

Abstract
RORγt is a master regulator of Th17 cells. Despite evidence linking RORγt deficiency/inhibition with metastatic thymic T cell lymphomas, the role of RORγt in lymphoma metabolism is unknown. Chronic alcohol consumption plays a causal role in many human cancers. The risk of T cell lymphoma remains unclear in humans with alcohol use disorders (AUD) after chronic RORγt inhibition. Here we demonstrated that alcohol consumption accelerates RORγt deficiency-induced lymphomagenesis. Loss of RORγt signaling in the thymus promotes aerobic glycolysis and glutaminolysis and increases allocation of glutamine carbon into lipids. Importantly, alcohol consumption results in a shift from aerobic glycolysis to glutaminolysis. Both RORγt deficiency- and alcohol-induced metabolic alterations are mediated by c-Myc, as silencing of c-Myc decreases the effects of alcohol consumption and RORγt deficiency on glutaminolysis, biosynthesis, and tumor growth in vivo. The ethanol-mediated c-Myc activation coupled with increased glutaminolysis underscore the critical role of RORγt-Myc signaling and translation in lymphoma.
Keywords: Thymus, lymphomagenesis, glycolysis, glutaminolysis, c-Myc
Introduction
Retinoic acid receptor-related orphan receptor γT (RORγt), encoded by Rorc gene, was identified as a lineage-specific transcription factor for Th17 cells[1]. The expression of RORγt is essential for the survival of double-positive (DP, CD4+ CD8+) cells in the thymus and its downregulation allows the maturation of CD4 single-positive (SP) and CD8 SP thymocytes[2]. Mice deficient in RORγt (referred to as Rorc−/− hereafter), have a shorter life expectancy, with 50% of Rorc−/− mice dying within 4 months due to metastasizing thymic T cell lymphomas[3]. RORγt antagonists are currently being developed to treat autoimmunity by blocking Th17 cell development and related cytokine production. However, it was recently reported that RORγt inhibitors recapitulate thymic aberrations seen in Rorc−/− mice[4, 5]. Furthermore, induction of RORγt deficiency in adult mice also leads to the development of thymic lymphoblastic lymphoma[6]. Thymic development of T cells, particularly the early stages, appears to be highly sensitive to metabolic perturbations[7], [8, 9]. However, to date the role of RORγt in thymic metabolism and lymphoma metabolism has remained unclear.
Alcohol use disorders (AUD) have damaging effects leading to fatty liver, diabetes, and certain cancers[10, 11]. However, the molecular mechanisms of alcohol-associated cancers remain poorly understood. Ethanol is mainly metabolized in hepatocytes by cytosolic alcohol dehydrogenase (Adh) to generate acetaldehyde, which is subsequently metabolized by mitochondrial acetaldehyde dehydrogenase 2 (Aldh2) to produce acetate[12]. Acetate is further converted to Acetyl-CoA by acetyl-CoA synthetase (Acss), where it enters the citric cycle or serves as a precursor for the biosynthesis of fatty acids. Thus, alcohol may directly and indirectly impact the aspects of lipid flux that ultimately leads to lipid accumulation, which appears to confer biosynthetic/bioenergetic advantages to proliferating tumor cells. One of the primary metabolic changes observed in thymic T cells or malignant T cells is increased catabolic glucose metabolism to use glucose as a carbon source for their increased biosynthetic demands[7, 13]. However, it has been reported that alcohol inhibits gluconeogenesis in the liver, which should decrease glucose concentrations[14]. Therefore, under alcohol consumption condition, highly proliferative cancer cells may require additional supplies of biosynthetic precursors not met by glucose metabolism, such as glutaminolysis and/or pyruvate carboxylation as well as fatty acids[15, 16]. Indeed, glutamine metabolism is upregulated by many oncogenic insults and mutations and glutaminolysis is found as a major component of Myc-driven oncogenesis in most settings[17]. T-cell acute lymphoblastic leukemias (T-ALL) have a high glucose metabolism[13] and c-Myc[18], mTORC1[19] and HIF1α[20] are important for their development. One important question is whether or how alcoholism regulates the metabolic reprogramming of malignant T cells?
Our recent report has found that RORγt deficiency reduces Th17 cells accumulation and ameliorates gut inflammation and liver steatosis in alcohol-associated liver disease (ALD)[21]. However, to our surprise, alcohol consumption was also associated with high incidences of thymic T cell lymphomas in Rorc−/− mice. In this study, we demonstrated that loss of RORγt signaling cooperates with c-Myc to induce alterations in glucose and glutamine metabolism, that accelerates lymphomagenesis. Interestingly, alcohol consumption results in inhibition of aerobic glycolysis and upregulation of glutaminolysis by suppression of HIF1α expression and increase of c-Myc expression, respectively. This metabolic change in Rorc−/− T lymphoma is characterized by inhibition of carbon flow toward lactate and increased flux of glutamine intermediates toward lipid biosynthesis after alcohol consumption. Induction of this metabolic shift is dependent of c-Myc, as silencing of c-Myc by shRNA reduces the effects of Rorc loss and alcohol consumption on glutaminolysis, biosynthesis, and tumor growth in vivo. Our findings indicate that RORγt is a metabolic sensor essential for the coordination of metabolic activities that support cell growth and proliferation in T lymphoma.
Materials and Methods
Animals and treatments
C57BL/6 and Rorc−/− mice were obtained from Jackson Laboratory. All animal procedures were approved by the University of Louisville Institutional Animal Care and Use Committee. For metabolic analysis of thymocytes, age and sex matched WT littermates were employed as controls for Rorc−/− mice in all of experiments. Rorc−/− mice without leukemogenesis were used for thymocytes analysis at the age of 5 weeks. Since RORγt is exclusively present in immature DP thymocytes[22], DP thymocytes were sorted and compared in both genotypes of mice in some experiments. The binge-on-chronic NIAAA (Gao) model with 5-week-old mice was used. Briefly, male or female mice were acclimated to the Lieber-DeCarli liquid control diet (F1259SP; Bio-Serv, Flemington, NJ) or gradually introduced to and increased on the ethanol diet (5% ethanol-w/v; F1258SP; Bio-Serv) for 3 days followed by further feeding with the liquid control (pair feeding, PF) or ethanol diet (alcohol feeding, AF) for additional 10 days (RNA-Seq and stable-isotope tracing experiments). For induction of thymic lymphoma, starting 1 month after 5% alcohol administration, Rorc−/− mice were received 3% ethanol diet for 10 days, followed by a 5-day rest period with liquid control (PF) diet, then another 10-day cycle of 3% ethanol diet by a second 5-day rest period and, subsequently, a cycle of 15-days of ethanol diet and resting continued until the termination of the experiment. The mice were monitor and sacrificed between 90–120 days.
Details of other methods used in this study are described in the Supplemental Materials and Methods.
Results
Alcohol consumption accelerates RORγt deficiency-driven lymphomagenesis
To better understand how changes in alcohol metabolism might contribute to the development of T lymphoma, we fed an alcohol (alcohol feeding [AF]) or an isocaloric control (pair feeding [PF]) diet to male and female Rorc−/− mice. Compared to control diet-fed (PF) Rorc−/− mice, alcohol-fed (AF) Rorc−/− mice displayed accelerated thymic lymphomagenesis with a median tumor onset of 7 weeks (Figures 1A–1B, and Figures S1A–S1B). 50% of the Rorc−/− mice had become moribund or died with thymic lymphoma by 80 days of age when under an alcohol-fed condition compared to 125 days of age when under the pair-fed condition (Figure 1A). Massive metastases to the lung (Figure 1C) and liver (data not shown) were observed in alcohol-fed Rorc−/− mice. In situ, the proportion of Ki67+ proliferating thymic lymphoma was significantly increased in AF mice compared to PF mice (Figure 1D). Proliferating T cells/thymomas require fatty acids for lipid and membrane synthesis. In accordance with proliferation, thymic lymphomas displayed increased neutral lipid staining in situ (Figure 1E) and contained higher levels of triglyceride (TG) (Figure 1F) in AF Rorc−/− mice compared to PF Rorc−/− mice. Alcohol treatment also led to a significant increase in the expression of Aldh2 and Acss1 and other alcoholism-related enzymes in the thymus of WT mice, which were further up-regulated in Rorc−/− mice (Figure 1G and Figure S1C). We next generated stable RORγt deficient thymic lymphoma lines from Rorc−/− mice (named Rorc−/− lymphoma)[3, 5]. RORγt was then ectopically expressed in these cell lines using a retrovirus bearing RORγt-GFP (named RorcRES lymphoma, RES: Rorc rescued). The levels of acetyl-CoA and ATP were measured in RorcRES and Rorc−/− lymphomas with/without alcohol treatment. Interestingly, the production of acetyl-CoA and the level of intracellular ATP was increased in resting Rorc−/− lymphoma compared to RorcRES lymphoma (Figure 1H). A much greater increase in ATP was observed in ethanol-treated Rorc−/− lymphoma (Figure 1H). Furthermore, we used retrovirus to knock down (KD) to inhibit RORγt expression in T lymphoma EL4 cells (named ShRorc) and exposed the EL4 cells to ethanol or its metabolite acetate. Rorc KD led to a significant increase in acetyl-CoA and free fatty acids production, particularly under alcohol culture conditions (Figure S1D). In addition, alcohol stimulation significantly upregulated carnitine palmitoyltransferase 1 (Cpt1a and Cpt1c) in EL4 cells, particularly in EL4 cells with Rorc KD (Figure 1I). Activation of acetate to acetyl-CoA can occur via enzymes ACSS1 in mitochondria or ACSS2 in the cytoplasm, respectively. We also observed that oxygen consumption rate (OCR) and ATP production were significantly enhanced by acetate treatment in EL4 cells, even more in EL4 cells with Rorc KD (Figure S1E). These results indicate that alcohol accelerates RORγt-related acetyl-CoA metabolism in lymphomas. Consistently, EL4 cells exposed to alcohol up-regulated OCR at both the baseline and maximal respiratory capacity as compared with the control (Figure 1J), and OCR were significantly enhanced by RORγt inhibitor MRL871 (Figure 1J). Similarly, exposure of the Rorc−/− lymphoma to alcohol also made the cells adopt a more energetic phenotype than the RorcRES lymphomas (Figure S1F). Taken together, these data suggest that alcohol metabolism may be promoting the lipogenesis in RORγt deficient lymphoma.
Figure 1. Impact of alcohol consumption on the development of RORγt deficiency-driven thymic lymphoma.

(A) Survival curves of Rorc−/− mice fed control (pair-fed, PF) or alcohol (alcohol-fed, AF) diet. The percentage of mice surviving versus the mice alive at the onset is plotted against age.
(B) Representative picture of thymic tumors observed at day 75 and a graph showing tumor volume. Mean ± SEM (A-B); n = 24, **p < 0.01.
(C) H&E-staining of lung sections and histologic fold increase for metastatic lesions.
D) Sections of Rorc−/− thymic lymphomas were examined by Ki67 (red) and CD8 (green) staining and a graph showing the percent of Ki67+ cells as a total CD8+ cells. Mean ± SEM, ***p < 0.001.
(E) Neutral lipid contents in sections of Rorc−/− thymic lymphomas were examined by BODIPY 493/503 (green) and CD8 (red) staining.
(F) The amount of triglyceride in the Rorc−/− thymic lymphomas.
(G) Real-time PCR analysis of Aldh1b1, Aldh2 and Acss1 mRNA in thymocytes from WT and Rorc−/− mice fed control (PF) or alcohol (AF) diet. Mean ± SEM; n = 8, *p < 0.05, **p < 0.01.
(H) Rorc−/− thymic lymphoma-derived cell lines (Rorc−/−) without or with Rorc overexpression (RorcRES) treated with 75 mM ethanol (EtOH) for 6 h. Production of intracellular Acetyl-CoA and ATP were examined. Mean ± SEM, n = 5, *p < 0.05, **p < 0.01.
(I) EL4 cells without (ShCtr) or with Rorc KD (ShRorc) treated with ethanol (75 mM) for 6 h. Real-time PCR analysis of the expression of indicated genes. Mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001.
(J) DMSO (Control)- or RORγt inhibitor MRL871 (20μM,36 h)-treated EL4 cells cultured for 72 h in the presence of PBS or ethanol (75 mM) and then the OCR of cells was measured by seahorse and in response to oligomycin (Oligo), FCCP, and rotenone + antimycin (Rot+Ant). Data represent three experiments shown as mean ± SEM.
Next, we determined thymocyte-intrinsic consequences of RORγt deficiency under alcohol consumption in vivo. WT and Rorc−/− mice were fed with/without alcohol over a 10-day periods. RNA sequenced from thymocytes of these four groups was used to compare transcriptomes. Analysis of differentially expressed genes (DEGs) showed that alcohol feeding under RORγt deficiency induced a combination of the ‘alcohol consumption’- and ‘RORγt’-induced genes (Figure 2A). We then performed KEGG enrichment analysis to determine whether an a priori defined set of genes showed statistically significant and concordant differences between the Rorc−/− and WT thymocytes with/without under alcohol consumption conditions. The expression of genes involved in the oncogenic and metabolic pathways, such as OXPHOS, glycolysis and the TCA cycle were significantly enriched (Figure 2B). Genes associated with RORγt deficiency under alcohol consumption conditions were predominantly driven by glycolysis and glutamate metabolism genes (Figures S2A–S2C). GSEA analysis suggested that shared gene clusters between the Rorc−/− and WT thymocytes were markedly associated with metabolic changes and oncogenic pathways including c-Myc, mTOR (mTORC1) and OXPHOS signaling (Figure 2C). The representative GSEA enrichment plots of OXPHOS, mTORC1 signaling, Myc targets, and glycolysis targets are shown in Figure 2C. Indeed, Myc proto-oncogene, which controls glycolysis and glutaminolysis[23], that was highly overexpressed in Rorc−/− thymocytes and elevated under alcohol treatment (Figure 2D). Increased c-Myc protein abundance may be caused by either increased c-Myc production or decreased decay. c-Myc protein stability can be regulated by two phosphorylation sites with opposing functions: serine 62 phosphorylation (pS62) stabilizes c-Myc whereas threonine 58 phosphorylation (pT58) promotes c-Myc degradation[24, 25]. We therefore tested the role of RORγt deficiency or prolonged ethanol treatment on the stabilization of c-Myc protein by examining the phosphorylation status of c-Myc at T58 or S62. Our data showed that the pS62/pT58 ratios of c-Myc is similar in thymocytes between WT and Rorc−/− mice (Figures S2D–S2E). Ethanol treatment also has no effect on the stabilization of c-Myc protein in EL4 with/without RORγt inhibition (Figures S2F–S2G). Taken together, mTORC1/c-Myc signaling components might be critical regulators for OXPHOS activity because these oncogenic pathways are directly linked to metabolic reprogramming in cancer cells[26, 27].
Figure 2. Transcriptomic profiling identifies metabolic reprogramming as a hallmark of RORγt deficiency and alcohol consumption.
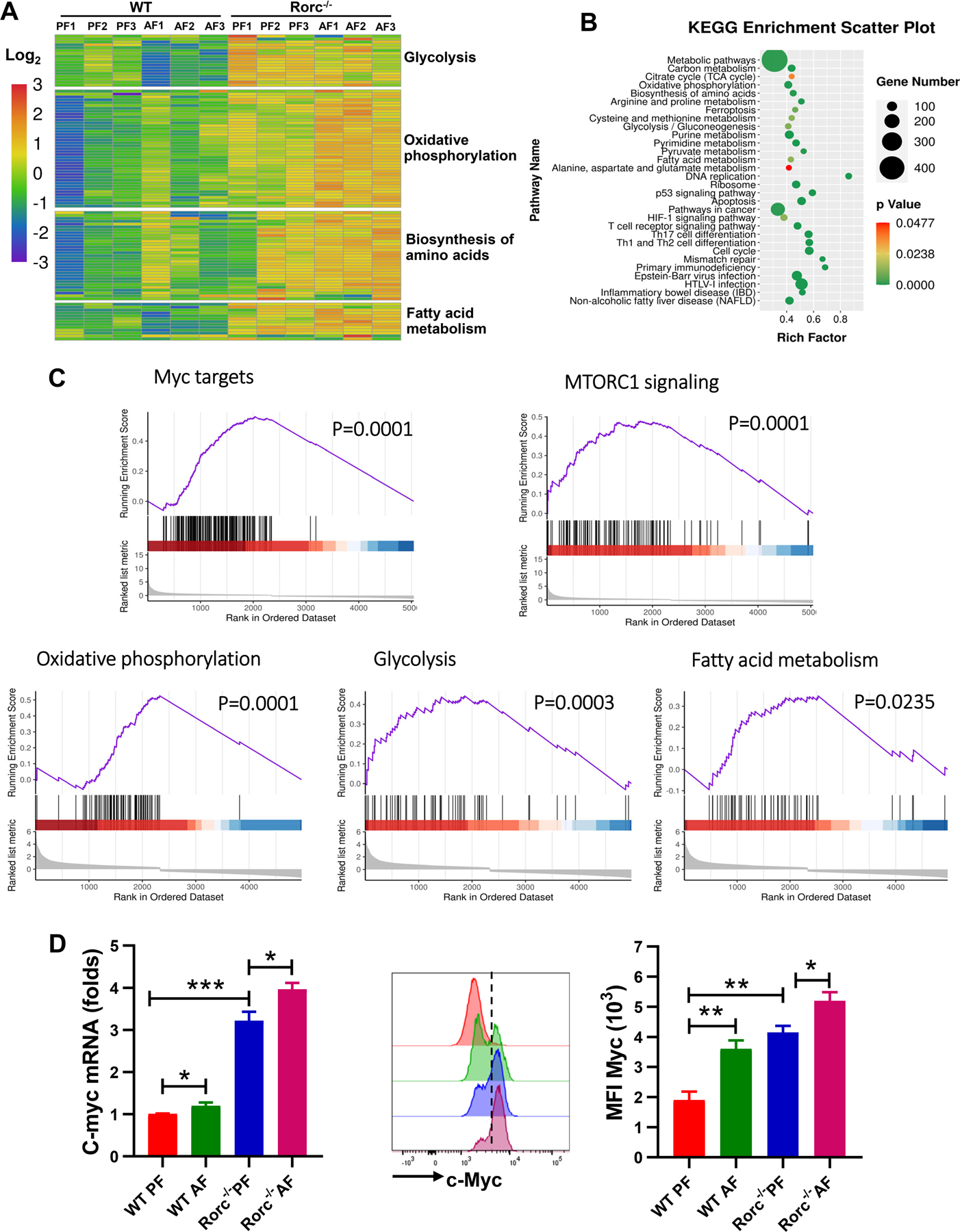
5–6 weeks old WT littermates and Rorc−/− mice were fed the control (PF) or alcohol (AF) diet for 10 days. Thymocytes were analyzed by RNA-sequence.
(A) Heatmap and unsupervised hierarchical clustering of the average expression of DEGs from pairwise comparisons (log2(fold-change) > 2, adjusted P < 0.05).
(B) KEGG enrichment analysis of genes defining each pathway is represented in the accompanying bubble plot.
(C) Representative enrichment plots for the hallmark OXPHOS pathway, mTORC1 signaling, and c-Myc targets and glycolysis pathways. The black vertical line at the bottom shows where the members of the gene set appear in the ranked list of genes. The graded red to blue bars on the x axis represents the DESeq2 statistical values (Rorc−/− PF mice versus WT PF mice), with red and blue denoting up-regulation and down-regulation, respectively.
(D) Real-time PCR (left) and FACS (right) analysis of the expression of c-Myc in thymocytes. Histogram shows the mean fluorescence intensity (MFI) of c-Myc. Mean ± SEM; n =8, *p < 0.05, **p < 0.01.
RORγt deficiency enhances mitochondrial activity and lipid storage and metabolism in the thymus
Given the central role of mitochondria in cellular metabolism, we assessed their activity as a way to determine if RORγt deficiency switches metabolic programming during thymocyte development. As previous report[2], there is a marked loss in thymic cellularity in the Rorc−/− mice that is characterized by a particularly striking loss in double positive (DP) thymocytes (Figure S3A). Notably, the size of Rorc−/− thymocytes were increased in the steady state (Figure 3A). An increase in cell size is an indicator of increased cell metabolism[28]. We found that Rorc−/− DP and CD8+ SP thymocytes displayed increased MitoTracker (Figure 3A, and Figure S3B) and tetramethylrhodamine ethyl ester (TMRE) staining (Figure 3A, and Figure S3C), indicating higher mitochondrial mass (normalized to cell size) and mitochondrial membrane potential (ΔΨm), respectively. Moreover, confocal analysis of Rorc−/− thymocytes stained with either MitoTracker or TMRE also revealed large and active mitochondria compared to WT thymocytes (Figure 3B). These data suggest that RORγt mainly regulates mitochondria activity on DP thymocytes before their positive selection, which is consistent with that RORγt is exclusively present in immature DP thymocytes[22]. Therefore, DP thymocytes in mice without leukemogenesis were sorted and compared in both genotypes of mice for the metabolic analysis. We first measured the mRNA levels of key mitochondrial and OXPHOS-related genes in DP thymocytes and found the expression of Cox6a1, Cox6a2, Cox15, Ndufa10, Ndufab1 and P32, which orchestrates mitochondrial DNA transcription, is enriched in Rorc−/− DP thymocytes (Figure 3C). Furthermore, Seahorse extracellular flux analysis showed that Rorc−/− DP thymocytes have both higher basal OCR and ATP production than those of WT DP thymocytes (Figure 3D). These data highlight mitochondrial and metabolic features that are associated with RORγt deficiency in the thymus. Using real-time PCR analysis, RORγt deficiency significantly upregulated thymocyte expression of key fatty acid (FA) synthesis genes- Fasn, Acc1 and the FA elongases Elovl5 and Elovl6, as well as Cpt1a (Figure 3E). Consistent with these, Rorc−/− thymocytes had a higher neutral lipid content (assessed by BODIPY493/503 staining) than WT thymocytes (Figure 3F). This differential lipid content was further increased upon alcohol stimulation in vivo (Figure S3D). When incubated with the long-chain FA oleate, Rorc−/− thymocytes exhibited an increase in BODIPY staining (Figure S3E–S3F). Lipid droplets store neutral lipids including triglycerides (TGs) and cholesterol esters. Rorc−/− thymocytes have higher TG content (Figure 3G) and free cholesterol, as determined by filipin III staining (Figure 3F). We next checked lipid uptake in Rorc−/− thymocytes, which could account for lipid storage. Using fluorescently labeled palmitate (BODIPY-FL-C16) and cholesterol ester (BODIPY CholEsteryl FL-C12), we found that Rorc−/− thymocytes displayed higher ability to take up fatty acids (Figure 3F and Figure S3G), but not cholesterol (Figure S3H). Corresponding to the upregulation of these lipid genes, FA oxidation stress test showed that Rorc−/− DP thymocytes acquired an elevated OCR, indicating increased oxidative phosphorylation, which was significantly suppressed by etomoxir, a CPT1a inhibitor (Figure 3H).
Figure 3. RORγt regulates mitochondrial and metabolic phenotypes of thymocytes. Thymocytes were analyzed from 5–6 weeks old male and female WT littermates and Rorc−/− mice.
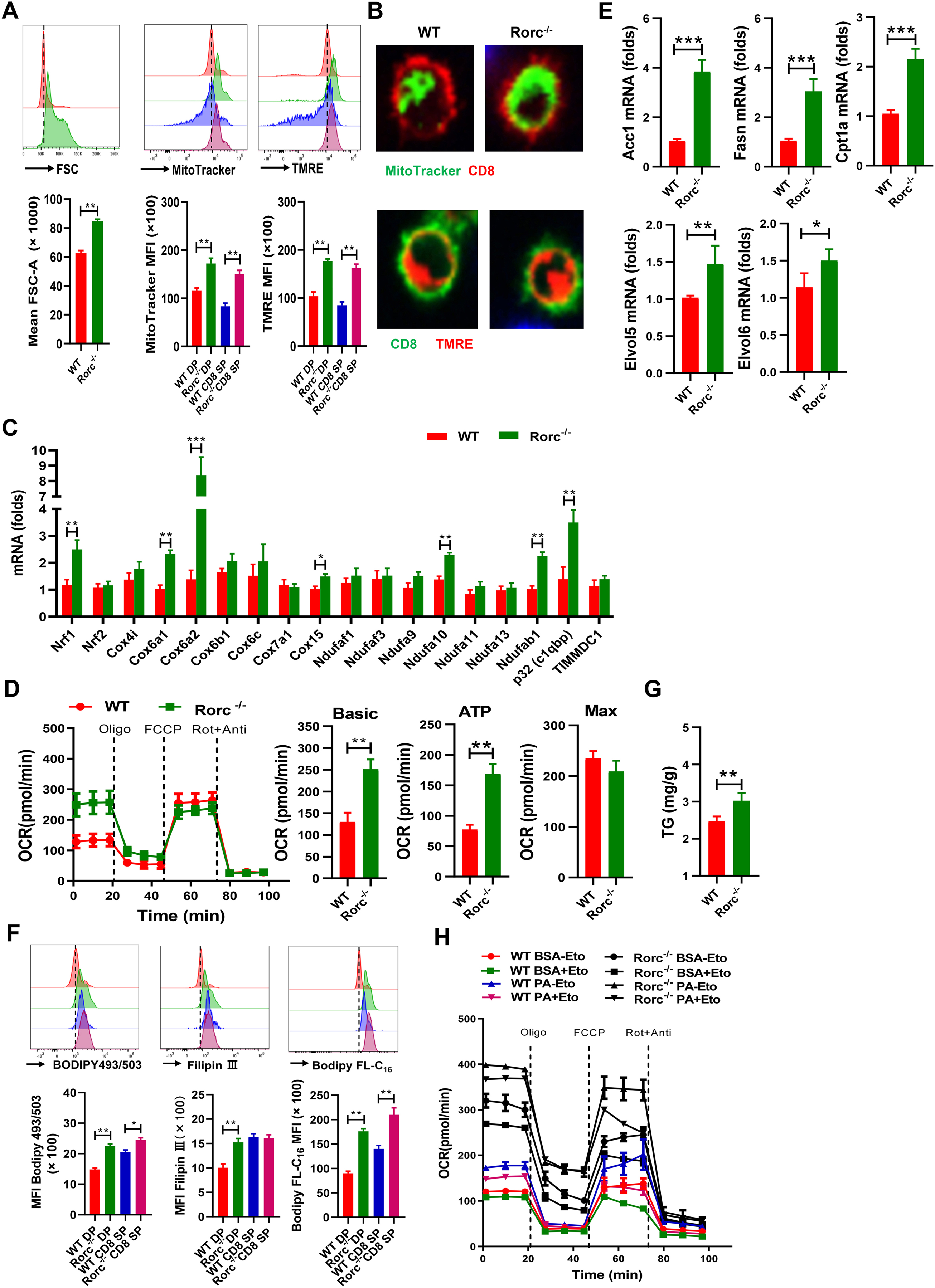
(A) Representative FACS plots (top) and summary graphs (bottom) of the MFI of cell size, MitoTracker and TMRE in thymocytes from WT and Rorc−/− mice. SP: single positive. DP: double positive. Mean ± SEM; n = 6, **p < 0.01.
(B) Representative confocal images of thymocytes stained with MitoTracker (green) and TMRE (red).
(C) Real-time PCR analysis of OXPHOS-related genes. DP thymocytes in mice without leukemogenesis were sorted and compared in both genotypes of mice for all of metabolic analysis unless notice. Mean ± SEM; n =8, *p < 0.05, **p < 0.01.
(D) OCR of fresh thymocytes from WT and Rorc−/− mice was measured by seahorse in real time. Histograms show basal OCR and ATP. Data are representative of 3 independent experiments. Mean ± SEM; **p < 0.01.
(E) Real-time PCR analysis of the genes related to lipid metabolism, Mean ± SEM; n =8, *p < 0.05, **p < 0.01.
(F-G) Representative plots (top) and summary graphs (bottom) of the MFI of BODIPY 493/503 staining, Filipin III staining or BODIPY-FL-C16 uptake, or the level of triglycerides (G) in thymocytes from WT and Rorc−/− mice. Mean ± SEM; n =6, *p < 0.05, **p < 0.01.
(H) OCR of thymocytes from WT and Rorc−/− mice was measured by seahorse under FA oxidation stress test conditions and in response to the indicated drugs. Eto: etomoxir. Data are representative of 3 independent experiments. Mean ± SEM.
Because RORγt is highly expressed in DP thymocytes, we next determined whether the changes in metabolic activity of DP thymocytes in Rorc−/− mice was a result of T cell extrinsic or intrinsic effects by bone marrow (BM) chimeras--transferring BM from Rorc−/− (CD45.2+) mice into WT recipients (CD45.1+) (KO →WT). WT BM (CD45.2+) were also transferred to WT (CD45.1+) recipient (WT →WT) as control. We found that DP thymocytes were significantly reduced in the KO →WT mice compared to that of WT →WT mice eight weeks after reconstitution (Figure S4A). Furthermore, DP thymocytes from KO →WT mice also acquired an elevated OCR (Figure S4B). Thus, the paucity of DP thymocytes and the mitochondrial dynamics in Rorc−/− mice were primarily due to T cell intrinsic effects.
Loss of RORγt in thymocytes induces aerobic glycolysis that is suppressed by alcohol consumption
Our RNA-seq analysis indicates that the genes involved in glycolysis and mitochondrial respiration in thymocytes are significantly enriched. We used real-time PCR to confirm that RORγt-deficient thymocytes displayed an upregulation of mRNAs encoding metabolic enzymes involved in glycolysis (Figure 4A) and the pentose phosphate pathway (PPP) (Figure S5A) including increased mRNA expression of Hk2, Gpi1, Tpi-1, Pgam1, Eno-1, Ldhα and Aldolase A (Aldoa) (Figure 4B). Consistent with the increase, the level of pyruvate dehydrogenase kinase 1 (Pdk1), which curbs pyruvate entry into the TCA cycle by antagonizing the action of the pyruvate dehydrogenase (Pdh) complex[29], significantly decreased in RORγt-deficient thymocytes (Figure 4B). A similar pattern of gene expression also displayed in Rorc−/− lymphomas when compared to RorcRES lymphoma cells (Figure S5B). Immunoblot analysis further confirmed the elevated HK2 and LDHα protein levels were detected in RORγt-deficient thymocytes (Figure 4C). We next compared the expression of genes involved in glycolytic regulation in thymocytes from mice that had been fed with the control or alcohol diet for 10 days prior to isolation. Interestingly, real-time PCR analysis revealed that alcohol treatment significantly downregulates the levels of the genes encoding glycolytic enzymes, but not glucose transporter glut1, in WT mice (Figure 4D, and Figure S5C). There was no difference in glucose uptake in thymocytes between PF mice and AF mice (data not shown). Of not, alcohol exposure markedly reduces the expression of HIF1α, which regulates the transcription of genes encoding glucose transporters and glycolytic enzymes, in both types of mice (Figure 4D) or in EL4 cells (Figure S5D), suggesting that alcohol inhibits the genes involved in aerobic glycolysis. To confirm the above findings, we conducted a glycolysis stress test on thymocytes from WT and Rorc−/− mice. Measurement of the extracellular acidification rate (ECAR) revealed that RORγt deficiency increased glycolysis in thymocytes in the steady state. However, alcohol consumption significantly decreased the glycolysis in thymocytes from both types of mice (Figure 4E). Notably, regulation of thymocyte glycolysis by RORγt is also T cells-intrinsic and inhibition of glycolysis by alcohol consumption is independent on RORγt, as evidenced by bone marrow chimeras (Figure S5E). We next performed in vivo 13C6-D-glucose tracing experiment in WT and Rorc−/− mice fed with/without alcohol over 10 days. The thymocyte extracts were analyzed by GC-MS to quantify glucose-derived 13C isotopologues of lactate (lactate with different numbers of 13C atoms). Deletion of RORγt resulted in an increase of glucose-derived 13C-pyruvate (m+1, m+2, m+3) and 13C-lactate production (m+1, m+2, m+3) (Figure 4F). However, under alcoholic conditions, the production of isotopologue of pyruvate (m+1, m+2, m+3), lactate (m+2, m+3) and total G6P were significantly decreased in both types of mice (Figure 4F). Thus, although RORγt deficiency increases aerobic glycolysis, alcohol feeding inhibits the aerobic glycolysis in thymocytes from both types of mice. These data are consistent with the recent finding that the alcohol metabolite acetaldehyde inhibits glucose metabolism in T cells by inhibiting aerobic glycolysis-related signaling pathways[30].
Figure 4. RORγt and alcohol consumption regulate aerobic glycolysis in thymocytes.
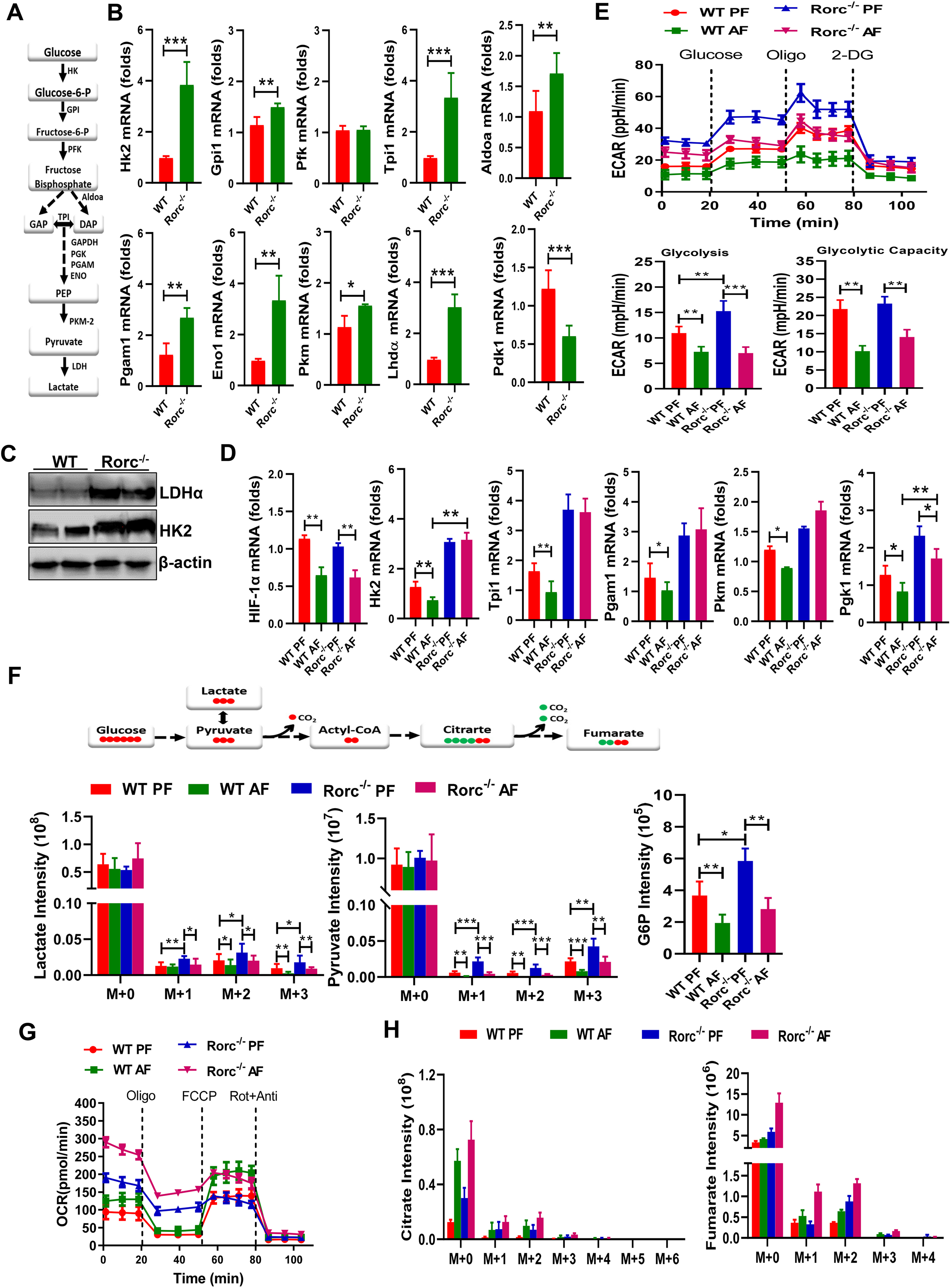
(A-B) The glycolytic pathway (A) and real-time PCR analyses (B) of metabolic genes in the glycolytic pathway in thymocytes from naïve WT and Rorc−/− mice. Mean ± SEM, n = 8, *p < 0.05, **p < 0.01, ***p < 0.001.
(C) Immunoblot analysis of HK2 and LDHα expression in thymocytes from naïve WT and Rorc−/− mice.
(D) Real-time PCR analyses of metabolic genes in the glycolytic pathway in thymocytes from WT and Rorc−/− mice fed the control (PF) or alcohol (AF) diet for 10 days. Mean ± SEM, n = 8, *p < 0.05, **p < 0.01.
(E) ECAR of fresh thymocytes from WT and Rorc−/− mice was measured by seahorse under glycolysis stress test condition and in response to the indicated drugs. Data represent three experiments shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
(F) The isotopologue distributions (Top) in pyruvate and lactate as well as total G6P (Bottom) were determined by GC-MS in thymocytes from WT and Rorc−/− mice that were fed the control (PF) or alcohol (AF) diet for 10 days and then i.v. injected with 13C6-D-glucose. Data are mean ± SEM (n=3–4). *p < 0.05, **p < 0.01, ***p < 0.001.
(G) OCR of fresh thymocytes from WT and Rorc−/− mice fed on the control (PF) or alcohol (AF) diet for 10 days. Data represent three experiments shown as mean ± SEM. *p < 0.05, **p < 0.01.
(H) The isotopologue distributions in citrate and fumarate were determined by GC-MS in thymocytes prepared as described in F above.
Glucose entry into the TCA cycle is induced by RORγt deficiency and heightened under alcoholic conditions
Even with high rates of glycolysis, most cells require intact mitochondrial function to proliferate[31]. We therefore asked whether the reduced glycolytic phenotype exhibited by chronically alcohol-stimulated thymocytes might reflect increased mitochondrial capacity and an altered glucose flux into the TCA cycle. OCR was assessed in thymocytes from mice on the PF or AF diets. A substantial basal OCR and ATP production in thymocytes was increased in AF WT mice compared to PF WT mice (Figure 4G, and Figure S5F), indicating that in these cells, alcohol enhances OXPHOS. We also showed that RORγt-deficient thymocytes had increased basal OCR/ ATP compared to WT thymocytes in the steady state. As expected, basal OCR and ATP was markedly increased in response to alcohol consumption in RORγt-deficient thymocytes (Figure 4G, and Figure S5F). Indeed, alcohol feeding increased the conversion of glucose to citrate (m+1, m+2, m+3) and to the TCA cycle intermediate, fumarate (m+2) (Figure 4H). RORγt deficiency also induced conversion of glucose to TCA cycle intermediates citrate (m+2) and fumarate (m+2), which are less dependent of alcohol treatment (Figure 4H). It is notable that RORγt deficiency and/or alcohol treatment induces a significant increase in a large fraction of citrate (m+0) that was not derived from the labeled glucose, suggesting an alcohol-related alternative source and/or prolonged half-lives of the metabolite that could have existed prior to the administration of labeled glucose.
RORγt regulates glutamine metabolism in thymocytes
CD98 is a heterodimeric amino acid transporter that is induced in activated T cells and crucial for glutamine uptake[32] and composed of a heavy-chain subunit (CD98hc, also known as Slc3a2) and a light chain Slc7a5. RORγt deficient thymocytes expressed higher surface CD98 (Figure 5A) and Slc7a5 mRNA than controls (Figure 5B). On the contrary, Slc7a11 (also commonly known as xCT), which is mainly responsible for the cellular uptake of cystine in exchange for intracellular glutamate, is significantly reduced more than 50-fold in Rorc−/− thymocytes (Figure 5B). We further found that deletion of RORγt increases the expression of glutaminolysis genes (carbamoyl-phosphate synthetase (Cad), phosphoribosyl pyrophosphate amidotransferase (Ppat), Pfas, and glutaminase 2 (Gls2)), which converts glutamine (Gln) to glutamate (Glu) (Figure 5B). Glutamate can generate the anaplerotic substrate α-ketoglutarate (α-KG), that can be metabolized through the TCA cycle. The higher expression of genes that convert glutamate to α-KG (glutamate dehydrogenase 1 (Glud1), glutamate oxaloacetate transaminase 2 (Got2), and ornithine aminotransferase (Oat) were also detected in RORγt deficient thymocytes (Figure 5B). Furthermore, metabolic enzymes in the TCA cycle including Citrate synthase (CS), isocitrate dehydrogenase (Idh2, Idh3), succinate dehydrogenase complex subunits (Sdh), malate dehydrogenase (Mdh) were increased, and malic enzyme 2 (Me2) were significantly decreased in RORγt deficient thymocytes (Figure 5C), suggesting that RORγt deficiency may promote glutamine anaplerosis into the TCA cycle via α-KG.
Figure 5. RORγt deficiency cooperates with alcohol consumption to promote glutaminolysis.
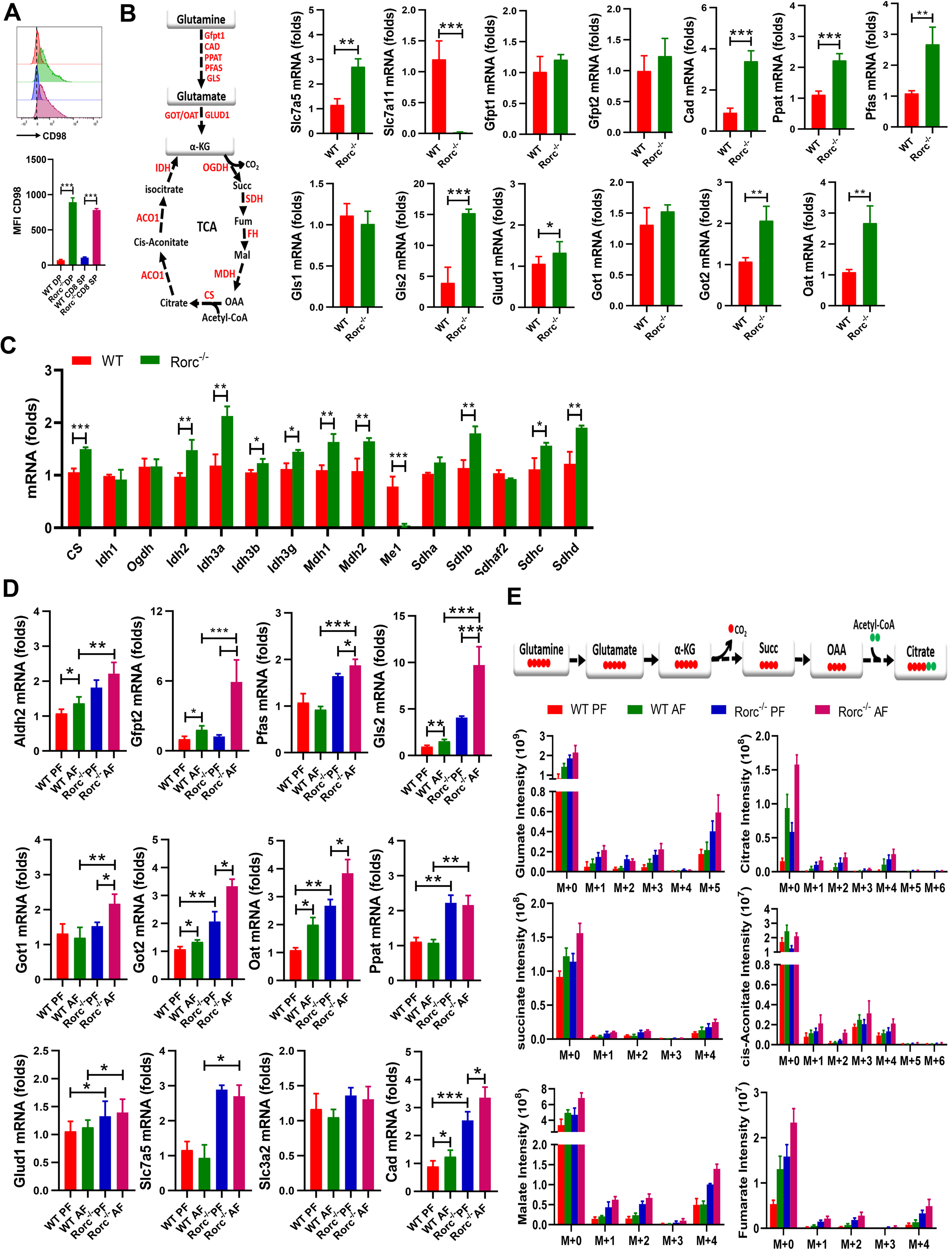
(A) Representative plots (top) and summary graphs (bottom) of the MFI of CD98 in thymocytes from naïve WT and Rorc−/− mice. Mean ± SEM; n = 5, ***p < 0.001.
(B-C) The glutamine pathway (B, left) and real-time PCR analyses of metabolic genes in the glutamine pathway (B, right) and TCA cycle (C) in thymocytes from naïve WT and Rorc−/− mice. Mean ± SEM; n =8, *p < 0.05, **p < 0.01, ***p < 0.001.
(D) Real-time PCR analyses of metabolic genes in the glutamate pathway in thymocytes from WT and Rorc−/− mice fed on the control (PF) or alcohol (AF) diet for 10 days Mean ± SEM; n =6–8, *p < 0.05, **p < 0.01, ***p < 0.001.
(E) The isotopologue distributions in TCA intermediates were determined by GC-MS in thymocytes from WT and Rorc−/− mice that were fed with control (PF) or alcohol (AF) diet for 10 days and then i.v. injected with 13C5-L-glutamine. Data are means ± SEM (n = 3–4).
Alcohol consumption promotes glutaminolysis via the TCA Cycle that is enhanced by RORγt deficiency
Because it was previously documented that glutamate signaling in hepatic stellate cells drives alcoholic liver steatosis[33], we sought to determine whether alcohol consumption would affect glutamine anaplerosis in thymocytes. Real-time PCR analysis revealed that alcohol consumption significantly increased mRNA expression of alcohol metabolic enzyme Aldh2, glutamine catabolism genes, and glutamine transporter genes, including Gls1, Gls2, Gpt2, Gfpt2, Oat, Got1, Got2 and Cad in the thymocytes of both WT mice and Rorc−/− mice when compared to paired feeding (Figure 5D). Importantly, RORγt deficiency greatly enhanced the effect of alcohol on glutamine metabolism in thymocytes (Figure 5D). In addition, alcohol apparently increases the expression of alcoholism enzyme Aldh2 and glutamine anaplerotic metabolic enzymes including Gls1, Gls2, Got1, Glud1 and Gpt2 mRNA in EL4 cells (Figure S6A) and Rorc−/− lymphoma (Figure S6B). Of note, RORγt inhibition or knockdown greatly enhanced the expression of genes involved in glutamine catabolism, whereas overexpression of RORγt abolished alcohol-induced expression of these glutamine catabolism genes (Figures S6A and S6B).
Using U-13C5-L-glutamine as a tracer, we determined whether glutamine entry into the TCA cycle would be regulated by alcohol and/or RORγt deficiency in vivo. GC-MS analysis revealed that intracellular glutamine was converted to glutamate (m+1, m+3, m+5 isotopologue) in an alcohol-dependent way and significantly upregulated by RORγt deficiency (Figure 5E). Although the relative flux of glutamine into citrate (m+1, m+2, m+4) in thymocytes from alcohol-fed mice with both genotypes was higher than the values from PF mice, this increase was more in RORγt-deficient thymocytes (Figure 5E). We also found the labeled TCA intermediates including succinate (M+4), fumarate (m+1, m+2, m+4), malate (m+1, m+2, m+4) and cis-aconitic acid (m+1, m+2, m+3, m+4) significantly increased after alcohol treatment (Figure 5E). Importantly, RORγt deficiency also enhanced the conversion of labeled glutamine to these labeled TCA intermediates, particularly under the condition of alcohol consumption (Figure 5E). Furthermore, GC-MS analysis of the same set of polar extracts showed that the levels of M+0 citrate, succinate, fumarate were significantly increased by RORγt deficiency and/or alcohol treatment. This is consistent with the result of M+0 citrate in Figure 4H. We further investigated if carbons from glutamine were incorporated into synthesized FAs. Tracing of 13C-L-glutamine in thymocytes in vivo via mass spectrometry (MS) 1 h after injection revealed that even in this short time, alcohol and/or RORγt deficiency can drive the incorporation of carbons into the FA chains of TG, PS and PC lipid (Figure S6C). In accordance with alcohol-induced oxidative stress and increased glutamate, a decrease in GSH in thymocytes was also found in WT mice, but not Rorc−/− mice after ethanol treatment (Figure S6D). These data suggest that, in contrast to alcohol inhibiting aerobic glycolysis, alcohol triggers catabolic glutamine metabolism, even more under RORγt deficient conditions.
Inhibition of RORγt promotes glutamine-dependent biosynthesis in lymphomas under glucose insufficient conditions.
In cancer cells, glutamine provides an alternate carbon source for lipid synthesis in low glucose conditions (LG)[15]. Since our data showed that alcohol consumption apparently suppresses aerobic glycolysis, we hypothesize that RORγt deficiency would enhance glutamine-dependent TCA cycle activity for lymphoma growth and survival under glucose insufficient conditions. To this end, EL4 cells were cultured in normal media A (10 mM glucose, 2 mM glutamine), low glucose (LG) media B (2 mM glucose, 2 mM glutamine), or low glucose with high glutamine media C (2 mM glucose, 8 mM glutamine) in the presence or absence of RORγt inhibitor. Addition of high glutamine (8mM) increase the OCR in LG media C when compared to LG media B, consistent with the view that glutamine supports OXPHOS (Figure 6A). Although inhibition of RORγt increased the OCR in EL4 cells cultured with the three types of media, RORγt inhibitor treatment induced maximal OCR differences in media C LG condition (Glu 2, Gln 8) compared to DMSO treatment (Figure 6A). To determine whether RORγt inhibition favored the promotion of glutamine entry into the TCA cycle in EL4 lymphoma under low glucose conditions, we used 13C5-L-glutamine as a tracer. We found m+4 citrate levels were elevated in Rorc-KD cells relative to control cells, particularly under glucose-insufficient conditions (Figure 6B). We also measured glucose-dependent and glutamine-dependent lipid biosynthesis by culturing EL4 cells with radioactive D-[U-14C]-glucose or [U-14C]-L-glutamine. Interestingly, RORγt inhibition dramatically promoted both glucose-dependent and glutamine-dependent lipogenesis in EL4 cells, as evidenced by increased 14C-labeling in lipids (Figure 6C). Together, these data indicate that RORγt deficient lymphoma prefer to use glutamine for biosynthesis under glucose insufficient or alcohol consumption condition.
Figure 6. Inhibition of RORγt promotes glutamine-dependent biosynthesis in lymphoma under glucose insufficient condition.
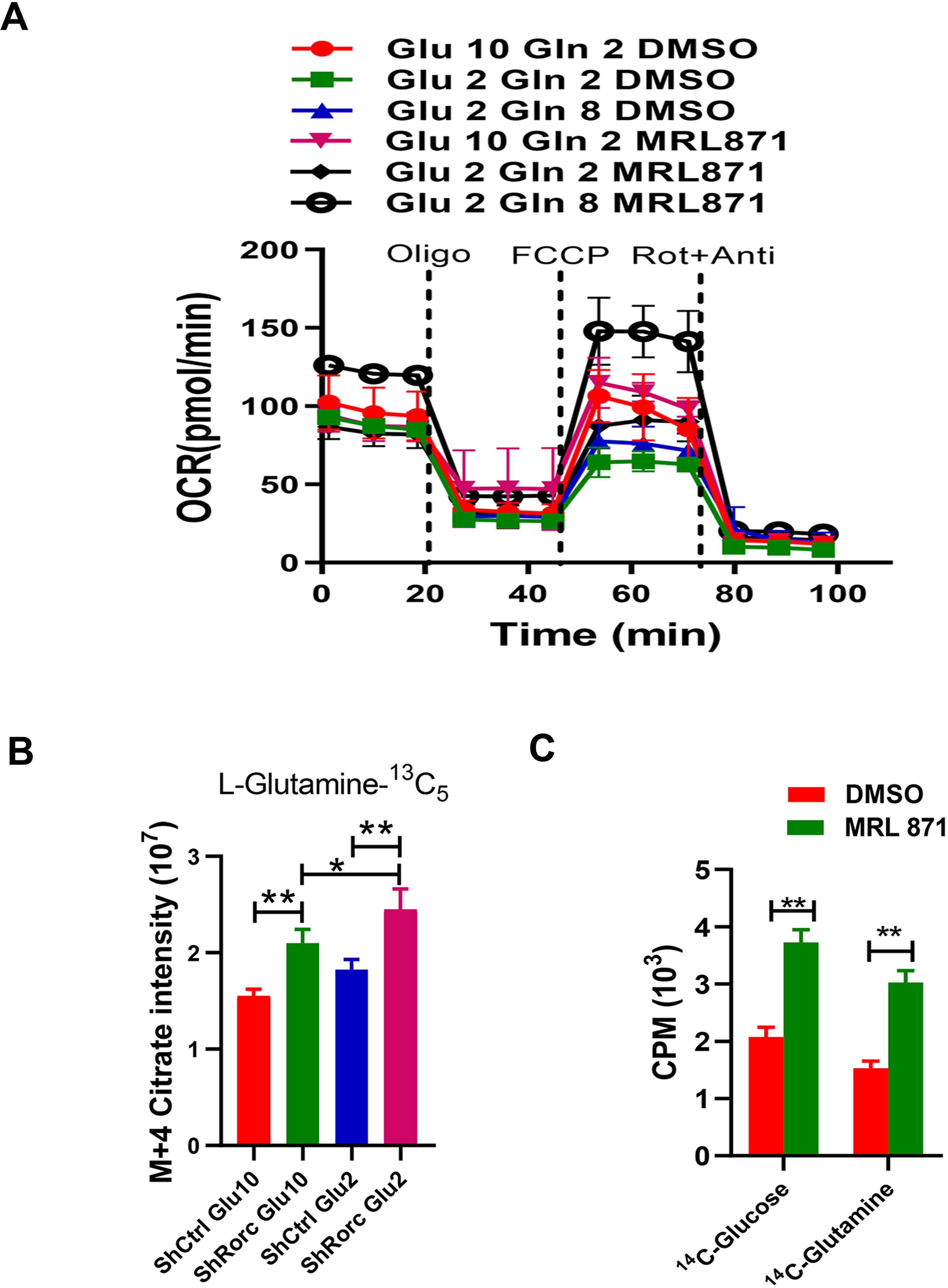
(A) OCR of cells was measured by seahorse and in response to the indicated drugs. EL4 cells were cultured under different glucose and glutamine conditions in the presence or absence of RORγt inhibitor MRL871. Mean ± SEM, n=3.
(B) The m+4 isotopologues of citrate were determined by GC-MS. EL4 cells or MRL871-treated EL4 cells were grown for 12 h in U-13C5-glutamine medium with normal (10 mM) or low glucose concentration (2 mM).
(C) Lipid biosynthesis in EL4 cells or EL4 cells with RORγt inhibition. EL4 cells or MRL871-treated EL4 cells were incubated for 12 hr with uniformly labeled 14C-glucose or 14C-glutamine, and radioactive counts in extracted lipids were measured.
mTOR/c-Myc signaling drives increased glutaminolysis and biosynthesis in RORγt-deficient lymphoma
mTOR responds to nutrient deficiency by stimulating the synthesis of proteins and lipids and has been linked to control of c-Myc expression[34]. Since c-Myc is highly induced at both the transcription and protein levels in RORγt-deficient thymocytes (Figure 2D), we speculated that RORγt deficiency in thymocytes might promotes glutamine utilization via mTOR/c-Myc activation. When compared to WT thymocytes, using flow cytometry we discovered that RORγt deficiency induced the phosphorylation of mTOR S2448 and mTORC1 target S6, and CD71 that is a c-Myc target and marker for T cell metabolism and activation (Figure 7A). Alcohol consumption increased the levels of the mTOR/c-Myc targets pS6 and CD98 in both genotypes of thymocytes (Figure S7A). BM chimaeras demonstrated that the changes in these mTOR-related metabolic markers are RORγt thymocytes-intrinsic (Figure S7B). To examine the contribution of mTOR signaling to c-Myc in Rorc−/− lymphoma, we treated cells with mTOR inhibitor rapamycin and found the levels of c-Myc mRNA (Figure S8A) and c-Myc protein were significantly reduced in both RorcRES and Rorc−/− lymphoma, with the latter demonstrating with a large decrease in c-Myc expression upon mTOR inhibition (Figure 7B). Moreover, mTOR inhibition reduced the expression of the c-Myc targets CD71 and CD98 and proliferation in cells (Figure 7B) regardless of RORγt expression, suggesting that rapamycin may exert its effects in part through regulation of c-Myc mRNA. Similar effects of rapamycin on the expression of c-Myc, CD71, CD98 and the proliferation were found for EL4 cells with/without RORγt knockdown (Figure S8B).
Figure 7. c-Myc mediates the effects of RORγt loss on glutaminolysis and biosynthesis.
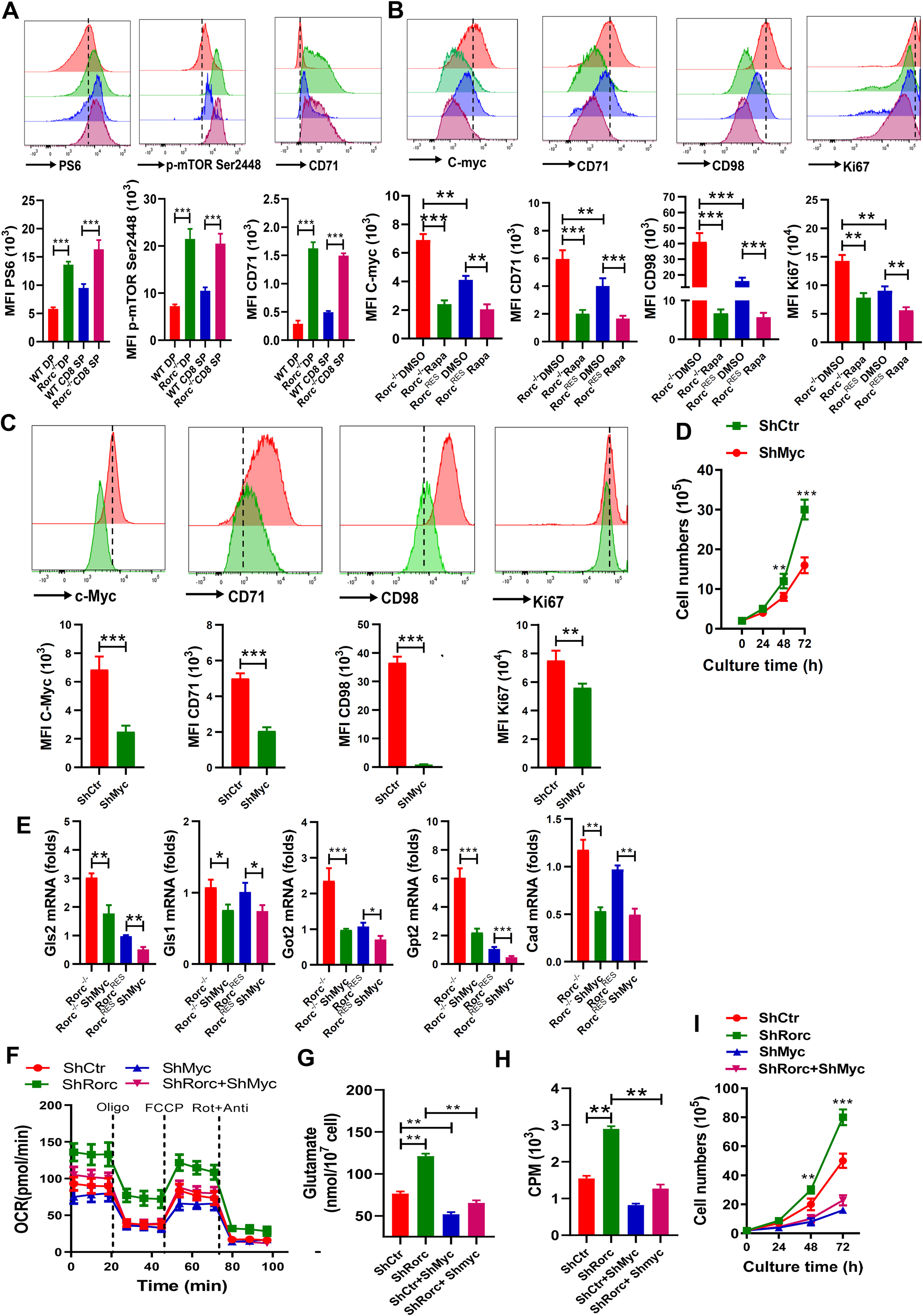
(A) Representative plots (top) and summary graphs (bottom) of the MFI of pS6, p-mTOR Ser2448, and CD71 in thymocytes from WT and Rorc−/− mice. Mean ± SEM; n = 5, ***p < 0.001.
(B-C) Representative plots (top) and summary graphs (bottom) of the MFI of c-Myc, CD71, CD98, and Ki67 in RorcRES and Rorc−/− lymphomas treated with/without the mTOR inhibitor rapamycin (Rapa) (B), or in Rorc−/− lymphomas without (ShCtr) / with c-Myc KD (ShMyc) (C).
(D) Growth curves of Rorc−/− lymphoma without (ShCtr) or with c-Myc KD (ShMyc).
(E) Real-time PCR analyses of metabolic genes in the glutamate pathway in RorcRES or Rorc−/− lymphoma without (ShCtr) or with c-Myc KD (ShMyc). (B-E) Mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001.
(F-G) OCR (F) or Intracellular glutamate production (G) of EL4 cells (ShCtr), EL4 cells with c-Myc KD (ShMyc), Rorc KD (ShRorc) or both, i.e., ShMyc+ShRorc. Mean ± SEM, n = 5, *p < 0.05, **p < 0.01.
(H) Lipid biosynthesis in EL4 cells (ShCtr), EL4 cells with c-Myc KD (ShMyc), Rorc KD (ShRorc) or both, i.e., ShMyc+ShRorc. Cells were incubated with uniformly labeled 14C-glutamine for 12 h, and radioactive counts in extracted lipids were measured. Mean ± SEM, n = 5 *p < 0.05, **p < 0.01.
(I) Growth curves of in EL4 cells (ShCtr), EL4 cells with c-Myc KD (ShMyc), Rorc KD (ShRorc) or both, i.e., ShMyc+ShRorc. Mean ± SEM, n = 5, **p < 0.01, ***p < 0.001.
Given that aberrant c-Myc expression drives both enhanced glutaminolysis and glutamine-derived lipogenesis, we reasoned that RORγt dependent changes in glutamine metabolism may be attributed to c-Myc. To this end, we transduced Rorc−/− lymphoma with a ShMyc-expressing retrovirus. Surface expression of the Myc and its target CD98 and CD71 was downregulated on ShMyc-expressing Rorc−/− lymphoma compared to transduced controls (Figure 7C). Silencing of c-Myc also reduced the proliferation of Rorc−/− lymphoma to that of control cells (Figure 7C–7D). To confirm the contribution of c-Myc to the glutaminolysis phenotype observed in RORγt deficient lymphoma, we also stably silenced c-Myc using a shRNA-Myc-expressing retrovirus in EL4 cells having a silenced RORγt. Silencing of c-Myc in RORγt shRNA-expressing cells reduced c-Myc protein expression to control levels (Figure S8C). Expression of protein levels of the c-Myc targets CD71 and CD98 expression was also decreased when c-Myc was silenced in RORγt shRNA-expressing EL4 cells (Figure S8C). Thus, RORγt controls mTOR activity and Myc production in thymocytes and lymphoma.
We next determined whether silencing Myc could reverse the glutaminolysis triggered by loss of RORγt activity. Expression of Myc shRNA reduced the metabolic genes Gls1, Gls2, Gpt2, Got2 and Cad mRNA in the glutamine catabolic pathways in Rorc−/− lymphoma to control levels (Figure 7E). We also found that silencing of c-Myc reduced the increase of OCR response (Figure 7F) and glutamate production (Figure 7G) in Rorc-knockdown EL4 cells. Moreover, suppression of c-Myc dramatically reduced glutamine-dependent lipogenesis (Figure 7H) and proliferation (Figure 7I) in Rorc-knockdown EL4 cells. Collectively, the data suggest that RORγt negatively regulates the metabolic and biosynthetic programs of proliferating cells through the inhibition of c-Myc function.
c-Myc-controlled glutaminolysis is required for the alcohol-mediated progression of Rorc−/− lymphomas
Our findings that RORγt deficiency-induced c-Myc expression enhances glutamine metabolism, particularly in alcoholic condition, led us to test whether targeting c-Myc and glutaminase is feasible for alcohol associated cancer therapy. We first determined if alcohol treatment has an impact on cellular biosynthesis and proliferation through c-Myc expression. Expression of Myc shRNA blocked alcohol-dependent induction of c-Myc targeted genes (Gls2) mRNA in both RORγt -deficient and control cells (Figure 8A). Suppression of c-Myc also had a significant suppressive effect on glutamate production in alcohol-treated cells (Figure 8B). While silencing of Myc in alcohol-treated EL4 led to slight reductions in m+4 citrate production by tracing 13C5 -L-Glutamine (Figure 8C) and in OCR (Figure 8D), Myc shRNA significantly lowered the m+4 citrate level and OCR in alcohol-treated EL4 cells with a Rorc knockdown (Figure 8C–8D). We also determined the consequences of Gls2 inhibitor BPTES treatment on the proliferation of EL4 cells expressing Rorc shRNA. BPTES decreased the proliferation in EL4 cells, even more under Rorc KD (Figure 8E).
Figure 8. c-Myc mediates the metabolic and tumorigenic effects induced by RORγt loss with alcohol consumption.
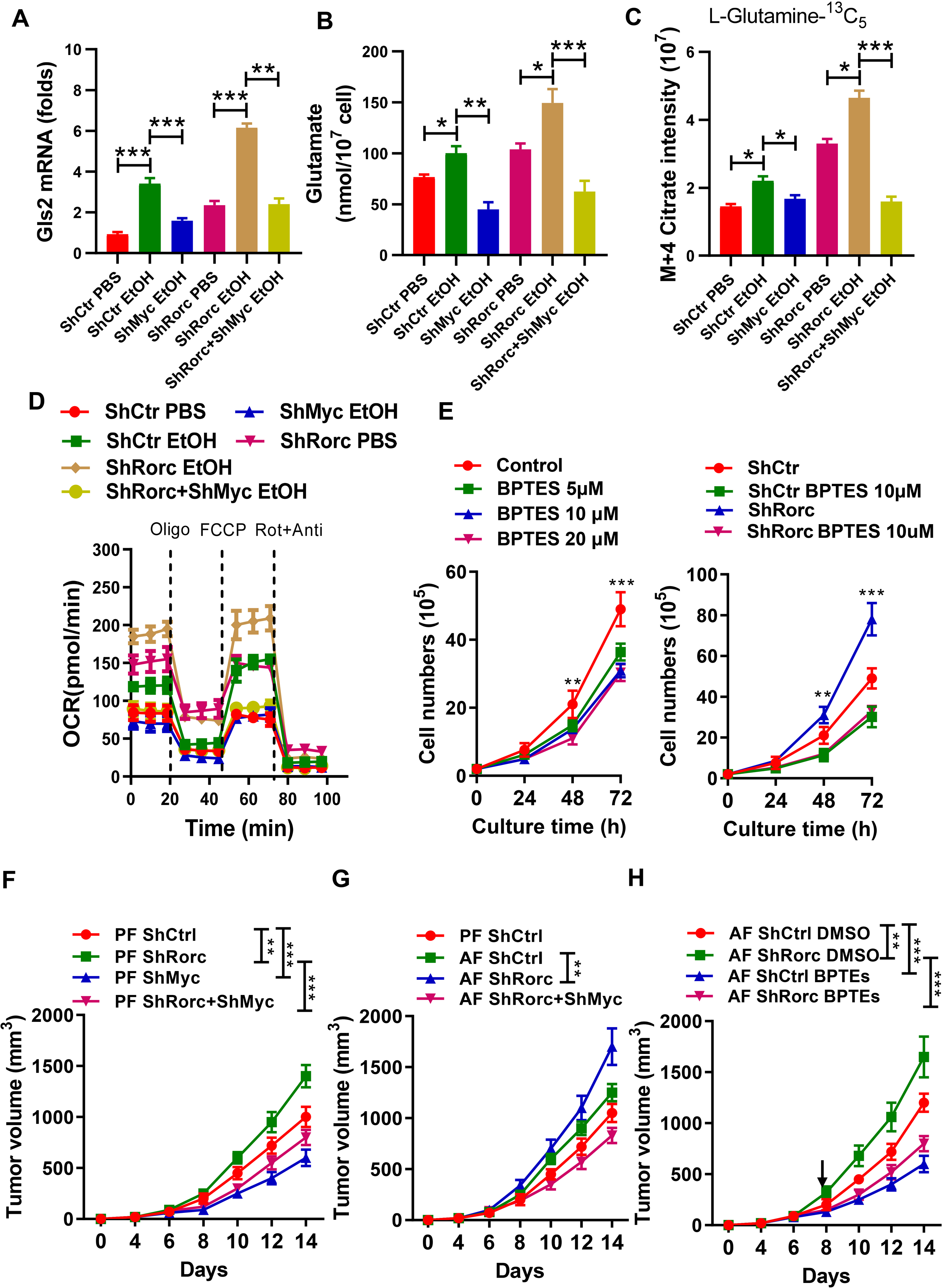
(A-D) Gls2 mRNA expression (A), intracellular glutamate production (B), m+4 citrate (C) or OCR (D) in EL4 cells (ShCtr), EL4 cells with c-Myc KD (ShMyc), Rorc KD (ShRorc) or both, i.e., ShMyc+ShRorc. Cells were cultured for 6h (A, B, C), for 72 h (D) in the presence of PBS or EtOH (75 mM). mean ± SEM, n = 5, **p < 0.01, ***p < 0.001.
(E) Effect of the glutaminase inhibitor, BPTES, on EL4 cell growth (left) and on EL4 cells without (ShCtr) or with Rorc KD (ShRorc) (right). mean ±SEM.
(F) The tumor growth was monitored every 2 d for 14 d after EL4-ShCtr, EL4-ShRorc, EL4-ShMyc, or EL4-ShRorc+ShMyc inoculation. Mice were fed the control diet (PF).
G) The tumor growth was monitored every 2 d for 14 d after EL4-ShCtr, EL4-ShRorc, or EL4-ShRorc+ShMyc inoculation. Mice were fed the control diet (PF) or alcohol diet (AF) as indicated.
(H) Tumor growth of EL4 or EL4-ShRorc-bearing mice treated every other day with 200 μg BPTES (i.p.) or vehicle alone. Mice were fed the alcohol diet and therapy was begun when the mice had a palpable tumor (day 8, arrow). Mean ± SEM, n=5, *P < 0.05, **P < 0.01, ***P < 0.001 (F-H).
Next we tested the requirement of c-Myc for alcohol-mediated tumor progression in vivo. Silencing of c-Myc in lymphomas promoted a general decrease in the tumor size (Figure 8F). Interestingly, the growth of lymphoma cells expressing c-Myc shRNA was significantly delayed under Rorc KD condition (Figure 8F). We also tested if alcohol consumption promoted tumor progression in vivo in EL4 cells or EL4 cells with ShRorc. While alcohol consumption led to a slight increase in EL4 tumor size, statistical significance was not reached. However, alcohol feeding significantly increased tumor size in the EL4 cells with ShRorc, but not EL4 cells with ShRorc+ShMyc (Figure 8G). To determine the significance of our findings, EL4 cells or EL4 cells with ShRorc tumor xenograft-bearing mice were fed alcohol and treated with intraperitoneal injections of BPTES. As compared with DMSO vehicle-treated mice, the BPTES-treated mice demonstrated a significantly diminished tumor progression (Figure 8H). Taken together, these data suggest that loss of RORγt signaling promotes a metabolic and growth advantage in lymphoma cells and that c-Myc-regulated glutaminolysis is required for the growth of RORγt null tumors in vivo, particularly under alcohol consumption condition.
Discussion:
Recent identification of small-molecule RORγ antagonists have been used to study the effector function of Th17 cells and to treat autoimmune diseases[35, 36]. However, how RORγt inhibition may impact thymocyte and lymphoma development, particularly under alcohol consumption conditions remains unclear. In this study, we provide genetic evidence that RORγt displays the regulatory activity of lymphoma metabolism in vivo and we define the possible translational risk of developing thymic T cell aberrations and lymphoma in patients with autoimmunity and AUD after prolonged therapy with RORγt inhibitors.
Previous study showed that RORγt deficiency results in the loss of Bcl-xL, an antiapoptotic protein, and increases apoptosis of preselection DP thymocytes[22, 37]. We demonstrated an uncharacterized function of RORγt in inhibiting cell metabolism and mitochondrial biogenesis in DP thymocytes. Defects in thymic development have been observed upon disruption of multiple pathways important for nutrient sensing and modulating cellular metabolism[38]. Our RNA-seq data showed that the RORγt-regulated genes in the thymus fall into multiple pathways: the glycolysis, glutaminolysis and OXPHOS pathways. We demonstrated that RORγt is a negative and direct regulator of both aerobic glycolysis and glutaminolysis in thymocytes and T lymphoma. Furthermore, we found that loss of RORγt resulted in increased anabolic metabolism and mTOR signaling, an indicator of increased cellular metabolic activity[39]. mTOR is able to control T-ALL metabolism by controlling expression of two key transcription factors c-Myc and HIF1α. Our study shows that c-Myc is critical for T-lymphoma development induced by a RORγt deletion. In this respect, we showed that mTOR inhibition significantly prevented the increases of c-Myc targeted genes and proliferation in Rorc−/− T lymphoma. One role for HIF1α is to regulate the transcription of genes encoding glucose transporters and glycolytic enzymes[40]. On the other hand, c-Myc controls the transcription of genes encoding glucose transporters and enzymes controlling glycolysis and glutaminolysis in T cells[23]. We found that c-Myc is a key mediator of RORγt-dependent effects on cellular metabolism and lymphomas growth in vivo. Thus, RORγt may act in lymphomas as a direct metabolic gatekeeper that functions to suppress lymphoma bioenergetics and metabolism, and its loss of function can enhance lymphomagenesis. Although our data showed that the role of RORγt on thymocyte metabolism is T cells-intrinsic, more in-depth mechanistic approaches including studies in mice with T-specific deletion of RORγt will be required to fully understand the mechanism of alcohol-mediated lymphomagenesis.
Excessive alcohol consumption is a serious health problem. Alcohol exposure may result in the overexpression of certain oncogenes in human cells thereby increasing the intracellular concentration of reactive oxygen species (ROS), and triggering disease initiation and promotion. Our data indicate that ethanol exposure, also its metabolite acetate, might promote the metabolic effects of RORγt, i.e. increased OXPHOS and lipid synthesis in T lymphoma. This discovery is consistent with previous studies showing that acetate supports cell proliferation, invasion, and metastases in other cancers[41, 42]. Surprisingly, alcohol treatment significantly reduced HIF1α expression and enhanced c-Myc expression in thymocytes in vivo and lymphoma in vitro, two key factors for controlling glycolysis and glutaminolysis, respectively. Indeed, decreased HIF1α and upregulated c-Myc were found in the liver of patients with advanced ALD and in alcohol-fed mice[43, 44]. Here, we provided evidence suggesting that alcohol inhibits glucose metabolism in thymocytes and lymphoma via the downregulation of Hk2 expression and inhibition of several metabolic signaling pathways. First, alcohol downregulated expression of two important genes involved in aerobic glycolysis, Hk2 and HIF1α in thymocytes and T lymphoma[45, 46]. Second, tracer labeled 13C6-D-glucose administered in vivo revealed that alcohol-treated cells display decreased aerobic glycolysis marked by reduced pyruvate and lactate production from glucose (Figure 4). Consistent with these, a recent study has showed that Aldh2 deficiency or increased acetaldehyde production is associated with suppression of aerobic glycolysis and T-cell functions post-alcohol consumption[30].
By contrast, our data further showed that alcohol exposure induces a metabolic shift to glutaminolysis in thymocytes and T lymphoma, which was evidenced by great increases of the genes involved in glutamine metabolism in vivo and in vitro. We also used uniformly labeled 13C5-L-glutamine and traced the fate of glutamine carbon in vivo in both normal and alcoholic conditions in WT and Rorc−/− mice. Our results uncovered a glutamine-dependent TCA cycle significantly increased by alcohol consumption, and further enhanced by RORγt deficiency (Figure 5). More intriguingly, alcohol consumption also promoted glutamine-dependent lipid synthesis via the TCA cycle. Our data show that the import rate of glutamine greatly exceeds that of glucose under alcoholic and RORγt deficient conditions. Additionally, RORγt deficient cells can synthesize more FA by using glutamine-derived carbon, and this suggests that glutamine is the major source for energy and anaplerosis after loss of RORγt. We also found that c-Myc is a key mediator of alcohol-dependent effects on glutamine metabolism. Combined with the fact that increased c-Myc is responsible for RORγt-mediated glutaminolysis, we propose that Rorc−/− lymphomas survive or even proliferate compared to RorcRES lymphomas under the alcoholic or glucose insufficient conditions encountered in the tumor microenvironment (Figure S9). Consist with our results, recent studies have found that expression of c-Myc and oxidative stress caused by chronic ethanol intake can increase hepatic excretion of glutamate and hepatic lipid synthesis and accelerate the progression of ALD[33].
According to these findings, we tested the effect of a glutaminase inhibitor (BPTES) on the growth of Rorc deficient EL4 cells and showed that blocking glutamine metabolism not only inhibited tumor cell growth under normal dietary conditions but also under alcoholic conditions and led to a reduction of tumor xenograft growth in vivo (Figure 8). Our finding that there is an alcohol-dependent glutamine-driven TCA cycle and Rorc deficiency-related metabolic pathway suggest that the translational risk of RORγt antagonists-induced thymic T cell aberrations and lymphomas should be considered in patients with autoimmunity who are also heavy drinker. Future studies delineating alcoholism and glutaminolysis in the development thymic T cells and lymphoma and whether alcohol-induced changes in nutritional status orchestrate these pathways to control cellular lifespan are needed.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH R21AA025724, R21AI128206, R21AI159194 and R01 DK115406 (Z.D.), and NIH P50AA024337, P20GM113226, (CJ.M.). We thank Dr. J. Ainsworth for editorial assistance.
Footnotes
Conflicts of Interest
The authors disclose no conflicts of interest.
Reference:
- 1.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006; 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 2.He YW, Beers C, Deftos ML, Ojala EW, Forbush KA, Bevan MJ. Down-regulation of the orphan nuclear receptor ROR gamma t is essential for T lymphocyte maturation. Journal of Immunology 2000; 164: 5668–5674. [DOI] [PubMed] [Google Scholar]
- 3.Ueda E, Kurebayashi S, Sakaue M, Backlund M, Koller B, Jetten AM. High incidence of T-cell lymphomas in mice deficient in the retinoid-related orphan receptor RORgamma. Cancer Res 2002; 62: 901–909. [PubMed] [Google Scholar]
- 4.Guntermann C, Piaia A, Hamel ML, Theil D, Rubic-Schneider T, Del Rio-Espinola A et al. Retinoic-acid-orphan-receptor-C inhibition suppresses Th17 cells and induces thymic aberrations. JCI Insight 2017; 2: e91127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Y, MacIsaac KD, Chen Y, Miller RJ, Jain R, Joyce-Shaikh B et al. Inhibition of RORgammaT Skews TCRalpha Gene Rearrangement and Limits T Cell Repertoire Diversity. Cell Rep 2016; 17: 3206–3218. [DOI] [PubMed] [Google Scholar]
- 6.Liljevald M, Rehnberg M, Soderberg M, Ramnegard M, Borjesson J, Luciani D et al. Retinoid-related orphan receptor gamma (RORgamma) adult induced knockout mice develop lymphoblastic lymphoma. Autoimmun Rev 2016; 15: 1062–1070. [DOI] [PubMed] [Google Scholar]
- 7.Jun S, Mahesula S, Mathews TP, Martin-Sandoval MS, Zhao Z, Piskounova E et al. The requirement for pyruvate dehydrogenase in leukemogenesis depends on cell lineage. Cell Metab 2021. [DOI] [PubMed] [Google Scholar]
- 8.Ramstead AG, Wallace JA, Lee SH, Bauer KM, Tang WW, Ekiz HA et al. Mitochondrial Pyruvate Carrier 1 Promotes Peripheral T Cell Homeostasis through Metabolic Regulation of Thymic Development. Cell Rep 2020; 30: 2889-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D et al. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab 2014; 20: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odera JO, Xiong ZH, Huang CZ, Gu N, Yang WJ, Githang’a J et al. NRF2/ACSS2 axis mediates the metabolic effect of alcohol drinking on esophageal squamous cell carcinoma. Biochemical Journal 2020; 477: 3075–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol 2020; 72: 946–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology 2007; 46: 2032–2039. [DOI] [PubMed] [Google Scholar]
- 13.Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab 2016; 23: 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siler SQ, Neese RA, Christiansen MP, Hellerstein MK. The inhibition of gluconeogenesis following alcohol in humans. Am J Physiol-Endoc M 1998; 275: E897–E907. [DOI] [PubMed] [Google Scholar]
- 15.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab 2012; 15: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. P Natl Acad Sci USA 2008; 105: 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458: 762–U100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Gene Dev 2006; 20: 2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evangelisti C, Ricci F, Tazzari P, Tabellini G, Battistelli M, Falcieri E et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia 2011; 25: 781–791. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1 alpha Eliminates Cancer Stem Cells in Hematological Malignancies. Cell Stem Cell 2011; 8: 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu S, Sun R, Gu X, Chen L, Liu M, Guo H et al. Inhibition of sphingosine-1-phosphate-induced Th17 cells ameliorates alcoholic steatohepatitis in mice. Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 2000; 288: 2369–2373. [DOI] [PubMed] [Google Scholar]
- 23.Wang RN, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011; 35: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu Y, Zhang J, Ma X, Kim BW, Wang H, Li J et al. Stabilization of the c-Myc Protein by CAMKIIgamma Promotes T Cell Lymphoma. Cancer Cell 2017; 32: 115–128 e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell 1999; 3: 169–179. [DOI] [PubMed] [Google Scholar]
- 26.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016; 16: 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S et al. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. P Natl Acad Sci USA 2007; 104: 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pollizzi KN, Waickman AT, Patel CH, Sun IH, Powell JD. Cellular Size as a Means of Tracking mTOR Activity and Cell Fate of CD4+T Cells upon Antigen Recognition. Plos One 2015; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–185. [DOI] [PubMed] [Google Scholar]
- 30.Gao YH, Zhou Z, Ren TY, Kim SJ, He Y, Seo W et al. Alcohol inhibits T-cell glucose metabolism and hepatitis in ALDH2-deficient mice and humans: roles of acetaldehyde and glucocorticoids. Gut 2019; 68: 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015; 162: 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017; 46: 675–689. [DOI] [PubMed] [Google Scholar]
- 33.Choi WM, Kim HH, Kim MH, Cinar R, Yi HS, Eun HS et al. Glutamate Signaling in Hepatic Stellate Cells Drives Alcoholic Steatosis. Cell Metab 2019; 30: 877–889 e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell 2012; 149: 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C et al. Small-molecule RORgammat antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 2014; 40: 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheepstra M, Leysen S, van Almen GC, Miller JR, Piesvaux J, Kutilek V et al. Identification of an allosteric binding site for RORgammat inhibition. Nat Commun 2015; 6: 8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurebayashi S, Ueda E, Sakaue M, Patel DD, Medvedev A, Zhang F et al. Retinoid-related orphan receptor gamma (RORgamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc Natl Acad Sci U S A 2000; 97: 10132–10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang K, Blanco DB, Chen X, Dash P, Neale G, Rosencrance C et al. Metabolic signaling directs the reciprocal lineage decisions of alphabeta and gammadelta T cells. Sci Immunol 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev 2012; 249: 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C et al. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 2012; 11: 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK et al. Acetate dependence of tumors. Cell 2014; 159: 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015; 27: 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nevzorova YA, Cubero FJ, Hu W, Hao F, Haas U, Ramadori P et al. Enhanced expression of c-myc in hepatocytes promotes initiation and progression of alcoholic liver disease. J Hepatol 2016; 64: 628–640. [DOI] [PubMed] [Google Scholar]
- 44.Shao T, Zhao C, Li F, Gu Z, Liu L, Zhang L et al. Intestinal HIF-1alpha deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J Hepatol 2018; 69: 886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–197. [DOI] [PubMed] [Google Scholar]
- 46.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol 2013; 31: 259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.