

Abstract
Background:
Hormonal therapy targeting the androgen receptor inhibits prostate cancer (PCa), but the tumor eventually recurs as castration-resistant prostate cancer (CRPC).
Objective:
To understand the mechanisms by which subclones within early PCa develop into CRPC.
Design, setting, and participants:
We isolated epithelial cells from fresh human PCa cases, including primary adenocarcinoma, locally recurrent CRPC, and metastatic CRPC, and utilized single-cell RNA sequencing to identify subpopulations destined to become either CRPC-adeno or small cell neuroendocrine carcinoma (SCNC).
Outcome measurements and statistical analysis:
We revealed dynamic transcriptional reprogramming that promotes disease progression among 23 226 epithelial cells using single-cell RNA sequencing, and validated subset-specific progression using immunohistochemistry and large cohorts of publically available genomic data.
Results and limitations:
We identified a small fraction of highly plastic CRPC-like cells in hormone-naïve early PCa and demonstrated its correlation with biochemical recurrence and distant metastasis, independent of clinical characteristics. We show that progression toward castration resistance was initiated from subtype-specific lineage plasticity and clonal expansion of pre-existing neuroendocrine and CRPC-like cells in early PCa.
Conclusions:
CRPC-like cells are present early in the development of PCa and are not exclusively the result of acquired evolutionary selection during androgen deprivation therapy. The lethal CRPC and SCNC phenotypes should be targeted earlier in the disease course of patients with PCa.
Patient summary:
Here, we report the presence of pre-existing castration-resistant prostate cancer (CRPC)-like cells in primary prostate cancer, which represents a novel castration-resistant mechanism different from the adaptation mechanism after androgen deprivation therapy (ADT). Patients whose tumors harbor increased pre-existing neuroendocrine and CRPC-like cells may become rapidly resistant to ADT and may require aggressive early intervention.
Keywords: Castration-resistant prostate cancer, Primary prostate cancer, Single-cell transcriptomes, Intratumor heterogeneity, Castration-resistant prostate cancer–like cells, Evolutionary trajectory, Critical transcription regulator, Neuroendocrine differentiation, Large population validation
1. Introduction
The vast majority of prostate cancer (PCa) cases are classified as adenocarcinoma with glandular formation and luminal differentiation including the expression of androgen receptor (AR) and prostate-specific antigen (PSA). Hormonal therapy, by inhibiting AR signaling, controls PCa initially but leads to the development of castration-resistant prostate cancer (CRPC) [1]. Most CRPCs are still histologically classified as adenocarcinoma (CRPC-adeno); however, a significant proportion of the clinical CRPC cases belong to a histologic variant form of PCa known as small cell neuroendocrine (NE) carcinoma (SCNC or CRPC-NE) [2], which is highly aggressive. In comparison with adenocarcinoma, SCNC is composed of tumor cells that have lost luminal differentiation and acquired NE differentiation through lineage plasticity [3,4]. The mechanisms responsible for the switch from one differentiated cell type to another are still debated.
Single-cell RNA sequencing (scRNA-seq) technology has been applied to PCa research using mouse prostate models [5,6] and human samples [7,8]. However, previous scRNA-seq studies on human biopsies were conducted using unsorted cells, and the nonepithelial contamination significantly impacts the ability to identify rare epithelial cell subtypes. We have developed a tumor procurement protocol to obtain single-cell preparations from fresh human PCa specimens [9], which allows us to study purified epithelial cells [10,11]. In this study, we delineated a nonuniform distribution of distinct tumor-cell subpopulations in both primary PCa and CRPC samples, and discovered a small fraction of CRPC-like cells in primary PCa that are responsible for therapy resistance in a validation set of 1582 PCa samples obtained from multiple public datasets.
2. Patients and methods
Detailed methods can be found in the Supplementary material.
2.1. Tissue collection
A total of six fresh primary PCa and CRPC/mCRPC specimens were collected in 2018 at Duke University Hospital, including three cases of primary prostate adenocarcinoma (Gleason score 4 + 3, both cancer and matched tumor-adjacent tissue were procured) obtained through radical prostatectomies; two posthormonal therapy locally recurrent CRPC with CRPC-adeno and SCNC histology, obtained through transurethral resection of the prostate (TURP); and one metastatic CRPC (mCRPC) to the soft tissue of pelvic sidewall obtained through surgical resection. A frozen section diagnosis was performed to identify cancer areas, and the corresponding fresh tissue was then used for the preparation of single cells as described previously [9]. The use of anatomic materials was approved by the Duke University’s Institutional Review Board.
Immunohistochemical (IHC) stains were performed using biopsies or surgically resected PCa tissue, including 18 hormone-sensitive PCa cases obtained through prostatectomies, 20 locally recurrent CRPC cases (after hormonal therapy) obtained through TURP, and 12 distant mCRPC cases obtained in a biopsy trial as reported previously [12].
2.2. Single-cell RNA sequencing and data analyses
Fluorescence-activated cell sorting (FACS) was performed on single-cell suspensions using flow cytometer (BD DiVa). Trop2+CD45-CXCR2− (luminal), Trop2+CD45-CD49f+ (basal), and Trop2+CD45-CXCR2+ (NE enriched) were collected (Supplementary Fig. 1), as described previously [9,11].
Suspensions of 5000 single cells from each FACS-isolated cell collections were encapsulated into single droplets using Chromium Controller, and libraries were prepared using Chromium Single Cell 3′ Reagent Kits v2 (10x Genomics). Intratumor heterogeneity was delineated using Seurat [13], and subtype signaling was assessed using the Molecular Signatures Database (MSigDB) [14] and ChIP-Seq data from Enrichr [15]. Probabilistic temporal transcriptional trajectories of CRPC/SCNC progression was defined using Monocle [16] and validated by cluster specific mRNA signatures, as well as mutation and copy number variation (CNV) patterns, measured by Souporcell [17] and HoneyBadger [18].
3. Results
3.1. Intratumor heterogeneity in primary PCa and CRPC
To characterize intratumor heterogeneity as it relates to therapy resistance and disease progression, we collected fresh tissue from the entire spectrum of PCa, including three cases of hormone-naïve primary adenocarcinoma (Gleason score 4 + 3), two cases of posthormonal therapy locally recurrent CRPC with CRPC-adeno and SCNC histology, and one mCRPC (mCRPC-adeno, to the soft tissue of pelvic sidewall; Table 1 and Fig. 1A). For the SCNC case, the patient’s original tumor was diagnosed as prostatic adenocarcinoma and treated with hormonal therapy, but the recurrent tumor showed classic SCNC histology. We isolated multiple epithelial cells (Trop2+CD45−) from each sample by FACS and performed droplet-based scRNA-seq (Supplementary Fig. 1). After removing cells with minimum and maximum thresholds for unique molecular identifier (UMI), nGene, and mitochondrial RNA genes, we obtained 24 385 single cells, and no significant batch effects were observed (Supplementary Fig. 2). Using the Seurat [13] feature of integrating data analyses from different conditions, we identified 18 distinct cell clusters (Fig. 1B) that correspond to subset cluster specific markers (Supplementary Table 1 and Supplementary Fig. 3A).
Table 1.
Clinical characteristics
| Case ID | PCa1 | PCa2 | PCa3 | CRPC1 | CRPC2 | CRPC3 |
|---|---|---|---|---|---|---|
|
| ||||||
| Age | 66 | 63 | 61 | 66 | 58 | 71 |
| PSA at diagnosis | 10.6 | 7.6 | NA | 60.6 | 10.4 | 11 |
| Gleason score | 4 + 3 | 4 + 3 | 4 + 3 | 4 + 4 | 5 + 5 | 4 + 3 |
| TNM at diagnosis | pT2N0M0 | pT2N0M0 | pT2N0M0 | NA | NA | pT3aN0M0 |
| Systemic therapy | NA | NA | NA | Leuprolide acetate, bicalutamide | Abiraterone, prednisone | Leuprolide acetate |
| Time from treatment to CRPC | NA | NA | NA | 2.5 yr | 4 mo | 4 yr |
| PSA at time of tissue collection | 10.6 | 7.6 | 7 | 481 | 0.04 | 26.1 |
| Site of collection | Prostate | Prostate | Prostate | Prostate | Prostate | Pelvic side wall |
| Histology at collection | AdenoCa | AdenoCa | AdenoCa | CRPC-adeno | SCNC | mCRPC-adeno |
| TNM at tissue collection | pT2N0M0 | pT2N0M0 | pT2N0M0 | pT3N1M1 | pT3N1M1 | pT3N1M1 |
AdenoCa = adenocarcinoma; CRPC = castration-resistant prostate cancer; mCRPC = metastatic CRPC; NA = not available; PCa = prostate cancer; PSA = prostate-specific antigen; SCNC = small cell neuroendocrine carcinoma; TNM = tumor, node, metastasis.
Fig. 1. Intratumoral heterogeneity in primary PCa and CRPC.
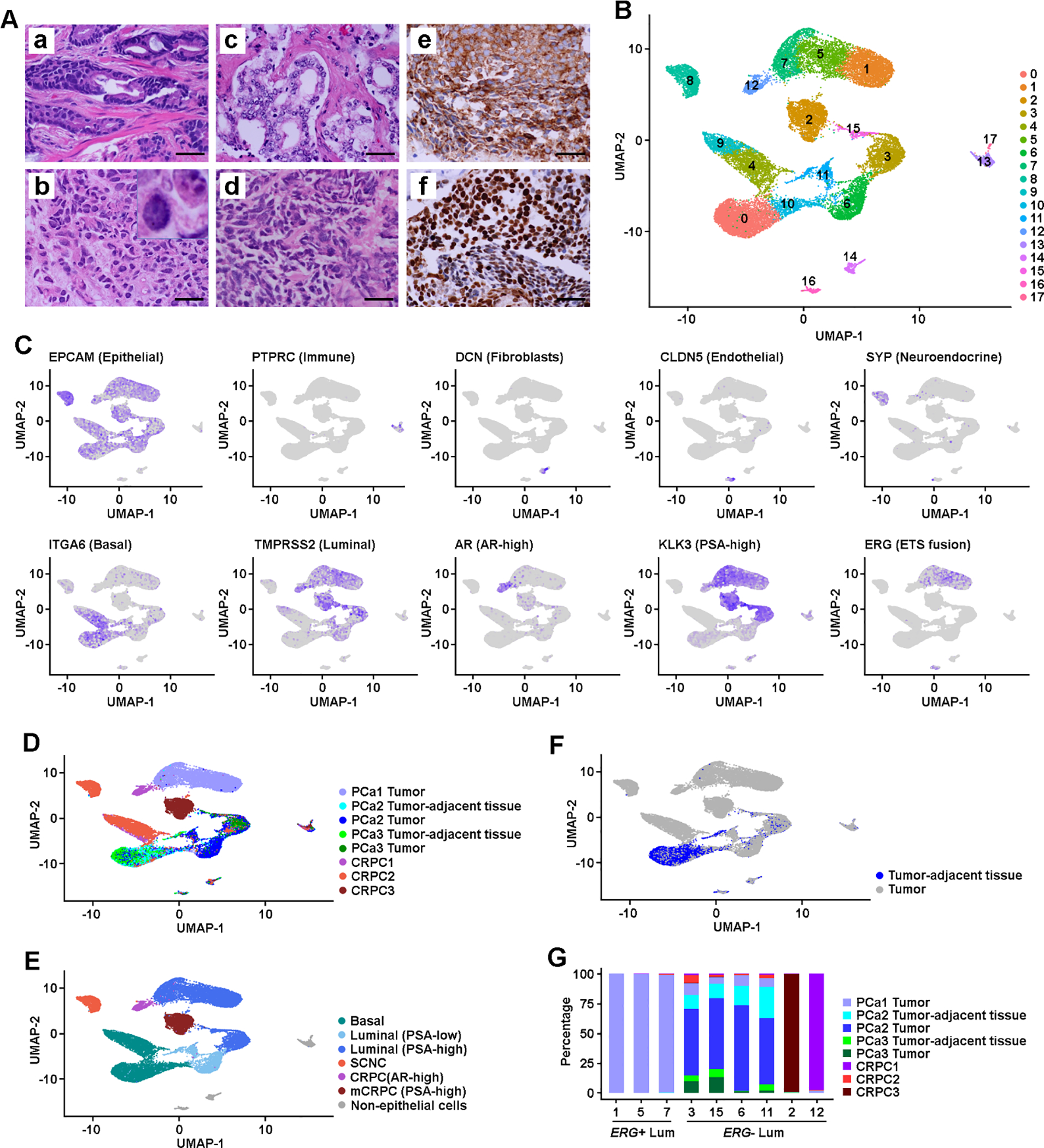
(A) Histology and IHC images: (a) Representative image of primary PCa (GS 4 + 3); (b) locally recurrent CRPC-adeno (the insert shows a higher magnification of tumor cells with nuclear features of adenocarcinoma); (c) Metastatic CRPC (mCRPC) to pelvic side wall, tumor shows classic features of CRPC-adeno; (d) locally recurrent PCa shows classic architectural and cytologic features of SCNC; (e) positive cytoplasmic staining of SCNC cells in (d) for the expression of an NE marker synaptophysin; and (f) positive nuclear staining of SCNC cells in (d) for the expression of an NE marker TTF-1. Scale bars in each panel are equal to 50 μm. (B) UMAP projection of expression profiles of 24 385 cells isolated from primary PCa and CRPC/SCNC samples. Dots represent single cells, colored by cell clusters. (C) Feature plots of cell type and lineage markers. Color represents expression level, from no expression (gray) to high level expression (dark blue). (D) UMAP view of cells colored by sample. (E) UMAP view of cells colored by lineage subtypes. Cell clusters were classified as basal, NE (in primary PCa), PSA-low and PSA-high luminal (in primary PCa), mCRPC with PSA-high, local CRPC with AR-high, SCNC, and lymphocyte. (F) UMAP view of tumor-adjacent tissue cell distribution among the cell clusters. (G) Distribution of cells isolated from tumor-adjacent tissue, PCa, and CRPC/SCNC samples among luminal clusters. AR = androgen receptor; CRPC = castration-resistant prostate cancer; GS = Gleason score; IHC = immunohistochemical; Lum = luminal; NE = neuroendocrine; PCa = prostate cancer; PSA = prostate-specific antigen; SCNC = small cell neuroendocrine carcinoma; UMAP = uniform manifold approximation and projection.
After removing clusters of leukocytes (C13 and C17), fibroblasts (C14), and endothelial cells (C16; Fig. 1C and Supplementary Fig. 4), we defined cellular lineage classes of each cluster using the expression of lineage markers and the origin of the cells (Fig. 1C and 1D, and Supplementary Fig. 5A). We identified multiple subpopulations of PSA-high luminal (C1, C3, C5, C7, and C15), PSA-low luminal (C6 and C11), and basal (C0 and C10) cells from primary PCa, as well as from mCRPC (C2), locally recurrent CRPC-adeno (AR-high, C12) or SCNC (C8), and basal cells from CRPC (C4 and C9; Fig. 1E). Interestingly, we found activation of multiple oncogenic signals in basal cells isolated from CRPC samples, indicating the potential for lineage plasticity in these cells (Supplementary Table 2).
We observed three PSA-high luminal clusters (C1, C5, and C7) that formed a separate cell population from other luminal clusters, and over 99% of cells in these clusters were from the PCa1 sample (Fig. 1D). Analyzing the status of gene fusion of ETS-family genes [19], we found that upregulated ERG expression in C1, C5, and C7 was associated with the positive TMPRSS2-ERG fusion signature (Fig. 1C and 2A, and Supplementary Fig. 3B and 5B) [20]. We confirmed that ERG was overexpressed using IHC analysis (Fig. 2C). We thus classified C1, C5, and C7 as ERG+ luminal clusters, and other luminal clusters as ERG− subtypes.
Fig. 2. Subtype-specific oncogenic signaling.
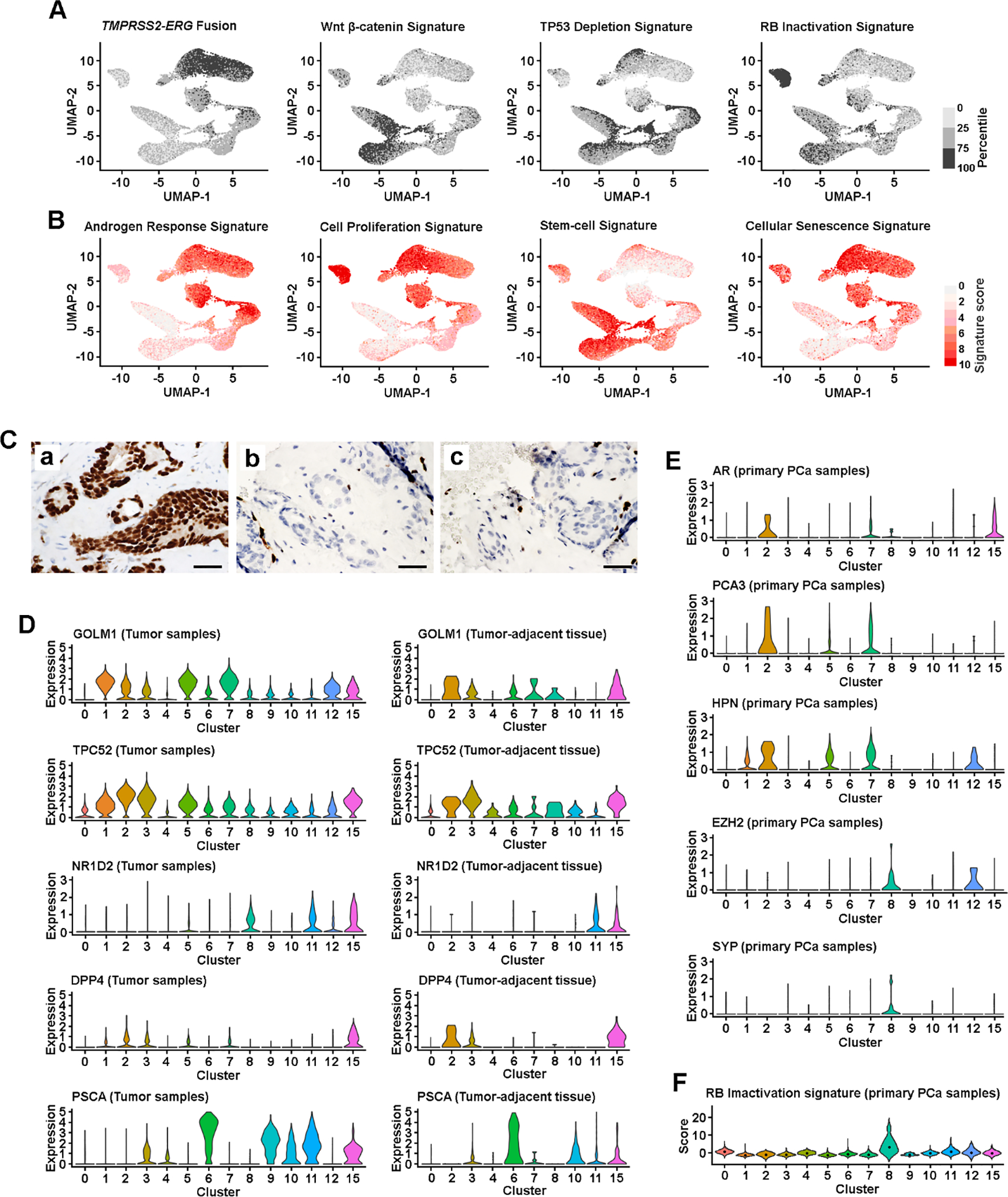
(A) UMAP view of cellular pathway signaling, colored by the quartiles of signature score. (B) UMAP view of cellular gene set signature scores. Color represents signature score level, from no expression (light gray) to high level expression (red). (C) Antibody-based IHC detection of ERG rearrangement: (a) PCa1 sample, (b) PCa2 sample, and (c) PCa3 sample. Scale bars in each panel are equal to 50 μm. (D) PCa markers defined subtype differences in tumor (both PCa and CRPC cells) and tumor-adjacent tissue. Violin plot visualized cellular gene expression within a cluster. Dot represents the median score level. (E) Lineage and PCa marker defined subtype differences among the cells isolated from primary PCa samples. (F) RB inactivation status among the cells isolated from primary PCa samples. Violin plot visualized the cellular signature score within a cluster. Dot represents the median score level. AR = androgen receptor; CRPC = castration-resistant prostate cancer; IHC = immunohistochemical; PCa = prostate cancer; SYP = synaptophysin.
Although the majority of the cells from tumor-adjacent tissue obtained from ERG− PCa2 and PCa3 cases were basal cells, 23% of these cells were luminal cells (Fig. 1F). Instead of forming distinct nonmalignant luminal clusters, luminal cells from tumor-adjacent tissue were mixed into each of the tumor cell clusters (Fig. 1G), suggesting that ERG− primary PCa shared similar patterns of cellular heterogeneity in tumor and adjacent tissue, while ERG+ luminal clusters tend to be patient specific.
3.2. Tumor cell heterogeneity is related to therapy resistance and disease progression
To identify potential functional differences among the cell clusters, we evaluated a wide series of RNA expression signatures using MSigDB that are upregulated in cancer including stem cell (n = 45), cellular proliferation (n = 36), and senescence (n = 7; Supplementary Table 3). We observed upregulated androgen response and proliferation signatures in PSA-high luminal clusters, compared with basal and PSA-low luminal clusters (Fig. 2B and Supplementary Fig. 3B). The higher proliferation in PSA-high luminal clusters was correlated with activated expression of TPD52 and GOLM1, oncogenes that promote PCa proliferation (Fig. 2D) [21]. The highest proliferation signatures were seen in cells from SCNC cluster (C8), consistent with the previous finding that SCNC cells are highly proliferative and extremely aggressive [10,12]. However, the PSA-low clusters (C6 and C11) are unlikely to be aged cells, as the lower proliferation was not correlated with upregulated senescence signatures (Fig. 2B and Supplementary Fig. 3B). Cells in cluster C11 possessed stem-cell features, and activated Wnt/β-catenin and EMT signaling (Fig. 2A and 2B, and Supplementary Fig. 3B). The TP53 deficient signaling [3] and upregulated NR1D2 expression [8] in C11 and C15 (Fig. 2A and 2D) suggested lineage plasticity in these two clusters.
A recent study of murine PCa identified two luminal populations (Dpp4+/Prom1+ and Psca+/Krt4+) that contributed to androgen-induced regeneration after castration [5]. A subtype of Psca+ luminal cells was also defined as a progenitor of PCa [6]. We found that the expression of both DPP4 and PSCA was highly enriched in C15, and upregulated PSCA in other luminal clusters (C3, C6, and C11) was progressively increased from tumor-adjacent tissue to PCa, indicating that each of these luminal cell subtypes may participate in tumorigenesis (Fig. 2D).
3.3. Critical signaling network of disease progression toward either CRPC-adeno or SCNC
We next studied potential correlations between intrinsic cellular heterogeneity and disease progression to CRPC/SCNC. Using the Monocle [16] reverse graph embedding approach, nonbasal CRPC/SCNC cells were decomposed into a trifurcated architecture of the cell trajectory, moving toward SCNC (C8), locally recurrent CRPC-adeno with AR-high (C12), and mCRPC with PSA-high clusters (C2; Fig. 3A). When both nonbasal PCa and CRPC cells were integrated into the evolutionary trajectory analysis, a bifurcated architecture of AR-dependent and AR-independent cell trajectory was observed, in which locally recurrent CRPC-adeno cells were located at the interphase on the branch progressing toward mCRPC cluster (Fig. 3B).
Fig. 3. Characterization of evolutionary trajectories and CRPC-like cells in primary PCa samples.
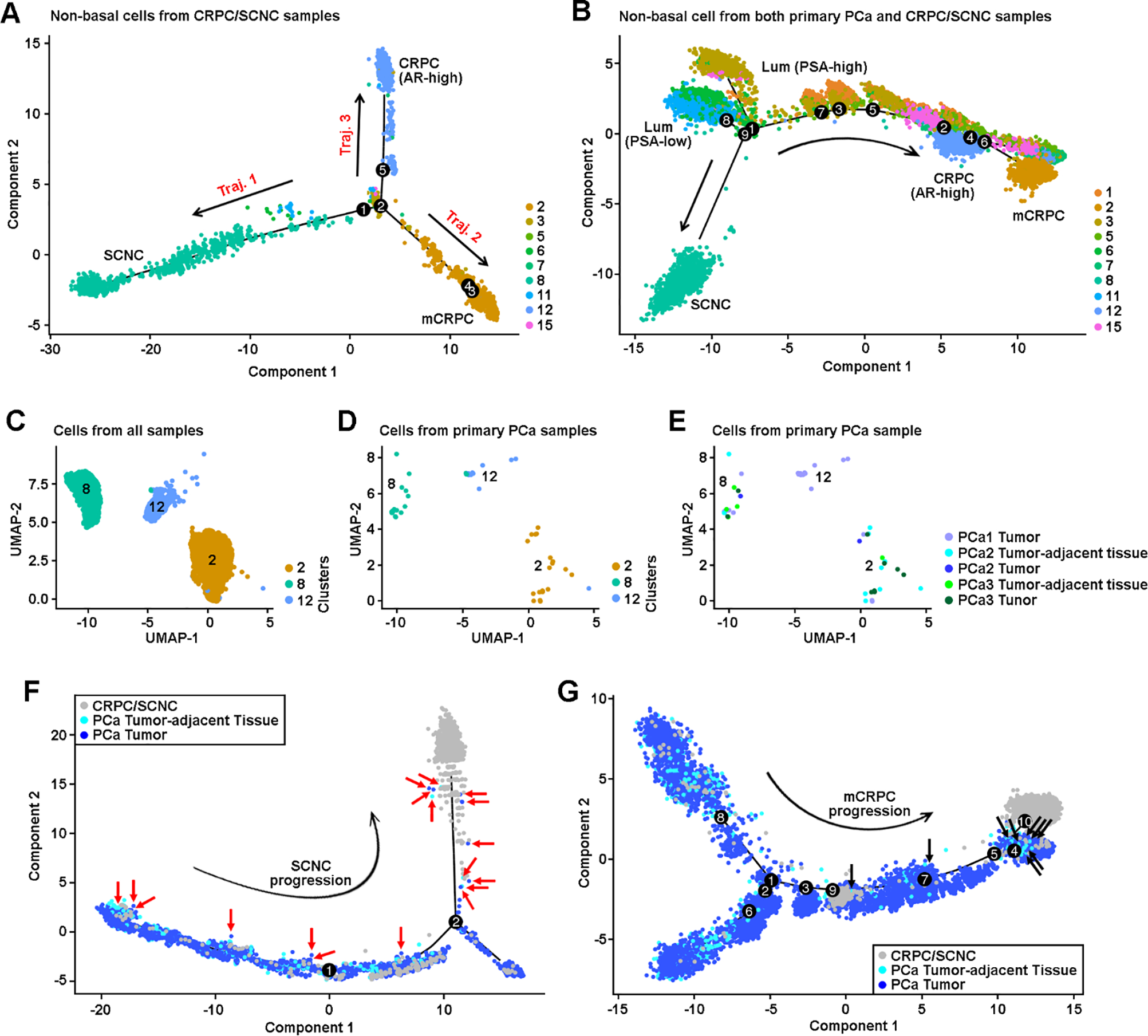
(A) Alignment of cells along trifurcating trajectories of CRPC progression. The trajectory analysis was performed using nonbasal clusters, and only cells isolated from CRPC samples were analyzed. Dots represent single cells. Solid lines represent distinct cell trajectories defined by single-cell transcriptomes. Color represents individual cell cluster. Arrow line represents trajectory direction. (B) Bifurcating trajectories of CRPC progression through AR-dependent and AR-independent mechanisms. Both PCa and CRPC cells from nonbasal clusters were included in this analysis. (C) UMAP view of cells in C2, C8, and C12 clusters isolated from both primary PCa and CRPC/SCNC samples, colored by cell clusters. (D) UMAP view of cells in C2, C8, and C12 clusters isolated from primary PCa samples, colored by cell clusters. (E) UMAP view of cells in C2, C8, and C12 clusters, isolated from primary PCa samples, colored by primary PCa tumor and tumor-adjacent tissue. (F) Distribution of NE cells on the trajectory of SCNC progression (trajectory 1), colored by samples. Nonbasal cells from both primary PCa and CRPC/SCNC samples were analyzed. Cells from mCRPC (C2) and CRPC-adeno (C12) clusters were excluded. We determined the NE cells by visualizing which C8 cells (Supplementary Fig. 7A) were isolated from primary PCa samples. Rad arrow indicates the NE cells. (G) Distribution of CRPC-like cells on the trajectory of mCRPC progression (trajectory 2), colored by samples. Nonbasal cells from both primary PCa and CRPC/SCNC samples were analyzed. Cells from SCNC (C8) and CRPC-adeno (C12) clusters were excluded. We determined the CRPC-like cells by visualizing which C2 or C12 cells (Supplementary Fig. 7C) were isolated from primary PCa samples. Black arrow indicates the CRPC-like cells. AR = androgen receptor; CRPC = castration-resistant prostate cancer; Lum = luminal; mCRPC = metastatic CRPC; NE = neuroendocrine; PCa = prostate cancer; SCNC = small cell neuroendocrine carcinoma; UMAP = uniform manifold approximation and projection.
To validate evolutionary trajectories, we performed receiver operating curve analyses to determine whether any of the clusters carries the CRPC/SCNC signature (Supplementary Table 4 and Supplementary Fig. 6A).
Hierarchical relationships among cell clusters were further assessed using genotype (SNP + mutation) and CNV patterns of the same CRPC/SCNC patients (Supplementary Fig. 6B–E). We found that a multipotent stem-like PSA-low luminal cluster (C11) gave rise to both treatment-emergent SCNC and CRPC-adeno, while progression of CRPC-adeno could also originate from multiple subpopulations including PSA-high luminal (C3 and C15) and PSA-low luminal cell (C6) clusters (Supplementary Fig. 6F).
By comparing the changes in relative gene expression over the five major dynamic processes (Supplementary Fig. 7 and 8), we revealed 1705 genes whose expression was associated with one or more trajectories (Supplementary Table 1) and identified 13 critical transcription factors that modulated progression to SCNC/CRPC (Supplementary Fig. 6G).
3.4. Presence of rare CRPC-like cells in primary PCa
By analyzing up to 7949 epithelial cells per patient sample (Supplementary Fig. 3C), we observed that a small number of primary PCa cells (52 cells) clustered into CRPC/SCNC clusters (C2, C8, and C12; Fig. 3C–E). Using the expression of well-established PCa markers, we confirmed that these were not misclassified noise cells (Fig. 2E). Based on positive expression of SYP and EZH2 [22] and inactivated RB1 signaling, we classified the primary PCa cells in the C8 cluster as NE cells (Fig. 2E and 2F). The primary PCa cells with upregulated expression of AR, HPN [23], and PCA3 [24] in CRPC-adeno clusters (C2 and C12) were defined as CRPC-like cells (Fig. 2E).
We found that NE cells from primary PCa were distributed on each state of NED trajectory toward SCNC (Fig. 3F), suggesting that the NE cells have the ability of self-renewal that can accelerate clonal expansion under the pressure of hormonal therapy. Unlike NE cells, 16 of 19 (84%) CRPC-like cells in cluster C2 resided next to the end state of mCRPC trajectory, indicating that these cells are fully progressed CRPC cells in primary PCa that will promote resistance to hormonal therapy (Fig. 3G).
To further determine the correlation between pre-exiting CRPC-like cells and castration resistance, we developed a novel signature called CRPCsig51 using 51 genes that were significantly upregulated in CRPC-like cells and associated with the CRPC/SCNC evolutionary trajectory (Supplementary Table 1 and Supplementary Fig. 9A and 9B). Using 897 PCa samples obtained from The Cancer Genome Atlas (TCGA) and multiple GEO data sets (Supplementary Table 5) [19, 25–28], we found that the CRPCsig51 signature was significantly upregulated in high grade, high stage, and metastatic PCa (Supplementary Fig. 10 and Supplementary Table 6). To assess the diagnostic ability of CRPCsig51 to predict survival, we used Youden’s index to dichotomize CRPCsig51 into low- and high-risk levels. A high CRPCsig51 score was associated with biochemical recurrence (GSE21034, p < 0.001) and progression (TCGA, p < 0.001; Fig. 4 and Supplementary Table 7). When modeled as a continuous score in Cox regression, CRPCsig51 remained a significant predictor of progression (clinical recurrence and distant metastasis, or biochemical recurrence) after adjusting clinical variables, such as Gleason grade, stage, and local and distant metastasis (Supplementary Table 7). Our results suggested that CRPC-like cells lead to disease progression independent of clinical variables of primary PCa.
Fig. 4. Prognosis of the CRPC-like cells related to CRPC/SCNC evolutionary signature (CRPCsig51).
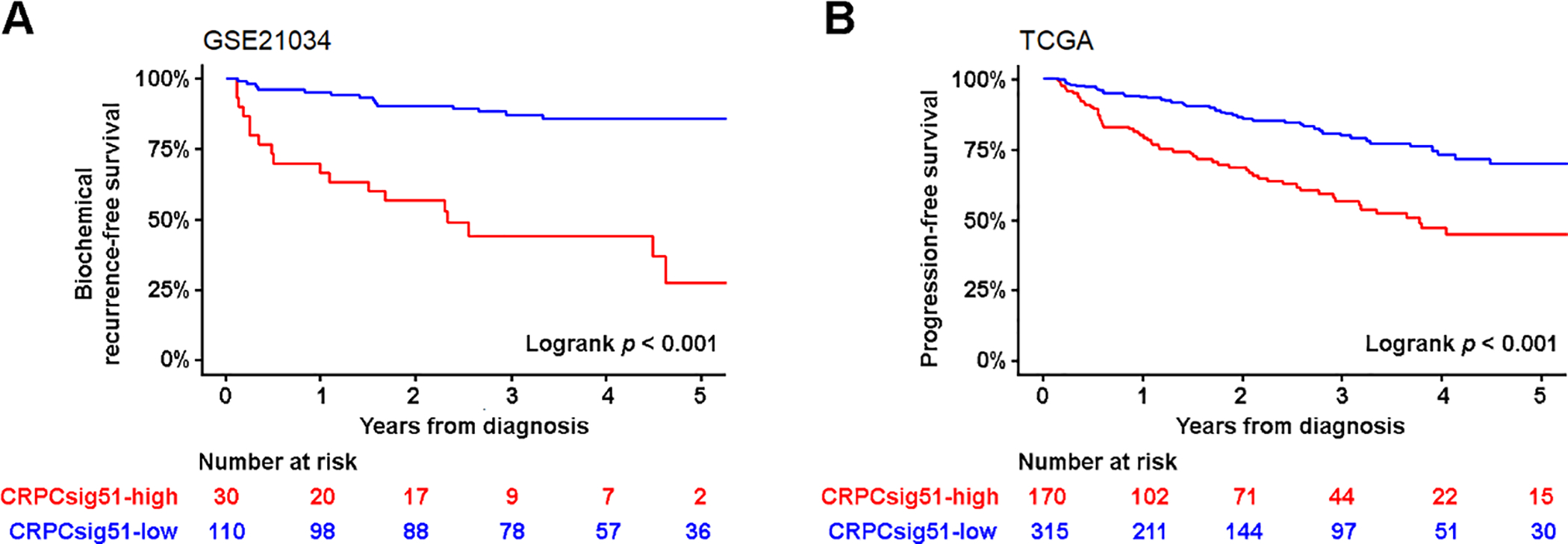
(A) Association between CRPCsig51 signature and biochemical recurrence (BCR)-free survival. Kaplan-Meier estimates of BCR-free survival in 140 PCa cases obtained from GSE21034 (p < 0.001, HR = 22.10, 95% CI: 8.70, 56.13). By assessing the diagnostic ability of CRPCsig51 for local and distant metastasis (N1 or M1, AJCC stage IV), Youden’s index was selected as the cut-point for CRPCsig51-high versus CRPCsig51-low samples. Red line represents CRPCsig51-high samples and blue line represents CRPCsig51-low samples. The p values were calculated using Mantel-Cox test. (B) CRPCsig51-high was significantly associated with a higher risk of PCa disease progression (both clinical recurrence and BCR). Kaplan-Meier estimates of progression-free survival in 485 PCa cases were obtained from TCGA (p < 0.001; HR = 3.10; 95% CI: 2.10, 4.58). AJCC = American Joint Committee on Cancer; CI = confidence interval; CRPC = castration-resistant prostate cancer; HR = hazard ratio; PCa = prostate cancer; SCNC = small cell neuroendocrine carcinoma; TCGA = The Cancer Genome Atlas.
3.5. Highly plastic CRPC-like cells were observed in multiple independent datasets
To assess the pre-existing CRPC-like cells, we reanalyzed cluster-specific markers using cells isolated from primary PCa samples and identified 16 markers of CRPC-like cells, including TOP2A [29], NUSAP1 [30], and PHGR1 (Supplementary Table 1 and Supplementary Fig. 11A). An IHC analysis revealed a small fraction of CRPC-like cells in hormone-sensitive PCa samples that are negative for NE marker (CHGA; Fig. 5A). CRPC-like cells were highly enriched in CRPC (5–40%) and mCRPC samples (10–80%), which is consistent with our results using multiple public datasets (Fig. 5B and Supplementary Fig. 11B).
Fig. 5. Distribution of CRPC-like cells in primary PCa and CRPC/mCRPC samples.
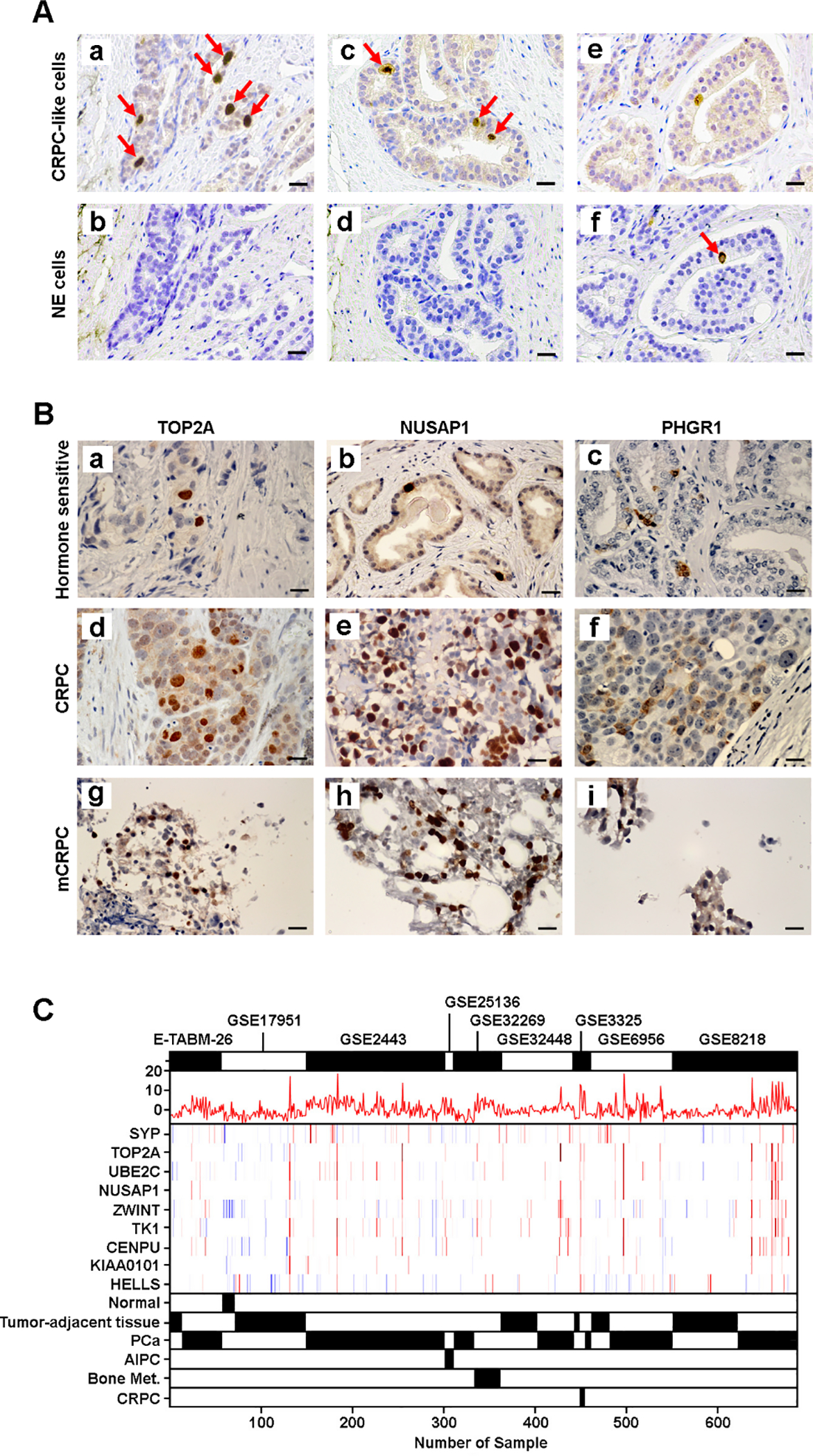
(A) Visualization of CRPC-like and NE cells in hormone-sensitive PCa samples. Representative IHC images of paired hormone-sensitive PCa slides staining with (a) TOP2A, (b) CHGA, (c) NUSAP1, (d) CHGA, (e) TOP2A, and (f) CHGA. CRPC-like cells are negative for NE marker, while NE cell is negative for CRPC-like markers. Red arrow indicates the positive staining cells. Scale bars in each panel are equal to 25 μm. (B) CRPC-like cells are highly enriched in CRPC and mCRPC samples. Representative IHC images of hormone-sensitive PCa staining with (a) TOP2A, (b) NUSAP1, and (c) PHGR1; CRPC staining with (d) TOP2A, (e) NUSAP1, and (f) PHGR1; and mCRPC staining with (g) TOP2A, (h) NUSAP1, and (i) PHGR1. Scale bars in each panel are equal to 25 μm. (C) Visualization of PCa samples with enriched CRPC-like cells in an independent dataset of 685 samples, using CRPCsig51 score, SYP expression, and the expression of eight CRPC-like cell markers. The combined dataset of 685 PCa samples was developed using nine datasets (E-TABM-26, GSE17951, GSE2443, GSE25136, GSE32269, GSE32448, GSE3325, GSE6956, and GSE8218) the gene expression profiles of which were measured using the Affymetrix U133A or U133 Plus 2.0 expression array. CRPC = castration-resistant prostate cancer; IHC = immunohistochemical; mCRPC = metastatic CRPC; Met. = metastasis; NE = neuroendocrine; PCa = prostate cancer.
We found that nine of 16 CRPC-like cell markers were upregulated in both NE and CRPC-adeno clusters (Supplementary Fig. 9C), and gene set enrichment analysis revealed that both FOXM1 [31] and SOX2 [4] signaling were activated in CRPC-like cells (Supplementary Fig. 9D). Unlike the cells from the CRPC-adeno sample, the C12 cells from primary PCa did not show overexpression AR, but had upregulated HPN [23] and EZH2 [22], indicating that these cells were in a stage of lineage plasticity (Supplementary Fig. 5C). We further validated the pre-existing CRPC-like cells using an independent cohort of 685 samples obtained from nine datasets (Supplementary Fig. 12), and distinguished between NE and CRPC-like cells using both CRPCsig51 score and SYP expression (Fig. 5C).
4. Discussion
We have identified a subset of multipotent stem-like PSA-low luminal cells (C11) in primary PCa that can give rise to both AR-dependent and AR-independent CRPC, while CRPC-adeno can also be derived from multiple subsets of luminal cells. We identified 13 critical transcription regulators (including SOX2 [4], AR, and FOXA1 [32]) that modulate the bifurcated disease progression toward SCNC or CRPC-adeno. This molecular interrogation and subtyping of PCa cells may guide more effective therapeutic strategies.
The discovery of pre-existing CRPC-like cells in early primary PCa and in tumor-adjacent tissue represents a novel castration-resistant mechanism different from the adaptation mechanism after androgen deprivation therapy (ADT) [33]. Although several studies combining mouse models and patient samples have previously reported the existence of CRPC-like cells during the PCa progression [34,35], isolation and characterization of these cells at single-cell resolution in hormone-naïve primary human PCa have not been reported. Unlike NE cells that have the capability of self-renewal and can accelerate clonal expansion under the pressure of hormonal therapy, we found that the CRPC-like cells in primary PCa are fully developed CRPC cells, with multiple upregulated PCa progression-related oncogenes and features of highly activated lineage plasticity. IHC stains revealed that CRPC-like cells were highly enriched in both locally recurrent CRPC and mCRPC, suggesting that CRPC-like cells that are present in early PCa may emerge during androgen deprivation as the dominant cell type.
Among the CRPC-like cell markers, TOP2A has been used to define an aggressive PCa subgroup with increased metastatic potential [29], and NUSAP1 expression was increased in recurrent PCa [30]. Both TOP2A and NUSAP1 were detected in a small fraction of isolated cells within primary PCa samples [30,36]. Although metastases often arise after long latency periods [37], early dissemination from the primary tumor can occur due to genetic diversity [38]. As CRPC-like cells share an identical transcriptome profile with CRPC/mCRPC, some of these cells could disseminate to distant sites prior to prostatectomy.
Using 1582 PCa and CRPC samples obtained from 14 datasets, we found that a subset of primary PCa samples is enriched with pre-exiting CRPC-like cells. To assess the prognosis of the CRPC-like cells, we developed a novel RNA expression signature (CRPCsig51) using a subset of specific markers of both NE and CRPC-like cells that were also associated with an evolutionary trajectory. We found that CRPCsig51 was significantly associated with high grade and distant metastatic PCa, and the predictive power of CRPCsig51 for biochemical recurrence was independent of clinical factors. Therefore, patients whose tumors have high CRPCsig51 scores likely harbor pre-existing NE and CRPC-like cells that may become rapidly resistance to ADT, for whom early aggressive interventions may be necessary. In contrast, patients whose tumors have low scores might be more appropriately managed by active surveillance. Future prospective studies are warranted to investigate this signature further.
4.1. Limitations
Although the number of cases in our scRNA-seq study is relatively small, they represent extremely rare and most precious fresh tumor samples, particularly because they cover the entire disease spectrum (primary PCa, CRPC-adeno, mCRPC, and SCNC). The genomic landscape of tumors can be elucidated from single tumor biopsy samples, when a large number of tumor cells are analyzed [39,40].
5. Conclusions
We found that a subset of PCa cells with intrinsic properties of castration resistance is present in hormone-naïve PCa before the initiation of ADT. These pre-existing castration-resistant cells can rapidly expand during ADT and lead to the development of CRPC, and should be sought out and targeted earlier in the evolution of PCa than they are presently.
Supplementary Material
Acknowledgments:
We thank Yanjing Li and members of the Biorepository and Precision Pathology Center at Duke University for assistance with tissue processing, and Duke Cancer Institute Flow Cytometry Shared Resource for performing FACS. We thank Helene Fradin Kirshner, Dan Somers, David Corcoran, and Hilmar Lapp from Duke Center for Genomic and Computational Biology, and Chaitanya R. Acharya from Department of Surgery for technical support.
Funding/Support and role of the sponsor:
This study was supported by National Institutes of Health (1R01CA205001, 1R01CA200853) and Prostate Cancer Foundation 2018 Movember Foundation – PCF Valor Challenge Award (principal investigator: Steven Patierno).
Footnotes
Financial disclosures: Jiaoti Huang certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Jiaoti Huang is a consultant for or owns shares in the following companies: Kingmed, MoreHealth, OptraScan, Genetron, Omnitura, Vetonco, York Biotechnology, Genecode, VIVA Biotech, and Sisu Pharma. None of these companies contributed to or directed any of the research reported in this article. The other authors declare that they have no competing interests.
Data sharing: The single-cell RNA-seq data have been deposited in NCBI’s SRA database and is accessible through SRA accession: PRJNA699369. Gene expression and clinical data of TCGA, GSE21034, GSE6919, GSE32269, GSE6752, E-TABM-26, GSE17951, GSE2443, GSE25136, GSE32269, GSE32448, GSE3325, GSE6956, and GSE8218 were obtained from TCGA (http://cancergenome.nih.gov/), Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and AEArrayExpress (https://www.ebi.ac.uk/arrayexpress/).
We discovered that rare tumor cells in primary prostate cancer possess genomic features of castration-resistant prostate cancer. This represents a novel resistance mechanism distinct from the widely held notion that prostate cancer becomes resistant to hormonal therapy through a therapy-induced adaptation mechanism.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA 2005;294:238–44 [DOI] [PubMed] [Google Scholar]
- [2].Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015;15:701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in. Science 2017;355:84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Karthaus WR, Hofree M, Choi D, et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 2020;368:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Guo W, Li L, He J, et al. Single-cell transcriptomics identifies a distinct luminal progenitor cell type in distal prostate invagination tips. Nat Genet 2020;52:908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen S, Zhu G, Yang Y, et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat Cell Biol 2021;23:87–98. [DOI] [PubMed] [Google Scholar]
- [8].He MX, Cuoco MS, Crowdis J, et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat Med 2021;27:426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Goldstein AS, Drake JM, Burnes DL, et al. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat Protoc 2011;6:656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Park JW, Lee JK, Sheu KM, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018;362:91–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li Y, He Y, Butler W, et al. Targeting cellular heterogeneity with CXCR2 blockade for the treatment of therapy-resistant prostate cancer. Sci Transl Med 2019;11:eaax0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aggarwal R, Huang J, Alumkal JJ, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol 2018;36:2492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heaton H, Talman AM, Knights A, et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Methods 2020;17:615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fan J, Lee HO, Lee S, et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res 2018;28:1217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].TCGA. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Setlur SR, Mertz KD, Hoshida Y, et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst 2008;100:815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature 2001;412:822–6. [DOI] [PubMed] [Google Scholar]
- [22].Zhang Y, Zheng D, Zhou T, et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun 2018;9:4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ma X, Guo J, Liu K, et al. Identification of a distinct luminal subgroup diagnosing and stratifying early stage prostate cancer by tissue-based single-cell RNA sequencing. Mol Cancer 2020;19:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Auprich M, Bjartell A, Chun FK, et al. Contemporary role of prostate cancer antigen 3 in the management of prostate cancer. Eur Urol 2011;60:1045–54. [DOI] [PubMed] [Google Scholar]
- [25].Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol 2004;22:2790–9. [DOI] [PubMed] [Google Scholar]
- [27].Chandran UR, Ma C, Dhir R, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 2007;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cai C, Wang H, He HH, et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest 2013;123:1109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Labbe DP, Sweeney CJ, Brown M, et al. TOP2A and EZH2 provide early detection of an aggressive prostate cancer subgroup. Clin Cancer Res 2017;23:7072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gulzar ZG, McKenney JK, Brooks JD. Increased expression of NuSAP in recurrent prostate cancer is mediated by E2F1. Oncogene 2013;32:70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aytes A, Mitrofanova A, Lefebvre C, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 2014;25:638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Parolia A, Cieslik M, Chu SC, et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019;571:413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zong Y, Goldstein AS. Adaptation or selection—mechanisms of castration-resistant prostate cancer. Nat Rev Urol 2013;10:90–8. [DOI] [PubMed] [Google Scholar]
- [34].Robinson JL, Hickey TE, Warren AY, et al. Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene 2014;33:5666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaushik AK, Shojaie A, Panzitt K, et al. Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat Commun 2016;7:11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Resende MF, Vieira S, Chinen LT, et al. Prognostication of prostate cancer based on TOP2A protein and gene assessment: TOP2A in prostate cancer. J Transl Med 2013;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shah SP, Morin RD, Khattra J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009;461:809–13. [DOI] [PubMed] [Google Scholar]
- [38].Klein CA. Selection and adaptation during metastatic cancer progression. Nature 2013;501:365–72. [DOI] [PubMed] [Google Scholar]
- [39].Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.