

Abstract
Autism spectrum disorder (ASD) affects ~2% of the population in the US, and monogenic forms of ASD often result in the most severe manifestation of the disorder. Recently, SCN2A has emerged as a leading gene associated with ASD, of which abnormal sleep pattern is a common comorbidity. SCN2A encodes the voltage-gated sodium channel NaV1.2. Predominantly expressed in the brain, NaV1.2 mediates the action potential firing of neurons. Clinical studies found that a large portion of children with SCN2A deficiency have sleep disorders, which severely impact the quality of life of affected individuals and their caregivers. The underlying mechanism of sleep disturbances related to NaV1.2 deficiency, however, is not known. Using a gene-trap Scn2a-deficient mouse model (Scn2atrap), we found that Scn2a deficiency results in increased wakefulness and reduced non-rapid-eye-movement (NREM) sleep. Brain region-specific Scn2a deficiency in the suprachiasmatic nucleus (SCN) containing region, which is involved in circadian rhythms, partially recapitulates the sleep disturbance phenotypes. At the cellular level, we found that Scn2a deficiency disrupted the firing pattern of spontaneously firing neurons in the SCN region. At the molecular level, RNA-sequencing analysis revealed differentially expressed genes in the circadian entrainment pathway including core clock genes Per1 and Per2. Performing a transcriptome-based compound discovery, we identified dexanabinol (HU-211), a putative glutamate receptor modulator, that can partially reverse the sleep disturbance in mice. Overall, our study reveals possible molecular and cellular mechanisms underlying Scn2a deficiency-related sleep disturbances, which may inform the development of potential pharmacogenetic interventions for the affected individuals.
Keywords: Voltage-gated sodium channel, Nav1.2, channelopathy, sleep disorder, circadian clock, autism spectrum disorder, neurodevelopmental disorder
Introduction
Autism spectrum disorder (ASD), a major neurodevelopmental disorder, affects approximately 1 out of 44 children in the US according to the report from the Centers for Disease Control and Prevention (CDC). Multiple genetic studies have identified a range of monogenetic mutations in patients with ASD (Hoischen et al., 2014; Johnson et al., 2016; Sanders et al., 2012; Wang et al., 2016). Most recently, a large-scale study has revealed that both protein-truncating variants and missense variants of SCN2A were among the largest set of variants identified in children with ASD (Satterstrom et al., 2020), highlighting an important role of the SCN2A gene in ASD etiology.
SCN2A encodes the voltage-gated sodium channel NaV1.2, a major sodium channel widely expressed in the central nervous system (CNS). NaV1.2 is known to mediate action potential (AP) firing and synaptic functions (Gazina et al., 2015; Hu et al., 2009). Previous work with mouse models has shown that the complete knockout of Scn2a is lethal (Planells-Cases et al., 2000). Studies of the heterozygous (HET) knockout of Scn2a (expressing ~50% of NaV1.2 protein) reveal minor to modest behavioral abnormalities in mice (Léna and Mantegazza, 2019; Ogiwara et al., 2018; Planells-Cases et al., 2000; Shin et al., 2019; Spratt et al., 2019; Tatsukawa et al., 2019). Our lab established a unique mouse model of Scn2a deficiency with a gene-trap strategy (homozygous for the gene-trap is referred to as Scn2a-deficient or Scn2atrap mice), which results in the expression of ~25% of the wild-type (WT) level of Scn2a protein in adulthood. These mice display major neurodevelopmental defects including impaired innate behavior and elevated anxiety, among others (Eaton et al., 2021), likely modeling the severe aspects of Scn2a deficiency-related disorders.
Sleep problem is known to be prevalent comorbidity in children with ASD (Glickman, 2010; Maxwell-Horn and Malow, 2017; Missig et al., 2020; Veatch et al., 2015). An estimated 40–80% of the ASD population experience sleep disturbances that do not improve with age (Lai et al., 2014). Additionally, sleep impairment is a strong predictor of the severity in core ASD symptoms (MacDuffie et al., 2020; Schreck et al., 2004). A clinical study of children with SCN2A deficiency suggests that they have difficulty falling and staying asleep (Crawford et al., 2021). According to Simons Searchlight Registry Update for SCN2A, about 76.9% SCN2A children have a high likelihood of sleep problems with “wakes during the night”, “sleeps too little”, “doesn’t fall asleep in 20 minutes” and “seems tired” as the top complaints (SimonsSearchLight, September 2021 report). The sleep problems of affected children reduce their quality of life and also pose a major burden on their caregivers (Berg et al., 2021). Despite the prevalence of sleep issues in these patients, whether a mouse model of Scn2a deficiency may recapitulate sleep abnormalities has not been determined, hindering the understanding of mechanisms underlying sleep disorders to advance drug discovery efforts.
Here, we performed a study to elucidate the sleep architecture of our Scn2a-deficient mice. We found that a global ~75% Scn2a deficiency resulted in increased wakefulness and reduced non-rapid-eye-movement (NREM) sleep, which can be partially recapitulated by Cre recombinase-induced brain region-specific Scn2a deficiency in the suprachiasmatic nucleus (SCN) containing hypothalamic region. Interestingly, SCN neurons with Scn2a deficiency display abnormal hyperexcitability and irregular firing intervals, suggesting a possible cellular mechanism underlying the sleep disturbance phenotype. Moreover, RNA-sequencing (RNA-seq) analysis found that Scn2a deficiency resulted in a widespread change of gene expression including the core clock genes Per1 and Per2. Using a transcriptome-based compound discovery approach, we further identified a putative glutamate receptor modulator HU-211 that partially reverses the sleep disturbances of the Scn2a-deficient mice. Together, our study may inform the development of pharmacogenetic therapeutics to intervene in sleep disturbances related to SCN2A deficiency.
Results
Scn2a-deficient mice display disrupted sleep patterns.
Clinical studies have found that children carrying SCN2A loss-of-function or protein-truncating variants (collectively known as SCN2A deficiency) often have sleep problems (Crawford et al., 2021). To understand how Scn2a deficiency affects sleep in mice, we utilized a Scn2a-deficient mouse model generated using a gene-trap strategy (homozygous for the gene-trap is referred to as Scn2a-deficient or Scn2atrap mice) (Eaton et al., 2021) (Figure S1A). We performed video-electroencephalogram (EEG)/electromyography (EMG) recording to continuously monitor freely moving male and female mice (Figure S2A) (Soltani et al., 2019). With video confirmation, EEG/EMG waveforms were classified in 10-sec epochs as i) wakefulness (low-voltage, high-frequency EEG; high-amplitude EMG) (Figure S2B); ii) non-rapid-eye-movement (NREM) sleep (high-voltage, mixed-frequency EEG; low-amplitude EMG) (Figure S2C); or iii) rapid-eye-movement (REM) sleep (low-voltage EEG with a predominance of theta activity [~4–8 Hz]; low-amplitude EMG) (Figure S2D) (Chen et al., 2018). Notably, we observed that Scn2a-deficient mice had abnormal sleep patterns with increased wakefulness and reduced NREM sleep (Figure 1A). In particular, we found that Scn2a-deficient mice had a significant increase in wakefulness during both light and dark cycles (Figure 1B). This increased wakefulness seems to be accompanied by the reduction of NREM sleep (Figure 1C). The REM sleep between WT and Scn2a-deficient mice, however, was largely unchanged (Figure 1D). Notably, heterozygous (HET) Scn2a-deficient mice (HET) with a reduction of Scn2a expression to ~50% (Figure S3A–B) of the WT level do not have an observable sleep disturbance phenotype (Figure S3C–E).
Figure 1. Scn2a-deficient mice display abnormal sleeping patterns.
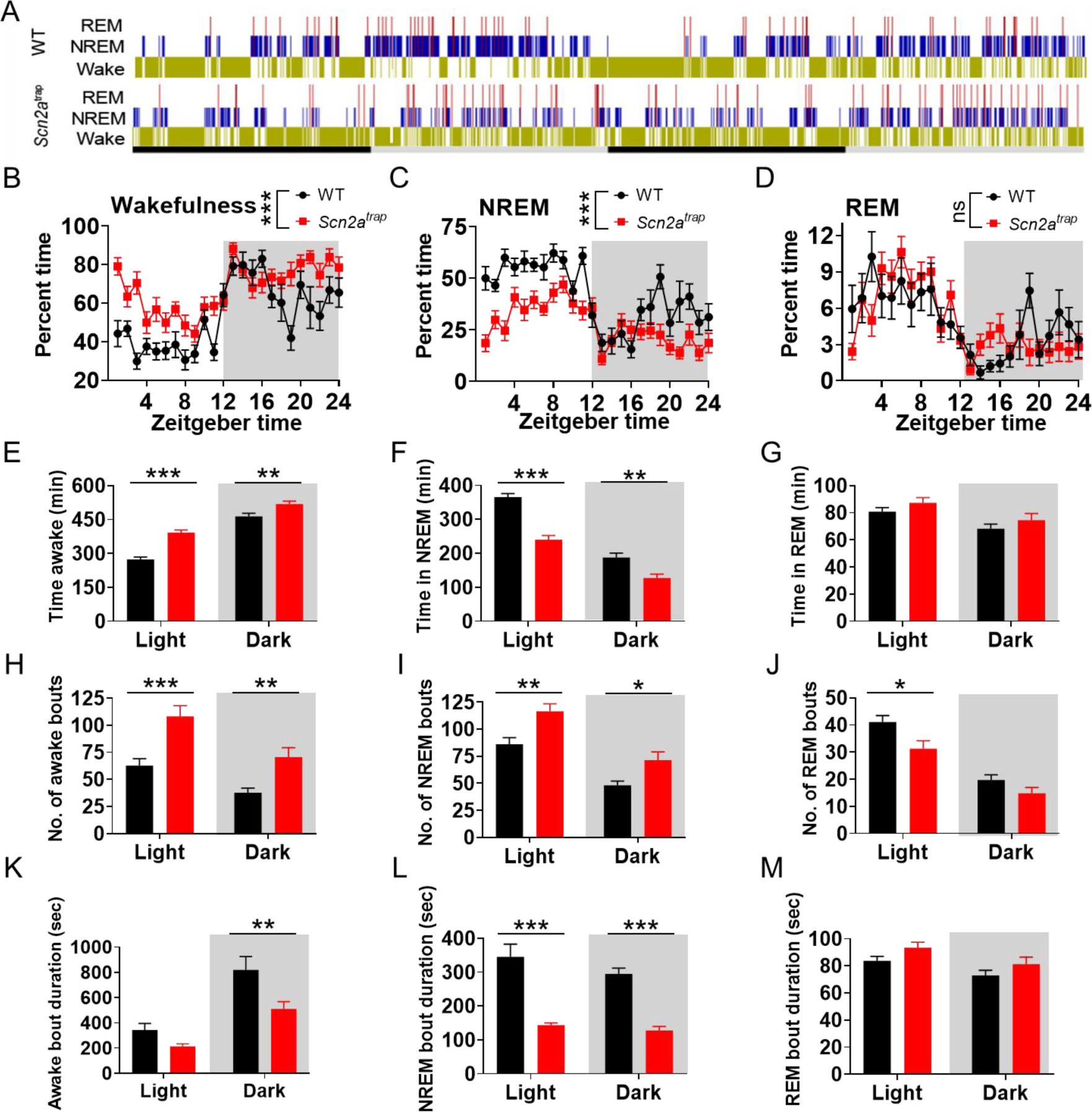
A, representative 48h long hypnograms of ultradian rhythms over the diurnal light/dark phase in freely moving WT and Scn2a-deficient (Scn2atrap) mice, yellow: wakefulness (Wake); blue: NREM; red: REM. B-D, Time spent in wakefulness (B), NREM (C), and REM (D) states showed as a percentage of recording time per hour. E-G, Time spent in wakefulness (E), NREM (F), and REM (G) in light (left) and dark (right) phases among WT and Scn2atrap mice, respectively. H-J, Number of bouts in wakefulness (E), NREM (F), and REM (G) in light (left) and dark (right) phases among WT and Scn2atrap mice, respectively. K-M, Duration of bouts in wakefulness (K), NREM (L), and REM (M) in light (left) and dark (right) phases among WT and Scn2atrap mice, respectively. No sex difference was found between males and females thus the data were combined for presentation. Data were analyzed by two-way ANOVA with Bonferroni’s multiple comparisons test. All data are presented as means ± SEM (*p<0.05; **p<0.01; ***p<0.001), n= minimum of 9 WT and 11 Scn2atrap mice.
To gain a deeper understanding of the sleep-wake profiles of Scn2a-deficient mice, we determined the percent of the time, epoch, number, and mean duration of bouts/episodes among wakefulness, NREM, and REM in both light and dark periods (Figure 1E–M, and S3C–E). Scn2a-deficient mice had significantly increased time/epochs in wakefulness and increased number of bouts in both light and dark cycles, while the duration of bouts was decreased (Figure 1E, 1H, 1K, S3C). On the other hand, the time and epochs of NREM in Scn2a-deficient mice were decreased, the number of bouts in NREM sleep was increased, and the duration of the bout was decreased (Figure 1F, 1I, 1L, and S3D). Changes in REM sleep were also observed, but at a smaller scale (Figure 1G, 1J, 1M, and S3E). These data collectively indicate that the sleep pattern of Scn2a-deficient mice had more fragmentation compared with that of WT mice. We also quantified the power density of wakefulness, NREM, and REM states (Figure S2E–G and Figure S3F–H). We identified an increase in the power density of the delta band (~0–4 Hz) in the awake state during the dark cycle and a decrease in the power density of the delta band in NREM sleep during the light cycle in Scn2a-deficient mice. Furthermore, we also observed an increase in the power density of the theta band (~4–8 Hz) in NREM and REM states during the light cycle, as well as an increase in the power density of the delta band in the REM state during the dark cycle (Figure S2E–G and Figure S3F–H). No notable difference was observed in the three vigilance states (wakefulness, NREM, REM) of heterozygous (HET) Scn2a-deficient mice compared with WT mice (Figure S3F–H). Our data indicate that only homozygous (but not heterozygous) Scn2atrap mice have major sleep disturbances.
The suprachiasmatic nucleus (SCN) of the hypothalamus is a major locus that regulates circadian rhythms and sleeps in the brain (Hastings et al., 2018; Mohawk et al., 2012; Saper et al., 2005). The expression of Scn2a has been found in SCN-containing regions (Wen et al., 2020; Zeisel et al., 2018). This led us to test whether the sleep abnormalities observed in Scn2a-deficient mice may be related to the dysregulation of Scn2a function specifically in the SCN-containing regions. To this end, we studied SCN-containing region-specific manipulation of Scn2a expression in our mice. In particular, we first obtained Scn2a “conditional ready” mice with Exon 2 and 3 of Scn2a floxed by LoxP sites (Scn2a-tm1c or Scn2afl/fl mice) (Figure S1A). We then achieved brain region-specific conditional deficiency of Scn2a with local AAV-Cre injection. Scn2a-tm1c mice were identified by genotyping (Figure S1B), and indeed had similar Scn2a expression levels compared to that of the WT mice (Figure S1C–D), validating this genetic modification strategy. AAV-Cre was microinjected into the SCN containing region, followed by a video-EEG/EMG recording (Figure 2A–B). Interestingly, we found that mice with Cre recombinase-induced Scn2a deficiency in the SCN-containing region had increased wakefulness and reduced NREM sleep (Figure 2C–H), partially mimicking the phenotypes observed in global Scn2a-deficient mice. Cre-mediated Scn2a-deficient mice also had significantly increased bouts of wakefulness and NREM in the light cycle with no major difference in REM sleep compared to mice with vehicle injection (Figure 2I–K). The average bout duration in NREM sleep was significantly decreased during both the light and dark cycles in Cre-mediated Scn2a-deficient mice compared to mice with vehicle injection, whereas those in wakefulness and REM remain largely unchanged (Figure 2L–N). Nesting is suggested to be related to sleep behavior (Eban-Rothschild et al., 2016). Cre-mediated Scn2a deficiency in the SCN-containing region of the mice also severely impaired nesting behaviors (Figure S4A–B), which has been previously observed in our global Scn2a-deficient mice (Eaton et al., 2021). Together, our data suggest an important role of Scn2a in the SCN-containing region, which regulations many aspects of sleep-related behaviors.
Figure 2. Cre recombinase-mediated Scn2a deficiency in SCN containing region produces sleep disturbance.
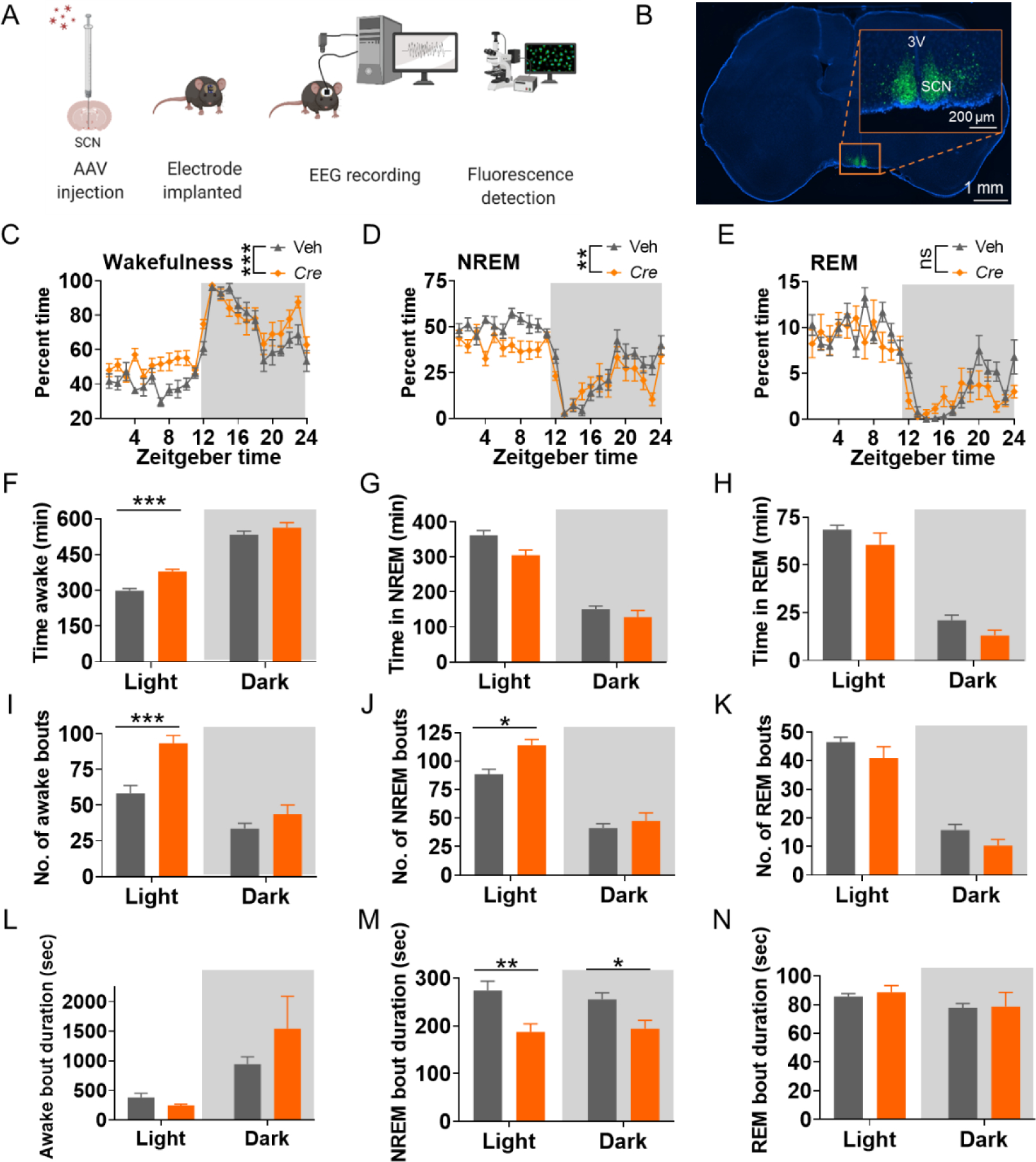
A, Scheme of the conditional knockout strategy and EEG recording procedure for the mice. B, brain slice imaging of Cre-virus injection into the SCN containing region and enlarged imaged showing SCN region (3V: third ventricle). C-E, Time spent in wakefulness (C), NREM (D), and REM (E) showed as a percentage of recording time per hour. F-H, Time spent in wakefulness (F), NREM (G), and REM (H) of tm1c mice with Cre-virus (Cre) or control virus (Veh) injection. I-K, Number of bouts in wakefulness (I), NREM (J), and REM (K) of tm1c mice with Cre-virus (Cre) or control virus (Veh) injection. L-N, Duration of bouts in wakefulness (L), NREM (M), and REM (N) of tm1c mice with Cre-virus (Cre) or control virus (Veh) injection. Data were analyzed by two-way ANOVA with post-hoc comparisons using Bonferroni’s correction. Veh: vehicle. Cre: mouse with Cre virus injection in SCN containing region to achieve brain region-specific Scn2a deficiency. Data are presented as means ± SEM (*p<0.05; **p<0.01; ***p<0.001), n= minimum of 12 vehicle and 14 Cre mice.
Scn2a-deficient mice show impaired wheel-running activity.
Disrupted sleeping patterns are often associated with an impairment of circadian rhythms (Borbely et al., 2016). To determine explicitly whether the circadian rhythm behavior is defective in Scn2a-deficient mice, we utilized a wheel-running test (Figure 3A), which measures the rhythmic locomotor activity over time (Ingiosi et al., 2019). A neurotypical WT mouse runs on the wheel during the dark cycle when they are awake, but does not run much during the light cycle when they are asleep. As expected, WT mice had a rhythmic change of running activity, peaking during the middle of the dark cycle, largely following a sine wave pattern (Figure 3B, 3D). Although the Scn2a-deficient mice still had higher running activity in the dark cycle per se, the running activity of the Scn2a-deficient mice in the light cycle was higher than that of WT with a decreased sine wave pattern (Figure 3C, 3E, 3F). Moreover, the R2 for sine-wave fitting in Scn2a-deficient mice was significantly smaller than that in WT mice, indicating a larger heterogeneous running activity and weaker circadian rhythm in the Scn2a-deficient mice (Figure 3G). JTK_CYCLE analysis was performed to calculate the circadian-related parameters such as the period and phase (Hughes et al., 2010). While the 24-hour circadian period remained similar between WT and Scn2atrap mice (Figure 3H), we observed that phase (peak of the relative wheel-running activity) of Scn2a-deficient mice was delayed for about 2 h, compared to a phase located to ~5.5 h for WT mice (Figure 3I). Furthermore, we also noted that heterozygous (HET) mice show no detectable differences in wheel-running behavior compared to WT mice (Figure S5A–B). Our data thus demonstrate that the circadian rhythms of the Scn2a-deficient mice are potentially disturbed.
Figure 3. Scn2a-deficient mice show impairment of wheel-running activity in light-dark conditions.
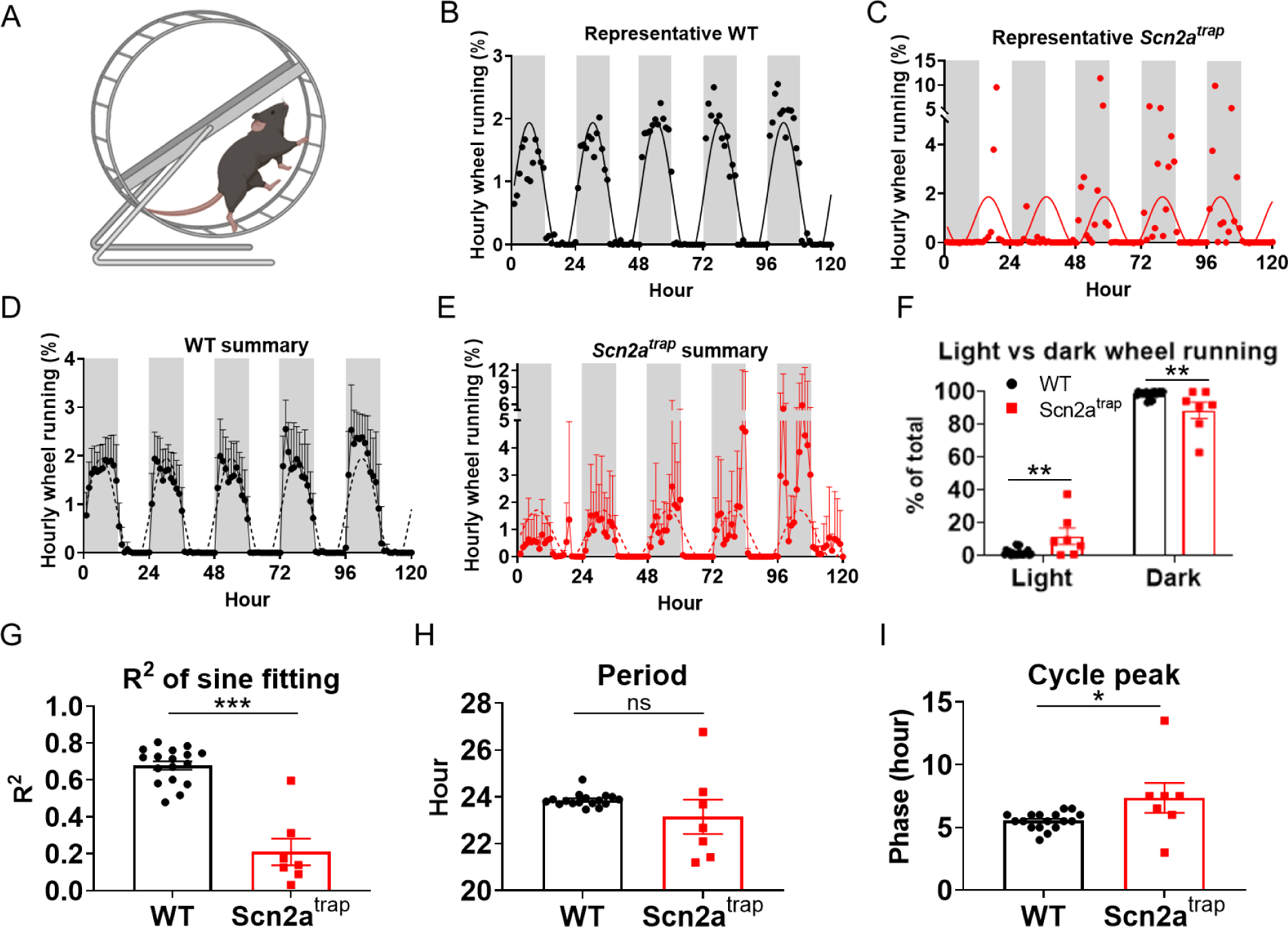
A, Illustration of the wheel running assay. B-C, Representative relative wheel-running activity and sine wave fitting for WT (B), and Scn2atrap mice (C). Individual dots show the normalized activity per hour for WT, and Scn2atrap mice, solid lines show the sine wave fitting of the data for WT and Scn2atrap mice. D-E, Averaged relative wheel-running activity and sine wave fitting for WT and Scn2atrap mice. Gray shading represents the dark period. Dash line shows the sine wave fitting. F, Distribution of wheel-running activity in the light and dark phases show an increased percentage in light phase wheel-running activities of Scn2atrap mice. G, the R2 comparison of sine wave fitting for WT and Scn2atrap mice was shown. H, Period of the wheel-running activity is not different between Scn2atrap and WT mice. I, Cycle peak or phase of the wheel-running activity shows a notable difference in Scn2atrap mice compared to WT mice. No sex difference was found between males and females thus the data were combined for presentation. Data were analyzed by unpaired student’s t-test and presented as means ± SEM (*p<0.05; **p<0.01; ***p<0.001; ns p>0.05), n= minimum of 17 WT and 7 Scn2atrap mice.
Scn2a deficiency causes drastic changes in the firing pattern of SCN neurons.
The SCN is a critical brain region involved in sleep and circadian rhythms (Borbely et al., 2016; Saper et al., 2005), and our data showed that the Cre recombinase-mediated Scn2a deficiency in SCN containing region resulted in an altered sleep pattern. Thus, we asked how the function of SCN neurons could be affected by Scn2a deficiency, while acknowledging that other brain regions can regulate the sleep process as well (Saper et al., 2005). As the majority of SCN neurons fire action potentials spontaneously (Harvey et al., 2020), we used a cell-attached configuration to record the activity of these neurons in the SCN core (Figure 4A). SCN neurons from WT mice displayed clear spontaneous firing, averaged at 2.42 ± 0.32 Hz (Figure 4B–C). Interestingly, SCN neurons in Scn2a-deficient mice fired at a significantly higher frequency of 3.61 ± 0.32 Hz (Figure 4B–C). Accordingly, the inter-spike interval was significantly reduced. Since we noticed that the spike firing pattern in Scn2a-deficient mice seemed to be less rhythmic (Figure 4B), we plotted the distribution of spike intervals for both WT and Scn2a-deficient mice. We noticed that the action potential spike interval of neurons in Scn2a-deficient mice was highly enriched in the <500 ms range, whereas the number of intervals at >500 ms range was reduced (Figure 4D–E). Our data thus suggest firing irregularities of Scn2a-deficient mice compared to WT mice, which may contribute to an irregular circadian rhythm and abnormal sleep patterns.
Figure 4. Scn2a deficiency disrupts the spontaneous firing of SCN neurons.

A, Illustration of the recording of SCN neurons. (P: pipette; 3V: The third ventricle; SCN: suprachiasmatic nucleus; scale bar, 200 μm). B, Representative cell-attached patch traces from neurons of WT or Scn2atrap mouse. Scale bar, 1s. C, Spontaneous AP frequencies of SCN neurons from WT and Scn2atrap mice during the light cycle (ZT2–8, WT: 6 slices from 3 mice; Scn2atrap: 6 slices from 3 mice). Scn2a deficiency resulted in an increased firing frequency. D, Illustration of two spontaneous firing events from a typical SCN neuron showing trough, peak, and interval; scale bar, 50 pA and 5 ms. E, Intervals distribution plot showing enrichment in low intervals (<500 ms) and reduction in high intervals (>500 ms) of neurons from Scn2atrap mice compared to those from WT mice. Data are presented as means ± SEM (student’s t-test; **p<0.01).
RNA-sequencing and qPCR analyses reveal disrupted circadian entrainment pathways and altered core clock genes expression.
To understand the possible molecular mechanisms underlying Scn2a deficiency-related phenotypes, we conducted RNA sequencing analysis (Zhang et al., 2021). Statistically significant differentially expressed genes (DEG) between WT and Scn2a-deficient mice show an enrichment of the circadian entrainment pathway (Figure 5A). A heatmap of the DEG from all four WT and Scn2a-deficient mice (Figure 5B, Figure S6A, S6B) shows segregation of gene expression. Of particular interest, we found that the expressions of core clock genes including Per1 and Per2 were significantly reduced (71% and 69% of the WT value, respectively). Moreover, the protein-protein interaction map of the DEGs revealed three clusters of pathways, including circadian rhythms, cGMP-PKG/MAPK signaling, and glutamatergic synapse clusters (Figure 5C).
Figure 5. Scn2a deficiency results in an altered circadian entrainment pathway and change of core clock genes expressions.
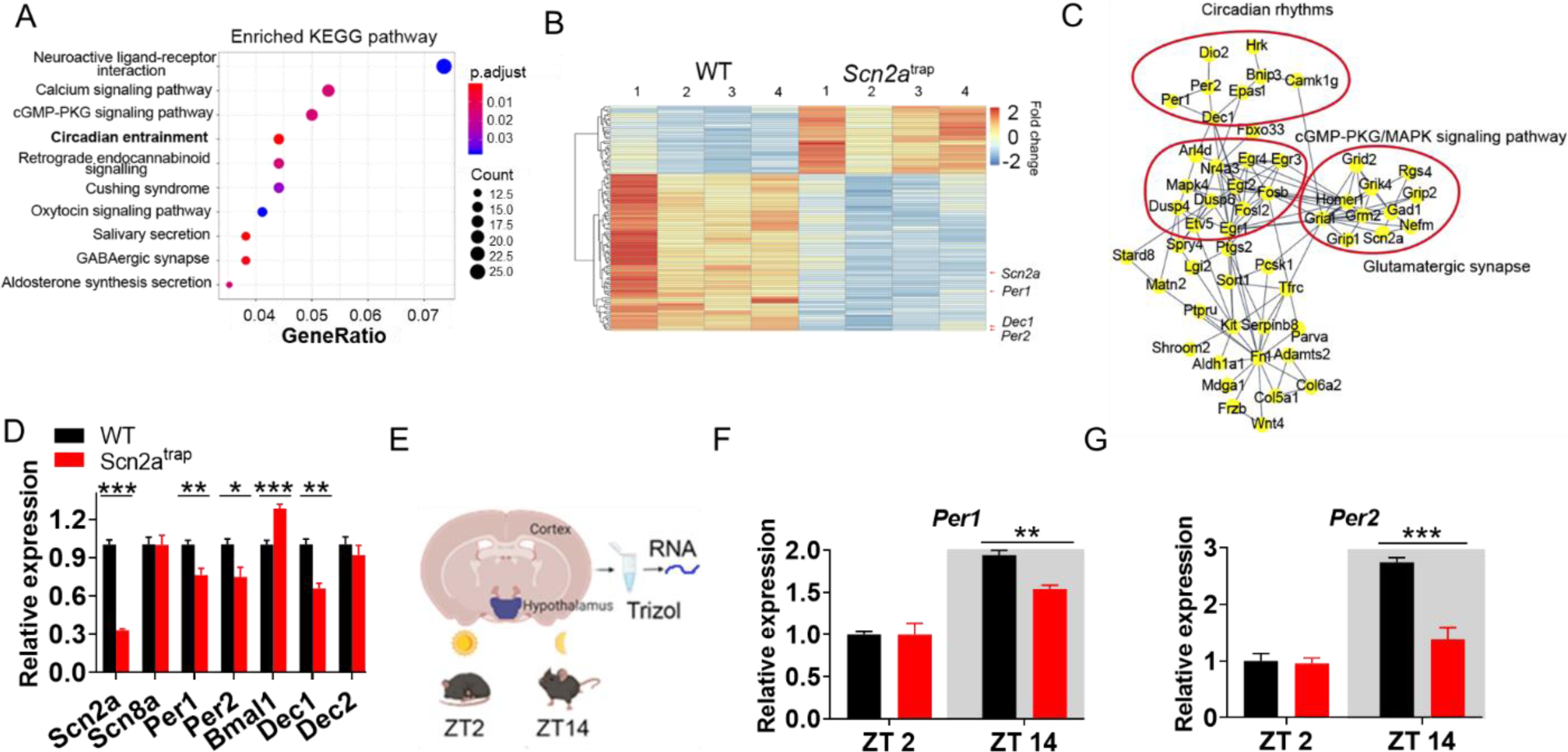
A, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showing altered pathways based on differentially expressed genes (DEG) between WT and Scn2atrap groups. Note that the circadian entrainment pathway is ranked fourth. B, Heatmap of the top 100 DEG between WT vs Scn2atrap groups (ranking by q-value), showing Scn2a, Per1, Per2, and Dec1 as highly downregulated genes. The heatmap also shows a significantly different expression signature between WT and Scn2atrap mice. C, Protein-protein interaction map of the DEG between WT and Scn2atrap, showing circadian rhythms, cGMP/PKG/MAPK signaling pathway, and glutamatergic synapse clusters. D, qPCR analysis of relative mRNA expression of Scn2a, Scn8a, Per1, Per2, Bmal1, Dec1, and Dec2 from both WT and Scn2atrap mice. E, Illustration of dissected brain regions for qPCR analysis at two different time points. F, The Per1 mRNA expression at ZT2 and ZT14 in the SCN-containing hypothalamus of WT and Scn2a-deficient mice. G, The Per2 mRNA expression at ZT2 and ZT14 in the SCN containing hypothalamus of WT and Scn2a-deficient mice. Unpaired student’s t-test. Data are presented as means ± SEM (*p<0.05; **p<0.01; ***p<0.001), n= minimum of 4 WT and 3 Scn2atrap mice.
To validate the RNA-seq results, especially the change of expression in core clock genes, we performed qPCR experiments in an independent set of samples that were different from the samples used for RNA sequencing. Our data confirmed that the expression of Scn2a was markedly reduced (sample collected at ZT12-ZT14, Figure 5D). NaV1.6 and NaV1.2 are two major sodium channels often working in a coordinated fashion in principal neurons of the CNS (Bunton-Stasyshyn et al., 2019; Lopez-Santiago et al., 2017). Interestingly, our result shows that the expression of Scn8a was largely unchanged in Scn2a-deficient mice. Notably, we revealed that the expressions of canonical core clock genes were significantly changed in Scn2a-deficient mice, with decreased expression in Per1, Per2, and Dec1, and increased expression in Bmal1 (Figure 5D). To understand whether the expressions of core clock genes may lose their signature rhythmic expression in Scn2a-deficient mice, we further tested the expression of Per1 and Per2 at two-time points (ZT2 in the light cycle and ZT14 in the dark cycle) (Figure 5E). These core clock genes are expected to have high expression levels in the dark cycle (ZT14) and low expression levels in the light cycle (ZT2) (Kohsaka et al., 2007). Interestingly, we found that the expressions of both Per 1 and Per 2 were reduced in the dark cycle of Scn2a-deficient mice compared to the WT mice (Figure 5F–G), suggesting that the rhythmic expressions of core clock genes were disrupted. As sleep patterns are closely associated with circadian entrainment, our data suggest that abnormal expression of core clock genes related to circadian rhythms could be a molecular mechanism underlying the altered sleep pattern of Scn2a-deficient mice.
Putative glutamate receptor modulator dexanabinol (HU-211) partially reverses the abnormal sleep phenotypes.
Transcriptome-based compound discovery approaches have emerged as a novel strategy for the identification of molecules with therapeutic potential via a connective map (CMAP) analysis (Dhindsa et al., 2021; Lamb et al., 2006; Musa et al., 2018; Paranjpe et al., 2019; Shukla et al., 2021; Subramanian et al., 2017). Analyzing the transcriptome of a “diseased transcriptome” and “reference transcriptome” obtained from the treatment of different molecules, this approach identifies compounds that may reverse the “diseased” transcriptome. Applying this algorithm to our RNA-seq data, we identified a list of perturbagen molecules that displayed a negative tau score of < −90, with a score close to −100 value indicating a better reversal of the “diseased” transcriptome (Figure 6A). As our RNA-seq analysis revealed that the endocannabinoid (Figure 5A) and glutamate-related pathways (Figure 5C) were altered, we were particularly interested in one of the compounds on the list, dexanabinol (HU-211). This cannabinoid derivative has been suggested to be a putative glutamate receptor modulator (Figure 6B), which was studied in clinical trials (Eshhar et al., 1993; Shohami et al., 1993; Striem et al., 1997). It does not appear to be sedative and has been shown to induce locomotor hyperactivity in rodents (Eshhar et al., 1993; Shohami et al., 1993).
Figure 6. Putative glutamate receptor modulator dexanabinol (HU-211) partially reverses the sleep disturbance phenotypes.
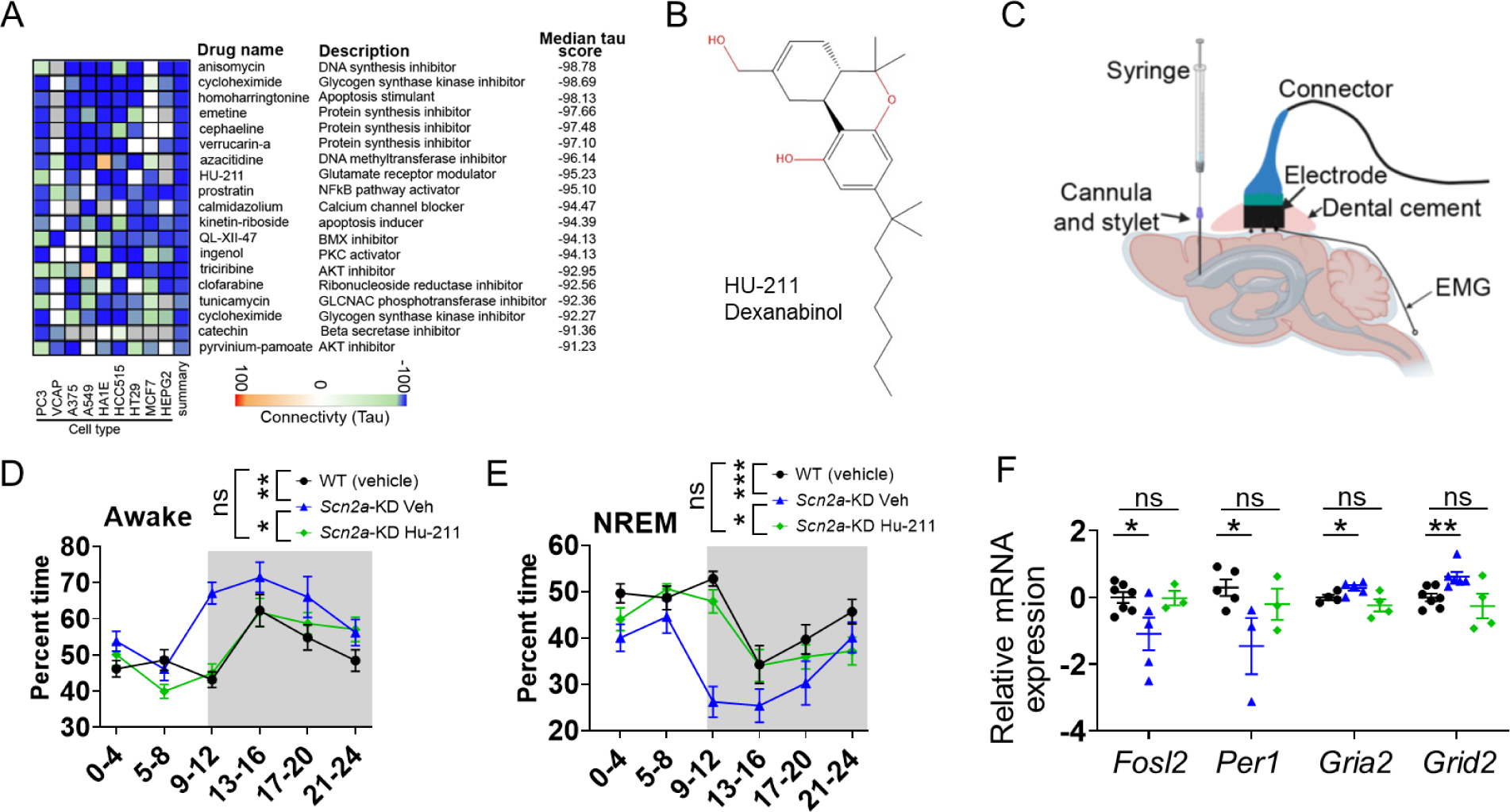
A, Connectivity map analysis of the top 100 DEGs identified by RNA-sequencing predicts the compounds that may reverse the altered transcriptome. B, Structure of the HU-211. C, Schematic for the intraventricular injection to administer HU-211 and implantation for EEG electrodes on the skull of the mice. D, Time spent every four hours for the wakefulness showing HU-211 was able to partially reduce wakefulness in Scn2a-KD mice towards the WT level. E, Time spent in every four hours for the NREM sleep showing HU-211 were able to partially increase NREM sleep of Scn2a-KD mice towards the WT level. Repeated measures two-way ANOVA with post-hoc pairwise comparisons using Bonferroni’s multiple comparisons test with n= minimum of 13 WT (vehicle), 11 Scn2a-KD (vehicle), and 10 Scn2a-KD (HU-211). F, qPCR assay of the expression patterns of Fosl2, Per1, Gria2, and Grid2 genes in hypothalamus samples of Scn2a-KD mice treated with vehicle, Scn2a-KD mice treated with HU-211, and WT mice treated with vehicle (student’s t-test). Black: WT control with vehicle injection; Blue: Scn2a-KD mice with vehicle injection; Green: Scn2a-KD mice with HU-211 injection. Data are presented as means ± SEM (*p<0.05; **p<0.01; ***p<0.001; ns p>0.05).
To experimentally test the effect of HU-211 on sleep behaviors, a head-mount for EEG/EMG recordings was implanted in the Scn2a-knockdown (KD mice achieved by virus injection) together with a cannula. HU-211 (2.5 mg/ml in 2 μl) was given to the mice via intraventricular injection into the cannula every 24 hours for 3 days (Shohami et al., 1993) (Figure 6C). After that, the sleep patterns of the mice were examined for two days. Since Scn2a deficiency mainly increases wakefulness and reduces NREM sleep, we focused our analysis on these two states. Interestingly, we found that HU-211 was able to reduce wakefulness of the Scn2a-KD mice towards WT level compared with vehicle treatment, especially during the light cycle when mice normally sleep (Figure 6D). Correspondingly, the NREM sleep was increased in Scn2a-KD mice with HU-211 treatment (Figure 6E). We also collected the SCN-containing tissues of these mice to quantify the expression of multiple genes identified by CMAP analysis using qPCR. We noted that the expressions of multiple circadian-related genes (Fosl2, Per1, Ptgs2, Egr3) that were differentially expressed in the Scn2a-deficient mice were shifted toward WT level after the HU-211 treatment (Figure 6F & Figure S7). Furthermore, we revealed that the HU-211 treatment could also affect the expression of glutamate receptors Gria1, Gria2, Grid2, and Grik4 at a transcriptional level (Figure 6F & Figure S7). Taken together, our data suggest that HU-211, a candidate molecule identified from transcriptome-based compound discovery, was able to partially rescue the sleep disturbance phenotypes and gene expression pattern of Scn2a-KD mice.
Discussion
Here we report that Scn2a deficiency in mice results in increased wakefulness and reduced NREM sleep, accompanied by increased fragmentations of sleep. These abnormal sleep phenotypes can be partially recapitulated by Cre recombinase-induced deficiency of Scn2a in the SCN-containing region, a main brain area associated with the regulation of circadian rhythms and sleep. Our data further reveal that spontaneously firing neurons in SCN with Scn2a deficiency have altered excitability and interval of firing, likely serving as a cellular mechanism underlying impaired sleep and circadian rhythm. Consistent with these findings, we identified circadian entrainment as one of the leading pathways altered in Scn2a-deficient mice using RNA-seq. Utilizing a transcriptome-based compound discovery approach, we demonstrated that HU-211, a putative glutamate receptor modulator, can partially rescue the sleep disturbance phenotypes.
Voltage-gated sodium channels NaV1.1, NaV1.2, and NaV1.6, encoded by Scn1a, Scn2a, and Scn8a, respectively, are the three main sodium channels expressed in the adult CNS that mediate action potential firing. Because of their important roles in neurons, not surprisingly mutations of these channels are involved in many types of neurodevelopmental disorders (Meisler et al., 2021). NaV1.2 and NaV1.6 are suggested to be predominantly expressed in principal neurons, whereas NaV1.1 plays important roles primarily in interneurons (Han et al., 2012a; Hu et al., 2009). Mice with Scn8a/NaV1.6 loss-of-function mutations display increased NREM sleep and impaired REM sleep generation and quality (Papale et al., 2010). On the other hand, Scn1a+/− mice have a fragmented rhythm of NREM sleep and an elongated circadian period (Sanchez et al., 2019). These mice display reduced theta power related to NREM sleep (Kalume et al., 2015) and have dramatic impairments in intercellular signaling in the SCN including circadian deficits (Han et al., 2012b). A study on the knockin mouse model carried R1648H loss-of-function mutation in Scn1a revealed increased wakefulness, as well as reduced NREM and REM sleep during the dark phase (Papale et al., 2013). It is worth noting that the impaired sleep phenotypes we identified from the Scn2a/NaV1.2 deficient mice are quite distinct. While Scn2a and Scn8a may have overlapping expression patterns in certain neurons, Scn8a deficiency led to an increased NREM duration of the total sleep, but Scn2a deficiency resulted in decreased NREM sleep. A reduced power density in NREM and REM sleep was reported in Scn1a+/− mice, but we noted an increase in the power density in Scn2a-deficient mice. These differences in sleep and circadian rhythms observed in mouse models of Scn1a, Scn2a, and Scn8a may suggest different roles of these sodium channels in sleep and circadian homeostasis. The availability of diverse models thus allows further comparisons to identify distinct or convergent mechanisms underlying neurodevelopmental disorder and its sleep comorbidity to develop targeted therapeutics.
The conventional paradigm suggests that Scn2a deficiency impairs neuronal excitability, but we and others recently reported that Scn2a-deficient principal neurons of the cortico-striatal circuit display an unexpected elevation of neuronal excitability (Spratt et al., 2021; Zhang et al., 2021), to an extent similar to the phenotypes of Scn2a gain-of-function variant (Que et al., 2021). In this current study, we found that SCN neurons in Scn2a-deficient mice, which are suggested to be the pacemaker of the circadian clock, display an increased firing frequency. It is worth pointing out that without proper genetic labeling, the exact identity of the neurons we patched is not known. However, as we were mainly focusing on the neurons in the SCN core region, these neurons we studied were likely to be vasoactive intestinal peptide (VIP) neurons (Abrahamson and Moore, 2001; Dibner et al., 2010; Kawamoto et al., 2003; Mohawk et al., 2012; Zhang et al., 2009). The finding of increased firing in these SCN neurons is quite remarkable, considering SCN neurons are functionally very different compared to cortical and striatal neurons. Additionally, our detailed analysis further reveals that the regularity of spike interval was altered in Scn2a-deficient SCN neurons. This is interesting as it was suggested that temporal coding (timing or pattern of firing) can affect the synaptic plasticity to control sleep (Harvey et al., 2020; Tabuchi et al., 2018). Indeed, NaV1.2 is suggested to contribute to the backpropagation of action potential in cortical pyramidal neurons, which could be essential for synaptic plasticity (Spratt et al., 2019). However, whether NaV1.2 also plays a similar role in SCN neurons remains to be determined. Regardless, the abnormal distribution of spike intervals observed in the Scn2a-deficient mice thus may indicate a temporal coding alteration, potentially contributing to the abnormal circadian rhythms and sleep disturbance phenotypes.
It is not intuitive how the deficiency of NaV1.2, a transmembrane protein, may result in the disrupted expression of core clock genes related to sleep and circadian rhythm. While we did not have direct evidence to elucidate a complete pathway, our interaction map analysis pointed to an interesting hypothesis that deficiency of the NaV1.2 may affect the expression of core clock genes via the MAPK/ERK pathways, which could phosphorylate factors that are known to transcriptionally regulate core clock genes (Hirayama et al., 2005; Karin, 1995; Tao et al., 2015; Yap and Greenberg, 2018). The MAPK/ERK pathway, interestingly, is found to be important in multiple mouse models related to neurodevelopmental disorders. Inhibition of MAPK-interacting kinases (NMKs) was shown to ameliorate behavioral abnormalities in an Nlgn3-loss mouse model related to ASD (Hornberg et al., 2020) as well as Fmr1 knockout mice related to Fragile X syndrome (Gkogkas et al., 2014). Notably, sleep disruption is also presented in these mouse models (Boone et al., 2018; Thomas et al., 2017). An altered MAPK/ERK signaling pathway may be a potential convergent mechanism underlying genetically distinct mouse models related to neurodevelopmental disorders that display sleep abnormalities.
Transcriptome-based compound discovery has emerged as a novel approach to identify promising molecules with therapeutic potential based on the “reversal” of the gene expression profile for many different types of disorders (Lamb et al., 2006; Paranjpe et al., 2019; Shukla et al., 2021; Siavelis et al., 2016; Subramanian et al., 2017; Szymczak et al., 2021). Here in this study, we applied this approach to study Scn2a deficiency-related phenotypes. We were able to observe a partial rescue of the abnormal sleep phenotypes by HU-211, suggesting the utility of such an approach for intervening in sleep disturbance. While the exact target and mechanism of HU-211 remain to be determined, we suspect that directly or indirectly modulation of glutamate receptor activities by HU-211 may impact sleep homeostasis, considering glutamate concentration and glutamate receptor activity are highly correlated with wakefulness and NREM sleep (Ferraro et al., 1997; Minzenberg and Carter, 2008). While the identification of molecules to alleviate the phenotype using this novel transcriptome-based compound discovery is interesting, there are potential limitations worth mentioning. First of all, the initial “reference” dataset of this analysis was generated with cell lines tailored for cancer research. Thus, the results are likely to be biased towards molecules that regulate signaling pathways associated with cancer biology, potentially limiting “matches” that can be calculated by the algorithm with transcriptome from brain tissues. Second, this method is solely based on gene expression and is disease type/phenotype agnostic. As Scn2a deficiency could cause a range of distinct cellular and behavioral abnormalities, it is not known to what extent the identified molecules may affect the wide array of cell types and disease phenotypes. Third, RNA-seq was performed on cortex samples due to practical consideration. As the SCN brain region is very small, it is hard to accurately target or obtain only the SCN region for study. However, we partially mitigated this issue by further validating the results using SCN-containing hypothalamic samples. Fourth, while HU-211 is likely to rescue the AP phenotype in the Scn2a-deficient neurons of SCN, our current study did not provide direct evidence regarding the effect of HU-211 on AP firing, which could be a possible future experiment. However, while limitations exist, our study provided proof-of-principle evidence that this transcriptome-based compound discovery approach might be useful in the study of monogenic neurodevelopmental disorders related to Scn2a deficiency. Our study calls for additional investigation to further demonstrate the utility of this approach in different neurological disorders and distinct disease models.
Conclusions
In summary, we revealed that Scn2a deficiency results in impaired sleep architecture and circadian rhythm in mice at the molecular, cellular, and behavioral levels. We further demonstrated that a transcriptome-based compound discovery approach was able to identify a molecule that partially reverses the abnormal sleep phenotypes. Our findings may provide insights into the development of novel disease-modifying agents to treat the neurodevelopmental phenotypes and co-morbidities related to Scn2a deficiency.
Materials and Methods.
Animals.
All experimental procedures were approved by the Purdue University Institutional Animal Care and Use Committee (IACUC) and Peking University Health Science Center. Mice were housed under a 12:12 hour light/dark photoperiod with ad libitum access to food and water and were maintained at a consistent temperature and humidity. C57BL/6N-Scn2a1tm1aNarl/Narl (here we refer to as Scn2atm1a/tm1a, Scn2atrap or Scn2a-deficient) mice were used in this study. Scn2aWT /tm1a (HET) mice were used as breeding pairs to provide offspring for testing. We crossed the Scn2aWT/tm1a with mice (B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ, Flpo mice, Jackson lab) in which codon-optimized flippase (FlpO) was expressed from preimplantation onward to achieve extensive gene recombination in the developing germline and generate the Scn2aWT/tm1c mice. Scn2aWT/tm1c were used as breeding pairs to get Scn2atm1c/tm1c mice. The resulting Scn2a-tm1c (Scn2afl/fl) mice were then backcrossed to C57BL6N mice for five or more generations before the study. Both male and female mice were used. No significant sex differences were observed in this study, thus data from both sexes were combined for statistical analysis. Experiments were conducted in a blind manner whenever possible.
Assessment of sleep-wake behavior.
Recording Implants.
Electroencephalograph (EEG) and electromyography (EMG) electrodes were implanted in mice by using a prefabricated headmount (Pinnacle Technologies), which was stabilized by four stainless steel epidural screws electrodes. The first two electrodes (frontal and ground) were located 0.5 mm posterior to bregma and 1.5 mm on either side of the central suture. The second two electrodes (parietal and common reference) were located 4.5 mm posterior to bregma and 1.5 mm on either side of the central suture. Electrical continuity between the screw electrode and head mount was achieved with silver epoxy. EMG activity was monitored using stainless-steel Teflon-coated wires inserted bilaterally into the nuchal muscle. The head mount (integrated 2×3 pin grid array) was secured to the skull with dental acrylic. Mice were allowed to recover for at least seven days before recording.
Stereotaxically guided Scn2a deficiency/knockdown virus injection and intracerebroventricular injection surgery.
For Scn2a conditional genetic manipulating experiments, injections of adeno-associated viruses (pENN.AAV.hSyn.HI.eGFP-Cre.WPRE.SV40, 105540, here referred to as Cre-virus) and saline (vehicle) were given to Scn2atm1c/tm1c mice. In brief, Scn2atm1c/tm1c mice were injected with the virus seven days before the electrode was implanted. A single craniotomy was made with the following stereotaxic coordinates: medial/lateral (ML): ±0.22 mm; anterior/posterior (AP): −0.5 mm; dorsal/ventral (DV): −5.3 mm from lambda. Injections were made with a Micro Syringe Pipette (RWD) and UMP3 UltraMicroPump system (World Precision Instruments) for a total volume of 250 nl.
For HU-211 experiments, mice (6–8 weeks) were deeply anesthetized by isoflurane (3–5% for induction and 1–1.5% for maintenance) and secured in the stereotaxic apparatus (RWD). 250 nl of AAV carrying Scn2a knockdown (Scn2a-KD) or control virus bilaterally injected into SCN regions: ML: ±0.22 mm; AP: −0.5 mm; DV: −5.3 mm. Guide cannulas were made of 26 g needles and 1 × 2 mm fixed plastic holders; dummy and injection cannulas were made of 33 g needles (RWD Ltd). Seven days after virus injection, the guide cannula was placed at the following position: ML= 0.5 mm; AP= 0.9 mm; DV= −2.75 mm.
Target identification by Connectivity Map.
Top 100 DEGs between the WT and Scn2atrap mice were processed with the Connectivity Map (CMAP) dataset (Lamb et al., 2006; Subramanian et al., 2017) using the cloud-based CLUE software platform (https://clue.io/). This allowed us to query the database for compounds that were driving down the input genomic signatures, revealing potential molecules that could be repurposed to treat the diseases/phenotype. Then, we focused on perturbagen molecules that displayed a negative median tau score and were retained as potential compounds with a median tau score < −90.
Compound administration.
Intracerebroventricular injections were performed by inserting injection cannulas into the guide cannulas 2.75 mm deep following a 7-day rehabilitation period after surgery. Injection cannulas were connected to 10 μl syringes by polyethylene tubing. Dexanabinol (HU-211, MedChemExpress) was dissolved in co-solvent Cremophor EL/ethanol to make a stock solution with concertation of 50 mg/ml. Then, the stock solution was diluted (1:20) in saline prior to injection (Shohami et al., 1995; Shohami et al., 1993). For each mouse, 2 μl HU-211 (2.5 mg/ml) (Shohami et al., 1993) was injected into the ventricles for 4 min at a speed of 0.5 μl/min using a syringe pump. Dexanabinol was given to mice every 24 hours for three days.
Video-based EEG/EMG recordings and power spectral analysis.
After at least one week of recovery after surgery, mice were moved to the recording chamber and connected to a lightweight tether attached to a low-resistance commutator mounted over the cage (Pinnacle Technologies, USA) (Cho et al., 2017; Clasadonte et al., 2017). This enabled the freely moving of the mice in the cage. Recording of EEG/EMG waveforms began at lights-off and ran for 48 hours. Data acquisition was performed on Sirenia Acquisition software (Pinnacle Technologies). Signals were amplified and high pass filtered (0.5 Hz) via a preamplifier. EEG signals were then further amplified, low pass filtered with a 25 Hz cutoff, and collected continuously at a sampling rate of 2000 Hz. EEG/EMG waveforms were classified in 10-sec epochs as “wakefulness” (low-voltage, high-frequency EEG; high-amplitude EMG); NREM sleep (high-voltage, mixed-frequency EEG; low-amplitude EMG); or rapid-eye-movement (REM) sleep (low-voltage EEG with a predominance of theta activity [~4–8 Hz]; low amplitude EMG) by a trained observer with Sirenia Sleep Pro software (Pinnacle Technologies). For power spectral analysis, signals were digitally filtered and spectrally analyzed by fast Fourier transformation using Sirenia software. The mean spectral density of all stimulated events per animal was sorted into successive 0.25 Hz frequency bands between 0 and 25 Hz. Each frequency band was normalized to the sum of the power over the entire range (0–25 Hz).
Nesting behavior.
We evaluated nest-building using a five-point scale modified from previous reports (Deacon, 2006; Neely et al., 2019). 1) Assign a score of 1 when the shredded paper or small squares remained scattered throughout the cage, or the cotton square or twist remained untouched; 2) Assign a score of 2 when some of the material was constructed into a nest, but over 50% of the material was not used for nest construction; 3) Assign a score of 3 when a noticeable nest was constructed, but several pieces were still scattered; 4) Assign a score of 4 when almost all the material was used for the nest, but few pieces of material remained scattered or were near the nest; 5) Assign a score of 5 when all material was used to make an identifiable nest.
Wheel running activity monitoring.
Mice were placed in a cage with running wheels (Lafayette Instruments). Food and water were available ad libitum. Mice were habituated for two days before recording for five days. Duration, speed, and distance of running on the wheel were recorded.
Non-linear sine wave fitting/JTK_CYCLE to calculate the circadian rhythms-related parameters.
Non-linear sine wave fitting was applied to fitting the raw wheel-running data (Refinetti et al., 2007) using the following formula:
JTK_CYCLE analysis was also performed to calculate the circadian-related parameters, such as the period, phase, and amplitude (Hughes et al., 2010).
Non-linear Gaussian fitting to calculate the amplitude/interval distribution of spontaneous firing.
Interval/amplitude distributions were calculated with the Gaussian components by the maximum likelihood method (Jones and Gibb, 2005).
Model as follows:
Amplitude is the height of the center of the distribution in Y units. Mean is the X value at the center of the distribution. SD is a measure of the width of the distribution, in the same units as X.
Immunofluorescence.
Mice were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.), and then transcardially perfused, and decapitated to dissect brains into ice-cold paraformaldehyde (PFA; 4%). The brain was fixed in PFA solution overnight and then transferred to the glucose solution (30%) for cryopreservation for two days. Brain sections (50-μm in thickness) were cut by using a Cryostat Microtome (Leica CM1860s). To image brain sections, we carried out fluorescence imaging.
Ex vivo electrophysiology.
All chemicals were obtained from Sigma-Aldrich unless otherwise specified. Electrophysiology was performed in slices prepared from 2–3-month-old Scn2atm1a/tm1a (Scn2a-deficient mice) and WT littermates. Mice were deeply anesthetized with ketamine/xylazine (100/10 mg/kg, i.p., 0.1 mL per 10 grams of body weight), and then transcardially perfused, and decapitated to dissect brains into ice-cold slicing solution containing the following (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 25 glucose, 0.6 sodium ascorbate, and 3.1 sodium pyruvate (bubbled with 95% O2 and 5% CO2, pH 7.4, 305–315 mOsm). Acute coronal slices containing suprachiasmatic nucleus (SCN) (250-μm in thickness) were cut by using a vibratome (Leica VT1200S) and transferred to normal artificial cerebrospinal fluid (aCSF) (in mM): 125 NaCl, 2.5 KCl, 2.0 CaCl2, 2.0 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 10 glucose (bubbled with 95% O2 and 5% CO2, pH = 7.4, 305–315 mOsm). Then, slices were incubated at 37°C for 20~30 minutes and stored at room temperature before use. Slices were transferred to an open recording chamber and continuously perfused with aCSF at a rate of 2 mL/min at 32–33°C, and were visualized under IR-DIC (infrared-differential interference contrast) using a BX-51WI microscope (Olympus) with an IR-2000 camera (Dage-MTI). All somatic recordings were performed from identified SCN neurons between ZT2 and 8. Glass pipettes (4–6 MegaΩ) were filled with filtered, internal solution (in mM: 122 KMeSO4, 4 KCl, 2 MgCl2, 0.2 EGTA, 10 HEPES, 4 Na2ATP, 0.3 Tris-GTP, 14 Tris-phosphocreatine, adjusted to pH = 7.25 with KOH, 295–305 mOsm). For recordings in which the primary goal was to examine neuronal firing rate, the loose cell-attached patch recording technique was chosen, given that it does not affect endogenous membrane properties either by the formation of a tight seal or by dialysis of intracellular milieu. The spontaneous firing was recorded for 3 min under the gap-free mode. Electrophysiological signals were processed with an Axon MultiClamp 700B amplifier (Molecular Devices, USA) and data were acquired using pClamp 11.1 software (Axon Instruments, USA), filtered at 2 kHz and sampling rate at 20 kHz with an Axon Digidata 1550B plus HumSilencer digitizer (Molecular Devices, USA). At least six slices from three mice were used between ZT2 to ZT8, between 10 to 20 neurons were recorded from each slice.
RNA sequencing.
Samples for RNA sequencing were collected from the neocortex of the mice. poly-A selected libraries were prepared (Novogene), and ~40 million paired-end 150 bp reads were sequenced on a NovaSeq6000 platform. Before library preparation, RNA quality was checked using an Agilent Nano RNA Chip; samples with RNA integrity numbers greater than 6.5 were sequenced.
FastX-Toolkit v. 0.0.14 was used to trim reads based on quality score. A trim score of 30 (the minimum quality score for trimming reads was 30), and a trim length of 50 (reads shorter than 50 bases will be discarded) was used. Unpaired reads were discarded after trimming and quality control. STAR v. 2.5.4b (Dobin et al., 2013) was used to align reads to the Ensembl Mus musculus genome database version GRCm38.p6. HTSeq v.0.7.0 (Anders et al., 2015) in “intersection-nonempty” mode, with Biopython v.2.7.3 was used to generate a count matrix. Relative log expression (RLE) normalization was performed followed by a differential expression analysis using the Bioconductor package DESeq2 (Love et al., 2014). The Benjamini-Hochberg false discovery rate correction is used to correct p-values for multiple testing in differential expression analyses and subsequent enrichment analyses. Raw and processed RNA sequencing data are publicly available at GEO through accession number: GSE179818.
Transcripts (ranked by the q-value), which were found to be significantly altered in WT mice and Scn2a-deficient mice at FDR<0.05, were examined using the DAVID ontology tools (Huang da et al., 2009) and clusterProfiler v. 4.0.2 (Yu et al., 2012), using the set of all genes with detected expression in Mus musculus as the background. These annotation charts were visualized using the R package (ggplot2), and the transcript cluster IDs were also used for the protein and protein interaction (PPI) network by using the STRING v11 web tools (Szklarczyk et al., 2019) and visualized by Cytoscape 3.8 software (Shannon et al., 2003).
Real-time RT-PCR (qPCR).
Total RNA was isolated from brain tissue samples with QIAzol Lysis Reagent (QIAGEN, Germany) and used for the first-strand cDNA synthesis with the Reverse Transcription System (Thermofisher, USA). Potential DNA contamination was removed by RNase-free DNase treatment using a cDNA synthesis kit. Relative quantitation was determined using the Bio-Rad CFX96 qPCR system (Bio-Rad Inc., USA) that measures real-time SYBR green fluorescence and then calculated using the comparative Ct method (2−ΔΔCt) with the expression of β-actin as an internal control (Schmittgen and Livak, 2008).
Western blot.
Samples were carefully collected and then were quickly frozen by liquid nitrogen. Then samples were transferred to −80°C for long-term storage. Brain samples were homogenized in radioimmunoprecipitation assay buffer (RIPA) buffer supplied with a proteinase inhibitor cocktail. For western blotting, membranes were incubated with NaV1.2 (ASC-002, 1:1000, Alomone Labs) and β-actin (3700S, 1:1000, Cell Signaling Technology) overnight at 4 °C followed by incubation with a proper Li-COR secondary antibody (AB_10956166 and AB_10956588, 1:5000, LI-COR Biosciences). Bands were visualized Li-COR system and quantified by densitometry with Image Studio Lite Ver 5.2 software (LI-COR Biosciences).
Statistical analysis.
Two-tailed Student’s t-test (parametric) or unpaired two-tailed Mann-Whitney U-test (non-parametric) was used for single comparisons between two groups. Other data were analyzed using either one-way or two-way ANOVA analysis with post-hoc Bonferroni’s multiple comparisons test (except as otherwise noted). Post-hoc comparisons were carried out only when the primary measure showed statistical significance. Data were expressed as mean ± SEM (except as otherwise noted), with statistical significance determined at p-values <0.05. p≥0.05 is indicated ns (no significance), p<0.05 is indicated as one asterisk (*), p<0.01 is indicated as two asterisks (**), and p<0.001 is indicated as three asterisks (***). Mice from different litters were randomly assigned to different treatment groups, and no other specific randomization was used for the animal studies.
Supplementary Material
Highlights.
Scn2a deficiency results in sleep disturbance in mice;
Scn2a deficiency-related sleep disturbance involves suprachiasmatic nucleus;
Scn2a deficiency disrupts neuronal firing in suprachiasmatic nucleus;
Scn2a deficiency results in a change of core clock gene expression;
Transcriptome-based molecule discovery identifies HU-211 to alleviate sleep disturbance.
ACKNOWLEDGMENTS.
We thank Dr. Amy Brewster of Southern Methodist University, and Dr. Lan Chen of the Chemical Genomics Facility of Purdue Institute for Drug Discovery for helpful discussion. This work is supported by Purdue startup funding, Ralph W. and Grace M. Showalter Research Trust, and Purdue Big Idea Challenge 2.0 on Autism to Y.Y. Yang lab gratefully acknowledges additional support from the Purdue University Institute for Drug Discovery and Institute for Integrative Neuroscience. Yang lab is grateful to the FamilieSCN2A foundation for the Action Potential Grant support. This project was funded, in part, with support from the Indiana Spinal Cord & Brain Injury Research Fund from the Indiana State Department of Health, and the Indiana Clinical and Translational Sciences Institute (CTSI) funded. The Yang lab appreciates the bioinformatics support of the Collaborative Core for Cancer Bioinformatics (C3B) with support from the Indiana University Simon Comprehensive Cancer Center, Purdue University Center for Cancer Research, and Walther Cancer Foundation. A portion of Y.Y.’s salary effort during the revision period of this paper was supported by the NINDS of the NIH (R01 NS117585 and R01 NS123154). M.E. is supported by the National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) and AAUW dissertation fellowship. This work is also partially supported by Beijing Natural Science Foundation (Z181100001518001 to Z.H.). Part of the publication cost of this article is supported by Purdue Libraries.
Abbreviations:
- AP
Action potential
- ASD
autism spectrum disorder
- CNS
central nervous system
- DEG
Differentially expressed genes
- EEG
electroencephalogram
- EMG
Electromyography
- HET
heterozygous
- HOM
homozygous
- NREM
non-rapid-eye-movement sleep
- REM
rapid-eye-movement sleep
- SCN
suprachiasmatic nucleus
- WT
Wild-type
Footnotes
DISCLOSURES.
Leah Schust is the president of FamilieSCN2A Foundation, which provided partial funding for Yang lab to perform this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AVAILABILITY.
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Raw and processed RNA sequencing data are publicly available at GEO through accession number GEO: GSE179818.
References.
- Abrahamson EE, Moore RY, 2001. Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 916, 172–91. [DOI] [PubMed] [Google Scholar]
- Anders S, et al. , 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 31, 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, et al. , 2021. SCN2A-Developmental and Epileptic Encephalopathies: Challenges to trial-readiness for non-seizure outcomes. Epilepsia. 62, 258–268. [DOI] [PubMed] [Google Scholar]
- Boone CE, et al. , 2018. Abnormal Sleep Architecture and Hippocampal Circuit Dysfunction in a Mouse Model of Fragile X Syndrome. Neuroscience. 384, 275–289. [DOI] [PubMed] [Google Scholar]
- Borbely AA, et al. , 2016. The two-process model of sleep regulation: a reappraisal. J Sleep Res. 25, 131–43. [DOI] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RKA, et al. , 2019. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain. 142, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KS, et al. , 2018. A Hypothalamic Switch for REM and Non-REM Sleep. Neuron. 97, 1168–1176 e4. [DOI] [PubMed] [Google Scholar]
- Cho JR, et al. , 2017. Dorsal Raphe Dopamine Neurons Modulate Arousal and Promote Wakefulness by Salient Stimuli. Neuron. 94, 1205–1219 e8. [DOI] [PubMed] [Google Scholar]
- Clasadonte J, et al. , 2017. Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron. 95, 1365–1380 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K, et al. , 2021. Computational analysis of 10,860 phenotypic annotations in individuals with SCN2A-related disorders. Genet Med. 23, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM, 2006. Assessing nest building in mice. Nat Protoc. 1, 1117–9. [DOI] [PubMed] [Google Scholar]
- Dhindsa RS, et al. , 2021. A Transcriptome-Based Drug Discovery Paradigm for Neurodevelopmental Disorders. Ann Neurol. 89, 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibner C, et al. , 2010. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 72, 517–49. [DOI] [PubMed] [Google Scholar]
- Dobin A, et al. , 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton M, et al. , 2021. Generation and basic characterization of a gene-trap knockout mouse model of Scn2a with a substantial reduction of voltage-gated sodium channel Nav 1.2 expression. Genes Brain Behav. 20, e12725. [DOI] [PubMed] [Google Scholar]
- Eban-Rothschild A, et al. , 2016. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat Neurosci. 19, 1356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshhar N, et al. , 1993. HU-211, a non-psychotropic cannabinoid, rescues cortical neurones from excitatory amino acid toxicity in culture. Neuroreport. 5, 237–40. [DOI] [PubMed] [Google Scholar]
- Ferraro L, et al. , 1997. The antinarcoleptic drug modafinil increases glutamate release in thalamic areas and hippocampus. Neuroreport. 8, 2883–7. [DOI] [PubMed] [Google Scholar]
- Gazina EV, et al. , 2015. ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum Mol Genet. 24, 1457–68. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, et al. , 2014. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 9, 1742–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman G, 2010. Circadian rhythms and sleep in children with autism. Neurosci Biobehav Rev. 34, 755–68. [DOI] [PubMed] [Google Scholar]
- Han S, et al. , 2012a. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 489, 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, et al. , 2012b. Na(V)1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci U S A. 109, E368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey JRM, et al. , 2020. Ion Channels Controlling Circadian Rhythms in Suprachiasmatic Nucleus Excitability. Physiol Rev. 100, 1415–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings MH, et al. , 2018. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat Rev Neurosci. 19, 453–469. [DOI] [PubMed] [Google Scholar]
- Hirayama J, et al. , 2005. Common pathways in circadian and cell cycle clocks: light-dependent activation of Fos/AP-1 in zebrafish controls CRY-1a and WEE-1. Proc Natl Acad Sci U S A. 102, 10194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoischen A, et al. , 2014. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci. 17, 764–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberg H, et al. , 2020. Rescue of oxytocin response and social behaviour in a mouse model of autism. Nature. 584, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, et al. , 2009. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 12, 996–1002. [DOI] [PubMed] [Google Scholar]
- Huang da W, et al. , 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hughes ME, et al. , 2010. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms. 25, 372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingiosi AM, et al. , 2019. Shank3 modulates sleep and expression of circadian transcription factors. Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MR, et al. , 2016. Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat Neurosci. 19, 223–32. [DOI] [PubMed] [Google Scholar]
- Jones S, Gibb AJ, 2005. Functional NR2B- and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J Physiol. 569, 209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, et al. , 2015. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol Dis. 77, 141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, 1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 270, 16483–6. [DOI] [PubMed] [Google Scholar]
- Kawamoto K, et al. , 2003. Two types of VIP neuronal components in rat suprachiasmatic nucleus. J Neurosci Res. 74, 852–7. [DOI] [PubMed] [Google Scholar]
- Kohsaka A, et al. , 2007. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 6, 414–21. [DOI] [PubMed] [Google Scholar]
- Lai MC, et al. , 2014. Autism. Lancet. 383, 896–910. [DOI] [PubMed] [Google Scholar]
- Lamb J, et al. , 2006. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 313, 1929–35. [DOI] [PubMed] [Google Scholar]
- Léna I, Mantegazza M, 2019. Na(V)1.2 haploinsufficiency in Scn2a knock-out mice causes an autistic-like phenotype attenuated with age. Sci Rep. 9, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, et al. , 2017. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci U S A. 114, 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, et al. , 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDuffie KE, et al. , 2020. Sleep Onset Problems and Subcortical Development in Infants Later Diagnosed With Autism Spectrum Disorder. Am J Psychiatry. 177, 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell-Horn A, Malow BA, 2017. Sleep in Autism. Semin Neurol. 37, 413–418. [DOI] [PubMed] [Google Scholar]
- Meisler MH, et al. , 2021. Sodium channelopathies in neurodevelopmental disorders. Nat Rev Neurosci. 22, 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzenberg MJ, Carter CS, 2008. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology. 33, 1477–502. [DOI] [PubMed] [Google Scholar]
- Missig G, et al. , 2020. Sleep as a translationally-relevant endpoint in studies of autism spectrum disorder (ASD). Neuropsychopharmacology. 45, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, et al. , 2012. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 35, 445–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa A, et al. , 2018. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform. 19, 506–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely CLC, et al. , 2019. Nest Building Behavior as an Early Indicator of Behavioral Deficits in Mice. J Vis Exp. [DOI] [PubMed] [Google Scholar]
- Ogiwara I, et al. , 2018. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol. 1, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, et al. , 2013. Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+). Epilepsia. 54, 625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, et al. , 2010. Dysfunction of the Scn8a voltage-gated sodium channel alters sleep architecture, reduces diurnal corticosterone levels, and enhances spatial memory. J Biol Chem. 285, 16553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjpe MD, et al. , 2019. Insights into Computational Drug Repurposing for Neurodegenerative Disease. Trends Pharmacol Sci. 40, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planells-Cases R, et al. , 2000. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J. 78, 2878–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que Z, et al. , 2021. Hyperexcitability and Pharmacological Responsiveness of Cortical Neurons Derived from Human iPSCs Carrying Epilepsy-Associated Sodium Channel Nav1.2-L1342P Genetic Variant. J Neurosci. 41, 10194–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refinetti R, et al. , 2007. Procedures for numerical analysis of circadian rhythms. Biol Rhythm Res. 38, 275–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez REA, et al. , 2019. Circadian regulation of sleep in a pre-clinical model of Dravet syndrome: dynamics of sleep stage and siesta re-entrainment. Sleep. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, et al. , 2012. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 485, 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, et al. , 2005. Hypothalamic regulation of sleep and circadian rhythms. Nature. 437, 1257–63. [DOI] [PubMed] [Google Scholar]
- Satterstrom FK, et al. , 2020. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 180, 568–584 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ, 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 3, 1101–8. [DOI] [PubMed] [Google Scholar]
- Schreck KA, et al. , 2004. Sleep problems as possible predictors of intensified symptoms of autism. Res Dev Disabil. 25, 57–66. [DOI] [PubMed] [Google Scholar]
- Shannon P, et al. , 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W, et al. , 2019. Scn2a Haploinsufficiency in Mice Suppresses Hippocampal Neuronal Excitability, Excitatory Synaptic Drive, and Long-Term Potentiation, and Spatial Learning and Memory. Front Mol Neurosci. 12, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohami E, et al. , 1995. Long-term effect of HU-211, a novel non-competitive NMDA antagonist, on motor and memory functions after closed head injury in the rat. Brain Res. 674, 55–62. [DOI] [PubMed] [Google Scholar]
- Shohami E, et al. , 1993. A nonpsychotropic cannabinoid, HU-211, has cerebroprotective effects after closed head injury in the rat. J Neurotrauma. 10, 109–19. [DOI] [PubMed] [Google Scholar]
- Shukla R, et al. , 2021. Signature-based approaches for informed drug repurposing: targeting CNS disorders. Neuropsychopharmacology. 46, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siavelis JC, et al. , 2016. Bioinformatics methods in drug repurposing for Alzheimer’s disease. Brief Bioinform. 17, 322–35. [DOI] [PubMed] [Google Scholar]
- Soltani S, et al. , 2019. Sleep-Wake Cycle in Young and Older Mice. Front Syst Neurosci. 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt PWE, et al. , 2021. Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep. 36, 109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt PWE, et al. , 2019. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron. 103, 673–685 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striem S, et al. , 1997. Interaction of dexanabinol (HU-211), a novel NMDA receptor antagonist, with the dopaminergic system. Eur J Pharmacol. 338, 205–13. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. , 2017. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 171, 1437–1452 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, et al. , 2019. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak F, et al. , 2021. Gene expression signatures of target tissues in type 1 diabetes, lupus erythematosus, multiple sclerosis, and rheumatoid arthritis. Sci Adv. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi M, et al. , 2018. Clock-Generated Temporal Codes Determine Synaptic Plasticity to Control Sleep. Cell. 175, 1213–1227 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, et al. , 2015. EGR1 regulates hepatic clock gene amplitude by activating Per1 transcription. Sci Rep. 5, 15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsukawa T, et al. , 2019. Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol Autism. 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, et al. , 2017. Sleep/Wake Physiology and Quantitative Electroencephalogram Analysis of the Neuroligin-3 Knockout Rat Model of Autism Spectrum Disorder. Sleep. 40. [DOI] [PubMed] [Google Scholar]
- Veatch OJ, et al. , 2015. Sleep in Autism Spectrum Disorders. Curr Sleep Med Rep. 1, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, et al. , 2016. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 7, 13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S, et al. , 2020. Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat Neurosci. 23, 456–467. [DOI] [PubMed] [Google Scholar]
- Yap EL, Greenberg ME, 2018. Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron. 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, et al. , 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 16, 284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, et al. , 2018. Molecular Architecture of the Mouse Nervous System. Cell. 174, 999–1014 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, et al. , 2009. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 139, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. , 2021. Severe deficiency of the voltage-gated sodium channel NaV1.2 elevates neuronal excitability in adult mice. Cell Rep. 36, 109495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Raw and processed RNA sequencing data are publicly available at GEO through accession number GEO: GSE179818.