106B doublet fibril and (B) the interface between the two protofilaments in the TMEM106B doublet mediated by a non-proteinaceous, anionic cofactor (purple mesh) that binds to the sidechains of residues K178 and R180 of each protofilament. See also Figure S5 and Table S2.
The protofilament core consists of residues 120 to 254, derived from the lumenal topological domain (Figures 4A and S5A), which is distinct from the stable N-terminally cleaved fragments from the cytoplasmic topological domain that have previously been reported (Brady et al., 2014). The 135 residues adopt a C-shaped protofilament with the N-terminus embedded in the center, surrounded by a long connecting strand which meanders back on itself to form a closely packed, three-layered C-terminal fold (Figures 4A and S5A). In total, 19 β strands were identified (β1-β19, Figures 5D, S5A, and S5C) and found to be linked with either short loops (Q138-R140, T166-A167, G177-A179, G188-P189, V199-P200, E206-S208, C214-T215, K220-N223, Q229-V232, F237-G238, Q245-V251), gentle turn (N154) or tight single residue turns (G127, Y132, N145, E161, N183, Q194). Diverse sidechain-sidechain interactions facilitate the packing or positioning of β strands (Figures 4A, S5A, and S5C).
Starting from the N-terminus, strands β1-β3 forms a Z-shape, with the tight junction Y132 forming a putative hydrogen bond with N154 that effectively hold neighboring strands β5 and β6 in place. A closely packed β-helix is formed by strands β3, β4, and β5. Following a gentle turn at N154, strand β6 is interrupted by E161, which forms a salt bridge with H239 that contributes to the positioning of strand β18. A short β7 strand leads into a long β8 strand that forms a short antiparallel sheet-sheet interface with strand β17. Strands β8-β10 form a large, enclosed hydrophobic cavity, involving residues I176, A179, L181, I184 and I186. The cavity is likely to contain small cofactors as detached densities are observed frequently in sharpened maps. Strand β11 marks the beginning of the C-terminal three-layer fold forming a tight convergence point with strand β16 and loops V230-V232 through the hydrophobic residues I186, L190, M227 and V230. Hydrophobic clustering also locks strands β12, β13 in place with strand β15 nestled in between through small, apolar residues V199, I217 and I219. A bulky, aromatic β14 strand may be stabilized by π-π interactions between Y209 and Y211, and between Y211 and F213, as the density indicates that two rotamers are accessible to Y211. Strand β15 packs against the extreme C-terminus through the formation of a disulfide bond between C214 and C253, connecting to strand β16 via the loop K220-N223 (a potential cation-π interaction is formed by K220 and H222). N223 also plays an important role in facilitating the interdigitation of sidechains between strands β16 and β18 by forming a hydrogen bond with E246. The potential strain is relieved by forming a L-shape pin turn with the short β17 strand. A multi-sheet, cross-β interface between strands β16→β18→β1 cements the center of the fold with a short β19 strand closing the protofilament structure at the potential cleavage site G254. The disulfide bond between C214 and C253 (Figure 4B) stabilizes the fibril structure. Ultrastructural polymorphism of the singlet and doublet fibrils is mediated by a non-proteinaceous density nestled between K178 and R180, connecting the two protofilaments of the doublet fibril (Figures 3 and S5C). In cross-section, this measures approximately 9 Å × 4 Å and is presumably anionic, given that it is bound to positively charged sidechains (Figures 3B and S5C).
Structural role of posttranslational modifications in TMEM106B amyloid fibrils
Post-translational modifications were preserved for the filament sample and allowed assignment of glycans in the atomic model that match with previous studies of the native protein. Native TMEM106B has six potential N-glycosylation sites all located on the lumenal domain, with the first three on residues N145, N151, and N164 (N1–N3) being non-complex glycosylation and the last three on N182, N183, and N256 (N4–N6) being complex N-glycosylation (Lang et al., 2012). In the protofilament structure, we found that these N-glycosylations were largely retained as densities consistent with NAG were identified at residues N183 (Figure 4C), N164 (Figure 4D), N151 (Figure 4E), and N145 (Figure 4F). All prominent non-proteinaceous densities are shown schematically in Figures S5A–C.
The structure of TMEM106B(182–185) is significant as this region has a disease-risk associated polymorphism (T185S, Table S1). It includes a potentially phosphorylated T185, a heavily glycosylated N183 (Figures 4A and 4C), and a N182 that also shows proximal, non-proteinaceous density in some maps. The density near T185 shows a putative phosphate density. The phosphorylation of T185 is thought to determine the glycosylation state of N183 (Nicholson et al., 2013) which shows a large, strong density connected to the Nδ2 atom corresponding to multiple glycans (Figure 4C). Interestingly, TMEM106B(120–254) fibrils were observed irrespective of the presence of the risk (T185) or protective (S185) haplotype (Table S1). All TMEM106B(120–254) fibril structures have been built using a threonine at residue 185 since it is not possible to distinguish between a serine and threonine sidechain (van der Waals volume of 73 Å3 and 93 Å3, respectively) or an average of both sidechains at a resolution of 2.7 Å.
Singlet TMEM106B(120–254) amyloid fibrils display molecular polymorphism
The protofilament structure (singlet fibril) formed by TMEM106B(120–154) is identical across many cases with a uniform inter-strand twist angle of −0.4° (Figure S3B, Tables S2 and S3). However, distinct misfolding minima exist with a similar, but structurally distinct, conformer displaying an inter-strand twist angle of −0.67°. The more twisted singlet fibril is also comprised of residues S120 to G254, adopting a C-shape similar to the less twisted singlet fibril and involving an embedded N-terminus and a three-layered C-terminus (Figures 5A and S5B). However, the C-terminal fold is less kinked and more compact, showing an ordered rectangular arrangement. Notably, in sharp contrast to the outward-pointing corner turn observed in the TMEM106B protofilament (Figures 4A, S5A, and S5C), in this instance the chain curves inward and forms an L-shape pointing directly towards the extreme C-terminus (Figures 5A and S5B). In total, 19 β strands were identified (β1-β19), matching the number of β strands we found in the less twisted singlet fibril. Although residues S120-G254 form both low- and high-twist singlet fibrils, comparison of the secondary structure motifs adopted by TMEM106B(120–254) in each molecular polymorph (Fitzpatrick and Saibil, 2019; Petkova et al., 2005) show differences with 21 in 93 residues having a β strand/loop mismatch (Figure 5D).
In general, β strands (β1-β6) at the N-terminus are identical in terms of amino acid composition and highly similar in spatial arrangements. However, from β7 onwards, the more twisted singlet fibril shows significant variation from its less twisted counterpart (Figures 5C, S5A, and S5B). Strand β8 is shorter, packed against β17 by forming a short, hydrophobic antiparallel sheet-sheet interface. However, this interface is not tightly-packed due to the lack of interaction between F237 and Q170. Strand β8 then leads into β9 with a flexible short loop consisting of S172-A179. There is a strong, large density visibly attached to K178 (Figures 5B and S5B) that could be an anionic cofactor or posttranslational modification (Arakhamia et al., 2020). Strands β9-β13 organize into a continuous long strand, with β10 marking the beginning of the C-terminal three-layered fold. These five strands (β9-β13) are arranged horizontally in a straight line, connected with short loops (G188-P189, V199-P200, E206-S208) and tight single residue turns (N183 and Q194). Strand β12 is packed closely against β16, possibly by hydrophobic clustering with the small hydrophobic residues I195, I224 and L226. Strand β13 continues into β14 with a near 90-degree turn, with A204 at the corner. We hypothesize that a more complex interaction occurs in this aromatic region as the density suggests two rotamers for Y209, which is able to form either a methionine-aromatic interaction with M207 or π-π interaction with Y211. At the C-terminal end of β14, C214 again forms a disulfide bond with C253, the same as observed in the less twisted singlet fibril. This disulfide bond results in a sharp tip that steers into a conserved region consisting of β15-β17 and positions β19 into forming a flattened and smooth strand following β18.
Comparison of TMEM106B in pathological and non-pathological human tissue
As the discovery of TMEM fibrils is a recent and surprising finding, antibodies that are specific to TMEM106B fibrillar signatures, such as PHF1 (pS396/404) or pS129 available for tau and α-synuclein filaments, respectively, have not yet been generated and validated. Antibodies that recognize fibrillar phospho-epitopes are necessary to unambiguously image TMEM106B fibrils in patient tissue using immunofluorescence and to quantitatively measure the amount of TMEM106B fibrils in insoluble protein fractions.
Despite the current lack of reagents that specifically bind to an exclusively fibrillar epitope, we performed western blot analysis on fractionated tissue pellets from 5 neurologically normal individuals aged between 68 and 73 years of age using a commercially available TMEM106B C-terminal antibody (Figure S6). For comparison, we also included a 48-year-old FTLD-TDP type A (case 5, TMEM106B fibrils solved by cryo-EM) in an adjacent lane and performed separate western blot analysis on fractionated tissue pellets from FTLD-TDP types A (case 4, TMEM106B fibrils solved by cryo-EM), B, and C along with five α-synucleinopathies (Figure S6). To probe for TMEM106B, we used a polyclonal antibody that binds to residues (204–253). As this antibody is not specific to a fibrillar epitope, all TMEM106B species are potentially detected. We observe bands consistent with monomeric TMEM106B(120–254), molecular weight of 15.49 kDa, at ~16 kDa (Figure S6) and multiple bands ~25 kDa (Figure S6) that may correspond to glycosylated TMEM106B(120–254) monomers. Notably, there are many higher molecular weight bands at 110 kDa, 135 kDa, and 190 kDa that are consistent with TMEM106B oligomerization (Jiang et al., 2022) (Figure S6). The general trend we observe is that these high molecular weight species are more abundant in pathological compared to non-pathological controls, although there are exceptions (e.g. FTLD-TDP case 4 is a sporadic case that shows a high concentration of TMEM fibrils by cryo-EM but not by western blot).
Importantly, we and others (Jiang et al., 2022) contend that western blots do not reliably detect TMEM106B aggregation. An antibody has recently been generated which detects non-fibrillar TMEM106B by binding to residues (239–250) which are buried in the fibril core (Schweighauser et al., 2021). Such an antibody can only measure accumulation of monomeric TMEM106B in the soluble or insoluble fractions and is therefore not suitable for detecting fibrils.
Discussion
Our study has identified previously undetected amyloid fibrils composed of aggregated TMEM106B(120–254) protein fragments in postmortem brain tissue from multiple TDP-43 proteinopathies, a tauopathy, and a synucleinopathy. These observations were both unexpected and surprising. We further note that TMEM106B(120–254) fibrils were not observed in our previous studies of Alzheimer’s disease (Arakhamia et al., 2020; Fitzpatrick et al., 2017) or CBD tau filaments (Arakhamia et al., 2020). Although this suggests that TMEM106B(120–254) fibrils may not be a consistent feature of all tauopathies, our absence of evidence could simply be due to the limited N-number and varying brain regions in our cryo-EM studies of other tauopathies. It has also recently been suggested that variations in tissue fractionation protocols may significantly affect TMEM106B fibril yield (Schweighauser et al., 2021).
Notably, the abundance of TMEM106B(120–254) fibrils in two of our PSP cases is very high. We find that in PSP case 1, which also contains the PSP tau fold (Shi et al., 2021) (Figures S3D, Table S3), TMEM106B(120–254) singlet fibrils are equally abundant as tau filaments. In PSP case 2, TMEM106B(120–254) singlet and doublet fibrils are approximately equal in number, with no PSP tau filaments observed in this sample (Figure 2, Table S3). This is perhaps not surprising as PSP tau filaments are extremely sparse in the majority of PSP tissue samples we have screened (See STAR Methods). In contrast, PSP case 3 only contained PSP tau filaments (Figure S3E) and TMEM106B(120–254) fibrils were not observed. Recently, two cases with FTLD-TDP type B were shown to contain narrow, highly twisted filaments formed by TDP-43 (cross section ~40 Å × 70 Å, helical pitch ~600 Å), although these fibrils were not found in all cortical regions and were not confirmed in other FTLD-TDP subtypes (Arseni et al., 2022). We observe TMEM106B(120–254) fibrils to be the predominant fibrillar species in all FTLD-TDP cases we studied (Figure 2, Table S2), irrespective of fractionation protocol (See STAR Methods). Finally, DLB is traditionally characterized by untwisted α-synuclein filaments with widths less than 100 Å (Schweighauser et al., 2020). In the present DLB case, we observe copious amounts of TMEM106B(120–254) singlet and doublet fibrils (Figure 2, Table S3) but narrow, untwisted α-synuclein filaments were not observed (presumably due to insufficient fibril yield, see STAR Methods).
Interestingly, TMEM106B fibril structures similar to those identified in our study in patients across a wide range of proteinopathies as well as neurologically normal aged individuals was recently reported (Schweighauser et al., 2021). Although the findings are similar, our study is distinct in focusing primarily on genetic and sporadic forms of FTLD-TDP (N=8, Table S1) whereas the work described in the preprint draws mainly from various tauopathies and synucleinopathies. The consensus finding that TMEM106B fibrils occur across diverse proteinopathies makes it difficult to determine whether TMEM106B fibrils are disease specific or are a general feature of neurodegeneration and aging. Importantly, the observation of increased TMEM106B fibrils in neurologically normal aged individuals (Schweighauser et al., 2021) does not preclude a role in disease etiology as a similar observation is seen for tau (Brier et al., 2016; Chatterjee et al., 2021) and α-synuclein filaments (Hatton et al., 2022). Additionally, it is unclear whether the formation of TMEM106B amyloid fibrils represents a primary pathogenic process or a non-specific secondary phenomenon (e.g. general downstream marker of lysosomal stress). The observation of TMEM106B fibrils in some, but not all, cases of PSP (Figures 2, S3D, and S3E) (Shi et al., 2021) and DLB (Figure 2) (Schweighauser et al., 2021) suggests that the degree of fibrillization is highly variable in neurodegenerative diseases. Nevertheless, the combined trend across the two studies is that in the majority of cases with known neurodegeneration, fibrillization of TMEM106B is substantially greater when compared to age-matched controls (Figure S6) (Schweighauser et al., 2021).
Despite the small number of cases, this trend is most pronounced in the FTLD-TDP spectrum of neurodegenerative disease. All eight cases of FTLD-TDP in our study, representing all major pathological and genetic subtypes (Table S1, Figure S1), contained large amounts of TMEM106B fibrils per gram of frontal cortex (Figure 2 and S6). One relatively young patient (5th decade at death) with FTLD-TDP type A was replete with TMEM106B fibrils (Figures 2 and S6), arguing against TMEM106B fibrillization representing a nonspecific feature of aging (Schweighauser et al., 2020). Furthermore, the observation that genetic variation in TMEM106B is a strong modifier of FTLD-TDP disease risk suggests that TMEM106B fibrilization may play a more central role in FTLD-TDP pathogenesis (Finch et al., 2011; Van Deerlin et al., 2010; van der Zee et al., 2011).
If TMEM106B(120–254) is a potential driver of neurodegeneration, it will be important to determine what triggers the formation of these amyloid fibrils. Cellular models have revealed that over-expression of TMEM106B is associated with proteolytic cleavage that generates one or more N-terminal fragments and a presumed (though not yet observed) C-terminal fragment (Brady et al., 2014). We speculate that cleavage of the native TMEM106B fold by protease(s) generates an aggregation-prone fragment TMEM106B(120–254). As the isoelectric point of this lumenal C-terminal fragment is pH 5.4, the acidic environment of the lysosome (pH 4.5–5.5) would favor charge-neutralization and possibly fibrillization (Smirnovas et al., 2005). The misfolding and aggregation of TMEM106B(120–254) may be influenced by posttranslational modifications (Arakhamia et al., 2020), such as glycosylation (N145, N151, N164, N183), and binding of cofactors (K178, R180) increasing the likelihood of molecular (Petkova et al., 2005; Zhang et al., 2019), and ultrastructural (Arakhamia et al., 2020; Fitzpatrick et al., 2013; Fitzpatrick et al., 2017), polymorphism which manifest as amyloid fibril subtypes.
Based on our TMEM106B(120–254) fibril structures, we predict that cleavage at S120 may be due to thrombin, granzyme A, kallikrein-related peptidase 4, or cathepsin P (Table S4, Figure S5D). Granzyme A is particularly intriguing due to its role in the inflammatory response and the biogenic link between lytic granules and lysosomes (van Daalen et al., 2020). The degree to which proteolytic cleavage of the C-terminal fragment occurs in vivo is uncertain; however, it raises the possibility that factors that increase lysosomal stress, such as aging or reduced levels of progranulin (PGRN), combined with the presence of the higher expressing TMEM106B risk haplotype may drive more TMEM106B into the proteolytic pathway, resulting in the generation of TMEM106B C-terminal fragments in excess of the critical aggregation concentration favoring fibril formation (Hortschansky et al., 2005).
Given the presence of these previously undetected amyloid fibrils in myriad proteinopathies, it is pertinent to ask what possible effect could the cleavage and aggregation of TMEM106B(120–254) have on the trafficking and degradation of TDP-43, tau, and α-synuclein filaments? For now, we can only speculate, but given our current knowledge we outline various possibilities based upon our structural observations (Figure 6). As TMEM106B is a lysosomal/endosomal protein, its loss-of-function via fibrillization could disrupt lysosomal trafficking on microtubules, impairing cellular clearance mechanisms (Brady et al., 2014; Lüningschrör et al., 2020; Schwenk et al., 2014). If TMEM106B(120–254) forms fibrils in the lumen, degradation of these protein aggregates places an extra burden on the lysosome. This will impede disposal of aberrantly aggregated filaments delivered to the lysosomes by autophagosomes or endosomes (Jiang and Bhaskar, 2020). If the lysosomal burden is increased, there may also be lysosomal leakage resulting in the release of TMEM106B(120–254) fragments and aggregates into the cytosol where they can propagate. Generally, we hypothesize that the fibrillization of TMEM106B(120–254) inside or outside the lysosome will trigger lysosomal dysfunction promoting the accumulation of TDP-43, tau, and α-synuclein filaments in susceptible individuals (Figure 6). If TMEM106B(120–254) fibrils were to accumulate intracellularly, the loss/gain-of-function arising from the fibrillization of TMEM106B(120–254) would lead to markedly less efficient removal of TDP-43, tau, and α-synuclein filaments through neuronal or glial lysosomal degradation. Ultimately, we hypothesize that TMEM106B(120–254) fibrils exacerbate lysosomal stress and amplify or accelerate the accumulation of pathological proteins (Monaco and Fraldi, 2020).
Figure 6. Structure-based model of a possible interrelationship between TMEM106B, aggregated filaments, and lysosomes in neurodegenerative diseases.

Under physiological conditions, TMEM106B spans the lysosomal/endosomal membrane. Upon cleavage of the C-terminal fragment, TMEM106B(120–254) fibrils may form. It is currently unknown whether TMEM106B(120–254) fibrillizes in the lumen or if lysosomal leakage occurs and TMEM106B(120–254) fragments aggregate into fibrils in the cytosol. Based on our set of TMEM106B fibril structures, we speculate that the aggregation of TMEM106B(120–254) into fibrils leads to lysosomal dysfunction, which promotes the accumulation of aberrantly aggregated amyloid fibrils such as those formed by TDP-43 (PDB:7PY2, green sticks), tau (PDB:7P65, blue sticks), or α-synuclein.
In conclusion, we have determined the structures of TMEM106B(120–254) fibrils identified in a range of phenotypically distinct proteinopathies using cryo-EM. This unknown fibrillization pathway in all major TDP-43 proteinopathies, a tauopathy, and a synucleinopathy raises many intriguing questions. While future experiments will determine whether TMEM106B(120–254) fibril formation represents a loss- or gain-of-function, our findings identify a common fibrillization pathway and widespread structural target that may be implicated in neurodegeneration.
Limitations of the study
At the time of publication, an antibody that binds exclusively to TMEM106B(120–254) fibrils does not exist. This has prevented immunohistochemistry experiments to directly visualize TMEM106B(120–254) fibrils in postmortem human tissue using immunofluorescence or immunogold electron microscopy.
Star Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anthony W. P. Fitzpatrick (Anthony.Fitzpatrick@columbia.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All cryo-EM maps have been deposited to the Electron Microscopy Data Bank (EMDB; www.ebi.ac.uk/pdbe/emdb/). All refined atomic models have been deposited to the Protein Data Bank (PDB; www.rcsb.org/). All EMDB and PDB accession numbers are listed in the key resources table. The mass spectrometry raw data has been deposited to MassIVE (massive.ucsd.edu) under the identifier MSV000088875.
The paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TMEM106B (204–253) | Novus Biologicals | Cat#NBP1–91311; RRID: AB_11019681; QC18333–42825 |
| IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat#926–32213; RRID: AB_621848; D11005-09 |
| Mouse monoclonal anti-TMEM106B (1–46) | Proteintech | Cat#60333-1-Ig; RRID: AB_2881442 |
| Rabbit polyclonal anti-TMEM106B (101–200) | Bioss | Cat#bs-11694R; RRID: AB_2905622 |
| Rabbit polyclonal anti-TMEM106B (111–190) | Biorbyt | Cat#orb158617; RRID: AB_2905623 |
| Rabbit polyclonal anti-TMEM106B (150–275) | Proteintech | Cat#20995-1-AP; RRID: AB_10694293 |
| Rabbit polyclonal anti-TMEM106B (204–253) | Sigma Aldrich | Cat#SAB2106773; RRID AB_2905624 |
| Rabbit polyclonal anti-TMEM106B (218–252) | LSBio | Cat#LS-C757550; RRID: AB_2905625 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser262 and Ser356) | P. Seubert, Elan Pharmaceuticals; San Francisco, CA; USA | 12E8 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser396 and Ser404) | P. Davies, Albert Einstein College of Medicine; New York, NY; USA | PHF1; RRID: AB_2315150 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser202) | P. Davies, Albert Einstein College of Medicine; New York, NY; USA | CP13; RRID: AB_2314223 |
| Rabbit polyclonal anti-Tau (Human-specific) | L. Petrucelli, Mayo Clinic; Jacksonville, FL; USA | E1; RRID: 2819185 |
| Rabbit polyclonal anti-TDP-43 | Proteintech | Cat#10782-2-AP; RRID: AB_615042 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser202 and Thr205) | Thermo Fisher | Cat#MN1020; RRID: AB_223647 |
| Mouse monoclonal anti-α-synuclein | Invitrogen | Cat# 180215; RRID: AB_86714 |
| Mouse monoclonal anti-beta amyloid | Dako | Cat#M0872; RRID: AB_2056966 |
| Rabbit polyclonal anti-ubiquitin | Dako | Cat#Z0458; RRID: AB_2315524 |
| Biological Samples | ||
| FTLD-TDP frontal cortex tissue | University of British Columbia (UBC) | https://neurology.med.ubc.ca/programs/alzheimer-disease-and-related-disorders-program/ |
| PSP Case 1 caudate tissue | Mayo Clinic Brain Bank | https://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank |
| PSP Cases 2 and 3 frontal cortex tissue | University of British Columbia (UBC) | https://neurology.med.ubc.ca/programs/alzheimer-disease-and-related-disorders-program/ |
| DLB frontal cortex tissue | Mayo Clinic Brain Bank | https://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Intercept Blocking Buffer | LI-COR Biosciencs | Cat#927–70001; 210816 |
| iBlot 2 PVDF Mini Stacks | Invitrogen/Thermo Fisher Scientific | Cat#IB24002; 2PM131221-01 |
| Novex WedgeWell 4–20% Tris-Glycine Gel | Invitrogen/Thermo Fisher Scientific | Cat#XP-4205BOX; 21030390 |
| SDS-PAGE Running Buffer [10X] | GBiosciences | Cat#786-029G; 181405 |
| LI-COR Odyssey CLx | LI-COR Biosciences | CLX-0707 |
| iBlot 2 Gel Transfer Device | Invitrogen/Thermo Fisher Scientific | Cat#IB21001 0302180117 |
| BLUEstain 2 Protein ladder 5–245 kDa | Gold Biotechnology | Cat#P008-500 051301P008 |
| Paraformaldehyde 32% Solution | Electron Microscopy Sciences | Cat#15714-S; 190107-10 |
| XCell Sure Lock Mini-Cell | Invitrogen/Thermo Fisher Scientific | Cat#EI0001 |
| cOmplete Mini, EDTA-free protease inhibitor cocktail tablets | Roche Diagnostics | Cat#11836170001 35440400 |
| Phenylmethylsulfonyl fluoride (PMSF) | Gold Biotechnology | Cat#P-470-10 0302.091819A |
| Polytron Homogenizer | Kinematica | Cat#11030012 |
| Deposited Data | ||
| Raw mass spectrometry data | This paper | MassIVE MSV000088875 |
| Cryo-EM density map of FTLD-TDP Type A (all cases combined) singlet | This paper | EMD-26296 |
| Cryo-EM density map of FTLD-TDP Type A (all cases combined) protofilament | This paper | EMD-26279 |
| Cryo-EM density map of FTLD-TDP Type A (case 1) singlet | This paper | EMD-26295 |
| Cryo-EM density map of FTLD-TDP Type A (case 1) doublet | This paper | EMD-26294 |
| Cryo-EM density map of FTLD-TDP Type A (case 1) protofilament | This paper | EMD-26274 |
| Cryo-EM density map of FTLD-TDP Type A (case 2) singlet | This paper | EMD-26275 |
| Cryo-EM density map of FTLD-TDP Type A (case 2) doublet | This paper | EMD-26293 |
| Cryo-EM density map of FTLD-TDP Type A (case 3) singlet | This paper | EMD-26292 |
| Cryo-EM density map of FTLD-TDP Type A (case 3) doublet | This paper | EMD-26291 |
| Cryo-EM density map of FTLD-TDP Type A (case 4) singlet | This paper | EMD-26276 |
| Cryo-EM density map of FTLD-TDP Type A (case 4) doublet | This paper | EMD-26290 |
| Cryo-EM density map of FTLD-TDP Type A (case 5) doublet | This paper | EMD-26289 |
| Cryo-EM density map of FTLD-TDP Type B case 1 (case 6) singlet | This paper | EMD-26288 |
| Cryo-EM density map of FTLD-TDP Type B case 2 (case 7) high-twist singlet | This paper | EMD-26278 |
| Cryo-EM density map of FTLD-TDP Type C (case 8) singlet | This paper | EMD-26277 |
| Cryo-EM density map of FTLD-TDP Type C (case 8) doublet | This paper | EMD-26287 |
| Cryo-EM density map of PSP case 1 singlet | This paper | EMD-26284 |
| Cryo-EM density map of PSP case 2 singlet | This paper | EMD-26285 |
| Cryo-EM density map of PSP case 2 doublet | This paper | EMD-26286 |
| Cryo-EM density map of PSP case 2 protofilament | This paper | EMD-26273 |
| Cryo-EM density map of PSP case 1 tau filament | This paper | EMD-26268 |
| Cryo-EM density map of PSP case 3 tau filament | This paper | EMD-26283 |
| Cryo-EM density map of DLB singlet | This paper | EMD-26281 |
| Cryo-EM density map of DLB doublet | This paper | EMD-26282 |
| Atomic model of FTLD-TDP Type A (all cases combined) protofilament | This paper | 7U16 |
| Atomic model of FTLD-TDP Type A (T185S) (all cases combined) protofilament | This paper | 7U18 |
| Atomic model of FTLD-TDP Type A (case 1) protofilament | This paper | 7U11 |
| Atomic model of FTLD-TDP Type A (case 2) singlet | This paper | 7U12 |
| Atomic model of FTLD-TDP Type A (case 4) singlet | This paper | 7U13 |
| Atomic model of FTLD-TDP Type B case 2 (case 7) high-twist singlet | This paper | 7U15 |
| Atomic model of FTLD-TDP Type B case 2 (case 7) (T185S) high-twist singlet | This paper | 7U17 |
| Atomic model of FTLD-TDP Type C (case 8) singlet | This paper | 7U14 |
| Atomic model of PSP case 1 tau filament | This paper | 7U0Z |
| Atomic model of PSP case 2 protofilament | This paper | 7U10 |
| Software and Algorithms | ||
| MotionCor2 | (Zheng et al., 2017) | https://msg.ucsf.edu/software |
| Gctf v1.06 | (Zhang, 2016) | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/ |
| RELION | (He and Scheres, 2017) | http://www2.mrc-lmb.cam.ac.uk/relion |
| Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix.real_space_refine (v1.17.1–3660) | (Afonine et al., 2018) | https://www.phenix-online.org/ |
| MolProbity | (Chen et al., 2010) | http://molprobity.biochem.duke.edu/ |
| CamSol method | (Sormanni et al., 2015) | http://www-vendruscolo.ch.cam.ac.uk/camsolmethod.html |
| PyMOL | www.pymol.org | |
| AlphaFold | (Jumper et al., 2021) | https://alphafold.ebi.ac.uk/ |
| TrRosetta | (Yang et al., 2020) | https://yanglab.nankai.edu.cn/trRosetta/ |
| Robetta | (Kim et al., 2004) | http://robetta.bakerlab.org/ |
| DeepEMHancer | (Sanchez-Garcia et al., 2021) | https://cosmic-cryoem.org/tools/deepemhancer/ |
| Deep Tracer | (Pfab et al., 2021) | https://deeptracer.uw.edu/home |
| Cryo-ID | (Ho et al., 2020) | https://github.com/EICN-UCLA/cryoID |
| findMySequence | (Chojnowski et al., 2022) | https://gitlab.com/gchojnowski/findmysequence |
| ProCleave server | (Li et al., 2020) | http://procleave.erc.monash.edu/ |
| COSMIC | (Cianfrocco MA, 2017) | https://github.com/cianfrocco-lab/COSMIC-CryoEM-Gateway |
| MEROPS database | (Rawlings et al., 2017) | https://www.ebi.ac.uk/merops/ |
| MaxQuant | (Cox and Mann, 2008) | https://www.maxquant.org/ |
| Other | ||
| Holey carbon grids (Au R1.2/1.3, 300 mesh) | Quantifoil | N/A |
| BioRender | https://biorender.com/ | |
| atom2svg | https://gist.github.com/biochem-fan/026ec2f191fee9285424d12fc2b84ce7 | |
Experimental Model and Subject Details
Acquisition of Human Tissues
The tissues used for this study were from the frontal cortices of brains with FTLD-TDP (N=11, 4 males, 7 females), PSP (N=3, 2 male, 1 female), DLB (N=6, 3 males, 3 females), and neurologically normal controls (N=5, 3 males, 2 females), with the exception of PSP case 1 tissue being taken from the caudate region. Patients had been followed at the University of British Columbia Clinic for Dementia and Related Disorders. Informed consent for autopsy and the use of tissue for research was obtained from the legal next-of-kin in accordance with institutional ethics guidelines. Human brain samples were collected under a Mayo Clinic IRB-approved autopsy protocol. Tissues were de-identified prior to use, with no identifiers to patient names linked to brain samples. Patient confidentiality is strictly kept. As such, the utilization of autopsy derived, de-identified human brain tissue in the current study is exempt from IRB regulations. All tissue was handled with universal precautions. For human postmortem brain tissue requests from the Mayo Clinic Brain Bank, see www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank.
Clinical history and pathology of FTLD-TDP Type A case 1
Filaments for cryo-EM analysis were extracted from fresh-frozen post-mortem brain tissue samples from eight cases with FTLD-TDP, including four with FTLD-TDP type A caused by different GRN mutations (one of which also carried the C9orf72 repeat expansion, Case 5) and four sporadic FTLD-TDP (one type A, two type B and one type C), in which GRN and C9orf72 mutations were excluded (Table S1). The TMEM106B haplotypes were determined for each case. The patient was a 60-year-old man with a five-year history of behavioral variant FTD (bvFTD) and mild parkinsonism. There was a family history of early-onset dementia and genetic sequencing of the patient identified a GRN mutation (c.1072C>T).
Clinical history and pathology of FTLD-TDP Type A case 2
The patient was a 55-year-old woman with a seven-year history of non-fluent variant primary progressive aphasia (nfvPPA) and subsequent behavioral abnormalities. Detailed family history was not available; however, genetic analysis identified a GRN mutation (c.1180-1G>A).
Clinical history and pathology of FTLD-TDP Type A case 3
The patient was a 60-year-old woman with a history of psychosis who developed progressive aphasia and behavioral changes at age 54. There was a family history of dementia and parkinsonism and genetic analysis identified a GRN mutation in the patient (c.102delC).
Clinical history and pathology of FTLD-TDP Type A case 4
The patient was an 89-year-old man with a 10-year history of sporadic nfvPPA.
Clinical history and pathology of FTLD-TDP Type A case 5
The patient was a 48-year-old woman who developed progressive non-fluent aphasia at age 44 and had evidence of mild bulbar weakness the year prior to her death. There was a strong family history of FTD and genetic analysis demonstrated a GRN mutation (c.87_90dup) as well as the abnormal C9orf72 repeat expansion in the patient and other affected family members. All five cases with FTLD-TDP type A pathology had abundant compact neuronal cytoplasmic inclusions (NCI), short dystrophic neurites (DN) and occasional lentiform neuronal intranuclear inclusions (NII) concentrated in the upper neocortical laminae (Figure S1). Case 1 and Case 3 also had tau-immunoreactive neurofibrillary tangles (NFT) restricted to the entorhinal cortex and/or hippocampus (Braak stage I and III, respectively). In addition to FTLD-TDP, Case 5 also had mild loss of lower motor neurons in the hypoglossal nucleus with rare TDP-43-positive inclusions and abundant dipeptide repeat immunoreactive neuronal inclusions, characteristic of the C9orf72 repeat expansion.
Clinical history and pathology of FTLD-TDP Type B case 1 (case 6)
The patient was a 62-year-old man with a four-year history of behavioral and cognitive changes followed by progressive weakness, consistent with amyotrophic lateral sclerosis (ALS). TDP-43 immunohistochemistry demonstrated abundant NCI throughout all layers of the neocortex, with most having a diffuse, granular morphology, typical of FTLD-TDP type B. There was also significant loss of spinal cord lower motor neurons with many containing TDP-43-immunoreactive inclusions. NFT were restricted to the hippocampus (Braak stage III).
Clinical history and pathology of FTLD-TDP Type B case 2 (case 7)
The patient was a 74-year-old Asian woman who developed progressive limb and bulbar weakness at age 72. Electrophysiological studies revealed denervation in multiple muscle groups, consistent with a diagnosis of ALS. Cognitive assessment at age 73 demonstrated significant problems with short-term memory, attention and abstract thought. Postmortem examination confirmed ALS with degeneration of the corticospinal tracts and loss of lower motor neurons with many of the remaining cells containing TDP-43 immunoreactive cytoplasmic inclusions. TDP-43 pathology in the primary motor cortex and prefrontal cortex consisted of abundant diffuse, granular neuronal cytoplasmic inclusions and small neuropil dots in all cortical laminae, consistent with FTLD-TDP type B. Neurofibrillary tangles were restricted to the hippocampus (Braak stage III/VI).
Clinical history and pathology of FTLD-TDP Type C case 1 (case 8)
The patient was a 69-year-old man with a 14-year history of progressive language problems with prominent word finding difficulties, characteristic of semantic variant PPA (svPPA), and mild behavioral problems. TDP-43 immunohistochemistry demonstrated numerous DN in the neocortex, many of which were long and tortuous, typical of FTLD-TDP type C. He also had a number of small chronic cerebral infarcts.
Clinical history and pathology of FTLD-TDP Type A case 6 (case 9)
The patient was a woman who developed bvFTD at age 53 and died at age 60. There was a strong family history of FTD and genetic analysis identified a GRN mutation (c.87_90dup). Post-mortem evaluation demonstrated FTLD-TDP type A as the only significant pathology.
Clinical history and pathology of FTLD-TDP Type B case 3 (case 10)
The patient was a 58-year-old woman who presented with nfvPPA followed by mild changes in personality and behavior. During the year prior to death, she developed swallowing difficulty and electrophysiological studies demonstrated lower motor dysfunction. There was no family history of neurological disease and no GRN or C9orf72 mutations were detected in the patient. Post mortem evaluation demonstrated typical changes of FTLD-TDP type B and ALS with TDP-43-positive inclusions in lower motor neurons. There were rare diffuse senile plaques in the neocortex and neurofibrillary tangles were restricted to the entorhinal/transentorhinal cortex (Braak II/VI).
Clinical history and pathology of FTLD-TDP Type C case 2 (case 11)
The patient was a 72-year-old woman who developed language difficulty consistent with svPPA, personality and behavioral changes and poor memory in her early sixties. There was no family history of neurological disease and no GRN or C9orf72 mutations were detected. Post mortem exam demonstrated FTLD-TDP type C pathology and neurofibrillary tangles restricted to the hippocampus (Braak IV/VI).
Clinical history and pathology of PSP case 1
The patient was a 68-year-old white man who developed double vision six years prior to death. He subsequently developed slurred speech, bradykinesia, difficulty walking and unexplained falls, clinically diagnosed as Parkinson-plus syndrome. Post-mortem evaluation revealed findings of cortical-predominant PSP with severe neuronal and glial tau pathology in the motor and premotor cortices, and marked white matter oligodendroglial pathology at the gray-white junction. There was no TDP-43 pathology.
Clinical history and pathology of PSP case 2
The patient was a 75-year-old man with a 10-year history of progressive speech apraxia, cognitive changes and ophthalmoplegia. Gallyas-positive, tau-immunoreactive NFT and glial inclusions, including tufted astrocytes, ramified astrocytes, threads and coil bodies were present throughout the neocortex and numerous subcortical regions, consistent with PSP. There was no TDP-43 or α-synuclein-immunoreactive pathology.
Clinical history and pathology of PSP case 3
The patient was a 59-year-old woman with eight years of progressive cognitive impairment, behavioral changes, supranuclear gaze palsy, gait problems, dysphagia and akinetic-rigidity, clinically diagnosed as PSP. Typical neuropathological features of PSP were present including numerous tau-positive NFT, tufted astrocytes and other glial inclusions throughout the neocortex and many other brain regions. There was no TDP-43 and α-synuclein pathology.
Clinical history and pathology of DLB case 1
This 68-year-old man had a 10-year history of parkinsonism, with subsequent cognitive impairment. His parkinsonism was characterized by tremors, stiffness, gait difficulties, falls and handwriting difficulties. His cognitive problems included change in personality, disorientation and visual hallucinations. Post mortem examination demonstrated degeneration of the substantia nigra and locus ceruleus. Histologic studies showed diffuse α-synuclein immunoreactive cortical Lewy bodies and minimal Alzheimer type pathology (Braak stage I, Thal phase 1, CERAD neuritic plaque score 0).
Clinical history and pathology of DLB cases used for western blot analysis
DLB cases used for western blot analysis had a pathological diagnosis of DLB or variants thereof. Namely, case 2 (male, 57 years old, DLB A53T), case 3 (male, 66 years old, familial DLB), case 4 (female, 62 years old, DLB), case 5 (female, 81 years old, severe DLB), case 6 (female, 71 years old DLB A53T).
Clinical history and pathology of neurologically normal cases (NN) used in western blot analysis
Neurologically normal cases NN1 (male, 70 years old), NN2 (male, 73 years old), NN3 (female, 73 years old), NN4 (male, 68 years old), and NN5 (female, 70 years old) had no significant neurodegenerative pathology apart from age-related senile plaques (sparse in NN5, moderate in NN3).
Method Details
Histology, Immunohistochemistry, Immunoblotting
FTLD-TDP
The post-mortem evaluation followed consensus recommendations for the neuropathological assessment of dementia (Montine et al., 2012) and FTLD (Cairns et al., 2007). FTLD-TDP subtypes were assigned in accordance with recent consensus criteria (Mackenzie et al., 2011). Five μm thick sections of formalin fixed, paraffin embedded tissue were stained using haematoxylin and eosin and Bielschowsky silver methods. Routine diagnostic Immunohistochemistry (IHC) was performed using the Ventana BenchMark XT automated immunostainer with primary antibodies against TDP-43, hyperphosphorylated tau (pS202, pT205), α-synuclein, beta-amyloid, and ubiquitin.
In an attempt to demonstrate TMEM106B in post-mortem tissue, we tested a number of commercially available antibodies raised against different regions of the protein, using a wide range of IHC protocols. A mouse monoclonal antibody against amino acids 1–46 of the N-terminal region showed a pattern of reactivity consistent with the expected lysosomal localization of physiological TMEM106B, consisting of small cytoplasmic dots in neuronal perikarya and oligodendrocytes. However, this antibody did not label any structures that resembled the pathological inclusions seen with TDP-43 IHC. Despite employing a number of different pretreatment and amplification steps, none of the antibodies against C-terminal epitopes showed any staining that was interpreted as being specific for either physiological or pathological forms of TMEM106B. These included rabbit polyclonal antibodies against TMEM106B epitopes (101–200), (111–190), (150–275), (204–253), and (218–252).
PSP
For Case 1 paraffin sections were immunostained for tau (CP13, 1:1,000) using a DAKO Autostainer. For Cases 2 and 3 sections of formalin fixed, paraffin embedded tissue were stained using haematoxylin and eosin, Bielschowsky and Gallyas silver methods. Immunohistochemistry was performed using the Ventana BenchMark XT automated immunostainer with primary antibodies against TDP-43, hyperphosphorylated tau (pS202, pT205), α-synuclein, beta-amyloid, and ubiquitin.
DLB
Immunohistochemistry for α-synuclein (NACP) was performed after formic acid with the DAKO Autostainer.
Genetic sequencing
DNA was extracted from frozen brain tissue using standard techniques. Sanger sequencing analysis of all exons of the GRN gene and expansion analysis of the GGGGCC repeat in C9orf72 using the 2-step protocol was performed in all FTLD-TDP cases as described previously (Baker et al., 2006; DeJesus-Hernandez et al., 2011). TMEM106B SNP rs3173615 (encoding T185S) was genotyped in all cases using an inventoried Taqman SNP genotyping assay C__27465458_10, as described (Nicholson et al., 2013).
Human brain tissue fractionation
All cases except PSP case 1
Added 10 volumes (w/v) of cold buffer I (10 mM Tris, 800 mM NaCl, 1 mM EGTA, 5mM EDTA, 10% sucrose, 1mM PMSF, Roche protease and phosphate inhibitor, pH 7.4) to frozen brain tissue from frontal cortex and homogenized by polytron (PT2500E, Kinematica, AG). The homogenized samples were centrifuged at 20,100 g for 20 min at 4°C and the supernatant was collected. The pellet was resuspended with 1 volume of cold buffer I, homogenized again and centrifuged at 21,100 g for 20 min at 4°C. The supernatant was then collected and combined with saved supernatant after the 1st centrifugation step. The supernatant was incubated with sarkosyl at a final concentration of 1% with gently shaking for 1h at room temperature. Subsequently, the samples were centrifuged at 100,000 g for 1h at 4°C. The final pellet was resuspended in 20 μl cold buffer II (5 mM Tris pH 7.4) and saved for cryo-EM grid preparation. With regard to α-synuclein fibrils in our DLB case, we note that the tissue fractionation protocol used in the present study is similar to a previous cryo-EM paper (Schweighauser et al., 2020). However, we did not observe thin (<100 Å wide), untwisted fibrils characteristic of α-synuclein filaments in our images. Similarly, despite tissue fractionating type B case 2 (case 7) using our protocol or a recently published protocol for obtaining TDP-43 fibrils (Arseni et al., 2022), we do not observe a significant number of TDP-43 fibrils in our images. Although we performed identical purification methods to isolate filaments from neurologically normal tissue samples (N=5), these cases all had markedly smaller sarkosyl-insoluble pellets than those derived from diseased tissue (2–3 times smaller per gram of frontal cortex).
PSP case 1
To obtain a sufficient yield of tau filaments from PSP tissue, we evaluated the amount of tau present within the sarkosyl-insoluble (P3) fraction from four different brain regions in three different PSP cases, without pronase-treatment to preserve posttranslational modifications (Arakhamia et al., 2020; Carlomagno et al., 2021). As tau levels were consistently highest in the P3 fractions from caudate and frontal cortex, we screened these two regions in an additional 12 PSP cases. Using EM, we observed a tractable filament yield (~0.006 mg/mL) with low background in the P3 fraction from the caudate of case #1 (Figure S2C). To further characterize the PSP case used for cryo-EM, we evaluated total and phosphorylated tau levels in the P3 fraction of case #1 and 3 additional PSP cases by western blot (Figure S2D). The similarities in positivity for common disease-associated phosphoepitopes indicates case #1 is a typical PSP case (Figure S2D), suggesting that the morphology of tau filaments in this case will be representative of PSP.
Mass spectrometry
A sample imaged by cryo-EM, FTLD-TDP type A case 1, was incubated in 4M guanidinium hydrochloride to disaggregate the amyloid fibrils. In solution, digestion was performed using LysC and trypsin, followed by desalting and LC-MS/MS for protein identification and quantification. The data were processed by MaxQuant (Cox and Mann, 2008). Mass spectrometry data was searched against Uniprot human protein database. The TMEM106B fragment that was identified by mass spectrometry in FTLD-TDP type A case 1 was TMEM106B(130–139) (Table S5A). Mass spectrometry was performed in an identical manner on a second sample, FTLD-TDP type A case 6 (case 9), to identify proteins and phosphorylation sites (Table S5). To identify phosphorylation sites, the digest was enriched with TiO2 for phosphopeptides before being subjected to LC-MS/MS. The TMEM106B fragments that were identified by mass spectrometry in FTLD-TDP type A case 6 (case 9) were TMEM106B(130–139) and TMEM106B(248–255) (Table S5B), with no phosphorylation sites detected on these peptides (Table S5C). Raw data was deposited on MassIVE (Project Accession # MSV000088875).
Western blots of sarkosyl-insoluble fractions from donor brains
Sarkosyl-insoluble pellets from fractionated tissue samples were resolved on Novex WedgeWell 4–20% Tris-Glycine gels (Invitrogen) and electrophoresis performed at 25 Milliamps, 2 hours. Proteins were dry transferred onto 0.2 μm PVDF membranes using iBlot 2 Mini Stacks in an iBlot 2 Gel Transfer Device (Invitrogen) at 23 Volts for 6 minutes. Before blocking in Intercept Blocking Buffer (LI-COR), membranes were fixed in 1% paraformaldehyde for 30 min at room temperature. Blots were incubated with primary antibody TMEM106B against epitope (204–253) overnight at 4 degrees. Membranes were washed three times (10 minutes each) in PBS-T, then incubated with secondary antibody IRDye 800CW Donkey anti-Rabbit at 1:10,000 for 1 hour at room temperature. Membranes were washed three times (10 minutes each) in PBS-T and imaged on LI-COR Odyssey CLx.
Predicted native fold of TMEM106B and aggregation propensity
The native structure of TMEM106B was predicted used AlphaFold (Jumper et al., 2021), TrRosetta (Yang et al., 2020), and Robetta (Kim et al., 2004). All three methods converged on highly similar structures of the full length-protein with the most variability predicted to be in the disordered N-terminus. Predicted structures of TMEM106B(120–254) using these tools have a backbone root mean square deviation <2 Å. The aggregation propensity of each TMEM106B(120–254) amino acid at pH 5 were predicted using CamSol (Sormanni et al., 2015) with an average insolubility of −1.39. Predictions of TMEM106B(120–254) aggregation propensity at lysosomal pH 5 and cytosolic pH 7 using the CAMSOL method are similar (integer pH values only possible).
Cryo-electron microscopy
All cases except PSP case 1
For cryoEM, 3 μl aliquots of purified TMEM106B filaments from human brain tissue was applied to glow-discharged holey carbon Quantifoil Au R1.2/1.3 grids (Electron Microscopy Science, Hatfield, PA), blotted with filter paper to create a thin film by removing excess sample using a FEI Vitrobot Mark III at 25°C and 100% humidity (blot force setting 0 and blotting time between 3.5–5.5 s) and plunge-frozen into liquid ethane. High-resolution data was collected primarily at the Zuckerman Institute, Columbia University with additional movies collected at the Columbia University Cryo-EM facility and New York Structural Biology Center (NYSBC), on Titan Krios microscopes equipped with BioQuantum-K3 imaging systems (see Tables S2 and S3 for details). All movies were collected in counting mode with a nominal pixel size of 1.07 Å/pixel, dose rate of 1.5 e/Å2/frame, over 40 frames. The underfocus range was set between 1.4 and 2.8 micrometers (Tables S2 and S3).
PSP Case 1
3 μL aliquots of purified tau filaments from PSP human brain tissue (~0.006 mg/mL) were applied to glow-discharged holey carbon grids (Quantifoil Au R1.2/1.3, 300 mesh), blotted with filter paper and plunge-frozen into liquid ethane using an FEI Vitrobot Mark IV. Images of the frozen grids were collected on three FEI Titan Krios microscopes on multiple occasions due to the paucity of the tau filaments. Imaging conditions were always identical (Table S3), but minute differences in Å/pixel had to be corrected for during image processing. All images were acquired at 300 kV and energy filtered (slit width of 20 eV) using a Gatan K2/K3 Summit detector in counting mode. All movies were collected at a nominal pixel size of 1.0605 Å, using a dose rate of 1.5 electrons per Å2 per frame and an underfocus range of 1 to 3 μm (Table S3).
Helical reconstruction
Movie frames were gain-corrected, aligned, dose-weighted, and summed using MotionCor (Zheng et al., 2017) implemented in RELION (Zivanov et al., 2018). Since amyloid fibrils extend over a few microns, spanning entire micrographs, during motion correction no patching was employed. Parameters for the contrast transfer function were estimated from each motion-corrected micrograph using Gctf (Zhang, 2016). All subsequent image processing was mainly performed using helical reconstruction in RELION (Zivanov et al., 2018), as previously described (Fitzpatrick et al., 2017). Singlet and doublet fibrils were manually picked and separated with initial segmentation at 10.7 Å/pixel and then 3.21 Å/pixel to determine helical parameters (Table S2). Initial models of the singlet and doublet fibrils were generated from 2D class averages of helical pitch-views of the fibrils using a cylinder as an initial model and helically reconstructed in C1 using 3D classification with a T regularization value of 20. The doublet initial model quickly converged using FTLD-TDP particles; the singlet fibril initial model was generated from a PSP dataset in the following manner.
Helical processing of TMEM106B(120–254) singlet fibrils from PSP case 1 began with segments extracted at 3.1815 Å/pixel and 2D classified to determine the helical parameters. The low twist angles hampered initial model generation and so a cylinder was used as a starting model in 3D classification of 8 classes with a T regularization value of 20. One class consistently showed a C-shaped protofilament, irrespective of starting model (which was low-pass filtered to 40 Å). Particles from this class were re-extracted at 1.0605 Å/pixel and 3D classification with local optimization of the helical twist and rise was performed with T=100, decreasing the angular and translational sampling as the rotational and translational accuracy of the helical 3D reconstruction improved. A final subset of particles was 3D auto-refined with T=100 and postprocessed in RELION (Zivanov et al., 2018). Per-particle estimates of ctf values were used to improve resolution during a final refinement with an overall resolution of 4.4 Å being attained. This map had a fairly even distribution of views and was used as an initial model for refinement of TMEM106B(120–254) singlet fibrils from all other cases.
To achieve a high-resolution doublet fibril map, particles from selected 3D classes were re-extracted at 1.07 Å/pixel and re-aligned and averaged to generate 2D classes. Optimal 2D class averages showing clear β strand separation were selected and used for helical reconstruction in C1 using 3D classification with a T regularization value of 100. A persistent 2-fold symmetry was apparent in the doublet fibril reconstructions and this was imposed in subsequent 3D auto-refinements. Other symmetries, such as a 2-start helix or a helix with opposite hand, were tested but failed to attain high-resolution. Helical twist and rise were optimized during 3D auto-refinements (Table S2) and once the resolution of the 3D reconstruction was better than 4 Å, particles were corrected using Bayesian polishing and local ctf parameter refinement in RELION (Zivanov et al., 2018) to improve resolution further. Due to the low average inter-strand twist of approximately −0.4°, many reconstructions appeared to have anisotropic cross-sections. The two-fold symmetry of the doublet fibril was exploited to correct for this anisotropy by invoking relion_particle_symmetry_expand and refining the two masked, subtracted, protofilaments of the doublet cross-section separately, then combining the aligned particles in a final 3D auto-refinement. This improved our reconstructions significantly and a combined, sharpened map using the best particles from each FTLD-TDP case was generated and used for model building. Postprocessing was performed in RELION (Zivanov et al., 2018) and DeepEMHancer (Sanchez-Garcia et al., 2021) via COSMIC (Cianfrocco MA, 2017). Local resolution estimates of the combined map were determined using RELION (Zivanov et al., 2018) and displayed using Chimera (Pettersen et al., 2004) (Figure S4). The final overall resolution estimates were calculated from Fourier shell correlations at 0.143 between the two independently refined half-maps in RELION (Zivanov et al., 2018) (Figure S4).
Helical processing of tau filaments from PSP case 1 was performed using RELION (He and Scheres, 2017), as previously described (Fitzpatrick et al., 2017). Twisted tau filaments were manually selected and segments were extracted at various Å/pixel (1×, 2×, 3×, 5× binned) and 2D classified to determine the helical parameters and ascertain whether - or not - the filament had C2 symmetry. Initial models were generated from 2D class averages of helical pitch views and used as a reference for helical reconstruction in C1 using 3D classification with a T regularization value of 100. When a clear chain trace emerged in the helical cross-section at 1.0605 Å/pixel, a final subset of particles was 3D auto-refined with T=20.
During the preparation of this manuscript, the structure of tau filaments from a PSP case was solved to a resolution of 2.7 Å. The public release of this high-resolution map (EMD-13218) and raw micrographs (EMPIAR-10765) helped in resolving continuous main-chain density in 3D reconstructions of tau filaments from PSP case 1 using our final subset of particles. Helical twist and rise were optimized in the range −0.7° to −0.9° and 4.7 Å to 4.8 Å, respectively. Once the helical parameters were optimized (Table S3), helical symmetry remained fixed in all subsequent refinements. Postprocessing was performed in RELION (He and Scheres, 2017), and DeepEMHancer (Sanchez-Garcia et al., 2021) via COSMIC (Cianfrocco MA, 2017) also aiding interpretation of the maps. The final overall resolution of 4.2 Å was calculated from Fourier shell correlations at 0.143 between the two independently refined half-maps in RELION (Zivanov et al., 2018) (Table S3). Tau filaments from PSP case 3 were extremely sparse, with only 3,942 particles at 3.22 Å/pixel being used in a final 3D auto-refinement with T=4 resulting in a map consistent in size and shape with EMD-13218 low-pass filtered to 6.5 Å (Figure S3E).
Model building
A continuous Cα chain was generated from the combined map using DeepTracer (Pfab et al., 2021). Tryptic digest-mass spectrometry of a fibril sample extracted from FTLD-TDP type A case 1 identified 600 proteins which we used to help identify the fibril’s constituent protein. By cross-referencing the proteins in the sample identified by mass spectrometry to the 2.7 Å density map using cryo-ID (Ho et al., 2020) and findMySequence (Chojnowski et al., 2022), we unambiguously determined that the fibril was composed of TMEM106B(120–254). TMEM106B(120–254) sidechains were manually added to the 135 amino acid Cα chain and iteratively improved using real-space refinement in COOT (Emsley et al., 2010). A 3-chain stack of identical rungs was generated in Chimera and multiple rounds of backbone and sidechain geometry refinement were performed manually followed by local real-space refinement in COOT (Emsley et al., 2010). For refinement of the atomic model built into the combined map (Figures 4 and S4), we used phenix.real_space_refine applying secondary structure, non-crystallographic symmetry, and hydrogen bond restraints (Afonine et al., 2018). Once we had a good atomic model of the TMEM106B(120–254) chain, additional glycans were added to match the density map in COOT (Emsley et al., 2010). A final refinement of the atomic model, including glycans was then performed and validated using MolProbity (Chen et al., 2010) (Table S2). Figures were made using Pymol (www.pymol.org), Chimera (Pettersen et al., 2004), atom2svg (atom2svg.py) and BioRender (biorender.com).
Predicted enzymatic cleavage
To investigate potential proteases responsible for TMEM106B cleavage at position 120 we utilized the ProCleave server (Li et al., 2020) and the MEROPS database (Rawlings et al., 2017). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46, D624-D632.) ProCleave results with scores above 0.5 at position TMEM106B(120) having the highest score are listed in Table S4: Granzyme A (single site), Thrombin (3 sites) and Kallikrein-related peptidase 4 (3 sites). Cathepsin P is not currently in the ProCleave suite but does have a predicted cleavage motif in MEROPS consistent with selective cleavage at position 120 of TMEM106B.
Quantification and Statistical Analysis
The 0.143 Fourier Shell Correlation (FSC) criterion was used to estimate resolution of cryo-EM density maps.
Supplementary Material
Figure S1. Immunohistochemistry of FTLD-TDP cases used for cryo-EM and mass spectrometry, Related to Figure 2 and STAR Methods. TDP-43 immunoreactive pathologic findings in frontal cortex of cases used for cryo-EM and mass spectrometry. Cases 1–5 and 9 (Type A case 6) showed changes typical of FTLD-TDP type A, with abundant compact neuronal cytoplasmic inclusions, short dystrophic neurites and occasional lentiform neuronal intranuclear inclusions in the upper cortical laminae. Cases 6 (Type B case 1) and 7 (Type B case 2) had FTLD-TDP type B pathology, characterized by diffuse, granular neuronal cytoplasmic inclusions in all cortical layers. Case 8 (Type C) had long, tortuous dystrophic neurites, diagnostic of FTLD-TDP type C. Scale bar, 20 μm.
Figure S2. Neuropathological and biochemical analysis of three PSP cases used for cryo-EM, Related to Figure 2 and STAR Methods. (A) and (B) The pigment-containing neurons of the substantia nigra have globus neurofibrillary tangles (hematoxylin and eosin), and tau immunohistochemistry shows neuronal and glial lesions (pretangles, tufted astrocytes, and oligodendroglial coiled bodies). Scale bar, 20 μm. (C) Sarkosyl-insoluble fraction extracted from the caudate/putamen of four different PSP cases as evaluated by immunoblotting for the tau antibodies 12E8 (pS262/356), CP13 (pS202), PHF1 (pS396/404), and E1 (human tau-specific). Tau filaments from (Mayo Clinic, Florida) PSP case #1 were subsequently used for cryo-EM analysis. (D–G) Pathologic findings in frontal cortex of PSP case 2 (D, E) and PSP case 3 (F, G) (both from UBC) had abundant neuronal and glial inclusions, including neurofibrillary tangles and tufted astrocytes, that were immunoreactive for hyperphosphorylated tau. (H, I) show immunohistochemistry of the DLB case used for cryo-EM. Frontal cortex with synuclein and also with thioflavin S fluorescent microscopy. Diffuse cortical LBs and minimal Alzheimer-type plaques are illustrated. (D, F) scale bar, 50 μm. (E, G) scale bar, 10 μm. (H, I) scale bar, 50 μm.
Figure S3. Comparison of the observed fibril types, Related to Figure 2 and STAR Methods. (A-C) Pitch views of the (A) TMEM106B doublet, (B) singlet, and (C) twisted singlet fibrils and their respective cross-sections (A-C) are on the same scale. (D, E) Pitch views of PSP tau filaments from PSP (D) case 1 and (E) case 3 and their respective cross-sections. (D, E) are on the same scale.
Figure S4. Cryo-EM resolution estimates of a combined cryo-EM protofilament map from multiple FTLD-TDP cases, Related to Figure 4. Local resolution estimates (top) and Fourier Shell Correlation (FSC) curve (bottom) between two independently refined half-maps for the TMEM106B(120–254) protofilament.
Figure S5. Schematic views of polymorphic TMEM106B(120–254) amyloid fibrils and fibril structure-based protease predictions, Related to Figures 3–5 and Table S4. Singlet fibrils with (A) low inter-strand twist and (B) high inter-strand twist are shown. Bold line denotes β strand secondary structure with experimentally detected glycosylations shown in cyan. There is an unknown density (orange) attached to K178 in the highly twisted singlet fibril. (C) Doublet fibril with the amino acids K178 and R180 binding to an anionic cofactor highlighted in red. (D) Enzymes predicted to cleave TMEM106B at site S120.
Figure S6. Western blots of sarkosyl-insoluble fractions from donor brains probed with a TMEM106B polyclonal that binds to residues (204–253), Related to Figure 2 and STAR Methods. (A) Equally-exposed western blots of sarkosyl-insoluble fractions from pathological and non-pathological tissues. (B) High contrast images of western blots in (A). Type A, Type B, Type C: FTLD-TDP subtypes. DLB: Dementia with Lewy Bodies. NN: Neurologically Normal.
Table S5. All mass spectrometry raw data on (A) FTLD-TDP type A case 1 and (B, C) FTLD-TDP type A case 6 (case 9), Related to Figures 3–5 and STAR Methods. (A) All peptides identified in sarkosyl-insoluble pellet fractionated from FTLD-TDP type A case 1. (B) All peptides identified in sarkosyl-insoluble pellet fractionated from FTLD-TDP type A case 6 (case 9). (C) All phosphorylation sites identified in sarkosyl-insoluble pellet fractionated from FTLD-TDP type A case 6 (case 9).
Highlights:
Cryo-EM structures of brain-derived TMEM106B fibrils from neurodegenerative diseases
Endolysosomal membrane protein TMEM106B C-terminal fragment forms amyloid fibrils
TMEM106B fibrillization is widespread among diverse neurodegenerative proteinopathies
Identification of fibrillization pathway potentially implicated in neurodegeneration
Acknowledgments:
The authors are grateful to Dr. R. S. Mann for helpful discussions, and to the patients and their families for their participation in this work. Human biological samples and associated data were obtained from the Mayo Clinic Brain Bank and UBC. This work was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorder and Stroke (U01NS110438 to L.P. and A.W.P.F, U54NS110435 to D.D. and A.W.P.F.); the Association for Frontotemporal Degeneration; Canadian Institutes of Health Research (74580 to I.R.A.M.); MCDB Neurodegenerative Disease Fund (to M.H.B.S.). The authors are grateful to Simon Cheung for performing IHC, Dr. H. Uryu for electron microscopy, Drs. X. Chen and K. Song for help collecting data on the Titan Krios at the University of Massachusetts Medical School Cryo-EM Core Facility; R. Grassucci and Dr. Z. Zhang for help collecting data at the Columbia University Cryo-Electron Microscopy Center including the Titan Krios housed at the Zuckerman Institute and the Titan Krios housed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy (New York Structural Biology Center). This paper is dedicated to the memory of Mr. Patrick Trainor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors declare that they have no competing interests.
References
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN, et al. (2020). Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 180, 633–644.e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arseni D, Hasegawa M, Murzin AG, Kametani F, Arai M, Yoshida M, and Ryskeldi-Falcon B (2022). Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601, 139–143. atom2svg.pygist.github.com/biochem-fan/026ec2f191fee9285424d12fc2b84ce7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. [DOI] [PubMed] [Google Scholar]
- Brady OA, Zhou X, and Hu F (2014). Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J Biol Chem 289, 19670–19680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, et al. (2016). Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med 8, 338ra366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch JI, Martinez-Lage M, Ashbridge E, Grossman M, Van Deerlin VM, Hu F, Lee VM, Trojanowski JQ, and Chen-Plotkin AS (2013). Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun 1, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, et al. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno Y, Manne S, DeTure M, Prudencio M, Zhang YJ, Hanna Al-Shaikh R, Dunmore JA, Daughrity LM, Song Y, Castanedes-Casey M, et al. (2021). The AD tau core spontaneously self-assembles and recruits full-length tau to filaments. Cell Rep 34, 108843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee P, Pedrini S, Stoops E, Goozee K, Villemagne VL, Asih PR, Verberk IMW, Dave P, Taddei K, Sohrabi HR, et al. (2021). Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Translational Psychiatry 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnowski G, Simpkin AJ, Leonardo DA, Seifert-Davila W, Vivas-Ruiz DE, Keegan RM, and Rigden DJ (2022). findMySequence: a neural-network-based approach for identification of unknown proteins in X-ray crystallography and cryo-EM. IUCrJ 9, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocco MA, W. M, Youn C, Wagner R, Leschziner AE (2017). COSMIC2: A Science Gateway for Cryo-Electron Microscopy Structure Determination. Practice & Experience in Advanced Research Computing 2, 1–5. [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, and Goedert M (2018). Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M, et al. (2019). Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Lacrampe A, and Hu F (2021). Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol 141, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Sheng RR, Solé-Domènech S, Ullah M, Zhou X, Mendoza CS, Enriquez LCM, Katz II, Paushter DH, Sullivan PM, et al. (2020). A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 143, 2255–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, et al. (2011). TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AW, and Saibil HR (2019). Cryo-EM of amyloid fibrils and cellular aggregates. Curr Opin Struct Biol 58, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Müller SA, et al. (2013). Atomic structure and hierarchical assembly of a cross-β amyloid fibril. P Natl Acad Sci USA 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton C, Ghanem SS, Koss DJ, Abdi IY, Gibbons E, Guerreiro R, Bras J, Consortium, I.D.G., Walker L, Gelpi E, et al. (2022). Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, and Scheres SHW (2017). Helical reconstruction in RELION. Journal of Structural Biology 198, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CM, Li X, Lai M, Terwilliger TC, Beck JR, Wohlschlegel J, Goldberg DE, Fitzpatrick AWP, and Zhou ZH (2020). Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat Methods 17, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortschansky P, Christopeit T, Schroeckh V, and Fändrich M (2005). Thermodynamic analysis of the aggregation propensity of oxidized Alzheimer’s beta-amyloid variants. Protein Sci 14, 2915–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, and Bhaskar K (2020). Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front Mol Neurosci 13, 586731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YX, Cao Q, Sawaya MR, Abskharon R, Ge P, DeTure M, Dickson DW, Fu JY, Ogorzalek Loo RR, Loo JA, et al. (2022). Amyloid fibrils in frontotemporal lobar degeneration with TDP-43 inclusions are composed of TMEM106B, rather than TDP-43. bioRxiv, 2022.2001.2031.478523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, and Walker LC (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametani F, Yoshida M, Matsubara T, Murayama S, Saito Y, Kawakami I, Onaya M, Tanaka H, Kakita A, Robinson AC, et al. (2020). Comparison of Common and Disease-Specific Post-translational Modifications of Pathological Tau Associated With a Wide Range of Tauopathies. Front Neurosci 14, 581936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DE, Chivian D, and Baker D (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32, W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, and Haass C (2012). Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 287, 19355–19365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, and Trojanowski JQ (2001). Neurodegenerative tauopathies. Annu Rev Neurosci 24, 1121–1159. [DOI] [PubMed] [Google Scholar]
- Li F, Leier A, Liu Q, Wang Y, Xiang D, Akutsu T, Webb GI, Smith AI, Marquez-Lago T, Li J, et al. (2020). Procleave: Predicting Protease-specific Substrate Cleavage Sites by Combining Sequence and Structural Information. Genomics Proteomics Bioinformatics 18, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüningschrör P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D, Wanner R, Lüllmann-Rauch R, Wefers B, Wurst W, et al. (2020). The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep 30, 3506–3519.e3506. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, and Lee VM (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco A, and Fraldi A (2020). Protein Aggregation and Dysfunction of Autophagy-Lysosomal Pathway: A Vicious Cycle in Lysosomal Storage Diseases. Front Mol Neurosci 13, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB 3rd, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R, et al. (2013). TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 126, 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, and Tycko R (2005). Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 307, 262–265. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pfab J, Phan NM, and Si D (2021). DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Nicholson AM, Ren Y, Koga S, Nguyen HP, Brooks M, Qiao W, Quicksall ZS, Matchett B, Perkerson RB, et al. (2021). Loss of Tmem106b leads to cerebellum Purkinje cell death and motor deficits. Brain Pathol 31, e12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, and Finn RD (2017). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46, D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia R, Gomez-Blanco J, Cuervo A, Carazo JM, Sorzano COS, and Vargas J (2021). DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Communications Biology 4, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighauser M, Arseni D, Huang M, Lövestam S, Shi Y, Yang Y, Zhang W, Kotecha A, Garringer HJ, Vidal R, et al. (2021). Age-Dependent Formation of TMEM106B Amyloid Filaments in Human Brain. bioRxiv, 2021.2011.2009.467923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, et al. (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature 585, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, Lichtenthaler SF, Hoogenraad CC, Capell A, Haass C, et al. (2014). The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. Embo j 33, 450–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, van Beers M, Tarutani A, Kametani F, Garringer HJ, et al. (2021). Structure-based classification of tauopathies. Nature 598, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnovas V, Winter R, Funck T, and Dzwolak W (2005). Thermodynamic Properties Underlying the α-Helix-to-β-Sheet Transition, Aggregation, and Amyloidogenesis of Polylysine as Probed by Calorimetry, Densimetry, and Ultrasound Velocimetry. The Journal of Physical Chemistry B 109, 19043–19045. [DOI] [PubMed] [Google Scholar]
- Sormanni P, Aprile FA, and Vendruscolo M (2015). The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol 427, 478–490. [DOI] [PubMed] [Google Scholar]
- Stagi M, Klein ZA, Gould TJ, Bewersdorf J, and Strittmatter SM (2014). Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci 61, 226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroobants S, D’Hooge R, and Damme M (2021). Aged Tmem106b knockout mice display gait deficits in coincidence with Purkinje cell loss and only limited signs of non-motor dysfunction. Brain Pathol 31, 223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Daalen KR, Reijneveld JF, and Bovenschen N (2020). Modulation of Inflammation by Extracellular Granzyme A. Frontiers in Immunology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, Santens P, Van den Broeck M, Joris G, Brys J, et al. (2011). TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134, 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, and Baker D (2020). Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences 117, 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Falcon B, Murzin AG, Fan J, Crowther RA, Goedert M, and Scheres SH (2019). Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T, et al. (2020). Novel tau filament fold in corticobasal degeneration. Nature 580, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC, Parsons TM, Castanedes-Casey M, Phillips V, Librero AL, et al. (2020). Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep 21, e50197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunohistochemistry of FTLD-TDP cases used for cryo-EM and mass spectrometry, Related to Figure 2 and STAR Methods. TDP-43 immunoreactive pathologic findings in frontal cortex of cases used for cryo-EM and mass spectrometry. Cases 1–5 and 9 (Type A case 6) showed changes typical of FTLD-TDP type A, with abundant compact neuronal cytoplasmic inclusions, short dystrophic neurites and occasional lentiform neuronal intranuclear inclusions in the upper cortical laminae. Cases 6 (Type B case 1) and 7 (Type B case 2) had FTLD-TDP type B pathology, characterized by diffuse, granular neuronal cytoplasmic inclusions in all cortical layers. Case 8 (Type C) had long, tortuous dystrophic neurites, diagnostic of FTLD-TDP type C. Scale bar, 20 μm.
Figure S2. Neuropathological and biochemical analysis of three PSP cases used for cryo-EM, Related to Figure 2 and STAR Methods. (A) and (B) The pigment-containing neurons of the substantia nigra have globus neurofibrillary tangles (hematoxylin and eosin), and tau immunohistochemistry shows neuronal and glial lesions (pretangles, tufted astrocytes, and oligodendroglial coiled bodies). Scale bar, 20 μm. (C) Sarkosyl-insoluble fraction extracted from the caudate/putamen of four different PSP cases as evaluated by immunoblotting for the tau antibodies 12E8 (pS262/356), CP13 (pS202), PHF1 (pS396/404), and E1 (human tau-specific). Tau filaments from (Mayo Clinic, Florida) PSP case #1 were subsequently used for cryo-EM analysis. (D–G) Pathologic findings in frontal cortex of PSP case 2 (D, E) and PSP case 3 (F, G) (both from UBC) had abundant neuronal and glial inclusions, including neurofibrillary tangles and tufted astrocytes, that were immunoreactive for hyperphosphorylated tau. (H, I) show immunohistochemistry of the DLB case used for cryo-EM. Frontal cortex with synuclein and also with thioflavin S fluorescent microscopy. Diffuse cortical LBs and minimal Alzheimer-type plaques are illustrated. (D, F) scale bar, 50 μm. (E, G) scale bar, 10 μm. (H, I) scale bar, 50 μm.
Figure S3. Comparison of the observed fibril types, Related to Figure 2 and STAR Methods. (A-C) Pitch views of the (A) TMEM106B doublet, (B) singlet, and (C) twisted singlet fibrils and their respective cross-sections (A-C) are on the same scale. (D, E) Pitch views of PSP tau filaments from PSP (D) case 1 and (E) case 3 and their respective cross-sections. (D, E) are on the same scale.
Figure S4. Cryo-EM resolution estimates of a combined cryo-EM protofilament map from multiple FTLD-TDP cases, Related to Figure 4. Local resolution estimates (top) and Fourier Shell Correlation (FSC) curve (bottom) between two independently refined half-maps for the TMEM106B(120–254) protofilament.
Figure S5. Schematic views of polymorphic TMEM106B(120–254) amyloid fibrils and fibril structure-based protease predictions, Related to Figures 3–5 and Table S4. Singlet fibrils with (A) low inter-strand twist and (B) high inter-strand twist are shown. Bold line denotes β strand secondary structure with experimentally detected glycosylations shown in cyan. There is an unknown density (orange) attached to K178 in the highly twisted singlet fibril. (C) Doublet fibril with the amino acids K178 and R180 binding to an anionic cofactor highlighted in red. (D) Enzymes predicted to cleave TMEM106B at site S120.
Figure S6. Western blots of sarkosyl-insoluble fractions from donor brains probed with a TMEM106B polyclonal that binds to residues (204–253), Related to Figure 2 and STAR Methods. (A) Equally-exposed western blots of sarkosyl-insoluble fractions from pathological and non-pathological tissues. (B) High contrast images of western blots in (A). Type A, Type B, Type C: FTLD-TDP subtypes. DLB: Dementia with Lewy Bodies. NN: Neurologically Normal.
Table S5. All mass spectrometry raw data on (A) FTLD-TDP type A case 1 and (B, C) FTLD-TDP type A case 6 (case 9), Related to Figures 3–5 and STAR Methods. (A) All peptides identified in sarkosyl-insoluble pellet fractionated from FTLD-TDP type A case 1. (B) All peptides identified in sarkosyl-insoluble pellet fractionated from FTLD-TDP type A case 6 (case 9). (C) All phosphorylation sites identified in sarkosyl-insoluble pellet fractionated from FTLD-TDP type A case 6 (case 9).
Data Availability Statement
All cryo-EM maps have been deposited to the Electron Microscopy Data Bank (EMDB; www.ebi.ac.uk/pdbe/emdb/). All refined atomic models have been deposited to the Protein Data Bank (PDB; www.rcsb.org/). All EMDB and PDB accession numbers are listed in the key resources table. The mass spectrometry raw data has been deposited to MassIVE (massive.ucsd.edu) under the identifier MSV000088875.
The paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TMEM106B (204–253) | Novus Biologicals | Cat#NBP1–91311; RRID: AB_11019681; QC18333–42825 |
| IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat#926–32213; RRID: AB_621848; D11005-09 |
| Mouse monoclonal anti-TMEM106B (1–46) | Proteintech | Cat#60333-1-Ig; RRID: AB_2881442 |
| Rabbit polyclonal anti-TMEM106B (101–200) | Bioss | Cat#bs-11694R; RRID: AB_2905622 |
| Rabbit polyclonal anti-TMEM106B (111–190) | Biorbyt | Cat#orb158617; RRID: AB_2905623 |
| Rabbit polyclonal anti-TMEM106B (150–275) | Proteintech | Cat#20995-1-AP; RRID: AB_10694293 |
| Rabbit polyclonal anti-TMEM106B (204–253) | Sigma Aldrich | Cat#SAB2106773; RRID AB_2905624 |
| Rabbit polyclonal anti-TMEM106B (218–252) | LSBio | Cat#LS-C757550; RRID: AB_2905625 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser262 and Ser356) | P. Seubert, Elan Pharmaceuticals; San Francisco, CA; USA | 12E8 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser396 and Ser404) | P. Davies, Albert Einstein College of Medicine; New York, NY; USA | PHF1; RRID: AB_2315150 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser202) | P. Davies, Albert Einstein College of Medicine; New York, NY; USA | CP13; RRID: AB_2314223 |
| Rabbit polyclonal anti-Tau (Human-specific) | L. Petrucelli, Mayo Clinic; Jacksonville, FL; USA | E1; RRID: 2819185 |
| Rabbit polyclonal anti-TDP-43 | Proteintech | Cat#10782-2-AP; RRID: AB_615042 |
| Mouse monoclonal anti-pTau (phosphorylated at Ser202 and Thr205) | Thermo Fisher | Cat#MN1020; RRID: AB_223647 |
| Mouse monoclonal anti-α-synuclein | Invitrogen | Cat# 180215; RRID: AB_86714 |
| Mouse monoclonal anti-beta amyloid | Dako | Cat#M0872; RRID: AB_2056966 |
| Rabbit polyclonal anti-ubiquitin | Dako | Cat#Z0458; RRID: AB_2315524 |
| Biological Samples | ||
| FTLD-TDP frontal cortex tissue | University of British Columbia (UBC) | https://neurology.med.ubc.ca/programs/alzheimer-disease-and-related-disorders-program/ |
| PSP Case 1 caudate tissue | Mayo Clinic Brain Bank | https://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank |
| PSP Cases 2 and 3 frontal cortex tissue | University of British Columbia (UBC) | https://neurology.med.ubc.ca/programs/alzheimer-disease-and-related-disorders-program/ |
| DLB frontal cortex tissue | Mayo Clinic Brain Bank | https://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Intercept Blocking Buffer | LI-COR Biosciencs | Cat#927–70001; 210816 |
| iBlot 2 PVDF Mini Stacks | Invitrogen/Thermo Fisher Scientific | Cat#IB24002; 2PM131221-01 |
| Novex WedgeWell 4–20% Tris-Glycine Gel | Invitrogen/Thermo Fisher Scientific | Cat#XP-4205BOX; 21030390 |
| SDS-PAGE Running Buffer [10X] | GBiosciences | Cat#786-029G; 181405 |
| LI-COR Odyssey CLx | LI-COR Biosciences | CLX-0707 |
| iBlot 2 Gel Transfer Device | Invitrogen/Thermo Fisher Scientific | Cat#IB21001 0302180117 |
| BLUEstain 2 Protein ladder 5–245 kDa | Gold Biotechnology | Cat#P008-500 051301P008 |
| Paraformaldehyde 32% Solution | Electron Microscopy Sciences | Cat#15714-S; 190107-10 |
| XCell Sure Lock Mini-Cell | Invitrogen/Thermo Fisher Scientific | Cat#EI0001 |
| cOmplete Mini, EDTA-free protease inhibitor cocktail tablets | Roche Diagnostics | Cat#11836170001 35440400 |
| Phenylmethylsulfonyl fluoride (PMSF) | Gold Biotechnology | Cat#P-470-10 0302.091819A |
| Polytron Homogenizer | Kinematica | Cat#11030012 |
| Deposited Data | ||
| Raw mass spectrometry data | This paper | MassIVE MSV000088875 |
| Cryo-EM density map of FTLD-TDP Type A (all cases combined) singlet | This paper | EMD-26296 |
| Cryo-EM density map of FTLD-TDP Type A (all cases combined) protofilament | This paper | EMD-26279 |
| Cryo-EM density map of FTLD-TDP Type A (case 1) singlet | This paper | EMD-26295 |
| Cryo-EM density map of FTLD-TDP Type A (case 1) doublet | This paper | EMD-26294 |
| Cryo-EM density map of FTLD-TDP Type A (case 1) protofilament | This paper | EMD-26274 |
| Cryo-EM density map of FTLD-TDP Type A (case 2) singlet | This paper | EMD-26275 |
| Cryo-EM density map of FTLD-TDP Type A (case 2) doublet | This paper | EMD-26293 |
| Cryo-EM density map of FTLD-TDP Type A (case 3) singlet | This paper | EMD-26292 |
| Cryo-EM density map of FTLD-TDP Type A (case 3) doublet | This paper | EMD-26291 |
| Cryo-EM density map of FTLD-TDP Type A (case 4) singlet | This paper | EMD-26276 |
| Cryo-EM density map of FTLD-TDP Type A (case 4) doublet | This paper | EMD-26290 |
| Cryo-EM density map of FTLD-TDP Type A (case 5) doublet | This paper | EMD-26289 |
| Cryo-EM density map of FTLD-TDP Type B case 1 (case 6) singlet | This paper | EMD-26288 |
| Cryo-EM density map of FTLD-TDP Type B case 2 (case 7) high-twist singlet | This paper | EMD-26278 |
| Cryo-EM density map of FTLD-TDP Type C (case 8) singlet | This paper | EMD-26277 |
| Cryo-EM density map of FTLD-TDP Type C (case 8) doublet | This paper | EMD-26287 |
| Cryo-EM density map of PSP case 1 singlet | This paper | EMD-26284 |
| Cryo-EM density map of PSP case 2 singlet | This paper | EMD-26285 |
| Cryo-EM density map of PSP case 2 doublet | This paper | EMD-26286 |
| Cryo-EM density map of PSP case 2 protofilament | This paper | EMD-26273 |
| Cryo-EM density map of PSP case 1 tau filament | This paper | EMD-26268 |
| Cryo-EM density map of PSP case 3 tau filament | This paper | EMD-26283 |
| Cryo-EM density map of DLB singlet | This paper | EMD-26281 |
| Cryo-EM density map of DLB doublet | This paper | EMD-26282 |
| Atomic model of FTLD-TDP Type A (all cases combined) protofilament | This paper | 7U16 |
| Atomic model of FTLD-TDP Type A (T185S) (all cases combined) protofilament | This paper | 7U18 |
| Atomic model of FTLD-TDP Type A (case 1) protofilament | This paper | 7U11 |
| Atomic model of FTLD-TDP Type A (case 2) singlet | This paper | 7U12 |
| Atomic model of FTLD-TDP Type A (case 4) singlet | This paper | 7U13 |
| Atomic model of FTLD-TDP Type B case 2 (case 7) high-twist singlet | This paper | 7U15 |
| Atomic model of FTLD-TDP Type B case 2 (case 7) (T185S) high-twist singlet | This paper | 7U17 |
| Atomic model of FTLD-TDP Type C (case 8) singlet | This paper | 7U14 |
| Atomic model of PSP case 1 tau filament | This paper | 7U0Z |
| Atomic model of PSP case 2 protofilament | This paper | 7U10 |
| Software and Algorithms | ||
| MotionCor2 | (Zheng et al., 2017) | https://msg.ucsf.edu/software |
| Gctf v1.06 | (Zhang, 2016) | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/ |
| RELION | (He and Scheres, 2017) | http://www2.mrc-lmb.cam.ac.uk/relion |
| Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix.real_space_refine (v1.17.1–3660) | (Afonine et al., 2018) | https://www.phenix-online.org/ |
| MolProbity | (Chen et al., 2010) | http://molprobity.biochem.duke.edu/ |
| CamSol method | (Sormanni et al., 2015) | http://www-vendruscolo.ch.cam.ac.uk/camsolmethod.html |
| PyMOL | www.pymol.org | |
| AlphaFold | (Jumper et al., 2021) | https://alphafold.ebi.ac.uk/ |
| TrRosetta | (Yang et al., 2020) | https://yanglab.nankai.edu.cn/trRosetta/ |
| Robetta | (Kim et al., 2004) | http://robetta.bakerlab.org/ |
| DeepEMHancer | (Sanchez-Garcia et al., 2021) | https://cosmic-cryoem.org/tools/deepemhancer/ |
| Deep Tracer | (Pfab et al., 2021) | https://deeptracer.uw.edu/home |
| Cryo-ID | (Ho et al., 2020) | https://github.com/EICN-UCLA/cryoID |
| findMySequence | (Chojnowski et al., 2022) | https://gitlab.com/gchojnowski/findmysequence |
| ProCleave server | (Li et al., 2020) | http://procleave.erc.monash.edu/ |
| COSMIC | (Cianfrocco MA, 2017) | https://github.com/cianfrocco-lab/COSMIC-CryoEM-Gateway |
| MEROPS database | (Rawlings et al., 2017) | https://www.ebi.ac.uk/merops/ |
| MaxQuant | (Cox and Mann, 2008) | https://www.maxquant.org/ |
| Other | ||
| Holey carbon grids (Au R1.2/1.3, 300 mesh) | Quantifoil | N/A |
| BioRender | https://biorender.com/ | |
| atom2svg | https://gist.github.com/biochem-fan/026ec2f191fee9285424d12fc2b84ce7 | |